

Abstract
Alzheimer's disease (AD) is now established as a progressive compromise not only of the neurons but also of the cerebral vasculature. Increasing evidence also indicates that cerebrovascular dysfunction may be a key or an aggravating pathogenic factor in AD, emphasizing the importance to properly control this deficit when aiming for effective therapy. Here, we report that simvastatin (3–6 months, 40 mg/kg/d) completely rescued cerebrovascular reactivity, basal endothelial nitric oxide synthesis, and activity-induced neurometabolic and neurovascular coupling in adult (6 months) and aged (12 months) transgenic mice overexpressing the Swedish and Indiana mutations of the human amyloid precursor protein (AD mice). Remarkably, simvastatin fully restored short- and long-term memory in adult, but not in aged AD mice. These beneficial effects occurred without any decreasing effect of simvastatin on brain amyloid-β (Aβ) levels or plaque load. However, in AD mice with recovered memory, protein levels of the learning- and memory-related immediate early genes c-Fos and Egr-1 were normalized or upregulated in hippocampal CA1 neurons, indicative of restored neuronal function. In contrast, the levels of phospholipase A2, enkephalin, PSD-95, synaptophysin, or glutamate NMDA receptor subunit type 2B were either unaltered in AD mice or unaffected by treatment. These findings disclose new sites of action for statins against Aβ-induced neuronal and cerebrovascular deficits that could be predictive of therapeutic benefit in AD patients. They further indicate that simvastatin and, possibly, other brain penetrant statins bear high therapeutic promise in early AD and in patients with vascular diseases who are at risk of developing AD.
Introduction
Alzheimer's disease (AD) is a devastating disease of aging characterized by progressive cognitive deterioration and memory loss. Other clinical or neuropathological landmarks include amyloid-β (Aβ) increases and plaque formation, neurofibrillary tangles, glial cell activation, synaptic dysfunction, neurodegenerative changes (Querfurth and LaFerla, 2010), progressive reduction in cerebral glucose utilization (Mosconi et al., 2008), and a cerebrovascular pathology (Iadecola, 2004). The latter is portrayed functionally by a chronic cerebral hypoperfusion at rest and reduced perfusion to activated brain areas or impaired neurovascular coupling. Of interest, the decreases in cerebral blood flow (CBF) have been correlated with disease progression (Hirao et al., 2005) and with response to therapy (Yoshida et al., 2007). Recent evidence suggests that statins, cholesterol-lowering drugs broadly used in the treatment of cardiovascular diseases, have therapeutic potential in AD. Although not undisputed (Butterfield, 2011), statins have been linked to a reduced prevalence of AD in statin-prescribed populations (Jick et al., 2000; Wolozin et al., 2000), improved cognition in normocholesterolemic patients (Simons et al., 2002), and slower cognitive decline in mild-to-moderate AD patients (Sparks et al., 2006).
Particularly, there is a relatively good consensus that statin therapy in early old age, but not in late age, is associated with a lower incidence of AD independent from the ApoE allele and, possibly more so, in patients with hypertension and cerebrovascular or coronary artery disease (Rockwood et al., 2002; Li et al., 2007, 2010; Cramer et al., 2008; Haag et al., 2009). It has been argued that the age-dependent benefits of statins in protecting against AD, which are reportedly dose dependent and related to statin pleiotropic effects rather than cholesterol-lowering effects, may be due to a critical time window when statins are able to prevent or delay neurodegenerative processes, which would not be achievable when neuronal damage has already occurred (Li et al., 2010; Butterfield, 2011).
We recently found that short, low-dose simvastatin (8 weeks, 20 mg/kg/d) partly restored cerebrovascular reactivity and brain hemodynamics, but failed to normalize memory in 12-month-old transgenic AD mice that overexpress the Swedish (K670N, M671L) and Indiana (V717F) mutations of human amyloid precursor protein (APP mice, line J20) (Tong et al., 2009). In the present study, we investigated whether cerebrovascular and memory deficits would be rescued by earlier and longer simvastatin therapy at a higher dose (40 mg/kg/d), and if such treatment would impact on key pathogenic markers at endpoint in adult (6-month-old, treated for 3 months) and aged (12-month-old, treated for 6 months) APP mice. The study demonstrates that simvastatin is fully effective against both cerebrovascular and memory deficits only if initiated early in the disease process. The data also establish that simvastatin beneficial effects are independent from the amyloid pathology, and that memory rescue occurs concurrently with normalization of proteins critical to synaptic function and memory processes. These findings establish that the therapeutic benefit of statins depends on a critical time window when it is still possible to revert the AD pathogenic process, hence favoring their use in early AD patients.
Materials and Methods
Animals.
Experiments were approved by the Animal Ethics Committee of the Montreal Neurological Institute (McGill University, Montréal, Québec, Canada) and complied with local and national regulations in accordance to the Canadian Council on Animal Care. Heterozygous transgenic C57BL/6 mice that express the APP carrying the human Swedish (K670N, M671L; APPSwe) and Indiana (V717F; APPInd) familial AD mutations directed by the PDGF β-chain promoter (APP mice, J20 line) (Mucke et al., 2000) and wild-type (WT) mice were used, with males and females in about equal number. APP J20 mice display early (2–4 months) cerebrovascular deficits (Tong et al., 2005) and increased levels of soluble Aβ species (Mucke et al., 2000) that precede the formation (5–6 months) of diffuse (Mucke et al., 2000) and dense-core Aβ plaques (Aucoin et al., 2005), including cerebral amyloid angiopathy. The latter, present in small arterial branches, is not detected in main arteries of the circle of Willis even at 12 months of age (Tong et al., 2009). Furthermore, APP J20 mice develop progressive cognitive deficits that are fully manifested by 6 months of age (Palop et al., 2003; deIpolyi et al., 2008).
Treatments.
Simvastatin (Enzo Life Sciences International) was activated by alkaline lysis, per the manufacturer's protocol, and was added to the drinking water such that mice received simvastatin at a dose of ∼40 mg/kg of body weight/d, which is similar to or lower than that used in other studies looking at systemic or central effects of simvastatin (Li et al., 2006; Lloyd et al., 2009; Yudoh and Karasawa, 2010). The animals were divided into two different age groups: adult (6 months old at endpoint, treated at 3 months of age for a period of 3 months) and aged (12 months old at endpoint, treated at 6 months for 6 months). Controls received the same drinking solution without simvastatin. Simvastatin was administered at 20 mg/kg/d for 3 d, increased to 30 mg/kg/d for 4 d, and then increased to 40 mg/kg/d for the rest of the treatment. Two treatment groups were formed for both adult and aged APP mice (7–30 mice/group); simvastatin-treated WT mice were included in only one cohort of each age group. Total cholesterol levels were measured in one of each age cohort with an Accutrend GC meter (Roche Diagnostic) and were similar in all groups as follows (in mmol/L): adult (n = 4/group): WT, 4.78 ± 0.04; WT treated, 4.68 ± 0.02; APP, 4.76 ± 0.04; APP treated, 4.81 ± 0.01; aged: WT (n = 11), 4.39 ± 0.05; WT treated (n = 9), 4.38 ± 0.04; APP (n = 9), 4.36 ± 0.07; APP treated (n = 8), 4.35 ± 0.04. Gain in body weight, measured in aged mice treated for the longest period (6 months), did not differ between groups, as follows (in grams): WT (n = 11), 1.89 ± 0.55; WT treated (n = 9), 1.55 ± 0.55; APP (n = 9), 2.13 ± 1.13; APP treated (n = 8), 0.52 ± 0.78.
Morris water maze.
Spatial memory was tested in the Morris water maze adapted for transgenic APP mice (deIpolyi et al., 2008), except for the first group of aged mice that was submitted to the classic 5-d-long Morris water maze, as detailed for the low-dose simvastatin treatment (Tong et al., 2009). For the adapted maze, mice first received a 3 d training session whereby they were required to swim to a visible platform in a room with visual wall cues. Then, after switching wall cues and platform location, the platform was submerged and training started for 5 consecutive days in which the mice had to find the hidden platform using the visuospatial cues. The probe trial (platform removed) was performed on day 9 for all groups, and a second probe trial was performed in one cohort of adult mice on day 17 (8 d after the first one). All parameters were recorded and analyzed (2020 Plus tracking system and Water 2020 software, HVS Image). All experiments started at the same time every day.
Fluorodeoxyglucose-positron emission tomography.
A subset of aged mice were scanned for cerebral glucose uptake (CGU) of [18F] fluorodeoxyglucose (FDG) in the somatosensory cortex following whisker stimulation, as detailed recently (Nicolakakis et al., 2008, 2011). Under isoflurane sedation (1–2% in medical air), mice received a bolus injection of 100–150 μCi (100 μl) of [18F]FDG into the tail vein and underwent 45 min of continuous unilateral whisker stimulation (8–10 Hz) followed by a 25 min positron emission tomography (PET) acquisition in an R4 microPET scanner (Siemens Preclinical Solutions; 15 min emission/10 min transmission scan using a 57Co point source). Physiological parameters (heart rate, respiration, and body temperature) were monitored (Biopac Systems), and stability was maintained throughout the procedure. Fasting glycemia was measured before scanning with a commercial glucometer (One Touch Ultra, LifeScan) using blood collected from the tail tip, and was comparable among all groups. Image processing and analysis were conducted using a dedicated imaging computational environment developed at the Montreal Neurological Institute (http://www.bic.mni.mcgill.ca/ServicesSoftware/MINC) (Pedersen et al., 2007). Images were acquired using a list mode and were reconstructed via maximum a posteriori probability algorithms (Kornblum et al., 2000) into dynamic (5 min) and static images. Dynamic images were used for assuring the absence of movement or artifacts during the acquisition. After quality control, static images followed a standard analytical procedure. Brain radioactivity standard uptake values (SUV) were obtained by dividing individually static tissue–radioactivity images by body mass and injected [18F]FDG dose. Spatial normalization (coregistration) to high-resolution anatomical MRI templates, acquired in separate groups of 10–11-month-old mice (WT, n = 5; APP, n = 4) with a 7 T Pharmascan system (Bruker BioSpin) (Nicolakakis et al., 2011), was conducted with a two-step semi-interactive method based on affine rigid body registration (six parameters) using the program Register (Montreal Neurological Institute). Volumes of interest on the maximally activated contralateral somatosensory cortex and corresponding area of the ipsilateral cortex were designed guided by SUV images and the MRI templates using a mouse brain atlas (Franklin and Paxinos, 1997). Volumes of interest were applied to the SUV images to obtain regional SUV values. The magnitude of activation was expressed as the ratio and the percentage change of [18F]FDG SUV values in the contralateral relative to the ipsilateral cortex.
Laser Doppler flowmetry.
Three days after the water maze, mice were used for laser Doppler flowmetry (LDF) measurements of CBF (Transonic Systems), as described previously (Tong et al., 2009). Mice were anesthetized with ketamine (85 mg/kg, i.m.; Bioniche) and xylazine (3 mg/kg, i.m.; Haver), and were placed in a stereotaxic frame on a heating blanket for maintaining stable body temperature (37°C). The bone over the barrel cortex (between 0 and 1 mm posterior, 3.5–4.5 mm lateral from bregma) (Ayata et al., 2004) was thinned to translucency for the LDF probe placement. Contralateral CBF was recorded and averaged (four to six stimulations every 30–40 s) before, during, and after unilateral whisker stimulation (20 s at 8–10 Hz). The probe was moved within the delineated area until a “hot spot” corresponding to the highest CBF increase was reliably found upon repeated stimulations. The hot spot was in the laterocaudal part of the barrel field, and virtually no CBF increases were obtained outside this area. Stimulus-evoked CBF was expressed as the percentage change relative to baseline. In a separate group of adult mice, the femoral artery was catheterized under isoflurane anesthesia (2 min, 5% in medical air for induction, 1.5–2% during surgery); anesthesia was then switched to ketamine/xylazine, and CBF, heart rate, and mean arterial blood pressure (MAP) were acquired before and during whisker stimulation (Powerlab, ADInstruments). Blood gases (pO2 and pCO2) and pH were measured at the end of the experiment (RapidLab 348, Bayer). Whisker stimulation or simvastatin did not alter any of these parameters, particularly MAP, as follows (in millimeters mercury, before/during stimulation): WT (n = 4), 72.2 ± 5.8/71.5 ± 5.7; WT treated (n = 3), 68.6 ± 13.3/68.6 ± 12.7; APP (n = 3), 82.6 ± 8.5/82.9 ± 8.9; and APP treated (n = 3), 65.2 ± 2.3/64.7 ± 2.2. The respective evoked CBF responses (percentage from baseline) were 19.0 ± 2.8, 15.7 ± 1.1, 9.4 ± 2.0, and 18.9 ± 1.2 (p < 0.05 for APP vs WT and APP treated).
Anatomical stainings and detection of superoxide ions (O−2).
Some mice were perfused through the heart 3–22 d after the last probe trial (n = 6–8/group) with 4% PFA, and their brains were post-fixed overnight. One hemibrain was processed for paraffin sectioning (5-μm-thick horizontal sections), and the other for sectioning on a freezing microtome (25-μm-thick coronal sections). Mature, dense-core Aβ plaques were histochemically stained on frozen sections with 1% thioflavine S (8 min) (Tong et al., 2009). Free-floating frozen sections were incubated (overnight, room temperature) with a primary rabbit antibody against either c-Fos (1:200, Santa Cruz Biotechnology), cytoplasmic phospholipase A2 (PLA2) (1:300; N-216, Santa Cruz Biotechnology), or Egr-1 (also known as Zif268; 1:200; Santa Cruz Biotechnology), followed by species-specific biotinylated IgGs (1h30, Vector Laboratories) and the AB Complex (1h15, Vector Laboratories). The reaction was visualized with DAB (brown precipitate; cPLA2, Vector Laboratories) or the SG kit (gray precipitate; c-Fos and Egr-1, Vector Laboratories). Dewaxed paraffin sections were used for enkephalin (ENK) (1:200; Millipore) staining, and the reaction was visualized with DAB. Detection of superoxide (O2−) ions was performed on cryostat-cut 25-μm-thick fresh frozen sections stained (15 min) with 2 μm hydroethidine in 0.1 m PBS, pH 7.4, and observed under epifluorescence (Tong et al., 2009). Quantification was performed as described below.
Cerebrovascular reactivity.
After functional studies, some mice (n = 4/group) were used for testing arterial reactivity, as described before (Tong et al., 2005). Briefly, posterior cerebral artery (PCA) segments (∼2 mm long), devoid of cerebral amyloid angiopathy (Tong et al., 2009), were cannulated, pressurized (60 mmHg), and superfused with a Krebs' solution in a chamber for on-line videomicroscopy. Dilatations to acetylcholine (ACh; Sigma) and calcitonin gene-related peptide (CGRP; American Peptide) were tested with increasing concentrations of agonist on vessels slightly preconstricted with phenylephrine (2 × 10−7 m; Sigma). The tonic production of nitric oxide (NO) was tested on vessels at resting tone by superfusion with the NO synthase (NOS) inhibitor Nω-nitro-l-arginine (l-NNA; 10−5 m, 40 min; Sigma). ATP-sensitive potassium (KATP) channel function was assessed in preconstricted vessels from adult and aged WT, APP, and simvastatin-treated APP mice with the selective KATP channel opener levcromakalim (LEV; 10−9–10−4 m; Tocris Bioscience). The remaining cerebral arteries and attached pial membrane from one hemisphere were dissected on ice, frozen on dry ice, and stored (−80°C) for subsequent Western blot assays. The other cerebral hemisphere was snap frozen on dry ice (−45°C) and stored (−80°C) until cryostat sectioning for detection of O2− ions.
Western blot.
Cerebral cortex, hippocampus, and cerebral arteries (excluding the PCA) with attached pial membrane from mice that underwent vascular reactivity testing were homogenized in lysis buffer (or Laemmli buffer for vessels) (Tong et al., 2005) and were cleared by centrifugation. Samples were loaded onto a 15% or, for the gradient gel, an 8–20% Tris/tricine SDS-PAGE gel and were transferred to nitrocellulose membranes (Tong et al., 2009). Total soluble Aβ levels were detected in brain extracts with a mouse anti-Aβ1–16 antibody (1:1000; 6E10, Covance) (overnight), whereas levels of superoxide dismutase 2 (SOD2 1:3000; Stressgen) were measured in pial vessel extracts. All proteins were quantified with enhanced chemiluminescence (ECL plus kit, GE Healthcare) using PhosphorImager (Scanner STORM 860, GE Healthcare). Subcellular fractions were prepared as described by Gu et al. (2009). Tissues were homogenized (one stroke of a glass-Teflon pestle) in ice-cold lysis buffer [Tris HCl 15 mm, pH 7.6, sucrose 0.25 mm, phenylmethylsulfonyl fluoride 1 mm, EDTA 2 mm, EGTA 1 mm, sodium orthovanadate 10 mm, sodium fluoride 25 mm, sodium pyrophosphate 10 mm, and protease inhibitor cocktail tablet (Roche diagnostic)], and centrifuged (800 × g, for 5 min). The supernatants were sequentially centrifuged (10,000 × g, for 10 min; and 165,000 × g, for 30 min). The resulting pellet was suspended in lysis buffer containing 1% Triton X-100 and 300 mm NaCl, homogenized, and centrifuged (16,000 × g, for 30 min) to obtain Triton-soluble (supernatant, P1 fraction) and Triton-insoluble (pellet, P2 fraction) fractions. The P2 fraction, which contains primarily membrane-associated proteins from synaptosomes, was dissolved in 1% SDS. Equal protein amounts from each sample were loaded into 10% SDS-PAGE and transferred to nitrocellulose membranes. The blots were blocked by 5% skim milk (40 min); incubated overnight with antibodies against PSD-95 (1:2000; Stressgen), synaptophysin (1:2000; Dako), and NMDA receptor type 2B (NR2B; 1:2000; Abcam), then with horseradish peroxidase-conjugated secondary antibodies; and visualized with ECL.
Measurements of Aβ.
Levels of total (soluble and insoluble) Aβ1–40 and Aβ1–42 were measured by ELISA, which was performed as before (Tong et al., 2009) and as recommended by Invitrogen. Briefly, mouse hemibrains were weighted and homogenized in 5 m guanidine Tris HCl with a glass Teflon tissue grinder. Homogenates were diluted with a cold reaction buffer. The supernatants were used for analysis after centrifugation at 16,000 × g (20 min).
Data analysis.
Vascular responses (percentage change in diameter from basal or preconstricted tone) were plotted as a function of agonist concentration or duration of NOS inhibition. Concentration-dependent and maximal (EAmax) responses and the agonist concentration eliciting half the EAmax (EC50 value or pD2 ≤ log EC50) were used to compare agonist efficacy and potency, respectively. The number of and percentage area occupied (plaque load) by cortical and hippocampal thioflavine S-positive plaques were measured on digital pictures with the MetaMorph 6.1r3 program (Universal Imaging). For immunostaining, the numbers of positive c-Fos and Egr-1 nuclei were counted in 0.067 mm2 surface area on digital pictures (one picture from each section and three sections per mouse, four mice per group). The intensity of Egr-1-positive nuclei or cPLA2 staining in the hippocampus CA1 region was measured as total gray level using MetaMorph. For Western blot, bands were analyzed by densitometry using actin (mouse anti-β-actin; 1:1000; Sigma) to normalize for loading (Tong et al., 2005). LDF measurements of CBF were performed blind to the identity of the mice. For other in vivo experiments, the investigator was partially blinded (Morris water maze, mice being numbered and tested by gender) or only one of the investigators was totally blinded (FDG-PET) to the genotype. Data were expressed as mean ± SEM and compared by one-way ANOVA followed by Newman–Keuls post hoc multiple-comparison test or by Student's t test for two-group comparisons (GraphPad Prism 4). A p value <0.05 was taken as significant.
Results
Simvastatin restored cerebrovascular reactivity in APP mice
Cerebral arteries from adult and aged APP mice displayed impaired dilatations to ACh and CGRP at increasing and maximal agonist concentrations compared with WT controls (Fig. 1; Table 1). These deficits were not attributed to receptor desensitization since ACh and CGRP pD2 values did not differ between groups at any age (Table 1). The tonic production of the vasodilator NO, evaluated by inhibiting NOS activity with l-NNA, was also reduced in APP compared with WT arteries, the decrease being significant only in adult APP mice (Fig. 1, Table 1). In contrast to the low-dose short simvastatin treatment (20 mg/kg/d for 8 weeks) that failed to rescue ACh-induced dilatations in aged APP mice (Tong et al., 2009), a daily 40 mg/kg dose of simvastatin fully restored ACh- and CGRP-induced dilatations, and basal NO levels in adult or aged APP mice treated for 3 or 6 months (Fig. 1, Table 1). These findings suggest that dose and length of treatment are determinant for full recovery of cerebrovascular function.
Figure 1.

Simvastatin (SV) rescued cerebrovascular reactivity in adult and aged APP mice. The impaired dilatations to ACh and CGRP in APP mice (▴) compared with WT controls (●) were normalized by SV treatment (▵), which had no effect in WT mice (○). Dilatation to the KATP channel opener levcromakalim (LEV) was unaltered in adult APP mice, but was reduced in aged APP mice and completely restored by SV. Contractile responses to NOS inhibition (l-NNA, 10−5 m) were significantly reduced only in adult APP mice, and were normalized by SV. *p < 0.05; **p < 0.01; ***p < 0.001 for APP vs WT or APP (SV) mice. ANOVA and Newman–Keuls test, n = 4/group.
Table 1.
Effects of simvastatin on cerebrovascular reactivity in adult (6 months) and aged (12 months) APP mice
| Age (months) | WT | WT (SV) | APP | APP (SV) | |
|---|---|---|---|---|---|
| ACh | |||||
| EAmax | 6 | 57.1 ± 4.2 | 66.1 ± 3.4 | 32.5 ± 4.6** | 56.3 ± 3.9 |
| pD2 | 6 | 7.07 ± 0.14 | 7.10 ± 0.12 | 6.81 ± 0.19 | 7.10 ± 0.14 |
| EAmax | 12 | 56.1 ± 5.6 | 60.5 ± 10.4 | 30.1 ± 3.3** | 65.7 ± 7.9 |
| pD2 | 12 | 6.65 ± 0.16 | 7.74 ± 0.29** | 6.83 ± 0.17 | 7.02 ± 0.15 |
| CGRP | |||||
| EAmax | 6 | 55.8 ± 1.2 | 58.4 ± 2.1 | 24.5 ± 2.9*** | 61.2 ± 5.9 |
| pD2 | 6 | 8.39 ± 0.07 | 8.16 ± 0.08 | 8.04 ± 0.12 | 8.31 ± 0.13 |
| EAmax | 12 | 44.5 ± 3.9 | 48.5 ± 2.4 | 30.0 ± 6.1*** | 48.4 ± 2.8 |
| pD2 | 12 | 8.11 ± 0.12 | 8.40 ± 0.06 | 8.45 ± 0.25 | 8.12 ± 0.05 |
| l-NNA | |||||
| EAmax | 6 | 43.3 ± 2.9 | 53.6 ± 4.0 | 23.2 ± 2.1*** | 43.0 ± 3.4 |
| EAmax | 12 | 44.5 ± 4.2 | 41.2 ± 4.9 | 33.5 ± 8.9 | 53.6 ± 2.7 |
| LEV | |||||
| EAmax | 12 | 56.6 ± 8.4 | 72.9 ± 7.7 | 16.7 ± 13.2** | 69.4 ± 2.4 |
| pD2 | 12 | 5.80 ± 0.21 | 6.50 ± 0.21 | 6.20 ± 0.22 | 5.80 ± 0.12 |
Data are means ± SEM and are expressed as the maximal agonist response (EAmax) or agonist potency (pD2). EAmax is the percentage maximal dilatation to ACh, CGRP, or the KATP channel opener LEV, or corresponds to the percentage maximal diameter decrease after 40 min inhibition of NOS with 10−5 m l-NNA. n = 4 per group.
**p < 0.01,
***p < 0.001 when compared to WT, WT (SV), or APP (SV) by ANOVA. SV, Simvastatin.
Lack of NO bioavailability due to its trapping by NADPH oxidase-derived reactive oxygen species (ROS) (Iadecola et al., 1999; Tong et al., 2005; Park et al., 2008) and impaired cerebrovascular KATP channel function (Tong et al., 2009) have been identified as culprits of the altered dilatory responses. Consistently, in both adult and aged APP mice, O2− production was increased in the pial membrane and intracortical blood vessels, as were also the protein levels of the inducible antioxidant enzyme SOD2, a marker of increased oxidative stress (Wong and Goeddel, 1988) (Fig. 2). Simvastatin normalized both O2− production and SOD2 levels (Fig. 2), as previously reported for the low-dose simvastatin (Tong et al., 2009). In contrast, whereas the low-dose simvastatin had no benefit on KATP channel function in aged APP mice (Tong et al., 2009), the current treatment regimen completely restored the impaired dilatations to the selective KATP channel opener levcromakalim (16.7 ± 13.2% in APP vs 69.4 ± 2.4% of the induced tone in simvastatin-treated APP mice, p < 0.001) (Fig. 1). This may have allowed the recovery of CGRP-induced dilatations that are mediated by both NO and KATP channels (Kitazono et al., 1993). However, these channels were unimpaired in adult APP mice (Fig. 1) suggesting that, at that age, the dilatory dysfunctions likely resulted from ROS-induced sequestration of vasodilators or the impairment of other K+ channels, such as the calcium-dependent K+ (KCa) channels (Hong et al., 1996), which are also responsive to statin therapy, as shown in peripheral arteries (Miller et al., 2004).
Figure 2.
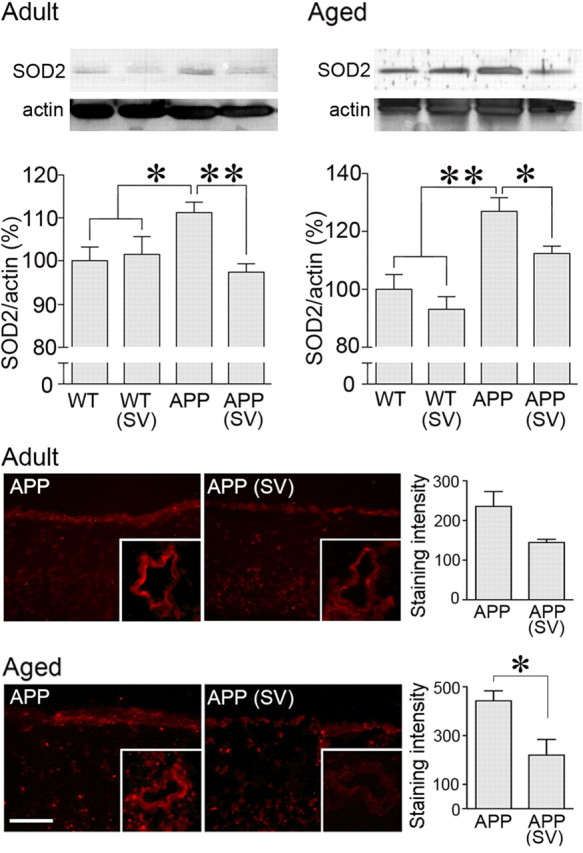
Simvastatin (SV) decreased oxidative stress in brain vessels and pial membranes from APP mice. In both adult and aged APP mice, SV normalized the increased SOD2 protein levels in cerebral arteries measured by Western blot (top) (n = 5–13 mice/group in adult and n = 4 mice/group in aged mice). SV also reduced the staining intensity of the O2− marker hydroethidine in the pial membrane (bottom) and intracortical blood vessels (not quantified). *p < 0.05; **p < 0.01 (n = 4–5 mice /group at both ages). Scale bar, 30 μm.
Simvastatin rescued neurovascular and neurometabolic coupling in APP mice
The localized CBF response to increased neuronal activity that ensures adequate oxygen and glucose supply to activated brain areas is impaired in APP mice at all ages (Niwa et al., 2002; Nicolakakis et al., 2008; Tong et al., 2009). Similarly, cerebral glucose uptake is decreased in young APP mice at rest (Niwa et al., 2002) and in elderly APP mice (∼16 months) during increased cortical neuronal activity evoked by whisker stimulation (Nicolakakis et al., 2008). Consistent with these findings, the hyperemic response to sensory stimulation was reduced in the somatosensory cortex of adult (−35.6 ± 7%, p < 0.01) and aged (−32.4 ± 11%, p < 0.01) APP mice relative to WT controls. Simvastatin fully normalized this response in both age groups (Fig. 3A). Likewise, the increase in CGU induced by whisker stimulation in the somatosensory cortex was significantly reduced in aged APP mice (1.1 ± 1.2% in APP vs 7.1 ± 2.6% in WT mice, p < 0.05) and fully recovered in simvastatin-treated mice (6.4 ± 1.5%, p < 0.05 vs APP), as determined by [18F]FDG microPET (Fig. 3B).
Figure 3.

Simvastatin (SV) normalized neurovascular and neurometabolic coupling in APP mice. A, SV normalized the impaired CBF response to whisker stimulation (shaded area) in adult and aged APP mice relative to WT controls. Curves represent 1 s average of the evoked CBF, expressed as a percentage change from 30 s prestimulus baseline [black, WT; blue, WT (SV), orange, APP; red, APP (SV)]. *p < 0.05; **p < 0.001. B, The impaired increase in glucose uptake after whisker stimulation in aged APP mice was restored by SV. The percentage specific activation was calculated from the [18F]FDG SUV in the activated contralateral and analogous ipsilateral somatosensory cortex (as indicated by arrows on the merged images). Average PET images were overlaid over average anatomical MRI images obtained from similarly aged WT and APP mice. *p < 0.05 compared with WT and APP(SV) mice. ANOVA and Newman–Keuls test, n = 4–7 per group.
Simvastatin preserved learning and memory in adult but not in aged APP mice
We found severe behavioral impairments in the Morris water maze at both ages, in agreement with previous studies in adult (Palop et al., 2003; deIpolyi et al., 2008) and aged (Nicolakakis et al., 2008; Tong et al., 2009) APP mice. APP mice overall had no difficulty learning to escape to the visible platform (days 1–3; Fig. 4). Importantly, time latencies to find the hidden platform were higher in APP mice compared with WT mice, indicating impaired learning at both ages (days 4–8; p < 0.01–0.001) (Fig. 4). Similarly, in the spatial memory component of the test, the time spent in the target quadrant or the number of crossings over the platform location was significantly reduced in adult and aged APP mice compared with their WT controls (probe trial, day 9; Fig. 4), despite no alteration in swim speed (data not shown). Simvastatin normalized learning and memory in adult APP mice (Fig. 4A), and this effect was both reproducible and persistent, as seen in the adult cohort tested in the probe trial on day 17 (Fig. 4B). However, as previously observed with the short low-dose simvastatin treatment in aged APP mice (Tong et al., 2009), simvastatin failed to rescue the learning and memory deficits of aged APP mice (Fig. 4C). Simvastatin had no effect on cognitive performance in either adult (Fig. 4B) or aged (data not shown) WT mice. Together, these findings show that pathological stage determines treatment efficacy and that timely therapeutic intervention is crucial for memory recovery.
Figure 4.

Simvastatin (SV) rescued learning and memory in adult but not in aged APP mice. APP mice of both ages (orange triangles) displayed longer latencies to reach the hidden platform (days 4–8), as well as decreased time and number of platform crossings in the target quadrant during the probe trial compared with WT mice (black circles). SV treatment (red triangles) rescued learning performance in both cohorts of adult APP mice. A, B, Further, SV restored memory tested in the early (day 9, A) and delayed probe trials (day 17, B). In aged APP mice, SV had no beneficial effect. One cohort of adult treated and untreated APP mice needed longer time to reach the visible platform (days 1–3) compared with WT mice (†p < 0.001). SV had no effect in adult (blue circles) or aged (data not shown) WT controls. ANOVA and Newman–Keuls test. *p < 0.05; **p < 0.01; ***p < 0.001.
Simvastatin did not reduce amyloidosis
To investigate whether simvastatin acted upon amyloidosis, a proposed but debatable role for statins (Tamboli et al., 2010), we measured soluble Aβ levels and Aβ plaque load in both age groups. Total soluble Aβ levels were detected in hippocampus of adult APP and in both cortex and hippocampus of aged APP mice by Western blot, but simvastatin exerted no reducing effect at either age (Fig. 5A). Similarly, levels of the different Aβ monomers or oligomers, including the 56 kDa assembly that has been linked to memory deficit (Lesné et al., 2006), were detected in adult APP mice with preserved memory using the anti-Aβ 6E10 antibody on a gradient gel, but no reducing effect of treatment was found in either cortex or hippocampus (data not shown). Simvastatin also had no significant effect on total Aβ1–40 and Aβ1–42 levels measured by ELISA in hemibrains of adult and aged APP mice (Fig. 5B). We then examined the Aβ deposits in cortex and hippocampus that, as expected, were smaller and less numerous in 6-month-old compared with 12-month-old APP mice (Fig. 5C). Simvastatin did not reduce Aβ plaque number or Aβ plaque load in cortex or hippocampus in treated APP mice at any age (Fig. 5C). The findings unequivocally demonstrate the ability of simvastatin to preserve memory in adult APP mice despite increased soluble Aβ species and ongoing plaque formation.
Figure 5.

Simvastatin (SV) did not reduce amyloidosis in adult and aged APP mice. A, B, SV failed to reduce the levels of cortical (Co) and hippocampal (Hi) soluble Aβ oligomers (4 kDa) detected by Western blot (A), as well as those of total soluble Aβ1–40 and Aβ1–42 measured by ELISA in hemibrains (B). C, Similarly, the number of thioflavine S-positive plaques and the area they occupied in cortex or hippocampus (plaque load) did not differ between treated and nontreated adult or aged APP mice. Student's t test, n = 4–6 per group.
Simvastatin upregulated c-Fos and Egr-1 in the hippocampus CA1 region
Reduced levels of c-Fos in the hippocampus of APP mice have been suggested to contribute to cognitive deficits (Palop et al., 2003). Similarly, Egr-1 (Zif268), a member of the immediate–early gene family of transcription factors essential for long-term potentiation (LTP) and memory formation (Jones et al., 2001), was decreased in various models of APP mice either at baseline (Dickey et al., 2003) or following memory testing (Blanchard et al., 2008). We found that both adult and aged APP mice had lower baseline c-Fos and Egr-1 immunoreactivity in the hippocampus compared with their respective WT controls (Figs. 6, 7). Specifically, in the CA1 area of APP mice at both ages, there was a decrease in the number of c-Fos-positive nuclei (Fig. 6; difference not significant) and a highly significant decrease in the staining intensity and number of Egr-1-positive nuclei (−37.7 ± 6.2%, p < 0.05 and −50.9 ± 3.9%, p < 0.001 in adult and aged APP mice, respectively) (Fig. 7). In simvastatin-treated adult APP mice with restored memory, c-Fos and Egr-1 immunoreactivity was upregulated, and the number of immunostained nuclei or staining intensity compared with or exceeded that of WT controls (Figs. 6, 7). In contrast, in aged APP mice that failed to recover memory, simvastatin did not significantly improve c-Fos expression (−51 ± 12.8%, p < 0.05 vs WT controls) and had no effect on Egr-1 immunostaining (−47.5 ± 0.7%, p < 0.001) (Figs. 6, 7). Interestingly, simvastatin had a small promoting effect on the number of c-Fos nuclei in aged WT mice, normalizing them to the levels of adult WT mice (Fig. 6). These data demonstrate that simvastatin can rescue critical proteins underlying hippocampus-mediated memory processes in adult but not aged APP mice.
Figure 6.

Simvastatin (SV) upregulated basal c-Fos expression in adult, but not aged, APP mice. Baseline immunostaining of c-Fos expression in the dorsal hippocampus indicated a smaller, albeit not significant, number of nuclei in adult and aged APP mice, as quantified in the CA1 area (delineated by arrows). In adult APP mice, SV treatment upregulated c-Fos expression, the number of c-Fos nuclei being higher than in WT mice. Whereas such an enhancing effect did not occur in aged APP mice, SV normalized the number of c-Fos nuclei in aged WT mice to that seen in adult WT mice. *p < 0.05, **p < 0.01. ANOVA and Newman–Keuls test, n = 9–13 per group (adult) and n = 4 per group (aged). Scale bar, 50 μm.
Figure 7.
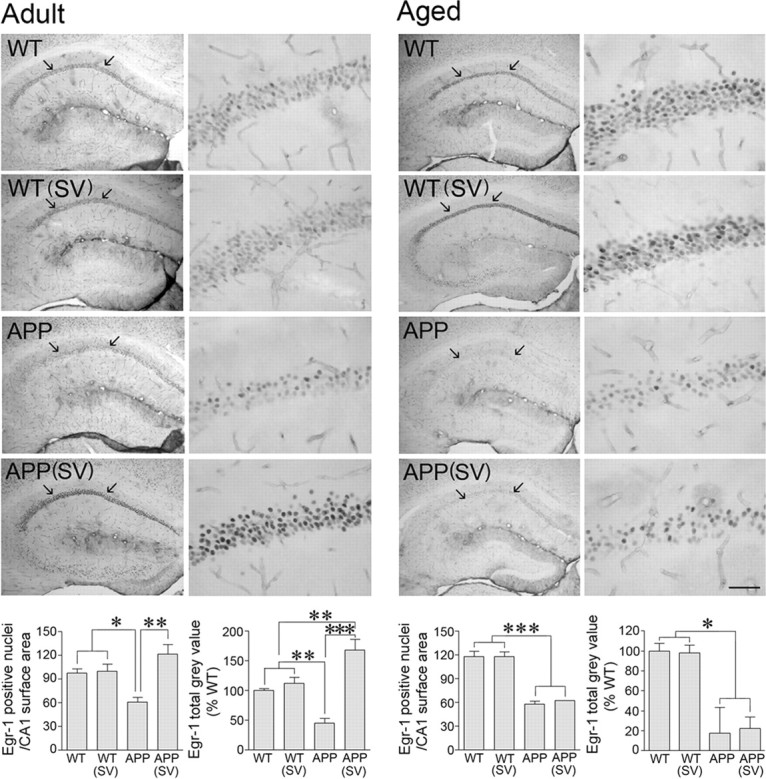
Simvastatin (SV) normalized basal Egr-1 protein levels in adult, but not aged, APP mice. Egr-1 immunostaining in the dorsal hippocampus revealed a drastic reduction in the number of nuclei and staining intensity in the CA1 area of both adult and aged APP mice (delineated by arrows in left panels and magnified in right panels). In adult APP mice, SV normalized the number of Egr-1-positive nuclei and increased staining intensity (total gray value) to levels higher than in WT mice treated or nontreated with SV. *p < 0.05; **p < 0.01; ***p < 0.001. ANOVA and Newman–Keuls test, n = 4 per group. Scale bar, 50 μm.
Simvastatin did not act upon other synaptic or memory-related proteins
Memory deficit in APP mice has also been imputed to increased levels of ENK (Meilandt et al., 2008) and PLA2 (Sanchez-Mejia et al., 2008), and to decreases in synaptic proteins and NMDA receptor subunits or their downstream signaling pathways (Yamin, 2009). We confirmed upregulation of ENK immunoreactivity in the dentate gyrus and mossy fiber area, and of PLA2 in the stratum radiatum of the CA1 area in the hippocampus of both adult and aged APP mice (Fig. 8). These increased protein levels, however, were not normalized by simvastatin at any age and despite memory recovery in the adult cohorts (Fig. 8). Furthermore, we found no change in synaptic markers PSD-95, synaptophysin, or the NMDA receptor subunit NR2B in synaptosomal P2 fractions from cortex and hippocampus in adult and aged APP mice compared with WT controls (Fig. 9). These findings agree with unchanged levels of presynaptic and postsynaptic markers in 6- or 18-month-old APPSwe (Gu et al., 2009) or APP/PS1 (Dickey et al., 2003) mice, but not with those reporting decreased density of synaptophysin-immunoreactive terminals in APPInd mice between 2 and 28 months of age (Hsia et al., 1999) or aged APPSwe,Ind mice (Mucke et al., 2000). Simvastatin did not affect these proteins in adult APP or WT mice (Fig. 9).
Figure 8.
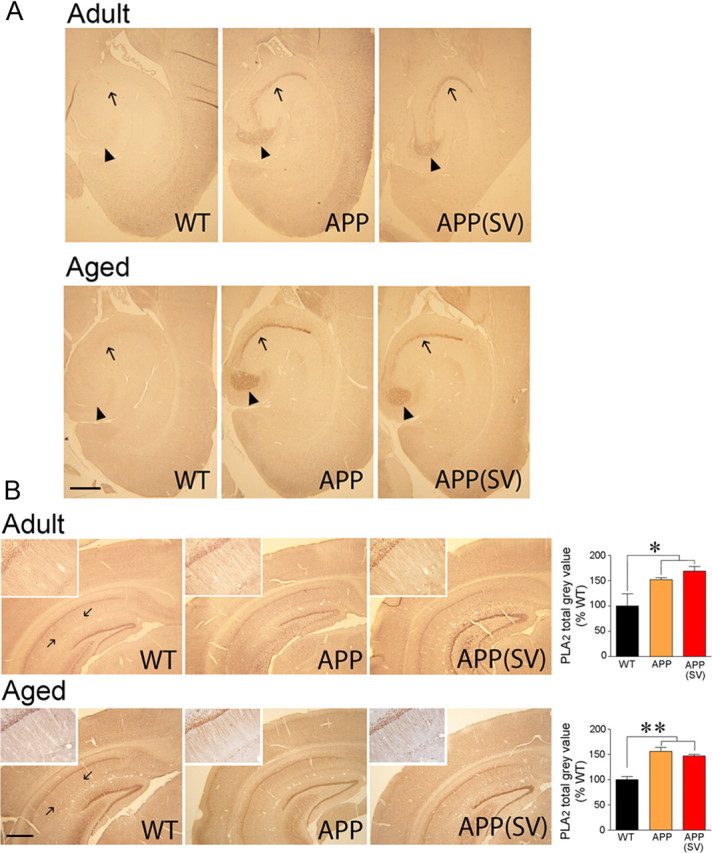
Simvastatin (SV) did not normalize the increased hippocampal ENK or phosphorylated PLA2 immunoreactivity in APP mice. A, Horizontal paraffin brain sections from adult and aged APP mice showed a marked increased in ENK immunoreactivity in the hilar region of the dentate gyrus (arrowheads) and in the mossy fiber area (arrows) compared with WT controls. The increase was stronger in aged APP mice. SV had no reducing effect at any age, despite memory recovery in adult APP mice. N = 3–5 per group. B, The intensity of PLA2 immunoreactivity was increased significantly in the stratum radiatum of the hippocampal CA1 area (delineated by arrows and magnified in insets) in both adult and aged APP mice (orange) compared with WT controls (black). SV (red) had no significant effect at either age. *p < 0.05; **p < 0.01. ANOVA and Newman–Keuls test, n = 4–5per group. Scale bars, 500 μm.
Figure 9.

Synaptic markers were unaltered in adult and aged APP mice and by simvastatin (SV). Representative Western blots for synaptic density proteins PSD-95, synaptophysin (SYN), and the NR2B subunit of NMDA receptors measured in the synaptosomal (P2) fraction prepared from cortex and hippocampus from WT, APP, and APP mice treated with SV. No change was observed between APP and WT mice at any age, and SV did not affect these proteins in adult APP mice. n = 4–5 per group.
Discussion
Using cognitive and cerebrovascular measures as indexes of clinical outcome in APP mice, we report that simvastatin showed full efficacy in restoring cerebrovascular function in adult and aged APP mice, but that therapeutic benefit on learning and memory was restricted to adult mice. We found that recovery of vasodilatory capacity concurred with restoration of NO bioavailability and KATP channel function, and that memory recovered concomitantly with upregulation of baseline c-Fos- and Egr-1-expressing cells in the CA1 area of the hippocampus, notwithstanding a progressing Aβ pathology. These findings demonstrate clear benefits for simvastatin on two main landmarks of AD pathology, and on key proteins required for hippocampal synaptic function through which memory could be preserved.
We performed all reactivity studies on proximal PCA branches to avoid the rare, albeit likely, presence of Aβ deposits in most distal segments of the artery in aged animals (Tong et al., 2009), as these could further impair dilatory capacity through mechanical damage (Park et al., 2008, Han et al., 2008). Hence, the cerebrovascular deficits reported here are strictly imputed to soluble Aβ species that impair cerebrovascular function essentially through increased production of NADPH oxidase-derived ROS (Iadecola et al., 1999; Tong et al., 2005, 2009; Park et al., 2008, 2011). In this respect, the antioxidant capacity of the high-dose simvastatin was evidenced in the cerebral vasculature of APP mice at both ages, but was particularly striking in aged mice with prominent oxidative stress. Indeed, simvastatin reduced O2− production and normalized the levels of the antioxidant SOD2. This likely resulted in sufficient NO bioavailability to restore basal vessel tone and ACh-mediated dilatations that necessitate the synthesis of additional endothelial NOS-derived dilators (Drouin et al., 2007). The functional recovery achieved with the high-dose, as opposed to the low-dose, simvastatin that failed to rescue the ACh dilatation in aged mice (Tong et al., 2009) was analogous to that obtained with pharmacologic or genetic ROS-countering strategies targeting the production (Kitazono et al., 1993; López et al., 2008) or elimination of O2− with SOD (Iadecola et al., 1999; Miller et al., 2004; Tong et al., 2009), or with the SOD mimetic Tempol (Nicolakakis et al., 2008). Hence, the greater antioxidant properties of a higher-dose simvastatin likely accounted for normalized KATP channel-mediated dilatation in aged APP mice, since O2− can inhibit BK, KATP, and inwardly rectifying potassium channels in cerebral arteries, and this impairment can be restored by topical SOD application (Erdös et al., 2004). Although not measured in the current study, myogenic tone of cerebral arteries is also highly sensitive to ROS (Maneen and Cipolla, 2007), and simvastatin may have acted at this level. Yet, an activating effect of simvastatin on brain endothelial NOS activity (Sugawara et al., 2008) and a direct relaxant effect through KCa channel activation, as reported for rosuvastatin in peripheral arteries (López et al., 2008), could also contribute to the functional recovery.
Simvastatin antioxidant and anti-inflammatory properties may be credited for the normalized stimulus-induced increases in glucose metabolism and CBF during whisker stimulation. Both responses require astrocytes as intermediaries either for glucose uptake (Chuquet et al., 2010) or calcium signaling and release of vasodilatory mediators (Koehler et al., 2009). Simvastatin attenuated astrocyte activation in the cortex where the CGU and CBF responses were recorded, and reduced the number of ROS-producing NADPH/p22phox subunit-positive glial cells (Tong et al., 2009). Similar rescue of neurometabolic and neurovascular coupling responses was observed in elderly APP mice following pharmacologic (Nicolakakis et al., 2008) or genetic (Park et al., 2008) anti-inflammatory or antioxidant therapy. Together, these findings indicate that simvastatin counters Aβ-induced deleterious effects by targeting inflammatory and oxidative reactions. With the latter being detrimental to astrocytes, neurons, and the brain vasculature (Yang et al., 2010), our data identify multiple cell types where simvastatin can act to rescue brain energetics and hemodynamics. Whereas such benefits were not exclusive to simvastatin, as pure antioxidants or other drugs with pleiotropic effects, such as peroxisome proliferator-activated receptor-γ (PPAR-γ) agonists, were as effective (Nicolakakis et al., 2008), they clearly point to the high efficacy of simvastatin in rescuing changes in brain metabolism and perfusion induced by neuronal activity.
On the contrary, one benefit unique to simvastatin, and not reproduced by antioxidants or PPAR-γ agonists, was the restoration of learning and memory, and most strikingly of long-term memory, in adult APP mice. Behavioral recovery occurred with no reduction in brain levels of soluble Aβ or Aβ plaque load, in agreement with a previous study that found cognitive improvement in Tg2576 APP mice independently from decreased amyloidosis (Li et al., 2006). These findings also match exquisitely well with those in asymptomatic middle-aged adults at risk for AD, in whom working memory improvements occurred without changes in CSF Aβ42 (Carlsson et al., 2008). What is more, we found that simvastatin-treated adult APP mice with restored memory displayed upregulated c-Fos and Egr-1 protein levels in the CA1 region of the hippocampus when tested up to 2 weeks after the last probe trial, demonstrating an effect of simvastatin on basal levels of these memory-related proteins or on hippocampal neuron function. Interestingly, a positive correlation between spatial learning and constitutive Egr-1 mRNA expression, selectively in CA1 pyramidal neurons, has been documented (Yau et al., 1996), and increased basal Egr-1 expression induced by transgenesis was reported to enhance long-term memory (Baumgärtel et al., 2008). Several studies have also related increased c-Fos or Egr-1 expression to various forms of learning and memory, particularly long-term memory (Jones et al., 2001; Davis et al., 2003; Fleischmann et al., 2003).
Deletion of the cAMP response element-binding protein (CREB), which is upstream of c-Fos and Egr-1, resulted in learning and long-term memory deficits (Pittenger et al., 2002). Activation of CREB has been associated with the MAPK/ERK and the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathways, both implicated in LTP and memory processes (Davis et al., 2000; Jones et al., 2001; Bozon et al., 2003; Mizuno et al., 2003). Most relevant to the present findings, these pathways are activated by simvastatin (Li et al., 2006; Beretta et al., 2011), and a body of evidence has recently accumulated for a role of the PI3K pathway in the positive effect of simvastatin on memory. Particularly, chronic simvastatin treatment increased brain levels of total and phosphorylated Akt (P-Akt) in WT mice with enhanced learning and memory performance (Li et al., 2006). In hippocampal slices from WT mice, prolonged simvastatin treatment increased P-Akt in the CA1 area concomitantly with improved LTP at CA3–CA1 synapses, an effect suppressed by inhibition of Akt phosphorylation (Mans et al., 2010). Together, these findings support that simvastatin rescued memory in adult APP mice by restoring the ability of hippocampal CA1 neurons to respond to synaptic input, possibly by restoring the PI3K/Akt → P-CREB → Egr-1/c-Fos signaling cascade that is crucial for spatial memory. This would also agree with the simvastatin-mediated activation of the PI3K/Akt pathway associated with neuroprotection after hypoxic-ischemic brain damage (Carloni et al., 2009). Simvastatin also improved CBF and behavioral outcome, and decreased hippocampal degeneration after brain injury (Wang et al., 2007; Wu et al., 2008), and was the most protective statin against kainate-induced excitotoxicity and memory impairment (Ramirez et al., 2011).
We conclude that the remarkable benefit of simvastatin on both cerebrovascular and neuronal deficits in adult AD mice results from its combined pleiotropic properties as a cerebrovascular antioxidant and neuronal regulator of memory-linked c-Fos and Egr-1 proteins. The results in aged mice further demonstrate that full recovery of cerebrovascular function does not necessarily translate into normalized cognitive performance, suggesting that simvastatin efficacy on neuronal pathways is protective rather than restorative even though there might be a time window where these two mechanisms overlap. Since the kind of statins, onset of treatment, and clinical status of the patients may be critical for efficacy in AD (Wolozin et al., 2000; Haag et al., 2009), our results of an age-related benefit for simvastatin on memory pathways establish simvastatin as a highly valuable statin to consider for the treatment of early AD and of patients at risk for AD due to vascular diseases.
Footnotes
This work was supported by the Canadian Institutes of Health Research Grant MOP-84275 (to E.H.) and a fellowship from the Heart & Stroke Foundation of Canada/Canadian Stroke Network (to C.L.). We thank Dr. L. Mucke (Gladstone Institute of Neurological Disease and Department of Neurology, University of California, San Francisco, San Francisco, CA) and the J. David Gladstone Institutes for the APP transgenic mouse breeders. We are indebted to Ms. P. Fernandes [Montreal Neurological Institute (MNI), Montréal, Québec, Canada] for the laser Doppler flowmetry experiments; J. Cakiroglu and Dr. B. Bedell (Small Animal Imaging Laboratory, MNI) for the MRI templates; Dr. P. Rosa-Neto (Brain Imaging Centre, MNI) for his help with the PET data analysis; Dr. N. Nicolakakis (MNI) for reviewing the manuscript; and P. Papadopoulos (MNI) for help with some treatments.
The authors declare no competing financial interests.
References
- Aucoin JS, Jiang P, Aznavour N, Tong XK, Buttini M, Descarries L, Hamel E. Selective cholinergic denervation, independent from oxidative stress, in a mouse model of Alzheimer's disease. Neuroscience. 2005;132:73–86. doi: 10.1016/j.neuroscience.2004.11.047. [DOI] [PubMed] [Google Scholar]
- Ayata C, Dunn AK, Gursoy-OZdemir Y, Huang Z, Boas DA, Moskowitz MA. Laser speckle flowmetry for the study of cerebrovascular physiology in normal and ischemic mouse cortex. J Cereb Blood Flow Metab. 2004;24:744–755. doi: 10.1097/01.WCB.0000122745.72175.D5. [DOI] [PubMed] [Google Scholar]
- Baumgärtel K, Genoux D, Welzl H, Tweedie-Cullen RY, Koshibu K, Livingstone-Zatchej M, Mamie C, Mansuy IM. Control of the establishment of aversive memory by calcineurin and Zif268. Nat Neurosci. 2008;11:572–578. doi: 10.1038/nn.2113. [DOI] [PubMed] [Google Scholar]
- Beretta S, Pastori C, Sala G, Piazza F, Ferrarese C, Cattalini A, de Curtis M, Librizzi L. Acute lipophilicity-dependent effect of intravascular simvastatin in the early phase of focal cerebral ischemia. Neuropharmacology. 2011;60:878–885. doi: 10.1016/j.neuropharm.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Blanchard J, Martel G, Guillou JL, Noguès X, Micheau J. Impairment of spatial memory consolidation in APP(751SL) mice results in cue-guided response. Neurobiol Aging. 2008;29:1011–1021. doi: 10.1016/j.neurobiolaging.2007.02.010. [DOI] [PubMed] [Google Scholar]
- Bozon B, Kelly A, Josselyn SA, Silva AJ, Davis S, Laroche S. MAPK, CREB and zif268 are all required for the consolidation of recognition memory. Philos Trans R Soc Lond B Biol Sci. 2003;358:805–814. doi: 10.1098/rstb.2002.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA. Atorvastatin and Aβ(1−40): not as simple as cholesterol reduction in brain and relevance to Alzheimer disease. Exp Neurol. 2011;228:15–18. doi: 10.1016/j.expneurol.2010.12.014. [DOI] [PubMed] [Google Scholar]
- Carloni S, Girelli S, Buonocore G, Longini M, Balduini W. Simvastatin acutely reduces ischemic brain damage in the immature rat via Akt and CREB activation. Exp Neurol. 2009;220:82–89. doi: 10.1016/j.expneurol.2009.07.026. [DOI] [PubMed] [Google Scholar]
- Carlsson CM, Gleason CE, Hess TM, Moreland KA, Blazel HM, Koscik RL, Schreiber NT, Johnson SC, Atwood CS, Puglielli L, Hermann BP, McBride PE, Stein JH, Sager MA, Asthana S. Effects of simvastatin on cerebrospinal fluid biomarkers and cognition in middle-aged adults at risk for Alzheimer's disease. J Alzheimers Dis. 2008;13:187–197. doi: 10.3233/jad-2008-13209. [DOI] [PubMed] [Google Scholar]
- Chuquet J, Quilichini P, Nimchinsky EA, Buzsáki G. Predominant enhancement of glucose uptake in astrocytes versus neurons during activation of the somatosensory cortex. J Neurosci. 2010;30:15298–15303. doi: 10.1523/JNEUROSCI.0762-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer C, Haan MN, Galea S, Langa KM, Kalbfleisch JD. Use of statins and incidence of dementia and cognitive impairment without dementia in a cohort study. Neurology. 2008;71:344–350. doi: 10.1212/01.wnl.0000319647.15752.7b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S, Vanhoutte P, Pages C, Caboche J, Laroche S. The MAPK/ERK cascade targets both Elk-1 and cAMP response element-binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. J Neurosci. 2000;20:4563–4572. doi: 10.1523/JNEUROSCI.20-12-04563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S, Bozon B, Laroche S. How necessary is the activation of the immediate early gene zif268 in synaptic plasticity and learning? Behav Brain Res. 2003;142:17–30. doi: 10.1016/s0166-4328(02)00421-7. [DOI] [PubMed] [Google Scholar]
- deIpolyi AR, Fang S, Palop JJ, Yu GQ, Wang X, Mucke L. Altered navigational strategy use and visuospatial deficits in hAPP transgenic mice. Neurobiol Aging. 2008;29:253–266. doi: 10.1016/j.neurobiolaging.2006.10.021. [DOI] [PubMed] [Google Scholar]
- Dickey CA, Loring JF, Montgomery J, Gordon MN, Eastman PS, Morgan D. Selectively reduced expression of synaptic plasticity-related genes in amyloid precursor protein + presenilin-1 transgenic mice. J Neurosci. 2003;23:5219–5226. doi: 10.1523/JNEUROSCI.23-12-05219.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin A, Thorin-Trescases N, Hamel E, Falck JR, Thorin E. Endothelial nitric oxide synthase activation leads to dilatory H2O2 production in mouse cerebral arteries. Cardiovasc Res. 2007;73:73–81. doi: 10.1016/j.cardiores.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Erdös B, Snipes JA, Miller AW, Busija DW. Cerebrovascular dysfunction in Zucker obese rats is mediated by oxidative stress and protein kinase C. Diabetes. 2004;53:1352–1359. doi: 10.2337/diabetes.53.5.1352. [DOI] [PubMed] [Google Scholar]
- Fleischmann A, Hvalby O, Jensen V, Strekalova T, Zacher C, Layer LE, Kvello A, Reschke M, Spanagel R, Sprengel R, Wagner EF, Gass P. Impaired long-term memory and NR2A-type NMDA receptor-dependent synaptic plasticity in mice lacking c-Fos in the CNS. J Neurosci. 2003;23:9116–9122. doi: 10.1523/JNEUROSCI.23-27-09116.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KB, Paxinos G. The mouse brain in stereotaxic coordinates. San Diego: Academic; 1997. [Google Scholar]
- Gu Z, Liu W, Yan Z. β-Amyloid impairs AMPA receptor trafficking and function by reducing Ca2/calmodulin-dependent protein kinase II synaptic distribution. J Biol Chem. 2009;284:10639–10649. doi: 10.1074/jbc.M806508200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag MD, Hofman A, Koudstaal PJ, Stricker BH, Breteler MM. Statins are associated with a reduced risk of Alzheimer disease regardless of lipophilicity. The Rotterdam study. J Neurol Neurosurg Psychiatry. 2009;80:13–17. doi: 10.1136/jnnp.2008.150433. [DOI] [PubMed] [Google Scholar]
- Han BH, Zhou ML, Abousaleh F, Brendza RP, Dietrich HH, Koenigsknecht-Talboo J, Cirrito JR, Milner E, Holtzman DM, Zipfel GJ. Cerebrovascular dysfunction in amyloid precursor protein transgenic mice: contribution of soluble and insoluble amyloid-β peptide, partial restoration via γ-secretase inhibition. J Neurosci. 2008;28:13542–13550. doi: 10.1523/JNEUROSCI.4686-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao K, Ohnishi T, Hirata Y, Yamashita F, Mori T, Moriguchi Y, Matsuda H, Nemoto K, Imabayashi E, Yamada M, Iwamoto T, Arima K, Asada T. The prediction of rapid conversion to Alzheimer's disease in mild cognitive impairment using regional cerebral blood flow SPECT. Neuroimage. 2005;28:1014–1021. doi: 10.1016/j.neuroimage.2005.06.066. [DOI] [PubMed] [Google Scholar]
- Hong KW, Yoo SE, Yu SS, Lee JY, Rhim BY. Pharmacological coupling and functional role for CGRP receptors in the vasodilation of rat pial arterioles. Am J Physiol. 1996;270:H317–H323. doi: 10.1152/ajpheart.1996.270.1.H317. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Niwa K, Eckman C, Turner SK, Fischer E, Younkin S, Borchelt DR, Hsiao KK, Carlson GA. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci. 1999;2:157–161. doi: 10.1038/5715. [DOI] [PubMed] [Google Scholar]
- Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet. 2000;356:1627–1631. doi: 10.1016/s0140-6736(00)03155-x. [DOI] [PubMed] [Google Scholar]
- Jones MW, Errington ML, French PJ, Fine A, Bliss TV, Garel S, Charnay P, Bozon B, Laroche S, Davis S. A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat Neurosci. 2001;4:289–296. doi: 10.1038/85138. [DOI] [PubMed] [Google Scholar]
- Kitazono T, Heistad DD, Faraci FM. Role of ATP-sensitive K+ channels in CGRP-induced dilatation of basilar artery in vivo. Am J Physiol. 1993;265:H581–H585. doi: 10.1152/ajpheart.1993.265.2.H581. [DOI] [PubMed] [Google Scholar]
- Koehler RC, Roman RJ, Harder DR. Astrocytes and the regulation of cerebral blood flow. Trends Neurosci. 2009;32:160–169. doi: 10.1016/j.tins.2008.11.005. [DOI] [PubMed] [Google Scholar]
- Kornblum HI, Araujo DM, Annala AJ, Tatsukawa KJ, Phelps ME, Cherry SR. In vivo imaging of neuronal activation and plasticity in the rat brain by high resolution positron emission tomography (microPET) Nat Biotechnol. 2000;18:655–660. doi: 10.1038/76509. [DOI] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Li G, Larson EB, Sonnen JA, Shofer JB, Petrie EC, Schantz A, Peskind ER, Raskind MA, Breitner JC, Montine TJ. Statin therapy is associated with reduced neuropathologic changes of Alzheimer disease. Neurology. 2007;69:878–885. doi: 10.1212/01.wnl.0000277657.95487.1c. [DOI] [PubMed] [Google Scholar]
- Li G, Shofer JB, Rhew IC, Kukull WA, Peskind ER, McCormick W, Bowen JD, Schellenberg GD, Crane PK, Breitner JC, Larson EB. Age-varying association between statin use and incident Alzheimer's disease. J Am Geriatr Soc. 2010;58:1311–1317. doi: 10.1111/j.1532-5415.2010.02906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Cao D, Kim H, Lester R, Fukuchi K. Simvastatin enhances learning and memory independent of amyloid load in mice. Ann Neurol. 2006;60:729–739. doi: 10.1002/ana.21053. [DOI] [PubMed] [Google Scholar]
- Lloyd DJ, Helmering J, Cordover D, Bowsman M, Chen M, Hale C, Fordstrom P, Zhou M, Wang M, Kaufman SA, Véniant MM. Diabetic effects of 11β-HSD1 inhibition in a mouse model of combined diabetes, dyslipidemia and atherosclerosis. Diabetes Obes Metab. 2009;11:688–699. doi: 10.1111/j.1463-1326.2009.01034.x. [DOI] [PubMed] [Google Scholar]
- López J, Mendoza R, Cleva Villanueva G, Martínez G, Castillo EF, Castillo C. Participation of K+ channels in the endothelium-dependent and endothelium-independent components of the relaxant effect of rosuvastatin in rat aortic rings. J Cardiovasc Pharmacol Ther. 2008;13:207–213. doi: 10.1177/1074248408321716. [DOI] [PubMed] [Google Scholar]
- Maneen MJ, Cipolla MJ. Peroxynitrite diminishes myogenic tone in cerebral arteries: role of nitrotyrosine and F-actin. Am J Physiol Heart Circ Physiol. 2007;292:H1042–H1050. doi: 10.1152/ajpheart.00800.2006. [DOI] [PubMed] [Google Scholar]
- Mans RA, Chowdhury N, Cao D, McMahon LL, Li L. Simvastatin enhances hippocampal long-term potentiation in C57BL/6 mice. Neuroscience. 2010;166:435–444. doi: 10.1016/j.neuroscience.2009.12.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meilandt WJ, Yu GQ, Chin J, Roberson ED, Palop JJ, Wu T, Scearce-Levie K, Mucke L. Enkephalin elevations contribute to neuronal and behavioral impairments in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2008;28:5007–5017. doi: 10.1523/JNEUROSCI.0590-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AW, Tulbert CD, Busija DW. Rosuvastatin treatment reverses impaired coronary artery vasodilation in fructose-fed, insulin-resistant rats. Am J Physiol Regul Integr Comp Physiol. 2004;287:R157–R160. doi: 10.1152/ajpregu.00647.2003. [DOI] [PubMed] [Google Scholar]
- Mizuno M, Yamada K, Takei N, Tran MH, He J, Nakajima A, Nawa H, Nabeshima T. Phosphatidylinositol 3-kinase: a molecule mediating BDNF-dependent spatial memory formation. Mol Psychiatry. 2003;8:217–224. doi: 10.1038/sj.mp.4001215. [DOI] [PubMed] [Google Scholar]
- Mosconi L, Pupi A, De Leon MJ. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer's disease. Ann N Y Acad Sci. 2008;1147:180–195. doi: 10.1196/annals.1427.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolakakis N, Aboulkassim T, Ongali B, Lecrux C, Fernandes P, Rosa-Neto P, Tong XK, Hamel E. Complete rescue of cerebrovascular function in aged Alzheimer's disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor γ agonist. J Neurosci. 2008;28:9287–9296. doi: 10.1523/JNEUROSCI.3348-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolakakis N, Aboulkassim T, Aliaga A, Tong XK, Rosa-Neto P, Hamel E. Intact memory in TGF-β1 transgenic mice featuring chronic cerebrovascular deficit: recovery with pioglitazone. J Cereb Blood Flow Metab. 2011;31:200–211. doi: 10.1038/jcbfm.2010.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa K, Kazama K, Younkin SG, Carlson GA, Iadecola C. Alterations in cerebral blood flow and glucose utilization in mice overexpressing the amyloid precursor protein. Neurobiol Dis. 2002;9:61–68. doi: 10.1006/nbdi.2001.0460. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc Natl Acad Sci U S A. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, Younkin L, Younkin S, Carlson G, McEwen BS, Iadecola C. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci U S A. 2008;105:1347–1352. doi: 10.1073/pnas.0711568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Wang G, Zhou P, Zhou J, Pitstick R, Previti ML, Younkin L, Younkin SG, Van Nostrand WE, Cho S, Anrather J, Carlson GA, Iadecola C. Scavenger receptor CD36 is essential for the cerebrovascular oxidative stress and neurovascular dysfunction induced by amyloid-β. Proc Natl Acad Sci U S A. 2011;108:5063–5968. doi: 10.1073/pnas.1015413108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen K, Simonsen M, Østergaard SD, Munk OL, Rosa-Neto P, Olsen AK, Jensen SB, Møller A, Cumming P. Mapping the amphetamine-evoked changes in [11C]raclopride binding in living rat using small animal PET: modulation by MAO-inhibition. Neuroimage. 2007;35:38–46. doi: 10.1016/j.neuroimage.2006.11.038. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Huang YY, Paletzki RF, Bourtchouladze R, Scanlin H, Vronskaya S, Kandel ER. Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus-dependent spatial memory. Neuron. 2002;34:447–462. doi: 10.1016/s0896-6273(02)00684-0. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Ramirez C, Tercero I, Pineda A, Burgos JS. Simvastatin is the statin that most efficiently protects against kainate-induced excitotoxicity and memory impairment. J Alzheimers Dis. 2011;24:161–174. doi: 10.3233/JAD-2010-101653. [DOI] [PubMed] [Google Scholar]
- Rockwood K, Kirkland S, Hogan DB, MacKnight C, Merry H, Verreault R, Wolfson C, McDowell I. Use of lipid-lowering agents, indication bias, and the risk of dementia in community-dwelling elderly people. Arch Neurol. 2002;59:223–227. doi: 10.1001/archneur.59.2.223. [DOI] [PubMed] [Google Scholar]
- Sanchez-Mejia RO, Newman JW, Toh S, Yu GQ, Zhou Y, Halabisky B, Cissé M, Scearce-Levie K, Cheng IH, Gan L, Palop JJ, Bonventre JV, Mucke L. Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer's disease. Nat Neurosci. 2008;11:1311–1318. doi: 10.1038/nn.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Schwärzler F, Lütjohann D, von Bergmann K, Beyreuther K, Dichgans J, Wormstall H, Hartmann T, Schulz JB. Treatment with simvastatin in normocholesterolemic patients with Alzheimer's disease: a 26-week randomized, placebo-controlled, double-blind trial. Ann Neurol. 2002;52:346–350. doi: 10.1002/ana.10292. [DOI] [PubMed] [Google Scholar]
- Sparks DL, Sabbagh M, Connor D, Soares H, Lopez J, Stankovic G, Johnson-Traver S, Ziolkowski C, Browne P. Statin therapy in Alzheimer's disease. Acta Neurol Scand Suppl. 2006;185:78–86. doi: 10.1111/j.1600-0404.2006.00689.x. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Ayer R, Jadhav V, Chen W, Tsubokawa T, Zhang JH. Simvastatin attenuation of cerebral vasospasm after subarachnoid hemorrhage in rats via increased phosphorylation of Akt and endothelial nitric oxide synthase. J Neurosci Res. 2008;86:3635–3643. doi: 10.1002/jnr.21807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamboli IY, Barth E, Christian L, Siepmann M, Kumar S, Singh S, Tolksdorf K, Heneka MT, Lütjohann D, Wunderlich P, Walter J. Statins promote the degradation of extracellular amyloid β-peptide by microglia via stimulation of exosome-associated Insulin-degrading enzyme (IDE) secretion. J Biol Chem. 2010;285:37405–37414. doi: 10.1074/jbc.M110.149468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong XK, Nicolakakis N, Kocharyan A, Hamel E. Vascular remodeling versus amyloid β-induced oxidative stress in the cerebrovascular dysfunctions associated with Alzheimer's disease. J Neurosci. 2005;25:11165–11174. doi: 10.1523/JNEUROSCI.4031-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong XK, Nicolakakis N, Fernandes P, Ongali B, Brouillette J, Quirion R, Hamel E. Simvastatin improves cerebrovascular function and counters soluble amyloid-beta, inflammation and oxidative stress in aged APP mice. Neurobiol Dis. 2009;35:406–414. doi: 10.1016/j.nbd.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Wang H, Lynch JR, Song P, Yang HJ, Yates RB, Mace B, Warner DS, Guyton JR, Laskowitz DT. Simvastatin and atorvastatin improve behavioral outcome, reduce hippocampal degeneration, and improve cerebral blood flow after experimental traumatic brain injury. Exp Neurol. 2007;206:59–69. doi: 10.1016/j.expneurol.2007.03.031. [DOI] [PubMed] [Google Scholar]
- Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439–1443. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- Wong GH, Goeddel DV. Induction of manganous superoxide dismutase by tumor necrosis factor: possible protective mechanism. Science. 1988;242:941–944. doi: 10.1126/science.3263703. [DOI] [PubMed] [Google Scholar]
- Wu H, Lu D, Jiang H, Xiong Y, Qu C, Li B, Mahmood A, Zhou D, Chopp M. Simvastatin-mediated upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway, and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J Neurotrauma. 2008;25:130–139. doi: 10.1089/neu.2007.0369. [DOI] [PubMed] [Google Scholar]
- Yamin G. NMDA receptor-dependent signaling pathways that underlie amyloid beta-protein disruption of LTP in the hippocampus. J Neurosci Res. 2009;87:1729–1736. doi: 10.1002/jnr.21998. [DOI] [PubMed] [Google Scholar]
- Yang X, Askarova S, Lee JC. Membrane biophysics and mechanics in Alzheimer's disease. Mol Neurobiol. 2010;41:138–148. doi: 10.1007/s12035-010-8121-9. [DOI] [PubMed] [Google Scholar]
- Yau JL, Olsson T, Morris RG, Noble J, Seckl JR. Decreased NGFI-A gene expression in the hippocampus of cognitively impaired aged rats. Brain Res Mol Brain Res. 1996;42:354–357. doi: 10.1016/s0169-328x(96)00220-3. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Ha-Kawa S, Yoshimura M, Nobuhara K, Kinoshita T, Sawada S. Effectiveness of treatment with donepezil hydrochloride and changes in regional cerebral blood flow in patients with Alzheimer's disease. Ann Nucl Med. 2007;21:257–265. doi: 10.1007/s12149-007-0022-2. [DOI] [PubMed] [Google Scholar]
- Yudoh K, Karasawa R. Statin prevents chondrocyte aging and degeneration of articular cartilage in osteoarthritis (OA) Aging. 2010;2:990–998. doi: 10.18632/aging.100213. [DOI] [PMC free article] [PubMed] [Google Scholar]