

Abstract
17-β-Estradiol (E2) is a steroid hormone involved in neuroprotection against excitotoxicity and other forms of brain injury. Through genomic and nongenomic mechanisms, E2 modulates neuronal excitability and signal transmission by regulating NMDA and non-NMDA receptors. However, the mechanisms and identity of the receptors involved remain unclear, even though studies have suggested that estrogen G-protein-coupled receptor 30 (GPR30) is linked to protection against ischemic injury. In the culture cortical neurons, treatment with E2 and the GPR30 agonist G1 for 45 min attenuated the excitotoxicity induced by NMDA exposure. The acute neuroprotection mediated by GPR30 is dependent on G-protein-coupled signals and ERK1/2 activation, but independent on transcription or translation. Knockdown of GPR30 using short hairpin RNAs (shRNAs) significantly reduced the E2-induced rapid neuroprotection. Patch-clamp recordings revealed that GPR30 activation depressed exogenous NMDA-elicited currents. Short-term GPR30 activation did not affect the expression of either NR2A- or NR2B-containing NMDARs; however, it depressed NR2B subunit phosphorylation at Ser-1303 by inhibiting the dephosphorylation of death-associated protein kinase 1 (DAPK1). DAPK1 knockdown using shRNAs significantly blocked NR2B subunit phosphorylation at Ser-1303 and abolished the GPR30-mediated depression of exogenous NMDA-elicited currents. Lateral ventricle injection of the GPR30 agonist G1 (0.2 μg) provided significant neuroprotection in the ovariectomized female mice subjected to middle cerebral artery occlusion. These findings provide direct evidence that fast neuroprotection by estradiol is partially mediated by GPR30 and the subsequent downregulation of NR2B-containing NMDARs. The modulation of DAPK1 activity by GPR30 may be an important mediator of estradiol-dependent neuroprotection.
Introduction
The steroid hormone estradiol [17-β-estradiol (E2)] regulates many functionally unrelated processes in numerous tissues. Estradiol may act through different estrogen receptors (ERs) and activate distinct secondary messenger pathways with different time courses and downstream mechanisms (Heldring et al., 2007; Woolley, 2007). In the classical mechanism, estradiol acts via soluble intracellular α or β receptors (ERα or ERβ). Once activated, these receptors translocate to the nucleus where they function as ligand-dependent transcription factors (Hall et al., 2001). In contrast, fast nongenomic effects are mediated by classic receptors (ERα and ERβ) and specific G-protein-coupled receptors [G-protein-coupled receptor 30 (GPR30) and ER-X] (Revankar et al., 2005; Abe et al., 2008) that regulate ligand-gated ion channels and neurotransmitter transporters (Carrer et al., 2003; Dominguez et al., 2007; Chu et al., 2009; Grassi et al., 2009).
The GPR30 receptor is reported to be a novel estrogen receptor uniquely localized to the endoplasmic reticulum (Funakoshi et al., 2006). This receptor is widely distributed and has numerous physiologic or pathologic functions in the CNS (Dominguez et al., 2007; Chu et al., 2009; Grassi et al., 2009). Through genomic and nongenomic mechanisms, E2 modulates neuronal excitability and rapidly increases signal transmission by regulating both NMDA and non-NMDA glutamate receptors (NMDARs) (Wong and Moss, 1992; Foy et al., 1999). The GPR30 receptor may act together with intracellular estrogen receptors to activate cell-signaling pathways to promote neuron survival after global ischemia (Hazell et al., 2009; Gingerich et al., 2010; Lebesgue et al., 2010). However, the role of GPR30 in neuroprotection against excitotoxic neuronal death induced by NMDA exposure remains unknown.
The accumulation of glutamate and excessive stimulation of glutamate receptors induce potent excitotoxicity in CNS (Du et al., 1997). The NMDA subtype of glutamate receptor plays a key role in mediating glutamate excitotoxicity because of its high calcium permeability (Arundine and Tymianski, 2003; Hardingham and Bading, 2003; Brennan et al., 2009; Stanika et al., 2009). Differences in the subunit composition of the NMDA receptor accounts for neuronal survival or death in some models (Liu et al., 2007). NR2A- and NR2B-containing NMDARs have been linked to different intracellular cascades and have different roles in synaptic plasticity, and the receptor isoforms have different cytotoxic potentials. Indeed, excitotoxicity is triggered by the selective activation of NMDARs containing the NR2B subunit (Krapivinsky et al., 2003; Zhou and Baudry, 2006; Liu et al., 2007; Stanika et al., 2009). In addition, overactivation of extrasynaptic NMDARs is linked to excitotoxic death (Sattler et al., 2000; Hardingham et al., 2002; Wahl et al., 2009), which imply that their postsynaptic location plays an important role in determining neuronal survival or death (Stanika et al., 2009). This study aims to investigate the possible protective efficacy of GPR30 in neuronal apoptosis induced by NMDAR activation and to elucidate the underlying mechanisms. The fast activation of GPR30 attenuated the glutamate neurotoxicity by depressing the phosphorylation of NR2B-containing NMDARs.
Materials and Methods
Materials.
All chemicals were purchased from Sigma unless stated otherwise. G1 was purchased from Cayman Chemical. (R)-N-(3,4-difluoro-2-(2fluoro-4-iodophenylamino)-6-methoxyphenyl)-1-(2,3-dihydroxypropyl)cyclopropane-1-sulfonamide (RDEA119) and JTP-74057 were purchased from Selleck. Anti-NR1, anti-NR2A, anti-NR2B, and anti-phosphorylated NR2B at S1303 were purchased from Millipore. Anti-phosphorylated death-associated protein kinase 1 (DAPK1) at Ser308 (p-Ser308) and anti-cadherin were purchased from Sigma. Anti-DAPK1 and anti-phosphorylated NR2B at Tyr1472 were purchased from Cell Signaling Technology. Anti-pERK1/2, anti-caspase-3, and anti-Bcl-2 were purchased from Santa Cruz Biotechnology. Anti-GPR30 was purchased from Abcam. All of the chemicals and reagents used were commercially available and of standard biochemical quality.
Primary mouse cortical neuronal culture.
The experiments were performed on C57BL/6J mice (embryonic, 18-d-old mice of both genders). The Animal Care and Use Committee of the Fourth Military Medical University approved all of the animal protocols used. Cultured prefrontal cortex neurons were prepared as described previously (Wang et al., 2008). Briefly, the prefrontal cortex was dissected, minced, and trypsinized for 15 min using 0.125% trypsin (Invitrogen). The cells were seeded onto either 24-well plates containing glass coverslips (Fisher Scientific), 100 mm dishes, or 96-well plates precoated with 50 μg/ml poly-d-lysine (Sigma) in water and grown in Neurobasal-A medium (Invitrogen) supplemented with B27 and 2 mm GlutaMax (Invitrogen). In the B27/Neurobasal medium, glial growth was reduced to <0.5% of the nearly pure neuronal population, as assessed using immunocytochemistry for glial fibrillary acidic protein and neuron-specific enolase (Brewer et al., 1993). The cultures were incubated at 37°C in 95% air/5% carbon dioxide with 95% humidity. The cultures were used for experiments on the 10th day in vitro (DIV 10). These cultures were washed twice using Mg2+-free extracellular solution containing the following (in mm): 140 NaCl, 3 KCl, 2 CaCl2, 10 HEPES, and 10 glucose, adjusted to pH 7.2–7.3 with NaOH and osmotic pressure 290 ± 5 with sucrose (Shen et al., 2008). To detect the protective effects mediated by specific ER subtypes, the cultures were pretreated with E2 or ER-specific agonists for 15 min. NMDA (200 μm) and glycine (20 μm) were simultaneously added to the same medium with the specific estrogen receptor regulators for another 30 min. The cells were washed twice and returned to the original culture medium for 24 h.
Cell viability analysis.
A 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyl tetrazolium bromide (MTT) assay was used to detect cell viability as described previously (Shen et al., 2008). Neurons were cultured in 96-well plates at a density of 8 × 104 cells per well. MTT was dissolved in DMEM and added to each well for incubation at 37°C for 4 h at a final concentration of 0.5 mg/ml. Then, the medium was replaced with 150 μl of dimethyl sulfoxide (DMSO). The optical density (OD) was recorded on a Universal Microplate Reader (Elx 800; Bio-TEK Instruments) at 570 nm (630 nm as a reference). Cell viability was expressed as a percentage of the control value. All data are expressed as the mean ± SEM of three separate experiments with six wells in each experiment.
Flow cytometric analysis.
Apoptosis analyses were performed as described previously (Behbahani et al., 2005). Apoptosis and cell viability were measured using annexin V-FITC (Assay Designs) and propidium iodide (PI) double staining. Primary cultures were harvested and washed with Ca2+ and Mg2+-free PBS. The cells were diluted in 100 μl of 1× annexin-binding buffer per assay and incubated with annexin V-FITC and PI for 15 min in the dark at room temperature. Right after, 400 μl of 1× binding buffer was added. The stained cells were immediately analyzed using flow cytometry. Annexin V-FITC versus PI was used to identify viable cell populations (annexin V−, PI−), early-phase apoptotic cells (annexin V+, PI−), late-phase apoptotic cells (annexin V+, PI+), and necrotic cells (annexin V−, PI+). Cell fluorescence was analyzed on a FACScan flow cytometer using CELLQuest software (Becton Dickinson).
Immunocytochemistry.
The cultured cortical neurons were fixed with ice-cold 4% paraformaldehyde in PBS, pH 7.4, for 30 min, blocked with 5% BSA in PBS for 2 h, incubated overnight with primary anti-GPR30 (1:300) or anti-DAPK1 antibodies (1:200) at 4°C, and then incubated with Cy3-conjugated goat anti-rabbit IgG (Sigma) diluted to 1:100 in blocking solution. Coverslips were mounted onto slides with 50% glycerin. The stained samples were photographed and were analyzed using an Olympus Fluoview FV100.
Western blot analysis.
Western blot analysis was performed as described previously (Chen et al., 2008). Equal amounts of protein (50 μg) from the cultures were separated and electrotransferred onto PDVF membranes (Invitrogen), which were probed with antibodies against pERK1/2 (dilution ratio, 1:200), ERK1/2 (dilution ratio, 1:400), NR1 (dilution ratio, 1:100), NR2A (dilution ratio, 1:200), and NR2B (dilution ratio, 1:500). NR2B phosphorylation at Ser1303 (dilution ratio, 1:1000), NR2B phosphorylation at Tyr1472 (dilution ratio, 1:500), DAPK1 (dilution ratio, 1:500), and pDAPK1 (dilution ratio, 1:500) were also assayed, with β-actin (dilution ratio, 1:10000) or cadherin (dilution ratio 1:500) as the loading control. The amount of proteins (100 μg) were loaded for detection of caspase-3 (dilution ratio, 1:400) and Bcl-2 (dilution ratio, 1:400). For data quantification, band intensity was expressed relative to the loading control (β-actin). The membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (anti-rabbit/anti-mouse IgG for the primary antibodies), and bands were visualized using an ECL system (PerkinElmer).
Surface biotinylation assay.
Surface NR2A and NR2B receptors were detected using a biotinylation assay, followed by Western blot analysis for antibodies were previously described (Wang et al., 2008). Cell monolayers were washed, and the cells were incubated in sulfo-NHS-LC-biotin (0.3 mg/ml in cold PBS; Pierce) for 30 min. Surface biotinylation was stopped by removal of the solution, and the samples were incubated in 10 mm ice-cold glycine in PBS for 20 min. The cells were then washed three times with cold PBS and lysed with RIPA buffer containing (in mm) 20 HEPES, pH 7.4, 100 NaCl, 1 EGTA, 1 Na3VO4, 50 NaF, and 1 mm 4-(2-aminoethyl)-benzenesulfonylfluoride hydrochloride, with 1% NP-40, 1% deoxycholate, 0.1% SDS, 10 mg/ml leupeptin, and 1 mg/ml aprotinin. The biotinylated proteins were precipitated with 100 ml of ImmunoPure Immobilized Streptavidin (Pierce), separated on 4–12% SDS-PAGE gels, and transferred onto polyvinylidene fluoride membranes. The membranes were probed with anti-NR2A (dilution ratio, 1:200), NR2B (dilution ratio, 1:500), and NR2B phosphorylation at Ser1303 (dilution ratio, 1:1000) antibodies. The membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (anti-rabbit/anti-mouse IgG for the primary antibodies), and bands were visualized using an ECL system (PerkinElmer).
Transfection of short hairpin RNAs.
Lentivirus (pGLV-U6-GFP)-encoding short hairpin RNAs (shRNAs) for GPR30 and DAPK1 were prepared by GenePharma. The shRNA sequences were designed based on the GPR30 (Hsieh et al., 2007) and DAPK1 (Tu et al., 2010) sequences as reported previously. The shRNA sequence for GPR30 is 5′-CATGTACAGCAGCG TCTTC-3′, whereas, the shRNA sequence for DAPK1 is 5′-CATCATG CAGAGTGAGACA-3′. A negative control shRNA was purchased from Shanghai Genepharma. Cells were transfected with the lentivirus for 24 h according to the directions provided by the manufacturer. The staining was photographed at 96 h and was analyzed using an Olympus Fluoview FV100. The knockdown efficiency at the protein level was assessed at 96 h using Western blot analysis.
Electrophysiological recording on the cultured neurons.
The cultured cortical neurons were placed in a recording chamber for visualization of whole-cell patch-clamp recordings using an Axopatch-200B amplifier. The cultures were superfused with Mg2+-free artificial CSF (ACSF). NMDA receptor-mediated currents were induced by cell superfusion of NMDA in Mg2+-free ACSF containing glycine (1 μm), CNQX (20 μm), and picrotoxin (100 μm). TTX (1 μm) was added into the ACSF during culture recordings to block synaptic activity. The neurons were clamped at −30 mV. The NMDA currents were analyzed by measuring the peak amplitude. The gravity-fed multibarrel perfusion system (ALA-VM4; Scientific Instruments) was controlled by the pClamp 10.0 (Molecular Devices) software. The tip of the multibarrel was positioned 200–300 μm away from the neuronal soma, allowing the flow from one of the barrels to bathe the entire neuron continuously. When a test solution is not applied, the neuron was continuously superfused with extracellular solution. The access resistance at 15–30 MΩ was monitored throughout the experiment. Data were discarded if access resistance changed by >15% during an experiment.
Middle cerebral artery occlusion and lateral cerebral ventricle infusion.
Female C57BL/6J mice 6 to 8 weeks old were used for the middle cerebral artery occlusion (MCAO). Mice were housed with food and water available ad libitum in a colony room with controlled temperature (24 ± 2°C) and humidity (50 to 60%), and a 12 h light/dark cycle. The experimental protocol was approved by the Animal Care and Use Committee of the Fourth Military Medical University. One week after ovariohysterectomy, the animals were subjected to MCAO as described previously (Liu et al., 2011). Briefly, the mice were anesthetized with a nitrous oxide/oxygen/isoflurane mixture (69/30/1%) administered through an inhalation mask. The right carotid bifurcation was exposed, and a silicone-coated 8-0 filament was inserted through the common carotid artery and advanced (9.0–10.0 mm) gently to occlude the middle cerebral artery. After 1 h of transient occlusion, cerebral blood flow was restored by removing the nylon suture. The common carotid arteries of the mice in the sham group were surgically exposed without subsequent MCAO. Drugs were administered immediately upon reperfusion. Lateral cerebral ventricle infusion was performed as described previously (Stennicke et al., 1998). A burr hole was drilled in the pericranium, 1.5 mm lateral to the sagittal suture and 0.5 mm posterior to the coronal suture. A 33-gauge needle attached to a 10 μl Hamilton syringe was stereotactically inserted into the left lateral ventricle, 2.5 mm under the surface of cortex. For lateral ventricle infusion, 2 μl of G1 (0.1 μg/μl) or 2 μl of 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one (PD98059; 0.25 μg/μl) was injected at a rate of 1 μl/min. The needle was withdrawn after 2 min. The hole was sealed with bone wax, the wound was sutured, and the animals were allowed to recover. Temperature was maintained at 37 ± 0.5°C throughout the surgery.
Neurobehavioral evaluation.
Neurobehavioral evaluation and infarct volume assessment were performed at 24 h after reperfusion. The neurological deficit was determined according to the previous report (Longa et al., 1989). The neurological evaluation was scored using a five-point scale: 0, no neurological deficit; 1, failure to extend left forepaw fully; 2, circling to the left; 3, inability to bear weight on the left; 4, no spontaneous walking with depressed level of consciousness.
Infarct volume assessment.
After neurological evaluation, the mice were reanesthetized with an overdose of pentobarbital sodium and then decapitated. The brains were rapidly removed and cooled in ice-cold saline for 10 min. Coronal sections (2 mm) were cut and immersed in 2% 2,3,5-triphenyltetrazolium chloride at 37°C for 30 min and then transferred to 10% buffered formalin solution for 24 h fixation. The brain slices were photographed using a digital camera (Kodak DC240) connected to a computer. The unstained areas were defined as infarcts and were measured using image analysis software (Adobe Photoshop 7.0 CS for Windows). The infarct volume was calculated by measuring the unstained area in each slice and multiplied by the slice thickness (2 mm) and then summing all six slices.
Nissl staining.
After neurological evaluation, the brains (n = 6 in each group) were perfused with cold 4% paraformaldehyde in 0.01 m PBS, pH 7.4. Brain blocks containing dorsal hippocampal and the prefrontal cortex area were embedded in paraffin. Coronal sections (5 μm) were cut using a Leica CM1800 Cryostat (Leica Microsystems) and then stained with 0.1% cresyl violet for 2 min. The sections were evaluated using light microscopy.
Results
Activation of ERα, but not ERβ, showed protective effect on NMDA-induced cell loss
To test the involvement of specific ER subtypes in neuroprotection, dissociated cortical neuronal cultures were pretreated first with agonists to various ER subtypes for 15 min, followed by NMDA (200 μm) and glycine (20 μm) treatment in the same medium for 30 min. The cells were returned to the original culture medium for another 24 h (Fig. 1A). As expected, exposure to NMDA caused a significant loss of cultured neurons (F(10, 77) = 10.73, p < 0.01, Tukey's test; p < 0.01, NMDA vs control; Fig. 1B). Cytotoxicity evoked by NMDA was blocked by pretreatment with the NR2B-selective antagonist (αR,βS)-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidinepropanol maleate (Ro25-6981) (0.3 μm; F(10, 77) = 10.73, p < 0.01, Tukey's test; p < 0.01, Ro25–6981 vs NMDA; Fig. 1B). Consistent with previous findings (Bae et al., 2000; Dominguez et al., 2009), the nonspecific ER agonist, E2 (1, or 10 nm), attenuated the neuronal death based on the dose induced by NMDA (F(10, 77) = 10.73, p < 0.05, Tukey's test; p = 0.025, 1 nm E2 vs NMDA; p < 0.01, 10 nm E2 vs NMDA; Fig. 1B). In addition, the ERα-specific agonist propyl pyrazole triol (PPT, 10 nm), and not the ERβ-specific agonist diarylpropiolnitrile (DPN, 1 or 10 nm), reduced the NMDA-induced cell death, which indicates that the ERα-receptor is involved in E2 neuroprotection (F(10, 77) = 10.73, p < 0.01, Tukey's test; p = 0.033, 10 nm PPT vs NMDA; Fig. 1B). AN observation was made wherein the treatment of E2 (1 nm), PPT (10 nm), or G1 (1 nm) alone had no effect on cell viability without NMDA exposure (Fig. 1C). Furthermore, the neuroprotective effect of E2 (1 nm) was partially reduced by pretreatment with 7α, 17β-[9-[(4,4,5,5,5-pentafluoropentyl)sulfinyl]nonyl]estra-1,3,5(10)-triene-3,17-diol (ICI182780, 10 min, 1 μm), an estrogen receptor antagonist used to block the effects mediated by classical ERα and ERβ receptor/transcription factors, On the other hand, ICI182780 blocked the neuroprotection of ERα agonist (F(8, 63) = 6.77, p < 0.01, Tukey's test; p = 0.152, 1 nm E2 plus ICI vs 1 nm E2; p = 0.038, PPT plus ICI vs PPT; Fig. 1D). ICI182780 marginally affected cell viability (Fig. 1D). These data indicate that other ER subtypes, such as the GPR30 receptor, may mediate the fast neuroprotective action of E2.
Figure 1.

Activation of ER subtypes contribute to the neuron viability. Cultures were used for experiments on DIV 10 and cell viability was determined by MTT assay. A, Cells were pretreated with E2, G1, PPT, DPN, or Ro25–6981 for 15 min and then treated with NMDA (200 μm) and glycine (20 μm) for another 30 min. One day later, MTT assay was performed. B, Cytotoxic effects of NMDA were prevented by pretreatment with E2 (1 or 10 nm), G1 (1 or 10 nm), PPT (10 nm), or Ro25–6981 (0.3 μm), but not DPN. C, Treatment of E2 (1 nm), PPT(10 nm), or G1(1 nm) alone without NMDA did not affect cell viability. D, Pretreatment of ICI182780 (10 min, 1 μm) partially reduced neuroprotection of E2 (1 nm), but totally blocked the effect of PPT (10 nm). However, ICI182780 did not affect the neuroprotection of G1. ICI182780 itself had marginal effect on the cell viability. E, Protective effects of E2 (1 nm), PPT (10 nm), or G1 (1 nm) were not blocked by actinomycin-D (act) or anisomycin (ani). $p < 0.05, $$p < 0.01 compared with control; *p < 0.05, **p < 0.01 compared with NMDA treatment alone. NS, No significance.
Activation of GPR30 mediated the fast protective effect on NMDA-induced cell loss
The neuroprotective efficacy of GPR30 activation was tested. Application of G1 (1 or 10 nm), an agonist of GPR30, induced a significant protective action against NMDA-induced neurotoxicity (F(10, 77) = 10.73, p < 0.01, Tukey's test; p < 0.01, 1 and 10 nm G1 vs NMDA; Fig. 1B), whereas the classical ER antagonist ICI182780 failed to block this GPR30 receptor-mediated neuroprotection (Fig. 1D). To investigate whether the genomic actions of GPR30 were involved in neuroprotection, we tested the effects of actinomycin-D, a transcription inhibitor, and anisomycin, a translation inhibitor, on G1-induced neuroprotection. The neurons were pretreated with actinomycin-D (20 μm) or anisomycin (10 μm) for 30 min to suppress transcription or protein synthesis before the addition of G1. As shown in the Figure 1E, treatment with E2 (1 nm), PPT (10 nm), or G1 (1 nm) suppressed the neuronal death induced by NMDA, which suggests that the neuroprotection does not depend solely on the transcription or translation.
To further test the involvement of GPR30 in neuroprotection, we investigated the effect of the treatments on the expression and trafficking of GPR30 in cortical neurons. As shown in Figure 2, the cytoplasm of cultured neurons was immunopositive for GPR30. Furthermore, exposure to NMDA or G1 did not result in the translocation of GPR30 to the nucleus.
Figure 2.
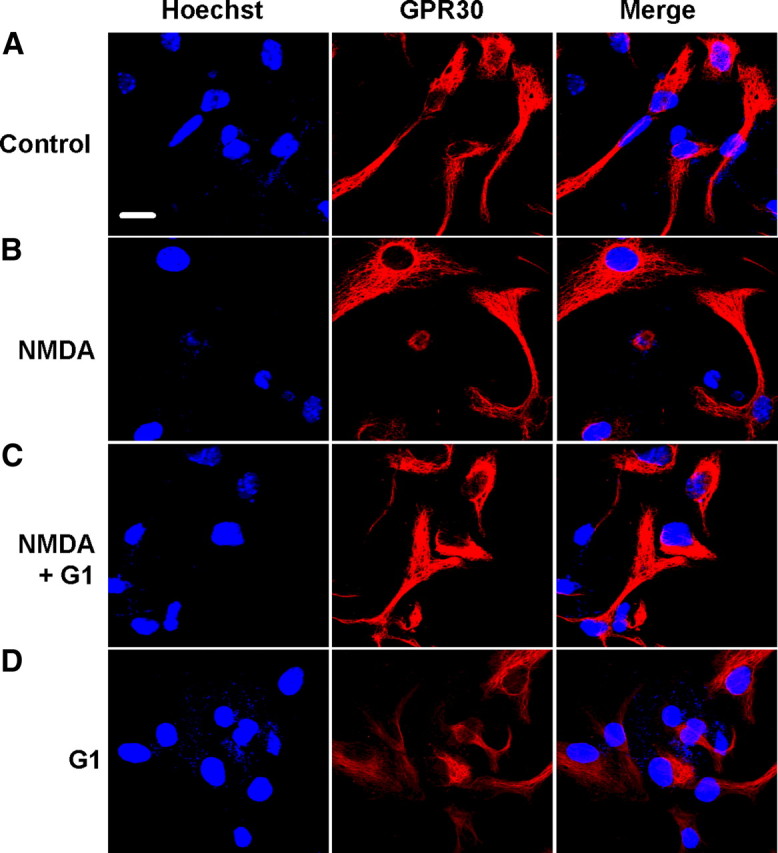
Expression of GPR30 in the cortical neurons. One-day-old NMDA treatment cultures were stained with Hoechst (blue) and anti-GPR30 (red) to identify GPR30 and cellular localization. A–D, Representative confocal images showed the distribution of GPR30 immunoreactivity in the control (A), NMDA (200 μm, B), NMDA plus G1 (200 μm and 1 nm[scap], respectively; C), and G1 (1 nm, D) treatment. GPR30 was localized in the intracellular cytoplasm, including the nuclear envelope in some cases. Expression and distribution of GPR30 were not changed in the NMDA- and G1-treated neurons. Scale bar: 10 μm.
Attenuation of apoptosis and necrosis by G1
To distinguish further the features of apoptotic versus necrotic cells, flow cytometry analyses were performed to compare the viability of the primary cultures with NMDA stimuli (250 μm) in the absence or presence of G1. Annexin V and PI double staining was used to separate the viable, early-phase apoptotic, late-phase apoptotic, and necrotic cells. As shown in Figure 3A, a typical representative dot plot analysis of the primary neuron cultures revealed 3.2% necrotic (annexin V−, PI+), 2.2% late-phase apoptotic (annexin V+, PI+), and 2.1% early-phase apoptotic (annexin V+, PI−) events. However, the analysis of cells cultured with NMDA stimuli showed a significant increase in the number of necrotic cells (23.4%), late-phase apoptotic cells (9.2%), and early-phase apoptotic cells (11.2%) compared with the saline controls (p < 0.01, t test; Fig. 3B–D). Treatment with 1 nm G1 significantly attenuated neuronal injury by NMDA stimuli. As shown in Figure 3, a marked decrease in the number of necrotic cells was observed (9.8%), late-phase apoptotic cells (6.2%), and early-phase apoptotic cells (4.5%) compared with the NMDA treated group (p < 0.05, t test; Fig. 3B–D). Therefore, ICI182780 (1 μm) did not block G1 activity. The data show that neuronal death is the prevailing form of necrosis and apoptosis under the present in vitro conditions and that G1 protects the cells by attenuation of necrosis, late-phase, and early-phase apoptosis induced by NMDA.
Figure 3.

Flow cytometry analyses of the apoptosis and necrosis. Cells were pretreated with G1 (1 nm) or G1 (1 nm) plus ICI182780 (1 μm) for 15 min, and then treated with NMDA (250 μm) and glycine (20 μm) for another 30 min. Annexin V and PI double staining was used to separate viable, early-apoptotic, late-apoptotic, and necrotic cells by flow cytometric analysis in 1-d-old cultures. A, Typical representative dot plots of the distribution of viable (annexin V−, PI−), necrotic (annexin V−, PI+), late-phase apoptotic (annexin V+, PI+), and early-phase apoptotic (annexin V+, PI−) cells are shown in control (top left), NMDA (top right), NMDA plus G1 (bottom left), and NMDA plus G1 plus ICI182780 (bottom right) treatments. B–D, Percentages of necrotic (B), late-apoptotic (C) and early-apoptotic (D) neurons were calculated from the total neurons. G1 (1 nm) significantly attenuated the necrosis, late-phase apoptosis, and early-phase apoptosis by NMDA stimuli, whereas ICI182780 (1 μm) could not block G1 activity. Values represent mean ± SEM of three independent experiments. $p < 0.05, $$p < 0.01 compared with control; *p < 0.05 compared with NMDA treatment alone.
To exclude the possible nonspecific action of G1 and to confirm GPR30 involvement, we knocked down the GPR30 via shRNA transfection. The neurons expressed the green fluorescent protein (GFP) when they were transfected successfully with GPR30 shRNA. As shown in the Figure 4A, the GFP-positive neurons were 89.4 ± 6.2% after 96 h of shRNA transfection. This resulted in a 71.9 ± 6.0% reduction in the GPR30 protein band intensity (p < 0.01, t test; Fig. 4B). Reduction in GPR30 levels prevented G1-induced neuroprotection and slightly inhibited the protective effects of E2 (F(7, 56) = 26.09, p < 0.01, Tukey's test; p = 0.011, G1 treated, shRNA vs negative; p = 0.591, E2 treated, shRNA vs negative; Fig. 4C). In contrast, transfection with a nonsilencing RNA sequence (negative control) did not block the protective efficacy of G1 and E2 (Fig. 4C). These data demonstrate that GRP30 mediates G1-induced neuroprotection and part of the E2-mediated neuroprotective effect.
Figure 4.
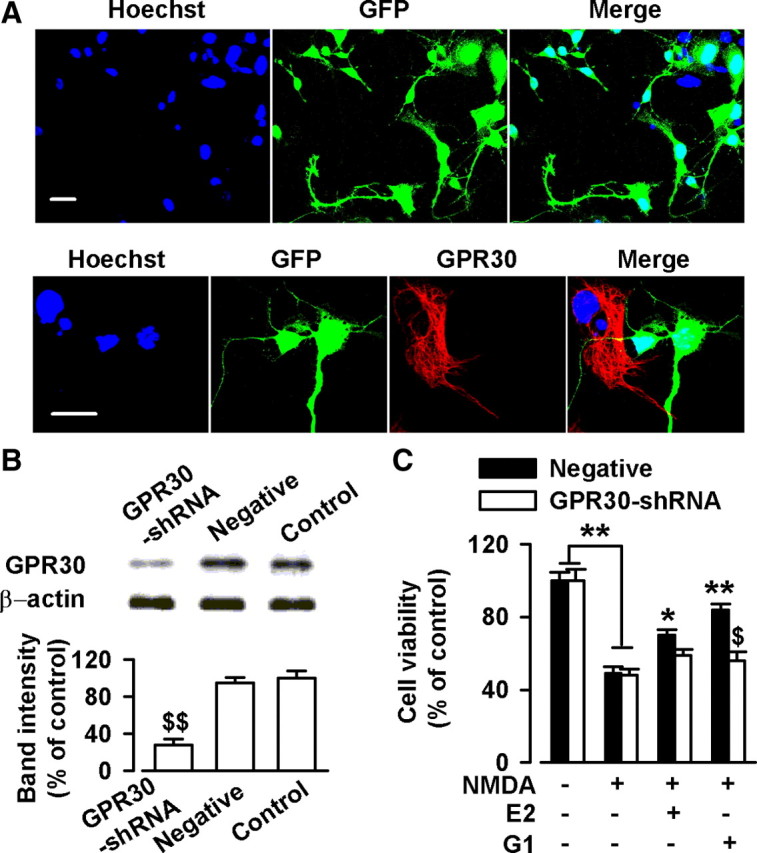
Knockdown of GPR30 depressed estrogen-induced neuroprotection. Cells were treated with GPR30-shRNA for 24 h. A, Confocal images showing the transfection rate of the GPR30-shRNA. Top, Nuclei were stained with Hoechst33258 (blue). GFP-positive neurons indicated the successful knockdown of GPR30 by transfection with GPR30-shRNA. Bottom, Typical neurons showing GPR30-shRNA transfection (green) and nontransfection (red). Scale bars: 20 μm. B, Transfection of GPR30-shRNA resulted in a reduction in GPR30 protein levels; the negative shRNA did not lead to the change of GPR30 expression. $$p < 0.01 compared with the control and negative shRNA. C, Transfection of GPR30-shRNA blocked the neuroprotection of G1 and slightly inhibited the effects of E2. $p < 0.05 compared with G1 treated negative shRNA; *p < 0.05, **p < 0.01 compared with NMDA treatment alone.
The ERK1/2-mediated pathway is involved in the neuroprotective effects of GPR30
ERK activation contributes to neuronal cell survival in certain neurotoxicity models (Han and Holtzman, 2000; Jin et al., 2002). To further characterize the cellular mechanisms of GPR30-mediated neuroprotection, we treated cultured cortical neurons with G1 in the presence of Bordetella pertussis toxin (PTX; 100 ng/ml), a G-protein blocker, and PD98059 and 1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene (U0126), the two ERK signaling inhibitors. As shown in Figure 5A, blockage of G-protein signaling by PTX completely blocked the protective effect of G1 (F(8, 63) = 9.59, p < 0.01, Tukey's test; p = 0.036, NMDA plus G1 plus PTX vs NMDA plus G1; Fig. 5A). In addition, treatment of the cultures with PD98059 (25 μm) or U0126 (10 μm) also inhibited the protective effect of G1 (F(8, 63) = 9.59, p < 0.01, Tukey's test; p = 0.033, NMDA plus G1 plus PD vs NMDA plus G1; p = 0.042, NMDA plus G1 plus U0126 vs NMDA plus G1; Fig. 5A), whereas PTX (100 ng/ml), PD98059 (25 μm), or U0126 (10 μm) alone had no effects on cell viability (Fig. 5A).
Figure 5.

Activation of ERK1/2 by G1. A, One day after NMDA treatment, MTT assay was performed to detect cell viability. Cotreated cultures with G1, PTX (100 ng/ml), PD98059 (25 μm), or U0126 (10 μm) blocked the protective effect of G1 on the cell viability. PTX, PD98059, or U0126 itself had no effects on the cell viability. B, Thirty minutes after NMDA treatment, the cells were lysed and Western blot analysis was performed using antibodies to detect ERK1/2 and p-ERK1/2 levels. C, E2 (1 nm) and G1 (1 nm) significantly increased phosphorylated level of ERK1/2. ICI182780 (1 μm) had no effects on the ERK1/2 phosphorylation induced by G1; however, ICI182780 slightly depressed ERK1/2 phosphorylation induced by E2. D, E2 (1 nm) and G1 (1 nm) significantly increased the ratio of phosphorylated ERK1/2 to total ERK1/2 levels. ICI182780 (1 μm) had no effects on the change in ratio induced by G1. The data were pooled from five independent experiments. E, Thirty minutes after the E2, G1, and NMDA treatments, the cells were lysed. Western blot analysis was performed using antibodies to detect ERK1/2 and p-ERK1/2 levels. F, NMDA treatment alone had no effects on the p-ERK1/2 levels; however, E2 (1 nm) or G1 (1 nm) alone significantly increased the p-ERK1/2 and ratio of phosphorylated ERK1/2 to total ERK1/2 levels compared to the NMDA treatment alone and the control group. The data were pooled from five independent experiments. #p < 0.05 compared with G1 and NMDA cotreatment; $p < 0.05, $$p < 0.01 compared with control; *p < 0.05, **p < 0.01 compared with NMDA treatment alone.
Furthermore, treatment with G1 (1 nm) or E2 (1 nm) significantly increased the levels of phosphorylated ERK1/2 (p-ERK1/2; F(5, 24) = 6.72, p < 0.01, Tukey's test; p < 0.01, NMDA plus G1 vs NMDA; p = 0.021, NMDA plus E2 vs NMDA; Fig. 5B,C) and the ratio of p-ERK1/2 to total ERK1/2 within 30 min of NMDA administration (F(5, 24) = 4.82, p < 0.01, Tukey's test; p = 0.018, NMDA plus G1 vs NMDA; p = 0.032, NMDA plus E2 vs NMDA; Fig. 5D), but did not change the total ERK1/2 levels (Fig. 5B). These results are consistent with the previous reports that indicated that GPR30 causes the ERK1/2 activation (Filardo et al., 2000; Chu et al., 2009). ICI182780 application (1 μm) did not block the ERK1/2 phosphorylation induced by G1 and slightly depressed the ERK1/2 phosphorylation induced by E2 (Fig. 5B,D). NMDA exposure (200 μm, 30 min) alone did not affect the ERK1/2 phosphorylation level. However, E2 (1 nm) or G1 (1 nm) alone significantly increased the p-ERK1/2 (F(3, 16) = 17.82, p < 0.01, Tukey's test; p < 0.01, G1 vs control; p < 0.01, G1 vs NMDA; Fig. 5E,F) and the ratio of phosphorylated ERK1/2 to the total ERK1/2 levels compared with that in the NMDA treatment alone and the control group (F(3, 16) = 9.46, p < 0.01, Tukey's test; p < 0.01, G1 vs control; p < 0.01, G1 vs NMDA; Fig. 5E,F). These data indicate that GPR30 stimulation might mediate neuroprotection through ERK phosphorylation and downstream ERK-dependent signaling.
GPR30-mediated antiapoptotic effects in culture
The possible relationship between NMDA-induced cell death and potential intracellular mediators was also assessed by determining the levels of the antiapoptotic protein Bcl-2 and the proapoptotic precursor protein procaspase-3. Procaspase-3 is an inactive form of active caspase-3 and a major physiologic target of caspase-8 and caspase-9 (Stennicke et al., 1998). Initiator caspase-8 directly activates procaspase-3 into active caspase-3, which causes the dialysis of Bcl-2 (Kirsch et al., 1999). Thus, high procaspase-3 levels indicate low apoptotic activity. Neurons were pretreated with anisomycin (10 μm) for 30 min to suppress protein synthesis before the addition of G1. Western blot analysis revealed a significant decrease in Bcl-2 (F(5, 24) = 8.23, p < 0.01, Tukey's test; p < 0.01, NMDA vs control; p < 0.01, NMDA plus G1 vs NMDA; p = 0.013, NMDA plus E2 vs NMDA; Fig. 6B) and procaspase-3 levels (F(5, 24) = 7.58, p < 0.01, Tukey's test; p < 0.01, NMDA vs control; p = 0.015, NMDA plus G1 vs NMDA; p = 0.048, NMDA plus E2 vs NMDA; Fig. 6C), and increase in the active caspase-3 levels (F(5, 24) = 31.24, p < 0.01, Tukey's test; p < 0.01, NMDA vs control; p < 0.01, NMDA plus G1 vs NMDA; p < 0.01, NMDA plus E2 vs NMDA; Fig. 6D) in cultured neurons after exposure to NMDA. However, both of E2 (1 nm) and G1 (1 nm) significantly reversed these alterations following NMDA stimulation (Fig. 6A–D). ICI182780 treatment (1 μm) slightly attenuated the protective effect of E2, but did not alter G1-mediated neuroprotection (Fig. 6A–D). To exclude the possibility that G1 may have nonspecific effects, we performed Western blot analysis on GPR30-shRNA-transfected cells. The results show that GPR30-shRNA transfection significantly decreased the effects of G1 and E2 on Bcl-2 and procaspase-3 levels compared with those in cultured neurons in the negative control after exposure to NMDA (Bcl-2, F(3, 16) = 12.1, p < 0.01, Tukey's test; p < 0.01, G1 treated, shRNA vs negative; p = 0.046, G1 vs E2 in shRNA treated; procaspase-3, F(3, 16) = 10.29, p < 0.01, Tukey's test; p < 0.01, G1 treated, shRNA vs negative; p = 0.027, G1 vs E2 in shRNA treated; caspase-3, F(3, 16) = 14.65, p < 0.01, Tukey's test; p < 0.01, G1 treated, shRNA vs negative; p < 0.01, G1 vs E2 in shRNA treated; Fig. 6E, right column, F). However, G1 and E2 marginally affected the expression of Bcl-2, procaspase-3, and caspase-3 in the cells without exposure to NMDA (Fig. 6E, left column). These data demonstrate that the antiapoptotic activity of GPR30 is associated with inhibiting the degradation of Bcl-2 and procaspase-3 in response to the excitotoxicity.
Figure 6.
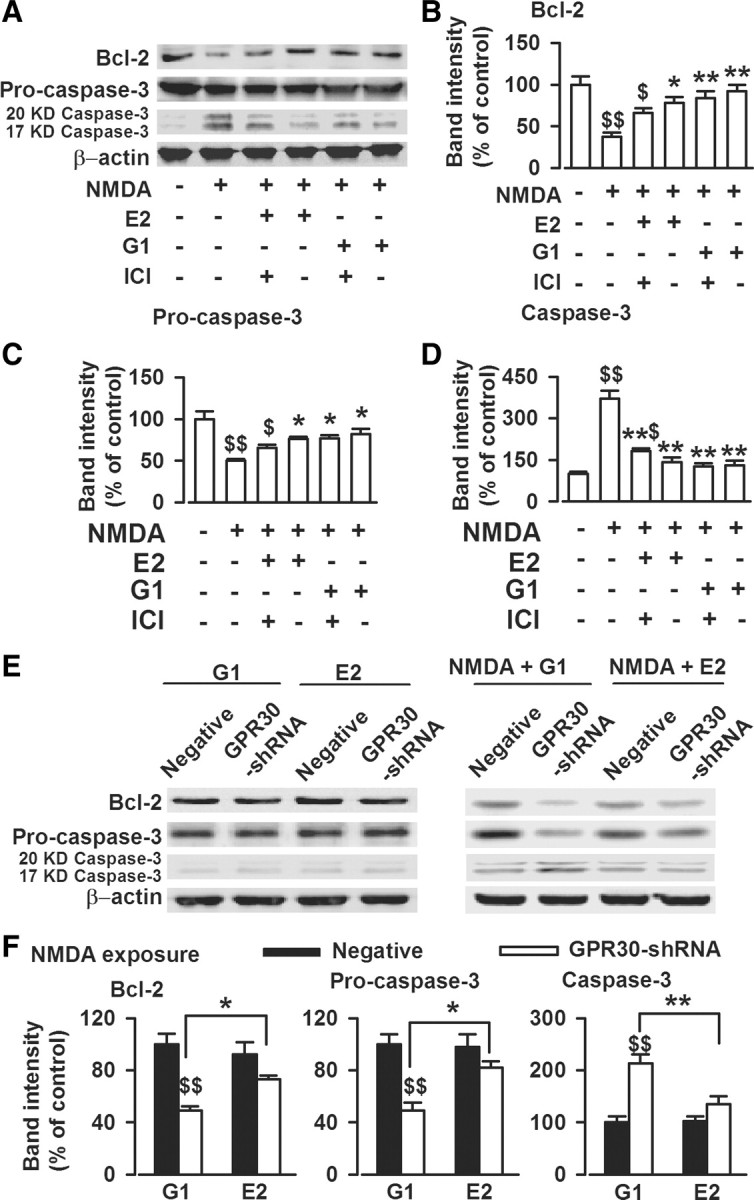
Activation of GPR30 on the expression of apoptosis-related proteins. A, One day after NMDA treatment, the cells were lysed, and Western blot analysis was performed using antibodies to detect Bcl-2, procaspase-3, and caspase-3 (20 and 17 kDa) levels. B, E2 (1 nm) or G1 (1 nm) significantly reversed the decreased Bcl-2 levels caused by NMDA exposure. ICI182780 (1 μm) slightly attenuated the effects of E2, whereas it did not alter the actions of G1. C, E2 (1 nm) and G1 (1 nm) significantly reversed the decreased procaspase-3 levels caused by NMDA exposure. D, E2 (1 nm) and G1 (1 nm) significantly reversed the increased caspase-3 levels caused by NMDA exposure. ICI182780 (1 μm) slightly attenuated the effects of E2, whereas it could not alter the actions of G1. B–D, $p < 0.05, $$p < 0.01 compared with control; *p < 0.05, **p < 0.01 compared with NMDA treatment alone. E, GPR30-shRNA transfection significantly decreased the effects of G1 or E2 on Bcl-2, procaspase-3, and active caspase-3 (20 and 17 kDa) compared with nonsilencing RNA transfection in cultured neurons exposed to NMDA (right). However, G1 and E2 had marginal actions on the expression of Bcl-2, procaspase-3, and active caspase-3 in the cells not exposed to NMDA (left). F, The summary data from the right column of E. GPR30-shRNA transfection significantly attenuated the protection of G1 in a larger scale than that of E2, as shown by greater reduction of in Bcl-2 and procaspase-3. The data were pooled from five independent experiments. $$p < 0.01 compared with G1 treated negative shRNA; *p < 0.05, **p < 0.01 compared between G1 and E2 treatments.
GPR30-mediated depression of NMDA-elicited currents
NMDARs play a key role in mediating glutamate excitotoxicity because of their high calcium permeability (Arundine and Tymianski, 2003; Hardingham and Bading, 2003; Brennan et al., 2009; Stanika et al., 2009), which suggests that the neuroprotective effect of G1 is partly due to GPR30-mediated modulation of NMDA receptor signaling. To test this concept, we examined the effect of G1 on the electrophysiologic response to exogenous NMDA superfusion at the soma of cultured neurons. As shown in Figure 7, exogenous NMDA (50 μm) elicited an inward current that was reduced to 72% of control in the presence of E2 (1 nm, 15 min; p < 0.05, t test; Fig. 7A3,B). The experiment was repeated on neurons treated with 1 nm G1 for 15 min. Similar to E2, G1 depressed the peak NMDA-elicited current to 76% that of the control (p < 0.05, t test; Fig. 7A2,B). This inhibitory effect on NMDA-elicited currents by G1 was abolished in the presence of the NR2B antagonist Ro25–6981 (0.3 μm; Fig. 7A4,C). This recording paradigm was repeated on neurons transfected with GPR30-shRNA. The depression of NMDA currents by G1 was completely blocked in these neurons (Fig. 7A5,D). Furthermore, G1-induced depression of NMDA currents was abolished in the presence of the ERK1/2 inhibitor PD98059 (25 μm; Fig. 7A6,E). The data indicate that ERK1/2 activation mediates the G1-induced depression of NR2B-containing NMDARs.
Figure 7.

Activation of GPR30 blocks NMDA-elicited currents. A, Whole-cell patch-clamp recordings were performed to detect the direct effect of G1 on the NMDA-elicited currents; the recording was stable for at least 30 min (A1). A2–A6, Representative recordings of currents elicited by a 0.5 min superfusion of 50 μm NMDA at baseline and in the presence of G1 (A2), E2 (A3), pretreatment with of Ro25–6981 (A4), GPR30-shRNA transfection (A5), and PD98059 (A6) from the culture neurons. B, Either 1 nm E2 (n = 6) or 1 nm G1 (n = 8) depressed exogenous NMDA-elicited currents. *p < 0.05 compared with the control. C, Pretreatment with Ro25–6981 (0.3 μm) totally blocked the effects of G1 on NMDA-elicited currents (n = 7). D, GPR30-shRNA transfection blocked the effects of G1 on NMDA-elicited currents (n = 8). E, PD98059 (25 μm) blocked the effects of G1 on NMDA-elicited currents (n = 6).
Attenuation of phosphorylation of NR2B by GPR30
Evidence suggests that NR2A- and NR2B-containing NMDARs have different roles in NMDAR-mediated excitotoxicity (Liu et al., 2007; von Engelhardt et al., 2007; Xu et al., 2009). The identity of the NMDAR isoforms involved in GPR30-induced survival was confirmed using Western blot analysis. As shown in Figure 8, short-term GPR30 activation by G1 (1 nm, 45 min) did not affect the total NR1, NR2A, and NR2B subunit expression (Fig. 8A,B). The function of NMDARs is regulated through their phosphorylation (Lu et al., 1998; Tu et al., 2010). Neither NMDA nor G1 treatment altered the levels of phosphorylated-Tyr-1472 of the NR2B (p-Tyr-1472) subunit in the culture system. However, G1 significantly depressed the elevated NR2B subunit phosphorylation at Ser-1303 (p-Ser1303) induced by exposure to NMDA (F(3, 16) = 7.57, p < 0.01, Tukey's test; p < 0.01, NMDA vs control; p < 0.01, G1 vs NMDA; p = 0.037, NMDA plus G1 vs NMDA; Fig. 8C,D). Increased surface insertion strengthens NMDAR function. Detection of the membrane surface expression of NMDAR subunits revealed that the surface expression of NR2A and NR2B was unaffected by G1 and NMDA treatment. However, G1 (1 nm) reversed the surface levels of NR2B-p-Ser1303 induced by NMDA treatment (F(3, 16) = 8.96, p < 0.01, Tukey's test; p < 0.01, NMDA vs control; p = 0.010, G1 vs NMDA; p = 0.031, NMDA plus G1 vs NMDA; Fig. 8E,F).
Figure 8.

Depression of NR2B subunit phosphorylation by GPR30. Cells were pretreated with G1 (1 nm) for 15 min and then cotreated with NMDA (200 μm) and glycine (20 μm) for another 30 min. A, Six hours after NMDA treatment, Western blot analysis was performed using antibodies against NR1, NR2A, and NR2B. B, GPR30 activation by G1 (1 nm, 45 min) did not affect the total NR1, NR2A, and NR2B expression. C, Western blot analysis was performed to detect the phosphorylated NR2B. D, GPR30 activation by G1 (1 nm, 45 min) depressed the levels of phosphorylated NR2B at Ser-1303 (p-Ser1303-NR2B), but not at (p-Tyr1472-NR2B). E, Detection of the membrane surface expression of NMDARs subunits. F, Only the membrane surface expression of NR2B-p-Ser1303 and not the total levels of NR2A and NR2B were affected by the G1 and NMDA treatments. The data were pooled from five independent experiments. $$p < 0.01 compared with control; *p < 0.05, **p < 0.01 compared with NMDA treatment alone.
Role of DAPK1 in GPR30-mediated depression of NR2B
DAPK1 is identified as a key component in the neuronal death signaling cascade, which phosphorylates the NR2B subunit at Ser-1303, resulting in irreversible neuronal death (Tu et al., 2010). DAPK1 is predominantly phosphorylated on Ser308 at the basal state (p-DAPK1), and p-DAPK1 dephosphorylation allows DAPK1 to bind with the NR2B subunit (Shohat et al., 2001; Tu et al., 2010). As shown in the Figure 9A, exposure to NMDA resulted in a significant increase in the p-DAPK1 dephosphorylation (F(4, 20) = 27.39, p < 0.01, Tukey's test; p < 0.01, NMDA vs control) and an increase in NR2B subunit phosphorylation at Ser-1303 (F(4, 20) = 22.94, p < 0.01, Tukey's test; p < 0.01, NMDA vs control; Fig. 9A). G1 (1 nm) significantly reversed this alteration following NMDA stimulation and kept the DAPK1 in the phosphorylated basal state (p-DAPK1, F(4, 20) = 27.39, p < 0.01, Tukey's test; p < 0.01, NMDA plus G1 vs NMDA; NR2B, F(4, 20) = 22.94, Tukey's test; p < 0.01, NMDA plus G1 vs NMDA). However, PD98059 (25 μm) abolished the effects of G1 on p-DAPK1 dephosphorylation (F(4, 20) = 27.39, p < 0.01, Tukey's test; p < 0.01, NMDA plus G1 plus PD vs NMDA plus G1) and NR2B subunit phosphorylation at Ser-1303 (F(4, 20) = 22.94, p < 0.01, Tukey's test; p < 0.01, NMDA plus G1 plus PD vs NMDA plus G1; Fig. 9A). To avoid the nonspecific effects of PD980059, we also tested the two novel selective MEK inhibitors, RDEA119 and JTP-74057, and found that RDEA119 (1 μm) or JTP-74057 (0.1 μm) abolished the effects of G1 on the DAPK1 (F(5, 24) = 26.53, p < 0.01, Tukey's test; p = 0.017, NMDA plus G1 plus RDEA vs NMDA plus G1; p < 0.01, NMDA plus G1 plus JTP vs NMDA plus G1) and NR2B phosphorylation (F(5, 24) = 19.35, p < 0.01, Tukey's test; p = 0.014, NMDA plus G1 plus RDEA vs NMDA plus G1; p = 0.046, NMDA plus G1 plus JTP vs NMDA plus G1; Fig. 9B). This result suggests that the decreased DAPK1 dephosphorylation caused by G1 through ERK activation contributes to the neuroprotective efficacy of GPR30.
Figure 9.

Depression of DAPK1 dephosphorylation by GPR30. A, Thirty minutes after NMDA treatment, the cells were lysed, and Western blot analysis was performed to detect the p-DAPK1 and DAPK1 levels. NMDA exposure resulted in the significant dephosphorylation of p-DAPK1 into DAPK1. G1 (1 nm) inhibited p-DAPK1 dephosphorylation, whereas PD98059 (25 μm) abolished the effects of G1 on DAPK1 and NR2B phosphorylation. The data were pooled from five independent experiments. $$p < 0.01 compared with the control; **p < 0.01 compared with NMDA treatment alone; &&p < 0.01 compared with G1 plus NMDA. B, RDEA119 (1 μm) or JTP-74057 (0.1 μm) abolished the effects of G1 on DAPK1 and NR2B phosphorylation. The data were pooled from five independent experiments. $$p < 0.01 compared with the control; *p < 0.05, **p < 0.01 compared with NMDA treatment alone; &p < 0.05, &&p < 0.01 compared with G1 plus NMDA. C, Confocal images showing the transfection rate of the DAPK1-shRNA. Top, Nuclei were stained with Hoechst33258 (blue). GFP-positive neurons indicated the successful knockdown of DAPK1 by transfection of DAPK1-shRNA. Bottom, Typical neurons showing DAPK1-shRNA transfection (green) and nontransfection (red). Scale bars: 20 μm. D, DAPK1-shRNA transfection resulted in a reduction in DAPK1 protein levels; the negative shRNA did not change of DAPK1 expression. **p < 0.01 compared with the control and negative shRNA. E, DAPK1 knockdown significantly depressed the phosphorylation of the NR2B subunit at Ser-1303 induced by NMDA exposure. F, DAPK1-shRNA transfection prevented the neurotoxicity of NMDA exposure. $$p < 0.01 compared with the control; **p < 0.01 compared with NMDA treatment alone and negative shRNA. G–H, DAPK1-shRNA transfection abolished the effects of G1 on NMDA-elicited currents (n = 7).
To further explore the signaling from GPR30 to NMDARs, we knocked down the DAPK1 using shRNA transfection. As shown in the Figure 9C, the GFP-positive neurons were 91.2 ± 7.5% after 96 h of transfection with shRNAs, which resulted in a 79 ± 4.6% reduction in DAPK1 protein band intensity (p < 0.01, t test; Fig. 9D). DAPK1 knockdown significantly blocked the phosphorylation of the NR2B subunit at Ser-1303 by NMDA (Fig. 9E), attenuated the excitotoxicity induced by NMDA exposure (F(5, 24) = 13.32, p < 0.01, Tukey's test; p < 0.01, DAPK1-shRNA plus NMDA vs NMDA; Fig. 9F), and abolished the GPR30-mediated depression of exogenous NMDA-elicited currents (Fig. 9G,H).
Prevention of the ischemia injury by GPR30 in vivo
The neuroprotective efficacy of GPR30 against ischemia-reperfusion injury by MCAO was evaluated through infarct volume, neurological deficit, and Nissl staining. As shown in Figure 10A, MCAO resulted in large infarcts in the brain. Lateral cerebral ventricle infusion of G1 (2 μl, 0.1 μg/μl) significantly reduced the infarct volume (F(3, 16) = 15.10, p < 0.01, Tukey's test; p < 0.01, G1 vs vehicle; p < 0.01, G1 plus PD vs G1; Fig. 10A) and neurological deficit score (F(3, 16) = 6.66, p < 0.01, Tukey's test; p < 0.01, G1 vs vehicle; p = 0.046, G1 plus PD vs G1; Fig. 10B). Nissl staining showed neuronal damage in the prefrontal cortex and hippocampus CA1 region after MCAO (Fig. 10C). The injured neurons showed shrunken cell bodies accompanied by shrunken and pyknotic nuclei. G1 (2 μl, 0.1 μg/μl) significantly reduced neuronal damage and preserved morphology (prefrontal cortex, F(4, 20) = 176.72, p < 0.01, Tukey's test; p < 0.01, G1 vs vehicle; p < 0.01, G1 plus PD vs G1; hippocampus, F(4, 20) = 164.12, p < 0.01, Tukey's test; p < 0.01, G1 vs vehicle; p < 0.01, G1 plus PD vs G1; Fig. 10C–E). The GPR30-mediated protection was blocked by simultaneous PD98059, an ERK1/2 inhibitor (Fig. 10A–E).
Figure 10.

Neuroprotection by GPR30 in vivo. A, Representative photographs of infarct volume after the MACO and treatments with vehicle (10% DMSO, 2 μl), G1 (0.2 μg), and PD98059 (0.5 μg). n = 6 in each group. B, G1 (0.2 μg) significantly reduced the neurological deficit score after MCAO. PD98059 (0.5 μg) reversed the effect of G1. C, Nissl staining showing the morphologic neuronal changes in the hippocampus CA1 region and prefrontal cortex. Scale bars: 50 μm. D, G1 (0.2 μg) significantly reduced the injury in the hippocampus CA1 region. E, G1 (0.2 μg) significantly reduced the injury in the prefrontal cortex. n = 6 in each group. &&p < 0.01 compared with the sham; $$p < 0.01compared with the vehicle; *p < 0.05, **p < 0.01 compared with G1.
Discussion
The present study yielded four novel findings: (1) GPR30 activation markedly attenuated NMDA excitotoxicity by activating the G-protein-coupled ERK signaling cascade. (2) GPR30 activation significantly depressed NMDAR-mediated currents. (3) Short-term GPR30 activation depressed NR2B subunit phosphorylation at Ser-1303 by inhibiting p-DAPK1 dephosphorylation. (4) Lateral ventricle infusion of GPR30 agonist prevented the ischemia-reperfusion injury induced by MCAO and activation of ERK1/2 signaling involved in GPR30 neuroprotection.
Activation of GPR30 attenuates excitotoxicity by exposure to NMDA
Acute estradiol application was sufficient to attenuate the neuronal injury induced by NMDA receptor stimulation. However, the classic ER (ERα and ERβ) antagonist ICI182780 partially blocked the E2-mediated neuroprotection, which suggests that other ER subtypes are also involved. GPR30 is a transmembrane G-protein-coupled receptor localized in the endoplasmic reticulum that binds estradiol with high affinity and activates multiple intracellular signaling pathways (Revankar et al., 2005). We used G1, a nonsteroidal, high-affinity (Ki of 11 nm), selective agonist of GPR30 and shRNA knockdown to evaluate the actions of GPR30 on cell viability. In this study, 1 nm G1 is sufficient to reach maximal GPR30 activation. Furthermore, the effects of G1 were greater than that of estradiol at a similar concentration. This may be explained by the highly selective affinity of G1 toward GPR30 compared with ERα and ERβ (Bologa et al., 2006), and the faster actions of G1 were compared with the effects of estradiol in the short time treatment (45 min). We also verified the involvement of GPR30 in neuroprotection by shRNA knockdown to exclude the possible nonspecific action of G1. Furthermore, the neuroprotection mediated by GPR30 was rapid and independent of transcription and translation. First, the effects were limited to ∼45 min and were not accompanied by the nuclear translocation of receptors. Second, the neuroprotection by G1 and E2 was not abolished by pretreatment with transcription and translation inhibitors.
Requirements of ERK signaling in GPR30 neuroprotection
GPR30-mediated rapid signaling events were sensitive to pertussis toxin, implying the GPR30 coupling to particular members of the G-protein superfamily (Revankar et al., 2005; Ding et al., 2009). Depending on the duration, magnitude, and subcellular localization of ERK activation, this kinase pathway controls or modulates a plethora of cell responses, which includes proliferation, migration, differentiation, and cell death (Murphy and Blenis, 2006). In the present study, four ERK signaling inhibitors were used. The significant neuroprotection by GPR30 was associated with increased ERK activation and was abolished by the blockade of the upstream mitogen-activated protein kinase cascade, thereby demonstrating that ERK activation by GPR30 is required to protect the neurons against NMDA insult.
GPR30 acts independently from the classic ER in stimulating adenylyl cyclase via Gαs (Filardo et al., 2002) and ERK1/2 via the PTX-sensitive Gβγ pathway (Filardo et al., 2000). The current findings suggest that GPR30 signals may take effect through the Gβγ pathway and ERK1/2 signaling cascades in cortical neurons. However, possible “cross talk” between cAMP and ERK cascades following GPR30 or ERs stimulation is not well documented (Prossnitz et al., 2008). Subsequent studies are required to reveal the complete GPR30-dependent signaling pathway and the roles of adenylyl cyclase and ERK activation in the GRP30-mediated protection of cortical neurons.
Depression of dephosphorylation of DAPK1 by GPR30
NMDAR overactivation is a critical step in the glutamate-induced excitotoxicity of CNS neurons. Both NR2A- and NR2B-containing NMDARs are linked to different intracellular cascades and have distinct roles in cell survival and excitotoxicity. The membrane distribution of NMDARs also influences excitotoxicity. Synaptic NMDARs have antiapoptotic activities, whereas extrasynaptic NMDARs reportedly activate signs that promote neuronal death following ischemia (Hardingham et al., 2002; Tu et al., 2010). Furthermore, NR1/NR2B receptors are known to be expressed predominantly at extrasynaptic sites (Tovar and Westbrook, 1999). In the present study, GPR30 activation depressed the NMDA-elicited currents, and the NR2B antagonist, Ro25–6981, eliminated its inhibitory function. Thus, GPR30 may reduce NR2B receptor conductance or depress the recruitment of NR2B subunits to reduce injurious Ca2+ influx. Further examination showed that GPR30 activation depressed NR2B subunit phosphorylation at Ser-1303. DAPK1 is identified as a key component of the neuronal death signaling cascade that the phosphorylates NR2B subunit at Ser-1303, resulting in irreversible neuronal death (Tu et al., 2010).
Ca2+/calmodulin (CaM)-dependent protein kinase II (CaMKII) is also one of the key kinases that bind to the NR2B subunit at Ser-1303 (Omkumar et al., 1996). The interaction between CaMKII and NR2B may be critical for synaptic plasticity (Omkumar et al., 1996; Bayer et al., 2001). LTP-inducing NMDA-receptor stimulation also leads to CaMKII translocation to excitatory synapses (Shen and Meyer, 1999; Shen et al., 2000; Bayer et al., 2001), which is thought to be mediated by Ca2+/CaM-induced direct binding of CaMKII to the NMDA-receptor subunit GluN2B. However, DAPK1 is considered a signal amplifier of NMDA receptors at extrasynaptic sites for mediating brain damage in stroke (Tu et al., 2010). Thus, the location of synaptic or extrasynaptic NMDARs determines their antiapoptotic activities or promotion of neuronal death (Hardingham et al., 2002; Tu et al., 2010).
DAPK1 is predominantly phosphorylated on Ser308 at the basal state (p-DAPK1), and p-DAPK1 dephosphorylation allows DAPK1 to bind with the NR2B subunit (Shohat et al., 2001; Tu et al., 2010). DAPK1 activation occurs via dephosphorylation at Ser-308 (Jin et al., 2006). A constitutively active DAPK1 phosphorylates the NR2B subunit at Ser-1303, which in turn enhances NR1/NR2B receptor channel conductance (Tu et al., 2010). GPR30 activation depresses p-DAPK1 dephosphorylation, and subsequently depresses NR2B subunit phosphorylation. Thus, GPR30 can be considered a signaling depressor of extrasynaptic NMDAR-mediated excitotoxicity.
Depression of NMDARs by GPR30
Numerous studies reported that 17-β-estradiol pretreatment increases NMDAR-mediated synaptic currents in CA1 pyramidal cells (Woolley et al., 1997; Smith and McMahon, 2005, 2006; Barneda-Zahonero et al., 2009; Snyder et al., 2011). Estradiol may enhance NMDAR function in the hippocampus, increasing the number of synaptic NMDARs at CA3–CA1 synapses (Zamani et al., 2004) or resulting from the recruitment of NR2B-containing NMDARs to synapses (Snyder et al., 2011). These studies were performed in vivo, and the time of E2 pretreatment is from 32 h to 1 week. The enhanced NMDAR-mediated EPSP or EPSC by E2 reflects the intrasynaptic NMDAR number or function. Studies have found that E2 must be elevated for >24 h for the heightened LTP to occur (Smith and McMahon, 2005). In the present study, a cultured cortical neuron system was used in vitro, and the E2 and G1 treatment lasted only 45 min. Short-term GPR30 activation by G1 (1 nm, 45 min) did not affect total NR1, NR2A, and NR2B subunit expression (Fig. 8A,B). Instead, it depressed the elevated NR2B subunit phosphorylation at Ser-1303 (p-Ser1303) induced by NMDA exposure (Fig. 8C,D). This result is consistent with the reports that acute treatment with E2 inhibits NMDA-induced intracellular Ca2+ increases (Kurata et al., 2004). Together, the discrepancy of NMDARs regulation by E2 can be attributed to the following reasons: (1) Different protocols were used in the experiments, i.e., in vivo or in vitro. (2) The time of the E2 treatment varied from 45 min to weeks. (3) Synaptic or extrasynaptic NMDAR functions are involved in the process. (4) Recordings are performed to detect NMDAR-mediated synaptic transmission in brain sections or the exogenous NMDA-elicited current in the cultures.
Proposed neuroprotective signaling through GPR30
GPR30 is a G-protein-coupled receptor that mediates ERK1/2 phosphorylation (Filardo et al., 2000, 2002). Upon agonist binding, GPR30 activates heterotrimeric G-proteins, which activates multiple effectors, including adenylyl cyclase and Src. The latter appears to be involved in the transactivation of epidermal growth factor receptors (EGFRs) via a G-protein-dependent pathway (Rozengurt, 2007). As shown in the present study, the ERK inhibitors inhibited the protective effect of G1. ERK inhibition prevented GPR30-mediated NMDA current reduction (Fig. 7A6,E) and depression of NR2B ser-1303 phosphorylation (Fig. 9A). Furthermore, the knockdown of DAPK1 prevented GPR30-mediated NMDA current reduction (Fig. 9F,G). Given its autoinhibitory mechanism, DAPK1 activation requires the dephosphorylation of Ser308, and the Ca2+/calmodulin-binding ERK-signaling pathways are involved in DAPK1 action regulation (Anjum et al., 2005; Chen et al., 2005). Together, the activation of ERK1/2 by GPR30 keeps the DAPK1 in the inactive phosphorylated form (p-DAPK1) within the signaling pathway. This leads to less active DAPK1 binding with NR2B, and then phosphorylates NR2B at Ser-1303 (Fig. 11). The decrease in NR2B subunits relieves the calcium influx as the phosphorylated NR2B at Ser-1303 enhances NMDAR function, which causes the Ca2+ overload. However, it does not exclude that GPR30 directly activates the PI3K/Akt survival signaling cascades (Lebesgue et al., 2009).
Figure 11.

Schematic of GPR30-mediated signaling. GPR30, a G-protein-coupled receptor, activates heterotrimeric G-proteins, which in turn activates multiple effectors, including adenylyl cyclase (resulting in cAMP production), the phosphorylation of ERK1/2, and Src. The latter appears to be involved in the activation of transactivation of EGFRs via a G-protein-dependent pathway. The activated ERK by GPR30 agonist inhibits p-DAPK1 from dephosphorylating to the active form (DAPK1). This leads to the inhibition of active DAPK1 binding with the NR2B, and then phosphorylated NR2B at Ser-1303. The phosphorylated NR2B at Ser-1303 enhances the function of NMDARs, which causes the overload of Ca2+. The GPR30 is also shown to directly activate the PI3K/Akt survival signaling cascades.
In summary, the current study uncovers a novel mechanism underlying GPR30-mediated neuroprotection that is associated with decreased NR2B-containing NMDARs. GPR30 stimulation by estradiol represents a rapid signal transduction mechanism necessary for neuronal function under both physiologic and pathologic conditions.
Footnotes
This work was supported by NSF of China Grants 31070923, 2008ZXJ09004-023, and 2009ZX09103-111, and the Program for New Century Excellent Talents in University.
The authors declare no competing financial interests.
References
- Abe H, Keen KL, Terasawa E. Rapid action of estrogens on intracellular calcium oscillations in primate luteinizing hormone-releasing hormone-1 neurons. Endocrinology. 2008;149:1155–1162. doi: 10.1210/en.2007-0942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anjum R, Roux PP, Ballif BA, Gygi SP, Blenis J. The tumor suppressor DAP kinase is a target of RSK-mediated survival signaling. Curr Biol. 2005;15:1762–1767. doi: 10.1016/j.cub.2005.08.050. [DOI] [PubMed] [Google Scholar]
- Arundine M, Tymianski M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium. 2003;34:325–337. doi: 10.1016/s0143-4160(03)00141-6. [DOI] [PubMed] [Google Scholar]
- Bae YH, Hwang JY, Kim YH, Koh JY. Anti-oxidative neuroprotection by estrogens in mouse cortical cultures. J Korean Med Sci. 2000;15:327–336. doi: 10.3346/jkms.2000.15.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barneda-Zahonero B, Miñano-Molina A, Badiola N, Fadó R, Xifró X, Saura CA, Rodríguez-Alvarez J. Bone morphogenetic protein-6 promotes cerebellar granule neurons survival by activation of the MEK/ERK/CREB pathway. Mol Biol Cell. 2009;20:5051–5063. doi: 10.1091/mbc.E09-05-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- Behbahani H, Rickle A, Concha H, Ankarcrona M, Winblad B, Cowburn RF. Flow cytometry as a method for studying effects of stressors on primary rat neurons. J Neurosci Res. 2005;82:432–441. doi: 10.1002/jnr.20634. [DOI] [PubMed] [Google Scholar]
- Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, Oprea TI, Prossnitz ER. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. 2006;2:207–212. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- Brennan AM, Won Suh S, Joon Won S, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci. 2009;12:857–863. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Carrer HF, Araque A, Buño W. Estradiol regulates the slow Ca2+-activated K+ current in hippocampal pyramidal neurons. J Neurosci. 2003;23:6338–6344. doi: 10.1523/JNEUROSCI.23-15-06338.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Wang WJ, Kuo JC, Tsai HC, Lin JR, Chang ZF, Chen RH. Bidirectional signals transduced by DAPK-ERK interaction promote the apoptotic effect of DAPK. EMBO J. 2005;24:294–304. doi: 10.1038/sj.emboj.7600510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Liu JC, Zhang XN, Guo YY, Xu ZH, Cao W, Sun XL, Sun WJ, Zhao MG. Down-regulation of NR2B receptors partially contributes to analgesic effects of Gentiopicroside in persistent inflammatory pain. Neuropharmacology. 2008;54:1175–1181. doi: 10.1016/j.neuropharm.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Chu Z, Andrade J, Shupnik MA, Moenter SM. Differential regulation of gonadotropin-releasing hormone neuron activity and membrane properties by acutely applied estradiol: dependence on dose and estrogen receptor subtype. J Neurosci. 2009;29:5616–5627. doi: 10.1523/JNEUROSCI.0352-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Gros R, Limbird LE, Chorazyczewski J, Feldman RD. Estradiol-mediated ERK phosphorylation and apoptosis in vascular smooth muscle cells requires GPR 30. Am J Physiol Cell Physiol. 2009;297:C1178–C1187. doi: 10.1152/ajpcell.00185.2009. [DOI] [PubMed] [Google Scholar]
- Dominguez R, Liu R, Baudry M. 17-Beta-estradiol-mediated activation of extracellular-signal regulated kinase, phosphatidylinositol 3-kinase/protein kinase B-Akt and N-methyl-D-aspartate receptor phosphorylation in cortical synaptoneurosomes. J Neurochem. 2007;101:232–240. doi: 10.1111/j.1471-4159.2006.04360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez R, Hu E, Zhou M, Baudry M. 17beta-estradiol-mediated neuroprotection and ERK activation require a pertussis toxin-sensitive mechanism involving GRK2 and beta-arrestin-1. J Neurosci. 2009;29:4228–4238. doi: 10.1523/JNEUROSCI.0550-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Bales KR, Dodel RC, Hamilton-Byrd E, Horn JW, Czilli DL, Simmons LK, Ni B, Paul SM. Activation of a caspase 3-related cysteine protease is required for glutamate-mediated apoptosis of cultured cerebellar granule neurons. Proc Natl Acad Sci U S A. 1997;94:11657–11662. doi: 10.1073/pnas.94.21.11657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Bland KI, Frackelton AR., Jr Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14:1649–1660. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Frackelton AR, Jr, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. 2002;16:70–84. doi: 10.1210/mend.16.1.0758. [DOI] [PubMed] [Google Scholar]
- Foy MR, Xu J, Xie X, Brinton RD, Thompson RF, Berger TW. 17beta-estradiol enhances NMDA receptor-mediated EPSPs and long-term potentiation. J Neurophysiol. 1999;81:925–929. doi: 10.1152/jn.1999.81.2.925. [DOI] [PubMed] [Google Scholar]
- Funakoshi T, Yanai A, Shinoda K, Kawano MM, Mizukami Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem Biophys Res Commun. 2006;346:904–910. doi: 10.1016/j.bbrc.2006.05.191. [DOI] [PubMed] [Google Scholar]
- Gingerich S, Kim GL, Chalmers JA, Koletar MM, Wang X, Wang Y, Belsham DD. Estrogen receptor alpha and G-protein coupled receptor 30 mediate the neuroprotective effects of 17beta-estradiol in novel murine hippocampal cell models. Neuroscience. 2010;170:54–66. doi: 10.1016/j.neuroscience.2010.06.076. [DOI] [PubMed] [Google Scholar]
- Grassi S, Frondaroli A, Dieni C, Scarduzio M. Effects of 17beta-estradiol on synaptic plasticity in the rat medial vestibular nuclei. Acta Otolaryngol. 2009;129:390–394. doi: 10.1080/00016480802566287. [DOI] [PubMed] [Google Scholar]
- Hall JM, Couse JF, Korach KS. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem. 2001;276:36869–36872. doi: 10.1074/jbc.R100029200. [DOI] [PubMed] [Google Scholar]
- Han BH, Holtzman DM. BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J Neurosci. 2000;20:5775–5781. doi: 10.1523/JNEUROSCI.20-15-05775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci. 2003;26:81–89. doi: 10.1016/S0166-2236(02)00040-1. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hazell GG, Yao ST, Roper JA, Prossnitz ER, O'Carroll AM, Lolait SJ. Localisation of GPR30, a novel G protein-coupled oestrogen receptor, suggests multiple functions in rodent brain and peripheral tissues. J Endocrinol. 2009;202:223–236. doi: 10.1677/JOE-09-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Ström A, Treuter E, Warner M, Gustafsson JA. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007;87:905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- Hsieh YC, Yu HP, Frink M, Suzuki T, Choudhry MA, Schwacha MG, Chaudry IH. G protein-coupled receptor 30-dependent protein kinase A pathway is critical in nongenomic effects of estrogen in attenuating liver injury after trauma-hemorrhage. Am J Pathol. 2007;170:1210–1218. doi: 10.2353/ajpath.2007.060883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Mao XO, Zhu Y, Greenberg DA. MEK and ERK protect hypoxic cortical neurons via phosphorylation of Bad. J Neurochem. 2002;80:119–125. doi: 10.1046/j.0022-3042.2001.00678.x. [DOI] [PubMed] [Google Scholar]
- Jin Y, Blue EK, Gallagher PJ. Control of death-associated protein kinase (DAPK) activity by phosphorylation and proteasomal degradation. J Biol Chem. 2006;281:39033–39040. doi: 10.1074/jbc.M605097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch DG, Doseff A, Chau BN, Lim DS, de Souza-Pinto NC, Hansford R, Kastan MB, Lazebnik YA, Hardwick JM. Caspase-3-dependent cleavage of Bcl-2 promotes release of cytochrome c. J Biol Chem. 1999;274:21155–21161. doi: 10.1074/jbc.274.30.21155. [DOI] [PubMed] [Google Scholar]
- Krapivinsky G, Krapivinsky L, Manasian Y, Ivanov A, Tyzio R, Pellegrino C, Ben-Ari Y, Clapham DE, Medina I. The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron. 2003;40:775–784. doi: 10.1016/s0896-6273(03)00645-7. [DOI] [PubMed] [Google Scholar]
- Kurata K, Takebayashi M, Morinobu S, Yamawaki S. beta-estradiol, dehydroepiandrosterone, and dehydroepiandrosterone sulfate protect against N-methyl-d-aspartate-induced neurotoxicity in rat hippocampal neurons by different mechanisms. J Pharmacol Exp Ther. 2004;311:237–245. doi: 10.1124/jpet.104.067629. [DOI] [PubMed] [Google Scholar]
- Lebesgue D, Chevaleyre V, Zukin RS, Etgen AM. Estradiol rescues neurons from global ischemia-induced cell death: multiple cellular pathways of neuroprotection. Steroids. 2009;74:555–561. doi: 10.1016/j.steroids.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebesgue D, Traub M, De Butte-Smith M, Chen C, Zukin RS, Kelly MJ, Etgen AM. Acute administration of non-classical estrogen receptor agonists attenuates ischemia-induced hippocampal neuron loss in middle-aged female rats. PLoS One. 2010;5:e8642. doi: 10.1371/journal.pone.0008642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Shang J, Tian F, Nishi H, Abe K. In vivo optical imaging for evaluating the efficacy of edaravone after transient cerebral ischemia in mice. Brain Res. 2011;1397:66–75. doi: 10.1016/j.brainres.2011.04.038. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- Lu YM, Roder JC, Davidow J, Salter MW. Src activation in the induction of long-term potentiation in CA1 hippocampal neurons. Science. 1998;279:1363–1367. doi: 10.1126/science.279.5355.1363. [DOI] [PubMed] [Google Scholar]
- Murphy LO, Blenis J. MAPK signal specificity: the right place at the right time. Trends Biochem Sci. 2006;31:268–275. doi: 10.1016/j.tibs.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Omkumar RV, Kiely MJ, Rosenstein AJ, Min KT, Kennedy MB. Identification of a phosphorylation site for calcium/calmodulindependent protein kinase II in the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem. 1996;271:31670–31678. doi: 10.1074/jbc.271.49.31670. [DOI] [PubMed] [Google Scholar]
- Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu Rev Physiol. 2008;70:165–190. doi: 10.1146/annurev.physiol.70.113006.100518. [DOI] [PubMed] [Google Scholar]
- Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- Rozengurt E. Mitogenic signaling pathways induced by G protein-coupled receptors. J Cell Physiol. 2007;213:589–602. doi: 10.1002/jcp.21246. [DOI] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, MacDonald JF, Tymianski M. Distinct roles of synaptic and extrasynaptic NMDA receptors in excitotoxicity. J Neurosci. 2000;20:22–33. doi: 10.1523/JNEUROSCI.20-01-00022.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Yuan Y, Ding F, Liu J, Gu X. The protective effects of Achyranthes bidentata polypeptides against NMDA-induced cell apoptosis in cultured hippocampal neurons through differential modulation of NR2A- and NR2B-containing NMDA receptors. Brain Res Bull. 2008;77:274–281. doi: 10.1016/j.brainresbull.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- Shen K, Teruel MN, Connor JH, Shenolikar S, Meyer T. Molecular memory by reversible translocation of calcium/calmodulin-dependent protein kinase II. Nat Neurosci. 2000;3:881–886. doi: 10.1038/78783. [DOI] [PubMed] [Google Scholar]
- Shohat G, Spivak-Kroizman T, Cohen O, Bialik S, Shani G, Berrisi H, Eisenstein M, Kimchi A. The pro-apoptotic function of death-associated protein kinase is controlled by a unique inhibitory autophosphorylation-based mechanism. J Biol Chem. 2001;276:47460–47467. doi: 10.1074/jbc.M105133200. [DOI] [PubMed] [Google Scholar]
- Smith CC, McMahon LL. Estrogen-induced increase in the magnitude of long-term potentiation occurs only when the ratio of NMDA transmission to AMPA transmission is increased. J Neurosci. 2005;25:7780–7791. doi: 10.1523/JNEUROSCI.0762-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CC, McMahon LL. Estradiol-induced increase in the magnitude of long-term potentiation is prevented by blocking NR2B-containing receptors. J Neurosci. 2006;26:8517–8522. doi: 10.1523/JNEUROSCI.5279-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder MA, Cooke BM, Woolley CS. Estradiol potentiation of NR2B-dependent EPSCs is not due to changes in NR2B protein expression or phosphorylation. Hippocampus. 2011;21:398–408. doi: 10.1002/hipo.20756. [DOI] [PubMed] [Google Scholar]
- Stanika RI, Pivovarova NB, Brantner CA, Watts CA, Winters CA, Andrews SB. Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc Natl Acad Sci U S A. 2009;106:9854–9859. doi: 10.1073/pnas.0903546106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stennicke HR, Jürgensmeier JM, Shin H, Deveraux Q, Wolf BB, Yang X, Zhou Q, Ellerby HM, Ellerby LM, Bredesen D, Green DR, Reed JC, Froelich CJ, Salvesen GS. Pro-caspase-3 is a major physiologic target of caspase-8. J Biol Chem. 1998;273:27084–27090. doi: 10.1074/jbc.273.42.27084. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM, Balel C, Wang M, Jia N, Zhang W, Lew F, Chan SL, Chen Y, Lu Y. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell. 2010;140:222–234. doi: 10.1016/j.cell.2009.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Engelhardt J, Coserea I, Pawlak V, Fuchs EC, Köhr G, Seeburg PH, Monyer H. Excitotoxicity in vitro by NR2A- and NR2B-containing NMDA receptors. Neuropharmacology. 2007;53:10–17. doi: 10.1016/j.neuropharm.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Wahl AS, Buchthal B, Rode F, Bomholt SF, Freitag HE, Hardingham GE, Rønn LC, Bading H. Hypoxic/ischemic conditions induce expression of the putative pro-death gene Clca1 via activation of extrasynaptic N-methyl-d-aspartate receptors. Neuroscience. 2009;158:344–352. doi: 10.1016/j.neuroscience.2008.06.018. [DOI] [PubMed] [Google Scholar]
- Wang H, Wu LJ, Kim SS, Lee FJ, Gong B, Toyoda H, Ren M, Shang YZ, Xu H, Liu F, Zhao MG, Zhuo M. FMRP acts as a key messenger for dopamine modulation in the forebrain. Neuron. 2008;59:634–647. doi: 10.1016/j.neuron.2008.06.027. [DOI] [PubMed] [Google Scholar]
- Wong M, Moss RL. Long-term and short-term electrophysiological effects of estrogen on the synaptic properties of hippocampal CA1 neurons. J Neurosci. 1992;12:3217–3225. doi: 10.1523/JNEUROSCI.12-08-03217.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley CS. Acute effects of estrogen on neuronal physiology. Annu Rev Pharmacol Toxicol. 2007;47:657–680. doi: 10.1146/annurev.pharmtox.47.120505.105219. [DOI] [PubMed] [Google Scholar]
- Woolley CS, Weiland NG, McEwen BS, Schwartzkroin PA. Estradiol increases the sensitivity of hippocampal CA1 pyramidal cells to NMDA receptor-mediated synaptic input: correlation with dendritic spine density. J Neurosci. 1997;17:1848–1859. doi: 10.1523/JNEUROSCI.17-05-01848.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Kurup P, Zhang Y, Goebel-Goody SM, Wu PH, Hawasli AH, Baum ML, Bibb JA, Lombroso PJ. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J Neurosci. 2009;29:9330–9343. doi: 10.1523/JNEUROSCI.2212-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamani MR, Levy WB, Desmond NL. Estradiol increases delayed, N-methyl-d-aspartate receptor-mediated excitation in the hippocampal CA1 region. Neuroscience. 2004;129:243–254. doi: 10.1016/j.neuroscience.2004.06.082. [DOI] [PubMed] [Google Scholar]
- Zhou M, Baudry M. Developmental changes in NMDA neurotoxicity reflect developmental changes in subunit composition of NMDA receptors. J Neurosci. 2006;26:2956–2963. doi: 10.1523/JNEUROSCI.4299-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]