

Abstract
Peptidyl arginine deiminase-4 (PAD4) catalyzes the conversion of peptidylarginine residues to peptidylcitrulline. We have previously shown that kidney ischemia-reperfusion (I/R) injury increases renal proximal tubular PAD4 expression and activity. Furthermore, kidney PAD4 plays a critical role in ischemic acute kidney injury (AKI) by promoting renal tubular inflammation, neutrophil infiltration, and NF-κB activation. However, the mechanisms of PAD4-mediated renal tubular inflammation and NF-κB activation after I/R remain unclear. Here, we show that recombinant PAD4 preferentially citrullinates recombinant IKKγ [also called NF-κB essential modulator (NEMO)] over recombinant IKKα or IKKβ. Consistent with this finding, PAD4 citrullinated renal proximal tubular cell IKKγ and promoted NF-κB activation via IκBα phosphorylation in vitro. NEMO inhibition with a selective NEMO-binding peptide attenuated PAD4-mediated proinflammatory cytokine mRNA induction in HK-2 cells. Moreover, NEMO inhibition did not affect proximal tubular cell survival, proliferation, or apoptosis, unlike global NF-κB inhibition. In vivo, NEMO-binding peptide treatment protected against ischemic AKI. Finally, NEMO-binding peptide attenuated recombinant PAD4-mediated exacerbation of ischemic AKI, renal tubular inflammation, and apoptosis. Taken together, our results show that PAD4 exacerbates ischemic AKI and inflammation by promoting renal tubular NF-κB activity and inflammation via NEMO citrullination. Targeting NEMO activation may serve as a potential therapy for this devastating clinical problem.
Keywords: acute kidney injury, apoptosis, citrullination, inflammation, ischemia, necrosis, neutrophil, NF-κB essential modulator, reperfusion
INTRODUCTION
Renal ischemia and reperfusion (I/R) injury is a major cause of clinical acute kidney injury (AKI) (44, 54). Indeed, patients subjected to major surgery including cardiac, aortovascular, or abdominal organ transplantation frequently (~50–80%) develop ischemic AKI (24, 36). Unfortunately, despite many decades of intense research, AKI remains a major clinical problem without effective therapy (25, 31). Pathological renal tubular inflammation and neutrophil infiltration that occur after the termination of ischemia result in renal tubular cell death. Therefore, attenuating the renal tubular inflammatory process after renal I/R would be a useful therapeutic target to protect against ischemic AKI.
Peptidylarginine deiminases (PADs) are Ca2+-dependent enzymes that catalyze the conversion of peptidylarginine residues to peptidylcitrulline (52, 58). Protein citrullination is a posttranslational modification that results in virtually no changes in molecular mass, and loss of a positive charge, however, leads to drastic changes in protein function (8, 58). PAD4, a member of the PAD family, is primarily a nuclear enzyme found in various tissues, including the kidney. Posttranslational citrullination by PAD4 is linked to various autoimmune and inflammatory pathologies (2, 3, 58).
We have previously shown that renal I/R injury not only increased renal tubular PAD4 activity but also led to cytosolic translocation of PAD4 (22, 49). Furthermore, we demonstrated that renal PAD4 plays a critical role in ischemic AKI as PAD4 inhibitors or genetic deletion of PAD4 protected against ischemic AKI (49). We also showed that recombinant PAD4 treatment exacerbated ischemic AKI by promoting NF-κB activation, renal tubular inflammation, and neutrophil infiltration. However, the mechanisms of PAD4-mediated exacerbation of renal tubular inflammation after ischemic AKI remain unknown.
As described in other cell types, we showed that PAD4 is primarily localized in the nucleus of renal proximal tubules in sham-operated mice (49). Surprisingly, we discovered that ischemic AKI increased cytosolic PAD4 translocation and expression in renal proximal tubules. Consistent with this, PAD4 wild-type mice had increased cytosolic peptidyl-citrullination after ischemic AKI (49). In contrast, mice deficient in PAD4 failed to show increased cytosolic peptidyl-citrullination after renal I/R. These findings allowed us to hypothesize that renal I/R induces cytosolic PAD4 activity, leading to an increased inflammatory response and renal tubular injury. Since recombinant PAD4 caused nuclear translocation of NF-κB in renal proximal tubule cells (22) and since NF-κB exists in the cytosol before its activation via dissociation with IκBα (61), we hypothesized that induction of cytosolic PAD4 after renal I/R promotes the activity of IκB kinases (IKKs; enzymes that phosphorylate IκBα) via citrullination, leading to the dissociation of IκBα from NF-κB complexes, allowing their nuclear translocation. Here, we show that PAD4 directly and selectively citrullinates IKKγ [NF-κB essential modulator (NEMO), a regulatory subunit of the IKK complex] over IKKα or IKKβ subtypes. IKKγ citrullination promotes renal tubular proinflammatory NF-κB signaling, leading to exacerbation of ischemic AKI. We also show here that a selective NEMO-binding peptide attenuates renal tubular NF-κB activation, proinflammatory gene induction, and renal tubular neutrophil infiltration, resulting in significantly reduced kidney injury after I/R. Importantly, selective IKKγ inhibition is devoid of the negative effects of global NF-κB inhibition (reduced cell survival, proliferation, or increased apoptosis).
MATERIALS AND METHODS
Murine renal I/R injury.
After approval from the Columbia University Institutional Animal Care and Use Committee (New York, NY), adult male C57BL/6 mice (Charles River, Wilmington, MA) were subjected to 20- or 30-min renal ischemia or to sham surgery as previously described (23, 49). For pain management, all mice received 0.5–1 mg/kg sc buprenorphine SR before surgery. Sham-operated animals underwent anesthesia followed by laparotomy, right nephrectomy, bowel manipulations, and wound closure without renal ischemia. The body temperature of all mice was sustained at ~37°C using a surgical heating pad during surgery as well as during recovery from anesthesia. Plasma and kidneys were collected 24 h after renal I/R injury to examine renal dysfunction (plasma creatinine and histology), inflammation (neutrophil infiltration and cytokine mRNAs), and apoptosis (TUNEL staining). To test whether exogenous PAD4 supplementation exacerbates renal I/R injury, mice were pretreated with recombinant human PAD4 (10 μg iv, Cayman Chemical, Ann Arbor, MI) 15 min before renal I/R injury. To determine whether NEMO inhibition prevents the exacerbation of ischemic AKI with recombinant PAD4 treatment, mice were pretreated with NEMO-binding peptide (5 mg/kg iv, EMD Millipore, Darmstadt, Germany) 15 min before recombinant PAD4 administration. Finally, to test whether NEMO inhibition protects against renal I/R injury, some mice were pretreated with NEMO-binding peptide 15 min before 30-min renal I/R injury.
Measurement of kidney injury after I/R.
Plasma creatinine was measured 24 h after reperfusion using an enzymatic creatinine reagent kit according to the manufacturer’s protocol (ThermoFisher Scientific, Waltham, MA). Kidney hematoxylin and eosin-stained sections were blindly assessed using a grading scale of kidney necrotic I/R injury to the proximal tubules (renal injury score of 0–4) as outlined by Jablonski et al. (28).
Detection of kidney tubular apoptosis and neutrophil infiltration.
Twenty-four hours after sham surgery or renal I/R injury, TUNEL staining detected fragmented DNA as described using a commercially available kit (Roche, Indianapolis, IN) (23, 39). Apoptotic TUNEL-positive cells were quantified in five to seven randomly chosen × 200 microscope images fields in the corticomedullary junction, and results were expressed as apoptotic cells counted per ×200 field. Neutrophil immunohistochemistry was performed as previously described (23) using rat anti-mouse Ly6G monoclonal antibody against polymorphonuclear leukocytes (AbD Serotec, Raleigh, NC) and primary antibody recognizing IgG2a (MCA1212, AbD Serotec) as a negative isotype control in all experiments. Quantification of kidney infiltrating neutrophils was performed using five to seven randomly chosen ×200 microscope image fields (corticomedullary junction for kidney neutrophils), and results were expressed as neutrophils counted per ×200 field.
Proximal tubule cell culture.
Immortalized human proximal tubular cells (HK-2, American Type Culture Collection, Manassas, VA) were grown as previously described (33, 34). Mouse kidney proximal tubules were isolated using Percoll density gradient separation and grown as previously described (57). Mouse and human renal proximal tubule cells were treated with recombinant human PAD4 (1–10 μg/ml for 1–6 h) when confluent. HK-2 cells were also treated with NEMO-binding peptide or with NF-κB inhibitors {2-amino-6-[2-(cyclopropylmethoxy)-6-hydroxyphenyl]-4-(4-piperidinyl)-3-pyridinecarbonitrile (ACHP) or caffeic acid phenethyl ester (CAPE), Tocris Bio-Techne, Minneapolis, MN} to determine the effects of NF-κB signaling modulators on cellular proliferation, survival, and apoptosis.
Quantitative RT-PCR.
Mouse kidney inflammation after I/R was also assessed by measuring proinflammatory mRNA markers, including monocyte chemoattractant protein-1 (MCP-1), chemokine macrophage inflammatory protein-2 (MIP-2), and keratinocyte-derived cytokine (KC) with quantitative RT-PCR as previously described with the primers shown in Table 1 (23). mRNAs isolated from HK-2 cells were also subjected to quantitative RT-PCR for MCP-1 and MIP-2 with the primers shown in Table 1. Primer design was based on published GenBank sequences. To confirm equal RNA input, GAPDH mRNA and relative expression of proinflammatory mRNA was calculated with the ΔΔCt method (where Ct is threshold cycle). Quantitative RT-PCR was performed using the MyiQ Real Time Detection System (Bio-Rad, Hercules, CA) using FastStart Universal SYBR Green Master (ROX, Roche, Indianapolis, IN).
Table 1.
Primers used in quantitative RT-PCR to amplify cDNAs based on published GenBank sequences for mouse and human cytokines and annealing temperatures
| Primers | Sequence | Annealing Temperature, °C |
|---|---|---|
| Mouse MCP-1 | 60 | |
| Sense | 5′-ACCTGCTGCTACTCATTCAC-3′ | |
| Antisense | 5′-TTGAGGTGGTTGTGGAAAAG-3′ | |
| Mouse MIP-2 | 60 | |
| Sense | 5′-CCAAGGGTTGACTTCAAGAAC-3′ | |
| Antisense | 5′-AGCGAGGCACATCAGGTACG-3′ | |
| Mouse KC | 60 | |
| Sense | 5′-CAATGAGCTGCGCTGTCAGTG-3′ | |
| Antisense | 5′-CTTGGGGACACCTTTTAGCATC-3′ | |
| Human MCP-1 | 64 | |
| Sense | 5′-AGCAAGTGTCCCAAAGAAGC-3′ | |
| Antisense | 5′-CTCAAAACATCCCAGGGGTA-3′ | |
| Human MIP-2 | 64 | |
| Sense | 5′-CTTGCCAGCTCTCCTCCTC-3′ | |
| Antisense | 5′-GCTTTCTGCCCATTCTTGAG-3′ | |
| Human/mouse GAPDH | 65 | |
| Sense | 5′-ACCACAGTCCATGCCATCAC-3′ | |
| Antisense | 5′-CACCACCCTGTTGCTGTAGCC-3′ |
KC, keratinocyte-derived cytokine; MCP-1, monocyte chemoattractant protein-1; MIP-2, macrophage inflammatory protein-2.
Immunoblot analysis.
We performed immunoblot analysis in human and mouse renal proximal tubule cells as previously described (46, 47). HK-2 cell nuclear fractions were prepared and subjected to the p65 subunit of NF-κB with antibodies from Santa Cruz Biotechnology (Santa Cruz, CA) as previously described (22). We also probed HK-2 cell lysates for caspase-3, poly (ADP-ribose) polymerase (PARP), IKKγ, phospho-IκBα, and total IκBα (Cell Signaling Technology, Beverly, MA). Human and mouse kidney proximal tubule cells were probed for citrullininated and total IKKγ (Abcam, Cambridge, MA) after immunoprecipitation as described below. All blots were imaged using ECL detection reagent (Thermo Scientific) and exposure using UVP Auto Chemi Darkroom and Vision Works LS acquisition and analysis software (Vision Works LS, Upland, CA).
Immunoprecipitation.
Renal proximal tubule cells or recombinant IKKγ were treated with recombinant PAD4 for 2 and 3 h, respectively. Immunoprecipitation of citrullinated IKKγ was performed followed by immunoblot analysis. In brief, citrulline was immunoprecipitated from samples using rabbit anti-citrulline antibody (Abcam) and incubated for 1 h at 4°C. The antibody-citrulline protein immunocomplex was captured from solution using Protein A/G PLUS agarose beads (Santa Cruz Biotechnology) and incubated overnight at 4°C. Samples were centrifuged at 1,000 g, and the pellet was washed with PBS followed by resuspension and boiling in Laemmli buffer (Bio-Rad Laboratories, Hercules, CA). Samples were then subjected to immunoblot analysis.
Statistical analysis.
Data were analyzed with one-way ANOVA plus Tukey’s post hoc multiple comparison test or Mann-Whitney nonparametric U-test to analyze renal injury scores (GraphPad Software, La Jolla, CA). All data are expressed throughout the text as means ± SE.
RESULTS
Recombinant PAD4 selectively citrullinates NEMO (IKKγ).
To determine whether PAD4 directly citrullinates IKK subtypes, we incubated 0.5 μg recombinant glutathione S-transferase (GST)-tagged IKKα, IKKβ, or IKKγ (NEMO) with 0.5 μg recombinant PAD4 for 3 h and probed for citrullinated IKK (immunoprecipitation for citrulline and immunoblotted for IKK subtypes) and total IKK subtypes. We found that recombinant PAD4 preferentially citrullinated IKKγ (NEMO) over other IKK subtypes (IKKα and IKKβ) (Fig. 1A, representative of 4 experiments). Note that IKKα-GST and IKKβ-GST showed larger bands at ~100 kDa, whereas IKKγ-GST was smaller with a molecular mass of ~75 kDa.
Fig. 1.

A: recombinant human peptidyl arginine deiminase-4 (PAD4) preferentially citrullinates IKKγ-glutathione S-transferase (GST) [NF-κB essential modulator (NEMO)]. Recombinant human IKKα-GST, IKKβ-GST, or IKKγ-GST (0.5 μg) was incubated with vehicle (saline) or with recombinant human PAD4 (0.5 μg) for 3 h and probed for citrullinated NEMO and total NEMO (representative images from 4 experiments). Note that IKKα-GST and IKKβ-GST showed larger bands at ~100 kDa, whereas IKKγ-GST was smaller with a molecular mass of ~75 kDa. B and C: recombinant human PAD4 citrullinates NEMO in human proximal tubule cells (HK-2; B) and mouse proximal tubule cells (C). HK-2 cells or freshly isolated mouse proximal tubule cells were treated with vehicle or with recombinant human PAD4 (10 μg/ml) for 3 h and probed for citrullinated NEMO and total NEMO. Bar graphs show band intensity quantifications demonstrating increased NEMO citrullination by recombinant human PAD4 in both experimental conditions. *P < 0.05 vs. vehicle (n = 4–6 experiments). Error bars represent 1 SE.
We next tested for PAD4-mediated citrullination of NEMO in cultured human and mouse renal proximal tubule cells. HK-2 cells (Fig. 1B) and mouse proximal tubule cells (Fig. 1C) were treated with 1 μg/ml recombinant PAD4 for 3 h, immunoprecipitated with IKKγ antibody, and subjected to either citrulline or IKKγ immunoblot analysis. Proximal tubule cells treated with recombinant human PAD4 (1 μg/ml) had a significantly higher amount of citrulllinated NEMO after PAD4 treatment compared with vehicle.
NEMO neutralization with NEMO-binding peptide inhibits PAD4-induced NF-κB nuclear translocation in HK-2 cells.
We have previously demonstrated that recombinant PAD4 induced NF-κB p65 subunit nuclear translocation in human renal proximal tubular cells (22). We tested whether inhibition of NEMO would prevent PAD4-mediated NF-κB activation. Figure 2 shows the increased nuclear translocation of NF-κB p65 subunit with recombinant PAD4 treatment (10 μg/ml for 4 h), which was attenuated with NEMO-binding peptide pretreatment (10 μM for 30 min).
Fig. 2.
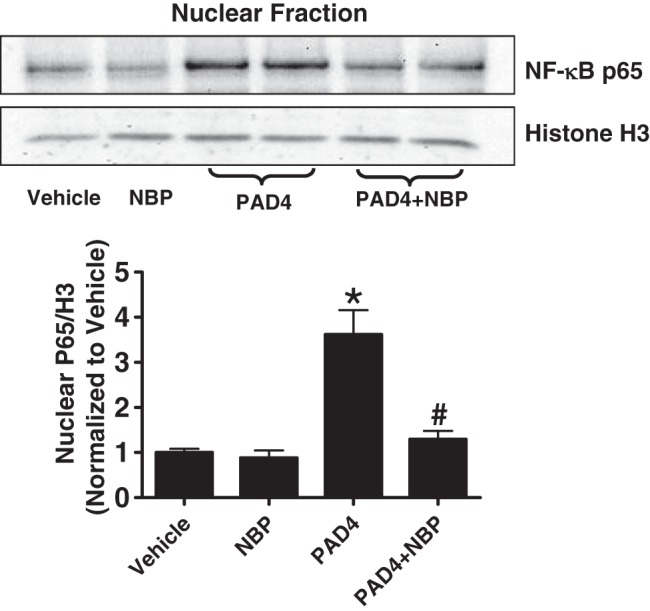
NF-κB essential modulator (NEMO) neutralization with NEMO-binding peptide (NBP) inhibits peptidyl arginine deiminase-4 (PAD4)-induced nuclear translocation of NF-κB in HK-2 cells. The nuclear p65 subunit of NF-κB increased with human recombinant PAD4 treatment (10 μg/ml for 4 h) in HK-2 cells. NEMO NBP (10 μM, 30-min pretreatment) blocked the increase in p65 subunit of NF-κB nuclear translocation in HK-2 cells. Top: histone 3 immunoblot analysis was performed to normalize nuclear protein loading. Bottom: bar graph shows band intensity quantifications demonstrating increased p65 nuclear translocation that was blocked by NBP treatment. *P < 0.05 vs. vehicle; #P < 0.05 vs. PAD4 (n = 6 experiments). Error bars represent 1 SE.
NEMO neutralization attenuates PAD4-mediated proinflammatory gene expression in HK-2 cells.
We also tested whether NEMO neutralization would attenuate PAD4-induced upregulation of proinflammatory cytokines in HK-2 cells. Recombinant PAD4 (10 μg/ml for 6 h) treatment profoundly induced MCP-1 and MIP-2 (two critical chemokines involved in leukocyte infiltration after renal I/R) in HK-2 cells. NEMO-binding peptide (10 μM) pretreatment attenuated the PAD4-mediated induction of these two chemokines in HK-2 cells (Fig. 3).
Fig. 3.
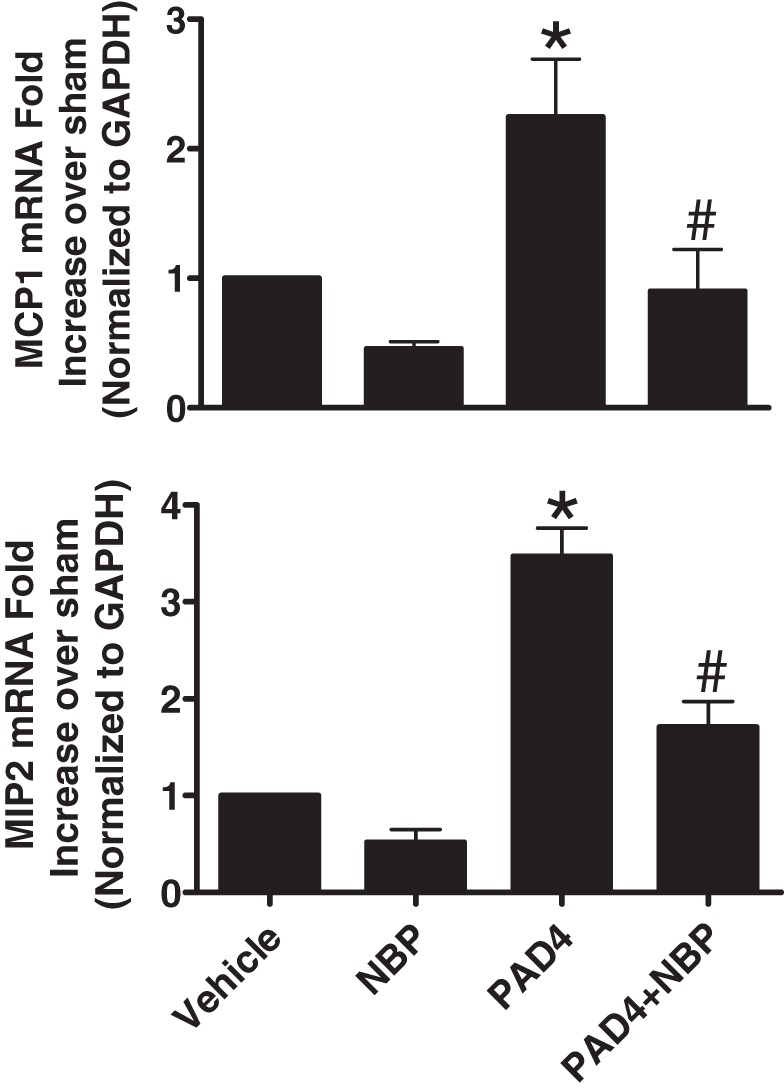
NF-κB essential modulator (NEMO) neutralization attenuates peptidyl arginine deiminase-4 (PAD4)-mediated proinflammatory gene expression in HK-2 cells. Recombinant human PAD4 (10 μg/ml for 6 h) treatment significantly induced monocyte chemoattractant protein-1 (MCP-1; top) and macrophage inflammatory protein-2 (MIP-2; bottom) mRNA in HK-2 cells (measured by quantitative RT-PCR). NEMO-binding peptide (NBP; 10 μM for 30 min) pretreatment attenuated the PAD4-mediated induction of these two chemokines in HK-2 cells. *P < 0.05 vs. vehicle; #P < 0.05 vs. PAD4 (n = 5–6 experiments). Error bars represent 1 SE.
PAD4 treatment causes phosphorylation of IκBα in HK-2 cells.
We next tested the hypothesis that PAD4 treatment causes phosphorylation of IκBα in HK-2 cells, leading to its dissociation from the p65 subunit and promoting nuclear translocation. Consistent with our hypothesis, we observed a significant increase in phosphorylated IκBα after 60 min of recombinant PAD4 treatment (5 μg/ml) compared with vehicle treatment (Fig. 4), suggesting that PAD4-induced NF-κB activation occurs via phosphorylation of IκBα.
Fig. 4.
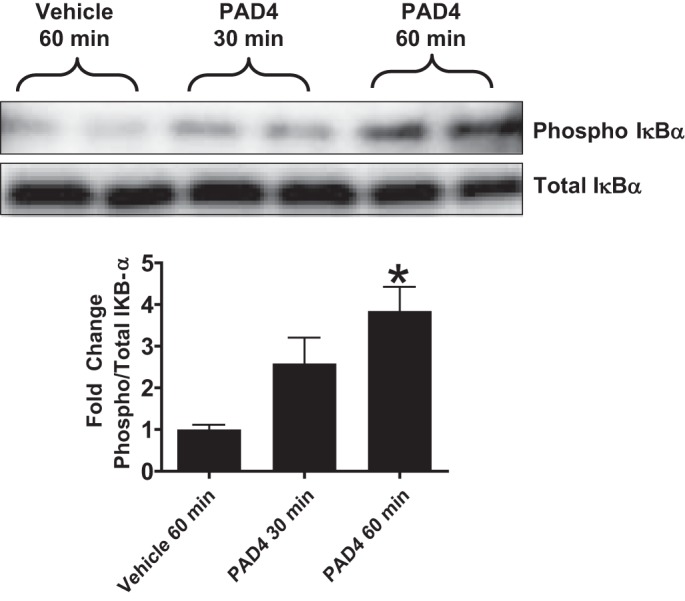
Top: peptidyl arginine deiminase-4 (PAD4) treatment causes phosphorylation of IκBα in HK-2 cells. HK-2 cells were treated with recombinant human PAD4 (5 μg/ml for 30–60 min). Bottom: bar graph shows band intensity quantifications demonstrating increased IκB-α phosphorylation. PAD4 significantly increased IκBα phosphorylation in HK-2 cells at 60 min compared with vehicle treatment. *P < 0.05 vs. vehicle treatment at 60 min (n = 4 experiments). Error bars represent 1 SE.
Ischemic AKI and PAD4-mediated exacerbation of renal injury, apoptosis, and inflammation are attenuated by NEMO inhibition.
We have previously demonstrated that ischemic AKI led to the induction of renal tubular PAD4 and that recombinant PAD4 treatment exacerbated ischemic AKI in mice (22, 49). Next, we tested whether NEMO blockade protects against renal I/R injury as well as recombinant PAD4-mediated exacerbation of ischemic AKI. Figure 5A shows that mice subjected to 30-min renal I/R injury increased plasma creatinine significantly. Mice treated with NEMO-binding peptide (5 mg/kg iv 15 min before renal 30-min ischemia) were protected against 30-min renal I/R injury with reduced plasma creatinine. Recombinant PAD4 treatment (10 μg iv 15 min before renal ischemia) exacerbated injury in mice subjected to 20-min renal I/R. NEMO-binding peptide (given 15 min before PAD4 treatment) prevented the PAD4-mediated exacerbation of ischemic AKI in mice (n = 4–6).
Fig. 5.

Ischemic acute kidney injury (AKI) and peptidyl arginine deiminase-4 (PAD4)-mediated exacerbation of renal injury, neutrophil infiltration, and apoptosis is attenuated by NF-κB essential modulator (NEMO) inhibition. A: mice subjected to 30-min renal ischemia-reperfusion (RIR) injury developed severe AKI with increased plasma creatinine. Mice treated with NEMO-binding peptide (NBP; 5 mg/kg iv 15 min before renal 30-min ischemia) were protected against 30-min RIR injury with reduced plasma creatinine. Recombinant PAD4 treatment (10 μg iv 15 min before renal ischemia) exacerbated injury in mice subjected to 20-min RIR. NBP given 15 min before PAD4 prevented the PAD4-mediated exacerbation of ischemic AKI in mice (n = 4–6). *P < 0.05 vs. vehicle-sham; #P < 0.05 vs. vehicle 30-min RIR; $P < 0.05 vs. PAD4 20-min RIR. Error bars = 1 SE. B and C: representative hematoxylin and eosin (H&E)-stained images (from 4−6 experiments; B) and renal injury scores (C) assessing the degree of renal tubular necrosis (scale of 0–4; Ref. 32) of mice subjected to 20-min renal ischemia and 24-h reperfusion (magnification: ×200). The kidneys of mice treated with recombinant PAD4 (10 μg iv 15 min before renal ischemia) and subjected to RIR showed increased tubular necrosis and proteinaceous casts as well as increased tubular dilatation and congestion compared with vehicle-treated mice. Mice treated with NBP 15 min before PAD4 treatment had significantly decreased renal tubular necrosis, congestion, and cast formation. D and E: representative images (from 4−6 experiments; D) of immunohistochemistry for neutrophils (dark brown) and counts of infiltrating kidney neutrophils (E; n = 4–6) in the kidneys in the kidneys of mice subjected to 20-min renal ischemia and 24-h reperfusion (magnification: ×200). Neutrophil infiltration markedly increased in mice treated with recombinant PAD4 at 10 μg iv 15 min before renal ischemia (concentrated near the corticomedullary junction). Mice treated with NBP 15 min before PAD4 treatment had significantly decreased neutrophil infiltration. F and G: representative images of TUNEL staining indicative of renal tubular apoptosis (F) and counts of TUNEL-positive kidney cells (G; n = 4). Increased renal tubule cell apoptosis occurred in mice treated with recombinant PAD4 at 10 μg iv 15 min before 20-min RIR injury. Mice treated with NBP 15 min before PAD4 treatment had significantly decreased TUNEL-positive renal tubule cells 24 h after reperfusion.
Figure 5B shows representative hematoxylin and eosin-stained images (from 4−6 experiments) of mice subjected to 20-min renal ischemia and 24-h reperfusion (magnification: ×200). As expected, the kidneys of mice treated with 10 μg iv recombinant PAD4 and subjected to renal I/R showed increased tubular necrosis and proteinaceous casts as well as increased tubular dilatation and congestion compared with vehicle-treated mice. Consistent with the plasma creatinine data, mice treated with NEMO-binding peptide 15 min before PAD4 treatment had decreased renal tubular necrosis, congestion, and cast formation with significantly reduced renal tubular injury scores compared with mice treated with PAD4 and subjected to renal I/R (Fig. 5C).
Figure 5D shows representative images (from 4−6 experiments) of immunohistochemistry for neutrophils (dark brown) in the kidneys of mice subjected to 20-min renal ischemia and 24-h reperfusion (magnification: ×200). Neutrophil infiltration markedly increased in mice treated with PAD4 (10 μg iv) 15 min before renal ischemia (concentrated near the corticomedullary junction). Mice treated with NEMO-binding peptide 15 min before PAD4 treatment had significantly decreased neutrophil infiltration (Fig. 5E).
TUNEL staining detected fragmented DNA suggestive of apoptosis in renal tubular cells in the kidneys of mice (from 4−6 experiments, magnification: ×200; Fig. 5F). Increased renal tubule cell apoptosis occurred in mice treated with recombinant PAD4 (10 μg iv) 15 min before 20-min renal I/R injury. Mice treated with NEMO-binding peptide 15 min before PAD4 treatment had significantly decreased TUNEL-positive renal tubule cells 24 h after reperfusion (Fig. 5G).
NEMO-binding peptide prevents recombinant PAD4-mediated increases kidney proinflammatory cytokine induction after renal I/R injury.
We measured the expression of proinflammatory chemokine mRNAs in the kidneys (KC, MCP-1, and MIP-2) 24 h after 20-min renal I/R by quantitative RT-PCR (primer sequences shown in Table 1). Mild renal I/R injury did not significantly increase any of the chemokine mRNAs measured (n = 4–6 experiments; Fig. 6). However, mice treated with recombinant PAD4 and subjected to renal I/R had significantly increased chemokine mRNAs. These exogenous PAD4-mediated inductions in chemokine mRNAs were significantly attenuated by pretreatment with NEMO-binding peptide.
Fig. 6.
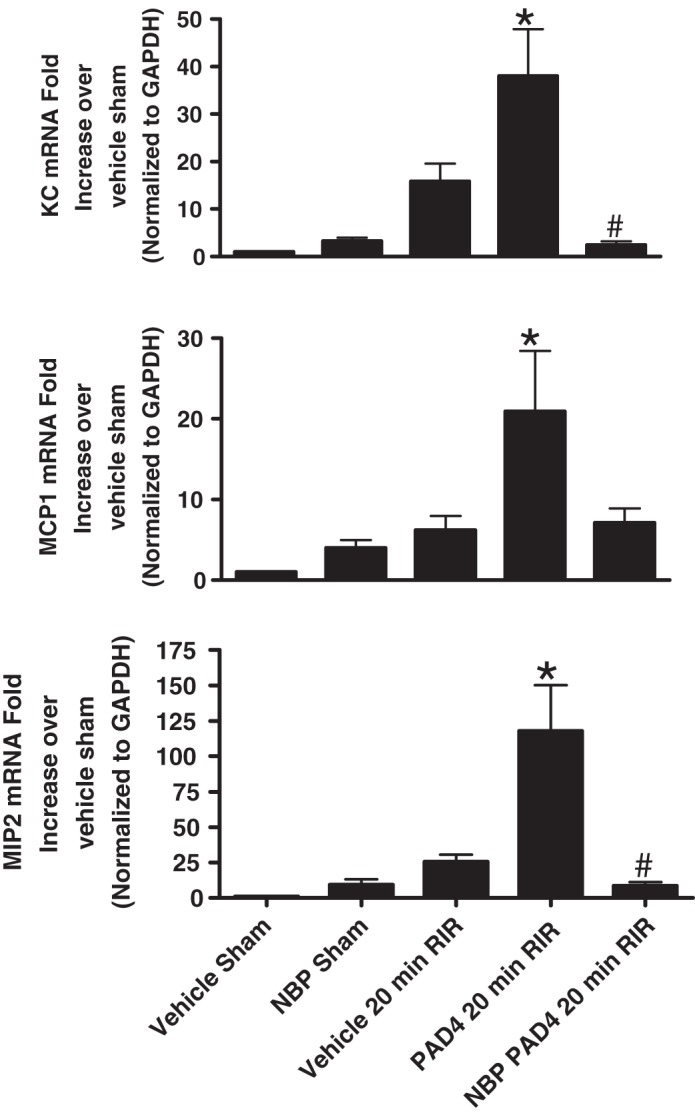
NF-κB essential modulator (NEMO)-binding peptide (NBP) attenuates peptidyl arginine deiminase-4 (PAD4)-mediated increases in neutrophil macrophage-attracting chemokines in the kidney after ischemia-reperfusion (I/R). With quantitative RT-PCR, we measured the expression of proinflammatory chemokine mRNAs in the kidney [keratinocyte-derived cytokine (KC), monocyte chemoattractant protein-1 (MCP-1), and macrophage inflammatory protein-2 (MIP-2)] 24 h after sham surgery or 20-min renal ischemia. Fold increases in proinflammatory mRNAs normalized to GAPDH from quantitative RT-PCRs for each indicated mRNA (n = 4–6) are shown. Mice were subjected to 20-min renal ischemia and 24-h reperfusion after vehicle (saline) or 10 μg human recombinant PAD4 treatment. Some mice were treated 5 mg/kg NBP 15 min before sham operation or renal I/R. For statistical analysis, one-way ANOVA plus Tukey’s post hoc multiple comparison test were used to detect significant changes. *P < 0.05 vs. vehicle-treated sham-operated mice; #P < 0.05 vs. PAD4-treated mice subjected to 20-min RIR injury. Error bars represent 1 SE.
NEMO blockade does not affect renal tubular cell proliferation, survival, or apoptosis.
Since NF-κB is a critical transcription factor for cell survival, proliferation and differentiation (41–43), global NF-κB inhibition may reduce the renal inflammatory response after I/R but also may have detrimental effects on renal cell proliferation and survival. We tested the hypothesis that selective IKKγ (NEMO) blockade while attenuating the proinflammatory effects of NF-κB activation but will be devoid of the detrimental effects on cell proliferation, survival, and apoptosis, unlike global NF-κB inhibition. We treated HK-2 cells with one of the NF-κB signaling inhibitors (10 μM NEMO-binding peptide, a selective IKKγ inhibitor; 25 μg/ml CAPE, a selective NF-κB inhibitor; or 10 μg/ml ACHP, a selective IKKα and IKKβ inhibitor) or with vehicle (1% DMSO) for 16 h. Cell survival was assessed with a MTT formazan absorbance assay, and cellular proliferation was assessed with a bromodeoxyuridine (BrdU) incorporation assay, as previously described (Fig. 7A) (12, 56). We detected apoptosis in human proximal tubule cells by performing caspase-3 and PARP immunoblot analysis. We found that NF-κB inhibition with CAPE significantly reduced HK-2 cell proliferation (BrdU incorporation) and survival (MTT assay, n = 4 experiments; Fig. 7A). NF-κB inhibition with either ACHP or with CAPE significantly increased apoptosis (caspase-3 and PARP fragmentation, representative of 4 experiments; Fig. 7B). In contrast, selective IKKγ inhibition with NEMO-binding peptide treatment did not significantly attenuate cell proliferation or survival or induce apoptosis in HK-2 cells.
Fig. 7.
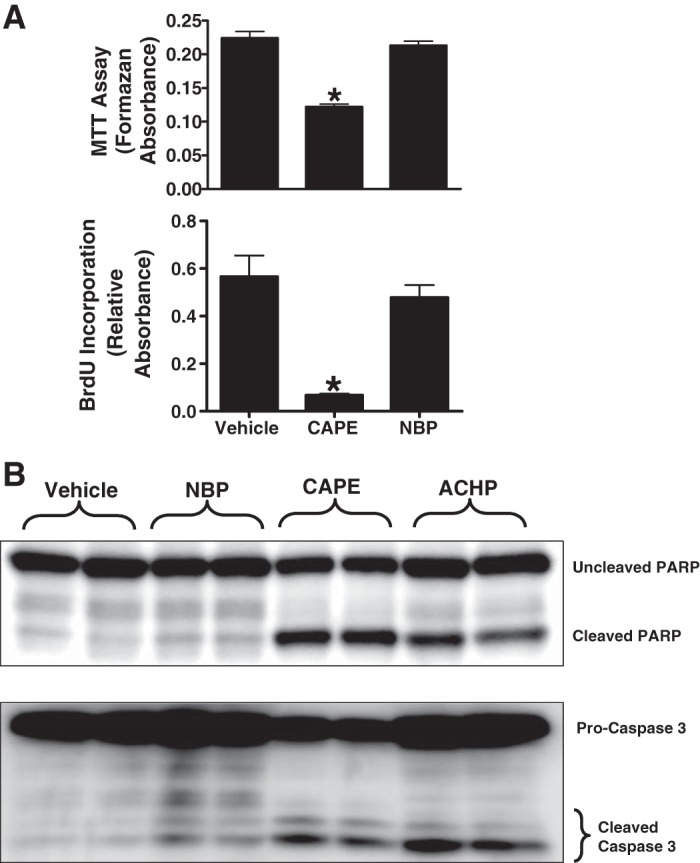
NF-κB essential modulator (NEMO) blockade does not affect renal tubular cell proliferation, survival, or promote apoptosis. HK-2 cells were treated with one of the NF-κB signaling inhibitors {10 μM NEMO-binding peptide (NBP; selective IKKγ inhibitor), 25 μg/ml caffeic acid phenethyl ester (CAPE; selective NF-κB inhibitor), or 10 μg/ml 2-amino-6-[2-(cyclopropylmethoxy)-6-hydroxyphenyl]-4-(4-piperidinyl)-3-pyridinecarbonitrile (ACHP; selective IKKα and IKKβ inhibitor)} or with vehicle (1% DMSO) for 16 h. A: NF-κB inhibition with CAPE significantly reduced HK-2 cell survival (MTT assay, n = 4; top) and proliferation [bromodeoxyuridine (BrdU) incorporation into DNA, n = 4; bottom]. B: NF-κB inhibition with either ACHP or CAPE significantly increased apoptosis [caspase-3 and poly (ADP-ribose) polymerase (PARP) fragmentation, n = 4]. In contrast, selective IKKγ inhibition with NBP treatment did not significantly attenuate cell proliferation, survival, or induce apoptosis in HK-2 cells. *P < 0.05 vs. vehicle.
DISCUSSION
AKI due to ischemia and reperfusion is a major and frequent clinical problem during the perioperative period (24, 59). Unfortunately, there is no effective therapy or preventive measures for this devastating clinical problem. Pathobiology of ischemic AKI is characterized not only by acute necrosis due to ischemic insult but also by a profound inflammatory response after the restoration of blood flow (30, 35). Indeed, induction of chemokines and cytokines during and after renal ischemia followed by infiltration and activation of resident and remote leukocytes are major contributors to renal tubular cell injury and death (29). Therefore, modulation of the hyperactive inflammatory response after renal I/R may attenuate the severity and may provide potential therapy for ischemic AKI.
The Ca2+-dependent class of PAD enzymes catalyzes the posttranslational modification by citrullination that promotes the conversion of peptidylarginine to peptidylcitrulline (15, 58). Of all PAD subtypes, PAD4 has been extensively studied due to its link to autoimmune diseases such as rheumatoid arthritis, colitis, and lupus (2, 3, 58). We have previously shown that renal I/R injury induced renal proximal tubular PAD4 synthesis and activity (22, 49). Our studies linked both renal proximal tubular as well as leukocyte PAD4 to critical roles in ischemic AKI as mice deficient in renal proximal tubular PAD4 as well as leukocyte-derived PAD4 were protected against ischemic AKI (40). Interestingly, renal tubular PAD4 appears to modulate not only the renal inflammatory response but also renal tubular apoptosis after renal I/R. The mechanism of renal tubular induction of PAD4 after renal I/R is via ATP released from necrotic renal cells activating cell surface P2X7 receptors (48). Moreover, we showed that PAD4 causes nuclear translocation of NF-κB, a critical transcription factor involved in inflammation and cell survival (22). However, the mechanism in which PAD4 induced renal inflammation and injury due to ischemic AKI has not been elucidated. It remained unclear how PAD4, a predominant nuclear enzyme, activates the translocation of NF-κB, which is bound in the cytosol until the inhibitor factor (IκB) gets phosphorylated and dissociates from NF-κB subunits (20, 41).
The impetus for the central hypothesis of this study was based on the observation that ischemic AKI causes induction and cytosolic translocation of PAD4 from the nucleus (49). We hypothesized that renal tubular PAD4 may translocate to the cytosol after I/R, causing dissociation of the IκB-NF-κB complex. Our findings are consistent with this hypothesis as we show here that PAD4 likely citrullinates one of the IκB-phosphorylating enzymes (IKKγ or NEMO) to facilitate IκBα phosphorylation and dissociation from the NF-κB complex, causing its nuclear translocation. We demonstrate here that neutralization of NEMO with a binding peptide attenuated PAD4-mediated NF-κB nuclear translocation and the inflammatory response in vivo and in vitro. Furthermore, NEMO blockade protected against severe ischemic AKI as well as recombinant PAD4-mediated exacerbation of ischemic AKI.
NF-κB is found in most animal cell types and is a critical transcription factor in response to stress and injury by promoting cytokine release and inflammation (20, 41, 43). NF-κB is found in the cytosol in the inactive state bound to IκBα/β. The IκB kinase complex (composed of α-, β-, and γ-subtypes) phosphorylates the IκB complex, releasing it from NF-κB. IKKγ is a part of the IKK complex, along with IKKα and IKKβ, responsible for phosphorylation of IκB proteins that results in ubiquitination and degradation. IκB phosphorylation and degradation are critical events that cause NF-κB activation via nuclear translocation (41). This results in activated NF-κB translocation into the nucleus, subsequently leading to proinflammatory cytokine induction.
IKKγ is also known as NEMO (21, 53). We show here that recombinant PAD4 citrullinates recombinant as well as renal proximal tubular NEMO (IKKγ) without significantly affecting other IKK subtypes (IKKα and IKKβ) (Fig. 1A). Indeed, NEMO has multiple peptidylarginine residues that can be citrullinated by PAD4. It is unclear why PAD4 preferentially citrullinates IKKγ over other IKK subtypes. It appears that NEMO citrullination leads to activation of its activity, IκBα phosphorylation, and NF-κB nuclear translocation with subsequent inflammatory cytokine induction. Our findings that NEMO blockade attenuates PAD4-mediated NF-κB nuclear translocation and inflammatory cytokine induction support this hypothesis.
After renal I/R, renal tubular NF-κB activation leads to the transcription of several proinflammatory chemokines including neutrophil-attracting IL-8/KC and MIP-2 (also called CXCL8 and CXCL2, respectively) (1, 4). Although NF-κB plays a critical role in generating the inflammatory response as a key transcription factor for several proinflammatory chemokines, NF-κB is also a critical transcription factor for cell survival, proliferation, and differentiation (41, 61). Therefore, global NF-κB inhibition may reduce the renal inflammatory response after I/R but also may have detrimental effects on renal cell proliferation and survival (55). Therefore, selective inhibition of the proinflammatory pathway would be required to protect against the inflammatory response after renal I/R. Our results suggest that PAD4 induces renal inflammation and exacerbates renal I/R injury by citrullination of NEMO, and inhibition of NEMO will selectively attenuate proinflammatory gene expression and improve kidney injury. Our data support the hypothesis that blockade of NEMO (IKKγ) selectively reduced the renal tubular inflammation without decreasing cell proliferation or survival.
In this present study, we showed that blockade of NEMO activity with NEMO-binding peptide protected against ischemic AKI. Previous studies have also implicated NEMO as a mediator of the kidney podocyte inflammatory response. Selective ablation of NEMO in kidney podocytes showed faster recovery, improved podocyte morphology, and showed less proteinuria in a mouse model of glomerulonephritis (10) compared with wild-type mice. They also observed TNF-α- and IL-1-induced inflammation, and secretion of proinflammatory chemokines from podocytes was inhibited in NEMO knockdown podocyte cells. NEMO-binding peptide also protects against hepatic I/R injury in a rat model of liver transplantation and rat cerebral I/R injury (11, 13). These previous findings combined with our present data support the hypothesis that NEMO inhibition selective attenuates I/R injury and the inflammatory response without promoting apoptosis or decreasing cellular proliferation.
Tissue hypoxia during and after I/R rapidly induces hypoxia-inducible factor (HIF), which is an endogenous and powerful tissue protective factor in many cell types (9, 16, 18). Interestingly, HIF induction directly regulates neutrophil extracellular trap formation, suggesting a potential link between HIF and PAD4 (9). Indeed, PAD4 is a critical regulator of neutrophil extracellular trap formation by directly promoting histone condensation by citrullination (11). Furthermore, the PAD4 NEMO-mediated inflammatory response may promote inflammation by controlling IKKβ activity, promoting its inflammatory signaling. Inflammation-driven tissue hypoxia, additionally driving HIF activation. There are endogenous break pathways, however. For example, hypoxia-mediated ATP degradation into anti-inflammatory adenosine signaling may provide anti-inflammatory signaling to counterbalance the proinflammatory signaling due to hypoxia (26, 27, 60). It remains unclear whether the HIF transcription factor is directly involved in PAD4 induction after renal I/R injury.
Hypoxia and inflammation are intimately linked, as hypoxia can induce inflammation and inflammation can lead to tissue hypoxia (1, 4, 14). The NF-κB pathway is a master regulator of inflammation and is indeed tightly regulated by hypoxia-driven signaling (5). Our present and previous studies (22, 48, 49) demonstrated that ischemic AKI induces PAD4 activation and induction, which promote proinflammatory NF-κB signaling via NEMO activation/citrullination. However, additional regulatory mechanisms of NF-κB activation during tissue hypoxia may exist. During renal ischemia, hypoxia-driven activation of prolyl hydroxylases plays a critical role in hypoxia-mediated (5).
In our previous study, we showed that ATP released from necrotic renal tubules after I/R directly induced renal tubular PAD4 to promote kidney inflammatory response and injury via P2X7 receptor activation (48). Furthermore, Riegel et al. (51) have elegantly shown that selective induction of endothelial P2Y6 receptor promotes vascular inflammation. Taken together, it appears that P2 purinergic signaling promotes the inflammatory response in several tissue and cell types (27). It is interesting to note that P1 purinergic receptor activation driven by breakdown products of ATP (P2 receptor agonist) can counteractive the P2 receptor-driven inflammatory responses (19, 37, 38, 45). Indeed, P1 adenosine receptor activation protects against ischemic AKI as well as septic AKI (7, 19). Therefore, it appears that adenosine, generated by sequential dephosphorylation of ATP, appears to counterbalance or place a “break” on the inflammatory and injury-promoting role of ATP released by necrotic cells.
Clinically, even mild ischemic AKI with a small increase in plasma creatinine results in large increased in mortality during the perioperative period (6). For example, plasma creatinine increases of <0.3 mg/dl after surgery was associated with two- to fivefold increases in mortality. This extremely high morbidity and mortality from AKI are in part due to the high incidence of extrarenal complications (17, 59). We believe that the inflammatory response brought on by PAD4 → NEMO citrullination not only results in increased ischemic AKI but also leads to exacerbated remote organ injury. Indeed, we showed in our previous study that mice deficient in PAD4 were not only protected against renal I/R injury but also had reduced hepatic dysfunction after AKI (49). Therefore, our findings that NEMO blockade lead to protection against ischemic AKI not only have implications for kidney injury but also have potential protection against remote organ injury after AKI.
In summary, we demonstrate in this study that PAD4 promotes NEMO citrullination that results in NF-κB activation after I/R. NEMO blockade after renal I/R resulted in improved kidney function and decreased proinflammatory cytokine stimulation. Our previous and present findings allow us to propose a detailed mechanism of PAD4-mediated renal tubular inflammation and exacerbation of ischemic AKI (Fig. 8). Necrotic renal tubular cells after I/R releases intracellular ATP, which subsequently induces PAD4 in neighboring renal proximal tubular cells via P2X7 receptor activation and PKC signaling. Renal I/R also causes cytosolic translocation of PAD4 in addition to its upregulation. We hypothesize that PAD4 preferentially citrullinates NEMO, which, in turn, drives proinflammatory NF-κB signaling with increased cytokine/chemokine synthesis and neutrophil infiltration. NEMO blockade with NEMO-binding peptide attenuates this detrimental inflammatory signaling induced by PAD4 upregulation. Overall, our results allow us to hypothesize that PAD4-mediated citrullination of NEMO is crucial in generating ischemic AKI via proinflammatory cytokine activation and that selective inhibition of the NF-κB pathway of by selectively targeting NEMO activity may reduce complications and mortality arising from ischemic AKI.
Fig. 8.

Overview of peptidyl arginine deiminase-4 (PAD4)-mediated inflammation via NF-κB essential modulator (NEMO) activation. Necrotic renal tubular cells after ischemia-reperfusion (I/R) releases intracellular ATP, which subsequently induces PAD4 in neighboring renal proximal tubular cells via P2X7 receptor activation and PKC signaling (48). Renal I/R also causes cytosolic translocation of PAD4 in addition to its upregulation (49). We hypothesized that PAD4 preferentially citrullinates NEMO, which, in turn, drives proinflammatory NF-κB signaling with increased cytokine/chemokine synthesis and neutrophil infiltration. NEMO blockade after renal I/R resulted in improved kidney function and decreased proinflammatory cytokine stimulation. MIP-2, macrophage inflammatory protein-2.
GRANTS
This work was supported by the Department of Anesthesiology, Columbia University, and in part by National Institute of Diabetes and Digestive and Kidney Diseases Grants R01-DK-109544 and DK-115694 (to H. T. Lee).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.M.R., S.J.H., M.K., and H.T.L. conceived and designed research; M.M.R., S.J.H., M.K., and V.D.D. performed experiments; M.M.R., S.J.H., M.K., V.D.D., and H.T.L. analyzed data; M.M.R., S.J.H., M.K., V.D.D., and H.T.L. interpreted results of experiments; M.M.R., S.J.H., M.K., and H.T.L. prepared figures; M.M.R., S.J.H., M.K., and H.T.L. drafted manuscript; M.M.R., S.J.H., M.K., and H.T.L. edited and revised manuscript; M.M.R., S.J.H., M.K., V.D.D., and H.T.L. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the technical assistance provided by Kevin M. Brown and Dr. Ahrom Ham.
REFERENCES
- 1.Akcay A, Nguyen Q, Edelstein CL. Mediators of inflammation in acute kidney injury. Mediators Inflamm 2009: 137072, 2009. doi: 10.1155/2009/137072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anzilotti C, Pratesi F, Tommasi C, Migliorini P. Peptidylarginine deiminase 4 and citrullination in health and disease. Autoimmun Rev 9: 158–160, 2010. doi: 10.1016/j.autrev.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Auger I, Charpin C, Balandraud N, Martin M, Roudier J. Autoantibodies to PAD4 and BRAF in rheumatoid arthritis. Autoimmun Rev 11: 801–803, 2012. doi: 10.1016/j.autrev.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 4.Bajwa A, Kinsey GR, Okusa MD. Immune mechanisms and novel pharmacological therapies of acute kidney injury. Curr Drug Targets 10: 1196–1204, 2009. doi: 10.2174/138945009789753174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartels K, Grenz A, Eltzschig HK. Hypoxia and inflammation are two sides of the same coin. Proc Natl Acad Sci USA 110: 18351–18352, 2013. doi: 10.1073/pnas.1318345110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartels K, Karhausen J, Clambey ET, Grenz A, Eltzschig HK. Perioperative organ injury. Anesthesiology 119: 1474–1489, 2013. doi: 10.1097/ALN.0000000000000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bauerle JD, Grenz A, Kim JH, Lee HT, Eltzschig HK. Adenosine generation and signaling during acute kidney injury. J Am Soc Nephrol 22: 14–20, 2011. doi: 10.1681/ASN.2009121217. [DOI] [PubMed] [Google Scholar]
- 8.Bicker KL, Thompson PR. The protein arginine deiminases: structure, function, inhibition, and disease. Biopolymers 99: 155–163, 2013. doi: 10.1002/bip.22127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowser JL, Lee JW, Yuan X, Eltzschig HK. The hypoxia-adenosine link during inflammation. J Appl Physiol (1985) 123: 1303–1320, 2017. doi: 10.1152/japplphysiol.00101.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brähler S, Ising C, Hagmann H, Rasmus M, Hoehne M, Kurschat C, Kisner T, Goebel H, Shankland S, Addicks K, Thaiss F, Schermer B, Pasparakis M, Benzing T, Brinkkoetter PT. Intrinsic proinflammatory signaling in podocytes contributes to podocyte damage and prolonged proteinuria. Am J Physiol Renal Physiol 303: F1473–F1485, 2012. doi: 10.1152/ajprenal.00031.2012. [DOI] [PubMed] [Google Scholar]
- 11.Cheng MX, Gong JP, Chen Y, Liu ZJ, Tu B, Liu CA. NBD peptides protect against ischemia reperfusion after orthotopic liver transplantation in rats. J Surg Res 176: 666–671, 2012. doi: 10.1016/j.jss.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 12.Darzynkiewicz Z, Juan G.. Analysis of DNA content and BrdU icorporation . In: Current Protocols in Cytometry. New York: Wiley, chapt. 7, Unit 7. 7, 2001. doi: 10.1002/0471142956.cy0707s02. [DOI] [PubMed] [Google Scholar]
- 13.Desai A, Singh N, Raghubir R. Neuroprotective potential of the NF-κB inhibitor peptide IKK-NBD in cerebral ischemia-reperfusion injury. Neurochem Int 57: 876–883, 2010. doi: 10.1016/j.neuint.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 14.Donnahoo KK, Meldrum DR, Shenkar R, Chung CS, Abraham E, Harken AH. Early renal ischemia, with or without reperfusion, activates NFkappaB and increases TNF-alpha bioactivity in the kidney. J Urol 163: 1328–1332, 2000. doi: 10.1016/S0022-5347(05)67772-5. [DOI] [PubMed] [Google Scholar]
- 15.Dreyton CJ, Anderson ED, Subramanian V, Boger DL, Thompson PR. Insights into the mechanism of streptonigrin-induced protein arginine deiminase inactivation. Bioorg Med Chem 22: 1362–1369, 2014. doi: 10.1016/j.bmc.2013.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eckle T, Brodsky K, Bonney M, Packard T, Han J, Borchers CH, Mariani TJ, Kominsky DJ, Mittelbronn M, Eltzschig HK. HIF1A reduces acute lung injury by optimizing carbohydrate metabolism in the alveolar epithelium. PLoS Biol 11: e1001665, 2013. doi: 10.1371/journal.pbio.1001665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elapavaluru S, Kellum JA. Why do patients die of acute kidney injury? Acta Clin Belg 62, Suppl 2: 326–331, 2007. doi: 10.1179/acb.2007.074. [DOI] [PubMed] [Google Scholar]
- 18.Eltzschig HK, Bratton DL, Colgan SP. Targeting hypoxia signalling for the treatment of ischaemic and inflammatory diseases. Nat Rev Drug Discov 13: 852–869, 2014. doi: 10.1038/nrd4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallos G, Ruyle TD, Emala CW, Lee HT. A1 adenosine receptor knockout mice exhibit increased mortality, renal dysfunction, and hepatic injury in murine septic peritonitis. Am J Physiol Renal Physiol 289: F369–F376, 2005. doi: 10.1152/ajprenal.00470.2004. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh G, Wang VY, Huang DB, Fusco A. NF-κB regulation: lessons from structures. Immunol Rev 246: 36–58, 2012. doi: 10.1111/j.1600-065X.2012.01097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghosh S, Febin Prabhu Dass J. Non-canonical pathway network modelling and ubiquitination site prediction through homology modelling of NF-κB. Gene 581: 48–56, 2016. doi: 10.1016/j.gene.2016.01.025. [DOI] [PubMed] [Google Scholar]
- 22.Ham A, Rabadi M, Kim M, Brown KM, Ma Z, D’Agati V, Lee HT. Peptidyl arginine deiminase-4 activation exacerbates kidney ischemia-reperfusion injury. Am J Physiol Renal Physiol 307: F1052–F1062, 2014. doi: 10.1152/ajprenal.00243.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han SJ, Li H, Kim M, Shlomchik MJ, Lee HT. Kidney proximal tubular TLR9 exacerbates ischemic acute kidney injury. J Immunol 201: 1073–1085, 2018. doi: 10.4049/jimmunol.1800211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hilmi IA, Damian D, Al-Khafaji A, Planinsic R, Boucek C, Sakai T, Chang CC, Kellum JA. Acute kidney injury following orthotopic liver transplantation: incidence, risk factors, and effects on patient and graft outcomes. Br J Anaesth 114: 919–926, 2015. doi: 10.1093/bja/aeu556. [DOI] [PubMed] [Google Scholar]
- 25.Hoste EA, Kellum JA, Katz NM, Rosner MH, Haase M, Ronco C. Epidemiology of acute kidney injury. Contrib Nephrol 165: 1–8, 2010. doi: 10.1159/000313737. [DOI] [PubMed] [Google Scholar]
- 26.Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature 509: 310–317, 2014. doi: 10.1038/nature13085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Idzko M, Ferrari D, Riegel AK, Eltzschig HK. Extracellular nucleotide and nucleoside signaling in vascular and blood disease. Blood 124: 1029–1037, 2014. doi: 10.1182/blood-2013-09-402560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jablonski P, Howden BO, Rae DA, Birrell CS, Marshall VC, Tange J. An experimental model for assessment of renal recovery from warm ischemia. Transplantation 35: 198–204, 1983. doi: 10.1097/00007890-198303000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Jang HR, Rabb H. Immune cells in experimental acute kidney injury. Nat Rev Nephrol 11: 88–101, 2015. doi: 10.1038/nrneph.2014.180. [DOI] [PubMed] [Google Scholar]
- 30.Kaczorowski DJ, Tsung A, Billiar TR. Innate immune mechanisms in ischemia/reperfusion. Front Biosci (Elite Ed) 1: 91–98, 2009. [DOI] [PubMed] [Google Scholar]
- 31.Kellum JA, Hoste EA. Acute kidney injury: epidemiology and assessment. Scand J Clin Lab Invest Suppl 68: 6–11, 2008. doi: 10.1080/00365510802144813. [DOI] [PubMed] [Google Scholar]
- 32.Khalid U, Pino-Chavez G, Nesargikar P, Jenkins RH, Bowen T, Fraser DJ, Chavez R. Kidney ischaemia reperfusion injury in the rat: the EGTI scoring system as a valid and reliable tool for histological assessment. J Histol Histopathol 3: 1, 2016. doi: 10.7243/2055-091X-3-1. [DOI] [Google Scholar]
- 33.Kim JY, Kim M, Ham A, Brown KM, Greene RW, D’Agati VD, Lee HT. IL-11 is required for A1 adenosine receptor-mediated protection against ischemic AKI. J Am Soc Nephrol 24: 1558–1570, 2013. doi: 10.1681/ASN.2013010114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim M, Ham A, Kim JY, Brown KM, D’Agati VD, Lee HT. The volatile anesthetic isoflurane induces ecto-5′-nucleotidase (CD73) to protect against renal ischemia and reperfusion injury. Kidney Int 84: 90–103, 2013. doi: 10.1038/ki.2013.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kinsey GR, Okusa MD. Expanding role of T cells in acute kidney injury. Curr Opin Nephrol Hypertens 23: 9–16, 2014. doi: 10.1097/01.mnh.0000436695.29173.de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kork F, Balzer F, Spies CD, Wernecke KD, Ginde AA, Jankowski J, Eltzschig HK. Minor postoperative increases of creatinine are associated with higher mortality and longer hospital length of stay in surgical patients. Anesthesiology 123: 1301–1311, 2015. doi: 10.1097/ALN.0000000000000891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee HT, Gallos G, Nasr SH, Emala CW. A1 adenosine receptor activation inhibits inflammation, necrosis, and apoptosis after renal ischemia-reperfusion injury in mice. J Am Soc Nephrol 15: 102–111, 2004. doi: 10.1097/01.ASN.0000102474.68613.AE. [DOI] [PubMed] [Google Scholar]
- 38.Lee HT, Kim M, Joo JD, Gallos G, Chen JF, Emala CW. A3 adenosine receptor activation decreases mortality and renal and hepatic injury in murine septic peritonitis. Am J Physiol Regul Integr Comp Physiol 291: R959–R969, 2006. doi: 10.1152/ajpregu.00034.2006. [DOI] [PubMed] [Google Scholar]
- 39.Lee HT, Park SW, Kim M, Ham A, Anderson LJ, Brown KM, D’Agati VD, Cox GN. Interleukin-11 protects against renal ischemia and reperfusion injury. Am J Physiol Renal Physiol 303: F1216–F1224, 2012. doi: 10.1152/ajprenal.00220.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H, Han SJ, Kim M, Cho A, Choi Y, D’Agati V, Lee HT. Divergent roles for kidney proximal tubule and granulocyte PAD4 in ischemic AKI. Am J Physiol Renal Physiol 314: F809–F819, 2018. doi: 10.1152/ajprenal.00569.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitchell S, Vargas J, Hoffmann A. Signaling via the NFκB system. Wiley Interdiscip Rev Syst Biol Med 8: 227–241, 2016. doi: 10.1002/wsbm.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 1: a000034, 2009. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol 12: 695–708, 2011. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 44.Okusa MD. The changing pattern of acute kidney injury: from one to multiple organ failure. Contrib Nephrol 165: 153–158, 2010. doi: 10.1159/000313754. [DOI] [PubMed] [Google Scholar]
- 45.Okusa MD, Linden J, Huang L, Rieger JM, Macdonald TL, Huynh LP. A2A adenosine receptor-mediated inhibition of renal injury and neutrophil adhesion. Am J Physiol Renal Physiol 279: F809–F818, 2000. doi: 10.1152/ajprenal.2000.279.5.F809. [DOI] [PubMed] [Google Scholar]
- 46.Park SW, Kim M, Brown KM, D’Agati VD, Lee HT. Inhibition of sphingosine 1-phosphate receptor 2 protects against renal ischemia-reperfusion injury. J Am Soc Nephrol 23: 266–280, 2012. doi: 10.1681/ASN.2011050503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park SW, Kim M, Kim JY, Brown KM, Haase VH, D’Agati VD, Lee HT. Proximal tubule sphingosine kinase-1 has a critical role in A1 adenosine receptor-mediated renal protection from ischemia. Kidney Int 82: 878–891, 2012. doi: 10.1038/ki.2012.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rabadi M, Kim M, Li H, Han SJ, Choi Y, D’Agati V, Lee HT. ATP induces PAD4 in renal proximal tubule cells via P2X7 receptor activation to exacerbate ischemic AKI. Am J Physiol Renal Physiol 314: F293–F305, 2018. doi: 10.1152/ajprenal.00364.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rabadi M, Kim M, D’Agati V, Lee HT. Peptidyl arginine deiminase-4-deficient mice are protected against kidney and liver injury after renal ischemia and reperfusion. Am J Physiol Renal Physiol 311: F437–F449, 2016. doi: 10.1152/ajprenal.00254.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Riegel AK, Faigle M, Zug S, Rosenberger P, Robaye B, Boeynaems JM, Idzko M, Eltzschig HK. Selective induction of endothelial P2Y6 nucleotide receptor promotes vascular inflammation. Blood 117: 2548–2555, 2011. doi: 10.1182/blood-2010-10-313957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rohrbach AS, Slade DJ, Thompson PR, Mowen KA. Activation of PAD4 in NET formation. Front Immunol 3: 360, 2012. doi: 10.3389/fimmu.2012.00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schröfelbauer B, Polley S, Behar M, Ghosh G, Hoffmann A. NEMO ensures signaling specificity of the pleiotropic IKKβ by directing its kinase activity toward IκBα. Mol Cell 47: 111–121, 2012. doi: 10.1016/j.molcel.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Srisawat N, Hoste EE, Kellum JA. Modern classification of acute kidney injury. Blood Purif 29: 300–307, 2010. doi: 10.1159/000280099. [DOI] [PubMed] [Google Scholar]
- 55.Sun B, Karin M. NF-kappaB signaling, liver disease and hepatoprotective agents. Oncogene 27: 6228–6244, 2008. doi: 10.1038/onc.2008.300. [DOI] [PubMed] [Google Scholar]
- 56.van de Loosdrecht AA, Beelen RH, Ossenkoppele GJ, Broekhoven MG, Langenhuijsen MM. A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J Immunol Methods 174: 311–320, 1994. doi: 10.1016/0022-1759(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 57.Vinay P, Gougoux A, Lemieux G. Isolation of a pure suspension of rat proximal tubules. Am J Physiol Renal Physiol 241: F403–F411, 1981. doi: 10.1152/ajprenal.1981.241.4.F403. [DOI] [PubMed] [Google Scholar]
- 58.Wang S, Wang Y. Peptidylarginine deiminases in citrullination, gene regulation, health and pathogenesis. Biochim Biophys Acta 1829: 1126–1135, 2013. doi: 10.1016/j.bbagrm.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yap SC, Lee HT. Acute kidney injury and extrarenal organ dysfunction: new concepts and experimental evidence. Anesthesiology 116: 1139–1148, 2012. doi: 10.1097/ALN.0b013e31824f951b. [DOI] [PubMed] [Google Scholar]
- 60.Yuan X, Lee JW, Bowser JL, Neudecker V, Sridhar S, Eltzschig HK. Targeting hypoxia signaling for perioperative organ injury. Anesth Analg 126: 308–321, 2018. doi: 10.1213/ANE.0000000000002288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-κB: a blossoming of relevance to human pathobiology. Cell 168: 37–57, 2017. doi: 10.1016/j.cell.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]