

Abstract
20-Hydroxyeicosatetraenoic acid (20-HETE) was recently identified as a novel contributor of ischemia-induced neovascularization based on the key observation that pharmacological interferences of CYP4A/20-HETE decrease ischemic neovascularization. The objective of the present study is to examine whether the underlying cellular mechanisms involve endothelial progenitor cells (EPCs) and preexisting endothelial cells (ECs). We found that ischemia leads to a time-dependent increase of cyp4a12 expression and 20-HETE production, which are endothelial in origin, using immunofluorescent microscopy, Western blot analysis, and LC-MS/MS. This is accompanied by increases in the tissue stromal cell-derived factor-1α (SDF-1α) expressions as well as SDF-1α plasma levels, EPC mobilization from bone marrow, and subsequent homing to ischemic tissues. Pharmacological interferences of CYP4A/20-HETE with a 20-HETE synthesis inhibitor, dibromo-dodecenyl-methylsulfimide (DDMS), or a 20-HETE antagonist, N-(20-hydroxyeicosa-6(Z), 15(Z)-dienoyl) glycine (6, 15-20-HEDGE), significantly attenuated these increases. Importantly, we also determined that 20-HETE plays a novel role in maintaining EPC functions and increasing the expression of Oct4, Sox2, and Nanog, which are indicative of increased progenitor cell stemness. Flow cytometric analysis revealed that pharmacological interferences of CYP4A/20-HETE decrease the EPC population in culture, whereas 20-HETE increases the cultured EPC population. Furthermore, ischemia also markedly increased the proliferation, oxidative stress, and ICAM-1 expression in the preexisting EC in the hindlimb gracilis muscles. We found that these increases were markedly negated by DDMS and 6, 15-20-HEDGE. Taken together, CYP4A/20-HETE regulates ischemia-induced compensatory neovascularization via its combined actions on promoting EPC and local preexisting EC responses that are associated with increased neovascularization.
NEW & NOTEWORTHY CYP4A/20-hydroxyeicosatetraenoic acid (20-HETE) was recently discovered as a novel contributor of ischemia-induced neovascularization. However, the underlying molecular and cellular mechanisms are completely unknown. Here, we show that CYP4A/20-HETE regulates the ischemic neovascularization process via its combined actions on both endothelial progenitor cells (EPCs) and preexisting endothelial cells. Moreover, this is the first study, to the best of our knowledge, that associates CYP4A/20-HETE with EPC differentiation and stemness.
Keywords: CYP4A/20-HETE, endothelial cells, endothelial progenitor cells, ischemia, neovascularization
INTRODUCTION
Neovascularization is the process of new blood vessel formation under both physiological and pathological conditions. Insufficient angiogenesis is a hallmark of pathological ischemic diseases. Postnatal compensatory neovascularization is well known as a vital process for tissue recovery in response to ischemia, occurring by remodeling existing blood vessels through both local stimulation of endothelial cells (ECs) in the process of angiogenesis (39) and recruitment of bone marrow (BM)-derived endothelial progenitor cells (EPCs) in the process of vasculogenesis (3, 51). Thus, identifying novel factors that contribute to increases of new collateral vessel formation in ischemic tissues and their underlying cellular mechanisms is crucial for developing targeted novel treatments of cardiovascular diseases.
20-Hydroxyeicosatetraenoic acid (20-HETE) is the ω-carbon hydroxylation product of arachidonic acid, a reaction catalyzed by cytochrome-P450 (CYP) monooxygenases of the CYP4A and 4F subfamilies (17, 38, 43). 20-HETE is found in many tissues and organs, including the kidney, liver, lung, and brain (7, 41). It is a primary eicosanoid in the renal and cerebral microcirculation and commonly functions as a vasoconstrictor. 20-HETE depolarizes vascular cells through the inhibition of large conductance calcium-activated K channels (BKCa). It also has mitogenic properties and plays a well-known role in hypertension and renal function (40, 41, 49). The CYP4A/F genes encode several enzymes capable of hydroxylating the terminal ω-carbon of arachidonic acid. Isoforms of the CYP4A subfamily are the major 20-HETE synthesizing enzymes. In humans, CYP4A11 has been identified as a prominent 20-HETE synthase (5), although CYP4F2 can also synthesize 20-HETE in humans (38). In mice, highly homologous members of the CYP4A subfamily have been identified, including cyp4a10, cyp4a12a, cyp4a12b, and cyp4a14, among which cyp4a12 is the most prominent 20-HETE-producing enzyme (42, 47).
An emerging role of CYP4A/20-HETE in neovascularization is becoming evident on the basis of the studies by us and others. 20-HETE has been identified to be an important proangiogenic factor (1, 10, 13, 21–23, 50) via its action on vascular ECs and/or vascular smooth muscle cells by inducing their proliferation, migration, survival, and tube formation as well as the secretion of proangiogenic growth factors (16, 21, 23, 28). We were the first to show that 20-HETE induces neovascularization in an in vivo model using the rat cornea pocket angiogenesis assay (13). We also demonstrated that 20-HETE stimulates vessel formation using a Matrigel plug angiogenesis assay (11). Thus, 20-HETE is angiogenic under a normoxic setting in vivo. Most importantly, our recent publication demonstrated that endogenous 20-HETE also contributes to neovascularization following ischemic/hypoxic injury (an ischemic and hypoxic setting) using a mouse hindlimb angiogenesis model (12), which is well documented to involve BM EPC mobilization and homing (51). In parallel, 20-HETE has been shown to upregulate angiogenic functions of EPCs in vitro and EPC-mediated angiogenesis in Matrigel (11, 22). However, we do not know whether 20-HETE is an essential vasculogenic regulator of the EPC mobilization and homing mechanisms in response to ischemia. Moreover, we also do not know whether 20-HETE can act on a preexisting EC network in local ischemic tissue to facilitate angiogenesis. The proposed studies will help us uncover the cellular mechanisms and further substantiate the notion that CYP4A/20-HETE is a crucial player in the regulation of ischemia-induced compensatory neovascularization and uncover its relationship to EPCs and ECs.
The present study aims to test the hypothesis that CYP4A/20-HETE promotes ischemic neovascularization via its collective actions on both BM-derived EPCs and local preexisting EC networks. To test this hypothesis, we first characterized the kinetics of cyp4a12 expression and 20-HETE production postischemia. We also explored the underlying cellular mechanisms by which CYP4A/20-HETE regulates ischemia-induced neovascularization. Here, we demonstrated that: 1) a transient endothelial increase of cyp4a12 expression and 20-HETE production drives neovascularization secondary to ischemia; 2) ischemia induces stromal cell-derived factor-1α (SDF-1α) expression in the gracilis muscle where neovascularization is taking place; 3) pharmacological inhibition of CYP4A/20-HETE by dibromo-dodecenyl-methylsulfimide (DDMS; a 20-HETE synthesis inhibitor) or 6, 15-20-HEDGE (a synthetic 20-HETE antagonist) decreases the mobilization of EPCs from the BM and subsequent homing to ischemic tissues during ischemic neovascularization; 4) CYP4A/20-HETE plays a novel role in regulation of EPC stemness, a vital characteristic of stem cell functions; and 5) pharmacological interferences of CYP4A/20-HETE also locally inhibited the preexisting EC responses secondary to ischemia. Taken together, these findings implicate that the BM-derived EPC and local preexisting EC network are the essential cellular targets of CYP4A/20-HETE during ischemia-induced compensatory neovascularization.
MATERIALS AND METHODS
Cell culture.
Human EPCs were collected and isolated from human umbilical cord blood under Institutional Review Board for human subject research approval at New York Medical College. EPC cell isolation, culturing, and characterization studies were performed as previously described (2, 11, 22, 29). Human microvascular endothelial cells were grown in Endothelial Cell Basal Medium 2 supplemented with Endothelial Cell GM MV2 (PromoCell, Heidelberg, Germany) as previously described (21, 23). Passages 5 were used for the experiments using human microvascular ECs. All cells were maintained at 37°C in a humidified incubator containing 5% CO2.
Mouse hindlimb ischemia angiogenesis model.
Twelve-week-old male Balb/c mice were purchased from the Jackson Laboratory (Bar Harbor, MA). The animal care and experimental protocols were approved by the Institutional Animal Care and Use Committee at New York Medical College. The mouse hindlimb ischemia model was established to study ischemia-induced neovascularization processes (9, 15, 20, 51) by unilateral (right) femoral artery ligation. The experimental protocol has been previously described in detail (12). The contralateral left side served as nonligated and nonischemic control.
LC-MS/MS analysis.
For in vivo experiments, gracilis muscles from both nonischemic and ischemic hindlimb were surgically extracted on days 0, 1, 3, 7, and 21 postligation. Tissues were homogenized in oxygenated Krebs buffer with glass homogenizer on ice. Homogenates were then incubated with 1 mM NADPH (Calbiochem, San Diego, CA) for 30 min at 37°C in Krebs buffer. For in vitro experiments, cultured human cord blood-derived EPCs were allowed to undergo spontaneous differentiation in culture. Cells were harvested at days 9, 12, 14, 21, and 28 after isolation from the cord blood to represent the different stages of their spontaneous differentiation. Both cell media and cell lysate were collected. Protein (mg) from cell lysate was measured using a Pierce BCA protein assay kit (Thermo Fisher Scientific, Rockford, IL).
Tissue and cell samples were acidified to pH 4.0 using 10% acetic acid. Protein amount in each muscle sample was also measured for normalization. The lipids were extracted 2 times with ethyl acetate in the presence of d6-20-HETE as an internal standard (0.5 ng) (Cayman Chemical, Ann Arbor, MI). The organic phase was collected and dried under nitrogen. 20-HETE production was quantified with a Shimadzu UFMS Triple Quadrupole Mass Spectrometer LCMS-8050 combined with a Nexera UHPLC using a negative ionization multiple reaction monitoring mode. This ultrasensitive method achieves 1 pg 20-HETE as the limit of quantitation. The details of analytical conditions are described previously (12). Final 20-HETE quantitation was normalized by 30-min reaction time and the total amount of mg protein/sample.
Pharmacological 20-HETE interventions.
The 20-HETE synthesis inhibitor, DDMS, and the 20-HETE antagonist, N-(20-hydroxyeicosa-6(Z), 15(Z)-dienoyl) glycine (6, 15-20-HEDGE), were used to interfere with the synthesis or action of 20-HETE in the mouse ischemic hindlimb angiogenesis model. For local administration of DDMS and 6,15-20-HEDGE to hindlimb gracilis muscle, an Alzet Mini-Osmotic pump (model 2002) was used to deliver the drugs (5 mg·kg−1·day−1) as previously described (12). For systemic administration, mice were treated with vehicle (DMSO), DDMS (10 mg·kg−1·day−1 ip), or 6, 15-20-HEDGE (10 mg·kg−1·day−1 ip) for 2 days before the unilateral femoral ligation. Treatment continued daily throughout the next 21 days.
SDF-1α ELISA.
Peripheral blood was obtained from mice treated with/without DDMS and 6,15-20-HEDGE at day 7 postischemia. Blood was centrifuged to collect plasma. SDF-1α determination was performed by SDF-1α Quantikine enzyme-linked immunosorbent assay kit (cat. no. DSA00; R&D Systems, Minneapolis, MN) according to the manufacturer's instructions using 100 μl undiluted plasma.
Flow cytometric analysis of EPC mobilization.
The level of BM-derived EPCs present in the peripheral blood was previously shown to peak at day 7 postischemia (51). Thus, we collected peripheral blood (500 μl) through heart puncture on day 7 postligation in animals treated with/without DDMS and 6,15-20-HEDGE. Mononuclear cells in the blood were separated by Ficoll density gradient separation. Cells were then incubated with P-phycoerythrin (PE)-conjugated anti-Sca-1 (1:100) (cat. no. FAB1226P; R&D Systems), FITC-conjugated anti-CD117 antibody (1:100) (cat. no. 135115; BioLegend, San Diego, CA), and PerCP-conjugated anti-VEGFR2 antibody (1:250) (cat no. FAB357C; R&D Systems) at 4°C overnight. Flow cytometric quantitation of the total number of circulatory EPCs (Sca-1+ CD117+VEGFR2+) per 2 × 105 cells per blood sample were performed by the New York Medical College Flow Cytometry Core.
Immunofluorescent microscopy.
For microvessel density analysis, hindlimb gracilis muscles from control, DDMS-, and 6,15-20-HEDGE-treated mice were surgically excised on days 3, 7, and 21. Samples were then frozen sectioned, and microvessels were identified and labeled by double immunofluorescent (IF) staining of CD31 and tomato lectin as previously described (12). The number of microvessels were counted in randomly chosen 6–8 fields and normalized to the number of muscle fibers per field.
For 20-HETE synthase cyp4a12 and CD31 analysis, ischemic hindlimb gracilis muscles were harvested on day 3 postligation and frozen sectioned (7 µm). Sections were incubated with rat anti-cyp4a12 (mouse 20-HETE synthase) (1:500; 4°C overnight) (kindly gifted by Dr. Wolf-Hagen Schunck, Max Delbrueck Center for Molecular Medicine, Berlin, Germany) followed by anti-rat FITC-conjugated secondary antibodies (1:1,000) (cat. no. 712-095-153; Jackson Immunoresearch, West Grove, PA) for 3 h at room temperature (RT). Rat anti-mouse CD31 (endothelial markers) (1:100) (cat. no. 14-0311-82; Thermo Fisher Scientific, Waltham, MA) was also used, followed by anti-rat Cy3-conjugated secondary antibodies (1:500) (cat. no. 712-165-253; Jackson Immunoresearch) for 3 h at RT.
For EPC homing to ischemic gracilis muscle, ischemic and control gracilis muscle sections were harvested on day 7 postligation as for the EPC mobilization study. Samples were then frozen sectioned and incubated with mouse PE-conjugated anti-Sca-1 (1:250) (R&D Systems) overnight at 4°C followed by tomato lectin (10 μg/ml; Vector Laboratories, Burlingame, CA) for 30 min at RT. DAPI was used as nuclei counter stain.
For postischemic EC proliferative index in vivo, ischemic and control gracilis muscle sections were incubated with mouse anti-Ki 67 (1:250) (cat. no. AB8191; Abcam), mouse anti-phosphoSer1179eNOS (1:500) (cat. no. AB184154; Abcam), and mouse anti-ICAM-1 antibodies (1:500) (cat. no. AB2213; Abcam), respectively. Anti-mouse FITC-conjugated antibodies (1:1,000) were used as secondary. Concurrently, rat anti-mouse CD31 (endothelial markers) (1:100) was also used followed by anti-rat Cy3-conjugated secondary antibodies (1:500) (Jackson Immunoresearch) for 3 h at RT. DAPI was used as counter stain for nuclei.
For TUNEL apoptosis assay, frozen ischemic and control gracilis muscle sections were harvested on day 3 postligation, and EC apoptosis was assessed using the TACS TDT in situ-fluorescein TUNEL assay kit (cat. no. 4812-30-K; R&D Systems) according to the manufacturer’s recommendations. Colocalization with CD31 was performed as described above.
For dihydroethidium (DHE) assay, frozen sections were first incubated with DHE (10 μM; Sigma-Aldrich, St. Louis, MO) in oxygenated PBS in a light-protected humidified incubator at 37°C for 15 min, followed by rat anti-mouse CD31 (endothelial markers) (1:100) (Abcam), then by anti-rat FITC-conjugated secondary antibodies (1:1,000) (Jackson Immunoresearch) for 3 h at RT.
All sections were washed, and the IF colocalization of various markers with CD31 were captured using the Zeiss AXIO Imager.M1 fluorescence microscope and quantitated. Six fields were randomly chosen to ensure the objectivity of the sampling.
RNA extraction and real-time PCR.
RNA was extracted from 9-day-old EPCs using TRIzol reagent and treated with DNase. RNA concentration was measured and quantified by reading absorbance at 260 nm. Then 1 μg RNA was reverse transcribed using a BluePrintTM1st Strand cDNA Synthesis Kit (TaKaRa Biotechnology, RR#6115A, Dalian, China). Real-time PCR was performed using the Mx3000p Real-Time PCR System (Agilent Technology, La Jolla, CA) as previously described (11). Primers used for PCR were: Oct4, forward 5′-CCCCTGGTGCCGTGAA-3′ and reverse 5′-GCAAATTGCTCGAGTTCTTTCTG-3′; Sox2, forward 5′-GGATAAGTACACGCTGCCCG-3′ and reverse 5′-ATGTGCGCGTAACTGTCCAT-3′; Nanog, forward 5′-GTCCCAAAGGCA AACAACCC-3′ and reverse 5′-GCTGGGTGGAAGAGAACACA-3′; β-actin, forward 5′-AAGATCATTGCTCCTC-CTGA-3′ and reverse 5′-CTCGTCATAC TCCTGCTTGCT-3′; and 18S rRNA, forward 5′-GATGGGC-GGCGGAAAATAG-3′ and reverse 5′-GCGTGGATTCTGCATAATGGT-3′. The reactions were incubated at 95°C for 10 min, followed by 40 cycles of 95°C for 10 s, 55°C for 5 s, and 72°C for 15 s. The threshold cycle (Ct) data were determined using default threshold settings and defined as the fractional cycle number at which the fluorescence passes the fixed threshold. The 2−ΔΔCt method was used to calculate the relative changes in the gene expression.
Colony forming unit-Hill assay.
EPC functional Hill assay was performed using the colony forming unit (CFU)-Hill Liquid Medium Kit (Stem Cell Technology, Cambridge, MA) using a modified protocol based on the manufacturer’s recommendations. In brief, EPCs were harvested from human umbilical cord blood and cultured for 9, 12, 14, 21, and 28 days before resuspending them in CFU-Hill liquid medium and being placed on fibronectin-coated plates. CFU colonies were counted 3 days later. A parallel experiment was performed in EPCs from day 9 in culture and then placed onto fibronectin-coated plates in the presence and absence of 20-HETE (10 nM) and/or 20-HETE+6,15-20-HEDGE (10 nM). Total numbers of small and large EPC-CFU-derived colonies were counted blindly under a transmitted light microscope from all experimental groups and quantitated.
Western blot analysis.
We first validated the cyp4a12 antibody (gifted by Dr. Wolf-Hagen Schunck) by using a positive control sample obtained from the renal interlobar artery of cyp4a12-overexpressing mice. Then, for in vivo studies, nonischemic control and ischemic gracilis muscles were extracted from mice on days 3, 7, and 21 postligation and homogenized using radioimmunoprecipitation assay buffer as previously described (12). Similarly, nonischemic control and ischemic gracilis muscles were also extracted from vehicle-, DDMS-, and 6,15-20-HEDGE-treated animals on day 3 postligation. For in vitro EC study, ECs were treated with 20-HETE (10 nM) for 0, 4, 8, and 24 h. For in vitro EPC study, EPC cultures were harvested at days 9, 12, 14, 21, and 28 from human cord blood. All cells were then lysed with radioimmunoprecipitation assay buffer. Equal amounts of protein (40 µg) were separated on a 10% Tris-glycine gel, transferred to a PVDF membrane, and incubated with either anti-cyp4a12 (1:500), anti-SDF-1α (1:1,000) (AB155090; Abcam), or anti-CYP4A11 (1:1500) (SAB2702082; Sigma-Aldrich) at 4°C overnight, respectively. Membranes were then incubated with anti-rabbit IRDye 800CW secondary antibody (1:10,000) (925-32212; LI-COR, Lincoln, NE) at RT for 2 h and scanned with the LI-COR Odyssey Infrared Imaging system. β-Actin (1:100,000) (SC47778; Santa Cruz Biotechnology, Santa Cruz, CA) was used as loading control. All Western blots were quantitated using National Institutes of Health ImageJ software by comparing the density ratios of control band/β-actin to experimental band/β-actin.
Statistical analysis.
In all cell culture experiments, data were expressed as means ± SD. In all animal experiments, data were expressed as means ± SE. Significance of difference in mean values was determined using standard t-test and one-way ANOVA analysis, followed by the Newman-Keuls post hoc test. P < 0.05 was considered to be significant.
RESULTS
Ischemia induces time-dependent and endothelial-originated increases in 20-HETE production.
We previously reported that 20-HETE production increases postischemic injury (12). However, the precise kinetics of 20-HETE generation postischemia remains unknown. We assessed the production of 20-HETE in gracilis muscles from the ischemic and the nonischemic hindlimbs at days 0, 3, 7, and 21 postligation using LC-MS/MS analysis. Muscles from the nonischemic limb were included as controls. Fig. 1A demonstrates that ischemic hindlimb muscles produce high levels of 20-HETE on days 3 and 7 postischemic ligation, whereas nonischemic control muscles produce negligible basal amounts of 20-HETE (73 ± 10 vs. 8 ± 2 pg/mg of protein; lowest limit of detection, pg). 20-HETE production at day 21 postischemia was returned to the comparable level as in the control tissue. Microvessel density analysis revealed that both DDMS and 6,15-20-HEDGE significantly inhibited ischemia-induced angiogenesis at days 3, 7, and 21 (Fig. 1B). We next validated the cyp4a12 antibody (gifted by Dr. Wolf-Hagen Schunck) by using renal interlobar artery harvested from cyp4a12-overexpressing mice as a positive control. Fig. 1C shows that the antibody produced a specific single band at ~50 kDa. We then performed Western blot analysis and quantitation of cyp4a12 expression in ischemic muscles harvested on days 3, 7, and 21 postligation. Cyp4a12 expression is significantly increased by 2.6 ± 0.33- and 1.51 ± 0.2-fold on days 3 and 7, respectively (Fig. 1D), compared with nonischemic controls. Further examination of ischemic gracilis muscles harvested from mice on day 3 postischemia (Fig. 1E) showed that murine 20-HETE synthase cyp4a12 was not expressed in nonischemic control gracilis muscles, whereas it is highly increased in the ischemic muscles. Furthermore, cyp4a12 was coexpressed with the endothelial marker CD31.
Fig. 1.

Time-dependent increases in 20-hydroxyeicosatetraenoic acid (20-HETE) secondary to ischemia is endothelial in origin. A: femoral artery ligation was performed in 12-wk-old Balb/c mice, and ischemic gracilis muscles were extracted on days 0, 3, 7, and 21. Contralateral nonischemic gracilis muscles were harvested as controls. All muscle samples were lipid extracted, and the kinetics of 20-HETE production in gracilis muscles postischemia were measured using LC-MS/MS analysis and normalized to total protein (pg·30 min−1·mg protein−1) (mean ± SD; n = 6; *P < 0.05 vs. nonischemic control). B: ischemic gracilis muscles were also isolated from mice treated with/without 10 mg·kg−1·day−1 dibromo-dodecenyl-methylsulfimide or 6,15-20-HEDGE. Assessment of microvessel density (MVD) was done by counting CD31+tomato lectin+, which then were normalized to the number of muscle fibers in the field and expressed as capillary-to-fiber ratio. Dark gray bars represent capillary-to-fiber ratio counts in the nonischemic control muscles of each treatment group (mean ± SD; #P < 0.05 vs. nonischemic limb and *P < 0.05 vs. ischemic limb). C: renal interlobar arteries were isolated from cyp4a12-overexpressing mice, and Western blot analysis was performed to validate cyp4a12 antibody. D: Cyp4a12 expression and quantitation in ischemic gracilis muscles on days 3, 7, and 21 postligation (mean ± SD; n = 3; *P < 0.05 vs. nonischemic controls). E: gracilis muscles from ischemic and nonischemic limb were harvested on day 3 postischemia and frozen sectioned. Immunofluorescent staining of 20-HETE synthase cyp4a12 (green) and microvessel marker CD31 (red) were then performed and merged using National Institutes of Health ImageJ. Representative images (×1,000) from 3 animals are shown.
Pharmacological intervention of CYP4A/20-HETE alters BM-derived EPC mobilization and homing during ischemic neovascularization.
SDF-1α is a member of the CXC chemokine family that plays a crucial role in stem cell BM mobilization, migration, homing, and survival (8, 9, 15, 36, 37). We first determined whether the expression of SDF-1α is altered in day 3 ischemic gracilis muscle, in which it shows the highest 20-HETE production. Western blot analysis showed that SDF-1α expression in ischemic gracilis muscles increased by 2.4 ± 0.21-fold compared with the nonischemic controls on day 3 postligation (Fig. 2A), whereas the 20-HETE synthesis inhibitor DDMS and the 20-HETE antagonist 6,15-20-HEDGE markedly negated this increase. Since 20-HETE is likely to originate from the endothelium, we asked whether increased 20-HETE production secondary to ischemia may be responsible for the increased SDF-1α production and release from ECs. As shown in Fig. 2B, ECs treated with 10 nM 20-HETE induced a time-dependent increase in SDF-1α expression by 2.8 ± 0.18- and 1.7 ± 0.21-fold at 4 h and 8 h, respectively. Additionally, we detected a marked increase of SDF-1α concentrations in the plasma of ischemic mice (1,165 ± 124 pg/ml) on day 3 postligation compared with sham control mice (552 ± 104 pg/ml), whereas both DDMS and 6,15-20-HEDGE significantly attenuated the increases of SDF-1α in mice subjected to ischemic injury (Fig. 2C).
Fig. 2.

Pharmacological interferences of cyp4a/20-hydroxyeicosatetraenoic acid (20-HETE) inhibit stromal cell-derived factor-1α (SDF-1α) expression, endothelial progenitor cell (EPC) mobilization, and homing during ischemic neovascularization. Mice were pretreated 2 days before femoral artery ligation surgery with dibromo-dodecenyl-methylsulfimide (DDMS; 20-HETE synthesis inhibitor, 10 mg·kg−1·day−1 ip) or 6,15-20-HEDGE (20-HETE antagonist; 10 mg·kg−1·day−1 ip). DMSO was used as solvent control. A: hindlimb gracilis muscles were harvested 3 days later and homogenized. Nonischemic muscle was used as control. Western blot analysis of SDF-1α expression and densitometric quantification in nonischemic limb vs. ischemic limb were performed (n = 6; *P < 0.05 vs. nonischemic controls). B: human microvascular endothelial cells were first treated with 20-HETE (10 nM) for 0, 4, 8, and 24 h, and SDF-1α expression was determined by Western blot analysis (n = 3; *P < 0.05 vs. time 0). C: peripheral blood was collected on day 3 postligation, and plasma was extracted by centrifugation. SDF-1α ELISA assay was performed according to the manufacturer’s recommended protocols (n = 3; *P < 0.05 vs. sham control; #P < 0.05 vs. DMSO control). D: peripheral blood was again collected on day 7 postligation, and mononuclear cells were separated and harvested by Ficoll gradient fractionation. Sham mice were used as control. Flow cytometry analysis was performed to quantitate the number of circulatory Sca-1+ CD117+ EPCs. Data were normalized to the number of EPC/2 × 105 cells (mean ± SD; n = 6–8; *P < 0.05 vs. sham control; #P < 0.05 vs. DMSO control). E: representative images of corresponding frozen gracilis muscle sections stained for Sca-1+ EPCs homed to the sites of ischemic neovascularization where they colocalized with tomato lectin+ endothelial cells, and DAPI was used to counter stain for nuclei. F: quantitation of EPC homing (mean ± SD; n = 6; *P < 0.05 vs. sham control; #P < 0.05 vs. DMSO control).
To determine whether CYP4A/20-HETE plays a role in EPC mobilization in response to ischemia, peripheral blood was drawn from the following groups of 12-wk-old Balb/c mice on day 7 postligation by heart puncture: 1) baseline nonsurgical sham control; 2) mice treated with vehicle DMSO intraperitoneally; 3) mice treated with 10 mg·kg−1·day−1 of DDMS ip; and 4) mice treated with 10 mg·kg−1·day−1 6,15-20-HEDGE ip. We chose day 7 for these experiments because the number of EPCs have been previously shown to peak in peripheral blood 7 days postischemic challenge (51). Flow cytometry was performed to quantify the numbers of murine Sca-1+ CD117+ VEGFR2+-positive circulatory EPCs. We found that the numbers of circulatory EPCs markedly increased on day 7 postischemia by 5.5-fold and that DDMS and 6,15-20-HEDGE (antagonist) inhibited the ischemia-induced increase in circulatory EPCs by 32% and 68%, respectively (Fig. 2D). We also examined the ability of BM-derived EPCs to home to the site of ischemic neovascularization in vivo on day 7 postligation (Fig. 2E). Finally, ischemic gracilis muscle from vehicle-, DDMS-, and 6,15-20-HEDGE-treated (5 mg·kg−1·day−1 sc osmotic pump) mice were excised on day 7 postligation, frozen sectioned, and stained with P-PE-conjugated anti-Sca-1 (1:100) and tomato lectin (10 μg/ml; EC marker). Sca-1+ EPCs that homed to gracilis muscle, which colocalized with ECs, were counted under an IF microscope. Data shown in Fig. 2F revealed that ischemia resulted in an increase of the numbers of BM-derived EPCs at the sites of ischemic neovascularization by ~20-fold compared with nonischemic controls. Both DDMS and 6,15-20-HEDGE significantly reduced the numbers of EPCs homed to the microvessels in ischemic muscles.
CYP4A/20-HETE is associated with the degree of EPC-EC differentiation and EPC functions in vitro.
EPCs undergo natural processes of differentiation in vitro and in vivo. Using flow cytometry analysis of a panel of cell surface markers (29), we have previously reported that human CB-derived EPCs predominantly exist as progenitor cells up to 14 days in culture (high stemness; high self-renewal) based on their positive expression of the progenitor cell surface markers CD133 and CD34. As the cells continue to differentiate spontaneously in culture, they lose CD133 progenitor markers and gain endothelial lineage cell surface markers including kinase insert domain receptor and vascular endothelial-cadherin by day 25 (low stemness; low self-renewal), as they turn into mature and differentiated ECs (22, 29). EPCs were harvested from cord blood and cultured for 9, 12, 14, 21, and 28 days. CYP4A11 expression and 20-HETE production were assessed by Western blot and LC-MS/MS analysis. As shown in Fig. 3, A and B, spontaneous EPC-EC differentiation is accompanied by a time-dependent decrease in both the CYP4A11 expression and 20-HETE production, whereas fully differentiated EPCs on day 28 produce negligible amounts of CYP4A11 expression and nondetectable amounts of 20-HETE production. Furthermore, EPC functional Hill assays (26) were performed in EPCs on days 9, 12, 14, 21, and 28 after cord blood isolation. Interestingly, we observed that spontaneous differentiation of EPCs in culture is correlated with a gradual decrease in their colony formation abilities (Fig. 3C). In contrast, when Hill assay was performed with 21-day-old EPCs in the presence and absence of pure 20-HETE (10 nM), we found that 20-HETE significantly increased the total numbers of EPC colony formation compared with the DMSO controls (15.1 ± 2.2 vs. 8.4 ± 1.5), indicative of improved EPC functions. Equally important, the addition of 20-HETE antagonist 6,15-20-HEDGE (10 nM) attenuated the EPC colony formation improved by 20-HETE.
Fig. 3.

CYP4A/20-hydroxyeicosatetraenoic acid (20-HETE) is associated with endothelial progenitor cell (EPC) differentiation and functions. Human umbilical cord blood-derived EPCs were isolated and seeded to allow spontaneous differentiation in culture. 20-HETE synthase CYP4A11 (A) and 20-HETE production (B) were determined on days 9, 12, 14, and 21 after EPC isolation, along with fully differentiated endothelial cells (ECs) from these EPCs (on day 28), by Western blot and LC-MS/MS, respectively (mean ± SD; n = 4; *P < 0.05 vs. day 9 EPCs; #P < 0.05 vs. differentiated ECs). EPC cultures (3 × 106) were harvested on days 9, 12, 14, 21, and 28, and colony formation Hill assay was performed according to the manufacturer’s recommendations (C; mean ± SD; n = 3; *P < 0.05 vs. day 9 EPCs; #P < 0.05 vs. differentiated ECs). Representative EPC colony is shown. EPCs (3 × 106) were harvested on day 21, and colony formation Hill assay was again performed according to the manufacturer’s recommendations in the presence and absence of 10 nM 20-HETE and/or 10 nM 6,15-20-HEDGE (D; mean ± SD; n = 3; *P < 0.05 vs. control). N.D., not detectible.
CYP4A/20-HETE regulates EPC stemness.
EPCs are known to participate in neovascularization by producing various angiogenic cytokines at their target sites (6, 53). Factors that are capable of maintaining the EPC stemness can promote EPC self-renewal and self-replication and thus increase their angiogenic functions. We then incubated 9-day-old EPC cultures in the presence and absence of DDMS (10 μM) or 6,15-20-HEDGE (10 μM) for 5 days. Fig. 4A showed that DDMS and 20-HEDGE both decrease the number of EPC-bearing primitive progenitor markers CD133 and CD34. Thus, inhibition of 20-HETE production or action decreased the number of progenitor cells compared with vehicle-treated controls (decreased stemness). In contrast, the addition of 20-HETE (1 and 10 nM) markedly increased the number of cells expressing primitive progenitor cell markers (high stemness) (Fig. 4B). Oct4, Sox2, and Nanog are self-renewal genes that are commonly associated with EPCs with high characteristics of stemness (4), and thus we assessed the effects of exogenous 20-HETE (1 nM) on the expression of these stemness genes in cord blood-derived EPCs using real-time PCR. Our data showed that 20-HETE significantly increased the expression of Oct4, Sox2, and Nanog by 1.5-, 2-, and 8.2-fold, respectively (Fig. 4). This finding further supports the notion that 20-HETE may play a role in promoting/maintaining EPC stemness.
Fig. 4.
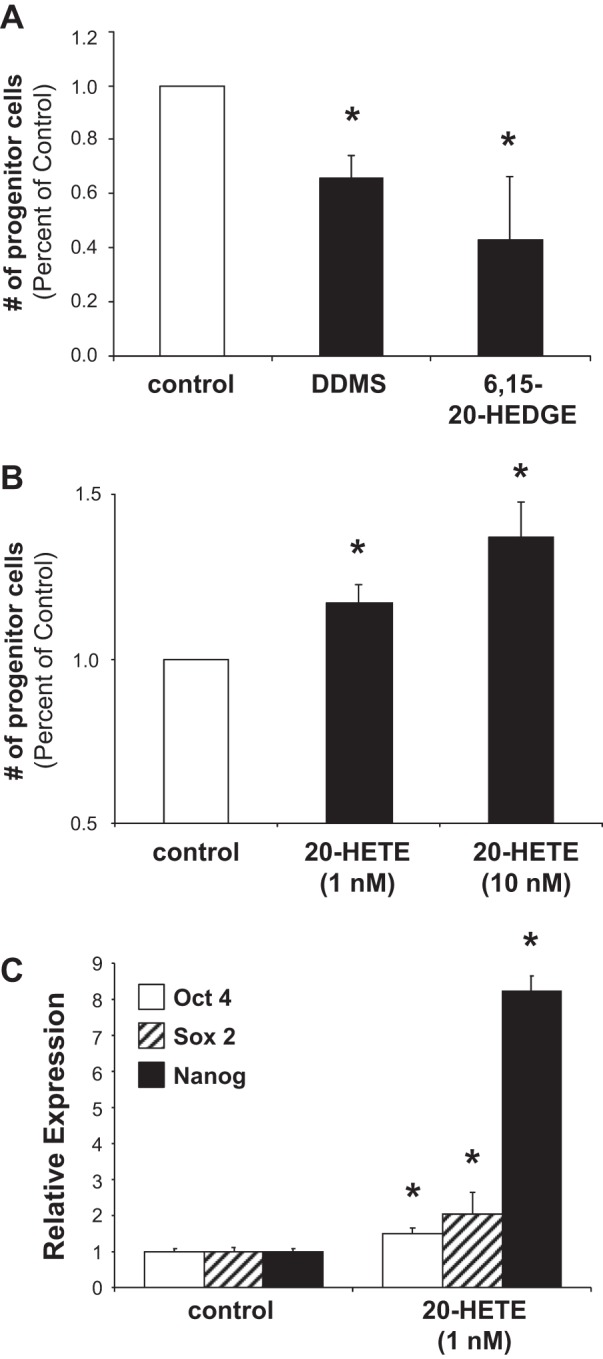
Pharmacological interferences of CYP4A/20-hydroxyeicosatetraenoic acid (20-HETE) alter endothelial progenitor cell (EPC) stemness. Human cord blood-derived EPCs were harvested and cultured for 9 days before treating with dibromo-dodecenyl-methylsulfimide (DDMS; 10 μM) and 6,15-20-HEDGE (10 μM) (A), or 20-HETE (1 and 10 nM) (B) for an additional 7 days. DMSO was used as solvent control. Cells were then fixed and stained with anti-CD133 (1:500) and anti-CD34 (1:500) antibodies (indicative of EPC population with high stemness). Flow cytometry analysis was performed in day 16 culture to quantify the number of progenitor cells based on CD133+CD34+ cell surface marker (mean ± SD; n = 3; *P < 0.05 vs. solvent controls). In a parallel experiment, another batch of 9-day-old EPCs were treated in the presence and absence of 20-HETE (1 nM) for 4 h (C). Real-time PCR was carried out for gene expression of Oct 4, Sox2, and Nanog (mean ± SD; n = 3 in triplicates; *P < 0.05 vs. corresponding controls).
Preexisting EC responses during ischemia-induced neovascularization is CYP4A/20-HETE-dependent.
It is well known that preexisting EC responses to ischemia drive compensatory angiogenesis. EC proliferation is one of the essential components of the angiogenic process. We first determined the effects of ischemia on EC proliferation in hindlimb gracilis muscle in vivo. Gracilis muscles from nonischemic control and ischemic hindlimb were harvested on day 3 postligation and frozen sectioned for IF staining of CD31 (EC marker; represented in green), Ki-67 (mitotic marker; represented in red), and DAPI (nuclei; represented in blue). Ischemic gracilis muscle under angiogenic stimuli revealed a marked increase in EC proliferation as indicated by the number of triple positive cells in the merged image compared with the nonischemic control (Fig. 5A). In parallel experiments, gracilis muscles from nonischemic control and ischemic hindlimb in the presence and absence of 5 mg·kg−1·day−1 DDMS were harvested 3 days postligation and frozen sectioned for IF staining of CD31 (EC marker) colocalization with various EC response indexes that have been previously associated with angiogenesis (21–23, 27, 28, 34, 44), including Ki-67 (mitotic marker), TUNEL (apoptosis marker), DHE [reactive oxygen species (ROS) marker], phosphorylated Ser1179 eNOS (eNOS marker), and ICAM-1 (EC adhesion marker). DAPI was used to stain cell nuclei. The number of triple-positive cells in all cases were counted from six blindly chosen fields and quantitated. Fig. 5B shows that ischemic gracilis muscle displays increased EC proliferation, decreased apoptosis, increased ROS formation, increased eNOS activation, and increased expression of ICAM-1. On the other hand, inhibition of 20-HETE synthesis by DDMS markedly attenuated these increases in EC proliferation, ROS formation, and ICAM-1 expression, whereas it had no significant effects on apoptosis and eNOS activation. Thus, specific EC responses associated with angiogenesis under ischemic stimuli are CYP4A/20-HETE-dependent.
Fig. 5.
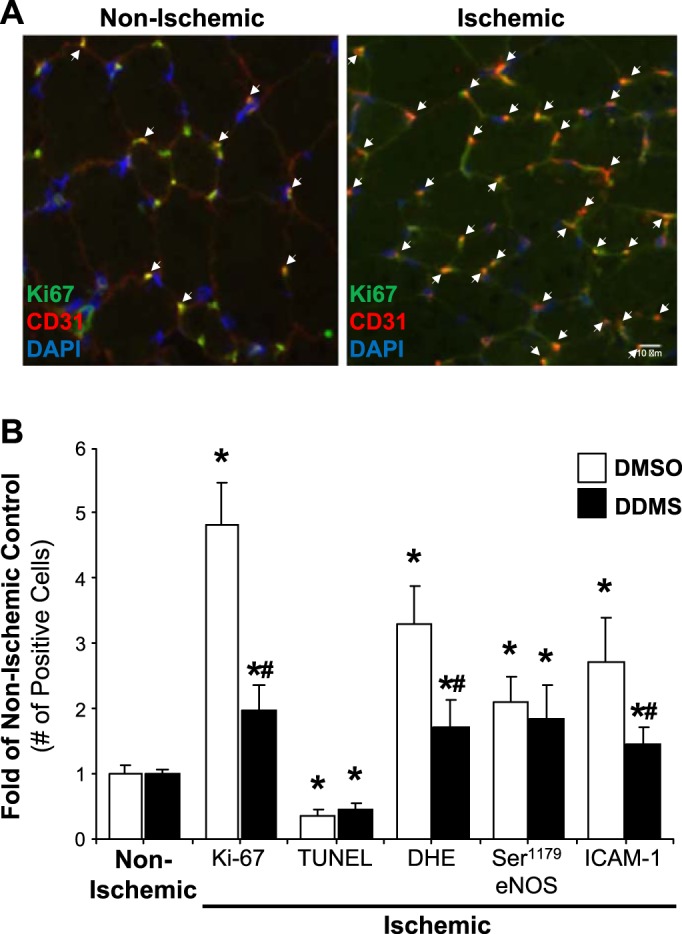
Preexisting endothelial cell (EC) responses to ischemia is CYP4A/20-hydroxyeicosatetraenoic acid-dependent. Femoral artery ligation was performed in animals pretreated with/without dibromo-dodecenyl-methylsulfimide (DDMS) (5 mg·kg−1·day−1; osmotic pump) for 2 days. Ischemic gracilis muscles were extracted 3 days postischemia for immunofluorescent (IF) microscopy of CD31 colocalization with various EC response indexes. Nonischemic contralateral hindlimbs were used as controls. A: representative images of IF staining of proliferative index Ki-67 (green) that colocalize with vessel marker CD31 (red) and DAPI staining of nuclei (blue). White arrows indicate proliferating ECs. B: IF microscopy of CD31 colocalization with Ki67, TUNEL, DHE, Ser1179 eNOS, and ICAM-1 was carried out and quantitated by counting. Data were normalized to the folds of nonischemic controls (mean ± SE; n = 6; *P < 0.05 vs. nonischemic control; #P < 0.05 vs. ischemic DMSO controls).
DISCUSSION
Newly emerging roles of CYP4A/20-HETE in regulation of pathological angiogenesis (10, 27, 46) highlight the importance of further understanding its underlying molecular and cellular mechanisms. Based on our recent report that 20-HETE contributes to ischemia-induced neovascularization (12), the current study is the first to explore the role of CYP4A/20-HETE in regulation of neovascularization in tissue ischemia via its collective actions on BM-derived EPCs and preexisting ECs. Our findings strongly support the conclusion that neovascularization under ischemic settings relies on a mechanism involving an early increase of 20-HETE at local endothelium, which upregulates BM-EPC mobilization, homing, vasculogenic functions, as well as enhances local preexisting EC angiogenic response. This conclusion is based on four crucial findings.
The first major finding of the current studies is that a transient increase of local endothelial cyp4a12 expression and 20-HETE production secondary to ischemia triggers an upregulation of SDF-1α, which in turn activates the recruitment of BM-derived EPCs to mobilize and home toward the sites of neovascularization. The comprehensive 20-HETE production kinetics using LC-MS/MS analysis in the current study revealed that the hindlimb gracilis muscles harvested on day 3 postischemia produced the highest levels of 20-HETE and gradually decreased on day 7 and eventually returned to the basal levels on day 21, thus a time-dependent process. We also showed that a time-dependent increase of cyp4a12 expression in ischemic gracilis muscles is consistent with 20-HETE production kinetics that we observed. Moreover, pharmacological inhibitions of CYP4A/20-HETE markedly reduced ischemia-induced angiogenesis on days 3, 7, and 21 postischemia. These results support that an early rise in 20-HETE production potentially served as a triggering and sustaining angiogenic signal to initiate and maintain neovascularization because of ischemia. As the ischemic angiogenesis approaches normalization, its production fades back to normal basal level. Identifying the precise cellular origin of increased 20-HETE in gracilis muscle postischemia is complex. 20-HETE may be derived from local ECs at the sites of neovascularization because hypoxic conditions such as ischemia can markedly increase 20-HETE production in ECs, which normally produce no measurable amount of 20-HETE (12). IF microscopy of the major murine 20-HETE-synthesizing enzyme cyp4a12 (42) and EC marker CD31 under high magnification clearly illustrated that they were coexpressed at the local endothelium within the ischemic gracilis muscle. This observation strengthens the notion that ischemia-induced 20-HETE increase may be endothelial in origin. Our group is in the process of specifically targeting the cyp4a-20-HETE in cellular components using cell-specific delivery of adenoviral and lentiviral shRNA constructs in vivo in our ischemic hindlimb experimental model. In parallel, development and genotyping of EC-specific Cre-Flox cyp4a12 knockout mice is currently ongoing. These new tools will enable us to further confirm that local endothelium is the cellular source of ischemia-induced 20-HETE in the future. However, this finding of 20-HETE that originated from local endothelium does not necessarily exclude the possibility of additional cellular sources such as systemic microcirculation (circulating inflammatory cells/EPCs) (14, 17, 24, 25, 28, 32, 41, 50, 56), and even skeletal muscle cells (1) can also contribute to the generation of ischemia-mediated 20-HETE at the local endothelium. After ischemic injury, production of inflammatory cytokines can quickly recruit inflammatory cells such as neutrophils to arrive at the site of injury, which then recruit macrophages through the release of additional cytokines. A recent study by Joseph et al. (30) suggested that elevated 20-HETE was associated with excessive expression of endothelial adhesion molecules and neutrophil infiltration in rats with metabolic syndrome. Whether inflammatory cells such as neutrophils/macrophages can potentially contribute to the increased 20-HETE production under ischemic settings remains completely unknown. Our group is currently studying the potential role that inflammatory cells play in 20-HETE generation postischemia. Thus, the precise cellular mechanisms by which ischemia induces the production of 20-HETE requires further elucidation.
The second major finding of our studies is that CYP4A/20-HETE plays a critical role in regulating the mobilization and homing of BM-derived EPCs during ischemia. EPCs are well known components of ischemia-induced neovascularization (9, 15, 31, 33, 35, 48, 51, 54, 55). SDF-1α mainly facilitates neovascularization via its synergistic effects with VEGF to stimulate local EC proliferation and tube formation as well as the mobilization, recruitment, and homing of BM-derived cells such as EPCs (45, 52). We demonstrated here that SDF-1α increase in ischemic tissues is CYP4A/20-HETE-dependent during ischemia, as well as that 20-HETE can induce SDF-1α production in EC cultures. These findings suggest that local production of SDF-1α at the ischemic tissue level is most likely the triggering signal for EPC mobilization and homing during ischemia. These new findings also further open the possibility that a positive feedback regulation of 20-HETE and VEGF may indeed exist, consistent with our previous reports (11, 12, 23). The precise underlying mechanism of 20-HETE and VEGF feed-forward interaction is ongoing in the laboratory. Moreover, our current findings that pharmacological interferences of CYP4A/20-HETE such as DDMS and 6,15–20-HEDGE markedly inhibit an ischemia-induced increase in the numbers of circulatory EPCs and EPCs homed to ischemic tissue is highly novel and significant. It implicates that EPC mobilization, recruitment, and homing during ischemia is CYP4A/20-HETE-dependent. Although pharmacological interferences of CYP4A/20-HETE-mediated decreases in circulatory EPCs postischemia are most likely due to effects on EPC mobilization from BM. However, it is also possible that such inhibition may result from the antiproliferative effects of DDMS and 6,15-20-HEDGE on circulatory EPCs in peripheral blood. Pilot studies using isolated circulatory EPCs harvested at various times postischemia in the presence of these inhibitors showed that their proliferation (Ki-67 indexing by flow cytometry) is not affected (unpublished data). Additionally, we found that the plasma SDF-1α concentration significantly increased postischemia to a level that is sufficient for mobilization of EPCs from the BM (15), and this increase is also CYP4A/20-HETE-dependent. Taken together, these results strongly suggest that CYP4A/20-HETE not only regulates EPC mobilization from the BM in response to ischemic challenge but also subsequently mediates EPCs to home toward vascular structures at the site of neovascularization, potentially via an SDF-1α-dependent mechanism.
The third major finding of the present study is that endogenous 20-HETE levels of EPCs are associated with vasculogenic functions of EPCs in vitro and in vivo, and an exogenous addition of 20-HETE can promote EPC stemness, a novel finding. We found that spontaneous differentiation of EPCs in culture is accompanied by a parallel decrease in CYP4A11 expression, 20-HETE production, and colony formation capacity of EPCs. These findings are further supported by our observations that pharmacological blockade of CYP4A/20-HETE leads to a decreased EPC population that displays primitive progenitor cell markers (high stemness). In contrast, addition of exogenous 20-HETE not only promotes colony formation by late differentiated EPCs, but it also increases the number of EPCs that are consistent with high stemness. It is interesting to note that while the action of 20-HETE can be reached with nM concentrations, μM concentrations of 20-HETE synthesis inhibitors and antagonists were needed for inhibition. This is likely due to a relatively high endogenous level of 20-HETE present in day 9 EPC culture, and thus it may require μM range of CYP4A/20-HETE inhibitors to fully exert their inhibitory action. Experiments in the current studies are performed using EPC cultures isolated from both human and murine origins. Since both 20-HETE and its pharmacological interfering agents have been shown to have differential effects on EPC proliferation, it is possible that these effects may partially attribute to some of our current observations. Although we have demonstrated that exogenous 20-HETE induces the expression of several EPC self-renewal genes, the precise mechanisms and epigenetics underlying these findings remain unknown and warrant further elucidation. In any regard, CYP4A/20-HETE may be a potential novel marker for evaluating the stemness characteristics of progenitor cells. We will continue to explore whether the changes induced by 20-HETE on EPC functions/markers can also be translated in vivo in our future studies. The association of endogenous 20-HETE levels and EPC functions as well as the positive effects of 20-HETE on EPC stemness revealed by our studies have significant potential clinical implications. The clinical trials using EPC-based cell therapy to promote neovascularization have been largely unsuccessful. These obscure clinical outcomes may have been contributed largely by impaired cellular functions of autologous EPCs harvested from “sick” patients with preexisting cardiovascular disease and risk factors. Thus, it will enable us to further develop and understand new pathways that can be used to “engineer” healthy EPCs ex vivo to improve their therapeutic capability. Moreover, identifying novel paracrine factors derived from EPCs, such as 20-HETE, will expedite stem cell-free therapeutics in treating pathological neovascularization without concerns arising from the use of dysfunctional EPCs from patients with preexisting cardiovascular conditions.
The last major finding of our studies is that ischemia-induced 20-HETE promotes various angiogenic functions of preexisting ECs at the site of neovascularization. ECs play an essential regulatory role in the capacity of the vasculature to adequately respond to injury or hypoxia. In tissue ischemia, newly formed capillaries are generated by proliferation and migration of ECs that sprout from preexisting capillaries, and thus ECs represent the critical cellular element responsible for new vessel formation (18, 19). We previously demonstrated that 20-HETE is proangiogenic by stimulating EC proliferation, migration, and tube formation via a mechanism involving ROS-mediated increases in VEGF production in vitro (21, 23). 20-HETE has also been shown by others to affect EC apoptosis (16), uncoupling of eNOS (14), and EC adhesion molecules (EC adhesion molecule changes during EC migration in response to angiogenic stimuli) (27, 28, 34, 44). Thus, we examined the role of CYP4A/20-HETE in the regulation of preexisting EC responses that are often associated with angiogenic stimuli such as ischemia in vivo, including EC proliferation, apoptosis, ROS formation, eNOS activation, and ICAM/E-selectin expression. The current study revealed that ischemia promotes EC proliferation, ROS formation, eNOS activation, and ICAM-1 expression, whereas it inhibited EC apoptosis. Furthermore, pharmacological interferences of CYP4A/20-HETE markedly inhibited the increases in EC proliferation, ROS formation, and ICAM-1 expression but had no effects on EC apoptosis and eNOS activation. Thus, most of the findings in our study are highly consistent with the effects of 20-HETE on ECs observed in vitro. Tissue and cell specificity as well as unique pathological conditions may contribute to the inaction of 20-HETE blockade on EC apoptosis and eNOS activation.
On the basis of the current findings, it is likely that CYP4A/20-HETE is involved in the regulation of ischemic neovascularization via a multistep process as illustrated in Fig. 6. Ischemia triggers an increase in local endothelial cyp4a12 expression and 20-HETE production (step 1), which in turn stimulates the production and release of chemokine SDF-1α (step 2). SDF-1α acts as an EPC mobilization signal from BM into peripheral circulation (steps 3 and 4). EPCs can then home toward ischemic gracilis muscle (the target sites of neovascularization) toward SDF-1α gradient (step 5), contributing to ischemia-induced vasculogenesis. In parallel, local endothelium 20-HETE increases secondary to ischemia can also influence specific preexisting EC responses that are associated with angiogenesis, such as EC proliferation in autocrine and paracrine manners (step 6). The hypoxic and ischemic conditions at the target sites may further reinforce the level of 20-HETE by amplifying additional angiogenic signals through preexisting ECs, contributing to angiogenesis. As a result, the combined vasculogenic effects via EPCs and angiogenic effects via preexisting ECs by 20-HETE collectively resulted in increased neovascularization in response to ischemia (step 7). Thus, the present findings further lead to a comprehensive understanding of the involvement of CYP4A/20-HETE in the regulation of vascular repair and consequently to novel therapeutic strategies.
Fig. 6.
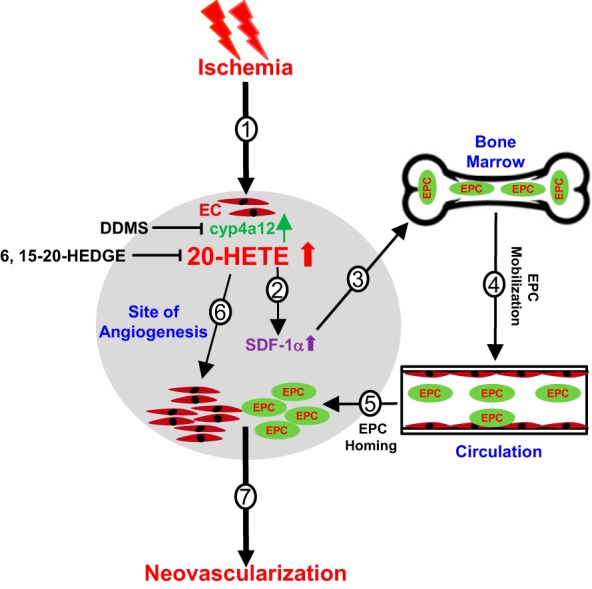
Schematics by which CYP4A/20-hydroxyeicosatetraenoic acid (20-HETE) regulates ischemia-induced neovascularization processes via endothelial progenitor cells (EPCs) and preexisting endothelial cells (ECs). The CYP4A/20-HETE axis regulates ischemic neovascularization via a multistep process. Ischemia induces an increase in cyp4a12 expression and 20-HETE production by injured endothelium (step 1); increases in 20-HETE stimulates the production and release of stromal cell-derived factor-1α (SDF-1α) (step 2); SDF-1α in turn signals EPC mobilization from bone marrow to circulation (steps 3 and 4); EPCs then home toward the target sites of angiogenesis (step 5). In parallel, 20-HETE increases secondary to ischemia at the local endothelium and leads to upregulated preexisting EC response, such as EC proliferation in autocrine and paracrine manners. The hypoxic and ischemic conditions at the target sites may further reinforce the level of 20-HETE by amplifying additional angiogenic signals through preexisting EC networks (step 6). Thus, the combined vasculogenic effects via EPC and angiogenic effects via a preexisting EC network by 20-HETE collectively resulted in increased neovascularization in response to ischemia.
GRANTS
This study was supported by the American Heart Association (Grants 11SDG6870004 and 17GRNT33430003 to A.M.G); the NIH (Grant HL-34300 to M.L.S and Grant DK-38226 to J.R.F); and the Robert A. Welch Foundation (Grant GL625910 to J.R.F).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.C., M.L.S., and A.M.G. conceived and designed research; L.C., S.T., F.F.Z., V.G., J.R.F., and A.M.G. performed experiments; L.C., J.R.F., and A.M.G. analyzed data; J.R.F., M.L.S., A.S.A., and A.M.G. interpreted results of experiments; L.C., S.T., V.G., and A.M.G. prepared figures; A.M.G. drafted manuscript; L.C., A.S.A., and A.M.G. edited and revised manuscript; J.R.F., M.L.S., and A.M.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Wolf-Hagen Schunck and Dr. Ramona Zummach for providing the mouse cyp4a12 antibodies. We also thank Katherine H. Gotlinger for technical assistance in the measurement of 20-HETE using LC-MS/MS.
REFERENCES
- 1.Amaral SL, Maier KG, Schippers DN, Roman RJ, Greene AS. CYP4A metabolites of arachidonic acid and VEGF are mediators of skeletal muscle angiogenesis. Am J Physiol Heart Circ Physiol 284: H1528–H1535, 2003. doi: 10.1152/ajpheart.00406.2002. [DOI] [PubMed] [Google Scholar]
- 2.Arbab AS, Janic B, Knight RA, Anderson SA, Pawelczyk E, Rad AM, Read EJ, Pandit SD, Frank JA. Detection of migration of locally implanted AC133+ stem cells by cellular magnetic resonance imaging with histological findings. FASEB J 22: 3234–3246, 2008. doi: 10.1096/fj.07-105676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, Kearne M, Magner M, Isner JM. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res 85: 221–228, 1999. doi: 10.1161/01.RES.85.3.221. [DOI] [PubMed] [Google Scholar]
- 4.Bayat H, Fathi F, Peyrovi H, Mowla SJ. Evaluating the expression of self-renewal genes in human endothelial progenitor cells. Cell J 14: 298–305, 2013. [PMC free article] [PubMed] [Google Scholar]
- 5.Bellamine A, Wang Y, Waterman MR, Gainer JV III, Dawson EP, Brown NJ, Capdevila JH. Characterization of the CYP4A11 gene, a second CYP4A gene in humans. Arch Biochem Biophys 409: 221–227, 2003. doi: 10.1016/S0003-9861(02)00545-3. [DOI] [PubMed] [Google Scholar]
- 6.Caiado F, Dias S. Endothelial progenitor cells and integrins: adhesive needs. Fibrogenesis Tissue Repair 5: 4, 2012. doi: 10.1186/1755-1536-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carroll MA, Balazy M, Huang DD, Rybalova S, Falck JR, McGiff JC. Cytochrome P450-derived renal HETEs: storage and release. Kidney Int 51: 1696–1702, 1997. doi: 10.1038/ki.1997.234. [DOI] [PubMed] [Google Scholar]
- 8.Cencioni C, Capogrossi MC, Napolitano M. The SDF-1/CXCR4 axis in stem cell preconditioning. Cardiovasc Res 94: 400–407, 2012. doi: 10.1093/cvr/cvs132. [DOI] [PubMed] [Google Scholar]
- 9.Ceradini DJ, Gurtner GC. Homing to hypoxia: HIF-1 as a mediator of progenitor cell recruitment to injured tissue. Trends Cardiovasc Med 15: 57–63, 2005. doi: 10.1016/j.tcm.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 10.Chen L, Ackerman R, Guo AM. 20-HETE in neovascularization. Prostaglandins Other Lipid Mediat 98: 63–68, 2012. doi: 10.1016/j.prostaglandins.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 11.Chen L, Ackerman R, Saleh M, Gotlinger KH, Kessler M, Mendelowitz LG, Falck JR, Arbab AS, Scicli AG, Schwartzman ML, Yang J, Guo AM. 20-HETE regulates the angiogenic functions of human endothelial progenitor cells and contributes to angiogenesis in vivo. J Pharmacol Exp Ther 348: 442–451, 2014. doi: 10.1124/jpet.113.210120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen L, Joseph G, Zhang FF, Nguyen H, Jiang H, Gotlinger KH, Falck JR, Yang J, Schwartzman ML, Guo AM. 20-HETE contributes to ischemia-induced angiogenesis. Vascul Pharmacol 83: 57–65, 2016. doi: 10.1016/j.vph.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen P, Guo M, Wygle D, Edwards PA, Falck JR, Roman RJ, Scicli AG. Inhibitors of cytochrome P450 4A suppress angiogenic responses. Am J Pathol 166: 615–624, 2005. doi: 10.1016/S0002-9440(10)62282-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng J, Ou JS, Singh H, Falck JR, Narsimhaswamy D, Pritchard KA Jr, Schwartzman ML. 20-hydroxyeicosatetraenoic acid causes endothelial dysfunction via eNOS uncoupling. Am J Physiol Heart Circ Physiol 294: H1018–H1026, 2008. doi: 10.1152/ajpheart.01172.2007. [DOI] [PubMed] [Google Scholar]
- 15.De Falco E, Porcelli D, Torella AR, Straino S, Iachininoto MG, Orlandi A, Truffa S, Biglioli P, Napolitano M, Capogrossi MC, Pesce M. SDF-1 involvement in endothelial phenotype and ischemia-induced recruitment of bone marrow progenitor cells. Blood 104: 3472–3482, 2004. doi: 10.1182/blood-2003-12-4423. [DOI] [PubMed] [Google Scholar]
- 16.Dhanasekaran A, Bodiga S, Gruenloh S, Gao Y, Dunn L, Falck JR, Buonaccorsi JN, Medhora M, Jacobs ER. 20-HETE increases survival and decreases apoptosis in pulmonary arteries and pulmonary artery endothelial cells. Am J Physiol Heart Circ Physiol 296: H777–H786, 2009. doi: 10.1152/ajpheart.01087.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fleming I. Cytochrome p450 and vascular homeostasis. Circ Res 89: 753–762, 2001. doi: 10.1161/hh2101.099268. [DOI] [PubMed] [Google Scholar]
- 18.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1: 27–30, 1995. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 19.Folkman J. Angiogenesis: initiation and control. Ann N Y Acad Sci 401, 1 Endothelium: 212–226, 1982. doi: 10.1111/j.1749-6632.1982.tb25720.x. [DOI] [PubMed] [Google Scholar]
- 20.Galasso G, De Rosa R, Ciccarelli M, Sorriento D, Del Giudice C, Strisciuglio T, De Biase C, Luciano R, Piccolo R, Pierri A, Di Gioia G, Prevete N, Trimarco B, Piscione F, Iaccarino G. β2-Adrenergic receptor stimulation improves endothelial progenitor cell-mediated ischemic neoangiogenesis. Circ Res 112: 1026–1034, 2013. doi: 10.1161/CIRCRESAHA.111.300152. [DOI] [PubMed] [Google Scholar]
- 21.Guo AM, Arbab AS, Falck JR, Chen P, Edwards PA, Roman RJ, Scicli AG. Activation of vascular endothelial growth factor through reactive oxygen species mediates 20-hydroxyeicosatetraenoic acid-induced endothelial cell proliferation. J Pharmacol Exp Ther 321: 18–27, 2007. doi: 10.1124/jpet.106.115360. [DOI] [PubMed] [Google Scholar]
- 22.Guo AM, Janic B, Sheng J, Falck JR, Roman RJ, Edwards PA, Arbab AS, Scicli AG. The cytochrome P450 4A/F-20-hydroxyeicosatetraenoic acid system: a regulator of endothelial precursor cells derived from human umbilical cord blood. J Pharmacol Exp Ther 338: 421–429, 2011. doi: 10.1124/jpet.111.179036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo AM, Scicli G, Sheng J, Falck JC, Edwards PA, Scicli AG. 20-HETE can act as a nonhypoxic regulator of HIF-1alpha in human microvascular endothelial cells. Am J Physiol Heart Circ Physiol 297: H602–H613, 2009. doi: 10.1152/ajpheart.00874.2008. [DOI] [PubMed] [Google Scholar]
- 24.Harder DR, Lange AR, Gebremedhin D, Birks EK, Roman RJ. Cytochrome P450 metabolites of arachidonic acid as intracellular signaling molecules in vascular tissue. J Vasc Res 34: 237–243, 1997. doi: 10.1159/000159228. [DOI] [PubMed] [Google Scholar]
- 25.Hill E, Murphy RC. Quantitation of 20-hydroxy-5,8,11,14-eicosatetraenoic acid (20-HETE) produced by human polymorphonuclear leukocytes using electron capture ionization gas chromatography/mass spectrometry. Biol Mass Spectrom 21: 249–253, 1992. doi: 10.1002/bms.1200210505. [DOI] [PubMed] [Google Scholar]
- 26.Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med 348: 593–600, 2003. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 27.Hoopes SL, Garcia V, Edin ML, Schwartzman ML, Zeldin DC. Vascular actions of 20-HETE. Prostaglandins Other Lipid Mediat 120: 9–16, 2015. doi: 10.1016/j.prostaglandins.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishizuka T, Cheng J, Singh H, Vitto MD, Manthati VL, Falck JR, Laniado-Schwartzman M. 20-Hydroxyeicosatetraenoic acid stimulates nuclear factor-kappaB activation and the production of inflammatory cytokines in human endothelial cells. J Pharmacol Exp Ther 324: 103–110, 2008. doi: 10.1124/jpet.107.130336. [DOI] [PubMed] [Google Scholar]
- 29.Janic B, Guo AM, Iskander AS, Varma NR, Scicli AG, Arbab AS. Human cord blood-derived AC133+ progenitor cells preserve endothelial progenitor characteristics after long term in vitro expansion. PLoS One 5: e9173, 2010. doi: 10.1371/journal.pone.0009173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joseph G, Soler A, Hutcheson R, Hunter I, Bradford C, Hutcheson B, Gotlinger KH, Jiang H, Falck JR, Proctor S, Schwartzman ML, Rocic P. Elevated 20-HETE impairs coronary collateral growth in metabolic syndrome via endothelial dysfunction. Am J Physiol Heart Circ Physiol 312: H528–H540, 2017. doi: 10.1152/ajpheart.00561.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalka C, Masuda H, Takahashi T, Kalka-Moll WM, Silver M, Kearney M, Li T, Isner JM, Asahara T. Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc Natl Acad Sci USA 97: 3422–3427, 2000. doi: 10.1073/pnas.97.7.3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalyankrishna S, Malik KU. Norepinephrine-induced stimulation of p38 mitogen-activated protein kinase is mediated by arachidonic acid metabolites generated by activation of cytosolic phospholipase A(2) in vascular smooth muscle cells. J Pharmacol Exp Ther 304: 761–772, 2003. doi: 10.1124/jpet.102.040949. [DOI] [PubMed] [Google Scholar]
- 33.Kawamoto A, Gwon HC, Iwaguro H, Yamaguchi JI, Uchida S, Masuda H, Silver M, Ma H, Kearney M, Isner JM, Asahara T. Therapeutic potential of ex vivo expanded endothelial progenitor cells for myocardial ischemia. Circulation 103: 634–637, 2001. doi: 10.1161/01.CIR.103.5.634. [DOI] [PubMed] [Google Scholar]
- 34.Kim I, Moon SO, Park SK, Chae SW, Koh GY. Angiopoietin-1 reduces VEGF-stimulated leukocyte adhesion to endothelial cells by reducing ICAM-1, VCAM-1, and E-selectin expression. Circ Res 89: 477–479, 2001. doi: 10.1161/hh1801.097034. [DOI] [PubMed] [Google Scholar]
- 35.Kocher AA, Schuster MD, Szabolcs MJ, Takuma S, Burkhoff D, Wang J, Homma S, Edwards NM, Itescu S. Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat Med 7: 430–436, 2001. doi: 10.1038/86498. [DOI] [PubMed] [Google Scholar]
- 36.Kucia M, Jankowski K, Reca R, Wysoczynski M, Bandura L, Allendorf DJ, Zhang J, Ratajczak J, Ratajczak MZ. CXCR4-SDF-1 signalling, locomotion, chemotaxis and adhesion. J Mol Histol 35: 233–245, 2004. doi: 10.1023/B:HIJO.0000032355.66152.b8. [DOI] [PubMed] [Google Scholar]
- 37.Kucia M, Reca R, Miekus K, Wanzeck J, Wojakowski W, Janowska-Wieczorek A, Ratajczak J, Ratajczak MZ. Trafficking of normal stem cells and metastasis of cancer stem cells involve similar mechanisms: pivotal role of the SDF-1-CXCR4 axis. Stem Cells 23: 879–894, 2005. doi: 10.1634/stemcells.2004-0342. [DOI] [PubMed] [Google Scholar]
- 38.Lasker JM, Chen WB, Wolf I, Bloswick BP, Wilson PD, Powell PK. Formation of 20-hydroxyeicosatetraenoic acid, a vasoactive and natriuretic eicosanoid, in human kidney. Role of Cyp4F2 and Cyp4A11. J Biol Chem 275: 4118–4126, 2000. doi: 10.1074/jbc.275.6.4118. [DOI] [PubMed] [Google Scholar]
- 39.Majesky MW. A little VEGF goes a long way. Therapeutic angiogenesis by direct injection of vascular endothelial growth factor-encoding plasmid DNA. Circulation 94: 3062–3064, 1996. doi: 10.1161/01.CIR.94.12.3062. [DOI] [PubMed] [Google Scholar]
- 40.Medhora M, Dhanasekaran A, Gruenloh SK, Dunn LK, Gabrilovich M, Falck JR, Harder DR, Jacobs ER, Pratt PF. Emerging mechanisms for growth and protection of the vasculature by cytochrome P450-derived products of arachidonic acid and other eicosanoids. Prostaglandins Other Lipid Mediat 82: 19–29, 2007. doi: 10.1016/j.prostaglandins.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 41.Miyata N, Roman RJ. Role of 20-hydroxyeicosatetraenoic acid (20-HETE) in vascular system. J Smooth Muscle Res 41: 175–193, 2005. doi: 10.1540/jsmr.41.175. [DOI] [PubMed] [Google Scholar]
- 42.Muller DN, Schmidt C, Barbosa-Sicard E, Wellner M, Gross V, Hercule H, Markovic M, Honeck H, Luft FC, Schunck WH. Mouse Cyp4a isoforms: enzymatic properties, gender- and strain-specific expression, and role in renal 20-hydroxyeicosatetraenoic acid formation. Biochem J 403: 109–118, 2007. doi: 10.1042/BJ20061328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nguyen X, Wang MH, Reddy KM, Falck JR, Schwartzman ML. Kinetic profile of the rat CYP4A isoforms: arachidonic acid metabolism and isoform-specific inhibitors. Am J Physiol 276, 6 Pt 2: R1691–R1700, 1999. doi: 10.1152/ajpregu.1999.276.6.R1691. [DOI] [PubMed] [Google Scholar]
- 44.Oh IY, Yoon CH, Hur J, Kim JH, Kim TY, Lee CS, Park KW, Chae IH, Oh BH, Park YB, Kim HS. Involvement of E-selectin in recruitment of endothelial progenitor cells and angiogenesis in ischemic muscle. Blood 110: 3891–3899, 2007. doi: 10.1182/blood-2006-10-048991. [DOI] [PubMed] [Google Scholar]
- 45.Petit I, Jin D, Rafii S. The SDF-1-CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis. Trends Immunol 28: 299–307, 2007. doi: 10.1016/j.it.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rocic P, Schwartzman ML. 20-HETE in the regulation of vascular and cardiac function. Pharmacol Ther 192: 74–87, 2018. doi: 10.1016/j.pharmthera.2018.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 82: 131–185, 2002. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 48.Schatteman GC, Hanlon HD, Jiao C, Dodds SG, Christy BA. Blood-derived angioblasts accelerate blood-flow restoration in diabetic mice. J Clin Invest 106: 571–578, 2000. doi: 10.1172/JCI9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spector AA, Norris AW. Action of epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell Physiol 292: C996–C1012, 2007. doi: 10.1152/ajpcell.00402.2006. [DOI] [PubMed] [Google Scholar]
- 50.Stec DE, Gannon KP, Beaird JS, Drummond HA. 20-Hydroxyeicosatetraenoic acid (20-HETE) stimulates migration of vascular smooth muscle cells. Cell Physiol Biochem 19: 121–128, 2007. doi: 10.1159/000099200. [DOI] [PubMed] [Google Scholar]
- 51.Takahashi T, Kalka C, Masuda H, Chen D, Silver M, Kearney M, Magner M, Isner JM, Asahara T. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med 5: 434–438, 1999. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- 52.Tang JM, Wang JN, Zhang L, Zheng F, Yang JY, Kong X, Guo LY, Chen L, Huang YZ, Wan Y, Chen SY. VEGF/SDF-1 promotes cardiac stem cell mobilization and myocardial repair in the infarcted heart. Cardiovasc Res 91: 402–411, 2011. doi: 10.1093/cvr/cvr053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Urbich C, Aicher A, Heeschen C, Dernbach E, Hofmann WK, Zeiher AM, Dimmeler S. Soluble factors released by endothelial progenitor cells promote migration of endothelial cells and cardiac resident progenitor cells. J Mol Cell Cardiol 39: 733–742, 2005. doi: 10.1016/j.yjmcc.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 54.Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res 95: 343–353, 2004. doi: 10.1161/01.RES.0000137877.89448.78. [DOI] [PubMed] [Google Scholar]
- 55.Urbich C, Heeschen C, Aicher A, Dernbach E, Zeiher AM, Dimmeler S. Relevance of monocytic features for neovascularization capacity of circulating endothelial progenitor cells. Circulation 108: 2511–2516, 2003. doi: 10.1161/01.CIR.0000096483.29777.50. [DOI] [PubMed] [Google Scholar]
- 56.Zhu D, Zhang C, Medhora M, Jacobs ER. CYP4A mRNA, protein, and product in rat lungs: novel localization in vascular endothelium. J Appl Physiol (1985) 93: 330–337, 2002. doi: 10.1152/japplphysiol.01159.2001. [DOI] [PubMed] [Google Scholar]