

Abstract
Tumor necrosis factor-α (TNFα) is a proinflammatory cytokine that is closely linked to the development of cardiovascular disease. TNFα activates NADPH oxidase 1 (Nox1) and reactive oxygen species (ROS), including superoxide (O2·−), production extracellularly is required for subsequent signaling in vascular smooth muscle cells (VSMCs). Apoptosis signal-regulating kinase 1 (ASK1) is a mitogen-activated protein kinase kinase kinase that is activated by oxidation of associated thioredoxin. The role of ASK1 in Nox1-mediated signaling by TNFα is poorly defined. We hypothesized that ASK1 is required for TNFα receptor endocytosis and subsequent inflammatory TNFα signaling. We employed a knockdown strategy to explore the role of ASK1 in TNFα signaling in VSMCs. siRNA targeting ASK1 had no effect on TNFα-induced extracellular O2·− production. However, siASK1 inhibited receptor endocytosis as well as phosphorylation of two endocytosis-related proteins, dynamin1 and caveolin1. Intracellular O2·− production was subsequently reduced, as were other inflammatory signaling steps including NF-κB activation, IL-6 production, inducible nitric oxide synthase and VCAM expression, and VSMC proliferation. Prolonged exposure to TNFα (24 h) increased tumor necrosis factor receptor (TNFR) subtype 1 and 2 expression, and these effects were also attenuated by siASK1. ASK1 coimmunoprecipitated with both Nox1 and the leucine rich repeat containing 8A anion channel, two essential components of the TNFR1 signaling complex. Activation of ASK1 by autophosphorylation at Thr845 occurs following thioredoxin dissociation, and this requires the presence of Nox1. Thus, Nox1 is part of the multiprotein ASK1 signaling complex. In response to TNFα, ASK1 is activated by Nox1-derived oxidants, and this plays a critical role in translating these ROS into a physiologic response in VSMCs.
NEW & NOTEWORTHY Apoptosis signal-regulating kinase 1 (ASK1) drives dynamin1 and caveolin1 phosphorylation and TNFα receptor endocytosis. ASK1 modulates TNFα-induced NF-κB activation, survival, and proliferation. ASK1 and NADPH oxidase 1 (Nox1) physically associate in a multiprotein signaling complex. Nox1 is required for TNFα-induced ASK1 activation.
Keywords: apoptosis signal-regulating kinase 1, endocytosis, inflammation, NADPH oxidase 1, tumor necrosis factor-α, vascular smooth muscle cells
INTRODUCTION
Tumor necrosis factor alpha (TNFα) is a proinflammatory cytokine that plays an important role in multiple chronic inflammatory diseases that profoundly affect the cardiovascular system (4, 6). TNFα signals through 2 receptors: the 55 kDa TNFα receptor 1 (TNFR1) and the 75 kDa TNFα receptor 2 (TNFR2). TNFR1, but not TNFR2, has the “death domain,” which can drive pathways leading to cell death by multiple mechanisms, including apoptosis. TNFR1 activation can also promote inflammation and proliferation, partly through nuclear factor-kappa B (NF-κB) activation (7, 42). The overall response to TNFα is clearly cell type-dependent. Death pathway activation seems to predominate in endothelial cells (11), whereas in vascular smooth muscle cells (VSMCs) the primary response is proliferation and inflammation (9).
TNFα induces production of reactive oxygen species (ROS) in both endothelial cells and vascular smooth muscle cells (VSMCs), and these ROS are required for activation of intracellular signaling pathways that balance cell growth, death, and survival (19, 25). In VSMCs, this signaling cascade begins with extracellular superoxide (O2·−) production by NADPH oxidase 1 (Nox1) in the plasma membrane (9, 27). Signaling continues in endosomes formed by TNFα receptor endocytosis. We previously reported that these “signaling” endosomes are the primary site where TNFα-induced O2·− production leads to NF-κB activation and proliferation in VSMCs (26–28). Importantly, extracellular O2·− produced by Nox1 is required for the triggering of TNFα receptor endocytosis (10).
Apoptosis signal-regulating kinase 1 (ASK1) is a mitogen-activated protein kinase kinase kinase that is activated by cytokines and various stress signals and stimulates downstream MAPK signaling (18). ASK1 has a prominent role in the control of apoptosis (18) and is an important modulator in vascular remodeling (21). Under resting conditions, ASK1 interacts with thioredoxin, which maintains it in an inactive state. Oxidation of thioredoxin triggers dissociation from ASK1, autophosphorylation at Thr845, and activation of kinase activity. TNFα induces formation of the “ASK1 signalosome,” a high molecular mass multiprotein complex (~2,000 kDa) that includes TNFR-associated factor 2 (TRAF2), a critical mediator of downstream TNFα signaling. It is recruited to the signaling complex following receptor occupation and thioredoxin dissociation and may physically link ASK1 to the complex (24, 31). TRAF2 recruitment to the TNFR1 complex is thus a hydrogen peroxide (H2O2)-dependent process (23). The precise role that ASK1 plays in the complex sequence of events that result from TNFR1 occupation remains unclear, and the source and nature of the redox signal that initiates this sequence of events have not been clearly defined.
TNFα induces phosphorylation of JNK, p38, and ERK MAPKs in murine VSMCs (9). It is well established that JNK and p38 can both be activated by ASK1 (18). We previously reported that JNK provides negative feedback modulation of Nox1-dependent O2·− production at the plasma membrane, which attenuates NF-κB activation, whereas a p38 inhibitor, SB203580, does not alter NF-κB activation in mouse VSMCs (9). In contrast, Watanabe et al. (43) identified p38 as an important upstream component of NF-κB activation in epithelial cells.
In the present study, we hypothesized that ASK1 is required for TNFα receptor endocytosis and therefore for many of the downstream inflammatory effects of TNFα. The findings demonstrate that ASK1 activation requires extracellular O2·− production by Nox1 and is facilitated by physical proximity, as both proteins are part of a TNFR1 “signalosome.” ASK1 is upstream of dynamin1 and caveolin1 phosphorylation and of TNFR1 endocytosis. Thus, the potent anti-inflammatory effects of ASK1 knockdown are in part the result of impaired TNFR1 endocytosis and therefore failure to form TNFR1 signaling endosomes. The impact of ASK1 knockdown on TNFα signaling may not be solely related to the expected impairment of JNK and p38 activation, as neither of these kinases is essential for TNFR endocytosis.
MATERIALS AND METHODS
Cell culture.
Primary aortic VSMCs were isolated from C57/BL6 mice. All procedures were performed in accordance with the “Guiding Principles in the Care and Use of Animals” and approved by the Vanderbilt University Institutional Animal Care and Use Committee. After mice were euthanized using CO2, thoracic aortas were excised, cleaned of fat and connective tissue, and the endothelial cell layer was removed by passing a pin through the lumen. The vessels were then cut into 2–3-mm2 sections in a cold phosphate-buffered saline, placed in a culture dish, and maintained in an incubator at 37°C in a humidified 5% CO2 atmosphere. After 7–10 days, the tissue sections were removed, and the VSMCs that migrated out of the explants were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, 1× minimum essential medium nonessential amino acids, 1X vitamin, and 25 mM HEPES. Nox1 knockout (KO) VSMCs (46) were a generous gift from Dr. Francis Miller (Duke University).
siRNA transfection.
siRNA (negative control; D-001210-01, ASK1; M-041179-01, Nox1; M-054651-00) was purchased from Dharmacon (Lafayette, CO). siRNA (100 nM) was incubated with Lipofectamine 2000 (Life Technologies) in serum-free medium for 15 min. The resultant complex of siRNA-Lipofectamine 2000 was added to the cells in DMEM containing 5% FBS and then maintained for 3 days before performing experiments.
Western blot analysis.
Cells were serum deprived (0.5% serum) for 3 h before TNFα (10 ng/ml) stimulation for the indicated time periods. Protein extracts (40–60 μg) were separated by electrophoresis on a polyacrylamide gel and transferred to nitrocellulose membranes. Nonspecific binding was inhibited with blocking buffer (LI-COR Biosciences, Lincoln, NE) for 1 h at room temperature. Membranes were then incubated with primary antibodies in Tris-buffered saline solution with Tween 20 (0.1%) overnight at 4°C. Antibodies were as follows: p-JNK (cat. no. 9255), JNK (cat. no. 9252), p-p38 (cat. no. 9216), p38 (cat. no. 9212), ASK1 (cat. no. 8662), TNFR1 (cat. no. 13377), p-caveolin 1 (tyrosine 14, cat. no. 3251), caveolin (cat. no. 3238), and dynamin (cat. no. 2342) from Cell Signaling Technology (Danvers, MA); p-dynamin 1 (Ser795, cat. no. sc-377568) from Santa Cruz Biotechnology (Dallas, TX); TNFR2 (cat. no. AF-426-PB) and VCAM (cat. no. AF643) from R&D Systems (Minneapolis, MN); inducible nitric oxide synthase (cat. no. 610328) from BD Transduction (San Jose, CA); PCNA (cat. no. 05-347) and Nox1 (cat. no. SAB2501686) from MilliporeSigma (Burlington, MA); leucine rich repeat containing 8A (LRRC8A; cat. no. A304-175A) from Bethyl (Montgomery, TX); p-ASK1 (cat. no. T845) from Bioss Antibodies (Woburn, MA); and Tubulin from Vanderbilt Antibody Core (Nashville, TN). After incubation with fluorescent secondary antibodies for 2 h, signals were developed using the Odyssey Imaging System (LI-COR Biosciences) and quantified densitometrically (Image Studio). Results were normalized to the indicated proteins and expressed as arbitrary units.
TNFα receptor endocytosis.
VSMCs were grown on chamber slides and incubated with siRNA for 3 days. In DMEM containing 0.5% FBS, human TNFα conjugated with biotin (R&D Systems) was incubated with FITC-labeled avidin (Life Technologies) for 1 h at 4°C and then exposed to cells for 2 h at 4°C. After washing with cold media, cells were warmed to 37°C in DMEM with 5% FBS for 15 min. Cells were then fixed in 3.7% formaldehyde, and nuclear counterstaining was performed with To-Pro-3 (Life Technologies) for 5 min. Cover slides were mounted with ProLong Gold antifade reagent (Life Technologies). Cells were imaged by fluorescence confocal microscopy and FITC signal quantified using ImageJ software.
Superoxide production.
For quantification of superoxide production, the membrane-impermeable electron spin resonance (ESR) probe 1-hydroxy-2,2,6,6-tetramethylpiperidin-4-yl-trimethylammonium chloride (CAT1H; Enzo Life Sciences, Farmingdale, NY) and cell-permeable spin probe 1-hydroxy-4-methoxy-2,2,6,6-tetramethylpiperidine (TMH; Enzo Life Sciences) were used. After siRNA transfection of VSMCs for 3 days, the cells were incubated with Krebs-HEPES containing CAT1H (0.5 mmol/l) or TMH (0.5 mmol/l) and TNFα (10 ng/ml) for 20 min. The cells were then washed, scraped, snap frozen, and placed in an ESR finger Dewar under liquid nitrogen. ESR spectra were recorded from the Krebs buffer (CAT1H) or from the cell samples (TMH) using the following ESR settings: field sweep, 80 G; microwave frequency, 9.39 GHz; microwave power, 2 mW; modulation amplitude, 5 G; conversion time, 327.68 ms; time constant, 5242.88 ms; 512 point resolutions; and receiver gain 1 × 104. The signal was normalized to protein concentration.
NF-κB activation.
Replication-deficient adenovirus-containing promoter region, which is induced by NF-κB binding and causes expression of luciferase, was used to measure NF-κB transcriptional activation. siRNA-transfected VSMCs were maintained for 2 days and then were infected with the adenovirus expressing a luciferase reporter for 24 h in DMEM containing 5% FBS followed by exposure to TNFα (10 ng/ml) in serum-free DMEM for 6 h. Luciferase activity (relative light units) was quantified according to the manufacturer’s protocol (Promega, Madison, WI) and normalized to protein concentration (bicinchoninic acid protein assay) with a FLUOstar Omega plate reader (BMG Labtech).
IL-6 production.
Media was collected at the completion of the TNFα exposure (24 h) and stored at −80°C. IL-6 concentrations were assessed using a commercially available enzyme-linked immunosorbent assay kit (eBioScience, San Diego, CA), according to the manufacturer’s specifications.
Cell viability.
Cell viability was measured by two independent methods: the sulforhodamine B (SRB) and MTS assays. VSMCs were grown on 96-well plates and were treated with TNFα (10 ng/ml) for 24 h after siRNA transfection for 3days. For the SRB assay, cells were fixed with 5% cold trichloroacetic acid for 1 h at 4°C, washed with water, air-dried at room temperature, stained with 0.057% SRB solution in 1% acetic acid for 30 min, and rinsed with 1% acetic acid. After again air-drying, 10 mM Tris base (pH 10.5) was added, and the optical density was measured at 510 nm in a microplate reader (FLUOstar Omega, BMG Labtech). For the MTS assay, the CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS; Promega) was used according to the manufacturer’s protocols. Briefly, assays were performed by adding the MTS reagent to culture wells, incubating for 2 h, and then recording absorbance at 490 nm in a microplate reader (FLUOstar Omega, BMG Labtech).
Immunoprecipitation.
VSMCs were stimulated with TNFα (10 ng/ml) then lysed (20 mM Tris·HCl, pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Nonidet P-40, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, protease inhibitor cocktail, and 1 mM phenylmethylsulfonyl fluoride) for 1 h with nutation at 4°C and centrifuged for 30 min at 20,000 g. Supernatants were precleared with protein-G sepharose beads for 1 h at 4°C. Cleared supernatants were incubated with antibody (3–5 µg) for 1.5 h, then incubated with protein-G sepharose for 1 h. Beads were washed with lysis buffer 3 times, resuspended in SDS sample buffer, boiled, and the associated proteins were then analyzed by Western blot analysis. Antibodies for immunoprecipitation were used as follows: ASK1 (cat. no. 8662; Cell Signaling Technology) and LRRC8A (cat. no. A304-175A, Bethyl).
Statistical analysis.
Data values are expressed as means ± SE, and n represents the number of individual experiments in cultured cells. Graphs were generated by GraphPad Prism 5.0 (GraphPad Software, San Diego, CA). Statistical differences were assessed by unpaired t-test or one-way ANOVA. Post hoc comparisons were performed using Newman-Keuls analysis to compare all groups. A P value < 0.05 was considered statistically significant.
RESULTS
To selectively investigate the role of ASK1 in TNFα-mediated signal transduction, we utilized ASK1 mRNA knockdown using siRNA. Targeting of ASK1 by siASK1 was quite effective, inducing an average 65.7 ± 3.8% (n = 16) decrease in protein abundance (Fig. 1A). Knockdown of ASK1 reduced JNK and p38 phosphorylation in response to TNFα (Fig. 1B), confirming that ASK1 is indeed an important upstream kinase that regulates both JNK and p38 signaling in response to TNFα in VSMCs.
Fig. 1.

A: ASK1 knockdown by siRNA is confirmed by Western blot analysis. Bar graphs show the relative abundance of ASK1 protein after normalization to tubulin expression. *P < 0.05 compared with siControl (n = 16). B: phosphorylation of JNK (p-JNK) and p38 (p-p38) in response to TNFα (10 ng/ml) was reduced by ASK1 siRNA. Results are representative of five independent experiments. ASK1, apoptosis signal-regulating kinase 1.
When TNFα binds to its receptor, signaling begins at the plasma membrane and continues in cytoplasmic vesicles after endocytosis (37). Extracellular O2·− production by Nox1 is a critical proximal event in this signaling cascade and is required for receptor endocytosis. Extracellular O2·− production in response to TNFα was reduced in Nox1 KO VSMCs (wild-type 1,139 ± 78, KO 653 ± 20, pmol/mg of protein), thus confirming that Nox1 is a critical source of these oxidants. As shown in Fig. 2A, siASK1 did not alter extracellular O2·− production as assayed by the membrane-impermeant CAT1H spin trap. Since receptor endocytosis is required to sustain TNFα-mediated inflammatory signaling (9), we evaluated TNFR endocytosis over 15 min using biotin-labeled TNFα and FITC-avidin (36). The formation of TNFR-containing endosomes was reduced by siASK1 (Fig. 2, B and C). We previously demonstrated that TNFR endocytosis was not altered by siJNK (10). Similarly, siRNA targeting of p38 also failed to disrupt TNFR endocytosis (data not shown). Reduced receptor endocytosis was anticipated to affect subsequent downstream signaling, including intracellular O2·− production. Intracellular O2·− was indeed reduced by siASK1 as assayed by the membrane-permeable TMH spin trap (Fig. 2D).
Fig. 2.

Effects of siASK1 on superoxide production and TNFR endocytosis. A: the CAT1H signal in the media following TNFα exposure was not altered by siASK1, demonstrating that extracellular superoxide production in response to TNFα is not ASK1 dependent (n = 3). B: endocytosis of TNFR in VSMCs was impaired by siASK1. Bar graphs reflect fluorescent dots per cell. *P < 0.05 vs. siControl 0 min. †P < 0.05. C: representative images show that TNFR endocytosis (white dots) is reduced by siASK1. Results are representative of at least three independent experiments. D: TNFα-induced intracellular superoxide production is attenuated by siASK1. Total intracellular superoxide production was quantified using TMH. Representative electron spin resonance spectra for TMH (left) and means ± SE data (right). *P < 0.05 vs. siControl. †P < 0.05 (n = 4). ASK1, apoptosis signal-regulating kinase 1; CAT1H, 1-hydroxy-2,2,6,6-tetramethylpiperidin-4-yl-trimethylammonium chloride; TMH, 1-hydroxy-4-methoxy-2,2,6,6-tetramethylpiperidine; TNFR, tumor necrosis factor receptor; VSMC, vascular smooth muscle cell.
To further delineate how ASK1 is associated with TNFR endocytosis, we assessed activation of the endocytosis-related proteins dynamin and caveolin. Caveolin 1 phosphorylation stimulates internalization of vesicles in endothelial cells (3). Dynamin is required for scission of newly formed endosomes from the plasma membrane (16, 37); however, the relationship between dynamin phosphorylation and endocytosis is not completely clear. We found that TNFα increased phosphorylation of both dynamin 1 and caveolin 1, and this response was attenuated by siASK1 (Fig. 3). This suggests that ASK1 contributes to the regulation of both dynamin 1 and caveolin 1 during the process of endocytosis.
Fig. 3.

ASK1 modulates phosphorylation of two endocytosis-related proteins, dynamin and caveolin. A: representative Western blot images compare the abundance of dynamin, caveolin and their phosphorylated forms (p-dynamin 1 and p-caveolin 1) after exposure to TNFα. B and C: relative abundance of phosphorylated proteins (normalization to total protein expression at each time point) following TNFα stimulation is ASK1 dependent. Results are presented as means ± SE in each experimental group after normalization to control (siControl, 0 min TNFα) levels. *P < 0.05 siControl vs. siASK1 at the same time point. †P < 0.05 compared with 0 min TNFα in the siControl group (n = 5 to 7). ASK1, apoptosis signal-regulating kinase 1.
Disruption of TNFR endocytosis profoundly modifies downstream TNFα signaling in VSMCs (9). We next assessed the impact of siASK1 on downstream TNFα-induced inflammatory signaling. ASK1 knockdown significantly decreased NF-κB activation in response to TNFα (Fig. 4A). TNFα also increased secretion of IL-6, a proinflammatory cytokine. We measured IL-6 in the media of TNFα-exposed VSMCs and observed that siASK1 reduced the amount of IL-6 produced over 24 h (Fig. 4B). Inducible nitric oxide synthase and VCAM-1 expression was increased following a 24-h exposure to TNFα, and these effects were also diminished by siRNA targeting of ASK1 (Fig. 4C). Finally, we investigated cell proliferation and viability using PCNA protein expression and SRB and MTS assays, respectively. TNFα increased the abundance of PCNA (Figs. 4C and 5A) as well as the number of VSMCs remaining alive at the end of a 24-h exposure to the cytokine (Fig. 5, B and C). All of these effects were blocked by siASK1.
Fig. 4.

siASK1 inhibits TNFα-induced inflammation in VSMCs. A: siASK1 reduced TNFα-mediated NF-κB activation (luciferase reporter assay). Results are expressed as means ± SE in each group after normalization to control. *P < 0.05 compared with control in siControl, †P < 0.05 (n = 8). B: siASK1 blocks IL-6 production in response to TNFα. *P < 0.05 compared with control in siControl, †P < 0.05 (n = 4). C: left: representative Western blot shows expression of iNOS, VCAM, and PCNA after 24 h of exposure to TNFα. Right: bar graphs show the relative abundance of proteins after normalization to tubulin expression. *P < 0.05 compared with control in siControl. †P < 0.05 (n = 3 to 4). ASK1, apoptosis signal-regulating kinase 1; iNOS, inducible nitric oxide synthase; VSMC, vascular smooth muscle cell.
Fig. 5.

ASK1 promotes VSMC viability following prolonged exposure to TNFα. A: siASK1 blocks TNFα-induced proliferation (see example Western blot in Fig. 4). Relative abundance of PCNA proteins was normalization to tubulin expression (n = 4). B and C: siASK1 decreased TNFα-mediated cell viability as measured by SRB (B; n = 10) or MTS (C; n = 10) assay. Results are presented as means ± SE in each experimental group after normalization to control (siControl) levels. *P < 0.05 compared with control in siControl, †P < 0.05. ASK1, apoptosis signal-regulating kinase 1; SRB, sulforhodamine B; VSMC, vascular smooth muscle cell.
We examined the impact of TNFα and siASK1 on the abundance of the two distinct TNFα receptors, TNFR1 and TNFR2. Surprisingly, TNFR1 and TNFR2 expression were both increased in response to 24 h of exposure to TNFα. Although siASK1 did not alter the resting expression of either receptor, it completely abrogated the increase in receptor abundance induced by prolonged exposure to TNFα (Fig. 6). These data demonstrate an ASK1-dependent positive feedback relationship between TNFα stimulation and the abundance of both of its receptor subtypes.
Fig. 6.

Expression of TNFRs is increased following 24 h of exposure to TNFα, and this effect is blocked by siASK1. A: representative Western blot images show expression of TNFR1 and TNFR2 in VSMCs. B and C: relative abundance of receptors after normalization to tubulin expression. *P < 0.05 compared with control in siControl. †P < 0.05 (n = 4). ASK1, apoptosis signal-regulating kinase 1; TNFR, tumor necrosis factor.
In VSMCs, Nox1 is part of a multiprotein TNFR1 complex (9, 10, 45). Our data indicate that ASK1 plays an important role in the induction of TNFR endocytosis, an event that by definition occurs at the plasma membrane. Based on the well-established redox-dependent regulation of ASK1 by thioredoxin, we considered that ASK1 activation might require it to be in close proximity to the Nox1-mediated O2·− production that is activated by TNFα at the plasma membrane. We therefore investigated the possibility that ASK1 physically associates with the TNFR1-Nox1 signaling complex by immunoprecipitation of ASK1 followed by Western blotting for Nox1 (Fig. 7). We showed previously that Nox1 also binds to LRRC8A, a component of volume-regulated anion channels (10). We observed association of both Nox1 and LRRC8A with ASK1 by immunoprecipitation of ASK1 followed by Western blot analysis for Nox1 and LRRC8A (Fig. 7). The association of these three proteins was enhanced by TNFα treatment. Immunoprecipitation of LRRC8A followed by Western blot analysis for Nox1 and ASK1 yielded similar results (data not shown). Thus, ASK1 is a recruitable component of the TNFR1-Nox1 complex of proteins, positioning it perfectly to modulate TNFα-dependent signal transduction.
Fig. 7.

ASK1 associates with Nox1 and LRRC8A. A: VSMCs were stimulated with TNFα (10 ng/ml) for the indicated times, immunoprecipitated with anti-ASK1, and immunoblotted with anti-Nox1, anti-LRRC8A, and anti-ASK1 antibodies. B and C: protein expression quantification of immunoprecipitated samples normalized to the abundance of the proteins in the cell lysate of each sample (n = 3). ASK1, apoptosis signal-regulating kinase 1; LRRC8A, leucine rich repeat containing 8A; Nox1, NADPH oxidase 1; VSMC, vascular smooth muscle cell. *P < 0.05 vs. 0 min exposure to TNFα.
Oxidation-dependent dissociation of thioredoxin from ASK1 is a prerequisite for activation of kinase activity by autophosphorylation at Thr845 in the activation loop. Autophosphorylation activity of ASK1 is also facilitated by H2O2 (40). We investigated the dependence of ASK1 activation on Nox1 using VSMCs obtained from wild-type and Nox1 KO mice and using siNox1. TNFα-induced ASK1 phosphorylation at Thr845 was attenuated in Nox1 KO cells (Fig. 8A). Surprisingly, ASK1 protein expression was remarkably enhanced in Nox1 KO VSMCs compared with wild-type cells (Fig. 8A). This may represent a compensatory response to loss of Nox1-dependent activation of the kinase. Acute knockdown of Nox1 using siRNA had a similar impact on ASK1 phosphorylation, as did Nox1 KO, but no compensatory overexpression of ASK1 was observed in these experiments (Fig. 8B). These data strongly support a model in which Nox1-derived ROS activate ASK1 in a manner that is facilitated by their colocalization.
Fig. 8.

TNFα-induced ASK1 phosphorylation at Thr845 is Nox1-dependent. A: representative Western blot images show ASK1 phosphorylation at Thr845 (p-ASK1) and total ASK1 protein after TNFα stimulation for 10 min in WT and Nox1 KO VSMCs. The top bar graphs show the relative abundance of p-ASK1 after normalization to total ASK1 protein expression. Lower bar graph depicts total ASK1 protein expression after normalization to tubulin and demonstrates increased ASK1 abundance in Nox1-null cells. B: representative Western blot images show ASK1 phosphorylation at Thr845 (p-ASK1) and total ASK1 protein after acute knockdown using Nox1 siRNA (siNox1) in WT VSMCs. Bar graphs show the relative abundance of p-ASK1 after normalization to total ASK1 expression. Results are presented as means ± SE in each experimental group after normalization to control levels in WT. *P < 0.05 vs. control in WT (A) or in siControl (B). †P < 0.05 (n = 4). ASK1, apoptosis signal-regulating kinase 1; KO, knockout; Nox1, NADPH oxidase 1; VSMC, vascular smooth muscle cell; WT, wild-type.
DISCUSSION
Knockdown of ASK1 does not affect the ability of TNFα to activate Nox1 in the plasma membrane but profoundly impairs subsequent TNFα-induced inflammatory signals. This is associated with reduced phosphorylation of caveolin and dynamin and failure of TNFR endocytosis. Subsequently, intracellular O2·− production, MAPK phosphorylation, NF-κB activation, and multiple transcription-dependent inflammatory responses, including proliferation and survival, are affected. Thus, ASK1 activation is an early and critical step in TNFα signal transduction. ASK1 is known to be part of a larger signaling complex that is activated by oxidative stress and can physically associate with TNFR1 via TRAF2 or 6 (31). The current work demonstrates that Nox1 and LRRC8A are components of a larger “signalosome” and suggests that physical proximity may link Nox1-derived ROS to ASK1 activation in VSMCs.
Following TNFR1 activation, Nox1 produces extracellular O2·−, and this event is not ASK1-dependent. This O2·− is required for multiple subsequent signaling events, including endocytosis of the receptor, which must occur in order for the intracellular, endosomal phase of TNFR1 signaling to proceed (10). Given the physical association between TNFR1, TRAF2, Nox1, and ASK1, it seems reasonable to suggest that this extracellular oxidant signal provides the trigger for localized, cytoplasmic ASK1 activation by TNFα. However, extracellular O2·− anion cannot directly cross membranes without the aid of an anion channel, a mechanism that has been proposed at both the plasma membrane (14) and in endosomes (29). Demonstration of LRRC8 anion channels (33) in the ASK1 signalosome raises the intriguing possibility that they might serve this purpose. O2·− rapidly and spontaneously dismutes to H2O2 in an aqueous environment. H2O2 can cross the plasma membrane, although this likely requires facilitation by aquaporin water channels (1, 2, 5). A response to exogenous H2O2 involving Nox1, aquaporins, and ASK1 activation has been shown to lead to rat aortic smooth muscle cell hypertrophy (1). Potential local mechanisms of ASK1 oxidation by Nox1-derived O2·− are diagrammed in Fig. 9.
Fig. 9.
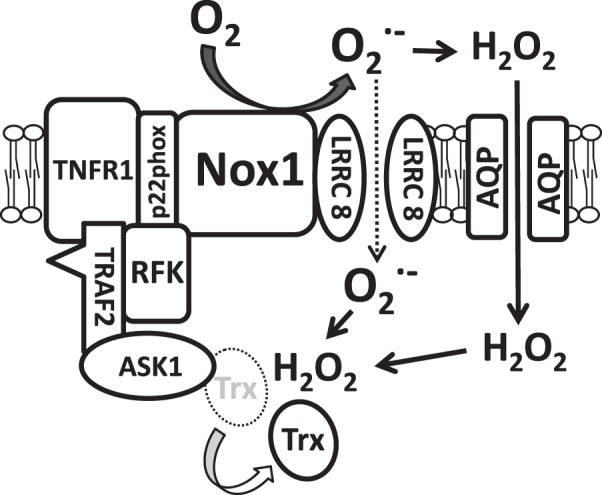
Schematic depiction of the relationship between ASK1, Nox1, and LRRC8A. TNFR1 has previously been shown to associate with ASK1 via TRAF2 (31). TNFR1 and Nox1 associate via p22phox and RFK (45). Finally, LRRC8A associates with Nox1. Here, we provide confirmatory evidence that LRRC8A and Nox1 associate with ASK1 in this multiprotein complex that has been termed the ASK1 “signalosome” (31). TNFα-induced extracellular O2·− is generated by Nox1, which is required for TNFα-induced ASK1 activation as reflected by phosphorylation. ASK1 is activated by oxidation and dissociation of Trx from ASK1. It remains to be directly determined how the extracellular oxidative signal originating from Nox1 crosses the plasma membrane. O2·− might cross via LRRC8 anion channels or dismute to H2O2 and across via AQP water channels (5). AQP, aquaporin; ASK1, apoptosis signal-regulating kinase 1; LRRC8A, leucine rich repeat containing 8A; Nox1, NADPH oxidase 1; O2·−, superoxide; RFK, riboflavin kinase; TNFR, tumor necrosis factor; TRAF, TNFR associated factor; Trx, thioredoxin.
Although it is clear that Nox1-mediated extracellular O2·− production is required for TNFR endocytosis (10), the mechanism by which O2·− initiates this process is completely unknown. Knockdown of ASK1 did not interfere with extracellular O2·− production but significantly blunted receptor endocytosis. Dynamin is an essential protein for scission of endocytic vesicles, separating them from the plasma membrane (37). It is a GTPase that can be phosphorylated by protein kinase C at Ser795 (34), by GSK3β at Ser774 (12), and by Cdk5 at Ser774 and Ser778 (39). Dynamin phosphorylation is required for TNFR1 endocytosis in VSMCs (9). Since protein kinase C is activated by TNFα via TNFR1 in VSMCs (35), we sought to determine if TNFα induced dynamin phosphorylation in VSMCs. We observed phosphorylation at Ser795, and this response was ASK1 dependent. Caveolin is the main structural component of caveolae and participates in TNFR1 internalization in human endothelial cells (13). Caveolin 1 phosphorylation by Src kinase at Tyr14 (3) induces caveolae-mediated endocytosis by translocating it from the plasma membrane to intracellular vesicles (3, 38). TNFα stimulated Tyr14 phosphorylation, and this was also ASK1 dependent. These data suggest that ROS-dependent ASK1 activation may represent a critical signaling step in the triggering of TNFR endocytosis.
Impairment of TNFR endocytosis is expected to disrupt intracellular signals that originate from signaling endosomes, and this was observed previously with respect to TNFα-mediated NF-κB activation in VSMCs (9). We therefore must ascribe a portion of the disruption of intracellular signaling pathways by siASK1 to reduced TNFR endocytosis. This includes reduced intracellular ROS production, as a proportion of this is made by Nox1 within these endosomes. Mitochondria, which are another important source of intracellular ROS, also exhibit ROS production that is dependent upon upstream Nox1-dependent signaling (41). The mechanism by which ASK1 knockdown inhibits signaling steps that are even further downstream, such as NF-κB activation and IL-6 secretion, may be related to reduction of both plasma membrane events such as JNK and p38 activation, and altered endocytosis-related signaling such as ERK phosphorylation (9). ASK1 knockdown potently reduced both basal and TNFα-induced IL-6 secretion, suggesting that the ASK1 signalosome has important effects on inflammatory signaling even under resting conditions.
ASK1 is a kinase involved in stress-induced apoptosis (18). Activated ASK1 results in death in many cell types. Downregulation of ASK1 activation is detected in several cancer cell types (20, 32). However, ASK1 has been implicated in cell proliferation and in the inflammatory response (15, 17). In this study, we demonstrated that ASK1 knockdown is associated with reduced TNFα-mediated proliferation. Thus, ASK1-dependent signaling has broad physiological implications that are dependent upon cell type and the nature of the stress responses.
The ability of prolonged TNFα exposure to increase TNFR protein abundance demonstrates an unusual positive feedback loop. Operating unchecked, this response has the potential to promote runaway inflammatory signaling. A similar phenomenon has been observed in breast cancer cells in which the mechanism was linked to p38 and NF-κB activation (8). It remains to be determined if the extra TNFα receptors are processed normally and are available to be activated at the plasma membrane. This positive feedback loop requires ASK1, but it remains to be determined how ASK1 affects TNFR expression induced by prolonged TNFα exposure.
Interestingly, ASK1 protein was more highly expressed in Nox1 KO VSMCs compared with wild-type cells. This may represent a compensatory mechanism in Nox1 KO cells and supports the concept that there is meaningful functional interaction between ASK1 and Nox1. Our data demonstrate a physical association between ASK1 and Nox1, but previous results suggest that this association may be indirect. TNFR1 is linked to Nox1 by binding to riboflavin kinase (45), which is the rate-limiting step in the synthesis of flavin mononucleotide, an essential prosthetic group of NADPH oxidases. It associates with both the death domain of TNFR1 and the p22phox subunit of NADPH oxidases. In addition, ASK1 associates with TRAF2 and 6, adapter proteins that also bind to the TNFR1 death domain and provide a platform for association of other key components of the TNFR1 complex. This association is known to be important in TNFα signaling (31). We previously showed that the TNFR1 signal complex includes both Nox1 and LRRC8A (10). We now directly establish for the first time that ASK1 colocalizes with Nox1 and LRRC8A (Fig. 9). This places it in close proximity to the redox active signaling molecules that are created by Nox1 in response to TNFα. The contribution of LRRC8A to signal generation remains to be established but might include charge compensation for Nox1 and/or provision of a pathway by which extracellular O2·− can enter the cytoplasm.
Whole animal studies clearly demonstrate the importance of ASK1 in the cardiovascular system. ASK1 dominant-negative in vivo gene transfer prevents neointimal formation after balloon injury (21). ASK1-null mice had a similar increase in blood pressure in response to angiotensin II infusion compared with wild-type; however, they demonstrated reduced cardiac hypertrophy and remodeling (22). Endothelial dysfunction and cardiac remodeling induced by Nω-nitro-l-arginine methyl ester treatment were absent in ASK1-null mice (44). ASK1 was also implicated in aldosterone/salt-induced cardiac inflammation and fibrosis caused by increased NADPH oxidase activity driven by the cardiac renin-angiotensin system (30). The current work sheds new light on the molecular mechanisms by which ASK1 plays these critical roles in vascular inflammation. Our results support the view that ASK1 is a promising therapeutic target in inflammation-associated cardiovascular disease.
GRANTS
This work was supported by American Heart Association Grant 16SDG-30610002 (to H. Choi) and National Institutes of Health Grants K08-GM-117367 (to R. J. Stark) and R01-HL-128386 (to F. S. Lamb).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
H.C. and F.S.L. conceived and designed research; H.C., R.J.S., B.S.R., and A.D. performed experiments; H.C., R.J.S., B.S.R., and A.D. analyzed data; H.C., R.J.S., A.D., and F.S.L. interpreted results of experiments; H.C., R.J.S., and A.D. prepared figures; H.C. drafted manuscript; H.C. and F.S.L. edited and revised manuscript; H.C., R.J.S., B.S.R., A.D., and F.S.L. approved final version of manuscript.
REFERENCES
- 1.Al Ghouleh I, Frazziano G, Rodriguez AI, Csanyi G, Maniar S, St Croix CM, Kelley EE, Egana LA, Song GJ, Bisello A, Lee YJ, Pagano PJ. Aquaporin 1, Nox1, and Ask1 mediate oxidant-induced smooth muscle cell hypertrophy. Cardiovasc Res 97: 134–142, 2013. doi: 10.1093/cvr/cvs295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Almasalmeh A, Krenc D, Wu B, Beitz E. Structural determinants of the hydrogen peroxide permeability of aquaporins. FEBS J 281: 647–656, 2014. doi: 10.1111/febs.12653. [DOI] [PubMed] [Google Scholar]
- 3.Aoki T, Nomura R, Fujimoto T. Tyrosine phosphorylation of caveolin-1 in the endothelium. Exp Cell Res 253: 629–636, 1999. doi: 10.1006/excr.1999.4652. [DOI] [PubMed] [Google Scholar]
- 4.Bäck M, Hansson GK. Anti-inflammatory therapies for atherosclerosis. Nat Rev Cardiol 12: 199–211, 2015. doi: 10.1038/nrcardio.2015.5. [DOI] [PubMed] [Google Scholar]
- 5.Bienert GP, Møller AL, Kristiansen KA, Schulz A, Møller IM, Schjoerring JK, Jahn TP. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J Biol Chem 282: 1183–1192, 2007. doi: 10.1074/jbc.M603761200. [DOI] [PubMed] [Google Scholar]
- 6.Brown DI, Griendling KK. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res 116: 531–549, 2015. doi: 10.1161/CIRCRESAHA.116.303584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cabal-Hierro L, Lazo PS. Signal transduction by tumor necrosis factor receptors. Cell Signal 24: 1297–1305, 2012. doi: 10.1016/j.cellsig.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 8.Cai X, Cao C, Li J, Chen F, Zhang S, Liu B, Zhang W, Zhang X, Ye L. Inflammatory factor TNF-α promotes the growth of breast cancer via the positive feedback loop of TNFR1/NF-κB (and/or p38)/p-STAT3/HBXIP/TNFR1. Oncotarget 8: 58338–58352, 2017. doi: 10.18632/oncotarget.16873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi H, Dikalova A, Stark RJ, Lamb FS. c-Jun N-terminal kinase attenuates TNFα signaling by reducing Nox1-dependent endosomal ROS production in vascular smooth muscle cells. Free Radic Biol Med 86: 219–227, 2015. doi: 10.1016/j.freeradbiomed.2015.05.015. [DOI] [PubMed] [Google Scholar]
- 10.Choi H, Ettinger N, Rohrbough J, Dikalova A, Nguyen HN, Lamb FS. LRRC8A channels support TNFα-induced superoxide production by Nox1 which is required for receptor endocytosis. Free Radic Biol Med 101: 413–423, 2016. doi: 10.1016/j.freeradbiomed.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi H, Nguyen HN, Lamb FS. Inhibition of endocytosis exacerbates TNF-α-induced endothelial dysfunction via enhanced JNK and p38 activation. Am J Physiol Heart Circ Physiol 306: H1154–H1163, 2014. doi: 10.1152/ajpheart.00885.2013. [DOI] [PubMed] [Google Scholar]
- 12.Clayton EL, Sue N, Smillie KJ, O’Leary T, Bache N, Cheung G, Cole AR, Wyllie DJ, Sutherland C, Robinson PJ, Cousin MA. Dynamin I phosphorylation by GSK3 controls activity-dependent bulk endocytosis of synaptic vesicles. Nat Neurosci 13: 845–851, 2010. doi: 10.1038/nn.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D’Alessio A, Al-Lamki RS, Bradley JR, Pober JS. Caveolae participate in tumor necrosis factor receptor 1 signaling and internalization in a human endothelial cell line. Am J Pathol 166: 1273–1282, 2005. doi: 10.1016/S0002-9440(10)62346-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hawkins BJ, Madesh M, Kirkpatrick CJ, Fisher AB. Superoxide flux in endothelial cells via the chloride channel-3 mediates intracellular signaling. Mol Biol Cell 18: 2002–2012, 2007. doi: 10.1091/mbc.e06-09-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayakawa T, Matsuzawa A, Noguchi T, Takeda K, Ichijo H. The ASK1-MAP kinase pathways in immune and stress responses. Microbes Infect 8: 1098–1107, 2006. doi: 10.1016/j.micinf.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 16.Henley JR, Krueger EW, Oswald BJ, McNiven MA. Dynamin-mediated internalization of caveolae. J Cell Biol 141: 85–99, 1998. doi: 10.1083/jcb.141.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ichijo H. From receptors to stress-activated MAP kinases. Oncogene 18: 6087–6093, 1999. doi: 10.1038/sj.onc.1203129. [DOI] [PubMed] [Google Scholar]
- 18.Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 275: 90–94, 1997. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 19.Irani K. Oxidant signaling in vascular cell growth, death, and survival: a review of the roles of reactive oxygen species in smooth muscle and endothelial cell mitogenic and apoptotic signaling. Circ Res 87: 179–183, 2000. doi: 10.1161/01.RES.87.3.179. [DOI] [PubMed] [Google Scholar]
- 20.Iriyama T, Takeda K, Nakamura H, Morimoto Y, Kuroiwa T, Mizukami J, Umeda T, Noguchi T, Naguro I, Nishitoh H, Saegusa K, Tobiume K, Homma T, Shimada Y, Tsuda H, Aiko S, Imoto I, Inazawa J, Chida K, Kamei Y, Kozuma S, Taketani Y, Matsuzawa A, Ichijo H. ASK1 and ASK2 differentially regulate the counteracting roles of apoptosis and inflammation in tumorigenesis. EMBO J 28: 843–853, 2009. doi: 10.1038/emboj.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Izumi Y, Kim S, Yoshiyama M, Izumiya Y, Yoshida K, Matsuzawa A, Koyama H, Nishizawa Y, Ichijo H, Yoshikawa J, Iwao H. Activation of apoptosis signal-regulating kinase 1 in injured artery and its critical role in neointimal hyperplasia. Circulation 108: 2812–2818, 2003. doi: 10.1161/01.CIR.0000096486.01652.FC. [DOI] [PubMed] [Google Scholar]
- 22.Izumiya Y, Kim S, Izumi Y, Yoshida K, Yoshiyama M, Matsuzawa A, Ichijo H, Iwao H. Apoptosis signal-regulating kinase 1 plays a pivotal role in angiotensin II-induced cardiac hypertrophy and remodeling. Circ Res 93: 874–883, 2003. doi: 10.1161/01.RES.0000100665.67510.F5. [DOI] [PubMed] [Google Scholar]
- 23.Li Q, Spencer NY, Oakley FD, Buettner GR, Engelhardt JF. Endosomal Nox2 facilitates redox-dependent induction of NF-kappaB by TNF-alpha. Antioxid Redox Signal 11: 1249–1263, 2009. doi: 10.1089/ars.2008.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu H, Nishitoh H, Ichijo H, Kyriakis JM. Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Mol Cell Biol 20: 2198–2208, 2000. doi: 10.1128/MCB.20.6.2198-2208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin KR, Barrett JC. Reactive oxygen species as double-edged swords in cellular processes: low-dose cell signaling versus high-dose toxicity. Hum Exp Toxicol 21: 71–75, 2002. doi: 10.1191/0960327102ht213oa. [DOI] [PubMed] [Google Scholar]
- 26.Matsuda JJ, Filali MS, Moreland JG, Miller FJ, Lamb FS. Activation of swelling-activated chloride current by tumor necrosis factor-α requires ClC-3-dependent endosomal reactive oxygen production. J Biol Chem 285: 22864–22873, 2010. doi: 10.1074/jbc.M109.099838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller FJ Jr, Filali M, Huss GJ, Stanic B, Chamseddine A, Barna TJ, Lamb FS. Cytokine activation of nuclear factor κ B in vascular smooth muscle cells requires signaling endosomes containing Nox1 and ClC-3. Circ Res 101: 663–671, 2007. doi: 10.1161/CIRCRESAHA.107.151076. [DOI] [PubMed] [Google Scholar]
- 28.Miller FJ Jr, Chu X, Stanic B, Tian X, Sharma RV, Davisson RL, Lamb FS. A differential role for endocytosis in receptor-mediated activation of Nox1. Antioxid Redox Signal 12: 583–593, 2010. doi: 10.1089/ars.2009.2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mumbengegwi DR, Li Q, Li C, Bear CE, Engelhardt JF. Evidence for a superoxide permeability pathway in endosomal membranes. Mol Cell Biol 28: 3700–3712, 2008. doi: 10.1128/MCB.02038-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakamura T, Kataoka K, Fukuda M, Nako H, Tokutomi Y, Dong YF, Ichijo H, Ogawa H, Kim-Mitsuyama S. Critical role of apoptosis signal-regulating kinase 1 in aldosterone/salt-induced cardiac inflammation and fibrosis. Hypertension 54: 544–551, 2009. doi: 10.1161/HYPERTENSIONAHA.109.135392. [DOI] [PubMed] [Google Scholar]
- 31.Noguchi T, Takeda K, Matsuzawa A, Saegusa K, Nakano H, Gohda J, Inoue J, Ichijo H. Recruitment of tumor necrosis factor receptor-associated factor family proteins to apoptosis signal-regulating kinase 1 signalosome is essential for oxidative stress-induced cell death. J Biol Chem 280: 37033–37040, 2005. doi: 10.1074/jbc.M506771200. [DOI] [PubMed] [Google Scholar]
- 32.Osaka N, Takahashi T, Murakami S, Matsuzawa A, Noguchi T, Fujiwara T, Aburatani H, Moriyama K, Takeda K, Ichijo H. ASK1-dependent recruitment and activation of macrophages induce hair growth in skin wounds. J Cell Biol 176: 903–909, 2007. doi: 10.1083/jcb.200611015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pedersen SF, Klausen TK, Nilius B. The identification of a volume-regulated anion channel: an amazing Odyssey. Acta Physiol (Oxf) 213: 868–881, 2015. doi: 10.1111/apha.12450. [DOI] [PubMed] [Google Scholar]
- 34.Robinson PJ, Sontag JM, Liu JP, Fykse EM, Slaughter C, McMahon H, Südhof TC. Dynamin GTPase regulated by protein kinase C phosphorylation in nerve terminals. Nature 365: 163–166, 1993. doi: 10.1038/365163a0. [DOI] [PubMed] [Google Scholar]
- 35.Roy S, Chakraborti T, Chowdhury A, Chakraborti S. Role of PKC-α in NF-κB-MT1-MMP-mediated activation of proMMP-2 by TNF-α in pulmonary artery smooth muscle cells. J Biochem 153: 289–302, 2013. doi: 10.1093/jb/mvs150. [DOI] [PubMed] [Google Scholar]
- 36.Schneider-Brachert W, Tchikov V, Neumeyer J, Jakob M, Winoto-Morbach S, Held-Feindt J, Heinrich M, Merkel O, Ehrenschwender M, Adam D, Mentlein R, Kabelitz D, Schütze S. Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity 21: 415–428, 2004. doi: 10.1016/j.immuni.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 37.Schütze S, Tchikov V, Schneider-Brachert W. Regulation of TNFR1 and CD95 signalling by receptor compartmentalization. Nat Rev Mol Cell Biol 9: 655–662, 2008. doi: 10.1038/nrm2430. [DOI] [PubMed] [Google Scholar]
- 38.Sverdlov M, Shajahan AN, Minshall RD. Tyrosine phosphorylation-dependence of caveolae-mediated endocytosis. J Cell Mol Med 11: 1239–1250, 2007. doi: 10.1111/j.1582-4934.2007.00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tan TC, Valova VA, Malladi CS, Graham ME, Berven LA, Jupp OJ, Hansra G, McClure SJ, Sarcevic B, Boadle RA, Larsen MR, Cousin MA, Robinson PJ. Cdk5 is essential for synaptic vesicle endocytosis. Nat Cell Biol 5: 701–710, 2003. doi: 10.1038/ncb1020. [DOI] [PubMed] [Google Scholar]
- 40.Tobiume K, Saitoh M, Ichijo H. Activation of apoptosis signal-regulating kinase 1 by the stress-induced activating phosphorylation of pre-formed oligomer. J Cell Physiol 191: 95–104, 2002. doi: 10.1002/jcp.10080. [DOI] [PubMed] [Google Scholar]
- 41.Tsai IC, Pan ZC, Cheng HP, Liu CH, Lin BT, Jiang MJ. Reactive oxygen species derived from NADPH oxidase 1 and mitochondria mediate angiotensin II-induced smooth muscle cell senescence. J Mol Cell Cardiol 98: 18–27, 2016. doi: 10.1016/j.yjmcc.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 42.Wajant H, Scheurich P. TNFR1-induced activation of the classical NF-κB pathway. FEBS J 278: 862–876, 2011. doi: 10.1111/j.1742-4658.2011.08015.x. [DOI] [PubMed] [Google Scholar]
- 43.Watanabe T, Jono H, Han J, Lim DJ, Li JD. Synergistic activation of NF-κB by nontypeable Haemophilus influenzae and tumor necrosis factor-α. Proc Natl Acad Sci USA 101: 3563–3568, 2004. doi: 10.1073/pnas.0400557101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamashita T, Yamamoto E, Kataoka K, Nakamura T, Matsuba S, Tokutomi Y, Dong Y-F, Ichijo H, Ogawa H, Kim-Mitsuyama S. Apoptosis signal-regulating kinase-1 is involved in vascular endothelial and cardiac remodeling caused by nitric oxide deficiency. Hypertension 50: 519–524, 2007. doi: 10.1161/HYPERTENSIONAHA.107.092049. [DOI] [PubMed] [Google Scholar]
- 45.Yazdanpanah B, Wiegmann K, Tchikov V, Krut O, Pongratz C, Schramm M, Kleinridders A, Wunderlich T, Kashkar H, Utermöhlen O, Brüning JC, Schütze S, Krönke M. Riboflavin kinase couples TNF receptor 1 to NADPH oxidase. Nature 460: 1159–1163, 2009. doi: 10.1038/nature08206. [DOI] [PubMed] [Google Scholar]
- 46.Zimmerman MC, Takapoo M, Jagadeesha DK, Stanic B, Banfi B, Bhalla RC, Miller FJ Jr. Activation of NADPH oxidase 1 increases intracellular calcium and migration of smooth muscle cells. Hypertension 58: 446s453, 2011. doi: 10.1161/HYPERTENSIONAHA.111.177006. [DOI] [PMC free article] [PubMed] [Google Scholar]