

Abstract
The “stress” kinases cAMP-dependent protein kinase (PKA) and calcium/calmodulin-dependent protein kinase II (CaMKII), phosphorylate the Na+ channel Nav1.5 subunit to regulate its function. However, how the channel regulation translates to ventricular conduction is poorly understood. We hypothesized that the stress kinases positively and differentially regulate conduction in the right (RV) and the left (LV) ventricles. We applied the CaMKII blocker KN93 (2.75 μM), PKA blocker H89 (10 μM), and broad-acting phosphatase blocker calyculin (30 nM) in rabbit hearts paced at a cycle length (CL) of 150-8,000 ms. We used optical mapping to determine the distribution of local conduction delays (inverse of conduction velocity). Control hearts exhibited constant and uniform conduction at all tested CLs. Calyculin (15-min perfusion) accelerated conduction, with greater effect in the RV (by 15.3%) than in the LV (by 4.1%; P < 0.05). In contrast, both KN93 and H89 slowed down conduction in a chamber-, time-, and CL-dependent manner, with the strongest effect in the RV outflow tract (RVOT). Combined KN93 and H89 synergistically promoted conduction slowing in the RV (KN93: 24.7%; H89: 29.9%; and KN93 + H89: 114.2%; P = 0.0016) but not the LV. The progressive depression of RV conduction led to conduction block and reentrant arrhythmias. Protein expression levels of both the CaMKII-δ isoform and the PKA catalytic subunit were higher in the RVOT than in the apical LV (P < 0.05). Thus normal RV conduction requires a proper balance between kinase and phosphatase activity. Dysregulation of this balance due to pharmacological interventions or disease is potentially proarrhythmic.
NEW & NOTEWORTHY We show that uniform ventricular conduction requires a precise physiological balance of the activities of calcium/calmodulin-dependent protein kinase II (CaMKII), PKA, and phosphatases, which involves region-specific expression of CaMKII and PKA. Inhibiting CaMKII and/or PKA activity elicits nonuniform conduction depression, with the right ventricle becoming vulnerable to the development of conduction disturbances and ventricular fibrillation/ventricular tachycardia.
Keywords: CaMKII, conduction and arrhythmia, phosphatases, PKA, right ventricle outflow tract
INTRODUCTION
Conventionally, the physiological generation of the electrical impulse has been considered to be an all-or-nothing response that operates well within a margin of safety, thus requiring no fine adjustment. Challenging this notion from a molecular level, Marionneau and Abriel (50) have well established that multiple phosphorylation sites populate the sodium channel (50), the main generator of the cardiac electrical impulse. From an intact heart level, various recent studies have suggested that cardiac conduction is regulated by constitutive activity of “stress” kinases, the cAMP-dependent protein kinase (PKA) and the calcium/calmodulin-dependent protein kinase II (CaMKII). In particular, longitudinal ventricular conduction velocity was reduced in adenyl cyclase knockout mice designed to limit PKA signaling through reduced cAMP production (75), and the atrio-ventricular conduction was compromised in a knockout model of the CaMKII (36). Conversely, adrenergic stimulation increased ventricular conduction velocity in open chest pigs subject to stellate ganglion stimulation (3) and in donor hearts (42). Taken together, these findings would suggest that the signaling through both “stress” kinases upregulates conduction or at least maintains normal conduction. This notion, however, is not universally accepted. In fact, the prevailing view is that CaMKII signaling confers loss-of-function on the sodium channel (81); hence, inhibition of CaMKII increases ventricular conduction velocity (73). If this is the case, then the signaling through CaMKII pathways would oppose that through PKA (75) in their effect on ventricular conduction. This seems to be counterintuitive, given that PKA and CaMKII synergistically upregulate Ca2+ cycling to enhance contractility under stress (67); thus one would expect that the pattern of dromotropic regulation by these signaling mechanisms would be concordant with the pattern of inotropic regulation. However, whereas previous studies suggest opposite effects of PKA and CaMKII on the conduction in the left ventricle (LV) (73, 75), the effects of the “stress” kinases on the right ventricular (RV) conduction are largely unstudied. In that regard, our previously published work (85) may remain the only study addressing effects of CaMKII on RV conduction. A signature finding of that study was that CaMKII inhibition by specific blocker KN93 induced a markedly nonuniform slowing of ventricular conduction with the epicenter of the effect occurring in the basal part of the RV, approximately corresponding to the RV outflow tract (RVOT) (85). In fact, the sheer heterogeneity of the effect was critical to induce reentrant excitation underlying the initiation of ventricular fibrillation (VF) under the additional stress of myocardial ischemia (85).
Given our previous findings and the unresolved fundamental question of interaction between PKA and CaMKII in their regulation of ventricular conduction, the goal of the current study was to ascertain chamber-specific effects of specific blockers for both kinases (H89 and KN93, respectively), applied alone and in combination. In addition, we tested the effect of the broad-spectrum phosphatase inhibitor calyculin (31), assuming that it should enhance phosphorylation and thus produce effects opposite to those caused by kinase inhibitors.
Using optical mapping of conduction in the RV and the LV, we show unequivocally that robust and uniform conduction across both chambers is indeed maintained by a precise physiological balance of the activities of CaMKII, PKA, and phosphatases yet to be determined. We assert a synergistic, not opposing, effect of PKA and CaMKII with regard to ventricular conduction. Disruption of CaMKII and/or PKA signaling leads to severe depression of conduction in the RV culminating in the initiation of VF due to abnormal impulse conduction. Conversely, inhibition of phosphatase activity accelerates conduction. In addition, we show that protein expression of both the CaMKII-δ isoform (CaMKII-δ) and the PKA-catalytic subunit-α (PKA-Cα) is significantly larger in the RVOT than in the apical LV, further supporting enhanced dependence of RVOT on “stress” kinase activity. Thus in the context of an altered kinase-phosphatase balance, the RV acquires a prominent role as the vulnerable epicenter for conduction disturbances elicited by suboptimal phosphorylation levels. This has to be considered when contemplating treatments involving manipulations on CaMKII or PKA signaling (4, 40) especially in vulnerable cohorts such as patients with Brugada syndrome (10) or Brugada phenocopies (6, 66).
MATERIALS AND METHODS
The study conformed to the National Institute of Health’s Guide for the Care and Use of Laboratory Animals (8th ed., 2011) and was approved by the Institutional Animal Care and Use Committee of the University of Utah (Protocol No. 17-08005).
Langendorff-perfused rabbit heart preparation.
Young adult New Zealand white rabbits (3–6 mo of age) of either sex (12 male and 18 female; weight: 2.3–3.0 kg) were euthanized by pentobarbital sodium (130 mg/kg iv) mixed with heparin (1 ml, 10.000 USP) to prevent blood clotting. Hearts were rapidly excised, cannulated on a Langendorff apparatus, and perfused retrogradely with a perfusion solution containing the following (in mM): 130 NaCl, 24 NaHCO3, 1.2 NaH2PO4, 1.0 MgCl2, 5.6 glucose, 4.0 KCl, 1.8 CaCl2, and 0.1 g/l albumin, gassed with an O2-CO2 mixture (adjusted to maintain pH at 7.4) at a fixed rate of 30 ml/min. The mitral valve was disrupted by inserting a drainage tube into the LV via a small cut in the left atrial appendage to prevent buildup of LV pressure due to venous efflux through Thebesian veins. Hearts were immersed in a perfusion solution filled chamber and temperature in the RV cavity, and the superfusate was maintained at 37.0 ± 0.5°C. To enable the control of ventricular excitation rate, the atrioventricular (AV) node was ablated by application of an electric shock from a defibrillator (3J) through a cathode put in contact with the AV node projection in the right atrium. Subsequent constant pacing at a cycle length (CL) of 300 ms was achieved by means of two pairs of Ag/AgCl custom designed disk-shaped (2-mm diameter) pacing electrodes that were positioned on the epicardial (cathode) and endocardial (anode) surfaces of the lateral LV and RV free walls, approximately at the half-distance between the base and the apex (85). The volume-conducted ECG was monitored continuously throughout the experiment.
Experimental protocol.
After the initial 20- to 25-min period of equilibration during which all necessary preparatory procedures were carried out, we started perfusing hearts with the electromechanical uncoupler blebbistatin (2.85 μM) to reduce motion artifact, which was maintained throughout the experiment. We allowed 25 min of blebbistatin perfusion, which was sufficient to completely remove all visible contraction. Following this period of time, we obtained baseline (predrug) optical recordings for the time point 0 min. After that we started delivering the drug of choice at 10 times the target concentration through a syringe pump connected to the perfusion line. The syringe pump and the main perfusion pump were set at the rates of 3 and 27 ml/min, respectively, enabling in-line mixing at 1:10 dilution of the delivered drugs. Optical recordings were obtained at 15, 30, 45, and 60 min of drug perfusion.
At each measurement time point, a sequence of movies was recorded during pacing at different CLs (400, 300, 260, 230, 200, 170, and 150 ms) first delivered through the RV pacing site, and then delivered through the LV pacing site. After that, an attempt was made to obtain a recording during pacing at the LV pacing site at the CL of 8,000 ms. These attempts were successful only in a subset of experiments and time points, due to unpredictable and uncontrollable occurrence of interbeat idioventricular activations.
Experimental groups.
A total of six experimental groups were used in this study that differed in the drug delivered through the syringe pump: sham (n = 4), calyculin (n = 6), KN93 (n = 6), H89 (n = 5), KN93 + H89 (n = 4), and TTX + KN93 (n = 3). In the sham group, DMSO (vehicle) was perfused at the largest concentration used for delivery of tested drugs in this study (1.27 mM). Calyculin (Santa Cruz Biotechnology), a broad-range phosphatase blocker (31, 45), was used at the final concentration of 30 nM (29). KN93 (Calbiochem), a CaMKII blocker (65), was used at the final concentration of 2.75 μM (85). H89 (Cayman), a PKA blocker (15), was used at the final concentration of 10 μM (90). In the TTX + KN93 group, a selective and specific sodium channel blocker TTX (EMD Biosciences) was used at a final concentration of 1 μM (16). In this group, the above-described experimental protocol was slightly modified such that the hearts were subjected to TTX alone for 15 min, followed by a combination of TTX and KN93 for 30 min. In preliminary experiments, we found that the TTX effects reach steady state after 10 min of perfusion, which is in good agreement with previous studies (16, 18, 38).
In two additional experiments in which the heart was perfused with either KN93 (2.75 μM) or H89 (10 μM), we simplified the pacing protocol to investigate solely the transition from CL of 400 to 8,000 ms or the shortest possible CL. Because of the distinct pacing applied to these hearts, the data generated from the experiments were analyzed separately.
Optical mapping.
Optical mapping was performed essentially as described in our previous publications (24, 85), and only the most salient features will be briefly mentioned here. The hearts were stained with the voltage sensitive dye di-4-ANEPPS (initial staining with 3-ml bolus of 10.4-μM stock; additional staining with 1 ml of stock 1–2 min before each measurement time point). The anterior aspects of the RV and the LV were imaged at a resolution of 64 × 64 pixels (0.5 mm/pixel) and a frame interval of 2.06 ms. Ambient light movies were recorded at each measurement time point at a resolution of 0.25 mm/pixel for precise demarcation of the boundary between the RV and the LV based on the position of the left anterior descending coronary artery (LAD). Data acquisition was performed using a custom software package written in Java and using libraries from Micro-Manager open source microscopy software (https://micro-manager.org/) and NetBeans open source program development framework (https://netbeans.org/).
Processing and analysis of optical mapping data.
Data processing of fluorescent signals was described in great detail in our previous publications (24, 85). Briefly, the individual fluorescent signals from each recorded pixel were first background-subtracted, inverted, and filtered in time and space. For each pixel and each optical action potential, we defined the activation time as the time of the fastest change in voltage-dependent fluorescence corresponding to dV/dtmax. For each paced activation captured in a fluorescence movie, we computed the local activation time and then color coded and projected the result on to each pixel of the imaged area to generate an activation map. The local delay of activation in a given pixel was computed as the magnitude of the local gradient of activation time. The local delay is the inverse of conduction velocity in the direction perpendicular to the activation wavefront and is presented in units of milliseconds per millimeters.
For each activation, computation and color coding of the local delay for each pixel generated a local delay map. In addition, for each pixel and each optical action potential, we computed the action potential duration (APD) as the time relative to the activation time at which the level of 80% repolarization was achieved. Projection of each data point to the corresponding pixel of the imaged area generated an APD map for each activation sequence. The space-averaged values of local delay or APD were computed for the RV and the LV part of the mapped area, as demarcated by the LAD, and were then averaged between different experiments in each experimental group for statistical analysis.
Western blot analysis.
Four additional young adult New Zealand white rabbits (2 male and 2 female; weight: 2.3 to 3.0 kg) were euthanized as described above to rapidly excise the heart and collect tissue samples from the RVOT and the anterior apical LV (AALV). The tissue samples were immediately frozen in liquid nitrogen and stored at −80°C until they were used for analysis.
Western blot analysis was carried out as previously described (84). Briefly, the tissue samples from RVOT and AALV were homogenized in 1% SDS buffer (50 mM Tris·HCl and 10 mM EDTA, pH7.4) with protease inhibitors (sodium butyrate, PMSF, sodium orthovanadate, sodium fluoride, no. 04693159001, and protease inhibitor cocktail tablet from Roche Applied Science). Protein extracts were resolved by SDS-PAGE (4–15% TGX Precast Protein Gel; Bio-Rad) and transferred to nitrocellulose membranes. Equal protein loading was confirmed by Ponceau S staining, which was imaged using the Image Laboratory software v6.0 (Bio-Rad). Western blotting was performed using antibodies against CaMKII-δ (no. MA5-27735; ThermoFisher), PKA-Cα (no. 610980; BD Bioscience), and PKA-RIα (no. 610609; BD Biosciences). Detection was performed by measuring the chemiluminescent signal as assayed by ProSignal Dura ECL Reagent (no. GSL-929-D10; Prometheus Protein Biology Products). The signal was normalized to the total protein concentration from the Ponceau signal (76).
Statistical analysis.
This study has a multidimensional design including factors of drug, ventricular chamber, time of drug perfusion, pacing CL, and the pacing cite. It would be practically impossible to analyze the effect of all factors in a straightforward manner. Therefore, we performed either paired t-test, or two-way ANOVA, or three-way ANOVA for selected subsets of factors as specified in the specific subsections of results. Tests were performed using XLSTAT (Addinsoft; https://www.xlstat.com/) and MATLAB Statistics Toolbox (MathWorks version 9.0). Specific post hoc tests are indicated with the presentation of respective results. Data are given as means ± SE. Differences of P < 0.05 were considered statistically significant.
RESULTS
Phosphatase inhibition accelerates conduction, whereas kinase inhibition depresses conduction.
The overall purpose of this study was to determine the pattern of regulation of conduction in intact ventricles by kinases and phosphatases. Figure 1 compares the effects of 15-min perfusion with vehicle (DMSO: sham group), the broad-acting phosphatase inhibitor calyculin, the CaMKII inhibitor KN93, and the PKA inhibitor H89. Figure 1, A–D, shows activation maps (Fig. 1, left) and respective local delay maps (right) from typical experiments in each group before (Fig. 1, top) and after (Fig. 1, bottom) perfusion with drug or vehicle. All depicted data were obtained during pacing in the lateral LV (white arrowheads) at the CL of 300 ms. One can see that in a representative sham experiment (Fig. 1A) a 15-min perfusion with the vehicle caused practically no change in activation or distribution of local conduction delays. In contrast to shams, perfusion with calyculin (Fig. 1B) produced a significant albeit subtle acceleration of conduction, which is illustrated by a reduction of the total activation time from 45 ms at baseline to 38 ms after 15 min of calyculin perfusion. Note that the corresponding conduction delay maps highlight the preferential effect of calyculin in the RV, where perfusion with the phosphatase inhibitor almost completely removed the red and yellow areas (delays > 2.4 ms/mm).
Fig. 1.
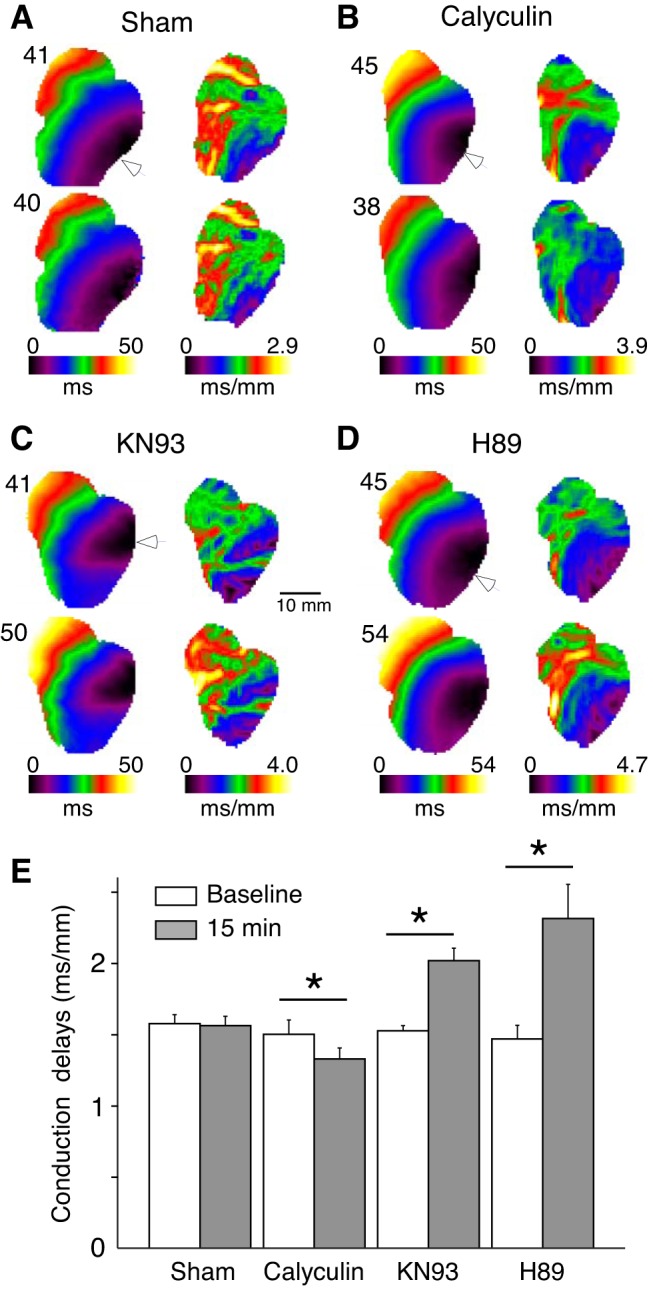
Regulation of ventricular conduction by kinases and phosphatases. A–D: representative activation maps (left) and the corresponding conduction delay maps (right) at baseline (top) and after 15 min drug/vehicle perfusion (bottom) in hearts treated with vehicle (DMSO; A), phosphatase inhibitor calyculin (B), CaMKII inhibitor KN93 (C), and PKA inhibitor H89 (D) experiments. Numbers above the activation maps indicate the latest activation time (ms) for each experimental condition. E: group wise means ± SE values of the local conduction delays averaged over the entire mapped surface at baseline and after 15 min of drug/vehicle perfusion. All data were obtained during left ventricle pacing at the locations indicated by white arrowheads, at cycle length (CL) = 300 ms. *P < 0.05 vs. baseline, paired Student’s t-test; sham: n = 4; calyculin: n = 6; KN93: n = 6; and H89: n = 5.
Figure 1, C and D, shows that in contrast to the sham and calyculin experiments, inhibition of both CaMKII and PKA caused a significant slowing of the impulse as it propagated across the ventricles. Note that the increase in the total activation time caused by 15-min drug perfusion is similar for both KN93 (41–50 ms, baseline vs. 15-min drug) and H89 (45 to 54 ms, baseline vs. 15-min drug). Note additionally that the increases in conduction delays induced by both kinase inhibitors are highly nonuniform, with the largest effect occurring at the base of the RV, as indicated by the increase in red/yellow pixels overlying this region.
Figure 1E depicts the group-wise quantification of the changes in conduction delays averaged over the entire mapped area (LV + RV). Consistent with the activation maps and conduction delay maps shown in Fig. 1, A–D, the group-wise mean conduction delays were unchanged in sham group (1.57 ± 0.06 to 1.56 ± 0.06 ms/mm, P = 0.3), significantly reduced in calyculin group (1.50 ± 0.10 to 1.33 ± 0.08 ms/mm, P = 0.003), and significantly increased in both KN93 (1.52 ± 0.04 to 2.02 ± 0.09 ms/mm, P = 0.002) and H89 (1.47 ± 0.10 to 2.31 ± 0.24 ms/mm, P = 0.03) groups.
Given the data shown in Fig. 1 and our previous findings (85), we hypothesized that regulation of conduction by kinases and phosphatases is different between the RV and the LV. Also, since previous patch-clamp studies showed that changes in kinase activity were associated with changes in the intermediate inactivation of the cardiac Na+ current (INa) (2, 89), we hypothesized that the effects of kinase and phosphatase blockers are rate dependent. Lastly, we thought it was important to determine when (and whether) the effects of drugs studied reached steady state. Therefore, the study was designed to determine the effect of ventricular chamber, pacing rate, and time of drug perfusion on local conduction delays in each experimental group. Figures 2–5 show representative examples from the sham, calyculin, KN93, and H89 groups, respectively. All data shown were obtained during LV pacing. In all images of Figs. 2–5, A, activation delay maps are shown for different pacing CLs (columns) and different time points (rows), whereas images in Figs. 2–5, B show the space-averaged values of conduction delays in the RV and the LV for each pacing CL and time point.
Fig. 2.
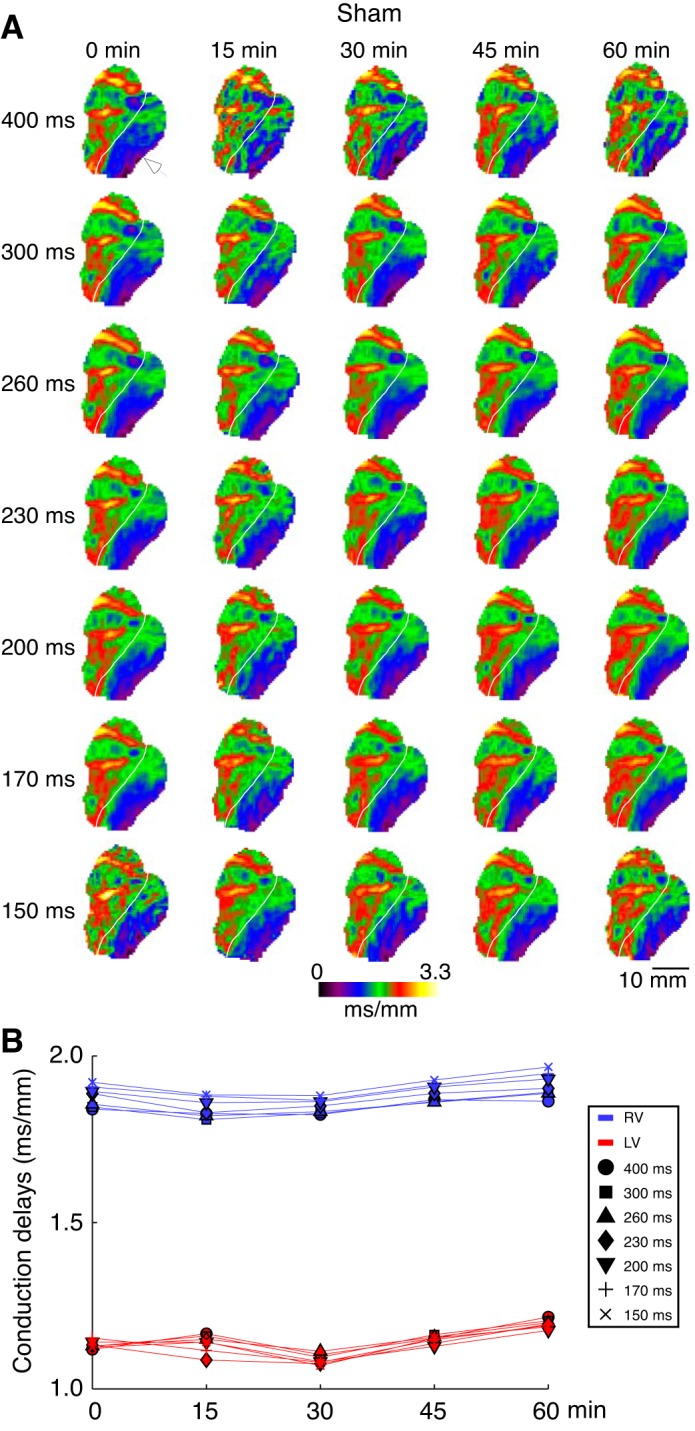
The right ventricle (RV) and left ventricle (LV) local conduction delays in a representative sham experiment as a function of pacing cycle lengths (CL) and time of the vehicle (DMSO) perfusion. A: conduction delay maps. Colors from dark violet to yellow indicate spatial distribution of local conduction delays in the range from 0 to 3.3 ms/mm. Columns (left to right) represent different time points (from 0 to 60 min). Rows (top to bottom) represent different pacing CLs (from 400 to 150 ms). White arrowhead indicates the site of pacing in the lateral LV. White line demarcates the RV and the LV. B: average RV (blue) and LV (red) conduction delay values computed for the maps shown in A. Symbols indicate values computed at different pacing CLs: circle (400 ms); square (300 ms); triangle (260 ms); diamond (230 ms); inverted triangle (200 ms); cross (170 ms); and tilted cross (150 ms). Overall, the ventricular conduction showed virtually no dependence on the pacing CL or the time of vehicle perfusion.
Fig. 5.
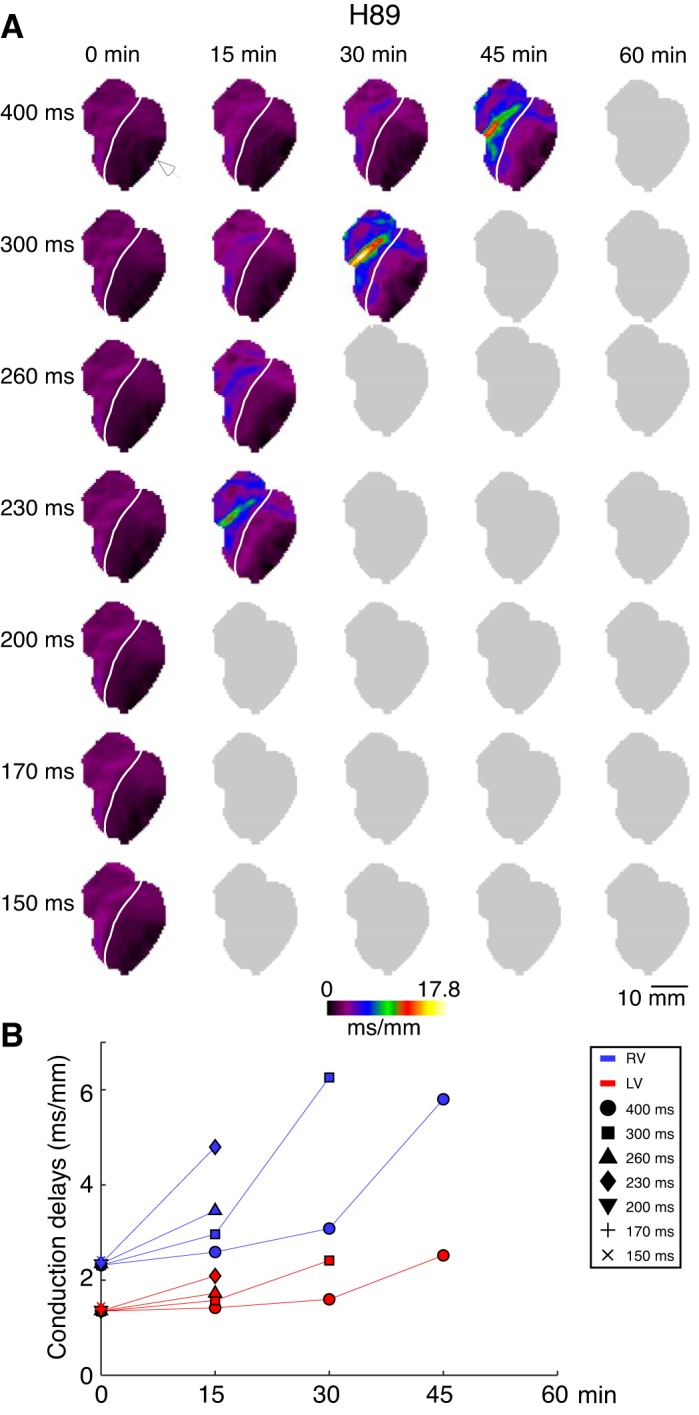
The right ventricle (RV) and left ventricle (LV) local conduction delays in a representative H89 experiment as a function of pacing cycle lengths (CL) and time of H89 perfusion. A: conduction delay maps. Colors from dark violet to yellow indicate spatial distribution of local conduction delays in the range from 0 to 17.8 ms/mm. Columns (left to right) represent different time points (from 0 to 60 min). Rows (top to bottom) represent different pacing CLs (from 400 to 150 ms). White arrowhead indicates the site of pacing in the lateral LV. White line demarcates the RV and the LV. B: average RV (blue) and LV (red) conduction delay values computed for the maps shown in A. Symbols indicate values computed at different pacing CLs: circle (400 ms); square (300 ms); triangle (260 ms); diamond (230 ms); inverted triangle (200 ms); cross (170 ms); and tilted cross (150 ms). Gray maps indicate inability to compute the conduction delays because pacing at the given condition did not result in 1:1 activation over the entire mapped surface. Similar to KN93, H89 caused progressive time- and CL-dependent slowing of conduction, which was most prominent in the basal anterior RV (approximately corresponding to the RVOT). However, the overall effect of H89 seemed to be stronger than that of KN93.
Figure 2 shows data from a representative sham experiment (same as in Fig. 1A). One can see that the distribution of conduction delays was very similar at different pacing CLs (in the range of 400–150 ms) and at different time points of the vehicle perfusion (in the range of 0–60 min). Note however that the conduction delays were consistently longer in the RV than in the LV (Fig. 2B), which we attribute to the chamber-specific difference between the direction of wave propagation and the direction of fiber orientation in the RV versus LV during LV pacing.
Figure 3 shows data from a representative calyculin experiment (same as in Fig. 1B). One can see that the conduction delays are reduced (hence, conduction is accelerated) already after 15 min of drug perfusion, with no major change between 15 and 30 min of the drug perfusion. As expected from the sham experiment shown in Fig. 2, there is almost no dependence of the conduction delays on the pacing CL at the baseline, and this was further reduced in the presence of calyculin (note the reduced dispersion of conduction delays at the corresponding time points). Note additionally, that the calyculin-induced change in conduction delays is somewhat larger in the RV (from 1.95 to 1.60 ms/mm at CL = 400 ms) than in the LV (from 1.43 to 1.25 ms/mm at CL = 400 ms), suggesting a higher sensitivity of RV conduction delays to the effect of calyculin.
Fig. 3.
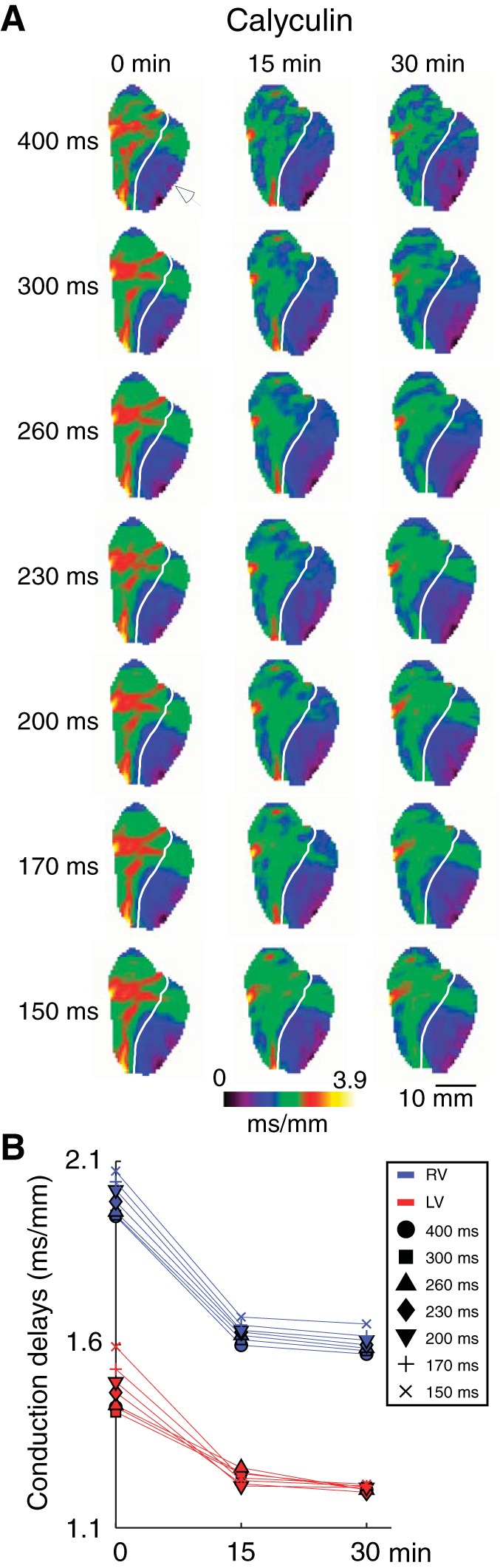
The right ventricle (RV) and left ventricle (LV) local conduction delays in a representative calyculin experiment as a function of pacing cycle lengths (CL) and time of calyculin perfusion. A: conduction delay maps. Colors from dark violet to yellow indicate spatial distribution of local conduction delays in the range from 0 to 3.9 ms/mm. Columns (left to right) represent different time points (from 0 to 30 min). Rows (top to bottom) represent different pacing CLs (from 400 to 150 ms). White arrowhead indicates the site of pacing in the lateral LV. White line demarcates the RV and the LV. B: average RV (blue) and LV (red) conduction delay values computed for the maps shown in A. Symbols indicate values computed at different pacing CLs: circle (400 ms); square (300 ms); triangle (260 ms); diamond (230 ms); inverted triangle (200 ms); cross (170 ms); and tilted cross (150 ms). Different from Fig. 2, only 30 min of drug perfusion is shown, which was sufficient to achieve maximal effect. Calyculin decreased conduction delays (hence, increased conduction velocity) in both chambers, the effect being slightly larger in the RV.
While calyculin typically reached maximum effect with respect to acceleration of conduction either at 15 or 30 min of drug perfusion, a prolonged calyculin perfusion (>40 min) caused a significant increase in perfusion pressure (not shown) followed by the development of a catastrophic APD shortening and frequent initiation of VF. Speculatively, we attribute this to metabolic stress secondary to vasoconstriction (35). While an interesting observation, it was beyond the scope of this study and was not further pursued, thus effectively limiting the calyculin series to 30 min of drug perfusion.
Figure 4 shows data from a representative KN93 experiment (same as in Fig. 1C). As expected, baseline distribution of conduction delays does not depend on the pacing CL in the 400–150 ms. However, with increasing time of KN93 perfusion, conduction delays progressively increase in a chamber (RV > LV)- and rate-dependent manner. In fact, the depression of conduction by KN93 culminates in failure of activation in 1:1 manner (precluding formal analysis of conduction delays), which occurs at progressively longer pacing CLs with increasing time of drug perfusion. The recordings in which 1:1 activation was lost in some parts of the mapped area are represented by gray “shadow” images. The overall progression of conduction abnormalities was as follows: at 15 min of KN93 perfusion, a dramatic increase in local delays (red-yellow pixels; >6.0 ms/mm) was observed in the basal-lateral anterior RV at pacing CL of 170 ms, and at pacing CL of 150 ms there was a 2:1 conduction block in the same region of the RV. At 30 min of KN93 perfusion, an exceedingly slow conduction in the RV was observed already at the pacing CL of 260 ms, and a 2:1 conduction block in the same region of the RV was observed at the pacing CL of 230 ms. Finally, after 45 min of KN93 perfusion 1:1 activation was observed only at the pacing CL of 400 ms, which is close to the upper bound of the physiological sinus node cycle length (CL) in the rabbit (47, 62); pacing at shorter CLs always elicited either lack of 1:1 capture, or turbulent conduction, or initiation of sustained VF. At 60 min of KN93 perfusion, the conduction delays at pacing CL of 400 ms further increased, reaching in the RV the value of 11.4 ms/mm.
Fig. 4.
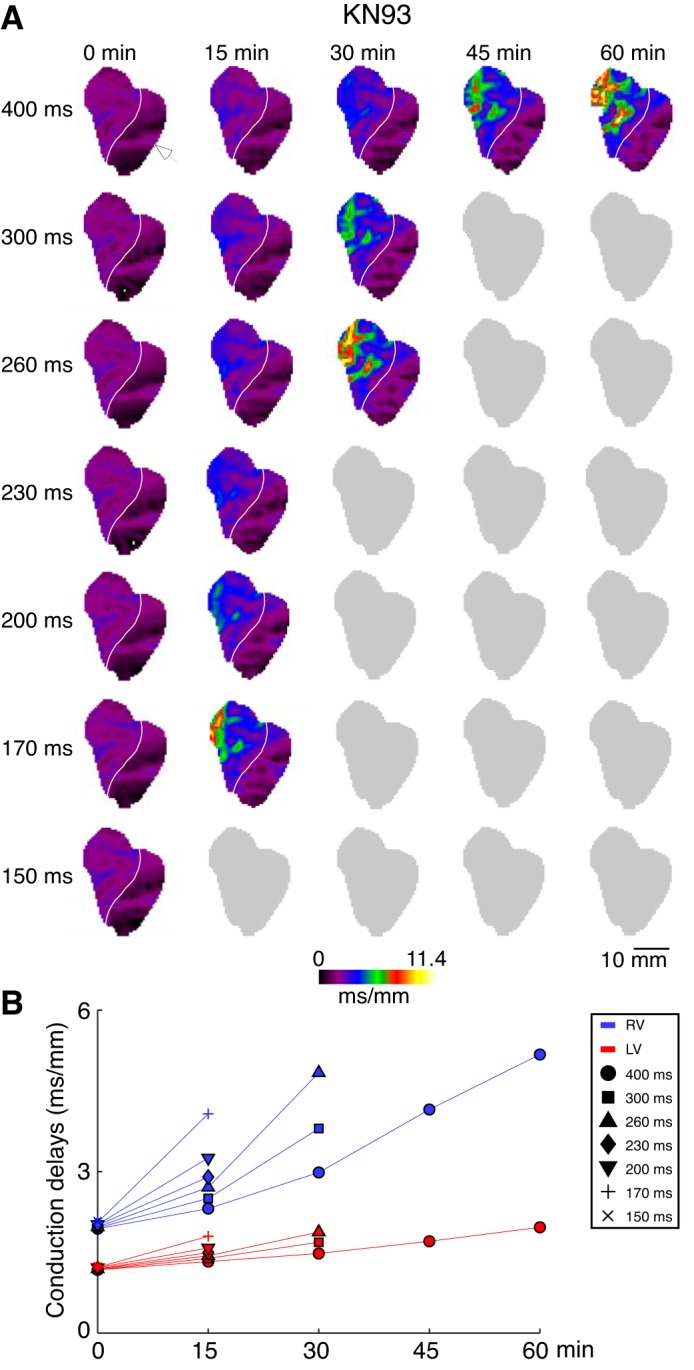
The right ventricle (RV) and left ventricle (LV) local conduction delays in a representative KN93 experiment as a function of pacing cycle lengths (CL) and time of KN93 perfusion. A: conduction delay maps. Colors from dark violet to yellow indicate spatial distribution of local conduction delays in the range from 0 to 11.4 ms/mm. Columns (left to right) represent different time points (from 0 to 60 min). Rows (top to bottom) represent different pacing CLs (from 400 to 150 ms). White arrowhead indicates the site of pacing in the lateral LV. White line demarcates the RV and the LV. B: average RV (blue) and LV (red) conduction delay values computed for the maps shown in A. Symbols indicate values computed at different pacing CLs: circle (400 ms); square (300 ms); triangle (260 ms); diamond (230 ms); inverted triangle (200 ms); cross (170 ms); and tilted cross (150 ms). Gray maps indicate inability to compute the conduction delays because pacing at the given condition did not result in 1:1 activation over the entire mapped surface. Note progressive time- and CL-dependent slowing of conduction, which is most prominent in the basal anterior RV (approximately corresponding to the RV outflow tract).
Figure 4B shows the space-averaged values of conduction delays in the RV and the LV for each pacing CL and time point. It highlights the differences between the chambers. Importantly, the changes in the RV conduction delays (blue symbols) increase much more prominently with time of drug perfusion than their LV conduction delay counterparts (red symbols). It is also easy to appreciate that the increase in conduction delays is highly CL dependent, with the most prominent increases occurring at the shorter pacing CLs. This is in stark contrast with the lack of rate dependence observed in experiments from the sham and calyculin groups where all measures formed compact groups at each time point.
Figure 5 illustrates the time and pacing CL-dependent changes of conduction delay distribution for a representative H89 experiment (same as in Fig. 1D). Overall, the effect of H89 on conduction patterns is similar to that observed in the KN93 experiment shown in Fig. 4. Namely, there is a progressive chamber- and pacing CL-dependent increase in conduction delays occurring with the time of H89 perfusion. It is interesting to note that even within the RV the effect of H89 is highly nonuniform, with the epicenter approximately corresponding to the lower part of the RVOT. Of note, at 60 min of H89 perfusion, in all hearts we were unable to elicit a 1:1 response even at the longest pacing CL (CL = 400 ms); this suggests a stronger depressant effect of 10 μM H89 as compared with 2.75 μM KN93.
Figure 6 presents a group-wise analysis of the changes in the RV (blue) and the LV (red) average conduction delays as the function of time of drug/vehicle perfusion. The data shown were obtained during pacing from either lateral RV (open symbols) or lateral LV (closed symbols) at CL = 400 ms. Figure 6, A–D, represents sham, calyculin, KN93, and H89 groups, respectively. In each group, two-way ANOVA was applied separately for data obtained at each pacing site, analyzing factors of time, chamber, and chamber-time interaction. In the sham group (Fig. 6A), the average RV and LV conduction delays remained constant throughout 60 min of vehicle perfusion. Hence, there was no significant effect of time. However, for each pacing site the average RV and LV conduction delays were clearly separated resulting in a significant effect of the chamber factor (P < 0.0001). For each pacing site (either lateral LV or RV), the distal chamber always developed the longer conduction delays. For example, at baseline during LV pacing, delays in the RV were larger than those in the LV (1.83 ± 0.05 vs. 1.31 ± 0.09, P < 0.0001). In contrast, during RV pacing, the LV delays were larger (1.64 ± 0.05 vs. 1.23 ± 0.08 in the RV, P < 0.0001). We attribute this finding to the differing angle at which the excitation wave hits the fibers depending on the combination of the pacing site and the chamber. This effect was consistently observed at all CLs tested.
Fig. 6.

Statistical analysis addressing the effect of chamber, time, and pacing location on the drug-induced conduction delay changes for each experimental group. A–D: group-wise means ± SE of the right ventricle (RV) (blue) and the left ventricle (LV) (red) conduction delay values in sham (A), calyculin (B), KN93 (C), and H89 (D) groups. For each heart, the RV and the LV conduction delay values were computed by averaging over all pixels in the respective regions of the local conduction delay maps. All data shown obtained at pacing cycle lengths (CLs) of 400 ms. Open and closed symbols represent conduction delays during pacing at the RV and the LV pacing site, respectively. A two-way ANOVA was designed to assess the role of chamber, drug perfusion time, and the chamber-time interaction in the distribution of mean RV and LV conduction delay values. The analysis was performed separately for each experimental group and each pacing location. For calyculin group, additional two-way ANOVA was applied to data normalized to the baseline values (B, inset). §P < 0.05 for chamber-time interaction during LV pacing; #P < 0.05 for chamber-time interaction during RV pacing. *P < 0.05 vs. 0 min (predrug) at the same chamber and pacing location. Multiple comparisons were corrected using the Tukey-Kramer post hoc test.
Figure 6B shows that calyculin induced a small reduction in the conduction delays (increase in conduction velocity) that was more prominent in the RV than in the LV. Two-way ANOVA analysis showed significant effect of chamber but no significant effect of calyculin perfusion time and no significant chamber-time interaction. This indicated that the variance due to interanimal differences exceeded the variance between time points (i.e., caused by calyculin perfusion). To unmask the rather subtle effect of calyculin, we analyzed conduction delay values normalized to the baseline values. Normalization revealed a significant effect of chamber, time, and chamber-time interaction in case of LV pacing (Fig. 6B, inset). Specifically, at 15 min of calyculin perfusion, during LV pacing normalized RV conduction delays were 0.85 ± 0.02 vs. 0.96 ± 0.03 in the LV (two-way ANOVA P < 0.05). Thus at least during LV pacing the sensitivity of the RV to the effect of calyculin was more marked than that of the LV.
Figure 6C summarizes the chamber-specific increase in conduction delays induced by KN93 perfusion. As expected from sham experiments, before the drug administration conduction delay values in the chamber distal from pacing site were larger than those in the proximal chamber. However, regardless of the pacing site, the RV local conduction delays steadily increased with time of KN93 perfusion, whereas the LV conduction delays remained relatively constant. Two-way ANOVA revealed significant effects of chamber, time, and chamber-time interaction, which held true for both pacing locations. The presence of a significant chamber-time interaction (P < 0.0002) indicates higher sensitivity of the RV to conduction slowing elicited by CaMKII inhibition.
Figure 6D summarizes the chamber-specific increase in conduction delays induced by H89 perfusion. The changes induced by PKA inhibition were qualitatively similar to those elicited by CaMKII inhibition. However, quantitatively, the effect of H89 appeared to be much larger than that of KN93. For example, after 45 min of H89 perfusion, the average RV conduction delays were at least two times larger than those observed after 45 min of KN93 perfusion. Note also, that in all experiments, 60-min H89 perfusion consistently lead to a failure of 1:1 propagation even at the longest pacing CL of 400 ms, which precluded the computation of conduction delays at this time point. Similarly to the KN93 group, in the H89 group two-way ANOVA revealed significant effects of chamber, time, and chamber-time interaction, which held true for both pacing locations. The presence of a significant chamber-time interaction (P < 0.0003) indicates higher sensitivity of the RV to conduction slowing elicited by PKA inhibition.
Overall, data in Fig. 6 indicate a higher sensitivity of conduction in the RV to drugs affecting phosphorylation/dephosphorylation balance as compared with the LV, at least for some pacing locations. Importantly, changes in the RV conduction delays caused by inhibition of the “stress” kinases were largely independent of the pacing location and far exceeded the dispersion of the RV conduction delays related to the pacing location.
Combined inhibition of CaMKII and PKA synergistically depresses RV conduction.
Given that the effects of the CaMKII blocker KN93 and PKA blocker H89 on conduction delay distribution are very similar (see Fig. 6, C and D) and given that previous studies indicated that CaMKII and PKA target distinct Nav1.5 phosphorylation sites (1, 5, 12, 25, 34, 55, 92), we hypothesized that regulation of conduction by the two kinases might be synergistic. Hence, we tested the effect of combined administration of KN93 and H89. Figure 7 depicts the average RV and LV conduction delays measured after 15-min perfusion with either KN93, H89, or KN93 + H89 during LV pacing. The data were analyzed using two-way ANOVA with factors of “chamber” and “intervention.” In all cases the conduction delays were asymmetrically distributed across the chambers, such that average values in the RV were larger than in the LV (for KN93, H89, and KN93 + H89, respectively: 2.31 ± 0.11 vs. 1.38 ± 0.07 ms/mm; 2.46 ± 0.12 vs. 1.20 ± 0.09 ms/mm; and 4.20 ± 0.44 vs. 1.80 ± 0.21 ms/mm; effect of “chamber” P < 0.0001). Additionally, the average delays after 15 min perfusion with the kinase inhibitors were sensitive to the type of drug present (effect of “intervention” P < 0.0001). More specifically, the RV conduction delays were significantly higher when both CaMKII and PKA were simultaneously inhibited (KN93 + H89) as compared with inhibition of CaMKII or PKA alone (both P < 0.05 using Tukey-Kramer post hoc test). As compared with baseline values, 15-min perfusion with KN93, H89, and KN93 + H89 induced 24.7, 29.9, and 114.2% increase in RV conduction delays, respectively. Clearly, the effect elicited by the combined use of the drugs is more than the sum of the effects elicited by each drug separately, which is indicative of a negative synergism of the two drugs. Such synergism was not observed in the effects of combined drugs on LV conduction delays.
Fig. 7.
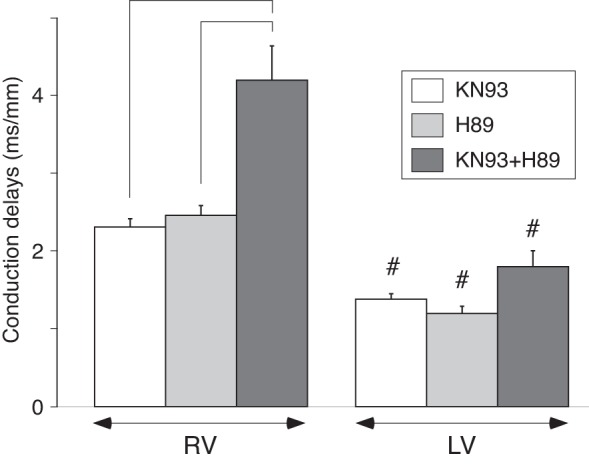
Synergistic effect of kinase inhibitors KN93 and H89 with respect to right ventricle (RV) conduction slowing. Values on the graph represent group-wise means ± SE of the average RV and left ventricle (LV) local conduction delays after 15 min of drug perfusion. All data obtained during pacing at cycle length (CL) = 400 ms from an LV location. Two-way ANOVA with the Tukey-Kramer post-hoc test was performed to assess how chamber and drug combination affected the mean conduction delays. #P < 0.05 vs. RV in the same experimental group; brackets indicate P < 0.05 vs. drug combination.
Spatial difference between effects of TTX and KN93.
Previous studies showed nonuniform distribution of Nav1.5, including reduced channel protein expression in RV basal regions (80). Additionally, our laboratory showed that the RVOT depolarization reserve was reduced as compared with the LV (85). In the light of these findings, there are two contrasting scenarios that may underlie the heterogeneous effect of stress kinase blockers on RV versus LV conduction demonstrated above. One possibility is that while kinase blockade causes regionally uniform reduction in INa, in this hypothetical scenario the intrinsic chamber-wise INa heterogeneity leads to a drug-induced differential response (RV > LV) because the relationship between the INa magnitude and conduction velocity follows a nonlinear saturating relationship, in which regions with reduced depolarization reserve subject to functional INa reduction will reach a threshold condition for conduction failure more readily (11, 71). Another possibility is that the function of stress kinases is chamber or region specific, thus leading to differential responses to kinase inhibition determined by the intrinsic distribution of the enzymes. (The 2 possibilities are not mutually exclusive.) To gain insight into the mechanism underlying nonuniform conduction depression following stress kinase inhibition, we compared the degree of RV-LV heterogeneity in the presence of sodium channel blocker TTX alone, in the presence of CaMKII inhibitor KN93 alone, and in the presence of their combination. The effects of TTX and TTX + KN93 were analyzed in the same hearts (TTX + KN93 group) and will be presented together (Fig. 8).
Fig. 8.
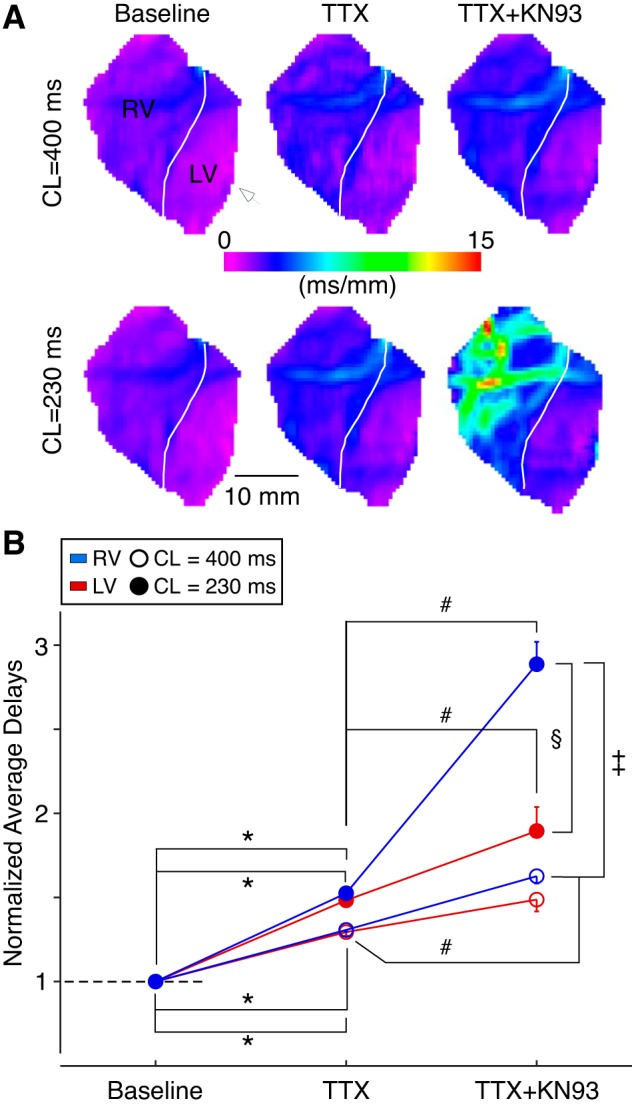
Effect of TTX alone and in combination with KN-93 on the right ventricle (RV) and the left ventricle (LV) conduction. A: conduction delay maps generated for LV paced activations at a cycle lengths (CL) of 400 ms (top) or 230 ms (bottom) in a representative experiment at baseline (left), after perfusion with TTX alone (middle), or after subsequent perfusion with TTX + KN93 (right). White arrowhead indicates the site of pacing in the lateral LV. White line demarcates the RV and the LV. B: changes in the mean chamber-specific normalized average delays for 3 hearts perfused initially with 1 μM TTX alone followed by a cocktail of 1 μM TTX and 2.75 μM KN93. Three-way ANOVA was used to assess the effect of intervention (no drug, TTX, and TTX + KN93), pacing CL (400 or 230 ms), and chamber (RV vs. LV). All factors exhibited significant contrasts (P < 0.0001) as determined by the statistical test. In addition, pairwise comparisons revealed significant differences (P < 0.05 by Newman-Keuls post hoc test) as labeled. *Significant differences between baseline and TTX comparing the same chamber and the same pacing CL. #Significant differences between TTX and TTX + KN93 comparing the same chamber and the same pacing CL. §Significant difference between the RV and the LV in the presence of TTX + KN93 at the CL of 230 ms. ‡Significant difference between the RV values at the CL of 400 vs. 230 ms in the presence of TTX + KN93.
The maps in Fig. 8A depict the distribution of conduction delays in response to LV pacing in a representative heart at baseline (left), after perfusion with TTX (middle), and after subsequent perfusion with TTX combined with KN93 (right). Note that at baseline, in line with other experimental data, conduction delays exhibited a narrow chamber-specific distribution with the chamber proximal to the pacing electrode (LV for this example) having smaller delays than the distal chamber (purple vs. blue/purple mix pixels in LV vs. RV locations). Perfusion with 1 μM TTX elicited a modest increase in the conduction delays (note pixels change to deep blue and purple/blue mix in RV and LV, respectively). Importantly, as compared with baseline, there was no appreciable increase in the RV-LV difference in conduction delays either during pacing at 400- or 230-ms CL. In contrast to the changes induced by TTX alone, TTX + KN93 elicited marked nonuniform conduction changes including a dramatic increase in the RV conduction delays (with an epicenter in the RVOT represented by the yellow/red pixels) amid a much more modest increase in the LV conduction delays (right bottom).
To effectively remove the baseline chamber-specific differences in conduction delays, we normalized the chamber-averaged conduction delays to the respective baseline values. We then performed a three-way ANOVA to assess the presence of significant chamber-specific conduction differences induced either by the factor of drug (no drug, TTX, or TTX + KN93), or by the factor of CL (400 vs. 230 ms), or by the factor of chamber (LV vs. RV). All factors exhibited significant contrasts (P < 0.0001) as determined by the statistical test (Fig. 8B). Pairwise comparisons using Newman-Keuls post hoc test allowed further insight into the combined effect of drugs, CL, and chamber. Only pairwise comparisons between values differing in just one factor were considered. As depicted in Fig. 8B, TTX alone significantly increased LV and RV mean normalized average delays both during pacing at CL of 400 and 230 ms (P < 0.05 for the corresponding 4 pairwise comparisons, labeled by an asterisk). However, pairwise analysis indicated that TTX alone did not separate the values of LV and RV conduction delays as a result of changing CL or because of their distinct anatomical location. Interestingly, following the addition of KN93 to TTX, normalized conduction delay values depicted further significant increases in both LV and RV measures (P < 0.05). Specifically, at the pacing CL of 230 ms both the LV and RV delays increased significantly, whereas at 400 ms CL only the increases in RV were significant (P < 0.05 for 3 pairwise comparisons, labeled by a number sign, #). Lastly, only the combination of TTX and KN93 induced a significant separation between the RV and the LV (observed only at the CL of 230 ms; P < 0.05, labeled by a section symbol) and also a significant separation between delay values in the same chamber measured at different pacing CLs (observed only in the RV, between CL of 400 and 230 ms, P < 0.05, labeled by a double dagger).
We also compared effect of TTX alone with an effect of KN93 alone using data obtained in the KN93 hearts. As shown above, the effect of KN93 was progressive with time. We deliberately chose the time point at which the effect of KN93 in the LV was close to that induced by TTX in the LV at the same pacing CL of 400 ms. Analyzing the average RV and LV conductions delays normalized to the predrug values in the same chamber with two-way ANOVA (Fig. 9), we found that when the effect of the two drugs was similar in the LV (1.41 ± 0.02 vs. 1.29 ± 0.02, NS), it was vastly different in the RV. Specifically, the effect of KN93 was much greater than the effect of TTX in this chamber (1.91 ± 0.13 vs. 1.31 ± 0.01; P < 0.05 by Tukey-Kramer post hoc test). Overall, these data suggest that region-specific conduction patterns caused by inhibition of protein kinases require region-specific differences in kinase expression or function.
Fig. 9.
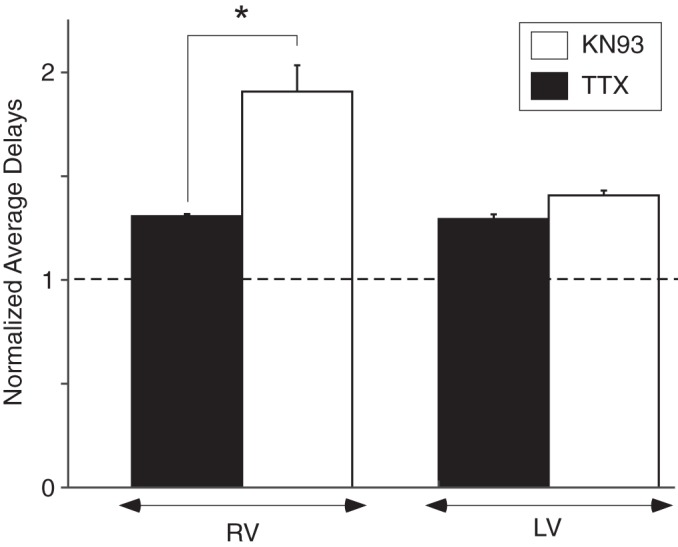
Distinct chamber specificity of TTX versus KN93 with respect to conduction slowing. Plot of the average right ventricle and left ventricle (LV) conduction delays normalized to respective baseline values during pacing from the LV at a cycle length of 400 ms. When the effect of the 2 drugs is similar in the LV, it is vastly different in the RV. Two-way ANOVA indicated that the effect of chamber, the effect of drug, as the effect of their interaction were significant (all P < 0.05). *P < 0.05 by Tukey-Kramer pairwise post hoc test.
Chamber specific protein expression of CaMKII and PKA.
To determine whether regional differences in CaMKII or PKA expression may underlie the RV-centric conduction vulnerabilities induced by the kinase blockers, we measured the expression levels of the principle proteins constituting either enzyme in the RVOT (RV region most vulnerable to kinase inhibition) versus the AALV (LV region most insensitive to kinase inhibition). CaMKII-δ, the predominant CaMKII isoform in ventricles (46), exhibited significantly higher expression levels in the RVOT as compared with the AALV (Fig. 10, A and B). Expression levels of the PKA subunit PKA-Cα, the catalytically active subunit of the PKA holoenzyme (37), also exhibited a similar differential distribution (Fig. 10, A and B). In contrast, The PKA regulatory subunit PKA-RIα, which is a known determinant of PKA activity (37, 88), was not differentially distributed among the ventricle regions that we investigated (Fig. 10, A and B). Overall, these findings suggest that the regional differences in stress kinase-mediated regulation of excitability underlie the increased conduction vulnerability to kinase block observed in the RV.
Fig. 10.
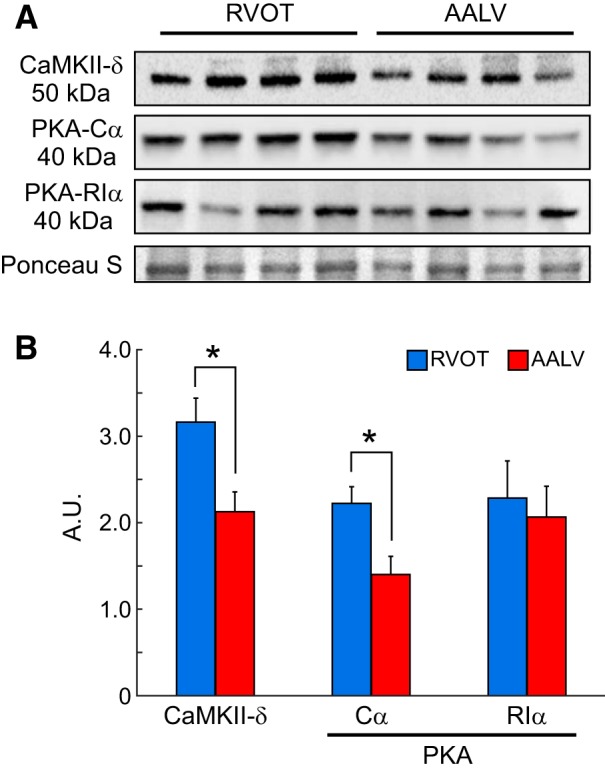
CaMKII-δ and PKA proteins are differentially expressed in the right ventricle (RV) and left ventricle (LV). A: representative Western blots of tissue extract from the RV outflow tract (RVOT) and the anterior apical LV (AALV). B: quantification of Western blotting results for CaMKII-δ, PKA-Cα, and PKA-RIα, showing that the expression levels of CaMKII-δ, PKA-Cα were significantly higher in the RVOT as compared with the AALV, whereas PKA-RIα exhibited a uniform regional distribution. Quantification of the enzymes’ regional expression levels was achieved by using the Ponceau S signals for normalization. A.U., arbitrary units. *P < 0.05, paired Student’s t-test; RVOT: n = 4; AALV: n = 4.
APD is uniformly altered in the RV and the LV by kinase and phosphatase inhibition.
In addition to Nav1.5, both CaMKII and PKA are known to regulate ion channels involved in repolarization of the action potential (8, 72), which has to be balanced by the action of phosphatases, although the identity of phosphatases involved in regulation of ion channels is not yet well established (31). Therefore, we tested the possibility that the chamber-specific effects on conduction elicited by kinase/phosphatase inhibitors are mediated, at least in part, by their chamber specific effect on the APD. Figure 11, A–D, shows APD at the level of 80% repolarization (APD80) values averaged over the RV (blue) and the LV (red) mapped regions as the function of drug/vehicle perfusion time in sham, calyculin, KN93, and H89 groups, respectively. All the data were obtained during pacing from the LV pacing site at CL of 400 ms. Two-way ANOVA testing the effect of chamber, time, and chamber-time interaction was applied separately in each experimental group. The results obtained during pacing from the RV pacing site were very similar (not shown). In sham experiments (Fig. 11A) the values of APD80 were almost identical in the RV and the LV and were constant over 60-min perfusion with vehicle (no significant effect by two-way ANOVA).
Fig. 11.

Statistical analysis addressing the effect of chamber and time on the drug-induced action potential duration (APD) changes for each experimental group. A–D: group-wise means ± SE of the average APD80 values in the right ventricle (RV) (blue) and the left ventricle (LV) (red) in sham (A), calyculin (B), KN93 (C), and H89 (D) groups. For each heart, the average RV and LV APD values were computed by averaging over all pixels in the respective regions of the APD maps. Black arrows indicate the time point from which the representative optical action potentials depicted in each panel were recorded. All data shown were obtained during pacing from an LV pacing site at a cycle length (CL) of 400 ms. For each group, a two-way ANOVA was performed to assess the role of chamber, time, and the chamber-time interaction in the distribution of mean conduction delay values. *P < 0.05 vs. baseline at the same chamber (Tukey-Kramer post hoc test).
Calyculin (Fig. 11B) induced a significant time-dependent monotonic APD shortening, which was almost the same in the two chambers (hence, no significant effect of chamber, no significant chamber-time interaction).
KN93 (Fig. 11C) induced a significant but similar increase in the mean APD values in the RV and LV. APD prolongation achieved a close to maximum value already at 15 min of perfusion with the CaMKII inhibitor (RV: 215 ± 5 vs. 181 ± 4 ms; LV: 207 ± 4 vs. 179 ± 3 ms; drug vs. baseline, respectively) and did not change much thereafter. There was a significant effect of time (P < 0.0001), but no significant effect of chamber and no significant chamber-time interaction.
Similar to KN93, the PKA inhibitor H89 (Fig. 11D) induced APD prolongation that was similar in the RV and the LV. However, in the presence of H89, the time course of APD prolongation was somewhat different, such that RV APD plateaued at 30 min of perfusion, whereas LV APD continued to rise up to 45 min of the drug perfusion. After 45 min of H89 perfusion, both RV and LV APD values converged on values that were somewhat higher than those achieved with 45-min perfusion of KN93 (compare Fig. 11, D and C). Two-way ANOVA analysis and Tukey-Kramer post hoc test indicated a significant effect of H89 perfusion time (P < 0.0001) but no significant effect of chamber and no significant chamber-time interaction. Note however that there was a trend for a separation between APD values in the RV and the LV at 30 min of H89 perfusion (RV vs. LV: 257 ± 7 vs. 234 ± 10 ms; two-way-ANOVA factor “chamber” P = 0.0296 but not significant after the Tukey-Kramer post hoc test correction). Note also that unlike all other interventions, H89 caused a dramatic change in the dynamics of repolarization, conferring a distinctly triangular shape on the action potential (see Fig. 11D, bottom inset).
In summary, kinase and phosphatase blockers had strong effects on APD, but those effects were rather uniform over the RV and the LV, which was in stark contrast to the effects the kinase manipulations conferred on conduction delays (see Fig. 6). Even though H89 tended to induce a greater APD prolongation in the RV at 30 min of the drug perfusion, there was no interchamber difference in APD at 45 min of the drug perfusion (see Fig. 11D), and yet there was an enormous interchamber difference in conduction delays at this time point (see Fig. 6D). Hence, we conclude that the differential regulation of conduction in the RV and the LV by kinase/phosphatase activity is independent on the regulation of the APD.
Dynamic nature of regulation of cardiac excitability by “stress” kinases.
The results presented above have demonstrated that blockade of CaMKII, PKA, or both, depress ventricular conduction (preferentially in the RV) in the range of heart rates (150–400 beats/min), which fully covers the physiological range of heart rates in the rabbit (~160–270 beats/min; see Ref. 62). However, previous studies demonstrated that altering CaMKII activity had a notable effect on the intermediate inactivation of INa (2, 89). Specifically, these studies indicated that intermediate inactivation is enhanced by CaMKII inhibition (89) and is reduced by increasing CaMKII activity (2). The time of recovery from intermediate inactivation is comparable to the normal sinus node pacemaker CL (62). We surmised that if the depression of ventricular conduction caused by CaMKII and/or PKA blockade is mainly due to an increase in intermediate inactivation of INa, then prolonging the pacing CL well beyond the physiological range should fully abolish the effects of the kinase blockers.
Data depicted in Fig. 12 obtained from two hearts respectively perfused with KN93 or H89 provide a strong support for this conjecture. At the designated time points, each heart was subject to a change in CL from 400 ms to the longest possible CL short of 8,000 ms. Figure 12A shows the activation maps for two consecutive activations recorded either at baseline or after 1 h of perfusion with KN93. Note that at baseline both the activation triggered at a CL of 400 ms and the subsequent one triggered at a CL of 8,000 ms elicit fast activation sequences across the ventricle surface (Fig. 12, A and B, top), which are associated with a uniform distribution of local conduction delay values (Fig. 12B, top). However, after 60-min perfusion with KN93, propagation of the impulse elicited by LV pacing at CL of 400 ms is markedly disturbed such that there is a region of conduction slowing that is most prominent in the basal part of the RV (Fig. 12A, bottom left). Accordingly, the associated conduction delay map exhibits markedly increased values especially in the RV (Fig. 12B, bottom left). Increasing the pacing CL from 400 to 4,000 ms eliminated the observed KN93-induced defects, thus effectively restoring the conduction pattern back to baseline levels on a beat-to-beat basis (Fig. 12, A and B, bottom right). Subsequent switch to the pacing CL of 400 ms immediately reverted the conduction patterns back to the highly abnormal pattern exhibiting RV conduction slowing (not shown).
Fig. 12.

Conduction disturbances elicited by KN93 or H89 are eliminated at very long pacing intervals. A: activation time maps computed for 2 consecutive activations [cycle length (CL) as identified by corresponding labels] generated by left ventricle (LV) pacing at baseline (top) and after 1-h perfusion with KN93 (bottom). White arrowheads indicate the site of pacing in the lateral LV. B: local delay maps depict the distribution of conduction delays for each corresponding activation map in A (one-to-one correspondence as indicated by CL labels). C and D: same layout as A and B but for H89 in place of KN93. White line demarcates the right ventricle (RV) and the LV. Prolongation of the pacing CL to an unphysiologically long interval (4,000–8,000 ms) almost fully eliminates severe slowing (and possibly full block) of conduction in the RV outflow tract induced by KN93 or by H89 at CL = 400 ms.
Figure 12, C and D, shows the results obtained for a similar experiment carried out with H89 in place of KN93. As with KN93, the drug-induced RV-centric slowing of activation fronts (Fig. 12C, bottom left) and associated local conduction delay increases (Fig. 12D, yellow/red pixels) were abolished on a beat-to-beat basis when the CL was switched from 400 to 8,000 ms (Fig. 12, C and D, bottom right).
These results strongly support the idea that the depressant effect exerted by kinase block on ventricular (especially RV) conduction is largely dynamic and is mediated via dysregulation of the intermediate inactivation of INa (2, 89).
Inhibition of either CaMKII or PKA leads to time- and rate-dependent conduction abnormalities and VF.
In all experimental groups, baseline pacing in the 150- to 400-ms CL range elicited fast and uniform ventricular conduction devoid of any arrhythmic events. In the sham group, this held true for the entire 60-min period of vehicle perfusion. In stark contrast, either CaMKII or PKA blockade was associated with an increased vulnerability to develop abnormal patterns of conduction, manifested as 1) the inability of some regions to follow 1:1 pacing; 2) a local turbulent conduction; and 3) reentrant ventricular tachycardia (VT)/VF. In the vast majority of cases, the epicenter of conduction abnormalities was located in the basal RV, approximately corresponding to the RVOT. In general, the incidence of these abnormal conduction episodes increased with the time of drug perfusion as well as with increases in the pacing rate. For example, after 30 min of drug delivery all hearts perfused with KN93 or H89 and paced at 400 ms CL from the LV exhibited 1:1 propagation of the impulse throughout the ventricles. However, when the CL was subsequently shortened to 300 ms only 66.6% (KN93) or 40% (H89) of the hearts exhibited completely unimpaired 1:1 propagation during LV pacing. After 60 min of drug perfusion, none of the hearts in KN93 and H89 groups were able to respond in a uniform 1:1 manner to pacing at the CL of 300 ms. At this time point, none of the hearts in the H89 group, and only four to six hearts in the KN93 group, could be effectively paced 1:1 even at the relatively long CL of 400 ms.
Figure 13 presents an example of rate-dependent conduction abnormalities leading to VF in a typical KN93 experiment (the data are obtained from Supplemental Movie S1: https://doi.org/10.5281/zenodo.2573179). Figure 13A, middle right, shows a time-space plot (TSP) constructed for the diagonal dashed line x-x′ crossing the RV and the LV as indicated in Fig. 13A, left. The TSP is generated by stacking, from left to right, images of the x-x′ line from consecutive frames of the movie recorded during LV pacing at CLs of 400, 300, and 260 ms. The optical action potential (OAP) sequences shown above and below the time-space plot in Fig. 13A, were obtained from points a and b located in the RV and the LV, respectively, and correspond to fluorescence profiles along the dashed red lines labeled a and b in the TSP. Note that during pacing at the CL of 400 ms the excitation waves propagate in a 1:1 manner across the LV and the RV, with normal and constant OAP shapes in points a and b. However, upon the transition to the CL of 300 ms, the OAP sequence in point a (in the RV) starts exhibiting a 2:1 pattern (alternans), including beat-to-beat changes in the slope of the upstroke as well as in the amplitude of the OAP (the 1st abnormal activation is identified). Upon the transition to the CL of 260 ms, the OAP sequence in point a becomes more complex and chaotic. In the spatial domain, this corresponds to a shifting position of a conduction discontinuity (interrupted white bands in the TSP). All the while, the OAP in point b (in the LV) maintains constant beat-to-beat shape, and the excitation bands in the TSP propagate continuously into the RV until they reach a vulnerable region somewhere near point a.
Fig. 13.

Development of abnormal conduction and arrhythmia during perfusion with KN93. A, left: snapshot of fluorescence recorded from a 30-min KN93-treated heart during a representative episode of left ventricle (LV)-paced activation at incremental cycle lengths (CLs) (400, 300, and 260 ms), which leads to the development of abnormal conduction in the right ventricle (RV). The solid white line indicates the location of the left anterior descending coronary artery (LAD) demarcating the RV and the LV. The dashed white line identifies the row of pixels between x and x′ positions used to construct the time-space-plot (TSP) shown in the rightmost section of A. Red pixels labeled a and b identify the location (RV and LV, respectively) of the optical action potential recordings depicted above (a) and below (b) the TSP. The white line on the TSP indicates the position of the LAD. The dashed red lines indicate positions of points a and b as labeled. Horizontal double-sided arrows identify the intervals during which a given pacing CL was applied as indicated by the corresponding labels. The asterisk identifies the first abnormal activation. B: distribution of the action potential phase at 0, 0.5, and 1 s of a sustained ventricular fibrillation elicited by a pacing protocol similar to that depicted in A during KN93 perfusion in the same heart. White circles identify the location of phase singularities.
Importantly, these types of events were ubiquitous in all KN93 experiments and shared a similar spatial organization regardless of the CL triggering the conduction abnormality or the time of drug perfusion. Often, paced-induced conduction abnormalities resulted in the initiation of self-sustained arrhythmia episodes. Figure 13B shows snapshots of a Hilbert-transformed phase movie at three different time points during self-sustained arrhythmia in the same heart as shown in Fig. 13A (Supplemental Movie S2, movie of phase: https://doi.org/10.5281/zenodo.2573181). Given the presence of multiple phase singularities that constitute the center of rotation of spiral waves, we classify this arrhythmia as VF.
Figure 14 presents an example of rate-dependent conduction abnormalities leading to an arrhythmia in a typical H89 experiment (the data were obtained from Supplemental Movie S3: https://doi.org/10.5281/zenodo.2573187). The layout of Fig. 14 is the same as that of Fig. 13. During pacing at the CL of 400 ms, the excitation waves propagate in a 1:1 manner across the LV and the RV, with constant OAP shapes in points a (RV) and b (LV). Upon the transition to the CL of 300 ms the activation becomes extremely slow (1st such beat is indicated), and then, eventually the OAP sequence in point a (in the RV) loses ability to follow excitation waves coming from the LV and settles on responding to every other excitation (2:1 block). This pattern persists after switching to the CL of 260 ms. The TSP shows a rather well-defined position of conduction block occurring every other beat, which is located in the RV close to the LAD. All the while, the LV maintains the ability to respond to pacing in 1:1 manner. As with KN93, perfusion with H89 was associated with the initiation of sustained arrhythmia such as VT or VF. Figure 14B shows snapshots of a Hilbert-transformed phase movie at three different time points during self-sustained arrhythmia in the same heart as shown in Fig. 14A (Supplemental Movie S4, movie of phase: https://doi.org/10.5281/zenodo.2573190). In this case, the arrhythmia was driven by a single rotor with the center of rotation located in the RV and hence was classified as a reentrant VT.
Fig. 14.

Development of abnormal conduction and arrhythmia during perfusion with H89. A, left: snapshot of fluorescence recorded from a 30-min H89-treated heart during a representative episode of left ventricle (LV)-paced activation at incremental cycle lengths (CLs) (400, 300, and 260 ms), which leads to the development of abnormal conduction in the right ventricle (RV). The solid white line indicates the location of the left anterior descending coronary artery (LAD) demarcating the RV and the LV. The dashed white line identifies the row of pixels between x and x′ positions used to construct the time-space-plot (TSP) shown in the rightmost section of A. Red pixels labeled a and b identify the location (RV and LV, respectively) of the optical action potential recordings depicted above (a) and below (b) the TSP. The white line on the TSP indicates the position of the LAD. The dashed red lines indicate positions of points a and b as labeled. Horizontal double-sided arrows identify the intervals during which a given pacing CL was applied as indicated by the corresponding labels. The asterisk identifies the first extremely slow activation. B: distribution of the action potential phase at 0, 0.5, and 1 s of a sustained ventricular tachycardia elicited by a pacing protocol similar to that depicted in A during H89 perfusion in the same heart. White circles identify the location of phase singularities. Note a 2:1 block developing in the right ventricle (RV) after reducing the pacing cycle length (CL) from 400 to 300 ms (A, right). Arrhythmias induced by H89 were typically more organized than those induced by H89, often with a single rotor located in the RV as seen in B.
DISCUSSION
This study establishes the following important physiological facts. First, the ventricular conduction is positively regulated by “stress” kinases PKA and CaMKII and is negatively regulated by phosphatases, whose identity is yet to be determined. Second, this regulation is differential between ventricular chambers, with the RV exhibiting a higher sensitivity to changes in kinase/phosphatase activity, and this is associated with RV-specific increased expression levels of both the PKA catalytic subunit PKA-Cα and the CaMKII isoform CaMKII-δ. Finally, reduced activity of PKA and/or CaMKII can lead to the initiation of conduction disturbances and arrhythmia originating in the RV. We will discuss the relevant prior knowledge and implications for basic and clinical cardiac electrophysiology.
Regulation of ventricular conduction by kinases and phosphatases: INa.
Entry of sodium ions through the cardiac sodium channels is responsible for generating the electric impulse that underlies tissue excitability in ventricular myocardium. Thus INa regulation by kinases/phosphatases directly impacts cellular excitability. It is well established that Nav1.5, the INa pore-forming α-subunit of the main cardiac sodium channel, contains various phosphorylation sites that are mostly located in the first intracellular linker loop of the channel (12, 30, 50, 51), some of which have been identified as targets for various protein kinases including CaMKII (5, 12, 25, 34), PKA (1, 55, 92), PKC (27), and others (20, 50). While an effective/optimal phosphorylation level is likely achieved by the combined action of protein kinases and phosphatases (14, 87), which phosphatases are responsible for this regulation and how dephosphorylation specificity for Nav1.5 is achieved remain largely unknown.
Conduction slowing and block induced by inhibition of CaMKII or PKA, or both, support the notion that the individual activity of each kinase upregulates INa, the principle generator of the ventricular excitation impulse. This notion is in agreement with those cellular studies that indicated that increasing/reducing either CaMKII or PKA activity was associated with increase/reduction of INa (2, 44, 52, 83, 85, 89) and is in contradiction with the cellular studies showing the opposite effect of either kinase (61, 70, 81).
The discrepant outcomes of the cellular studies mentioned above await explanation and resolution. The controversy regarding the regulation of cardiac excitability by CaMKII signaling pathway has recently translated to the intact heart level (74, 86) and could, at least in part, be due to off-target effects of the drugs used in whole heart experiments. However, taken together, our previous (85, 86) and current findings show that inhibition of CaMKII by KN93, inhibition of calmodulin (upstream regulator of CaMKII) with W7, and inhibition of PKA with H89 produce qualitatively similar pattern of conduction depression in the rabbit heart, with the epicenter of the effect in the basal RV approximately corresponding to the RVOT. The fact that these three drugs, dissimilar in chemical structure, have so similar a pattern of the effect, alleviates concerns regarding the drug specificity and reinforce the idea that both CaMKII and PKA positively regulate ventricular excitability.
The current work may be the first to investigate how PKA inhibition alters ventricular conduction in the intact heart. Our reported conduction depression induced by the PKA inhibitor H89 agrees well with previous studies showing that 1) increased adrenergic signals/activity induced by stellate ganglion stimulation leads to increases in conduction velocity in open chest pig hearts (3); 2) β-1 or β-2 stimulation increased conduction velocity in LV wedge preparations of human donor hearts (42); and 3) significant reduction in conduction velocity was observed in adenyl cyclase knockout mice designed to limit the activity of PKA through reduced production of cAMP (75).
The role of phosphatases in mediating/facilitating the reversible phosphorylation of numerous cellular proteins (up to 30% of all proteins) required for the regulation of the cell is well established (17, 31, 45). Previous studies designed to investigate the role of phosphatases in the regulation of cardiac contractility consistently reported that phosphatase inhibition using various drugs was associated with a positive inotropic response in cells and intact tissue (58–60). The reported effect is likely mediated, at least in part, by an increased calcium current induced by the phosphatase inhibition (22, 29, 59). However, there is a paucity of data addressing whether phosphatase activity controls cardiac excitability (45). To the best of our knowledge, our study is the first to report a positive dromotropic effect in intact ventricles following the inhibition of phosphatase activity using the broad-acting inhibitor calyculin. We should point out that calyculin exhibits similar potency for two major phosphatases, PP1 and PP2A (31). Dissecting the role of each of these phosphatases in the control of excitability remains a subject of future studies.
Considering that the depressant effect of kinase inhibition (observed using the CaMKII and PKA blockers KN93 and H89) likely is a consequence of reduced phosphorylation levels at the channel’s pore-forming protein, the improvement in conduction observed after perfusion with calyculin likely responds to an increased/protected level of Nav1.5 phosphorylation following the inhibition of phosphatase activity (i.e., the phosphorylation/dephosphorylation balance is shifted favoring phosphorylation). However, how the levels of Nav1.5 phosphorylation change in the presence of kinase/phosphatase inhibitors, as well as the effect that phosphatase blockers has on the biophysical properties of the ion channel, will require further studies. Additionally, it should be pointed out that inhibition of the phosphatases may potentially increase the activity of CaMKII, provided that the activated kinase has the capability to autophosphorylate. Such a self-induced modification then renders the kinase’s activity independent of calmodulin, the upstream activator of the kinase. Supporting this notion, the study by Huke et al. (33) showed that phosphatase inhibition by 1 μM calyculin increased phosphorylation at site 287, the established autophosphorylation site for cardiac CaMKII. Given the calyculin concentration used in that study was markedly greater than ours (1 μM vs. 30 nM), further studies specifically directed to determining how the phosphatase inhibitors alter the levels of CaMKII phosphorylation (vs. Nav1.5 phosphorylation levels) and how each of these changes modulates conduction have to be performed.
Regulation of ventricular conduction by kinases and phosphatases: other relevant channels.
In the cardiac syncytium, activation of INa initiates the circulation of local circuit currents that mediate transmission of the excitation pulse. Both the inward rectifying potassium channel IK1 and the cardiac gap junctions mediate the flow of local circuit currents, thus by their potential to modulate the magnitude/distribution of local circuit currents constitute key components of tissue excitability.
Of relevance, some studies suggest that connexin 43 (Cx43; primary channel protein constituting ventricle gap junctions) conductivity is upregulated by either PKA (13, 54) or CaMKII (32, 68); thus chamber-specific conduction slowing induced by KN93 and H89 may be mediated in part by reduced gap-junction conductance. Of note however, conductive properties of gap junctional channels are not time dependent, and thus the markedly CL-dependent phenomenon observed during inhibition of CaMKII and/or PKA cannot be readily attributed to changes in Cx43 conductance; altered recovery from intermediate inactivation of INa is a more likely candidate. It cannot be excluded, however, that regional differences in connexin distribution and subsequent differential response to the inhibitors may contribute in setting the stage for the increased RVOT vulnerability observed during perfusion with KN93/H89. On the other hand, data demonstrating that the phosphatase inhibitor okadaic acid increased Cx43 conductance (54) indicate that our reported improved conduction in response to calyculin perfusion may be in part due to changes in gap junctional conductance.
Adrenergic regulation of IK1 remains controversial, with some studies showing that adrenergic signals increased IK1 (23, 93), whereas others showed the reverse (41, 77). One study in rat myocytes showed that H89 inhibited IK1, reporting an IC50 for the blocker of 27.8 μM (64). Such drug-induced changes in the IK1 profile should theoretically have associated an increase in conduction velocity, which is opposite to our observations, suggesting that IK1 changes did not contribute to the H89-induced conduction slowing observed in our model. The same study showed that calyculin did not change the IK1 current-voltage profile, thus likely excluding a role for IK1 in calyculin-induced improved conduction in our model. On the other hand, inhibition of CaMKII with 1 μM KN93 attenuated calcium-dependent IK1 augmentation (57), which is in line with an CaMKII-induced IK1 increase reported by Wagner et al. (82). Again, reported kinase activity-induced changes in IK1 should theoretically result in conduction changes opposite to our observations suggesting that IK1 changes do not directly modulate conduction in our model.
The late sodium current (INaL) is an important component of INa, which has been implicated in arrhythmogenesis in various diseased states through its ability to prolong APD (7, 49). Of note, various studies agree upon the upregulation of INaL by CaMKII (2, 48, 81, 89), which includes the observation that KN93 perfusion reduced INaL (48, 89). Similarly, others showed that PKA increased INaL (83). Thus inhibition of either or both of the kinases should lead to reduced APD, which is contrary to our observations: this necessarily indicates that the net effect of kinase inhibition on all ion channels overrides the effect of the INaL changes. Importantly, our regional APD measurements indicate that there were no significant chamber specific differences, which as discussed above rules out the influence of APD on the observed chamber-specific conduction depression induced by kinase inhibition. We should also point out that theoretical APD reduction that resulted from INaL inhibition should/would in fact alleviate any reduction in INa caused by ineffective recovery from inactivation caused by the blockers. Thus kinase activity-induced changes in INaL are not responsible for conduction slowing in our model.
Mechanisms underlying enhanced sensitivity of conduction in the RV to kinase/phosphatase inhibition.
A novel finding of this work is the differential response of the RV and the LV conduction to the kinase and phosphatase inhibitors. For all drugs tested, the RV conduction was the most sensitive, exhibiting an increased vulnerability to conduction slowing in the case of the kinase inhibitors KN93 and H89, and a stronger acceleration of conduction when the phosphatase inhibitor calyculin was used.
The different sensitivity of the RV versus the LV conduction to kinase/phosphatase block could be related to chamber-specific differences in the repolarization, which would lead to differing diastolic intervals at a fixed pacing CL. However, under all experimental conditions, there were no significant differences in the APD between the RV and the LV, excluding this possibility.
As we discussed previously (85), it is possible that inherent chamber-specific differences in INa result in a reduced RV depolarization reserve, which is concealed under normal conditions due to a nonlinear (saturating) relationship between maximal INa and conduction velocity, creating a so-called safety margin for conduction (11, 71). Indeed, Veeraraghavan et al. (80) showed that Nav1.5 protein levels in the RV were reduced as compared with the LV in guinea pig hearts. In addition, we previously showed that the dV/dtmax of the action potential upstroke in rabbit myocytes isolated from the RVOT was reduced as compared with that measured in myocytes isolated from the LV (85), suggesting inherent differences in the peak INa between these two regions. Because of the existence of the safety margin, the underlying differences in depolarization reserve (RV < LV) may be unmasked only when INa is further reduced due to loss-of-function Nav1.5 channelopathy, or INa inhibition, or INa downregulation by signaling pathways. However, our experiments using TTX at the concentration of 1 μM failed to produce a chamber-specific effect on conduction. TTX caused a rather uniform increase in conduction delays (at CL = 400 ms, ~30% in each chamber, see Fig. 8).
TTX at the concentration of 1 μM is expected to reduce the maximal density of INa by ~50% (16). To put this into a pathophysiological perspective, such a degree of INa suppression is roughly equivalent to that caused by two genetic modifications in mice, Nav1.5 haploinsufficiency (63) and Nav1.5 1798InsD mutation (69). Both of these transgenic mice served the purpose of experimental modeling Brugada syndrome (BrS) (10). The signature electrophysiological abnormality of BrS is a preferential conduction slowing in the RVOT (91), and in both abovementioned transgenic mouse models epicardial mapping revealed exaggerated slowing of conduction in the RV/RVOT due to the loss-of-function INa mutation (69, 79). In our model, a supposedly similar reduction of INa by TTX was not sufficient to expose the RV/RVOT vulnerability. Clearly, the direct comparison between acute pharmacologic blockade of INa and genetically induced INa loss-of-function is difficult, especially given into account other factors, such as different species and different pacing/mapping configurations. However, it remains significant that in our study blockade of CaMKII by KN93 produced a relatively higher chamber-specific effect than estimated 50% blockade of INa by TTX. At a 400-ms CL, KN93 (45 min perfusion) caused a ~30% increase in LV conduction versus an ~85% increase in RV conduction delays. This outcome cannot be explained by the intrinsic heterogeneity in INa alone (80), because TTX, while having the same effect in the LV, could not match the effect of KN93 in the RV.
This brings us to an alternative/additional possibility that the differential sensitivity of the RV versus the LV to kinase/phosphatase block is due to regional differences in the expression and/or function of kinases or phosphatases. Supporting this, our study shows that the protein expression levels of CaMKII-δ (principle ventricular CaMKII isoform) and PKA-Cα (PKA catalytic subunit) are higher in the RVOT than in the apical LV (see Fig. 10). To the best of our knowledge, this is the first report of region-specific expression of these kinases in the ventricles. Interestingly, the study by DeGrande et al. (19) reported an increased PP2A catalytic and scaffolding subunit expression in the RV as compared with the LV of human hearts. Although a more detailed regional analysis of kinase/phosphatase expression in the ventricles is pending, it appears that either the entire RV, or specifically the RVOT, is more dependent on the dynamic phosphorylation/dephosphorylation activity than the LV. Additional layers of chamber- or region-specific regulation may arise from reported chamber-specific differences in the upstream signals for CaMKII (39) as well as for PKA (53).
Summarizing the currently available information, it appears that the reduced expression of Nav1.5 in at least the basal regions of the RV is naturally counterbalanced by increased expression of stress kinases and at least one major phosphatase. This fundamental physiological arrangement renders RV (and especially RVOT) more vulnerable to conduction failure in the context of reduced kinase activity (i.e., due to pharmacological inhibition) and may have other implications for various cardiac diseases that differentially affect ventricular chambers.
Rate dependence of the kinase block effects.
The effects of both CaMKII and PKA blockade were strongly rate dependent. Additionally, this detrimental rate-dependent conduction depression was much more prominent in the RV, which led to diverging conduction delay values between the chambers and ultimately underlain the formation of conduction abnormalities in the RV. Previous experiments in isolated rat cardiomyocytes suggested that the INa loss-of-function induced by CaMKII inhibition is likely due to a combination of changes in the kinetic properties of INa, including a reduction in the peak current, shift of the steady-state inactivation to more negative potentials, delayed recovery from slow inactivation, and enhancement of intermediate inactivation (89). Regarding the rate dependency of KN93 effect on ventricular (and especially RV) conduction, it is tempting to speculate that it is due to the strong prolongation of the recovery from the slow inactivation of INa induced by this drug (89). In the cited study, at baseline conditions the peak INa reached >90% of the prepulse level after 10–20 ms of return to the hyperpolarizing potential, whereas in the presence of KN93 the same level of recovery was achieved only after hyperpolarizing intervals lasting longer than 1,000 ms. Note that this interval goes well beyond the physiological range of diastolic intervals in both rats and rabbits (and humans, for that matter). These results agree well with our observations that at the baseline there was no change in conduction velocity even at diastolic intervals as short as 30 ms, whereas in the presence of KN93 conduction slowing was observed even at diastolic intervals as long as 200 ms. Yet, the conduction derangements caused by either CaMKII or PKA blockade virtually disappeared by simply prolonging the pacing CL from 400 ms to several seconds (see Fig. 12). Pending detailed patch-clamp studies addressing the effect of PKA inhibition on the INa kinetics, our findings predict that PKA regulates INa recovery from inactivation in a manner similar to that of CaMKII.
The progressive nature of conduction slowing induced by CaMKII and PKA inhibition.
Our experiments identified a time-dependent effect of both KN93 and H89 alone or in combination, such that conduction slowing was progressively worsened for at least 60 min of drug perfusion. This could be due to an altered Na+ channel redistribution/trafficking towards the plasma membrane. For example, Hallaq et al. (28) showed that PKA activation led to increased trafficking of cardiac Na+ channels to the plasma membrane in HEK 293 cells stably expressing Nav1.5 (28). However, the fact that severe conduction slowing caused by blockade of either PKA or CaMKII was virtually abolished just by switching the interbeat interval from 400 to 4–8 s (Figs. 12) speaks against this possibility. A more plausible explanation for the progressive deterioration in conduction is the persistent activity of phosphatases, which in the absence of a phosphorylation due to kinase blockade, gradually shifts the phosphorylation/dephosphorylation balance of Nav1.5 channels toward an unphosphorylated state.
Insights into the molecular mechanism underlying the adverse synergism of KN93 and H89.
It is noteworthy that blocking PKA or CaMKII in the intact rabbit hearts yielded a remarkably similar outcome regarding impulse conduction. Moreover, the combined effect of KN93 and H89 in the RV exceeded the sum of the individual effects (see Fig. 7). This finding would be in line with those cellular studies that showed that both CaMKII and PKA activities upregulate INa (2, 44, 52, 83, 85, 89). Studies from different groups revealed that both PKA and CaMKII phosphorylate the cardiac α-subunit (Nav1.5) at multiple residues (1, 5, 12, 25, 34, 55, 92), the majority located on the intracellular/cytosolic loop connecting domains 1 and 2 (1st intracellular linker loop) (12, 30, 50, 51). It remains undetermined whether there are targets in Nav1.5 shared by the two kinases. However, Ser571, a PKA phosphorylation site in the first intracellular linker loop predicted by the NetPhosK algorithm (9), was also identified as a principle substrate for CaMKII (34). Even if there are no shared targets, the close spatial association between the Nav1.5 residues phosphorylated either by PKA or CaMKII may result in similar conformational changes of the channel protein, underlying similar regulation of INa by the two kinases. In this regard, it is worth noting that the combined activity of CaMKII and PKA has been found to be an inherent part of Ca2+ cycling regulation (21, 67). Similarly, optimal fine-tuning of ventricular conduction may require dual function of PKA and CaMKII.
The synergistic detrimental effect observed with the dual CaMKII/PKA inhibition is intriguing, and various scenarios are plausible. On the one hand the negative synergy may be related to the close spatial association of the target residues and involves a nonlinear additive effect triggered by the dephosphorylation of the closely abutted CaMKII- and PKA-targeted sites; alternatively, we may speculate that the kinases target previously described distinct Nav1.5 populations (43), such that the combined downregulation of all channels following dual kinase inhibition is immediately catastrophic; yet a third possibility would be that the signaling path involving upregulation of INa by both CaMKII and PKA involves an organization of the signals such that CaMKII and PKA paths are (at least partially) intertwined as previously suggested (26).
Implications for basic and clinical electrophysiology.
This study supports a physiological regulatory scheme in which cardiac excitability is finely tuned by the combined action of protein kinases (CaMKII and PKA) and phosphatases (yet to be determined), which most likely target Nav1.5 to enable robust and uniform cardiac impulse conduction. While teleologically, it is difficult to accept that ventricular conduction needs to be regulated at all, it is important to understand that the constitutive, basal activity of both CaMKII and PKA is necessary for maintenance of normal conduction in the ventricles. Moreover, the two kinases appear to work in tandem, as the double blockade shows adverse synergism with respect to conduction slowing (see Fig. 7). In the age of personalized medicine, it is important to understand that the pharmacological blockade of the “stress” kinases, or any intervention or condition leading to the negative shifts in the phosphorylation/dephosphorylation balance, is potentially proarrhythmic due to the intrinsically reduced depolarization reserve in the basal RV/RVOT. This is especially relevant to the Brugada syndrome (10) and the growing array of Brugada phenocopies (6, 66) given that the blockade of the “stress” kinases reproduces the signature of the Brugada syndrome, the conduction slowing in the RVOT (78, 91). It is possible that shifts or fluctuations in the balance of phosphorylation/dephosphorylation activity contribute to intermittent manifestations of Brugada syndrome and Brugada-like conditions in vulnerable individuals. Further studies are clearly necessary to fully understand the complex, multidimensional regulation of cardiac excitation by the dynamic interaction of relevant kinases and phosphatases.
Limitations.
The data were obtained in the hearts immobilized with the electromechanical uncoupler blebbistatin. Although it could affect the outcomes of the study, we previously observed the same principal phenomenon, i.e., vulnerability of RV/RVOT conduction to CaMKII block, in beating hearts (86).
Whilst impulse propagation occurs across the ventricular wall thickness, optical mapping of voltage sensitive dyes is limited to the recording of excitation on the cardiac surface, which may impose some limitations to the interpretation of the recorded data. In particular, a significant transmural component of the activation vector will lead to overestimation of the conduction velocity as assessed with the surface mapping (reaching local infinity when the wavefront is exactly perpendicular to the ventricular wall). Thus the recorded slowing of conduction following CaMKII/PKA inhibition provides the lower bound of the effect induced by the kinase inhibitors (meaning that the actual degree of slowing in some locations could be even greater than estimated). With the same logic, we cannot exclude that the conduction acceleration observed with the calyculin may have been overestimated. However, given the fact that each heart served as its own control, if the observed acceleration at the surface was a consequence of increased transmural component, it would still require an increased conduction velocity at some layer beneath the surface, which would still be in line with the notion that phosphatase inhibition upregulates excitability in some parts of the ventricles. Further experiments are needed to address whether the effect of kinase/phosphatase inhibitors may vary across the different transmural layers of the ventricular myocardium.
In our previous publication we pointed out that KN93 has some reported off-target effects (85). While H89 is widely used as a PKA inhibitor, some effects independent of PKA have also been described (56). Nevertheless, it is highly unlikely that the common signature effects of both drugs, conduction depression in the RVOT, is due to a shared off-target effect of these two chemically unrelated compounds, especially given that qualitatively similar effect is also achieved by using another physiologically relevant but chemically unrelated compound, the calmodulin blocker W7 (86).
GRANTS
This work was supported by the Nora Eccles Treadwell Foundation Research Grant (to A. V. Zaitsev) and National Heart, Lung, and Blood Institute Grants 1R01-HL-103877 (to A. V. Zaitsev) and R01-HL-128752 [to M. Warren (D. Dosdall, Principal Investigator)].
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.V.Z., J.S.W., and M.W. conceived and designed research; A.V.Z., N.S.T., K.M.C., A.D.S., J.S.W., and M.W. performed experiments; A.V.Z., N.S.T., K.M.C., A.D.S., J.S.W., and M.W. analyzed data; A.V.Z. and M.W. interpreted results of experiments; A.V.Z., J.S.W., and M.W. prepared figures; A.V.Z., J.S.W., and M.W. drafted manuscript; A.V.Z. and M.W. edited and revised manuscript; A.V.Z. and M.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Wilson Lobaina and Bruce Steadman for invaluable help with various hardware and software necessary for this study.
REFERENCES
- 1.Aiba T, Farinelli F, Kostecki G, Hesketh GG, Edwards D, Biswas S, Tung L, Tomaselli GF. A mutation causing Brugada syndrome identifies a mechanism for altered autonomic and oxidant regulation of cardiac sodium currents. Circ Cardiovasc Genet 7: 249–256, 2014. [Erratum in Circ Cardiovasc Genet 7: 570, 2014]. doi: 10.1161/CIRCGENETICS.113.000480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aiba T, Hesketh GG, Liu T, Carlisle R, Villa-Abrille MC, O’Rourke B, Akar FG, Tomaselli GF. Na+ channel regulation by Ca2+/calmodulin and Ca2+/calmodulin-dependent protein kinase II in guinea-pig ventricular myocytes. Cardiovasc Res 85: 454–463, 2010. doi: 10.1093/cvr/cvp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ajijola OA, Lux RL, Khahera A, Kwon O, Aliotta E, Ennis DB, Fishbein MC, Ardell JL, Shivkumar K. Sympathetic modulation of electrical activation in normal and infarcted myocardium: implications for arrhythmogenesis. Am J Physiol Heart Circ Physiol 312: H608–H621, 2017. doi: 10.1152/ajpheart.00575.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol 51: 468–473, 2011. doi: 10.1016/j.yjmcc.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashpole NM, Herren AW, Ginsburg KS, Brogan JD, Johnson DE, Cummins TR, Bers DM, Hudmon A. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J Biol Chem 287: 19856–19869, 2012. doi: 10.1074/jbc.M111.322537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baranchuk A, Nguyen T, Ryu MH, Femenía F, Zareba W, Wilde AA, Shimizu W, Brugada P, Pérez-Riera AR. Brugada phenocopy: new terminology and proposed classification. Ann Noninvasive Electrocardiol 17: 299–314, 2012. doi: 10.1111/j.1542-474X.2012.00525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bengel P, Ahmad S, Sossalla S. Inhibition of late sodium current as an innovative antiarrhythmic strategy. Curr Heart Fail Rep 14: 179–186, 2017. doi: 10.1007/s11897-017-0333-0. [DOI] [PubMed] [Google Scholar]
- 8.Bers DM, Grandi E. Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J Cardiovasc Pharmacol 54: 180–187, 2009. doi: 10.1097/FJC.0b013e3181a25078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blom N, Sicheritz-Pontén T, Gupta R, Gammeltoft S, Brunak S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4: 1633–1649, 2004. doi: 10.1002/pmic.200300771. [DOI] [PubMed] [Google Scholar]
- 10.Brugada R, Campuzano O, Sarquella-Brugada G, Brugada J, Brugada P. Brugada syndrome. Methodist DeBakey Cardiovasc J 10: 25–28, 2014. doi: 10.14797/mdcj-10-1-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buchanan JW Jr, Saito T, Gettes LS. The effects of antiarrhythmic drugs, stimulation frequency, and potassium-induced resting membrane potential changes on conduction velocity and dV/dtmax in guinea pig myocardium. Circ Res 56: 696–703, 1985. doi: 10.1161/01.RES.56.5.696. [DOI] [PubMed] [Google Scholar]
- 12.Burel S, Coyan FC, Lorenzini M, Meyer MR, Lichti CF, Brown JH, Loussouarn G, Charpentier F, Nerbonne JM, Townsend RR, Maier LS, Marionneau C. C-terminal phosphorylation of NaV1.5 impairs FGF13-dependent regulation of channel inactivation. J Biol Chem 292: 17431–17448, 2017. doi: 10.1074/jbc.M117.787788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burt JM, Spray DC. Inotropic agents modulate gap junctional conductance between cardiac myocytes. Am J Physiol Heart Circ Physiol 254: H1206–H1210, 1988. doi: 10.1152/ajpheart.1988.254.6.H1206. [DOI] [PubMed] [Google Scholar]
- 14.Chiang DY, Heck AJ, Dobrev D, Wehrens XH. Regulating the regulator: Insights into the cardiac protein phosphatase 1 interactome. J Mol Cell Cardiol 101: 165–172, 2016. doi: 10.1016/j.yjmcc.2016.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem 265: 5267–5272, 1990. [PubMed] [Google Scholar]
- 16.Cohen CJ, Bean BP, Colatsky TJ, Tsien RW. Tetrodotoxin block of sodium channels in rabbit Purkinje fibers. Interactions between toxin binding and channel gating. J Gen Physiol 78: 383–411, 1981. doi: 10.1085/jgp.78.4.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen P. The structure and regulation of protein phosphatases. Annu Rev Biochem 58: 453–508, 1989. doi: 10.1146/annurev.bi.58.070189.002321. [DOI] [PubMed] [Google Scholar]
- 18.Coraboeuf E, Deroubaix E, Coulombe A. Effect of tetrodotoxin on action potentials of the conducting system in the dog heart. Am J Physiol Heart Circ Physiol 236: H561–H567, 1979. doi: 10.1152/ajpheart.1979.236.4.H561. [DOI] [PubMed] [Google Scholar]
- 19.DeGrande ST, Little SC, Nixon DJ, Wright P, Snyder J, Dun W, Murphy N, Kilic A, Higgins R, Binkley PF, Boyden PA, Carnes CA, Anderson ME, Hund TJ, Mohler PJ. Molecular mechanisms underlying cardiac protein phosphatase 2A regulation in heart. J Biol Chem 288: 1032–1046, 2013. doi: 10.1074/jbc.M112.426957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeMarco KR, Clancy CE. Cardiac Na channels: structure to function. Curr Top Membr 78: 287–311, 2016. doi: 10.1016/bs.ctm.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dries E, Santiago DJ, Johnson DM, Gilbert G, Holemans P, Korte SM, Roderick HL, Sipido KR. Calcium/calmodulin-dependent kinase II and nitric oxide synthase 1-dependent modulation of ryanodine receptors during β-adrenergic stimulation is restricted to the dyadic cleft. J Physiol 594: 5923–5939, 2016. doi: 10.1113/JP271965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erxleben C, Gomez-Alegria C, Darden T, Mori Y, Birnbaumer L, Armstrong DL. Modulation of cardiac Ca(V)1.2 channels by dihydropyridine and phosphatase inhibitor requires Ser-1142 in the domain III pore loop. Proc Natl Acad Sci USA 100: 2929–2934, 2003. doi: 10.1073/pnas.2628046100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gadsby DC. Beta-adrenoceptor agonists increase membrane K+ conductance in cardiac Purkinje fibres. Nature 306: 691–693, 1983. doi: 10.1038/306691a0. [DOI] [PubMed] [Google Scholar]
- 24.Garg V, Taylor T, Warren M, Venable P, Sciuto K, Shibayama J, Zaitsev A. β-Adrenergic stimulation and rapid pacing mutually promote heterogeneous electrical failure and ventricular fibrillation in the globally ischemic heart. Am J Physiol Heart Circ Physiol 308: H1155–H1170, 2015. doi: 10.1152/ajpheart.00768.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glynn P, Musa H, Wu X, Unudurthi SD, Little S, Qian L, Wright PJ, Radwanski PB, Gyorke S, Mohler PJ, Hund TJ. Voltage-gated sodium channel phosphorylation at Ser571 regulates late current, arrhythmia, and cardiac function in vivo. Circulation 132: 567–577, 2015. doi: 10.1161/CIRCULATIONAHA.114.015218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grimm M, Brown JH. Beta-adrenergic receptor signaling in the heart: role of CaMKII. J Mol Cell Cardiol 48: 322–330, 2010. doi: 10.1016/j.yjmcc.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hallaq H, Wang DW, Kunic JD, George AL Jr, Wells KS, Murray KT. Activation of protein kinase C alters the intracellular distribution and mobility of cardiac Na+ channels. Am J Physiol Heart Circ Physiol 302: H782–H789, 2012. doi: 10.1152/ajpheart.00817.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hallaq H, Yang Z, Viswanathan PC, Fukuda K, Shen W, Wang DW, Wells KS, Zhou J, Yi J, Murray KT. Quantitation of protein kinase A-mediated trafficking of cardiac sodium channels in living cells. Cardiovasc Res 72: 250–261, 2006. doi: 10.1016/j.cardiores.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 29.Hartzell HC, Hirayama Y, Petit-Jacques J. Effects of protein phosphatase and kinase inhibitors on the cardiac L-type Ca current suggest two sites are phosphorylated by protein kinase A and another protein kinase. J Gen Physiol 106: 393–414, 1995. doi: 10.1085/jgp.106.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herren AW, Weber DM, Rigor RR, Margulies KB, Phinney BS, Bers DM. CaMKII phosphorylation of Na(V)1.5: novel in vitro sites identified by mass spectrometry and reduced S516 phosphorylation in human heart failure. J Proteome Res 14: 2298–2311, 2015. doi: 10.1021/acs.jproteome.5b00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herzig S, Neumann J. Effects of serine/threonine protein phosphatases on ion channels in excitable membranes. Physiol Rev 80: 173–210, 2000. doi: 10.1152/physrev.2000.80.1.173. [DOI] [PubMed] [Google Scholar]
- 32.Huang RY, Laing JG, Kanter EM, Berthoud VM, Bao M, Rohrs HW, Townsend RR, Yamada KA. Identification of CaMKII phosphorylation sites in connexin43 by high-resolution mass spectrometry. J Proteome Res 10: 1098–1109, 2011. doi: 10.1021/pr1008702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huke S, Bers DM. Temporal dissociation of frequency-dependent acceleration of relaxation and protein phosphorylation by CaMKII. J Mol Cell Cardiol 42: 590–599, 2007. doi: 10.1016/j.yjmcc.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hund TJ, Koval OM, Li J, Wright PJ, Qian L, Snyder JS, Gudmundsson H, Kline CF, Davidson NP, Cardona N, Rasband MN, Anderson ME, Mohler PJ. A β(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest 120: 3508–3519, 2010. doi: 10.1172/JCI43621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ishihara H, Ozaki H, Sato K, Hori M, Karaki H, Watabe S, Kato Y, Fusetani N, Hashimoto K, Uemura D. Calcium-independent activation of contractile apparatus in smooth muscle by calyculin-A. J Pharmacol Exp Ther 250: 388–396, 1989. [PubMed] [Google Scholar]
- 36.Khoo MS, Kannankeril PJ, Li J, Zhang R, Kupershmidt S, Zhang W, Atkinson JB, Colbran RJ, Roden DM, Anderson ME. Calmodulin kinase II activity is required for normal atrioventricular nodal conduction. Heart Rhythm 2: 634–640, 2005. doi: 10.1016/j.hrthm.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 37.Kirschner LS, Yin Z, Jones GN, Mahoney E. Mouse models of altered protein kinase A signaling. Endocr Relat Cancer 16: 773–793, 2009. doi: 10.1677/ERC-09-0068. [DOI] [PubMed] [Google Scholar]
- 38.Kneller J, Kalifa J, Zou R, Zaitsev AV, Warren M, Berenfeld O, Vigmond EJ, Leon LJ, Nattel S, Jalife J. Mechanisms of atrial fibrillation termination by pure sodium channel blockade in an ionically-realistic mathematical model. Circ Res 96: e35–e47, 2005. doi: 10.1161/01.RES.0000160709.49633.2b. [DOI] [PubMed] [Google Scholar]
- 39.Kondo RP, Dederko DA, Teutsch C, Chrast J, Catalucci D, Chien KR, Giles WR. Comparison of contraction and calcium handling between right and left ventricular myocytes from adult mouse heart: a role for repolarization waveform. J Physiol 571: 131–146, 2006. doi: 10.1113/jphysiol.2005.101428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kotecha D, Flather MD, Altman DG, Holmes J, Rosano G, Wikstrand J, Packer M, Coats AJ, Manzano L, Böhm M, van Veldhuisen DJ, Andersson B, Wedel H, von Lueder TG, Rigby AS, Hjalmarson Å, Kjekshus J, Cleland JG; Beta-Blockers in Heart Failure Collaborative Group . Heart rate and rhythm and the benefit of beta-blockers in patients with heart failure. J Am Coll Cardiol 69: 2885–2896, 2017. doi: 10.1016/j.jacc.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 41.Koumi S, Backer CL, Arentzen CE, Sato R. beta-Adrenergic modulation of the inwardly rectifying potassium channel in isolated human ventricular myocytes. Alteration in channel response to beta-adrenergic stimulation in failing human hearts. J Clin Invest 96: 2870–2881, 1995. doi: 10.1172/JCI118358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lang D, Holzem K, Kang C, Xiao M, Hwang HJ, Ewald GA, Yamada KA, Efimov IR. Arrhythmogenic remodeling of β2 versus β1 adrenergic signaling in the human failing heart. Circ Arrhythm Electrophysiol 8: 409–419, 2015. doi: 10.1161/CIRCEP.114.002065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin X, Liu N, Lu J, Zhang J, Anumonwo JM, Isom LL, Fishman GI, Delmar M. Subcellular heterogeneity of sodium current properties in adult cardiac ventricular myocytes. Heart Rhythm 8: 1923–1930, 2011. doi: 10.1016/j.hrthm.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu T, Lee HC, Kabat JA, Shibata EF. Modulation of rat cardiac sodium channel by the stimulatory G protein alpha subunit. J Physiol 518: 371–384, 1999. doi: 10.1111/j.1469-7793.1999.0371p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lubbers ER, Mohler PJ. Roles and regulation of protein phosphatase 2A (PP2A) in the heart. J Mol Cell Cardiol 101: 127–133, 2016. doi: 10.1016/j.yjmcc.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res 92: 904–911, 2003. doi: 10.1161/01.RES.0000069685.20258.F1. [DOI] [PubMed] [Google Scholar]
- 47.Maltsev VA, Lakatta EG. Synergism of coupled subsarcolemmal Ca2+ clocks and sarcolemmal voltage clocks confers robust and flexible pacemaker function in a novel pacemaker cell model. Am J Physiol Heart Circ Physiol 296: H594–H615, 2009. doi: 10.1152/ajpheart.01118.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maltsev VA, Reznikov V, Undrovinas NA, Sabbah HN, Undrovinas A. Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: similarities and differences. Am J Physiol Heart Circ Physiol 294: H1597–H1608, 2008. doi: 10.1152/ajpheart.00484.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maltsev VA, Sabbah HN, Higgins RS, Silverman N, Lesch M, Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation 98: 2545–2552, 1998. doi: 10.1161/01.CIR.98.23.2545. [DOI] [PubMed] [Google Scholar]
- 50.Marionneau C, Abriel H. Regulation of the cardiac Na+ channel NaV1.5 by post-translational modifications. J Mol Cell Cardiol 82: 36–47, 2015. doi: 10.1016/j.yjmcc.2015.02.013. [DOI] [PubMed] [Google Scholar]
- 51.Marionneau C, Lichti CF, Lindenbaum P, Charpentier F, Nerbonne JM, Townsend RR, Mérot J. Mass spectrometry-based identification of native cardiac Nav1.5 channel α subunit phosphorylation sites. J Proteome Res 11: 5994–6007, 2012. doi: 10.1021/pr300702c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matsuda JJ, Lee H, Shibata EF. Enhancement of rabbit cardiac sodium channels by beta-adrenergic stimulation. Circ Res 70: 199–207, 1992. doi: 10.1161/01.RES.70.1.199. [DOI] [PubMed] [Google Scholar]
- 53.Molina CE, Johnson DM, Mehel H, Spätjens RL, Mika D, Algalarrondo V, Slimane ZH, Lechêne P, Abi-Gerges N, van der Linde HJ, Leroy J, Volders PG, Fischmeister R, Vandecasteele G. Interventricular differences in β-adrenergic responses in the canine heart: role of phosphodiesterases. J Am Heart Assoc 3: e000858, 2014. doi: 10.1161/JAHA.114.000858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moreno AP, Sáez JC, Fishman GI, Spray DC. Human connexin43 gap junction channels. Regulation of unitary conductances by phosphorylation. Circ Res 74: 1050–1057, 1994. doi: 10.1161/01.RES.74.6.1050. [DOI] [PubMed] [Google Scholar]
- 55.Murphy BJ, Rogers J, Perdichizzi AP, Colvin AA, Catterall WA. cAMP-dependent phosphorylation of two sites in the alpha subunit of the cardiac sodium channel. J Biol Chem 271: 28837–28843, 1996. doi: 10.1074/jbc.271.46.28837. [DOI] [PubMed] [Google Scholar]
- 56.Murray AJ. Pharmacological PKA inhibition: all may not be what it seems. Sci Signal 1: re4, 2008. doi: 10.1126/scisignal.122re4. [DOI] [PubMed] [Google Scholar]
- 57.Nagy N, Acsai K, Kormos A, Sebők Z, Farkas AS, Jost N, Nánási PP, Papp JG, Varró A, Tóth A. [Ca2+]i-induced augmentation of the inward rectifier potassium current (IK1) in canine and human ventricular myocardium. Pflugers Arch 465: 1621–1635, 2013. doi: 10.1007/s00424-013-1309-x. [DOI] [PubMed] [Google Scholar]
- 58.Neumann J, Boknik P, Herzig S, Schmitz W, Scholz H, Gupta RC, Watanabe AM. Evidence for physiological functions of protein phosphatases in the heart: evaluation with okadaic acid. Am J Physiol Heart Circ Physiol 265: H257–H266, 1993. doi: 10.1152/ajpheart.1993.265.1.H257. [DOI] [PubMed] [Google Scholar]
- 59.Neumann J, Bokník P, Herzig S, Schmitz W, Scholz H, Wiechen K, Zimmermann N. Biochemical and electrophysiological mechanisms of the positive inotropic effect of calyculin A, a protein phosphatase inhibitor. J Pharmacol Exp Ther 271: 535–541, 1994. [PubMed] [Google Scholar]
- 60.Neumann J, Herzig S, Boknik P, Apel M, Kaspareit G, Schmitz W, Scholz H, Tepel M, Zimmermann N. On the cardiac contractile, biochemical and electrophysiological effects of cantharidin, a phosphatase inhibitor. J Pharmacol Exp Ther 274: 530–539, 1995. [PubMed] [Google Scholar]
- 61.Ono K, Kiyosue T, Arita M. Isoproterenol, DBcAMP, and forskolin inhibit cardiac sodium current. Am J Physiol Cell Physiol 256: C1131–C1137, 1989. doi: 10.1152/ajpcell.1989.256.6.C1131. [DOI] [PubMed] [Google Scholar]
- 62.Opthof T, Coronel R, Rademaker HM, Vermeulen JT, Wilms-Schopman FJ, Janse MJ. Changes in sinus node function in a rabbit model of heart failure with ventricular arrhythmias and sudden death. Circulation 101: 2975–2980, 2000. doi: 10.1161/01.CIR.101.25.2975. [DOI] [PubMed] [Google Scholar]
- 63.Papadatos GA, Wallerstein PM, Head CE, Ratcliff R, Brady PA, Benndorf K, Saumarez RC, Trezise AE, Huang CL, Vandenberg JI, Colledge WH, Grace AA. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl Acad Sci USA 99: 6210–6215, 2002. doi: 10.1073/pnas.082121299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pearman C, Kent W, Bracken N, Hussain M. H-89 inhibits transient outward and inward rectifier potassium currents in isolated rat ventricular myocytes. Br J Pharmacol 148: 1091–1098, 2006. doi: 10.1038/sj.bjp.0706810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pellicena P, Schulman H. CaMKII inhibitors: from research tools to therapeutic agents. Front Pharmacol 5: 21, 2014. doi: 10.3389/fphar.2014.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peters CH, Abdelsayed M, Ruben PC. Triggers for arrhythmogenesis in the Brugada and long QT 3 syndromes. Prog Biophys Mol Biol 120: 77–88, 2016. doi: 10.1016/j.pbiomolbio.2015.12.009. [DOI] [PubMed] [Google Scholar]
- 67.Poláková E, Illaste A, Niggli E, Sobie EA. Maximal acceleration of Ca2+ release refractoriness by β-adrenergic stimulation requires dual activation of kinases PKA and CaMKII in mouse ventricular myocytes. J Physiol 593: 1495–1507, 2015. doi: 10.1113/jphysiol.2014.278051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Procida K, Jørgensen L, Schmitt N, Delmar M, Taffet SM, Holstein-Rathlou NH, Nielsen MS, Braunstein TH. Phosphorylation of connexin43 on serine 306 regulates electrical coupling. Heart Rhythm 6: 1632–1638, 2009. doi: 10.1016/j.hrthm.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Remme CA, Scicluna BP, Verkerk AO, Amin AS, van Brunschot S, Beekman L, Deneer VH, Chevalier C, Oyama F, Miyazaki H, Nukina N, Wilders R, Escande D, Houlgatte R, Wilde AA, Tan HL, Veldkamp MW, de Bakker JM, Bezzina CR. Genetically determined differences in sodium current characteristics modulate conduction disease severity in mice with cardiac sodium channelopathy. Circ Res 104: 1283–1292, 2009. doi: 10.1161/CIRCRESAHA.109.194423. [DOI] [PubMed] [Google Scholar]
- 70.Schubert B, VanDongen AM, Kirsch GE, Brown AM. Beta-adrenergic inhibition of cardiac sodium channels by dual G-protein pathways. Science 245: 516–519, 1989. doi: 10.1126/science.2547248. [DOI] [PubMed] [Google Scholar]
- 71.Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res 81: 727–741, 1997. doi: 10.1161/01.RES.81.5.727. [DOI] [PubMed] [Google Scholar]
- 72.Soni S, Scholten A, Vos MA, van Veen TA. Anchored protein kinase A signalling in cardiac cellular electrophysiology. J Cell Mol Med 18: 2135–2146, 2014. doi: 10.1111/jcmm.12365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takanari H, Bourgonje VJ, Fontes MS, Raaijmakers AJ, Driessen H, Jansen JA, van der Nagel R, Kok B, van Stuijvenberg L, Boulaksil M, Takemoto Y, Yamazaki M, Tsuji Y, Honjo H, Kamiya K, Kodama I, Anderson ME, van der Heyden MA, van Rijen HV, van Veen TA, Vos MA. Calmodulin/CaMKII inhibition improves intercellular communication and impulse propagation in the heart and is antiarrhythmic under conditions when fibrosis is absent. Cardiovasc Res 111: 410–421, 2016. doi: 10.1093/cvr/cvw173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Takanari H, Fontes MS, van der Heyden MA, Vos MA, van Veen TA. Response to the letter from Warren et al. Cardiovasc Res 113: 1799–1800, 2017. doi: 10.1093/cvr/cvx200. [DOI] [PubMed] [Google Scholar]
- 75.Tang T, Lai NC, Wright AT, Gao MH, Lee P, Guo T, Tang R, McCulloch AD, Hammond HK. Adenylyl cyclase 6 deletion increases mortality during sustained β-adrenergic receptor stimulation. J Mol Cell Cardiol 60: 60–67, 2013. doi: 10.1016/j.yjmcc.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thacker JS, Yeung DH, Staines WR, Mielke JG. Total protein or high-abundance protein: which offers the best loading control for Western blotting? Anal Biochem 496: 76–78, 2016. doi: 10.1016/j.ab.2015.11.022. [DOI] [PubMed] [Google Scholar]
- 77.Tromba C, Cohen IS. A novel action of isoproterenol to inactivate a cardiac K+ current is not blocked by beta and alpha adrenergic blockers. Biophys J 58: 791–795, 1990. doi: 10.1016/S0006-3495(90)82422-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tukkie R, Sogaard P, Vleugels J, de Groot IK, Wilde AA, Tan HL. Delay in right ventricular activation contributes to Brugada syndrome. Circulation 109: 1272–1277, 2004. doi: 10.1161/01.CIR.0000118467.53182.D1. [DOI] [PubMed] [Google Scholar]
- 79.van Veen TA, Stein M, Royer A, Le Quang K, Charpentier F, Colledge WH, Huang CL, Wilders R, Grace AA, Escande D, de Bakker JM, van Rijen HV. Impaired impulse propagation in Scn5a-knockout mice: combined contribution of excitability, connexin expression, and tissue architecture in relation to aging. Circulation 112: 1927–1935, 2005. doi: 10.1161/CIRCULATIONAHA.105.539072. [DOI] [PubMed] [Google Scholar]
- 80.Veeraraghavan R, Poelzing S. Mechanisms underlying increased right ventricular conduction sensitivity to flecainide challenge. Cardiovasc Res 77: 749–756, 2008. doi: 10.1093/cvr/cvm090. [DOI] [PubMed] [Google Scholar]
- 81.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest 116: 3127–3138, 2006. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wagner S, Hacker E, Grandi E, Weber SL, Dybkova N, Sossalla S, Sowa T, Fabritz L, Kirchhof P, Bers DM, Maier LS. Ca/calmodulin kinase II differentially modulates potassium currents. Circ Arrhythm Electrophysiol 2: 285–294, 2009. doi: 10.1161/CIRCEP.108.842799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang HW, Yang ZF, Zhang Y, Yang JM, Liu YM, Li CZ. Beta-receptor activation increases sodium current in guinea pig heart. Acta Pharmacol Sin 30: 1115–1122, 2009. doi: 10.1038/aps.2009.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Warren JS, Tracy CM, Miller MR, Makaju A, Szulik MW, Oka SI, Yuzyuk TN, Cox JE, Kumar A, Lozier BK, Wang L, Llana JG, Sabry AD, Cawley KM, Barton DW, Han YH, Boudina S, Fiehn O, Tucker HO, Zaitsev AV, Franklin S. Histone methyltransferase Smyd1 regulates mitochondrial energetics in the heart. Proc Natl Acad Sci USA 115: E7871–E7880, 2018. doi: 10.1073/pnas.1800680115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Warren M, Sciuto KJ, Taylor TG, Garg V, Torres NS, Shibayama J, Spitzer KW, Zaitsev AV. Blockade of CaMKII depresses conduction preferentially in the right ventricular outflow tract and promotes ischemic ventricular fibrillation in the rabbit heart. Am J Physiol Heart Circ Physiol 312: H752–H767, 2017. doi: 10.1152/ajpheart.00347.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Warren M, Zaitsev AV. CaMKII blockade, cardiac conduction, and arrhythmia. Cardiovasc Res 113: 1798–1799, 2017. doi: 10.1093/cvr/cvx199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weber S, Meyer-Roxlau S, Wagner M, Dobrev D, El-Armouche A. Counteracting protein kinase activity in the heart: the multiple roles of protein phosphatases. Front Pharmacol 6: 270, 2015. doi: 10.3389/fphar.2015.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yin Z, Jones GN, Towns WH 2nd, Zhang X, Abel ED, Binkley PF, Jarjoura D, Kirschner LS. Heart-specific ablation of Prkar1a causes failure of heart development and myxomagenesis. Circulation 117: 1414–1422, 2008. doi: 10.1161/CIRCULATIONAHA.107.759233. [DOI] [PubMed] [Google Scholar]
- 89.Yoon JY, Ho WK, Kim ST, Cho H. Constitutive CaMKII activity regulates Na+ channel in rat ventricular myocytes. J Mol Cell Cardiol 47: 475–484, 2009. doi: 10.1016/j.yjmcc.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 90.Yuan W, Bers DM. Protein kinase inhibitor H-89 reverses forskolin stimulation of cardiac L-type calcium current. Am J Physiol Cell Physiol 268: C651–C659, 1995. doi: 10.1152/ajpcell.1995.268.3.C651. [DOI] [PubMed] [Google Scholar]
- 91.Zhang J, Sacher F, Hoffmayer K, O’Hara T, Strom M, Cuculich P, Silva J, Cooper D, Faddis M, Hocini M, Haïssaguerre M, Scheinman M, Rudy Y. Cardiac electrophysiological substrate underlying the ECG phenotype and electrogram abnormalities in Brugada syndrome patients. Circulation 131: 1950–1959, 2015. doi: 10.1161/CIRCULATIONAHA.114.013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhou J, Shin HG, Yi J, Shen W, Williams CP, Murray KT. Phosphorylation and putative ER retention signals are required for protein kinase A-mediated potentiation of cardiac sodium current. Circ Res 91: 540–546, 2002. doi: 10.1161/01.RES.0000033598.00903.27. [DOI] [PubMed] [Google Scholar]
- 93.Zitron E, Kiesecker C, Lück S, Kathöfer S, Thomas D, Kreye VA, Kiehn J, Katus HA, Schoels W, Karle CA. Human cardiac inwardly rectifying current IKir2.2 is upregulated by activation of protein kinase A. Cardiovasc Res 63: 520–527, 2004. doi: 10.1016/j.cardiores.2004.02.015. [DOI] [PubMed] [Google Scholar]