

Abstract
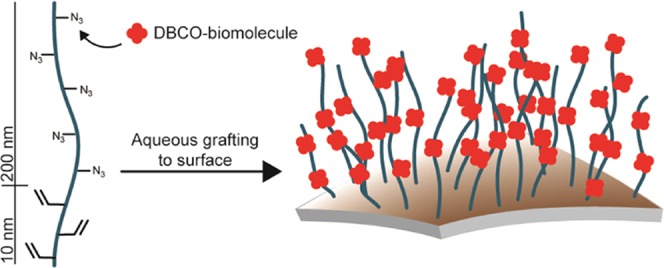
Polymer brushes are extensively used for the preparation of bioactive surfaces. They form a platform to attach functional (bio)molecules and control the physicochemical properties of the surface. These brushes are nearly exclusively prepared from flexible polymers, even though much stiffer brushes from semiflexible polymers are frequently found in nature, which exert bioactive functions that are out of reach for flexible brushes. Synthetic semiflexible polymers, however, are very rare. Here, we use polyisocyanopeptides (PICs) to prepare high-density semiflexible brushes on different substrate geometries. For bioconjugation, we developed routes with two orthogonal click reactions, based on the strain-promoted azide–alkyne cycloaddition reaction and the (photoactivated) tetrazole–ene cycloaddition reaction. We found that for high brush densities, multiple bonds between the polymer and the substrate are necessary, which was achieved in a block copolymer strategy. Whether the desired biomolecules are conjugated to the PIC polymer before or after brush formation depends on the dimensions and required densities of the biomolecules and the curvature of the substrate. In either case, we provide mild, aqueous, and highly modular reaction strategies, which make PICs a versatile addition to the toolbox for generating semiflexible bioactive polymer brush surfaces.
Introduction
Surfaces that interact specifically with biological systems are crucial for many applications, including tissue engineering,1 biosensing,2 medical implants,3 and nanomedicine.4,5 For example, growth factors can be attached to a surface to promote cell proliferation,6 antimicrobial peptides for bacterial killing,7 and RGD-based peptides may be used to increase cell adhesion.8 Polymer brushes are ideal designs for substrate biofunctionalization.9−11 Their chemical composition, polymer chain length, and grafting density control key parameters, including hydrophobicity, stiffness, and roughness. Moreover, bottom-up approaches allow for any desired two-dimensional (2D) pattern on the surface. In addition, brushes can be used to control the density and presentation of biomolecules, both of which are key factors that determine their activity. These biomolecules are commonly attached via physical adsorption, i.e., noncovalent interactions such as the biotin–streptavidin pair, or via covalent attachment.12
For typical polymer brushes, the (bio)polymers are tethered with one end to a surface at grafting densities high enough to crowd the polymers and force them to extend away from the surface due to entropic expulsion. In addition, the loose polymer ends of the brush have sufficient flexibility, which benefits the interaction between attached biomolecules and the biological system.13,7 Polymer brushes have been prepared using a wide range of polymers and substrates and the design of new polymers for novel brushes is still ongoing, for example, for the purpose of stimuli-responsive or -releasing brush surfaces.14−16 However, the current-generation polymer brushes are nearly exclusively prepared from flexible polymers.
In contrast, much stiffer brushes from semiflexible polymers are frequently found in biological interfaces.17,18 Examples include aggrecan brushes in cartilage tissue and the endothelial glycocalyx layer found on the inside of blood vessels.19,20 Computational modeling of the glycocalyx shows that the semiflexibility is crucial to control cell adhesion and leukocyte penetration.21,22 In addition, modeling predicts that semiflexible polymers reach extended conformations at much lower grafting densities, which enhances the presentation and accessibility of functional biological groups. Examples of brush surfaces from semiflexible polymers are limited to biopolymers such as DNA and hyaluronic acid and have not yet been used as bioactive surfaces to study cellular interactions.23,24 A primary reason why semiflexible brushes have been largely overlooked, so far, is the poor availability of synthetic, biocompatible, semiflexible polymers.
In this work, we present a route toward semiflexible polymer brushes from synthetic polymers that are easily biofunctionalized. As a semiflexible synthetic polymer, we use polyisocyanopeptides (PICs). With a stiffness that resembles that of natural protein-based filament, PICs have emerged as a new class of biocompatible polymers that closely mimic features of biological polymers, either as a hydrogel25 or in solution.26−30 PIC polymers can be up to 600 nm long, which allows for the introduction of multiple large biomolecules. For instance, PICs functionalized with antibodies and cytokines were already used as an artificial antigen-presenting cell (aAPC).26,27 Key factors in the high effectiveness of these aAPCs are the semiflexibility of the backbone, its length, and the density of functional groups. We note that the semiflexibility provides structure and allows for conformational changes to accommodate multivalent binding.
To maintain the largest possible versatility in our brushes, we developed a grafting-to approach, where we first synthesize the PIC with two orthogonal conjugation chemistries, where one is selectively introduced at one terminal end. We then either decorate the backbone with a model protein (bovine serum albumin (BSA)) via the strain-promoted azide–alkyne cycloaddition (SPAAC) and graft the construct to a substrate (“SPAAC first” strategy), or first graft the PIC polymer and decorate the surface with BSA subsequently (“graft first” strategy) (Figure 1). We find that, in contrast to common flexible polymers,31,32 our PIC chains need multiple surface binding groups for effective grafting. With the optimized design, we demonstrate that our brushes can be used on a variety of substrates, including iron oxide microbeads and nanoparticles (NPs) as well as glass coverslips. The polymers and grafting strategy described in this work facilitate the use of previously overlooked semiflexible brushes in the synthesis of bioactive surfaces and make it possible to study whether semiflexible brushes can be beneficial for bioactive applications.
Figure 1.
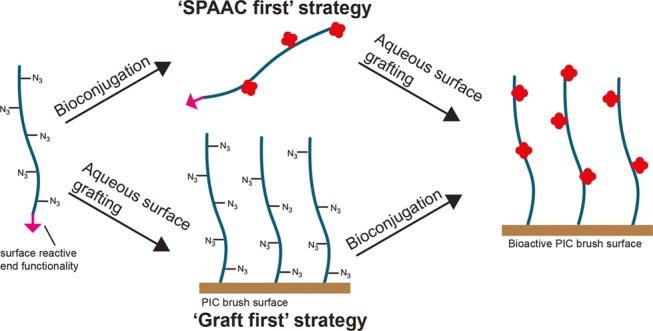
Schematic representation of the aim of this study. Azide-bearing semiflexible PICs with an orthogonal end-functionality are used to synthesize semiflexible PIC brushes via two strategies: first biofunctionalization of the azides using the SPAAC reaction, followed by surface grafting (SPAAC first), or biofunctionalization after surface grafting of the PIC (graft first).
Experimental Section
Materials, Instrumentation, and General Procedures
Unless stated otherwise, materials were obtained from Sigma-Aldrich (Merck) and used as obtained. Solvents were obtained from Fischer Scientific. The methoxy monomer 1 and azide monomer 2 (Figure 2a) were synthesized according to the procedures in the literature.26 Streptavidin beads MyOne C1 were bought from Thermo Fischer Scientific. Streptavidin nanoparticles were obtained from Ademtech. Dibenzocyclooctyne (DBCO)-poly(ethylene glycol) (PEG)4-N-hydroxysuccinimide (NHS) was obtained from Jena Bioscience. Click-iT DIBO-Alexa Fluor 647 was obtained from Thermo Fischer Scientific. The N-hydroxysuccinimide (NHS) ester of Atto 488 (NHS-Atto 488) was obtained from Atto-Tec. Streptavidin was bought from Sigma-Aldrich. For reactions of PIC with beads or nanoparticles (NPs), nonstick microfuge tubes from Ambion were used to reduce nonspecific binding of PIC. Magnetic microbeads and nanoparticles were washed using a magnetic rack from Westburg BV for magnetic separation. Thin-layer chromatography was performed on glass Silica gel 60 F254 plates from Merck. Flash chromatography was performed with SiliCycle silica gel 60 Å (40–63 μm). 1H and 13C NMR spectra were recorded with either Bruker Avance 500 or 400 NMR spectrometer. Electrospray liquid chromatography–mass spectrometry analysis was performed using a Finnigan LCQ Advantage IonTrap mass spectrometer. Infrared (IR) spectra were recorded on a Tensor 2700 spectrometer in attenuated total reflection (ATR) mode. Fluorescence was measured on a Spark M10 fluorescent plate reader using flat black 96-well plates form Corning. UV–vis measurements were performed on a Jasco V630 spectrometer. Flow cytometry was done using a BD Facs Verse cytometer and data were analyzed using FlowJo software. Confocal microscopy was performed on a Leica SP8 confocal microscope. Stochastic optical reconstruction microscopy (STORM) was performed on a custom-built low-drift inverted microscope setup (for details, see the Supporting Information).
Figure 2.
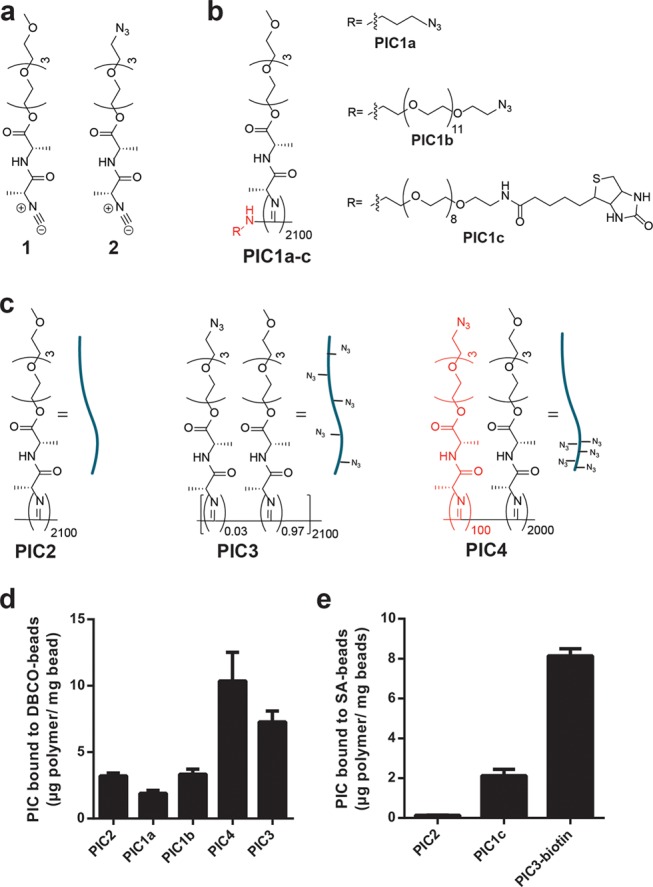
Structures of the monomer and polymers used to test binding to 1 μm beads and binding results. (a) Structure methoxy monomer 1 and azide monomer 2. (b) Structure of PIC1a–c and (c) structure of PIC2–4. (d) Binding of azide functional PIC to DBCO beads. (e) Binding of biotin functional PIC to streptavidin beads. In bar diagrams, average ± standard deviation of three independent experiments is shown. The part of the PIC structures in red indicates the part of the polymer that can bind the surface.
Synthesis of Semiflexible Polymers PIC1–5
General Procedure
The desired monomers were dissolved in dry toluene obtained from an MBraun SPS 800 solvent system (50 mg/mL), followed by addition of the desired catalyst solution with a catalyst-to-monomer ratio of 1:2000. The type of monomers and catalyst used per polymer type is given below. Polymerization was carried out overnight at room temperature. Isocyanide consumption was confirmed by disappearance of the characteristic IR absorbance at 2140 cm–1. The polymers were precipitated three times in diisopropyl ether and air-dried overnight to give off-white solids. The helical backbone of the PIC was confirmed by circular dichroism (CD) spectrometry of PIC solutions in phosphate-buffered saline (PBS) and the relative polymer length was determined by viscometry, as described in the literature, and is shown for each polymer in Table 1, Supporting Information.26 For PIC5b (PDI = 1.2), the length was also determined by atomic force spectroscopy (AFM), as described in the literature,30 using a Nanoscope IV instrument (Bruker) and NSG-10 tapping mode tips (NT-MDT).
Synthesis of PIC1a–c
For polymers PIC1a–c, different nickel carbene complexes were used as catalyst that were prepared by reacting tetrakis(tert-butyl isocyanide)nickel(II) perchlorate (Ni(CNR)4(ClO4)2) with an amine compound as described in the literature.33,34 The amine compounds used for the initiation are listed below for each polymer type. Tetrakis(tert-butyl isocyanide)nickel(II) perchlorate was synthesized according to the literature.35 The formation of the desired catalyst complexes was confirmed from the shift of the isocyanide stretching band in attenuated total reflection infrared spectroscopy (given per polymer below). The obtained catalyst solutions were then used without further purification. For the polymerizations of PIC1a–c, a monomer solution of methoxy monomer 1 (10 mg, 0.028 mmol, 40 mg/mL) was used.
PIC1a: 3-Azido-1-propaneamine was used as the amine for synthesis of the catalyst (N≡C stretch: 2210 cm–1). Yield: 7.33 mg (73%). PIC1b: Azido-PEG11-amine was used as the amine for catalyst synthesis (N≡C stretch: 2224 cm–1). Yield: 7.41 mg (74%). PIC1c: Biotin-PEG8-amine was used as the amine for catalyst synthesis (N≡C stretch: 2195 cm–1). Yield: 6.83 mg (68%).
Synthesis of PIC2
To 100 μL of a 27.7 mM solution of the azide monomer 2 (10 mg/mL) was added 6.94 μL of 4 mM catalyst solution Ni(ClO4)2·6H2O in 9:1 toluene/ethanol, monomer/catalyst ratio (100:1), and was reacted for 10 min. This reaction time was based on a colorimetric determination of the polymerization time (see below). Then, a 111 mM solution of methoxy monomer 1 in toluene was added (0.50 mL, 0.056 mmol) and the polymerization was continued as usual. Yield: 15.24 mg (76%) of PIC2.
Synthesis of PIC3
A monomer solution of methoxy monomer 1 (20 mg, 0.056 mmol) was used. A solution of Ni(ClO4)2·6H2O in toluene/ethanol (9:1) (1 mM) was used as catalyst solution. Yield: 15.6 mg (78%) of PIC3.
Synthesis of PIC4
A solution of azide monomer 2 (0.66 mg, 0.0018 mmol) and methoxy monomer 1 (19.34 mg, 0.054 mmol) was used. A solution of Ni(ClO4)2·6H2O in toluene/ethanol (9:1) (1 mM) was used as catalyst solution. Yield: 15.2 mg (75%) of PIC4.
Synthesis of PIC5a–e
A solution of the allyl monomer 3 and methoxy monomer 1 in toluene (100 μL) was prepared with the desired monomer ratio and an end concentration of 27.7 mM. To this mixture, 6.94 μL of 4 mM catalyst solution was added along with Ni(ClO4)2·6H2O in 9:1 toluene/ethanol with a monomer/catalyst ratio of 100:1, and was reacted for 10 min. This reaction time was based on a colorimetric determination of the polymerization time (see below). Then, a solution of azide monomer 2 (0.66 mg, 0.0018 mmol) and methoxy monomer 1 (19.34 mg, 0.054 mmol) in toluene (0.50 mL) was added and the polymerization was continued as usual.
PIC5a: First block: allyl monomer 3 only. Yield: 14.87 mg (89%). PIC5b: First block: monomer ratio 3:1 was 1:1. Yield: 15.35 mg (77%). PIC5c: First block: monomer ratio 3:1 was 1:5. Yield: 14.99 mg (75%). PIC5d: First block: monomer ratio 3:1 was 1:10. Yield: 13.84 mg (69%). PIC5e: First block: methoxy monomer 1 only. Yield: 14.45 mg (72%).
Synthesis of PIC5f
A solution of allyl monomer 3 (0.509 mg, 0.0013 mmol), azide monomer 2 (0.632 mg, 0.0017 mmol), and methoxy monomer 1 (18.86 mg, 0,052 mmol) was used. A solution of Ni(ClO4)2·6H2O in 9:1 toluene/ethanol (1 mM) was used as catalyst solution. Yield: 16.0 mg (80%) of PIC5f.
Tetrazine-Based Colorimetric Assay To Determine Polymerization Time
A polymerization with total volume 1 mL was performed using methoxy monomer at a final concentration of 10 mg/mL and 1 mM Ni(ClO4)2·6H2O in toluene/ethanol (9:1) as catalyst solution with a 1:100 catalyst-to-monomer ratio, as described above. At different time points, 10 μL of the polymerization mixture was taken out (0.278 μmol monomer) and diluted into 90 μL of a 1 mg/mL solution of benzylamino tetrazine in toluene (0.48 μmol, 1.73 equiv). The isocyanides that have not been incorporated in the polymer chain rapidly react with the excess tetrazine. The decrease in tetrazine signal was measured with UV–vis spectroscopy at 520 nm, and from this intensity, the concentration isocyanide and the corresponding conversion was calculated. All isocyanide was consumed after 10 min (Figure S1).
Binding Assay with PIC1–4
Synthesis of DBCO Beads
A 100 μL stock solution of streptavidin beads (MyOne C1 Streptavidin, Thermo Fischer Scientific) was suspended in 100 μL of borate buffer (50 mM, pH 8.5), and 1 μL of a 100 mM solution of NHS-PEG4-DBCO in dimethyl sulfoxide (DMSO) was added to react with the amine functionalities of the lysine residues of streptavidin. The beads were reacted overnight at room temperature and then washed five times with 200 μL of PBS and suspended in 100 μL of PBS before use. DBCO functionality on the beads was verified by reacting them with 3-hydroxy-7-azidocoumarin and measuring fluorescence by flow cytometry (Figure S2).
Binding Assay of PIC1–4 with Streptavidin and DBCO Beads
PIC1a–c, PIC3, PIC4, and PIC4-biotin were dissolved in PBS at a concentration of 0.5 mg/mL. Reactions with the beads were carried out by adding 10 μL of the polymer stock solution to 20 μL of a 10 mg/mL suspension of the corresponding beads. Control samples were made by adding 10 μL of the polymer stock solution to 20 μL of PBS. All samples were left rolling overnight at room temperature. The samples were then diluted with 90 μL of PBS, placed on a magnet for separation, and 100 μL of the supernatant was taken out to measure the amount of polymer by circular dichroism (peak at 272 nm). The amount of binding was determined by comparing bead samples with the nonbinding control. Average binding ± standard deviation of three independent experiments was determined for each condition.
Synthesis of Allyl Monomer
The allyl monomer was synthesized using a protocol similar to that of the methoxy monomer described in the literature,26 but instead of starting with tetraethylene glycol monomethyl ether, the synthesis was started with tetraethylene glycol mono allyl ether.
Synthesis of Tetraethylene Glycol Mono Allyl Ether
Sodium hydride in mineral oil (60%, 0.60 g, 14.9 mmol, 1 equiv) was dissolved in pentane (1 mL) on ice. The sodium hydride solution was then slowly added to a solution of tetraethylene glycol (56.25 g, 290 mmol, 19.4 equiv) in tetrahydrofuran (THF) (50 mL) on ice. After addition was complete, a cold solution of allyl bromide (1.81 g, 14.9 mmol, 1 equiv) in THF (2 mL) was slowly added and the reaction mixture was stirred on ice for 30 min at room temperature for 4 h. The reaction mixture was washed with water (3 × 50 mL) and brine. The organic fraction was dried over anhydrous sodium sulfate, filtered, and solvents were removed. The crude product was purified by column chromatography using ethyl acetate as eluent. The product was obtained as a colorless oil (2.65 g, 11.3 mmol, 76%). 1H NMR (400 MHz, CDCl3): δ = 5.90 (m, 1H), 5.27 (dm, 1H, J = 16 Hz), 5.18 (dm, 1H, J = 12 Hz), 4.02 (m, 2H), 3.75–3.57 (m, 14H). Mass spectrometry (MS) (electrospray ionization (ESI)) m/z: found 257.4 (M + Na+), calcd 257.29.
Further Synthesis of Allyl Monomer
For the rest of the synthesis of the monomer, the above-mentioned protocol from the literature was used. In short, the ester with Boc-l-alanine was prepared using N,N-dimethylaminopyridine and N,N′dicyclohexylcarbodiimide (DCC) as coupling agents. After Boc-deprotection with 4 M HCl in dioxane, a peptide coupling with Boc-d-alanine was performed using DCC and N-hydroxybenzotriazole (HOBt) as coupling agents. After deprotection, the free amine was formylated with ethyl formate and then dehydrated to the isocyanide using the Burgess reagent. The final product was obtained as a slightly yellow oil (0.316 g, 0.818 mmol, 34% over four steps). 1 H NMR (400 MHz, CDCl3): δ = 6.99 (bd, 1H), 5.90 (m, 1H), 5.26 (dm, 1H, J = 16 Hz), 5.17 (dm, 1H, J = 12 Hz), 4.58 (m, 1H), 4.32 (m, 2H), 4.20 (m, 1H), 4.01 (m, 2H), 3.73–3.53 (m, 14H), 1.64 (d, J = 8 Hz, 3H), 1.47 (d, J = 8 Hz, 3H). 13C NMR (500 MHz, CDCl3): δ = 171.98, 165.77, 134.71, 117.12, 72.22, 70.57, 69.40, 69.13, 68.82, 64.68, 63.54, 53.43, 48.58, 19.67, 18.01. MS (ESI) m/z: found 409.4 (M + Na+), calcd 409.45.
Synthesis of Tetrazoles
Synthesis of Tetrazole-PEG8-Biotin
Tetrazole-PEG8-biotin was synthesized by a peptide coupling of biotin-PEG8-NH2 (Click Chemistry Tools) to 4-(2-phenyl-2H-tetrazol-5-yl)benzoic acid (tetrazole-COOH). Tetrazole-COOH was synthesized according to the literature.36 To a solution of tetrazole-COOH (3.89 mg, 0.0146 mmol, 1 equiv) in dimethylformamide (0.5 mL) were added N,N-diisopropylethylamine (7.6 μL, 3 equiv), HOBt (5.9 mg, 0.044 mmol, 3 equiv), N,N′-diisopropylcarbodiimide (5.5 mg, 0.044 mmol, 3 equiv), and biotin-PEG8-NH2 (10 mg, 0.0146 mmol, 1 equiv). The reaction was stirred at room temperature for 4 h. Dichloromethane was added (2 mL) and the reaction mixture was washed 3× with 10% aqueous citric acid solution, 1× with water, and 1× with brine. The organic fraction was dried over anhydrous sodium sulfate, filtered, and solvents were removed. The crude product was purified by column chromatography over silica using ethyl acetate as eluent. Tetrazole-PEG8-biotin was obtained as an orange oil (11.7 mg, 0.0126 mmol, 86%). 1H NMR (400 MHz, CDCl3): δ = 8.33 (m, 2H), 8.21 (m, 2H), 8.02 (m, 2H), 7.62–7.58 (m, 3H), 4.50 (m, 1H), 4.33 (m, 1H), 3.71–3.54 (m, 40H), 2.92 (dd, 1H, J = 8, 4 Hz), 2.72 (d, 1H, 16 Hz), 2.22 (m, 2H), 1.46 (m, 4H), 1.31 (m, 2H). MS (ESI) m/z: found 932.4 (M + H+), calcd: 932.5.
Synthesis of Tetrazole-PEG4-Methyltetrazine for STORM Samples
Tetrazole-PEG4-methyltetrazine was prepared according to the above protocol using methyltetrazine-PEG4-NH2 (Click Chemistry Tools). Tetrazole-PEG4-methyltetrazine was obtained as a pink solid (10.55 mg, 17.2 μmol, 65%). 1H NMR (400 MHz, CDCl3): δ = 8.46 (m, 2H), 8.30 (m, 2H), 8.17 (m, 2H), 7.98 (m, 2H), 7.60–7.49 (m, 3H), 7.06 (bs, 1H), 7.02 (m, 2H), 4.17 (m, 2H), 3.86 (m, 2H), 3.76–3.86 (m, 12H), 3.03 (s, 3H). MS (ESI) m/z: found: 634.6 (M + H+), calcd: 634.7.
STORM Nanoscopy
Sample Preparation
PIC5b was reacted with tetrazole-PEG4-methyltetrazine using a similar protocol to that described for the biotinylation below. The double-functional PIC5b was diluted to 1 mg/mL in PBS and 100 μL of this solution was first reacted for 4 h with 1 mM DBCO-biotin (Jena Bioscience) in DMSO (1 μL, 1 nmol, 0.11 equiv to azides on polymer), and then for 2 h with 10 mM DIBO-Alexa Fluor 647 in DMSO (20 nmol, 2.27 equiv to azides on polymer). Afterward, 3.33 mM trans-cyclooctene (TCO)-Alexa Fluor 488 in DMSO with 27% PBS (see below) was added (6 μL, 20 nmol) and the polymer conjugation reaction was left overnight. Without further purification, the dual-labeled polymers were diluted to 10 μg/mL in PBS and 20 μL of the dilution was spotted on streptavidin-coated coverslips (#1.5 Micro Coverglass, Electron Microscopy Sciences, preparation described below). After incubation for 1 min, the coverslips were thoroughly rinsed with PBS to remove unbound polymer and unreacted dyes. The coverslip sample was mounted on a custom-made low-drift magnetic sample holder. Polymers were imaged in 1 mL of OxEA buffer.37 Imaging was performed in wide-field illumination mode. During data acquisition, sample plane excitation power densities of ca. 1.8–5.0 kW/cm2 were used for the 639 and 488 nm light sources. Optionally, for back-pumping purposes, simultaneous excitation with the 405 nm light source was used at gradually increasing excitation power densities up to maximum 0.05 kW/cm2 in the sample plane. Typically, 20 000–50 000 frames were acquired in a region of interest of 300 × 300 pixels (for each channel) with a pixel size of 0.111 μm at an exposure time of 10 ms.
Data Analysis and Image Reconstruction
Data sets were analyzed with Fiji ImageJ 1.52 g38 and the analysis module ThunderSTORM.39 A detection threshold of 200 photons was used; typical uncertainty mode values were 12 nm for the 639 channel and 18 nm for the 488 channel. Images were reconstructed using the averaged shifted histograms method, with a rendering pixel size of 10 nm. Software drift correction (ThunderSTORM) was applied using fiducial markers (100 nm Tetraspek, Life Technologies). Images were corrected for shifts due to chromatic aberrations, and channels were aligned using fiducial markers (100 nm, Tetraspek, Life Technologies).
Preparation of TCO-Alexa Fluor 488
A 10 mM solution of DIBO-Alexa Fluor 488 (Thermo Fischer Scientific) in DMSO (10 μL, 100 nmol) was reacted with a 10 mM solution of TCO-PEG8-azide in DMSO (12 μL, 120 nmol, 1.2 equiv) for 4 h. To quench the presence of unreacted TCO-azide, 50 nmol DBCO-NH2 (Jena Bioscience) was added and the solution was diluted with 8 μL of PBS to a final Alexa Fluor 488 concentration of 3.33 mM and allowed to react for 2 h. The resulting TCO-Alexa Fluor 488 was used without further purification.
Biotinylation of PIC5a–f with the Nitrile–Imine-Mediated Tetrazole–Ene Cycloaddition (NITEC) Reaction
A solution of tetrazole-PEG8-biotin in PBS (0.7 mM, 100 μL) was prepared in a 5 mL glass vial. The vial was placed under a 254 nm lamp (Camag) without cap at a distance of 10 cm and irradiated for 10 min. A 2 mg/mL solution (500 μL) of the desired polymer in PBS was added to the activated tetrazole solution and stirred overnight at room temperature. After reaction, the samples were diluted to 1 mg/mL with PBS before use. The solution (50 μL) was further diluted to 0.5 mg/mL with PBS and fluorescence was measured on Tecan Spark M10 plate reader to confirm the reaction (excitation, 368 nm; emission, 514 nm).
Binding of PIC5a–f to Streptavidin-Functionalized Beads and Nanoparticles
Before binding the biotinylated PIC to a surface, they were first labeled with a DIBO-Alexa Fluor 647 dye. To a solution of the PIC (1 mg/mL), the desired amount of the dye was added from a 10 mM stock in DMSO and reacted overnight on a roller bank at 4 °C. From the solution of labeled PIC (1 mg/mL), the desired volume was added to the desired amount of streptavidin-functionalized beads or nanoparticles (NPs) in a 0.5 mL Eppendorf tube. The bead or NP suspension was diluted to 4 × 107 beads/mL or 7 × 1010 NPs/mL with PBS and the reaction was left overnight on a roller bank. The beads/NP were washed 1× with 0.05% Tween (400 μL) and 4× with PBS (400 μL). The PIC-containing beads/NPs were suspended in 400 μL of PBS and analyzed by flow cytometry. For quantification of polymer density, a stripping assay was developed that is described below. The morphology of the beads/NPs is not changed after the conjugation of the polymer (Figure S3).
Stripping Assay To Quantify PIC Density on Beads and Nanoparticles
A 10 μL solution of the PIC beads or NPs was transferred to a new Eppendorf tube and supernatant was removed using magnetic separation. A 2% Tween solution in MQ (400 μL) was added and the sample was placed in a 90 °C water bath for 10 min and then on ice for 5 min. Of the supernatant, 150 μL was taken and diluted with 50 μL of PBS, after which fluorescence was measured on a Spark M10 plate reader (em, 637 nm; ex, 682 nm) and compared to that of a trend line of the same PIC treated under similar conditions to determine PIC concentration in the supernatant. From the amount of polymer found in the supernatant, the density that was originally on the beads/NPs was calculated (see Calculation 1 in the Supporting Information). The stripped beads were washed thrice with PBS and analyzed with flow cytometry to verify that all polymer was stripped from the beads.
BSA Conjugation to PIC Beads and Nanoparticles
Preparation of Labeled BSA
BSA (0.5 mg, 7.5 nmol) was dissolved in 0.05 M borate buffer pH 8.5 (0.5 mL) and 1.5 μL of a 10 mM solution of NHS-Atto 488 in DMSO was added (2 equiv). The reaction was stirred for 2 h at room temperature, and 250 μL of the reaction was transferred to another Eppendorf tube and 1 μL of a 10 mM solution of NHS-PEG4-DBCO (2.67 equiv) in DMSO was added. Then, the reaction was left for 2 h at room temperature to make the DBCO-Atto 488-BSA. Both the Atto 488-BSA and the DBCO-Atto 488-BSA were purified over a Zeba desalting column (5 mL, Thermo Fischer Scientific) using the manufacturer’s protocol. After purification, the protein concentration and degree of labeling were determined by UV–vis spectrometry on a NanoDrop 2000 spectrometer (Thermo Fischer Scientific). Samples were diluted to 0.1 mg/mL BSA before use. Obtained degree of labeling: Atto 488-BSA: 1.89 Atto 488/BSA. DBCO-Atto 488-BSA: 1.90 Atto 488/BSA, 2.2 DBCO/BSA.
BSA Conjugation of Beads and Nanoparticles
For the SPAAC first method, 50 μL of the desired polymer solution (1 mg/mL) was diluted with PBS (100 μL) and the desired BSA solution was added (50 μL, 5 mg/mL). The reaction was left rolling overnight at room temperature. The resulting BSA–polymer solution (10 μL) was then diluted with PBS (30 μL) and the desired bead of NP stock (as obtained from supplier) was added (10 μL). The beads/NPs were rolled overnight at room temperature and then washed as described above for polymer bead reactions.
For the graft first method, PIC beads and NP were prepared as described above. After washing, they were suspended in PBS (40 μL) and DBCO-BSA solution was added (10 μL, 5 mg/mL). The beads were left rolling at room temperature overnight and washed according to the protocol described above.
Grafting of PIC to Glass Coverslips
Preparation of Streptavidin-Coated Coverslips
Glass coverslips were cleaned by sonication in aqueous 10% NaOH (5 min), followed by sonication in isopropanol (5 min) and drying under nitrogen flow. The glass was then immersed in piranha solution for 30 min, washed five times with Milli-Q water, and dried under nitrogen flow. The coverslips were placed in a 2% (3-glycidyloxypropyl)trimethoxysilane solution in dry toluene for 30 min. The coverslips were washed twice with toluene and thrice with ethanol and placed in an oven at 80 °C for 30 min. Silanized coverslips were placed overnight in a 20 μg/mL solution of streptavidin (3 mL/coverslip) in borate buffer (50 mM, pH 8.5), corresponding to an excess of 1012 streptavidins/μm2. Streptavidin coverslips were placed in wells of a six-well plate (one per well), washed with PBS (7 × 3 mL, shaking 1 min per wash), and used immediately. To verify the surface coating steps, contact angles were measured using a home-built setup for piranha-cleaned glass, GOPTMS-silanized glass, and streptavidin-coated glass and were found to be 11, 50, and 26°, respectively (average of three coverslips per condition)
Binding of PIC to Streptavidin-Coated Coverslips
A 1 mg/mL solution of the desired PIC (500 μL) was placed on the desired coverslip and was then left at room temperature for 4 h. The liquid was then aspirated with a pipette and the coverslips were washed with PBS (7 × 3 mL) as described above. The PIC-coated coverslips were analyzed by confocal microscopy. Z-stacks of each sample were made measuring fluorescence of the Alexa Fluor 647 on the polymer. For each sample, some cells (peripheral blood myeloid cells) labeled with a violet dye were added and allowed to drop to the bottom before measuring to determine the z height of the coverslip surface.
Results and Discussion
Aqueous Grafting for Semiflexible PIC Brushes
Earlier, hydrophobic PIC brushes were grown from the surface in organic solvents.33 In this work, we designed an aqueous grafting-to approach that ensures optimum biocompatibility throughout the entire procedure. The grafting-to route requires PICs with one or more functional groups for surface binding that is only present at one end of the polymer. We studied two approaches: In the first approach, a single functional group is introduced at the terminus of a PIC chain through controlled initiation of the PIC polymerization reaction. The second route introduces multiple functional groups at one chain end through a block copolymer approach.
End-Group Functionalization Approach
For the first approach, we polymerized nonfunctional methoxy isocyanide monomer 1 (Figure 2a) from a functional catalyst–initiator complex.33 The complex is prepared by reacting tetrakis(tert-butyl isocyanide) nickel(II) complex with an amine nucleophile attached to a desired functional group. Subsequent polymerization of 1 with the nickel complex (monomer/initiator ratio 2100:1) yields long semiflexible polymers with a single functional group at one terminus of the polymer (Table 1, Supporting Information for polymer analysis). We introduced azide (PIC1a, PIC1b) and biotin as functional groups (PIC1c; see Figure 2b). Azides are compatible with the PIC polymerization,26 and both are readily conjugated to substrates functionalized with alkyne/cyclooctyne or streptavidin, respectively.
Grafting experiments with PIC1a–c were conducted with 1 μm magnetic beads surface-treated with DBCO for PIC1a, PIC1b, and streptavidin for PIC1c. As negative and positive controls, we used PIC2 that lacks any azide group and PIC3 that has many azides randomly distributed in the side chains (Figure 2c). For the streptavidin bead experiments, the azides of PIC3 were first converted into biotin functionalities through reaction with a DBCO-linked biotin. The amount of polymer grafted to the beads was determined by measuring the concentrations of PIC before and after incubation using circular dichroism spectroscopy following the literature procedure.30 We find that conjugation efficiency to the DBCO beads is of the same order of magnitude as the negative control and much lower than the positive control (Figure 2d) and we mainly see nonspecific binding to the hydrophobic beads. For the less hydrophobic streptavidin-covered beads, the results are slightly better because of reduced nonspecific interactions (Figure 2e), but the degree of loading on the beads remains very low. Apparently, a single functional end-group is insufficient to bind these long and relatively rigid polymers to a substrate and we designed an alternative approach that allows for the introduction of multiple polymer–substrate interactions.
Block Copolymer Approach
For the second grafting method, we developed a route to make PICs with a small block of functional monomers for substrate binding. Under typical polymerization conditions, we start the standard initiator/catalyst solution (NiClO4 in toluene/ethanol) to the azide-functionalized monomer 2 (Figure 2a), and after consumption of the majority of the monomer (∼10 min), we add the nonfunctional monomer 1. We aim for an average functional block length of 100 monomers and a second block of 2000 monomers (PIC4, Figure 2c).
Grafting studies with PIC4 showed significantly more grafting than the nonspecific binding from the negative control PIC2 and slightly more grafting than for positive control PIC3 (Figure 2d). The grafting efficiency of 10 μg PIC4 per mg beads corresponds to a polymer density of around 2600 polymers/μm2, which equals a spacing of ∼20 nm between polymers (Calculation 1, Supporting Information). The fact that binding is only observed for PIC4 and not for PIC1a–c suggests that at least for PICs, but maybe also for other long semiflexible polymers, multiple functional groups are needed, which is in line with our earlier work that showed that multiple biotins were required on a PIC chain for binding to monoavidin beads.30 Using the much more efficient block copolymer strategy, we next developed orthogonal functionalization strategies for the PIC polymers.
Orthogonal Conjugation Strategies for Biofunctionalization
For biofunctionalization of the PIC brush, we need a second orthogonal conjugation reaction. We selected the tetrazole–ene cycloaddition (NITEC) reaction between an allyl and a (photoactivated) tetrazole.40−42 The reaction offers many advantages, for instance, the photoactivation allows for patterning on 2D substrates, the pyrazoline product is fluorescent (easy analysis), and, important for this work, the allyl group is fully compatible with the PIC polymerization, as opposed to trans-cyclooctene (TCO) or tetrazine click handles that react with the nickel or isocyanides, respectively. As conjugation of (large) biomolecules through the SPAAC reaction is already well established,26,27,30 we choose to use the NITEC reaction for the grafting reaction following the block copolymer approach. As such, the analogous allyl-functionalized monomer 3 was synthesized (Figure 3a).
Figure 3.

Design of the PIC with new allyl monomers in the first block that can be further derivatized with the NITEC reaction. (a) Structure of the allyl monomer. (b) Reaction mechanism of the NITEC reaction. The fluorescent pyrazoline is depicted in red. (c) General structure of PIC5a–e. The part in red depicts the first block containing allyl functionality. (d) False color STORM images of PIC5b that was labeled with Alexa Fluor 647 (yellow) in the second block and Alexa Fluor 488 (blue) in the first block. Each 5 pixel cross represents the localization of a single dye with an accuracy of 12 nm (Alexa Fluor 647) or 20 nm (Alexa Fluor 488). The scale bar represents 50 nm.
The UV irradiation used for the NITEC reaction may be harmful to a broad range of biomolecules. To increase the biocompatibility of our approach, we designed a two-step conjugation protocol. In the first step, the NITEC reaction is used to convert the allyl groups at the start of the polymer to any desirable group. In our grafting work (see below), we use biotin, but any other (bio)molecule may be used as a conjugant. In the second step, this group (biotin) is then grafted to broadly commercially available streptavidin substrate. A strong advantage of this approach is that during the NITEC reaction, only the allyl-functionalized PIC polymer and desired functional group are present, which will prevent known side reactions of the nitrile–imine intermediate with nucleophiles in biomolecules.43 A final advantage of the two-step conjugation is that this approach will also work readily on surfaces that are sensitive to UV and in situations where homogeneous irradiation of UV light is difficult, for instance, particle suspensions or surfaces with three-dimensional structures.
Polymer Preparation and Characterization
We prepared a series of polymers analogous to PIC4, but with a first block containing the allyl monomer 3 only (PIC5a), both allyl and methoxy monomers in different ratios (PIC5b–d), or the methoxy monomer 1 only (PIC5e) and with a second block containing the azide 2 and methoxy monomer 1 in a ratio of 1:30 (Figure 3c, polymer analysis in Table 1, Supporting Information). In addition, a control was prepared with the same polymer length and monomer ratios as PIC5a, but with the allyl and azide monomer randomly distributed along the polymer (PIC5f).
To verify that the synthesis resulted in polymers with two differently functionalized blocks, we fluorescently labeled PIC5b. The azides (large block) were functionalized part with DBCO-biotin and part with an Alexa Fluor 647 dye; the allyl groups at the terminus were first converted to tetrazines using the NITEC reaction and subsequently functionalized with a trans-cyclooctene (TCO)-based Alexa Fluor 488 dye (see Figure S4). The resulting polymers were bound to a streptavidin surface and visualized using stochastic optical reconstruction microscopy (STORM). The results, indeed, confirm a two-block copolymer with an azide-containing block of around 200 nm visible with 647 nm excitation, and a small allyl-containing block of 10–30 nm visible by 488 nm excitation (Figure 3d).
Grafting of the Allyl-Containing PIC via the Two-Step Protocol
For grafting experiments of PIC5a–e, we converted the allyl groups to biotin. For this purpose, tetrazole was connected to biotin via a PEG8 linker to increase solubility in aqueous solution. The product tetrazole-PEG8-biotin was then reacted with PIC5a–e via the NITEC reaction (Figure 4a). To establish a biotinylation protocol, we used the increase in fluorescence of the reaction product as a measure for biotinylation and optimized the UV irradiation time for tetrazole activation and the reaction time with the allyl polymer (Figure S5). The reaction of photoactivated tetrazole-PEG8-biotin with PIC5a in an aqueous solution gave rise to a fluorescent signal with an emission maximum at 415 nm. Only a very weak fluorescence was observed when the tetrazole was reacted with a solution of PIC3, which contains no allyl groups.
Figure 4.
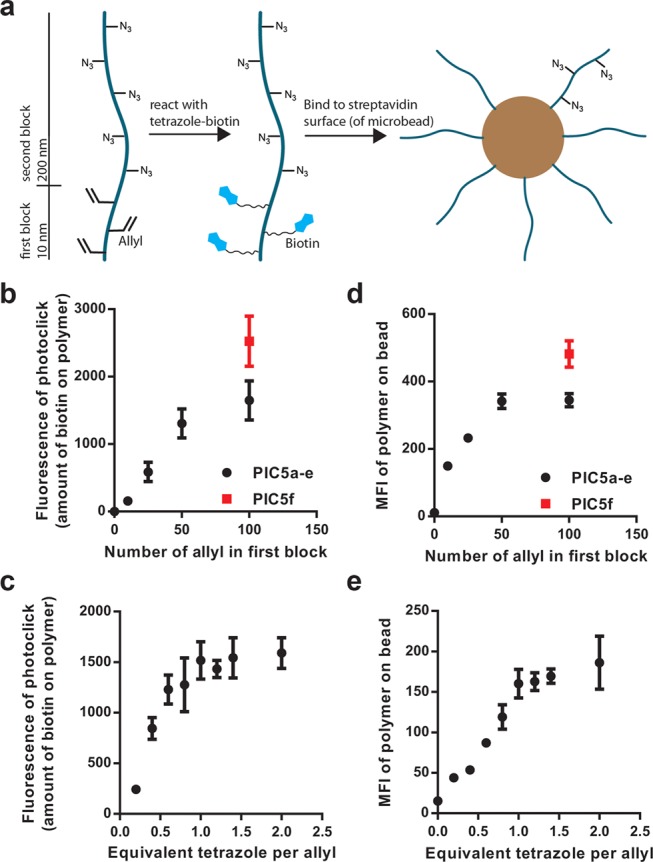
Biotin conjugation of PIC5a–f for grafting PIC brushes to streptavidin beads. (a) Schematic representation of the two-step binding protocol of the allyl polymers using tetrazole-PEG8-biotin. (b) Fluorescence intensity at λ = 415 nm of the conjugated biotin of PIC5a–f after reaction with an excess of tetrazole-PEG8-biotin. (c) Fluorescence intensity at λ = 415 nm of PIC5b when reacted with different equivalents of tetrazole-PEG8-biotin per allyl. (d) Binding of the biotinylated PIC5a–f (labeled with Alexa Fluor 647) to streptavidin-functionalized beads as measured by the mean fluorescence intensity of the beads by flow cytometry. (e) Binding of the biotinylated PIC5b from (c) (labeled with Alexa Fluor 647) to streptavidin-functionalized beads as measured by the mean fluorescence intensity of the beads by flow cytometry. Values plotted are mean ± standard deviation of three independent experiments.
Using this protocol, polymers PIC5a–e were reacted with an excess of photoactivated tetrazole-PEG8-biotin corresponding to 200 equiv per polymer. The resulting fluorescence of the NITEC reaction increases with the increasing (statistical) amount of allyl groups present in the polymer (Figure 4b). The linear correlation levels off for PIC5a with 100 allyl monomers in the first block, which we tentatively attribute to steric hindrance: as more biotins are clicked onto the polymer, they shield the availability of the remaining allyl groups. This assessment is supported by the reaction of PIC5f (same number of allyl groups as PIC5a, but distributed over the entire chain) with the tetrazole-PEG8-biotin for which the linear relation still holds.
To gain more insight into the efficiency of biotin conjugation with the NITEC reaction, PIC5b was reacted with different equivalents of tetrazole-PEG8-biotin. The fluorescence of these reactions was compared to control reactions with PIC3 that lacks the allyl groups. The fluorescence of the conjugated biotin increases with the amount of tetrazole added and reaches a plateau around 1 equiv of tetrazole (Figure 4c), which implies that the biotin conjugation with the NITEC is highly efficient and does not significantly suffer from any side reactions.
Next, the biotinylated products of PIC5a–f were labeled with an additional dye on the azides to allow flow cytometry characterization (DIBO-Alexa Fluor 647 dyes with on average two dye molecules per polymer chain) and added to 1 μm magnetic streptavidin beads. The amount of (labeled) polymer bound to the beads was quantified by flow cytometry and the mean fluorescent intensity (MFI) of the Alexa Fluor 647 signal on the beads was plotted against the number of allyl groups in the first block (Figure 4d). We find that the amount of polymer on the beads correlates well to the number of biotins in the first block and we observe no binding for PIC5e without allyl groups.
The polymers obtained from the biotinylation of PIC5b with different equivalents of tetrazole-biotin were similarly labeled with Alexa Fluor 647 (on average, one dye molecule per polymer) and bound to beads. We observe that the surface grafting of these polymers increases with increasing amounts of biotin in the first block (Figure 4e). These results are in line with our earlier findings that only limited binding was observed for polymers with a single conjugation group and underline our ideas that for these long and rigid polymers multiple surface binding groups are necessary for efficient grafting. Optimal biotinylation and grafting efficiencies were found for PIC5b that was reacted with 1 equiv of biotin per allyl. These polymers (biotin-PIC5-647) were used to quantify grafting densities.
Quantification of Grafting Densities
The maximum amount of polymer that can be grafted to the beads was determined by subjecting the beads to increasing amounts of biotin-PIC5b-647. We then quantify the amount of polymer by measuring the mean fluorescence intensity in a flow cytometry experiment. In addition, we release the polymers from the bead by breaking the biotin–streptavidin bond (2% sodium dodecyl sulfate, 90 °C, 10 min) and determine the fluorescence intensity of the supernatant, after magnetic separation, and calibrate the signal to known concentrations. The stripped beads were then measured by flow cytometry to verify that all bound polymer was removed.
From the flow cytometry experiment, we find that a maximum loading of the beads is reached at around 100 μg biotin-PIC5b-647 per mg bead (Figure 5a). The stripping essay (Figure 5b) gives a very similar binding curve, and the correlation between the two experiments (Figure 5c) is excellent. The maximum amount of bound polymer is around 6 μg polymer/mg beads, which corresponds to a density of ∼2174 polymers/μm2 and an average spacing of around 21.5 nm. This polymer density is much lower than the density of streptavidin on the surface, which means that the streptavidin density is not a limiting factor for PIC brush formation under these conditions. The obtained density implies that the surface is indeed in the brush regime with a spacing smaller than 2 times the radius of gyration (Calculation 2, Supporting Information).
Figure 5.

Binding of different amounts of labeled and biotinylated PIC5b to streptavidin microbeads. (a) MFI signal of the beads as a function of the amount of polymer added. (b) Grafted amount of polymer per bead as determined by the stripping assay plotted against amount of polymer added. (c) Correlation between MFI signal and calculated polymer density. Mean ± standard deviation of three independent experiments are shown.
Grafting PIC Brushes on Nanoparticles and Glass Coverslips
We demonstrate the versatility of our grafting approach by binding the polyisocyanopeptides to other surfaces, such as streptavidin-functionalized glass coverslips and nanoparticles (NPs) with a diameter of 200 nm. Analogous to the binding studies on the microbeads, we conjugated increasing amounts of biotin-PIC5b-647 to the substrates. For the NPs (Figure S6), the maximum observed polymer density was found to be 1600 polymers/μm2 (spacing of 25 nm), which is comparable to the density of PIC on the microbeads. To graft the polymers to glass coverslips, streptavidin-coated coverslips were first prepared and Biotin-PIC5b-647 was added. Surface conjugation results in a homogeneous polymer layer as is qualitatively confirmed by confocal microscopy (Figure S7), but has not been quantified. The absence of surface-bound PIC for the negative controls without streptavidin on the surface or biotin on the polymer confirms that surface binding takes place via the end-functionality. Just as for the microbeads, the streptavidin on both the NP and the coverslip surfaces is present in excess and does limit PIC grafting.
Protein Functionalization of the Semiflexible Brushes
Finally, we show that the obtained PIC brushes can be functionalized with biomolecules. To this end, Atto 488-labeled bovine serum albumin (BSA) containing DBCO click handles was linked to the azides in the side chains of the (Alexa Fluor 647-labeled) PIC polymers. The BSA-functionalized PIC brushes were prepared using two different strategies: In one, BSA is first conjugated to the polymer via the SPAAC reaction and the BSA–polymer conjugates are then grafted to the surface (SPAAC first, Figure 1, top route). In the alternative approach, the polymer is first grafted to a surface and BSA is then linked to PIC brushes (graft first, Figure 1, bottom route). PIC-NP and PIC microbeads were prepared via both methods. As controls that give levels of nonspecific binding, the labeled BSA was also added to the beads and particles lacking the PIC brushes.
BSA and PIC levels were determined by tracking the fluorescence of the Atto 488 and Alexa Fluor 647 by flow cytometry. For the microbead conjugations (Figure 6a), we find that the graft first strategy gives a good PIC density on the particles, but a BSA concentration that is only marginally higher than unspecific binding. In the SPAAC first strategy, the PIC loading is much lower, but since the BSA is already on the polymer, the ultimate BSA density is much higher. Considering the BSA dimensions, one can foresee that at high brush densities, the azides are insufficiently available. Along the same lines, the grafting density of the BSA-conjugated PIC will be much lower. In the case of the NP, both strategies yield similar biofunctional brushes (Figure 6b). For the NPs, the curvature of the particles is much higher (Figure 6c). For the (200 nm long) PIC chains, the azides on the polymers will be much better available for the graft first approach. Similarly, brush formation is much less hindered by the presence of the BSA in the SPAAC first strategy.
Figure 6.

Mean fluorescence intensity (MFI) of Atto 488-labeled BSA on and Alexa Fluor 647-labeled PIC on microbeads and nanoparticles. (a) MFIs for the different conditions used for binding to the streptavidin microbeads. (b) MFIs for the different conditions used for binding to the streptavidin nanoparticles. Mean ± standard deviation of three independent experiments is shown. (c) Schematic representation of curvature of the PIC brushes on microbeads, where NP, PIC, and surface dimensions are on scale.
Conclusions
In conclusion, we report the synthesis of semiflexible polymer brushes using end-functional PICs with lengths of around 200 nm. These polymers possess a small block of multiple allyl-containing monomers present only at one end of the polymer as well as azide groups present in the rest of the polymer chain. The allyl functionalities are efficiently reacted with tetrazoles to introduce new functional groups. We inserted biotin at one end of the PIC and grafted the polymers under aqueous conditions to a wide range of (commercially available) streptavidin substrates (coverslips, microbeads, and nanoparticles). On the particles and microbeads, polymer brushes with densities of around 2000 polymers/μm2 were reached. We stress that for these brushes, multiple surface anchor points are necessary to reach high grafting densities. For the synthesis of bioactive brush surfaces, often large biomolecules such as proteins need to be attached to the brush. We showed that this is readily achieved through conjugation of the azides in the brush with DBCO-functionalized biomolecules. Note that for optimum grafting densities the order of biomolecular conjugation and polymer grafting should be considered. Polyisocyanopeptides are long, easily functionalized synthetic polymers that are unique in their semiflexibility. The possibility to graft these semiflexible polymers to a wide range of surfaces under aqueous conditions and modify them with a variety of biological functions will be a useful addition to the toolbox of polymers for bioactive surfaces.
The synthetic route toward synthetic semiflexible bioactive surfaces presented here may be used to achieve biological responses that cannot be achieved by flexible polymers. For example, the more extended confirmation of semiflexible polymers is expected to provide higher multivalency and improved cell signaling compared to flexible polymers.26,29 It is also known that substrate stiffness and the stiffness observed by the cell upon nanoscale ligand reorganization influence cellular responses.44,45 Ligands attached via semiflexible brushes are expected to generate different forces upon reorganization and cellular interactions. We are currently investigating the effect of semiflexible brushes in cellular responses by comparison to commonly used flexible brushes.
Acknowledgments
The authors thank Chris Berkhout and Dr. Sergey Semin from the department of Spectroscopy of Solids and Interfaces for their help with measuring the contact angles. They also thank Gert-Jan Bakker for his contribution building the STORM microscope.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biomac.9b00385.
Viscometry and AFM data of polymers, colorimetric analysis of first block polymerization time, DBCO bead functionality, binding data of PIC1a–c, optimization of the NITEC reaction for biotinylation, calculations of obtained polymer densities, results of grafting to NP and coverslips, and details of the STORM setup (PDF)
Author Contributions
D.V. designed the end-functional PIC, as well as performed necessary synthesis and most binding experiments. M.S. helped design the stripping assay for quantification and helped with PIC conjugation to NP. J.W. helped with flow cytometry analysis and confocal microscopy of coverslips. B.J. performed the STORM nanoscopy. L.J.E. gave input on the experimental design. T.v.d.E. and B.I. helped with the synthesis and binding experiments of PIC1a–c. A.C. was involved in building the STORM setup. P.H.J.K., M.V., C.G.F., R.H., and A.E.R. supervised the project. The manuscript was written through contributions of all authors.
This work was supported by a grant from the Institute of Chemical Immunology (024.002.009) and ERC Advanced grant PATHFINDER (269019). C.G.F. is recipient of the NWO Spinoza award and KWO grant 2009-4402 of the Dutch Cancer Society. M.V. is recipient of an Institute of Chemical Immunology fellowship grant and ERC Starting grant CHEMCHECK (679921).
The authors declare no competing financial interest.
Supplementary Material
References
- Ma Z.; Mao Z.; Gao C. Surface Modification and Property Analysis of Biomedical Polymers Used for Tissue Engineering. Colloids Surf., B 2007, 60, 137–157. 10.1016/j.colsurfb.2007.06.019. [DOI] [PubMed] [Google Scholar]
- Demirci Uzun S.; Kayaci F.; Uyar T.; Timur S.; Toppare L. Bioactive Surface Design Based on Functional Composite Electrospun Nanofibers for Biomolecule Immobilization and Biosensor Applications. ACS Appl. Mater. Interfaces 2014, 6, 5235–5243. 10.1021/am5005927. [DOI] [PubMed] [Google Scholar]
- Meyers S. R.; Grinstaff M. W. Biocompatible and Bioactive Surface Modifications for Prolonged in Vivo Efficacy. Chem. Rev. 2012, 112, 1615–1632. 10.1021/cr2000916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendelman H. E.; Anantharam V.; Bronich T.; Ghaisas S.; Jin H.; Kanthasamy A. G.; Liu X.; McMillan J. E.; Mosley R. L.; Narasimhan B.; et al. Nanoneuromedicines for Degenerative, Inflammatory, and Infectious Nervous System Diseases. Nanomedicine 2015, 11, 751–767. 10.1016/j.nano.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischerhoff E.; Badi N.; Lutz J.-F.; Laschewsky A. Smart Bioactive Surfaces. Soft Matter 2010, 6, 705–713. 10.1039/B913594D. [DOI] [Google Scholar]
- Robinson D. E.; Smith L. E.; Steele D. A.; Short R. D.; Whittle J. D. Development of a Surface to Enhance the Effectiveness of Fibroblast Growth Factor 2 (FGF-2). Biomater. Sci. 2014, 2, 875–882. 10.1039/C4BM00018H. [DOI] [PubMed] [Google Scholar]
- Glinel K.; Jonas A. M.; Jouenne T.; Leprince J.; Galas L.; Huck W. T. S. Antibacterial and Antifouling Polymer Brushes Incorporating Antimicrobial Peptide Antibacterial and Antifouling Polymer Brushes Incorporating Antimicrobial Peptide. Bioconjugate Chem. 2009, 20, 71–77. 10.1021/bc800280u. [DOI] [PubMed] [Google Scholar]
- Fischer S. E.; Liu X.; Mao H. Q.; Harden J. L. Controlling Cell Adhesion to Surfaces via Associating Bioactive Triblock Proteins. Biomaterials 2007, 28, 3325–3337. 10.1016/j.biomaterials.2007.03.026. [DOI] [PubMed] [Google Scholar]
- Que Y.; Feng C.; Lu G.; Huang X. Polymer-Coated Ultrastable and Biofunctionalizable Lanthanide Nanoparticles. ACS Appl. Mater. Interfaces 2017, 9, 14647–14655. 10.1021/acsami.7b01452. [DOI] [PubMed] [Google Scholar]
- Xu F. J.; Neoh K. G.; Kang E. T. Bioactive Surfaces and Biomaterials via Atom Transfer Radical Polymerization. Prog. Polym. Sci. 2009, 34, 719–761. 10.1016/j.progpolymsci.2009.04.005. [DOI] [Google Scholar]
- Kim W.; Jung J. Polymer Brush: A Promising Grafting Approach to Scaffolds for Tissue Engineering. BMB Rep. 2016, 49, 655–661. 10.5483/BMBRep.2016.49.12.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard J. M.; Hotchkiss J. H. Polymer Surface Modification for the Attachment of Bioactive Compounds. Prog. Polym. Sci. 2007, 32, 698–725. 10.1016/j.progpolymsci.2007.04.002. [DOI] [Google Scholar]
- Coalson R. D.; Nasrabad A. E.; Jasnow D.; Zilman A. A Polymer-Brush-Based Nanovalve Controlled by Nanoparticle Additives: Design Principles. J. Phys. Chem. B 2015, 119, 11858–11866. 10.1021/acs.jpcb.5b02623. [DOI] [PubMed] [Google Scholar]
- Feng C.; Huang X. Polymer Brushes: Efficient Synthesis and Applications. Acc. Chem. Res. 2018, 51, 2314–2323. 10.1021/acs.accounts.8b00307. [DOI] [PubMed] [Google Scholar]
- Xu B.; Liu Y.; Sun X.; Hu J.; Shi P.; Huang X. Semifluorinated Synergistic Nonfouling/Fouling-Release Surface. ACS Appl. Mater. Interfaces 2017, 9, 16517–16523. 10.1021/acsami.7b03258. [DOI] [PubMed] [Google Scholar]
- Xu B.; Feng C.; Hu J.; Shi P.; Gu G.; Wang L.; Huang X. Spin-Casting Polymer Brush Films for Stimuli-Responsive and Anti-Fouling Surfaces. ACS Appl. Mater. Interfaces 2016, 8, 6685–6692. 10.1021/acsami.5b12820. [DOI] [PubMed] [Google Scholar]
- Römer F.; Fedosov D. A. Dense Brushes of Stiff Polymers or Filaments in Fluid Flow. EPL 2015, 109, 68001 10.1209/0295-5075/109/68001. [DOI] [Google Scholar]
- Egorov S. A.; Hsu H. P.; Milchev A.; Binder K. Semiflexible Polymer Brushes and the Brush-Mushroom Crossover. Soft Matter 2015, 11, 2604–2616. 10.1039/C4SM02862G. [DOI] [PubMed] [Google Scholar]
- Han L.; Dean D.; Ortiz C.; Grodzinsky A. J. Lateral Nanomechanics of Cartilage Aggrecan Macromolecules. Biophys. J. 2007, 92, 1384–1398. 10.1529/biophysj.106.091397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinbaum S.; Tarbell J. M.; Damiano E. R. The Structure and Function of the Endothelial Glycocalyx Layer. Annu. Rev. Biomed. Eng. 2007, 9, 121–167. 10.1146/annurev.bioeng.9.060906.151959. [DOI] [PubMed] [Google Scholar]
- Xu G. K.; Qian J.; Hu J. The Glycocalyx Promotes Cooperative Binding and Clustering of Adhesion Receptors. Soft Matter 2016, 12, 4572–4583. 10.1039/C5SM03139G. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Chien S.; Weinbaum S. Dynamic Contact Forces on Leukocyte Microvilli and Their Penetration of the Endothelial Glycocalyx. Biophys. J. 2001, 80, 1124–1140. 10.1016/S0006-3495(01)76090-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attili S.; Borisov O. V.; Richter R. P. Films of End-Grafted Hyaluronan Are a Prototype of a Brush of a Strongly Charged, Semiflexible Polyelectrolyte with Intrinsic Excluded Volume. Biomacromolecules 2012, 13, 1466–1477. 10.1021/bm3001759. [DOI] [PubMed] [Google Scholar]
- Bracha D.; Karzbrun E.; Shemer G.; Pincus P. A.; Bar-Ziv R. H. Entropy-Driven Collective Interactions in DNA Brushes on a Biochip. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 4534–4538. 10.1073/pnas.1220076110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouwer P. H. J.; Koepf M.; Le Sage V. A. A.; Jaspers M.; van Buul A. M.; Eksteen-Akeroyd Z. H.; Woltinge T.; Schwartz E.; Kitto H. J.; Hoogenboom R.; et al. Responsive Biomimetic Networks from Polyisocyanopeptide Hydrogels. Nature 2013, 493, 651–655. 10.1038/nature11839. [DOI] [PubMed] [Google Scholar]
- Mandal S.; Eksteen-Akeroyd Z. H.; Jacobs M. J.; Hammink R.; Koepf M.; Lambeck A. J. A.; van Hest J. C. M.; Wilson C. J.; Blank K.; Figdor C. G.; et al. Therapeutic Nanoworms: Towards Novel Synthetic Dendritic Cells for Immunotherapy. Chem. Sci. 2013, 4, 4168. 10.1039/c3sc51399h. [DOI] [Google Scholar]
- Mandal S.; Hammink R.; Tel J.; Eksteen-Akeroyd Z. H.; Rowan A. E.; Blank K.; Figdor C. G. Polymer-Based Synthetic Dendritic Cells for Tailoring Robust and Multifunctional T Cell Responses. ACS Chem. Biol. 2015, 10, 485–492. 10.1021/cb500455g. [DOI] [PubMed] [Google Scholar]
- Hammink R.; Mandal S.; Eggermont L. J.; Nooteboom M.; Willems P. H. G. M.; Tel J.; Rowan A. E.; Figdor C. G.; Blank K. G. Controlling T-Cell Activation with Synthetic Dendritic Cells Using the Multivalency Effect. ACS Omega 2017, 2, 937–945. 10.1021/acsomega.6b00436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermont L. J.; Hammink R.; Blank K. G.; Rowan A. E.; Tel J.; Figdor C. G. Cytokine-Functionalized Synthetic Dendritic Cells for T Cell Targeted Immunotherapies. Adv. Ther. 2018, 1800021 10.1002/adtp.201800021. [DOI] [Google Scholar]
- Hammink R.; Eggermont L. J.; Zisis T.; Tel J.; Figdor C. G.; Rowan A. E.; Blank K. G. Affinity-Based Purification of Polyisocyanopeptide Bioconjugates. Bioconjugate Chem. 2017, 28, 2560–2568. 10.1021/acs.bioconjchem.7b00398. [DOI] [PubMed] [Google Scholar]
- Ostaci R. V.; Damiron D.; Al Akhrass S.; Grohens Y.; Drockenmuller E. Poly(Ethylene Glycol) Brushes Grafted to Silicon Substrates by Click Chemistry: Influence of PEG Chain Length, Concentration in the Grafting Solution and Reaction Time. Polym. Chem. 2011, 2, 348–354. 10.1039/C0PY00251H. [DOI] [Google Scholar]
- Zdyrko B.; Luzinov I. Polymer Brushes by the “Grafting to” Method. Macromol. Rapid Commun. 2011, 32, 859–869. 10.1002/marc.201100162. [DOI] [PubMed] [Google Scholar]
- Lim E.; Tu G.; Schwartz E.; Cornelissen J. J. L. M.; Rowan A. E.; Nolte R. J. M.; Huck W. T. S. Synthesis and Characterization of Surface-Initiated Helical Polyisocyanopeptide Brushes. Macromolecules 2008, 41, 1945–1951. 10.1021/ma702531u. [DOI] [Google Scholar]
- Asaoka S.; Joza A.; Minagawa S.; Song L.; Suzuki Y.; Iyoda T. Fast Controlled Living Polymerization of Arylisocyanide Initiated by Aromatic Nucleophile Adduct of Nickel Isocyanide Complex. ACS Macro Lett. 2013, 2, 906–911. 10.1021/mz400410y. [DOI] [PubMed] [Google Scholar]
- Stephany R. W.; Drenth W. tetrakis(t-butyl isocyanide)nickel) perchlorate. Recl. Trav. Chim. Pays-Bas 1972, 91, 1453–1458. 10.1002/recl.19720911210. [DOI] [Google Scholar]
- Fan Y.; Deng C.; Cheng R.; Meng F.; Zhong Z. In Situ Forming Hydrogels via Catalyst-Free and Bioorthogonal “Tetrazole-Alkene” Photo-Click Chemistry. Biomacromolecules 2013, 14, 2814–2821. 10.1021/bm400637s. [DOI] [PubMed] [Google Scholar]
- Nahidiazar L.; Agronskaia A. V.; Broertjes J.; Van den Broek B.; Jalink K. Optimizing Imaging Conditions for Demanding Multi-Color Super Resolution Localization Microscopy. PLoS One 2016, 11, e0158884 10.1371/journal.pone.0158884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J.; Arganda-Carreras I.; Frise E.; Kaynig V.; Longair M.; Pietzsch T.; Preibisch S.; Rueden C.; Saalfeld S.; Schmid B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovesný M.; Křížek P.; Borkovec J.; Švindrych Z.; Hagen G. M. ThunderSTORM: A Comprehensive ImageJ Plug-in for PALM and STORM Data Analysis and Super-Resolution Imaging. Bioinformatics 2014, 30, 2389–2390. 10.1093/bioinformatics/btu202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z.; Ho L. Y.; Lin Q. Rapid, Photoactivatable Turn-on Fluorescent Probes Based on an Intramolecular Photoclick Reaction. J. Am. Chem. Soc. 2011, 133, 11912–11915. 10.1021/ja204758c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W.; Wang Y.; Qu J.; Lin Q. Selective Functionalization of a Genetically Encoded Alkene-Containing Protein via “Photoclick Chemistry” in Bacterial Cells. J. Am. Chem. Soc. 2008, 130, 9654–9655. 10.1021/ja803598e. [DOI] [PubMed] [Google Scholar]
- Song W.; Wang Y.; Qu J.; Madden M. M.; Lin Q. A Photoinducible 1,3-Dipolar Cycloaddition Reaction for Rapid, Selective Modification of Tetrazole-Containing Proteins. Angew. Chem., Int. Ed. 2008, 47, 2832–2835. 10.1002/anie.200705805. [DOI] [PubMed] [Google Scholar]
- Li Z.; Qian L.; Li L.; Bernhammer J. C.; Huynh H. V.; Lee J. S.; Yao S. Q. Tetrazole Photoclick Chemistry: Reinvestigating Its Suitability as a Bioorthogonal Reaction and Potential Applications. Angew. Chem., Int. Ed. 2016, 55, 2002–2006. 10.1002/anie.201508104. [DOI] [PubMed] [Google Scholar]
- Santos E.; Orive G.; Hernández R. M.; Pedraz J. L.. Cell-Biomaterial Interaction: Strategies To Mimic the Extracellular Matrix. On Biomimetics; IntechOpen, 2012. [Google Scholar]
- Huebsch N.; Arany P. R.; Mao A. S.; Shvartsman D.; Ali O. A.; Bencherif S. A.; Rivera-Feliciano J.; Mooney D. J. Harnessing Traction-Mediated Manipulation of the Cell/Matrix Interface to Control Stem-Cell Fate. Nat. Mater. 2010, 9, 518–526. 10.1038/nmat2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.