

Abstract
The type 2a sarco-/endoplasmic reticulum Ca2+-ATPase (SERCA2a) plays a key role in Ca2+ regulation in the heart. However, available techniques to study SERCA function are either cell destructive or lack sensitivity. The goal of this study was to develop an approach to selectively measure SERCA2a function in the cellular environment. The genetically encoded Ca2+ sensor R-CEPIA1er was used to measure the concentration of Ca2+ in the lumen of the endoplasmic reticulum (ER) ([Ca2+]ER) in HEK293 cells expressing human SERCA2a. Coexpression of the ER Ca2+ release channel ryanodine receptor (RyR2) created a Ca2+ release/reuptake system that mimicked aspects of cardiac myocyte Ca2+ handling. SERCA2a function was quantified from the rate of [Ca2+]ER refilling after ER Ca2+ depletion; then, ER Ca2+ leak was measured after SERCA inhibition. ER Ca2+ uptake and leak were analyzed as a function of [Ca2+]ER to determine maximum ER Ca2+ uptake rate and maximum ER Ca2+ load. The sensitivity of this assay was validated by analyzing effects of SERCA inhibitors, [ATP]/[ADP], oxidative stress, phospholamban, and a loss-of-function SERCA2a mutation. In addition, the feasibility of using R-CEPIA1er to study SERCA2a in a native system was evaluated by using in vivo gene delivery to express R-CEPIA1er in mouse hearts. After ventricular myocyte isolation, the same methodology used in HEK293 cells was applied to study endogenous SERCA2a. In conclusion, this new approach can be used as a sensitive screening tool to study the effect of different drugs, posttranslational modifications, and mutations on SERCA function.
NEW & NOTEWORTHY The aim of this study was to develop a sensitive approach to selectively measure sarco-/endoplasmic reticulum Ca2+-ATPase (SERCA) function in the cellular environment. The newly developed Ca2+ sensor R-CEPIA1er was used to successfully analyze Ca2+ uptake mediated by recombinant and native cardiac SERCA. These results demonstrate that this new approach can be used as a powerful tool to study new mechanisms of Ca2+ pump regulation.
Keywords: Ca2+ pump, Ca2+ signaling, HEK293 cells, sarcoplasmic reticulum, ventricular myocytes
INTRODUCTION
The sarco-/endoplasmic reticulum Ca2+-ATPase (SERCA) is an essential component of Ca2+ signaling (7). During the SERCA transport cycle, hydrolysis of one ATP molecule provides energy for translocation of two Ca2+ ions from the cytosol into the endoplasmic reticulum (ER) lumen. As a result of SERCA activity, the ER luminal Ca2+ concentration ([Ca2+]ER) can reach millimolar concentration, while cytosolic [Ca2+] ([Ca2+]i) is maintained at the submicromolar level. This large Ca2+ gradient is the foundation for Ca2+ signaling, as activation of ER Ca2+ release can produce a robust intracellular Ca2+ transient that is essential for activation of many cellular processes, including growth, migration, secretion, metabolism, neurotransmission, and contraction. Three SERCA genes with 10 isoforms have been identified (31). Despite a significant homology between SERCA types, they have specific differences in kinetics and Ca2+ sensitivity uniquely fitted to their function in different cell types. Here, we focus on the cardiac isoform SERCA2a.
As the muscle contraction-relaxation cycle is controlled by intracellular Ca2+ dynamics, Ca2+ transport is a particularly important point of cardiac regulation. The rate at which SERCA2a removes Ca2+ from the cytosol into the sarcoplasmic reticulum (SR) is a major determinant of the rate of cardiac muscle relaxation. SERCA2a activity also plays a key role in setting diastolic [Ca2+]i, which affects the compliance of the heart muscle as the ventricles fill with blood. SERCA2a also sets the total amount of Ca2+ in the SR (i.e., Ca2+ load), which determines the magnitude of SR Ca2+ release with each heartbeat. This latter function of SERCA2a is the primary determinant of the strength of the cardiac contraction. Thus SERCA2a activity must be closely regulated to match the varying demands of exercise and rest. This is accomplished through the action of the transmembrane peptide phospholamban (PLB). This peptide is the major regulator of the SR Ca2+ pump (24), inhibiting SERCA2a activity by lowering the apparent affinity of the pump for Ca2+ (20). Phosphorylation of PLB by PKA relieves this inhibition, increasing the Ca2+ pumping rate by several-fold (36). Stimulation of SR Ca2+ uptake by PLB phosphorylation is the major mechanism of acceleration of relaxation (lusitropy) and increased contraction strength (inotropy) during adrenergic stimulation (15). Because of the central role of SERCA2a in the cardiac contractile cycle, it is not surprising that impaired SERCA2a function has been reported in a number of pathological conditions, including heart failure (44). The impaired SERCA2a function in heart failure can be a consequence of SERCA2a downregulation at the mRNA and protein levels (13, 19, 30) or a result of posttranslational modifications of SERCA2a and PLB (1, 13, 22, 33). Moreover, it has been shown that several mutations of PLB contribute to cardiomyopathies (18). Consequently, the SERCA2a/PLB complex has attracted attention as a potential target for therapeutic approaches to improve heart function during diseases (17). Despite this interest in understanding SERCA-regulatory mechanisms, it remains problematic to selectively measure the SERCA-mediated Ca2+ uptake in living cells due to a contribution of different ion channels, exchangers, and pumps to Ca2+ homeostasis.
Several established experimental approaches are currently used for the quantification of SERCA2a function. In intact myocytes, SERCA-mediated Ca2+ uptake can be inferred from the decay of the cytosolic Ca2+ transient induced by an action potential (6) or from the total amount of Ca2+ stored in the SR (14, 40). Because the cytosolic measurements can be contaminated by plasmalemmal and mitochondrial Ca2+ transport, these indirect approaches are not selective for SERCA2a activity. Moreover, these measurements depend on high-affinity cytosolic Ca2+ indicators (e.g., Fluo-3) that can be easily saturated by SR Ca2+ release. In vitro assays of SERCA function based on ATP consumption (29) or Ca2+ uptake into ER/SR vesicles (34) are more direct and quantitative measures of SERCA2a function, but they require cell homogenization and relatively large amounts of protein. Recently, another approach has been developed which allows measuring the SR luminal [Ca2+] ([Ca2+]SR) directly in intact cells (11, 21, 35). This approach is based on the ability of low-affinity Ca2+ dyes, such as Fluo-5N, to measure SR luminal Ca2+. Thus SERCA2a-mediated Ca2+ uptake can be measured directly from the rate of [Ca2+]SR recovery during the Ca2+ transient (10, 46). However, this method also suffers from several limitations, including dye extrusion, nonspecific dye localization, and species limitation. Thus an alternative approach is needed to quantify SERCA2a transport kinetics in living cells.
The aim of this study was to develop an approach to selectively measure SERCA2a function. We used the genetically encoded ER Ca2+ sensor R-CEPIA1er to directly measure SR Ca2+ uptake mediated by recombinant and native SERCA2a. The approach was validated by testing effects of well-known SERCA inhibitors and regulators. The obtained results demonstrate that this new method can be used as a sensitive assay to study SERCA function in the cellular environment.
MATERIAL AND METHODS
Vector Production
The vector encoding the human RyR2 cDNA fused to green fluorescent protein (GFP) at the NH2-terminus domain was kindly provided by Dr. Christopher George (University of Cardiff). pCMV R-CEPIA1er was a gift from Dr. Masamitsu Iino (Addgene plasmid 58216; http://n2t.net/addgene:58216; RRID: Addgene_58216). The vector encoding human SERCA2a cDNA was kindly provided by Dr. David Thomas (University of Minnesota). The SERCA2a cDNA was cloned into the mCerulean-M1 modified plasmid (Addgene) using KpnI and NotI restriction enzymes, yielding a recombinant SERCA2a protein fused to a modified cerulean fluorescent protein (mCer) at the NH2-terminus. SERCA2a cDNA was also cloned into the inducible expression vector pcDNA5/FRT/TO using KpnI and EcoRV for the generation of a SERCA2a stable cell line. D426A/E429A/E435A (AAA) and E876Q/D877N/E882Q (QNQ) SERCA2a mutants were generated by site-directed mutagenesis (Q5 site-directed mutagenesis kit, NEB). The sequences were all verified by single-pass primer extension analysis (ACGT).
Generation of SERCA2a Stable Inducible Flp-In T-Rex-293 Cell Line
The stable inducible Flp-In T-Rex-293 cell line expressing SERCA2a was generated using the Flp-In T-REx Core Kit (Invitrogen). Briefly, Flp-In T-REx-293 cells were cotransfected with the pOG44 vector encoding the Flp recombinase and the expression vector pcDNA5/FRT/TO containing the SERCA2a cDNA (without a fluorescent tag) in a ratio of 9:1 using polyethylenimine (PEI; 1 μg/ml). Forty-eight hours after transfection, the growth medium was replaced with a selection medium containing 100 μg/ml hygromycin (Invitrogen). The selection medium was changed every 2 days until the stable line cell foci could be isolated. The hygromycin-resistant cell foci were selected and expanded. Stable line cells were cultured in high-glucose DMEM supplemented with 100 U/ml penicillin, 100 mg/ml streptomycin, and 10% fetal bovine serum at 5% CO2 and 37°C.
Characterization of hSERCA2a Stable Cell Line
Expression of SERCA2a in the stable cell line was verified by Western blot analysis. Briefly, samples were collected 48 h after induction of recombinant SERCA2a expression with 1 μg/ml tetracycline. Samples were lysed in a buffer containing 1% Triton and protease inhibitors. Equal amounts of protein were run on a SDS-PAGE and blotted on nitrocellulose using the turbo transfer system (Bio-Rad). Membranes were incubated with primary antibody anti-SERCA2 (IID8, Santa Cruz Biotechnology) and developed using with the horseradish peroxidase (HRP)-conjugated secondary antibody. Western blots were imaged in a ChemiDoc (Bio-Rad) apparatus after incubation with the HRP chemiluminescent substrate (Millipore).
Cell Line Transfection and Experimental Protocol
Two different cell lines (HEK293 and Flp-In T-Rex-293) were used to study the SERCA2a function. HEK293 cells at 60–80% confluence were transiently cotransfected with plasmids containing the cDNA of RyR2, SERCA2a, and R-CEPIA1er. Experiments were conducted 48 h after transfection to obtain the optimal level of recombinant proteins expression. Flp-In T-Rex-293 stably expressing SERCA2a were cotransfected with plasmids containing the cDNA of RyR2 and R-CEPIA1er, using the same conditions for the HEK293 cells. Before each experiment, cells cultured on a laminin-coated glass coverslip were washed with a solution containing (in mM) 150 K-aspartate, 0.25 MgCl2, 0.1 EGTA, 10 HEPES, pH 7.2. Afterward, cells were permeabilized with saponin (0.005%). Experiments were conducted after washout of saponin with a saponin-free experimental solution (in mM): 100 K-aspartate, 15 KCl, 5 KH2PO4, 5 MgATP, 0.35 EGTA, 0.22 CaCl2, 0.75 MgCl2, and 10 HEPES; 2% dextran (MW: 40,000), pH 7.2. Free [Ca2+] and [Mg2+] of this solution were 200 nM and 1 mM, respectively.
Adenovirus Production
The R-CEPIA1er was subcloned into pShuttle-CMV by standard methods and verified by sequencing. Adenoviral vectors of all the cDNAs were made using the Stratagene AdEasy XL System. Amplified viruses were purified by CsCl gradient ultracentrifugation for 2 h at 22,000 rpm. The resultant adenovirus band was dialyzed and spectrophotometrically read at 260 nm to determine the titer value before addition of 10% glycerol. The final adenovirus density was 5 × 1010 particles/ml.
In Vivo Gene Delivery of R-CEPIA1er
All animal husbandry and surgical procedures were performed under animal protocols approved by the Institutional Animal Care and Use Committee and conformed to the Guide for the Care and Use of Laboratory Animals (NIH Publication 85-23, Revised 1985). C57BL/6J mice (Jackson) were maintained on a 12-h:12-h light-dark cycle and were given ad libitum food and water throughout the experiments. Eight- to ten-week-old mice, weighing 20–25 g, were used in the experiments. Mice were anesthetized with isoflurane at a dose of 1–3% with oxygen (0.5%). Artificial respiration was maintained with a rodent ventilator. The heart was exposed upon opening of the left pleural cavity by cutting the intercostal muscles and by positioning a retractor between the left third and fourth ribs. The R-CEPIA1er adenovirus (5 × 1010 particles/ml) was administered by direct injection in the left ventricular wall of the myocardium (6 spots, 8 μl/site) using a syringe fitted with a 29-gauge needle. After the injection, the chest was closed in layers. Mice were carefully monitored for postoperative recovery and given free access to the analgesic carprofen. Expression of the R-CEPIA1er transgene was optimal at 6 days postinjection.
Myocyte Isolation
Six days after the injection with the R-CEPIA1er adenovirus, the mice underwent terminal surgery for cardiomyocytes isolation. Mice were anesthetized using isoflurane (1%). Following thoracotomy, hearts were quickly excised, immersed in Ca2+ free buffer, mounted on a Langendorff apparatus, and retrogradely perfused with liberase H (Roche)-containing solution at 37°C according to the procedure previously described (25). The left ventricle was excised from the digested heart, placed in stop buffer containing BSA (1 mg/ml), cut into several pieces (average size 1 mm), and gently triturated into single cells. Myocytes were pelleted by gravity (∼0.1 ml) and resuspended in low-Ca Tyrode’s buffer (in mM): 140 NaCl, 4 KCl, 0.1 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES; pH 7.4. [Ca2+] was gradually adjusted to 1 mM. Isolated cardiomyocytes were stored at room temperature (20°C). All chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Confocal Microscopy
Expression of recombinant proteins and changes in the luminal ER [Ca2+] ([Ca]ER) were measured with laser scanning confocal microscopy (Radiance 2000 MP, Bio-Rad) equipped with a ×40 oil-immersion objective lens (N.A. = 1.3).
Expression of recombinant proteins.
To verify and quantify expression of SERCA2a, mCer was excited with the 457-nm line of the argon laser and the signal was collected at >485 nm. To verify and quantify expression of RyR2, the GFP was excited with the 488-nm line of the argon laser, and the signal was collected at >515 nm. Two-dimensional images were collected at a speed of 6 ms/line.
[Ca2+]ER measurements.
[Ca2+]ER was recorded as changes in fluorescence intensity of the genetically encoded ER-targeted Ca2+ sensor R-CEPIA1er. R-CEPIA1er was excited with a 514-nm line of the argon laser, and the signal was collected at >560 nm. Line scan images were collected at a speed of 10 ms/line. The R-CEPIA1er signal (F) was converted to [Ca2+]ER by the following formula: [Ca2+]SE = Kd × [(F − Fmin)/(Fmax − F)]. Fmax was recorded in 5 mM Ca2+ and 5 μM ionomycin, and Fmin was recorded after ER Ca2+ depletion with 5 mM caffeine. For HEK293 cell experiments, the Kd (Ca2+ dissociation constant) was 564 μM (38). For myocyte experiments, the Kd was 609 μM (9). SERCA-mediated Ca2+ uptake was calculated as the first derivative of [Ca2+]ER refilling (d[Ca2+]ER/dt) after RyR2 inhibition with ruthenium red (15 μM) and tetracaine (1 mM). It has been shown the inhibitors used in these concentrations completely inhibit RyR2 activity in bilayer experiments (27, 45). RyR2-independent Ca2+ leak was analyzed as the first derivative of [Ca2+]ER decline (d[Ca2+]ER/dt) after inhibition of RyR2 and SERCA. The rates of SERCA2a uptake and Ca2+ leak were plotted as a function of [Ca2+]ER to analyze maximum ER Ca2+ uptake rate and maximum ER Ca2+ load. In intact ventricular myocytes, [Ca2+]SR dynamics were measured in Tyrode’s solution (in mM): 140 NaCl, 4 KCl, 1.5 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, pH 7.4. Action potentials were induced by electrical field stimulation using a pair of platinum electrodes, which were connected to a Grass stimulator (Astro-Med). R-CEPIA1er expression did not affect the SR Ca2+ buffer capacity (9). All two-dimensional images and line scan measurements for [Ca2+]SR were analyzed with ImageJ software (National Institutes of Health).
Statistics
Data are presented as means ± SE of n measurements. Statistical comparisons between groups were performed with Student's t-test for paired or unpaired data sets. Differences were considered statistically significant at P < 0.05. Significance between multiple groups was determined by two-way ANOVA followed by a Newman-Keuls post hoc test. Statistical analysis and graphical representation of averaged data were carried out with OriginPro7.5 software (OriginLab).
RESULTS
Model System to Study SERCA Function
The Ca2+ sensor R-CEPIA1er was used to directly measure [Ca]ER in HEK293 cells expressing human SERCA2a tagged with mCer. SERCA2a expression in HEK293 cells was verified by Western blot analysis, revealing an approximately fivefold higher expression over endogenous SERCA (Fig. 1). Endogenous RyR and PLB were undetectable in HEK293 cells (Fig. 1). To study ER Ca2+ uptake over the entire physiological range of [Ca2+]ER, we cotransfected these cells with cardiac RyR2 tagged with GFP (Fig. 2A). The plasma membrane of these cells was permeabilized with saponin to control cytosolic [Ca2+] at 200 nM and [ATP] at 5 mM. Coexpression of SERCA2a and RyR2 resulted in periodic Ca2+ waves due to spontaneous activation of RyR2 (Fig. 2B, cell #1). Ca2+ waves did not occur in HEK cells with low expression of SERCA2a (Fig. 2B, cell #2), suggesting a threshold level of SERCA activity is required to bring [Ca2+]ER to a critical level that can trigger “cardiac-like” Ca2+ waves. After recording spontaneous Ca2+ release events, full depletion of ER Ca2+ was accomplished by applying 5 mM caffeine (Fig. 2B, Caf). The caffeine action was rapidly reversed by exchanging the caffeine-containing solution with a solution containing RyR2 inhibitors (15 μM ruthenium red and 1 mM tetracaine) (Fig. 2B, RR+Tetr). With this approach, we were able to reliably switch RyR2-mediated Ca2+ release from full activation to full inhibition within a few seconds. With RyR2 blocked, SERCA-mediated Ca2+ uptake was measured as the rate of [Ca2+]ER refilling. ER Ca2+ uptake was analyzed as the first derivative of [Ca2+]ER refilling during RyR2 inhibition. Finally, the RyR2-independent Ca2+ leak was measured for each cell after inhibition of SERCA by thapsigargin (Fig. 2B) while maintaining RyR2 blockade. The RyR2-independent Ca2+ leak was analyzed as the first derivative of [Ca2+]ER decline after inhibition of RyR2 and SERCA. Figure 2C shows the dependence of ER Ca2+ uptake (black) and Ca2+ leak (gray) on [Ca2+]ER. RyR2-independent Ca2+ leak was subtracted from Ca2+ uptake to determine maximum ER Ca2+ uptake rate (Ca2+ uptake against a minimal Ca2+ gradient) and maximum ER Ca2+ load that SERCA could generate (Fig. 2D). For each cell, SERCA2a expression was estimated from the mCer fluorescence for normalization of the effects of different drugs and SERCA mutations on ER Ca2+ uptake.
Fig. 1.
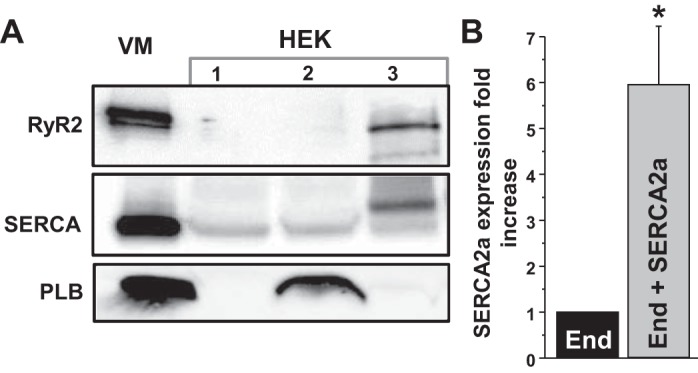
Expression levels of the Ca2+ release channel ryanodine receptor (RyR2), sarco-/endoplasmic reticulum Ca2+-ATPase (SERCA), and phospholamban (PLB) in mouse ventricular myocytes (VM) and HEK293 cells (HEK). A: RyR2, SERCA, and PLB expression levels in VM and HEK. HEK lane 1 is untransfected HEK293 cells; lane 2 is HEK293 cells transfected with PLB-modified cerulean fluorescent protein (mCer); lane 3 is HEK293 cells transfected with SERCA2a-mCer and RyR2-green fluorescent protein (GFP). B: SERCA expression in HEK293 cells before (endogenous; End) and after transfection with the SERCA2a-mCer plasmid (End+SERCA2a). These data were obtained by averaging results from 6 experiments. *P < 0.05 vs. End.
Fig. 2.

Model system to study SERCA function. A: images of HEK293 cells cotransfected with mCer-SERCA2a and GFP-RyR2. B: experimental protocol to measure SERCA-mediated endoplasmic reticulum (ER) Ca2+ uptake. Line-scan image of R-CEPIA1er during an initial 60-s recording is shown (marked by a gray box) and corresponding profile of changes in ER Ca2+ concentration ([Ca2+]ER; during entire experiment: 250 s). C: for each individual cell, ER Ca2+ uptake and RyR2-independent Ca2+ leak were analyzed as the first derivative (d[Ca2+]ER/dt) and plotted as a function of ER Ca2+ load ([Ca2+]ER). D: RyR2-independent Ca2+ leak was subtracted from ER Ca2+ uptake to determine maximum ER Ca2+ uptake rate and maximum ER Ca2+ load.
Effects of SERCA Inhibitors, [ATP]/[ADP] Ratio, and Oxidative Stress on ER Ca2+ Uptake
To quantify the contribution of the endogenous SERCA pump to ER Ca2+ uptake, we studied HEK293 cells that were not transfected with SERCA2a. Compared with SERCA2a, the maximum ER Ca2+ uptake rate by endogenous SERCA was about six times slower (Fig. 3, A and D), consistent with the lower expression level of endogenous SERCA (Fig. 1). Spontaneous Ca2+ waves were not detected in those cells, presumably because endogenous SERCA did not increase [Ca2+]ER to a level at which spontaneous Ca2+-induced Ca2+ release can occur. As expected, ER Ca2+ uptake by endogenous and SERCA2a was sensitive to thapsigargin and cyclopiazonic acid (CPA) (Fig. 3A and B). ER Ca2+ uptake in cells with endogenous and SERCA2a was completely blocked by 100 nM thapsigargin (n = 6) and 25 μM cyclopiazonic acid (n = 5). ER Ca2+ uptake was also sensitive to the cytosolic [ATP]/[ADP] ratio (Fig. 3B). Decreasing [ATP] from 5 mM to 2.5 mM and increasing [ADP] to 2.5 mM (ADP) slowed ER Ca2+ uptake by 69% (n = 8) (Fig. 3D). The changes in the [ATP]/[ADP] ratio also decreased the thermodynamic limit of SERCA (or maximum ER Ca2+ load) by 58% (n = 8; Fig. 3E). It is known that SERCA is sensitive to redox modifications (39, 42). We tested whether this approach can be used to study the effect of oxidative stress on SERCA function. SERCA2a oxidation with diamide (100 μM) decreased ER Ca2+ uptake rate by 38% (n = 12), without a significant effect on ER Ca2+ load (n = 12; Fig. 3, C and E). Low doses of diamide (≤50 μM), however, did not have a significant effect on SERCA function.
Fig. 3.

Effects of SERCA inhibitors, [ATP]/[ADP] ratio, and oxidative stress on ER Ca2+ uptake. A: representative traces of ER Ca2+ uptake mediated by endogenous and recombinant type 2a SERCA (SERCA2a) expressed in HEK293 cells. B: effect of the low [ATP]/[ADP] ratio (ADP) on ER Ca2+ uptake. Thapsigargin (TG; 100 nM) and cyclopiazonic acid (CPA; 20 μM) completely inhibited ER Ca2+ uptake by SERCA. C: effect of diamide (Diam; 100 μM) on ER Ca2+ uptake. Maximum [Ca2+]ER uptake rate (D) and maximum [Ca2+]ER load (E) mediated by endogenous SERCA (n = 6 cells), recombinant SERCA2a in control conditions (Ctrl; n = 20 cells), in the presence of the low [ATP]/[ADP] ratio (ADP; n = 8 cells), and Diam (n = 12 cells). *P < 0.05 vs. control. #P < 0.05 vs. control.
Effect of SERCA2a Mutations on ER Ca2+ Uptake
We also used this approach to quantify the function of two SERCA2a mutants (Fig. 4A). We have previously shown the functional significance of the SERCA cytosolic Nβ5–β6 loop, as mutation of three negatively charged amino acids to alanine (D426A/E429A/E435A “AAA-SERCA”) decreased ATPase and ER Ca2+ uptake (32) as a result of impaired headpiece closure kinetics (37). ER Ca2+ uptake by WT-SERCA2a and by the loss-of-function AAA-SERCA2a was only analyzed in cells that had a similar level of mCer fluorescence and, therefore, comparable SERCA2a expression levels. The mean mCer signal was 78 ± 4 arbitrary units (a.u.; n = 11) in cells expressing wild-type (WT) and 76 ± 7 a.u. (n = 10) in cells expressing the AAA mutant. We found that these cytosolic mutations decreased maximum ER Ca2+ uptake rate by 55% (n = 11) and maximum ER Ca2+ load by 15% (n = 10; Fig. 4, B and C), which is consistent with our previous observations (32). We also tested whether three negatively charged amino acids facing the ER lumen can regulate SERCA2a activity by a Ca2+-dependent mechanism. These amino acids were replaced with polar amino acid (E876Q/D877N/E882Q) to generate the QNQ-SERCA2a mutant. Analysis of ER Ca2+ uptake by WT and the QNQ mutant revealed that these mutations did not significantly affect maximal ER Ca2+ uptake and load (Fig. 4, B and C). The mCer signal was 86 ± 7 a.u. (n = 9) in WT cells and 79 ± 6 a.u. (n = 7) in cells expressing the QNQ mutant.
Fig. 4.

Effect of SERCA2a mutations on ER Ca2+ uptake. A: representative traces of ER Ca2+ uptake mediated by wild-type recombinant SERCA2a (WT) and 2 SERCA2a mutants (QNQ and AAA). Maximum [Ca]ER uptake rate (B) and maximum [Ca2+]ER load (C) mediated by WT (n = 20 cells), AAA (n = 13 cells), and QNQ (n = 10 cells) SERCA2a mutants. *P < 0.05 vs. WT.
Regulation of SERCA2a by PLB
We tested whether this approach could detect changes in SERCA function in response to physiological regulation. PLB is the most important regulator of SERCA2a function in ventricular myocytes (24). In these experiments, the effect of PLB on ER Ca2+ uptake was studied in the inducible human SERCA2a stable line (T-Rex-293 cells). Two days after SERCA2a gene induction with tetracycline, SERCA2a expression increased >10 times (Fig. 5A). PLB (tagged with yellow fluorescent protein) expression in these cells showed a similar pattern as CEPIA-1er (Fig. 5B), indicating that PLB is localized predominantly in the ER membrane. We found that PLB coexpression with SERCA2a decreased ER Ca2+ uptake and ER Ca2+ load (Fig. 5, D and E; Ctrl vs. PLB). Moreover, PLB decreased Ca2+ wave frequency by 43% (n = 7) and [Ca2+]ER recovery during Ca2+ waves by 35% (n = 15). The PKA catalytic subunit (10 U/ml) reversed the effect of PLB on SERCA2a function (Fig. 5, D and E; PLB vs. PLB+PKA). The PKA catalytic subunit, however, did not significantly affect ER Ca2+ uptake in HEK cells that didn’t express PLB (Fig. 5, D and E; Ctrl vs. +PKA).
Fig. 5.

Regulation of SERCA by phospholamban (PLB). A: SERCA2a expression in T-Rex-293 stable line cells after 24 and 48 h of SERCA2a gene induction with tetracycline. B: images of T-Rex-293 cell cotransfected with PLB-yellow fluorescent protein (YFP; left) and R-CEPIA1er (right). C: representative traces of ER Ca2+ uptake mediated by SERCA2a cotransfected with PLB in control conditions (PLB) and in the presence of the PKA catalytic subunit (PLB+PKA). Maximum [Ca]ER uptake rate (D) and maximum [Ca]ER load (E) mediated by SERCA2a in control conditions (Ctrl; n = 9 cells) and in the presence of PKA (+PKA; n = 6 cells), and by SERCA2a cotransfected with PLB (PLB; n = 11 cells) alone and in the presence of PKA (PLB+PKA; n = 5 cells). *P < 0.05 vs. Ctrl. #P < 0.05 vs. PLB.
SERCA2a Ca2+ Transport in Ventricular Myocytes
We assessed whether this approach can be used to study endogenous SERCA2a function in ventricular myocytes. We used in vivo gene delivery of the R-CEPIA1er cDNA with an adenovirus in mice hearts. Six days after adenovirus injection, myocytes were isolated from the left ventricle. Myocytes expressing R-CEPIA1er showed periodic peaks of fluorescence approximately every 1.8 μm, which corresponds to the sarcomere spacing of the junctional SR (Fig. 6A). First, we analyzed the effect of adrenergic receptor stimulation on SERCA2a function in intact ventricular myocytes (Fig. 6B). [Ca2+]SR was measured under control conditions and in the presence of isoproterenol (ISO; 0.1 μM). Myocytes were electrically stimulated at a constant frequency 0.5 Hz. Action potentials caused regular [Ca2+]SR depletion followed by a fast recovery of Ca2+ due to SERCA2a activity. ISO increased SR Ca2+ release amplitude (Fig. 6B), the rate of [Ca2+]SR recovery, and SR Ca2+ load (Fig. 6C). Adrenergic stimulation also increased the propensity of spontaneous Ca2+ waves. We next used a similar approach to study the effect of PKA activation on SR Ca2+ uptake in permeabilized ventricular myocytes. After SR Ca2+ depletion with caffeine, RyR2 inhibitors (RR+Tetr) were applied to measure SR Ca2+ uptake (Fig. 6D). Similar to experiments presented in Fig. 1, the same analysis was performed to calculate maximal SR Ca2+ uptake rate and maximal SR Ca2+ load generated by endogenous SERCA2a (Fig. 6E). We have shown previously that endogenous PKA can be activated by cAMP after membrane permeabilization (8). cAMP (10 μM) significantly increased maximal SR Ca2+ uptake and SR Ca2+ load (Fig. 6E). The effects of PKA activation on endogenous SERCA2a function were similar to those observed for recombinant SERCA2a (Fig. 5). Taken together, these results demonstrate that this new approach can be used as a sensitive tool to quantify SERCA function in expressing cell systems as well in cardiomyocytes.
Fig. 6.

SERCA2a properties in ventricular myocytes. A: transmitted light (left) and confocal (middle) images of left ventricular myocytes isolated from a mouse heart infected with the R-CEPIA1er adenovirus. Zoomed-out image of the R-CEPIA1er signal (right) is defined by the white dashed box in the middle image. Myocyte expressing R-CEPIA1er showed a periodic pattern of the SR network together with the nucleus envelope. B: changes in [Ca2+]SR during electrical filed stimulations (marked by triangles) in control conditions (Ctrl) and during isoproterenol (ISO) application. Ca2+ waves are marked by red asterisks. Caffeine (Caf; 10 mM) was applied to completely deplete [Ca2+]SR. C: effect of ISO on tau of [Ca2+]SR recovery and sarcoplasmic reticulum (SR) Ca2+ load (n = 13 myocytes, 4 animals). D: representative traces of SR Ca2+ uptake in left ventricular myocytes mediated by SERCA2a in control conditions (Ctrl) and in the presence of cAMP (cAMP). E: maximum [Ca2+]SR uptake rate and maximum [Ca]SR load mediated by SERCA2a in control conditions (Ctrl; n = 7 myocytes) and in the presence of cAMP (cAMP; n = 6 myocytes). *P < 0.05 vs. Ctrl.
DISCUSSION
Cardiac cytosolic Ca2+ dynamics are of great interest as they represent the central signals that orchestrate the cycle of contraction and relaxation. They have been intensively scrutinized using fluorescence microscopy and Ca2+-sensitive indicators that report the concentration of Ca2+ in the cytoplasm, yielding insight into the biophysical mechanisms that govern Ca2+ release from the SR during systole. These methods have also provided indirect assessment of the subsequent reuptake of Ca2+ into the SR during diastole. For example, SERCA transport activity can be inferred from the rate of decay of the cytosolic Ca2+ transient (4, 5, 41). SERCA activity could also be estimated from ATP consumption (29) or Ca2+ uptake by ER/SR vesicles (34). However, these semiquantitative measures suffer from several significant limitations, as addressed in the following paragraphs.
First, they do not control for changes in SR Ca2+ content, and how these changes might affect the transporter function. To address this issue, low-affinity Ca2+ indicators such as Fluo-5N have been used to directly measure [Ca2+]SR in myocytes (11, 46), but unfortunately this technique does not work in myocytes isolated from small rodents that are commonly used for transgenic studies. In the present study, we circumvented some of the drawbacks of previous approaches by using the genetically encoded ER Ca2+ sensor R-CEPIA1er to directly measure Ca2+ dynamics within the ER lumen in expressing cell systems as well as in freshly isolated cardiomyocytes. It has been previously established that R-CEPIA1er has an appropriate affinity and sufficient dynamic range to reliably track [Ca2+]ER in both non-muscle (38) and muscle cells (9). As an example of the versatility of R-CEPIA, we created a cardiomimetic Ca2+-handling system in non-muscle cells by coexpressing SERCA2a together with the cardiac Ca2+ release channel RyR2. This enabled fast, complete, and rapidly reversible depletion of ER Ca2+ load (with caffeine), so that the subsequent ER Ca2+ uptake could be measured over the entire physiological range of [Ca2+]ER, starting from fully empty to maximally loaded ER.
The second major limitation of the previous methods used to study SR Ca2+ uptake is that it is difficult to specifically separate Ca2+ transport by SERCA from Ca2+ fluxes mediated by other ion channels and pumps. One particularly challenging process to account for is the RyR2-independent Ca2+ leak. Due to the complex nature of the ER Ca2+ leak (12, 43), this pathway cannot be blocked completely or selectively. Instead of blocking it, we measured the RyR2-independent Ca2+ leak after RyR2 and SERCA inhibition (Fig. 2). We determined that the Ca2+ leak component was relatively small compared with ER Ca2+ uptake in HEK cells, suggesting that inositol 3-phosphate receptors and other Ca2+ leak channels are mainly inactive in the absence of appropriate agonists. Moreover, direct measurement of [Ca2+]ER separates the SERCA-dependent Ca2+ uptake from Ca2+ fluxes mediated by Ca2+ transporters on the surface membrane and other organelles, including the mitochondria.
In addition to overcoming the two limitations associated with previous methodologies outlined above, the advantage of the present approach enables access to two key biophysical parameters of SERCA function: maximum Ca2+ uptake rate and maximum Ca2+ load (Fig. 2D). These parameters reflect the kinetic and the thermodynamic efficacy of SERCA, respectively, and various interventions differentially modulated these properties. Lowering the cytosolic ATP/ADP ratio decreased both the maximum Ca2+ uptake rate and the maximum Ca2+ load (Fig. 3B). This can be explained if high [ADP] inhibits the SERCA kinetic efficacy (28), whereas low [ATP]/[ADP] decreases the thermodynamic limit, of the pump (23). SERCA2a oxidation (39, 42) had a stronger effect on ER Ca2+ uptake than on reduction of the ER Ca2+ load (Fig. 3C). Loss-of-function mutations of the SERCA2a cytoplasmic headpiece (32) also decreased the maximal Ca2+ uptake rate (Fig. 4), while several mutations of luminal residues were functionally benign.
We have previously designed a method to selectively measure [Ca2+]SR in intact ventricular myocytes using R-CEPIA1er (9). However, expression of the sensor requires >48 h of culturing, which significantly alters myocyte structure and function (for a review, see Ref. 26). To overcome this limitation, we used in vivo gene delivery of the R-CEPIA1er cDNA with an adenovirus in mice hearts. The same methodology to analyze SERCA2a function was used in freshly isolated myocytes (Fig. 6). The effects of PKA activation on endogenous SERCA2a were similar to those observed for recombinant SERCA2a (Fig. 5), suggesting that our heterologous expression system is a suitable model of Ca2+ pump regulation in cardiac myocytes. Moreover, using in vivo gene delivery of the R-CEPIA1er can open new perspectives to more directly characterize changes in SERCA2a function in different transgenic models of heart diseases.
However, the critical limitation of the in vivo gene delivery approach is that its success can be technically challenging, as it depends on the quality of animal survival surgery and myocyte isolation. In contrast to myocytes, HEK293 cells provided an ideal minimal cardiomimetic Ca2+-handling system for investigating SERCA2a-regulatory mechanisms. In HEK293 cells, the Ca2+ transients occur without eliciting contractions, which can cause motion artifacts in the myocyte experiments. Although myocyte contractility can be eliminated pharmacologically, blebbistatin is sensitive to the wavelengths used to excite fluorescence (16). and 2,3-butanedione 2-monoxime may alter Ca2+ handling (2). In contrast, the HEK cell model is readily and stably transfectable, it does not require generation of viral delivery vectors or transgenic animals, and it is without the confounding effects of endogenous PLB or other regulators of SERCA2a. This enabled us to observe PLB-dependent, PKA-reversible decreases in both the kinetic and thermodynamic efficacy of the pump (Fig. 5). We consider this well-defined model system to be ideal for testing the effects of disease-causing PLB mutations, or the effects of other regulatory micropeptides, some of which may specifically target SERCA catalytic efficiency (3).
In conclusion, the enhanced SERCA specificity of this new approach, together with differentiation of kinetic and thermodynamic parameters, may provide a more sensitive and discriminating assay for drugs, posttranslational modifications, and mutations that may alter human SERCA2a function.
GRANTS
This work was supported by the National Institutes of Health Grants HL-130231 (to A. V. Zima), HL-092321 (to S. L. Robia) and DK-101585 (I. Y. Kuo).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
E.B., R.N., S.B., D.K., Q.C., J.L.M., I.Y.K., S.R., and A.V.Z. conceived and designed research; E.B., R.N., S.B., D.K., Q.C., J.L.M., and A.V.Z. performed experiments; E.B., R.N., S.B., D.K., Q.C., J.L.M., I.Y.K., S.R., and A.V.Z. analyzed data; E.B., R.N., S.B., D.K., Q.C., J.L.M., I.Y.K., S.R., and A.V.Z. interpreted results of experiments; E.B., R.N., and A.V.Z. prepared figures; E.B., R.N., I.Y.K., S.R., and A.V.Z. drafted manuscript; E.B., R.N., S.B., D.K., Q.C., J.L.M., I.Y.K., S.R., and A.V.Z. edited and revised manuscript; E.B., R.N., S.B., D.K., Q.C., J.L.M., I.Y.K., S.R., and A.V.Z. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. David Thomas (Univ. of Minnesota) for providing the vector encoding the human hSERCA2a, Dr. Christopher George (Univ. of Cardiff) for providing the vector encoding the human RyR2, and Dr. Ino for donating the R-CEPIA1er vector.
REFERENCES
- 1.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res 97: 1314–1322, 2005. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 2.Allen TJ, Mikala G, Wu X, Dolphin AC. Effects of 2,3-butanedione monoxime (BDM) on calcium channels expressed in Xenopus oocytes. J Physiol 508: 1–14, 1998. doi: 10.1111/j.1469-7793.1998.001br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson DM, Makarewich CA, Anderson KM, Shelton JM, Bezprozvannaya S, Bassel-Duby R, Olson EN. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Sci Signal 9: ra119, 2016. doi: 10.1126/scisignal.aaj1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bassani JW, Bassani RA, Bers DM. Twitch-dependent SR Ca accumulation and release in rabbit ventricular myocytes. Am J Physiol Cell Physiol 265: C533–C540, 1993. doi: 10.1152/ajpcell.1993.265.2.C533. [DOI] [PubMed] [Google Scholar]
- 5.Bassani JW, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J Physiol 476: 279–293, 1994. doi: 10.1113/jphysiol.1994.sp020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bassani JW, Yuan W, Bers DM. Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am J Physiol Cell Physiol 268: C1313–C1319, 1995. doi: 10.1152/ajpcell.1995.268.5.C1313. [DOI] [PubMed] [Google Scholar]
- 7.Bers DM. Cardiac excitation-contraction coupling. Nature 415: 198–205, 2002. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 8.Bovo E, Huke S, Blatter LA, Zima AV. The effect of PKA-mediated phosphorylation of ryanodine receptor on SR Ca2+ leak in ventricular myocytes. J Mol Cell Cardiol 104: 9–16, 2017. doi: 10.1016/j.yjmcc.2017.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bovo E, Martin JL, Tyryfter J, de Tombe PP, Zima AV. R-CEPIA1er as a new tool to directly measure sarcoplasmic reticulum [Ca] in ventricular myocytes. Am J Physiol Heart Circ Physiol 311: H268–H275, 2016. doi: 10.1152/ajpheart.00175.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bovo E, Mazurek SR, Zima AV. The role of RyR2 oxidation in the blunted frequency-dependent facilitation of Ca2+ transient amplitude in rabbit failing myocytes. Pflügers Arch 470: 959–968, 2018. doi: 10.1007/s00424-018-2122-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brochet DX, Yang D, Di Maio A, Lederer WJ, Franzini-Armstrong C, Cheng H. Ca2+ blinks: rapid nanoscopic store calcium signaling. Proc Natl Acad Sci USA 102: 3099–3104, 2005. doi: 10.1073/pnas.0500059102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Camello C, Lomax R, Petersen OH, Tepikin AV. Calcium leak from intracellular stores—the enigma of calcium signalling. Cell Calcium 32: 355–361, 2002. doi: 10.1016/S0143416002001926. [DOI] [PubMed] [Google Scholar]
- 13.Currie S, Smith GL. Enhanced phosphorylation of phospholamban and downregulation of sarco/endoplasmic reticulum Ca2+ ATPase type 2 (SERCA 2) in cardiac sarcoplasmic reticulum from rabbits with heart failure. Cardiovasc Res 41: 135–146, 1999. doi: 10.1016/S0008-6363(98)00241-7. [DOI] [PubMed] [Google Scholar]
- 14.Díaz ME, O’Neill SC, Eisner DA. Sarcoplasmic reticulum calcium content fluctuation is the key to cardiac alternans. Circ Res 94: 650–656, 2004. doi: 10.1161/01.RES.0000119923.64774.72. [DOI] [PubMed] [Google Scholar]
- 15.El-Armouche A, Eschenhagen T. Beta-adrenergic stimulation and myocardial function in the failing heart. Heart Fail Rev 14: 225–241, 2009. doi: 10.1007/s10741-008-9132-8. [DOI] [PubMed] [Google Scholar]
- 16.Farman GP, Tachampa K, Mateja R, Cazorla O, Lacampagne A, de Tombe PP. Blebbistatin: use as inhibitor of muscle contraction. Pflügers Arch 455: 995–1005, 2008. doi: 10.1007/s00424-007-0375-3. [DOI] [PubMed] [Google Scholar]
- 17.Greenberg B, Yaroshinsky A, Zsebo KM, Butler J, Felker GM, Voors AA, Rudy JJ, Wagner K, Hajjar RJ. Design of a phase 2b trial of intracoronary administration of AAV1/SERCA2a in patients with advanced heart failure: the CUPID 2 trial (calcium up-regulation by percutaneous administration of gene therapy in cardiac disease phase 2b). JACC Heart Fail 2: 84–92, 2014. doi: 10.1016/j.jchf.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 18.Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan GC, Tsiapras D, Hahn HS, Adamopoulos S, Liggett SB, Dorn GW 2nd, MacLennan DH, Kremastinos DT, Kranias EG. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest 111: 869–876, 2003. doi: 10.1172/JCI17892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hasenfuss G, Reinecke H, Studer R, Meyer M, Pieske B, Holtz J, Holubarsch C, Posival H, Just H, Drexler H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca2+-ATPase in failing and nonfailing human myocardium. Circ Res 75: 434–442, 1994. doi: 10.1161/01.RES.75.3.434. [DOI] [PubMed] [Google Scholar]
- 20.Hicks MJ, Shigekawa M, Katz AM. Mechanism by which cyclic adenosine 3′:5′-monophosphate-dependent protein kinase stimulates calcium transport in cardiac sarcoplasmic reticulum. Circ Res 44: 384–391, 1979. doi: 10.1161/01.RES.44.3.384. [DOI] [PubMed] [Google Scholar]
- 21.Kabbara AA, Allen DG. The use of the indicator fluo-5N to measure sarcoplasmic reticulum calcium in single muscle fibres of the cane toad. J Physiol 534: 87–97, 2001. doi: 10.1111/j.1469-7793.2001.00087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E, Park WJ, Hajjar RJ. SUMO1-dependent modulation of SERCA2a in heart failure. Nature 477: 601–605, 2011. doi: 10.1038/nature10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kodama T. Thermodynamic analysis of muscle ATPase mechanisms. Physiol Rev 65: 467–551, 1985. doi: 10.1152/physrev.1985.65.2.467. [DOI] [PubMed] [Google Scholar]
- 24.Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res 110: 1646–1660, 2012. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuo IY, Kwaczala AT, Nguyen L, Russell KS, Campbell SG, Ehrlich BE. Decreased polycystin 2 expression alters calcium-contraction coupling and changes β-adrenergic signaling pathways. Proc Natl Acad Sci USA 111: 16604–16609, 2014. doi: 10.1073/pnas.1415933111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Louch WE, Sheehan KA, Wolska BM. Methods in cardiomyocyte isolation, culture, and gene transfer. J Mol Cell Cardiol 51: 288–298, 2011. doi: 10.1016/j.yjmcc.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lukyanenko V, Györke I, Subramanian S, Smirnov A, Wiesner TF, Györke S. Inhibition of Ca2+ sparks by ruthenium red in permeabilized rat ventricular myocytes. Biophys J 79: 1273–1284, 2000. doi: 10.1016/S0006-3495(00)76381-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Macdonald WA, Stephenson DG. Effects of ADP on sarcoplasmic reticulum function in mechanically skinned skeletal muscle fibres of the rat. J Physiol 532: 499–508, 2001. doi: 10.1111/j.1469-7793.2001.0499f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Madden TD, Quinn PJ, Chapman D. Cholesterol modulates activity of calcium-dependent ATPase of the sarcoplasmic reticulum. Nature 279: 538–541, 1979. doi: 10.1038/279538a0. [DOI] [PubMed] [Google Scholar]
- 30.O’Rourke B, Kass DA, Tomaselli GF, Kääb S, Tunin R, Marbán E. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure. I: Experimental studies. Circ Res 84: 562–570, 1999. doi: 10.1161/01.RES.84.5.562. [DOI] [PubMed] [Google Scholar]
- 31.Periasamy M, Kalyanasundaram A. SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve 35: 430–442, 2007. doi: 10.1002/mus.20745. [DOI] [PubMed] [Google Scholar]
- 32.Raguimova ON, Smolin N, Bovo E, Bhayani S, Autry JM, Zima AV, Robia SL. Redistribution of SERCA calcium pump conformers during intracellular calcium signaling. J Biol Chem 293: 10843–10856, 2018. doi: 10.1074/jbc.RA118.002472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmidt U, Hajjar RJ, Kim CS, Lebeche D, Doye AA, Gwathmey JK. Human heart failure: cAMP stimulation of SR Ca2+-ATPase activity and phosphorylation level of phospholamban. Am J Physiol 277: H474–H480, 1999. doi: 10.1152/ajpheart.1999.277.2.H474. [DOI] [PubMed] [Google Scholar]
- 34.Shannon TR, Bers DM. Assessment of intra-SR free [Ca] and buffering in rat heart. Biophys J 73: 1524–1531, 1997. doi: 10.1016/S0006-3495(97)78184-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shannon TR, Guo T, Bers DM. Ca2+ scraps: local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ Res 93: 40–45, 2003. doi: 10.1161/01.RES.0000079967.11815.19. [DOI] [PubMed] [Google Scholar]
- 36.Simmerman HK, Collins JH, Theibert JL, Wegener AD, Jones LR. Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. J Biol Chem 261: 13333–13341, 1986. [PubMed] [Google Scholar]
- 37.Smolin N, Robia SL. A structural mechanism for calcium transporter headpiece closure. J Phys Chem B 119: 1407–1415, 2015. doi: 10.1021/jp511433v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suzuki J, Kanemaru K, Ishii K, Ohkura M, Okubo Y, Iino M. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat Commun 5: 4153, 2014. doi: 10.1038/ncomms5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suzuki YJ, Ford GD. Redox regulation of signal transduction in cardiac and smooth muscle. J Mol Cell Cardiol 31: 345–353, 1999. doi: 10.1006/jmcc.1998.0872. [DOI] [PubMed] [Google Scholar]
- 40.Trafford AW, Díaz ME, Eisner DA. Measurement of sarcoplasmic reticulum Ca content and sarcolemmal fluxes during the transient stimulation of the systolic Ca transient produced by caffeine. Ann N Y Acad Sci 853: 368–371, 1998. doi: 10.1111/j.1749-6632.1998.tb08302.x. [DOI] [PubMed] [Google Scholar]
- 41.Trafford AW, Díaz ME, Sibbring GC, Eisner DA. Modulation of CICR has no maintained effect on systolic Ca2+: simultaneous measurements of sarcoplasmic reticulum and sarcolemmal Ca2+ fluxes in rat ventricular myocytes. J Physiol 522: 259–270, 2000. doi: 10.1111/j.1469-7793.2000.t01-2-00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res 71: 310–321, 2006. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 43.Zima AV, Bovo E, Bers DM, Blatter LA. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J Physiol 588: 4743–4757, 2010. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zima AV, Bovo E, Mazurek SR, Rochira JA, Li W, Terentyev D. Ca handling during excitation-contraction coupling in heart failure. Pflugers Arch 466: 1129–1137, 2014. doi: 10.1007/s00424-014-1469-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zima AV, Picht E, Bers DM, Blatter LA. Partial inhibition of sarcoplasmic reticulum Ca release evokes long-lasting Ca release events in ventricular myocytes: role of luminal Ca in termination of Ca release. Biophys J 94: 1867–1879, 2008. doi: 10.1529/biophysj.107.114694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zima AV, Picht E, Bers DM, Blatter LA. Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ Res 103: e105–e115, 2008. doi: 10.1161/CIRCRESAHA.107.183236. [DOI] [PMC free article] [PubMed] [Google Scholar]