

Abstract
The contributions of T lymphocytes to the pathogenesis of salt-sensitive hypertension has been well established. Under hypertensive stimuli, naive T cells develop into different subsets, including Th1, Th2, Th17, Treg, and cytotoxic CD8+ T cells, depending on the surrounding microenviroment in organs. Distinct subsets of T cells may play totally different roles in tissue damage and hypertension. The underlying mechanisms by which hypertensive stimuli activate naive T cells involve many events and different organs, such as neoantigen presentation by dendritic cells, high salt concentration, and the milieu of oxidative stress in the kidney and vasculature. Infiltrating and activated T subsets in injured organs, in turn, exert considerable impacts on tissue dysfunction, including sodium retention in the kidney, vascular stiffness, and remodeling in the vasculature. Therefore, a thorough knowledge of T-cell actions in hypertension may provide novel insights into the development of new therapeutic strategies for patients with hypertension.
Keywords: end-organ damage, hypertension, salt, T cell
INTRODUCTION
T-lymphocyte subsets, as important compartments of the adaptive immune system, play pivotal roles in eradicating invading viruses, bacteria, and other pathogens by infiltrating into injured tissues and producing cytokines and/or cytotoxic factors to combat specific pathogens. Over the years, accumulating evidence indicates that T-lymphocyte subsets significantly participate in blood pressure regulation, ranging from prohypertensive actions in the kidney that permit sodium homeostasis, endothelial dysfunction in the vasculature, sympathetic outflow from the nervous system, and newly described actions in the skin to regulate interstitial sodium storage. Salt can directly affect the differentiation of T cells. This scenario illustrates reciprocal regulatory mechanisms by which sodium and T-cell activation coordinately impact blood pressure. However, the molecular mechanisms and events for T-cell activation in the setting of hypertension are not fully understood. This review details the complex interactions between T-cell activation, salt and end-organ damage in the pathogenesis of essential hypertension, which may facilitate development of newer immunomodulatory strategies for hypertension.
T-CELL SUBSETS
CD3+ T lymphocytes can be mainly distinguished into CD4+ or CD8+ single-positive cells. There are also some T cells exhibiting CD4−CD8− double negativity. CD4+ T and CD8+ T cells migrate from the thymus and then acquire immunocompetence. Most naïve T lymphocytes need to obtain two signals from antigen-presenting cells (APCs) for activation: one is the T-cell receptor (TCR) activation signal through an antigen presented by APCs with their major histocompatibility (MHC) molecules. The second signal is costimulatory, typically mediated by the engagement-specialized molecules on APCs and the costimulatory molecule family on T cells. The CD4 and CD8 coreceptors on T cells bind MHC class II and I, respectively. When initially activated by a dendritic cell (DC), naive CD4+ T-helper cells undergo differentiation into Th1, Th2, Th17 cells, T regulatory (Treg) cells, Th9, or T-follicular helper (Tfh) cells. CD8+ T cells develop the phenotypes of cytotoxic CD8+ T cells. Each subset of differentiated T cells secretes its own panel of cytokines and exerts its own set of specialized functions (Fig. 1).
Fig. 1.

Distinct T-lymphocyte subsets are crucial in hypertension. Th1, Th17, γδ T, and T regulatory (Treg) subpopulations promote or suppress the genesis of hypertension through proinflammatory cytokines, including TNF-α, IFN-γ, and IL-17, or anti-inflammatory cytokines such as IL-10, respectively. The relationships between Th2, Th9, or Tfh cells and hypertension are not clear and need to be further investigated. Some differentiated T-effector cells convert to memory T cells, and memory T cells will become T-effector cells again in antigen-specific recall responses.
Of note, there is another subset of T cells, termed unconventional T cells, that do not recognize peptide antigens and exhibit a more limited TCR diversity. Unconventional T cells or non-MHC restricted T cells include specific subsets of αβ TCR T cells, and γδ T cells. Unconventional αβ TCR T cells are not known to modulate blood pressure, whereas the role of γδ T cells in hypertension is discussed further below. Meanwhile, memory T cells are also important to the development of hypertension. Itani et al. (20) found that an initial hypertensive challenge led to the development of memory T cells that predispose the host to the progression of hypertension in response to a second modest hypertensive challenge that would not otherwise result in hypertension (Fig. 1).
CD4+ T-HELPER LYMPHOCYTE
Each subset of differentiated CD4+ T-helper cells, including Th1, Th2, Th17, Treg, Th9, or Tfh, produces its own panel of cytokines and mediates distinct functions. Mice lacking CD4+ T cells were not protected from hypertension, belying the complexity of individual CD4+ T-cell subset functions.
Th1 LYMPHOCYTE
Th1 cells express the hallmark cytokines IFN-γ, TNF-α, and IL-2 through a specific transcriptional factor, T-bet. T-bet knockout (KO) mice developed a similar hypertensive response compared with wild-type (WT) controls but had attenuated kidney damage (63). The effects of IFN-γ on blood pressure elevation have been mixed in ANG II-induced hypertension (17). However, mice lacking TNF-α had a blunted hypertensive response and were protected from target organ damage. We further established that renal parenchymal cell-derived but not Th1-derived TNF-α was a major contributor to the ANG II-induced hypertensive response (63). These results suggest that Th1 cells may not significantly impact blood pressure but are responsible for end-organ damage.
Th2 LYMPHOCYTE
In contrast to cell-mediated immunity induced by Th1, Th2 cells secrete a variety of cytokines including IL-4, IL-5, IL-9, and IL-13, to promote humoral immunity. They are responsible for B-cell activation and subsequent antibody production. Additionally, Th2 cells predominate in the defense against extracellular parasites, including helminths. However, at present, little is known about the role of Th2 cells in hypertension, and further studies about Th2-cell functions in hypertension are needed (55).
Th17 LYMPHOCYTE
Similar to Th1 cells, Th17 cells produce a variety of proinflammatory cytokines, including IL-17, IL-21, and IL-22, that play important roles in defense against exogenous pathogens and in autoimmune disorders. Among the cytokines secreted by Th17 cells, IL-17 is one of the most extensively and intensively investigated in hypertension models. Th17 cells and IL-17 have multiple functions, including regulating sodium homeostasis in kidneys and orchestrating dysfunction of endothelial cells, culminating in exaggerated blood pressure elevation (30, 38).
Treg LYMPHOCYTE
Tregs, especially naturally arising CD25+CD4+ T cells, provide a major brake for overactive immune responses. Approximately 10% of CD4+ T cells are Treg cells. They function to inhibit innate and adaptive immune responses via production of anti-inflammatory cytokines IL-10, IL-35, and TGF-β1 (52). Treg cells are thought to modulate hypertension through IL-10, which is recognized as a key cytokine to normalize endothelial function and blood pressure. Repeated transfer of T regulatory cells that produced IL-10 could attenuate ANG II-induced hypertension (4). Recently, Chen et al. (7) revealed an intriguing interaction between the complement system and T regulatory cells and their roles in the pathogenesis of hypertension. The activation of C3aR and C5aR on Tregs suppressed Treg’s functions and then promoted ANG II-induced hypertension. Meanwhile, C3a and C5a were increased in hypertensive patients, and Tregs from these patients had increased C5aR. Therefore, blocking the complement system to preserve Treg’s functions may be protective and serve as a therapeutic strategy for hypertension.
Th9 AND Tfh CELLS
Th9 and Tfh cells are important specialized T-helper cell subpopulations. Th9 cells, characterized by their capacity to secrete IL-9 and IL-21, are found to play a key role in parasite infections and allergic diseases as well as anticancer processes (51). Tfh cells, marked by expression of CXCR5, support B-cell differentiation in germinal centers. They are critical players in chronic autoimmune disease and provide tumor surveillance (9). Studies to elucidate the roles of these T-cell subsets in hypertension will therefore clarify interactions between T and B cells in promoting blood pressure elevation.
CYTOTOXIC CD8+ T LYMPHOCYTE
Naive CD8+ T cells can be activated and subsequently mature into cytotoxic T cells that express IFN-γ, TNF-α, granzyme B, and perforin following a MHC-I-restricted antigen stimulation. These cytokines secreted by cytotoxic CD8+ T cells contribute to cell apoptosis and inflammation. CD8+T cells play an important role in the genesis of hypertension. They can release cytokines but also engage in direct cell-to-cell communication to impact the development of hypertension. CD8+ T cells are a primary source of IFN-γ and accumulate in the hypertension-challenged kidney. Mice with CD8+ T-cell deficiency had a decreased hypertensive response to ANG II, while mice lacking CD4+ cells did not get similar protection from hypertension. Additionally, infiltration of cytotoxic CD8+ T cells into the renal interstitium may also lead to capillary rarefaction. CD8+ T cell-mediated vascular rarefaction in turn promoted sodium retention and volume expansion. Thus the cytotoxic CD8+ T-cell functions as a key T-cell subset that drives blood pressure elevation (50). Moreover, Sun et al. (48) provided a new molecular mechanism related to CD8+ T-cell activation in hypertension. The activation of the mineralocorticoid receptor on CD8+ T cells via nuclear factor of activated T cell 1 (NFAT1) promoted CD8 T-cell activation, leading to the generation of prohypertensive cytokines (48). Finally, these T cells can directly contact the distal convoluted tubule (DCT) in the kidneys of DOCA-salt mice to propagate salt-sensitive hypertension. In an in vitro coculture system, CD8+ T cells interacted with mDCTs to upregulate mDCT expression of the Na-Cl cotransporter (NCC) via reactive oxygen species (ROS)-induced activation of Src kinase, activation of the Cl− channel, and upregulation of the K+ channel (27). Thus the CD8+ T cell is an important contributor to the pathogenesis of hypertension.
γδ T LYMPHOCYTE
γδ T cells, as a subtype of unconventional T cells, comprise ~0.5–10% of circulating lymphocytes. They mostly exhibit CD4 and CD8 double negativity and have a γδ TCR. γδ T cells are not a homogeneous subset of T cells with a single function. They don’t recognize specific antigens but can be activated by non-protein molecules without the help of APCs. They localize in non-lymphoid tissues and can respond faster via cytokine secretion and proliferation than MHC-restricted T cells following activation. Some γδ T cells, like Th17 cells, can release IL-17A following stimulation by the proinflammatory cytokines IL-1 and IL-23. Other γδ T-cell populations expressing CD27 produce IFN-γ (42). The absence of γδ T cells protected against ANG II infusion-induced blood pressure increase, vascular dysfunction, and remodeling (5). Thus proinflammatory γδ T cells significantly participate in the induction of hypertension.
MEMORY T LYMPHOCYTE
Memory T cells, derived from some activated naive T cells, have a characteristic of the capacity to engage previously identified antigens. They also respond more rapidly to a previously encountered antigen than naive T cells. The memory T-cell population is always differentiated into three subsets, central memory T (TCM) cells, effector memory T (TEM) cells, and resident memory T (TRM) cells, that can be distinguished by their differential markers and migratory patterns. The T memory populations are some antigen-specific T cells persistently expressing CD44 (CD45RO in humans) in mice in contrast to naive T cells expressing CD62L (CD45RA in humans) in mice and localizing to lymphoid tissues. TEM and TCM subsets are identified by CD62L and CCR7 homing receptors that allow entry of memory T cells into lymphoid tissues. TCM subsets are CD62L and CCR7 double positive and are found in lymphoid organs, whereas TEM subsets are CD62L and CCR7 double negative and are able to regress from the lymphoid organs into the circulation and injured tissues after chemokines are released during the inflammatory response. TEM cells can release inflammatory cytokines including IL-17A and IFNγ in autoimmune disorders. TRM subsets are CD62 and CCR7 double negative like TEM but CD69 and CD109 double positive and permanently localize to peripheral tissues.
As mentioned above, Itani et al. (20) found that TEM cells formed during an initial hypertensive challenge sensitized the host to a second mild hypertensive stimulus, leading to and elevation in blood pressure and kidney injury that did not occur without these cells. The interaction of CD27 on T cells with CD70 on activated APCs is necessary for TEM cell formation. Additionally, mice with CD70 deficiency did not experience TEM-cell accumulation in the target organ and manifested limited blood pressure elevation and renal damage to either high salt or the second ANG II challenge (20). These results reveal that CD70 as a costimulatory molecule plays a key role in TEM-cell formation and activation and the development of hypertension in response to reexposure to a mild hypertensive challenge. The actions of other T memory subsets on the pathogenesis of hypertension remain to be investigated.
MECHANISMS OF T-CELL ACTIVATION IN HYPERTENSION
The important role of T lymphocytes in the pathogenesis of experimental hypertension has been well established; however, the mechanisms by which the T cell is activated following hypertensive challenge are complex. Recently, several novel concepts have been introduced to explain how various hypertensive stimuli contribute to T-cell activation, including the production of neoantigens, salt, and the sympathetic nervous system (Fig. 2).
Fig. 2.

Proposed mechanisms by which hypertensive stimuli activate T lymphocytes. Hypertensive stimuli, such as salt or ANG II, promote T-cell activation through various mechanisms. Oxidant stress induced by hypertensive stimuli causes the generation of neoantigens, such as isoketal adducts in dendritic cells. T cells recognize modified autoantigens presented by dendritic cells and then mature and differentiate into activated T cells, producing a variety of cytokines, including IL-17, TNF-α, and IFN-γ, eventually leading to the development of hypertension. Additionally, hypertensive stimuli cause vascular endothelial cell and kidney damage, and augmented sympathetic outflow, which can further activate T cells. ROS, reactive oxygen species; DAMPs, damage-associated molecular patterns; PIGF, placental growth factor.
DC-MEDIATED T-CELL ACTIVATION
The full activation of T cells, as a part of the adaptive immune response, requires an antigen presented by MHC, and costimulatory signal provided by B7 ligands, CD80 or CD86, on APC. T-cell activation may be provoked via the loss of self-tolerance by mechanisms involving both humoral and cell-mediated immunity during essential hypertension, implying hypertension may be an autoimmune disorder.
DCs, recognized as unique APC, provoke a potent acquired immune response. They are specialized for the capture, processing, and presentation of antigens with MHC molecules to T cells. Following capture of antigen, DCs experience a process of “maturation” by upregulation of MHC molecules and induction of costimulatory molecules CD80 (B7-1) and CD86 (B7-2) which bind to CD28 on T cells. Concomitant signaling by the T-cell antigen receptor and the costimulatory receptor are necessary for activation and differentiation of naive T cells. Meanwhile, they migrate from injured tissues into lymph nodes where naive T cells are located and eventually activate naive T cells (47). In ANG II-and DOCA-salt-induced hypertension, Vinh et al. (53) found that blockade of costimulatory signaling with cytotoxic T-lymphocyte antigen-Ig (CTLA4-Ig) ameliorated hypertension, suppressing the perivascular infiltration of T cells and their production of cytokines. Ablation of B7 ligands in mice was similarly resistant to hypertension, suggesting that activation of T cells through costimulation was crucial in hypertension.
Additionally, investigators are committed to finding potential neoantigens that are produced to activate T cells in the pathogenesis of hypertension. A study conducted by the Rodriguez-Iturbe group (43) demonstrated that an oxidant stress-induced molecule, heat shock protein 70 (HSP70), was necessary for T-cell activation, regulation of IL-10 and IL-6 generation, and progression of salt-sensitive hypertension in vivo. In vitro studies indicate that HSP70 may act as a “signal 3” to stimulate T-cell proliferation rather than acting as a true neoantigen (12, 13). Soon afterward, a study published by the Harrison group demonstrated that hypertensive stimuli increased ROS production, formation of isoketals, and protein isoketal adducts in DCs. These isoketal-modified proteins functioned as neoantigens, provoking an autoimmune-like reaction driving T-cell proliferation and production of IFN-γ and IL-17A, leading to hypertension. Importantly, adoptive transfer of activated DCs obtained from mice treated with ANG II to recipient WT mice significantly induced activation, survival, proliferation, and cytokine production of T cells and subsequent hypertension. Moreover, the isoketal scavenger 2-hydroxybenzylamine (2-HOBA) retarded these events and hypertension. They also found that isoketal-modified proteins were markedly elevated in circulating monocytes and DCs from humans with hypertension (22). Collectively, in response to various hypertensive stimuli, T cells recognize modified autoantigens presented by DCs and then mature and differentiate into activated T cells, producing a variety of cytokines, such as IL-17, culminating in blood pressure elevation.
SODIUM
Sodium is suggested to activate T cells through direct or indirect mechanisms. Salt-sensitive hypertension is characterized by sodium retention. It has become evident that the concentration of sodium can be 40 mmol/l higher in the extravascular interstitium than that in blood plasma in hypertensive animals and humans (25, 29). T cells could be activated by such concentration of sodium to produce IL-17A via activation of the salt-sensing kinase serum and glucocorticoid-regulated kinase 1 (SGK1) (23, 56, 58). Norlander et al. (39) showed that SGK1 signaling in T cells exaggerated hypertension and promoted end-organ damage, whereas deletion of T-cell SGK1 retarded these events in response to either ANG II or DOCA-salt, implicating the contribution of SGK1 in T cells to hypertension. Meanwhile, the polarization of proinflammatory T cells that was augmented by mucosal sodium chloride further promoted sodium retention in the kidney, exacerbating hypertension in a reciprocal loop (38). Of note, high sodium concentrations not only directly activate T cells but also promote T-cell activation indirectly by activating DCs. Recently, Barbaro et al. (3) described that DCs activated by excess sodium produce increased IL-1β and promoted T cell production of cytokines IL-17A and IFN-γ. Thus salt stimulates Th17-cell formation not only directly but also indirectly through effects on APCs (Fig. 3).
Fig. 3.
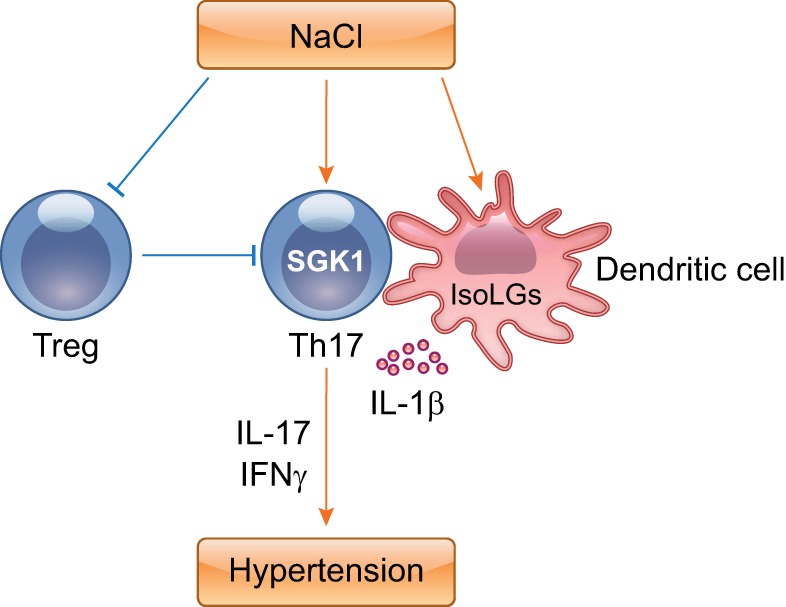
High salt concentrations modulate T-cell differentiation. Increased sodium promotes the development of IL-17-producing CD4+ T cells in a DC-dependent or -independent manner. Moreover, high salt impairs immunosuppressive functions of Tregs. These events coordinately underpin hypertension.
Tregs are known to suppress adaptive immunity and have been shown to alleviate hypertension and consequent target organ damage. The polarization of Tregs was also regulated by tonicity (4, 21). High concentrations of salt impaired immunosuppressive functions of Tregs (18). Thus chronically high levels of salt consumption as provided by a high-salt-diet (HSD) may accelerate hypertension by not only directly expanding intravascular volume but also shifting the T-cell repertoire toward the Th17 phenotype and away from the Treg lineage (Fig. 3).
THE VASCULATURE
In the setting of an initial hypertensive stimulus, the vascular wall is stretched by elevated blood pressure. Endothelial cells are thus exposed to excessive mechanical load, leading to the formation of ROS, chemokines, and proinflammatory mediators, such as IL-6 and IL-8, to direct immune cells into the vascular wall. Stretched endothelial cells can release cytokines that drive human monocyte differentiation and activation via STAT3, IL-6, and hydrogen peroxide, which in turn stimulate prohypertensive T-cell activation and proliferation. Through this cascade, endothelial cells activated by mechanical stretch can promote prohypertensive T-cell activation (28). Along with progression of hypertension, monocytes, DCs, and T cells can be activated and further augment damage to the vessel wall. Evans et al. (16) performed an elegant study to demonstrate that blood pressure elevation contributed to the initiation of renal T-cell infiltration into the kidney during the development of salt-sensitive hypertension by an electronic servo-control technique. This effect not only promoted infiltration of T cells but of B cells and monocytes/macrophages during the hypertensive challenge, with consequent renal injury and increased protein casts. Renal infiltration of T cells was not reversed even by the return of renal perfusion pressure to control levels (16). Together, increased blood pressure following hypertensive stimuli not only contributes to endothelial cell damage that promotes T-cell infiltration and activation in vascular walls, but also directly promotes T-cell accumulation in the kidney.
THE NERVOUS SYSTEM
The role of the nervous system in regulating blood pressure has been well recognized. Recent studies have found that sympathetic nerves contributed to the onset and progression of hypertension, possibly by activating T lymphocytes (40, 49, 60). Sympathetic nerves richly innervate lymphatic tissues (24) and can activate T lymphocytes through the adrenergic receptor on T cells. In the setting of hypertension, afferent signals to the central nervous system lead to enhanced sympathetic outflow, contributing to T-cell activation. Nerve disruption technology is one of the important techniques to study neuroimmune function. Renal denervation abolishes the infiltration of effector memory cells in the kidney, implicating the nervous system in T-cell functions during hypertension. Banek et al. (2) further demonstrated that renal sympathetic nerves were obligatory for kidney inflammation while renal afferent nerves contributed to salt-induced hypertension. The activation and egression of splenic T cells are dependent on splenic sympathetic nerve stimulation. This process requires cholinergic transmission upstream at the celiac ganglion through α7nAchR (60). Carnevale et al. (6) found that placental growth factor is a mediator linking the sympathetic nervous system and the splenic immune system in the pathogenesis of hypertension. Thus hypertension is truly a multisystem disorder.
ACTIVATED T CELL-MEDIATED ORGAN DAMAGE
The Kidney
Initial sodium retention is critical for the salt-induced increase in blood pressure. The kidney as a central controller of blood volume regulates a variety of ion and water channels throughout the nephron. The accumulation and activation of T cells in the kidney is a hallmark in multiple hypertension models, such as ANG II-induced and salt-induced hypertension (44, 62). Following activation in the kidney under hypertensive stimuli, T cells secrete a variety of cytokines to disrupt the nephron’s capacity to properly excrete sodium and water, which ultimately propagates blood pressure elevation. Moreover, T cells are observed surrounding glomeruli and the vasculature in the cortex and accumulating throughout the medullary interstitium. In transgenic animal models, T-cell deficiency protected against severe hypertension and maintained a natriuretic response to hypertension (11, 35, 45). Blocking T-cell accumulation and activation in the kidney with a lymphocyte-specific inhibitor during hypertension ameliorated kidney damage and lowered blood pressure (14, 34). These results underscore T cell-specific actions in the kidney that impair the natriuretic response and worsen hypertension.
Renal oxidative stress is prevalent in hypertensive models. Infiltrated T cells may also drive the production of ROS that promote sodium retention, renal injury, and vascular dysfunction (15). Increased infiltrating T lymphocytes induced by a high-salt diet was associated with an increase in superoxide production and renal damage in the Dahl salt-sensitive rat model. Moreover, administration of tacrolimus, which inhibited T-cell proliferation, reduced T-cell infiltration into the kidneys and blunted renal oxidative stress as measured by urinary thiobarbituric acid-reactive substances and renal p67phox expression (33). Adoptive transfer of T cells with an incomplete NADPH oxidase enzyme, a major source of free radicals, did not recapitulate ANG II-induced hypertension to similar levels of WT T cells in Rag1−/− lymphocyte-deficient mice (17). Thus T cells propagate hypertension at least in part through the generation of ROS.
The renin-angiotensin-aldosterone system is a critical player in the control of kidney function in both physiological and pathological conditions. T cells isolated from Dahl salt-sensitive hypertensive rat kidneys have detectable renin and angiotensin-converting enzyme activity. Thus T cells have the capacity to synthesize ANG II. Inversely, treatment with mycophenolate mofetil blunted T-cell accumulation in the kidney during hypertension and significantly reduced renal ANG II levels, indicating that infiltrating T cells may contribute to the production of intrarenal ANG II (15). To elucidate the effects of ANG II on T-cell function, we created a genetic model lacking the dominant murine AT1 receptor, AT1A, selectively on T lymphocytes and subjected these mice and their controls to an ANG II-induced hypertension model. We found that mice with T cells lacking AT1A had increased albumin excretion, exaggerated kidney injury, and augmented perivascular accumulation of CD4+ T lymphocytes in hypertensive kidney (64). Therefore, AT1 receptor activation on T cells ameliorates hypertensive kidney damage, likely by suppressing the Th1 immune response, and counteracts the tissue damage driven by blood pressure elevation related to renal AT1 receptor activation.
Th17 cells function as proinflammatory T cells to participate in autoimmune disorders by secreting cytokines IL-17 and IFN-γ (8). Th17-cell infiltration and activation are a significant manifestation of hypertensive kidney injury (38). However, the effects of IL-17 in hypertensive kidney injury are controversial. Blockade of IL-23 signaling or IL-17A blockade did not ameliorate or induce albuminuria in ANG II-induced hypertension (32), whereas deficiency of the IL-17/IL-23 axis aggravated albuminuria and renal damage in DOCA plus ANG II-induced hypertension, possibly by enhancing γδ T-cell infiltration (26). However, IL-17A−/− mice exhibited blunted hypertension, possibly through the decreased activation of distal tubule transporters, and were protected from glomerular and tubular injury (38). These discrepancies likely accrue from differential functions of the various IL-17 isoforms. These results underline the complexity of IL-17’s functions in hypertensive renal damage (Fig. 4).
Fig. 4.

Proposed roles of T cells in multiple organ dysfunction. Inflammatory-activated T cells infiltrate and accumulate in major target organs, including the kidney and vasculature, and then produce a variety of deleterious mediators to cause organ injury. The net effects of T-cell activation on sympathetic outflow in hypertension are complex and will require further study. Treg cells as anti-inflammatory cells normalize vascular and kidney function through anti-inflammatory cytokine IL-10. Another particular subset of CD4+ T cells, termed the CD4+ TChAT subset, plays a homeostatic role in vasorelaxation and regulation of blood pressure.
The Cardiovascular System
The cardiovascular system is one of the primary regulators and targets during hypertension. T cells participate in vascular dysfunction and subsequent development of hypertension (17). The infiltration of T cells into adventitial and perivascular adipose tissue is observed in animal models of hypertension. Once recruited, these T cells along with M2 macrophages can directly contribute to endothelial cell dysfunction, perivascular inflammation and fibrosis, and vascular stiffness and calcification, potentiating the blood pressure response to hypertensive challenge by directly generating a variety of bioactive mediators, including ROS and various cytokines such as IL-17, IFN-γ, and TNF-α (46).
Within the adaptive immune response, activated T lymphocytes in particular have the capacity to enhance levels of vascular oxidative stress via their natural function of eliminating host cells that have become infected by invading organisms. T cells expressed higher levels of the NADPH oxidase subunits p47phox, p22phox, and Nox2 to generate ROS within their milieu, possibly via TCR activation, leading to the development of hypertension induced by angiotensin or DOCA-salt (17, 57). Conversely, loss of NAPDH oxidase in T cells led to blunted superoxide generation in the aorta (17). Oxidative stress and ROS cause exaggerated vascular and endothelial dysfunction through several key proinflammatory signaling pathways, including Nrf2, NF-κB, Ets-1, and MK2 (10). The related inflammation in vessels causes deposition of collagen and increased vascular resistance and ultimately arterial remodeling and stiffening (59). Moreover, ROS could mediate the effects of matrix metalloproteinases (MMPs) on vascular remodeling. Although MMPs promote extracellular matrix turnover and potentially counteract vascular fibrosis and stiffness, they also drive endothelial cells and vascular smooth muscle cells toward secretory, migratory, proliferative, and senescent phenotypes by creating a proinflammatory milieu. MMPs inhibition attenuates vascular remodeling and stiffness and improves endothelial cell functions (54). Additionally, a cohort of cytokines such as TNF-α and IFN-γ secreted by T cells have considerable influence in vascular oxidative stress generation and stabilization. TNF-α and IFN-γ destabilize endothelial nitric oxide synthase (eNOS) mRNA (61) and collaboratively enhance oxidative stress by promoting NADPH oxidase subunit gp91phox expression (36). In contrast, NADPH oxidase activation in T cells also promotes generation of TNF-α (19). Blockade of TNF-α reduces production of superoxide in the vascular wall, lowers vascular stiffness, and preserves endothelial function (1, 31). Adoptive transfer of Tregs, which suppressed inflammation, reduced superoxide production and mononuclear cell accumulation in the vasculature (21). Collectively, ROS produced by T cells serve as a deleterious mediator in hypertensive vascular remodeling.
Th17 cells also provoke vascular dysfunction and remodeling during hypertension. Madhur et al. (30) found that mice of IL-17 had preserved vascular function, reduced T-cell infiltration in aortic media, and decreased superoxide production during hypertension. Injection of IL-17 causes endothelial dysfunction by activating RhoA/Rho-kinase (37). On the other hand, Krebs et al. (26) found that IL-17 was not necessary to elevate in DOCA plus ANG II challenge but mitigated hypertensive kidney injury. Thus, as mentioned above, the roles of Th17 cells and IL-17 may vary depending on isoforms, features of the selected model, or even the phase of experimental disease at which Th17 cells are studied. γδ T cells are another T-cell subset that propagates vascular injury and endothelial dysfunction during hypertension. Mice with γδ T-cell deletion or with an anti-TCR γδ antibody injection displayed improved endothelial dysfunction through decreased production of IL-17A and IFN-γ. In humans, an increase in γδ T-cell frequency was associated with hypertension (5). The aforementioned effects of γδ T cells on blood pressure could be the cause or result of vascular injury accruing from these unconventional T cells.
Recently, Olofsson et al. (41) have found that CD4+CD44hiCD62Llo T helpers were a distinct T-cell subset defined by choline acetyltransferase (ChAT) that are yet another mediator of vascular dysfunction and injury during hypertension. CD4 TChAT cells actively participate in vasorelaxation by increasing NOS in endothelial cells. Accordingly, mice with ChAT deficiency in CD4+ T cells exhibited exaggerated arterial blood pressure elevation. These results point to a novel therapeutic strategy to reduce vascular injury during hypertension by targeting T cell-mediated vasorelaxation (Fig. 4).
SUMMARY
As experimental evidence accumulates, we gain a deeper understanding of the T cell’s contributions to the pathogenesis of hypertension. The effects of T lymphocytes on blood pressure and consequent target organ damage depend on the T subset phenotype, T-cell localization, the participation of other inflammatory cell populations, and the experimental model. Thus the mechanisms through which T cells are activated, the interactions between distinct subsets of T cells, and the communication between T-cell subsets and end-organs require further investigation. Although there are a variety of medications for treating hypertension, treatment for up to half of hypertensive patients is not completely effective. In the absence of adequate treatment, the prognosis of these patients with “resistant” hypertension is poor. Considering hypertension as an autoimmune disorder will provide us a new perspective for the treatment of these “resistant” hypertension patients via the targeting of T cells.
GRANTS
This work was funded by National Institutes of Health Grants DK087893 and HL128355, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development Grant BX000893, and American Heart Association Award 18TPA34170047.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.R. prepared figures; J.R. and S.D.C. drafted manuscript; J.R. and S.D.C. edited and revised manuscript; J.R. and S.D.C. approved final version of manuscript.
REFERENCES
- 1.Angel K, Provan SA, Gulseth HL, Mowinckel P, Kvien TK, Atar D. Tumor necrosis factor-alpha antagonists improve aortic stiffness in patients with inflammatory arthropathies: a controlled study. Hypertension 55: 333–338, 2010. doi: 10.1161/HYPERTENSIONAHA.109.143982. [DOI] [PubMed] [Google Scholar]
- 2.Banek CT, Knuepfer MM, Foss JD, Fiege JK, Asirvatham-Jeyaraj N, Van Helden D, Shimizu Y, Osborn JW. Resting afferent renal nerve discharge and renal inflammation: elucidating the role of afferent and efferent renal nerves in deoxycorticosterone acetate salt hypertension. Hypertension 68: 1415–1423, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, Mernaugh RL, Itani HA, Loperena R, Chen W, Dikalov S, Titze JM, Knollmann BC, Harrison DG, Kirabo A. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Reports 21: 1009–1020, 2017. doi: 10.1016/j.celrep.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, Paradis P, Schiffrin EL. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension 57: 469–476, 2011. doi: 10.1161/HYPERTENSIONAHA.110.162941. [DOI] [PubMed] [Google Scholar]
- 5.Caillon A, Mian MOR, Fraulob-Aquino JC, Huo KG, Barhoumi T, Ouerd S, Sinnaeve PR, Paradis P, Schiffrin EL. γδ T cells mediate angiotensin II-induced hypertension and vascular injury. Circulation 135: 2155–2162, 2017. doi: 10.1161/CIRCULATIONAHA.116.027058. [DOI] [PubMed] [Google Scholar]
- 6.Carnevale D, Pallante F, Fardella V, Fardella S, Iacobucci R, Federici M, Cifelli G, De Lucia M, Lembo G. The angiogenic factor PlGF mediates a neuroimmune interaction in the spleen to allow the onset of hypertension. Immunity 41: 737–752, 2014. doi: 10.1016/j.immuni.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 7.Chen XH, Ruan CC, Ge Q, Ma Y, Xu JZ, Zhang ZB, Lin JR, Chen DR, Zhu DL, Gao PJ. Deficiency of complement C3a and C5a receptors prevents angiotensin II-induced hypertension via regulatory T cells. Circ Res 122: 970–983, 2018. doi: 10.1161/CIRCRESAHA.117.312153. [DOI] [PubMed] [Google Scholar]
- 8.Chen Z, O’Shea JJ. Regulation of IL-17 production in human lymphocytes. Cytokine 41: 71–78, 2008. doi: 10.1016/j.cyto.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity 41: 529–542, 2014. doi: 10.1016/j.immuni.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crowley SD. The cooperative roles of inflammation and oxidative stress in the pathogenesis of hypertension. Antioxid Redox Signal 20: 102–120, 2014. doi: 10.1089/ars.2013.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol 298: R1089–R1097, 2010. doi: 10.1152/ajpregu.00373.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Curtsinger JM, Mescher MF. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol 22: 333–340, 2010. doi: 10.1016/j.coi.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Curtsinger JM, Schmidt CS, Mondino A, Lins DC, Kedl RM, Jenkins MK, Mescher MF. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J Immunol 162: 3256–3262, 1999. [PubMed] [Google Scholar]
- 14.De Miguel C, Das S, Lund H, Mattson DL. T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 298: R1136–R1142, 2010. doi: 10.1152/ajpregu.00298.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Miguel C, Guo C, Lund H, Feng D, Mattson DL. Infiltrating T lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am J Physiol Renal Physiol 300: F734–F742, 2011. doi: 10.1152/ajprenal.00454.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evans LC, Petrova G, Kurth T, Yang C, Bukowy JD, Mattson DL, Cowley AW Jr. Increased perfusion pressure drives renal T-cell infiltration in the Dahl salt-sensitive rat. Hypertension 70: 543–551, 2017. doi: 10.1161/HYPERTENSIONAHA.117.09208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460, 2007. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hernandez AL, Kitz A, Wu C, Lowther DE, Rodriguez DM, Vudattu N, Deng S, Herold KC, Kuchroo VK, Kleinewietfeld M, Hafler DA. Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J Clin Invest 125: 4212–4222, 2015. doi: 10.1172/JCI81151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA, Gratze P, Weyand C, Harrison DG. Regulation of T-cell function by endogenously produced angiotensin II. Am J Physiol Regul Integr Comp Physiol 296: R208–R216, 2009. doi: 10.1152/ajpregu.90521.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Itani HA, Xiao L, Saleh MA, Wu J, Pilkinton MA, Dale BL, Barbaro NR, Foss JD, Kirabo A, Montaniel KR, Norlander AE, Chen W, Sato R, Navar LG, Mallal SA, Madhur MS, Bernstein KE, Harrison DG. CD70 exacerbates blood pressure elevation and renal damage in response to repeated hypertensive stimuli. Circ Res 118: 1233–1243, 2016. doi: 10.1161/CIRCRESAHA.115.308111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kasal DA, Barhoumi T, Li MW, Yamamoto N, Zdanovich E, Rehman A, Neves MF, Laurant P, Paradis P, Schiffrin EL. T regulatory lymphocytes prevent aldosterone-induced vascular injury. Hypertension 59: 324–330, 2012. doi: 10.1161/HYPERTENSIONAHA.111.181123. [DOI] [PubMed] [Google Scholar]
- 22.Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, Saleh MA, Trott DW, Itani HA, Vinh A, Amarnath V, Amarnath K, Guzik TJ, Bernstein KE, Shen XZ, Shyr Y, Chen SC, Mernaugh RL, Laffer CL, Elijovich F, Davies SS, Moreno H, Madhur MS, Roberts J 2nd, Harrison DG. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest 124: 4642–4656, 2014. doi: 10.1172/JCI74084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, Hafler DA. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496: 518–522, 2013. doi: 10.1038/nature11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kohm AP, Sanders VM. Norepinephrine and beta 2-adrenergic receptor stimulation regulate CD4+ T and B lymphocyte function in vitro and in vivo. Pharmacol Rev 53: 487–525, 2001. [PubMed] [Google Scholar]
- 25.Kopp C, Linz P, Wachsmuth L, Dahlmann A, Horbach T, Schöfl C, Renz W, Santoro D, Niendorf T, Müller DN, Neininger M, Cavallaro A, Eckardt KU, Schmieder RE, Luft FC, Uder M, Titze J. 23Na magnetic resonance imaging of tissue sodium. Hypertension 59: 167–172, 2012. doi: 10.1161/HYPERTENSIONAHA.111.183517. [DOI] [PubMed] [Google Scholar]
- 26.Krebs CF, Lange S, Niemann G, Rosendahl A, Lehners A, Meyer-Schwesinger C, Stahl RA, Benndorf RA, Velden J, Paust HJ, Panzer U, Ehmke H, Wenzel UO. Deficiency of the interleukin 17/23 axis accelerates renal injury in mice with deoxycorticosterone acetate+angiotensin ii-induced hypertension. Hypertension 63: 565–571, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02620. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Rafferty TM, Rhee SW, Webber JS, Song L, Ko B, Hoover RS, He B, Mu S. CD8+ T cells stimulate Na-Cl co-transporter NCC in distal convoluted tubules leading to salt-sensitive hypertension. Nat Commun 8: 14037, 2017. doi: 10.1038/ncomms14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loperena R, Van Beusecum JP, Itani HA, Engel N, Laroumanie F, Xiao L, Elijovich F, Laffer CL, Gnecco JS, Noonan J, Maffia P, Jasiewicz-Honkisz B, Czesnikiewicz-Guzik M, Mikolajczyk T, Sliwa T, Dikalov S, Weyand CM, Guzik TJ, Harrison DG. Hypertension and increased endothelial mechanical stretch promote monocyte differentiation and activation: roles of STAT3, interleukin 6 and hydrogen peroxide. Cardiovasc Res 114: 1547–1563, 2018. doi: 10.1093/cvr/cvy112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, Park JK, Beck FX, Müller DN, Derer W, Goss J, Ziomber A, Dietsch P, Wagner H, van Rooijen N, Kurtz A, Hilgers KF, Alitalo K, Eckardt KU, Luft FC, Kerjaschki D, Titze J. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med 15: 545–552, 2009. doi: 10.1038/nm.1960. [DOI] [PubMed] [Google Scholar]
- 30.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 55: 500–507, 2010. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mäki-Petäjä KM, Hall FC, Booth AD, Wallace SM, Yasmin, Bearcroft PW, Harish S, Furlong A, McEniery CM, Brown J, Wilkinson IB. Rheumatoid arthritis is associated with increased aortic pulse-wave velocity, which is reduced by anti-tumor necrosis factor-alpha therapy. Circulation 114: 1185–1192, 2006. doi: 10.1161/CIRCULATIONAHA.105.601641. [DOI] [PubMed] [Google Scholar]
- 32.Markó L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ, Bowman EP, Kleinewietfeld M, Fokuhl V, Dechend R, Müller DN. Interferon-γ signaling inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension 60: 1430–1436, 2012. doi: 10.1161/HYPERTENSIONAHA.112.199265. [DOI] [PubMed] [Google Scholar]
- 33.Mattson DL. Infiltrating immune cells in the kidney in salt-sensitive hypertension and renal injury. Am J Physiol Renal Physiol 307: F499–F508, 2014. doi: 10.1152/ajprenal.00258.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mattson DL, James L, Berdan EA, Meister CJ. Immune suppression attenuates hypertension and renal disease in the Dahl salt-sensitive rat. Hypertension 48: 149–156, 2006. doi: 10.1161/01.HYP.0000228320.23697.29. [DOI] [PubMed] [Google Scholar]
- 35.Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H. Genetic mutation of recombination activating gene 1 in Dahl salt-sensitive rats attenuates hypertension and renal damage. Am J Physiol Regul Integr Comp Physiol 304: R407–R414, 2013. doi: 10.1152/ajpregu.00304.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mir M, Asensio VJ, Tolosa L, Gou-Fabregas M, Soler RM, Lladó J, Olmos G. Tumor necrosis factor alpha and interferon gamma cooperatively induce oxidative stress and motoneuron death in rat spinal cord embryonic explants. Neuroscience 162: 959–971, 2009. doi: 10.1016/j.neuroscience.2009.05.049. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res 97: 696–704, 2013. doi: 10.1093/cvr/cvs422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Norlander AE, Saleh MA, Kamat NV, Ko B, Gnecco J, Zhu L, Dale BL, Iwakura Y, Hoover RS, McDonough AA, Madhur MS. Interleukin-17A regulates renal sodium transporters and renal injury in angiotensin II-induced hypertension. Hypertension 68: 167–174, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Norlander AE, Saleh MA, Pandey AK, Itani HA, Wu J, Xiao L, Kang J, Dale BL, Goleva SB, Laroumanie F, Du L, Harrison DG, Madhur MS. A salt-sensing kinase in T lymphocytes, SGK1, drives hypertension and hypertensive end-organ damage. JCI Insight 2: e92801, 2017. doi: 10.1172/jci.insight.92801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okusa MD, Rosin DL, Tracey KJ. Targeting neural reflex circuits in immunity to treat kidney disease. Nat Rev Nephrol 13: 669–680, 2017. doi: 10.1038/nrneph.2017.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Olofsson PS, Steinberg BE, Sobbi R, Cox MA, Ahmed MN, Oswald M, Szekeres F, Hanes WM, Introini A, Liu SF, Holodick NE, Rothstein TL, Lövdahl C, Chavan SS, Yang H, Pavlov VA, Broliden K, Andersson U, Diamond B, Miller EJ, Arner A, Gregersen PK, Backx PH, Mak TW, Tracey KJ. Blood pressure regulation by CD4+ lymphocytes expressing choline acetyltransferase. Nat Biotechnol 34: 1066–1071, 2016. doi: 10.1038/nbt.3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pang DJ, Neves JF, Sumaria N, Pennington DJ. Understanding the complexity of γδ T-cell subsets in mouse and human. Immunology 136: 283–290, 2012. doi: 10.1111/j.1365-2567.2012.03582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pons H, Ferrebuz A, Quiroz Y, Romero-Vasquez F, Parra G, Johnson RJ, Rodriguez-Iturbe B. Immune reactivity to heat shock protein 70 expressed in the kidney is cause of salt-sensitive hypertension. Am J Physiol Renal Physiol 304: F289–F299, 2013. doi: 10.1152/ajprenal.00517.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rucker AJ, Rudemiller NP, Crowley SD. Salt, hypertension, and immunity. Annu Rev Physiol 80: 283–307, 2018. doi: 10.1146/annurev-physiol-021317-121134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rudemiller N, Lund H, Jacob HJ, Geurts AM, Mattson DL; PhysGen Knockout Program . CD247 modulates blood pressure by altering T-lymphocyte infiltration in the kidney. Hypertension 63: 559–564, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Small HY, Migliarino S, Czesnikiewicz-Guzik M, Guzik TJ. Hypertension: focus on autoimmunity and oxidative stress. Free Radic Biol Med 125: 104–115, 2018. doi: 10.1016/j.freeradbiomed.2018.05.085. [DOI] [PubMed] [Google Scholar]
- 47.Steinman RM. Dendritic cells and the control of immunity: enhancing the efficiency of antigen presentation. Mt Sinai J Med 68: 160–166, 2001. [PubMed] [Google Scholar]
- 48.Sun XN, Li C, Liu Y, Du LJ, Zeng MR, Zheng XJ, Zhang WC, Liu Y, Zhu M, Kong D, Zhou L, Lu L, Shen ZX, Yi Y, Du L, Qin M, Liu X, Hua Z, Sun S, Yin H, Zhou B, Yu Y, Zhang Z, Duan SZ. T-cell mineralocorticoid receptor controls blood pressure by regulating interferon-gamma. Circ Res 120: 1584–1597, 2017. doi: 10.1161/CIRCRESAHA.116.310480. [DOI] [PubMed] [Google Scholar]
- 49.Touyz RM. The neuroimmune axis in the kidney: role in hypertension. Circ Res 117: 487–489, 2015. doi: 10.1161/CIRCRESAHA.115.307176. [DOI] [PubMed] [Google Scholar]
- 50.Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE, Wu J, Goldstein A, Arendshorst WJ, Madhur MS, Chen W, Li CI, Shyr Y, Harrison DG. Oligoclonal CD8+ T cells play a critical role in the development of hypertension. Hypertension 64: 1108–1115, 2014. doi: 10.1161/HYPERTENSIONAHA.114.04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Végran F, Apetoh L, Ghiringhelli F. Th9 cells: a novel CD4 T-cell subset in the immune war against cancer. Cancer Res 75: 475–479, 2015. doi: 10.1158/0008-5472.CAN-14-2748. [DOI] [PubMed] [Google Scholar]
- 52.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol 8: 523–532, 2008. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, Weyand CM, Harrison DG, Guzik TJ. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation 122: 2529–2537, 2010. doi: 10.1161/CIRCULATIONAHA.109.930446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang M, Kim SH, Monticone RE, Lakatta EG. Matrix metalloproteinases promote arterial remodeling in aging, hypertension, and atherosclerosis. Hypertension 65: 698–703, 2015. doi: 10.1161/HYPERTENSIONAHA.114.03618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wenzel U, Turner JE, Krebs C, Kurts C, Harrison DG, Ehmke H. Immune mechanisms in arterial hypertension. J Am Soc Nephrol 27: 677–686, 2016. doi: 10.1681/ASN.2015050562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, Haase S, Mähler A, Balogh A, Markó L, Vvedenskaya O, Kleiner FH, Tsvetkov D, Klug L, Costea PI, Sunagawa S, Maier L, Rakova N, Schatz V, Neubert P, Frätzer C, Krannich A, Gollasch M, Grohme DA, Côrte-Real BF, Gerlach RG, Basic M, Typas A, Wu C, Titze JM, Jantsch J, Boschmann M, Dechend R, Kleinewietfeld M, Kempa S, Bork P, Linker RA, Alm EJ, Müller DN. Salt-responsive gut commensal modulates TH17 axis and disease. Nature 551: 585–589, 2017. doi: 10.1038/nature24628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Williams RJ, Spencer JP, Rice-Evans C. Flavonoids: antioxidants or signalling molecules? Free Radic Biol Med 36: 838–849, 2004. doi: 10.1016/j.freeradbiomed.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 58.Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, Regev A, Kuchroo VK. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 496: 513–517, 2013. doi: 10.1038/nature11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L, Madhur MS, Chen W, Harrison DG. Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ Res 114: 616–625, 2014. doi: 10.1161/CIRCRESAHA.114.302157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xiao L, Kirabo A, Wu J, Saleh MA, Zhu L, Wang F, Takahashi T, Loperena R, Foss JD, Mernaugh RL, Chen W, Roberts J 2nd, Osborn JW, Itani HA, Harrison DG. Renal denervation prevents immune cell activation and renal inflammation in angiotensin II-induced hypertension. Circ Res 117: 547–557, 2015. doi: 10.1161/CIRCRESAHA.115.306010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yan G, You B, Chen SP, Liao JK, Sun J. Tumor necrosis factor-alpha downregulates endothelial nitric oxide synthase mRNA stability via translation elongation factor 1-alpha 1. Circ Res 103: 591–597, 2008. doi: 10.1161/CIRCRESAHA.108.173963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang J, Crowley SD. Role of T lymphocytes in hypertension. Curr Opin Pharmacol 21: 14–19, 2015. doi: 10.1016/j.coph.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang J, Patel MB, Griffiths R, Mao A, Song YS, Karlovich NS, Sparks MA, Jin H, Wu M, Lin EE, Crowley SD. Tumor necrosis factor-α produced in the kidney contributes to angiotensin II-dependent hypertension. Hypertension 64: 1275–1281, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang JD, Patel MB, Song YS, Griffiths R, Burchette J, Ruiz P, Sparks MA, Yan M, Howell DN, Gomez JA, Spurney RF, Coffman TM, Crowley SD. A novel role for type 1 angiotensin receptors on T lymphocytes to limit target organ damage in hypertension. Circ Res 110: 1604–1617, 2012. doi: 10.1161/CIRCRESAHA.111.261768. [DOI] [PMC free article] [PubMed] [Google Scholar]