

Abstract
Clonal cell culture is crucial for experimental protocols that require growth or selection of pure populations of cells. High-density derivation of neural progenitors from human embryonic stem cells (hESCs) can lead to incomplete differentiation, and transplantation of resulting heterogeneous cell mixtures can cause proliferation of tumorigenic clusters in vivo. We have identified the neural precursor that resides among normal hESC colonies as a TRA-1-60−/SSEA4−/SOX1+ cell and developed a method that allows for the clonal expansion of these FACS-selected progenitors to neural stem cells (NSCs) in serum-free conditions. Single TRA-1-60−/SSEA4−/SOX1+ cells grown in serum-free media give rise to multipotent NSCs with an efficiency of 0.7%. The fate of the TRA-1-60−/SSEA4−/SOX1+ neural precursor becomes specified in maintenance conditions by inhibition of BMP signaling. This clonal culture method can be scaled up to produce NSCs for differentiation and use in cell therapies.
Introduction
The developmental potential of human embryonic stem cells (hESCs) (Thomson et al., 1998) has triggered an explosion of research on directing differentiation to specific cell types required for cell replacement therapies. Neural stem cells (NSCs) are self-renewing, multipotent cells of the CNS that generate neurons and glia, and can be used as precursors for potential cell-based therapies, including dopaminergic neurons for parkinsonian patients.
Existing methods for deriving NSCs and their progenitors from hESCs are not clonal. Heterogeneous populations of cells produced by high-density culture can create problems for cell therapies: trials with transplanted hESC-derived NSCs in parkinsonian rats produced functional improvement; however, a subset of the heterogeneous mix of transplanted cells proliferated in undifferentiated tumorigenic clusters (Erdö et al., 2003; Roy et al., 2006; Cai et al., 2010). Uncontrolled differentiation techniques like embryoid body formation and high-density monolayer culture are used frequently in the simultaneous differentiation of multiple cell types from hESCs, including neural progenitors (Reubinoff et al., 2001; Schuldiner et al., 2001; Zhang et al., 2001; Nistor et al., 2005; Kang et al., 2007; Dhara and Stice, 2008; Elkabetz et al., 2008; Koch et al., 2009). Several groups have successfully used serum-free media (SFM) to generate neural cells from hESCs (Conti et al., 2005; Chambers et al., 2009; Vallier et al., 2009), but without a vigorous demonstration of clonality these methods also rely on high-density differentiation.
Addressing problems created by high-density propagation of mixed populations of cells in undefined conditions, we created a clonal method whereby single cells from hESC colonies were propagated at low densities to form NSCs in serum-free conditions. Interestingly, we found hESCs were primed to acquire a neural identity in maintenance conditions, unlike mouse ESCs (mESCs) that transition directly to a neural identity by default in minimal culture conditions that minimize transforming growth factor β (TGFB) signaling (Tropepe et al., 2001; Smukler et al., 2006).
We identified the NSC colony-forming cell among hESC colonies as TRA-1-60−/SSEA4−/SOX1+. This cell proliferated in defined conditions, forming clonal floating sphere colonies (neurospheres) containing NSCs and their progenitors. Quantitative PCR (Q-PCR) analyses of mRNA transcripts show that neurospheres express neural precursor markers but not early endodermal and mesodermal markers, indicating entirely neural colonies. Neurospheres generated from single NSCs were differentiated in vitro and in vivo to form neurons and glia [confirmed by immunocytochemical (ICC) staining and Q-PCR], illustrating multipotentiality. Neurospheres passaged long term (11 months), forming clonal descendent spheres, exhibiting self-renewal and confirming the presence of NSCs in addition to progenitors that can display multipotentiality but not passage repeatedly. Expansion of NSCs by passage was robust, thus scaleable for clinical needs.
The mechanism by which hESCs are primed to adopt a neural fate spontaneously in maintenance conditions relies on negative regulation of BMP signaling. BMP signaling was inhibited by addition of Noggin to maintenance cultures, generating an increase in NSCs produced by selection in serum-free conditions. While addition of anti-apoptotics like rho-associated kinase (ROCK) inhibitor leads to dramatic increase in frequency of colony production, however the cells in these colonies are not stably committed to a neural lineage and could be unsuitable for cell replacement therapies.
Materials and Methods
Propagation and maintenance of hESCs.
H9 (Thomson et al., 1998) (female, normal karyotype) and CA1 (male, normal karyotype) hESCs were used in all experiments. The CA1 hESCs are fully characterized and approved by the Stem Cell Oversight Committee at the Canadian Institute of Health Research. (Correspondence concerning the isolation and characterization of the CA1 hESC cells should be directed to Andras Nagy, Samuel Lunenfeld Research Institute, Mount Sinai Hospital, Toronto, Ontario M5G 1X5, Canada; nagy@mshri.on.ca.). Transgenic hESCs (provided by Jon Draper, McMaster University, Hamilton, Ontario, Canada) included H9-red fluorescent protein (RFP), CA1-RFP, and CA1-green fluorescent protein (GFP), which carry histone 2B/GFP, or histone 2B/RFP fusion proteins. hESCs were maintained on mitotically inactivated mouse embryonic fibroblast feeder layers in media containing 80% knock-out DMEM and 20% knock-out serum replacement (Invitrogen), 1% non-essential amino acids, 1 mm l-glutamine, 0.1 mm 2-mercaptoethanol, and 4 ng/ml basic fibroblast growth factor (FGF). Confluent colonies were disaggregated into ∼100 cell clumps using 0.1% type IV collagenase (Invitrogen) and passaged 1:3 or 1:4 every 4–7 d. For Noggin pretreatment, 100 ng/ml mouse recombinant Noggin/Fc chimera (R&D systems) was added to fresh media each day until colonies became confluent (4–7 d).
Neurosphere culture.
hESCs were incubated at 37°C for 15–20 min in TrypLE Express for dissociation and then quenched with mouse embryonic fibroblast (MEF)-conditioned media before centrifugation. Cells were resuspended and triturated with a small borehole Pasteur pipette before being filtered through a 40 μm cell strainer. The resulting single-cell suspension was plated at 10 cells/μl in chemically defined SFM with growth factors. SFM contained a 1:1 mixture of DMEM and F-12 nutrient (Invitrogen) with 0.6% d-glucose, 5 mm HEPES, 3 mm NaHCO3, 2 mm glutamine, 25 μg/ml insulin, 100 g/ml transferrin, 20 nm progesterone, 60 μm putrescine, and 30 nm sodium selenite (all Sigma). Growth factors added singly or in combination included 1400 U/ml human LIF (Millipore Bioscience Research Reagents), 10 ng/ml FGF2, 2 μg/ml heparin, and 20 ng/ml epidermal growth factor (EGF) (all Sigma), 1x B27 (Invitrogen), 100 ng/ml recombinant Noggin/Fc chimera (R&D systems), and rho-kinase inhibitor Y-27632 (Calbiochem), and were used at a final concentration of 10 μm. hESCs were plated in the center 24 wells of 48-well coated plates (Nunclon) and re-fed at 2 weeks with an equal volume of plating media (300 μl). Floating sphere colonies >75 μm were scored at 4 weeks. Neurospheres cultured with rho kinase inhibitor were collected every 11 d. To assess self-renewal of neurospheres, 4 week colonies were collected in bulk and floated in a 10 cm Petri dish where colonies >75 μm were picked for passage. Multiple spheres were dissociated into single cells (bulk dissociation) by incubation in trypsin/EDTA (Sigma) for 10 min, followed by manual trituration with a small borehole Pasteur pipette.
Single-neurosphere cell suspension was then plated at 10 cells/μl. Secondary sphere colonies were quantified after 2–4 weeks. For single-sphere passage, each sphere was dissociated separately, and the resulting single-cell suspension was replated into a single well. After 2–4 weeks, single neurospheres from a well were picked and dissociated individually, and the single-cell suspension from each colony was again plated into discreet wells. Karyotyping of the neurospheres was performed by WiCell. For differentiation, a single neurosphere per well was plated on matrigel (BD Bioscience)-coated plates in SFM supplemented with 1% fetal calf serum (FCS; Hyclone). Spheres were re-fed at 7 d and cultured for 14 d. Undifferentiated and differentiated neurospheres were collected for Q-PCR analysis or fixed for ICC.
Clonality assays.
A single H9-RFP cell per well was sorted into a minimum of 2016 wells (per experiment) of 96-well plates using the FACS Aria 3-laser system. After 3 h, all wells were scored for the presence of single cells. Wells containing single cells (80.4%) were scored at 2 weeks for the presence of a sphere colony >50 μm in diameter. A total of 19.6% of wells contained no cells.
For mixing experiments, H9-RFP and CA1-GFP cells were prepared for the neurosphere assay, as previously described. Equal proportions of two cell types were plated at final densities of 5, 10, 20, and 50 cells/μl. The following cell combinations were used: H9 and H9-RFP; CA1 and CA1-GFP; and H9-RFP and CA1-GFP. A control for frequency of sphere formation was performed for each cell type in each experiment. At 2 and 4 weeks, floating spheres were scored for chimerism. For single-cell-per-well experiments, H9-RFP cells were prepared for the neurosphere assay.
Transplant experiments.
On postnatal day 3 (P3) to P7, NOD/SCID mice were anesthetized using isoflurane and injected with 100,000 dissociated cells in 1.0 μl PBS using a 5.0 μl Hamilton syringe. Three cell types (CA1-RFP+) were tested: hESCs, and neurospheres grown in the presence or absence of ROCK inhibitor. Injections were made unilaterally, ∼1.5 mm lateral to bregma and 1.5 mm below the dura, to target the striatum. Animals were killed 3 weeks post-transplant. For tissue analysis, animals were perfused with 4% paraformaldehyde (Sigma); brains were removed, soaked in 4% paraformaldehyde overnight, and transferred to 30% sucrose for a minimum of 24 h. Brains were individually embedded in cryomatrix (Ted Pella) and frozen. Fifteen micrometer sections were taken using a Jencons OTF5000 cryostat and mounted on Superfrost Plus slides (Fisher Scientific). For immunohistochemical analysis, RFP+ transplanted cells were located around the injection site using 10× magnification and scored in random fields at 40×. Analysis and imaging were performed using an Olympus Fluoview Confocal Imaging System. See the “Immunocytochemistry” section for antibody information.
hESC minimal condition assays.
hESCs were dissociated into a single-cell suspension using TrypLE Express (Invitrogen) and plated at ≤10 cells/μl in defined serum-free media (described above) without growth factors. Cells were collected for Q-PCR analysis or fixed for immunocytochemical analysis at 4 h, 24 h, and 3 d time-points. For analysis by ICC, cells were plated at on laminin/poly-l-ornithine-coated culture plates (Nunclon). To assess the survival of individual hESCs, they were plated at 50 cells/well in 48-well plates. At 4 h, 24 h, and 3 d, viable cells were scored by trypan blue exclusion.
RNA isolation, cDNA preparation, and Q-PCR.
Total RNA was isolated using an RNeasy extraction kit (Qiagen) according to instructions with optional DNase treatment. Total RNA was quantified using a NanoDrop ND-1000 spectrophotometer. cDNA was prepared from 150–500 ng RNA using 200 U SuperScript III RNase H-Reverse Transcriptase (Invitrogen) and 0.2 ng random hexamer primer (6-mer, Fermentas). Q-PCR was performed using predesigned TaqMan gene expression assays (Table 1) with TaqMan universal PCR master mix (both Applied Biosystems) under universal cycling conditions (95°C for 10 min; 95°C for 15 s, 60°C for 1 min for 40 cycles) on a 7900HT Fast Real-Time PCR System (Applied Biosystems). Reactions were in triplicate and always included a negative control (no template). 18S was the endogenous control as it had consistent expression across all samples. A dilution curve of hESC RNA demonstrated that efficiencies of targets were similar to those of endogenous controls. The relative quantity was calculated by assigning the highest expressing sample of each gene as the calibrator to which the expression of that gene in other samples was related. In cases where the highest expresser was not consistent among experiments the value for the highest expresser is <1 and is represented as a mean ± SEM.
Table 1.
TaqMan gene expression assays
| Gene | Gene expression assay identification number |
|---|---|
| 18S | Hs99999901_s1 |
| β-Actin | Hs99999903_m1 |
| OCT4 | Hs01895601_u1 |
| LIN28 | Hs00702808_s1 |
| SOX2 | Hs00602736_s1 |
| NES | Hs00707120_s1 |
| SOX1 | Hs00534426_s1 |
| PAX6 | Hs00240871_m1 |
| TUBB3 | Hs00801390_s1 |
| FOXA2 | Hs00232764_m1 |
| HNF4A | Hs00766846_s1 |
| GATA4 | Hs00171403_m1 |
| GATA1 | Hs00231112_m1 |
| T | Hs00610080_m1 |
FACS analysis.
Fluorescence-activated cell sorting was performed on CA1 and H9 cells. hESC colonies were dissociated into single-cell suspensions (as above). Cells were suspended in blocking solution containing HBSS (Invitrogen) with 2% FCS (Hyclone) before adding primary antibodies TRA-1-60 (IgM) (Millipore Bioscience Research Reagents; 1:100) and SSEA4 (IgG) [Developmental Studies Hybridoma Bank (DSHB; 1:5)] at 4°C for 30 min. Goat anti-mouse R-phycoerythrin (RPE) conjugate and goat anti-mouse fluorescein isothiocyanate (FITC) conjugate (both at 1:100; Southern Biotech) secondary antibodies were applied for 30 min at 4°C. Cells were then washed twice and strained through a 40 μm cell strainer. 7-AAD (7-amino-actinomycin D) was added to mark dead and dying cells. Live cells were sorted at low pressure (20 psi) by a FACS Aria 3-laser system (Becton Dickinson) using FACS DiVa software version 5.02. Cells were sorted into four separate fractions (+/+, −/−, +/−, and −/+) based on RPE and FITC fluorescence. All fractions were kept on ice until plated in the neurosphere assay or collected for PCR. Unstained hESCs were used as negative controls. RPE-only-, FITC-only-, and RPE-plus-FITC-labeled cells were used as positive controls to set gates for sorting and to assess survival of sorted cells.
Immunocytochemistry.
Adherent cells, colonies and tissues were fixed with 4% paraformaldehyde in PBS, pH 7.3, for 20 min at room temperature and then washed with three times with PBS. Cells were permeabilized with 0.3% Triton X-100 for 5 min and then washed twice with PBS before being blocked with PBS containing 1% BSA and 10% normal goat serum (NGS) for 1 h at room temperature. Blocking solution was washed off with PBS before primary antibodies were applied. Antibodies were diluted in PBS containing 0.3% Triton and 1% NGS, and were applied to cultures overnight at 4°C. Primary antibodies used were as follows: mouse monoclonal human specific anti-NESTIN (NES) (IgG) at 1:200, mouse monoclonal human specific anti-NF (IgG) (Millipore) at 1:100, rabbit polyclonal anti-RFP at 1:100, mouse monoclonal anti-O4 (IgM), at 1:200, rabbit polyclonal anti-SOX1 at 1:100, mouse monoclonal anti-MAP2 (IgG) at 1:250, mouse monoclonal anti-NF (IgG) at 1:150, and rabbit polyclonal anti-TH at 1:1000, anti-GAD 65/67 at 1:1000 (all Millipore); mouse monoclonal anti-β3-tubulin (IgG) (Sigma) at 1:500; rabbit polyclonal anti-GFAP (Dako) at 1:1000; mouse monoclonal anti-OCT-3/4 (IgG) (BD Biosciences) 1:200; mouse monoclonal anti-FOXA2 (IgG) (DSHB) at 1:10; goat monoclonal anti-FOXA2 (IgG) (Santa Cruz Biotechnology) at 1: 200; and rabbit polyclonal anti-VGLUT1 at 1:1000 (Synaptic Systems). After overnight incubation, cultures were washed 3x with PBS before secondary antibodies were applied. Alexa Fluor secondary antibodies were used at a dilution of 1:400 and included: 488 goat anti-rabbit, 488 goat anti-mouse, 568 goat anti-rabbit, 568 goat anti-mouse, 568 donkey anti-goat, and 647 goat anti-mouse (Invitrogen). Secondary antibodies were suspended in PBS with 1% NGS and applied for 2 h at 37°C. Cultures were then washed three times before applying Hoechst dye 33258 (0.015 mg/ml stock solution diluted to 0.001 mg/ml in PBS; Sigma) to stain the nuclei of all cells. Hoechst was applied for 10 min at room temperature after which cells were washed three times with PBS. Positive, negative, and secondary only controls were used for each experiment. All controls were stained simultaneously using the same method, except for the omission of primary antibodies from secondary-only controls. Fluorescence was quantified only when negative and secondary only controls were negative for staining. Cells in the differentiation and neural default assays were quantified by assessing the number of positively stained cells as a percentage of Hoechst-positive nuclei in at least four photographed fields for a minimum four colonies in a minimum of three individual experiments. Cell fluorescence was visualized using a motorized inverted microscope (IX81; Olympus) and a Fluoview Confocal Microscope (Olympus). Fluorescent images were captured using Olympus MicroSuite version 3.2 image analysis software, MicroSuite Five Biological Suite (Soft Imaging System), and Olympus Fluoview Confocal Imaging System.
Embryoid body formation.
To confirm the pluripotent ability of CA1 and H9 hESCs, colonies were disaggregated into ∼100 cell clumps (as described previously) and cultured in hanging drops (in media containing 15% FCS (Hyclone), 100 μm β-mercaptoethanol (Invitrogen), 100 U/ml penicillin, 100 μg/ml streptomycin, 20% 5× high-glucose DMEM, 1 mm l-glutamine, 10 mm HEPES, 0.6% glucose, 0.1% NaCO3 (all Sigma). Thirty microliter drops containing ∼3000 cells were cultured for 2 d before being rinsed onto uncoated plates (Phoenix Biomedical) and cultured for an additional 3 to 10 d. Alternately, suspensions of single cells such as sorted cells needed to be aggregated using greater force; thus, a “spin embryoid body (EB)” method was used (Dang et al., 2002; Ungrin et al., 2008). EBs generated by both methods were collected for Q-PCR.
Statistical analysis.
Statistics were calculated using two-way ANOVA, one-way ANOVA, and Bonferroni post-tests. Error bars represent the SEM of three or more replicates. Data were pooled when no significant difference between cell lines was determined by two-way ANOVA. Single asterisks (*) indicate statistical significance, with p values reported in figure legends.
Results
Single cells from hESC colonies differentiate into colony-forming NSCs without serum or feeder cells
To investigate the ability of single cells from hESC colonies to proliferate in vitro to form neural colonies in the absence of serum, feeder layers, or EB formation, we used conditions originally used to isolate brain-derived NSCs that have since been adapted for the growth of clonal neurospheres from single mESCs (Reynolds and Weiss, 1996; Tropepe et al., 2001;). hESC colonies were dissociated to single cell suspensions and plated at low density in defined serum-free conditions. By 28 d in vitro, floating neurospheres had formed ranging from 50 to 300 μm. An average-sized colony of 150 μm contained ∼10,000 cells (Fig. 1a). As spheres derived from restricted progenitors tend to be smaller (Louis et al., 2008), only spheres ≥75 μm were included for analysis. Q-PCR showed that neurospheres expressed the neural markers SOX1, NES, PAX6, and TUBB3 but not the endodermal markers FOXA2, HNF4A, GATA4 nor mesodermal marker T (also known as brachyury), indicating that these colonies are neural in character (Fig. 1b). In addition, neurospheres do not express pluripotency genes OCT4 (also known as POU5F1) or LIN28, indicating the absence of cells that could be teratocarcinoma initiating in vivo (Roy et al., 2006).
Figure 1.
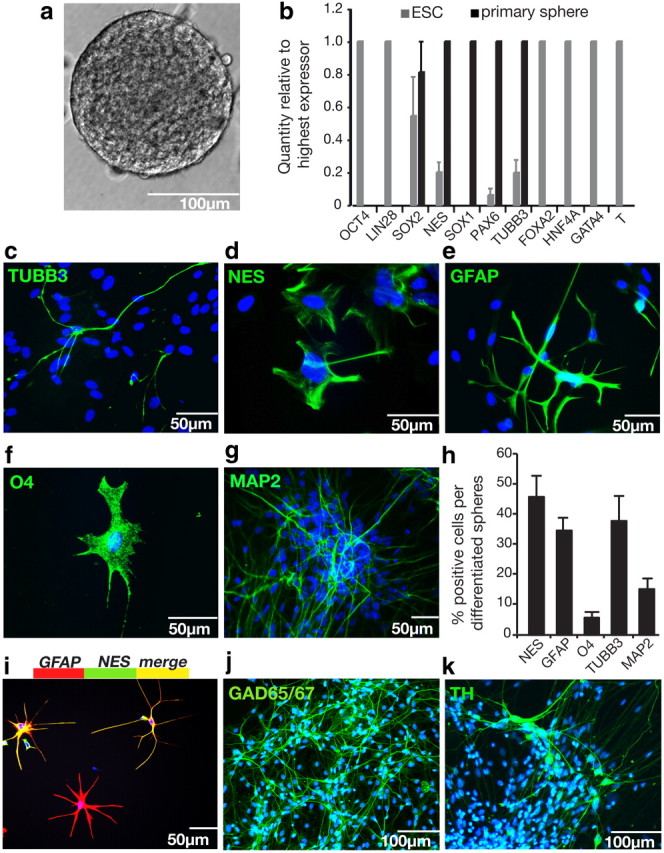
Clonal neurospheres grow in defined conditions over 4 weeks, express only neural markers, and could differentiate into neurons and glia. a, A floating clonal neurosphere at 28 d, grown from a single cell from an hESC colony. b, A comparison of mRNA transcript levels in hESCs versus primary neurospheres by Q-PCR revealed that clonal NSC colonies have an entirely neural phenotype: they express neural markers (NES, SOX1, PAX6, TUBB3) but not pluripotency genes (OCT4, LIN28), endodermal genes (FOXA2, HNF4A, GATA4), or mesodermal genes (GATA1, T), n = 4. c–g, Differentiation potential was assessed by plating single neurospheres on matrigel-coated plates in SFM with 1% FCS for 14 d. Differentiated neurospheres produced TUBB3+ (c) and MAP2+ (g) neurons (shown here growing out of a whole sphere), GFAP+ (e) astrocytes, and O4+ (f) oligodendrocytes, while many cells still expressed the neural precursor marker NES (d). All nuclei were counterstained with Hoechst stain (blue). h, Quantification of marker proteins by ICC show that differentiated cell types consisted of neural precursors (45.9% ± 6.9% NES+), neurons (37.9% ± 8.3% TUBB3+, and 15.2% ± 4.2% MAP2 +), astrocytes (34.7% ± 4.2% GFAP+), and oligodendrocytes (5.8% ± 1.9% O4+). i, A population of differentiating cells colocalize neural precursor marker NES, and astrocyte marker GFAP. Cells were double labeled with GFAP (red) and NES (green) to reveal a population of differentiating cells positive for both proteins (yellow). This overlapping expression accounts for combined marker expression in h being >100%. j, k, Differentiation to 5 weeks produced GABAergic (j) and dopaminergic (k) neurons. Data are represented as mean ± SEM; for Q-PCR, the highest expressing sample (value of “1”) was used as the calibrator.
To determine the potential of neurospheres to form neurons and glia, single colonies were placed on matrigel-coated plates and differentiated in the presence of serum for 14 d. By ICC, differentiating cells are positive for the neural precursor protein NES, early and mature neuronal proteins TUBB3 and MAP2, respectively, neural precursor and astrocyte marker GFAP, and oligodendrocyte marker O4 (Fig. 1c–g). All single colonies differentiated to contain proportions of neurons, astrocytes, and oligodendrocytes (Fig. 1h), and we found no unipotent or bipotent spheres. Immature astrocytes stain positively for neural precursor marker NES, as well as GFAP, and overlapping expression of GFAP and NES accounted for marker quantification being >100% (Fig. 1i). Further differentiation to 5 weeks generated GABAergic and dopaminergic neurons, but not glutamatergic neurons (Fig. 1j,k).
NSCs exhibit long-term self-renewal and are stably committed to the neural lineage
To assess long-term self-renewal, primary neurospheres were passaged at clonal density each month. Neurospheres passaged clonally for 11 months (Fig. 2a,b), indicating the continued generation of NSCs, in addition to progenitors that do not self-renew over multiple passages (Seaberg and van der Kooy, 2002). Moreover, there is enormous expansion of NSCs with continued culture. A single well containing 3000 dissociated neurosphere cells gives rise to an average of 79 ± 3 (n = 11) secondary colonies (Fig. 2a). This suggests that 2.6% of neurosphere cells are neural stem cells. With an average of 10,000 cells per clonal neurosphere, we estimate that each clonal sphere can give rise to 260 clonal secondary NSC-derived spheres. This 260-fold expansion from a single NSC with each passage suggests that a single NSC can expand to produce over 17 million NSCs within three passages.
Figure 2.

Neurospheres passaged clonally long term stably express neural markers after repeated passage, are committed to the neural lineage, and are karyotypically normal. a–c, Neurospheres passaged clonally long term and stably expressed neural markers. a, Primary neurospheres (passage 0) were dissociated and plated as single cells at clonal densities in SFM plus FGF each month for 11 months, n = 3. Over 11 passages, an average of 79 ± 3 new colonies form at each passage. b, Floating 11th passage neurospheres formed from single dissociated 10th passage neurosphere cells. c, Q-PCR analysis compares mRNA levels in passaged neurospheres versus cells from hESC colonies, n = 6. Passaged neurospheres stably express neural markers. d, Q-PCR analysis of mRNAs in late passage (P5–P7) neurospheres plated on MEFs shows that they do not reinitiate expression of endodermal, mesodermal, or pluripotency genes, n = 3. e, Karyotype for NSCs is normal under conditions of enzymatic dissociation of hESCs to single cells, n = 6. Data are represented as mean ± SEM; for Q-PCR, the highest expressing sample (value of “1”) was used as the calibrator.
Q-PCR of passaged neurospheres revealed that neural gene expression remained stable over time (Fig. 2c), indicating the sustained propagation of pure populations of NSCs and their progenitors. The ability of the progeny from late passage spheres to differentiate into neurons and glia was confirmed by ICC (data not shown).
To further investigate the commitment of hESC colony-derived NSCs to the neural lineage, we plated late-passage neurospheres on MEFs to test whether they could generate ES-like colonies or differentiate to endodermal or mesodermal cell types. This colony selection method is used in the derivation of induced pluripotent stem cells to identify clones that have been successfully reprogrammed (Takahashi and Yamanaka, 2006; Takahashi et al., 2007). After 7 d of cultivation in hESC maintenance conditions, cells were collected and analyzed by Q-PCR (Fig. 2d). This analysis revealed that extraneural gene expression was not initiated, further suggesting that hESC colony-derived NSCs are intractable to differentiation to other cell types.
Although there is some evidence that enzymatic cell dissociation can give rise to aneuploidy (Mitalipova et al., 2005), NSCs generated using our method (which includes enzymatic dissociation of hESCs) have a normal karyotype (Fig. 2e).
NSC colonies can be clonally derived from hESC colony cells
To establish that neurospheres can arise from a single cell and to investigate the frequency at which this occurs, we plated a single cell per microwell and checked for sphere formation at 2 weeks. After plating, all wells were assessed for the presence of a single cell, and only wells containing single cells were tracked for colony growth. In total, five colonies formed from 3881 single cells representing 0.11% of the starting population (Fig. 3a,e).
Figure 3.
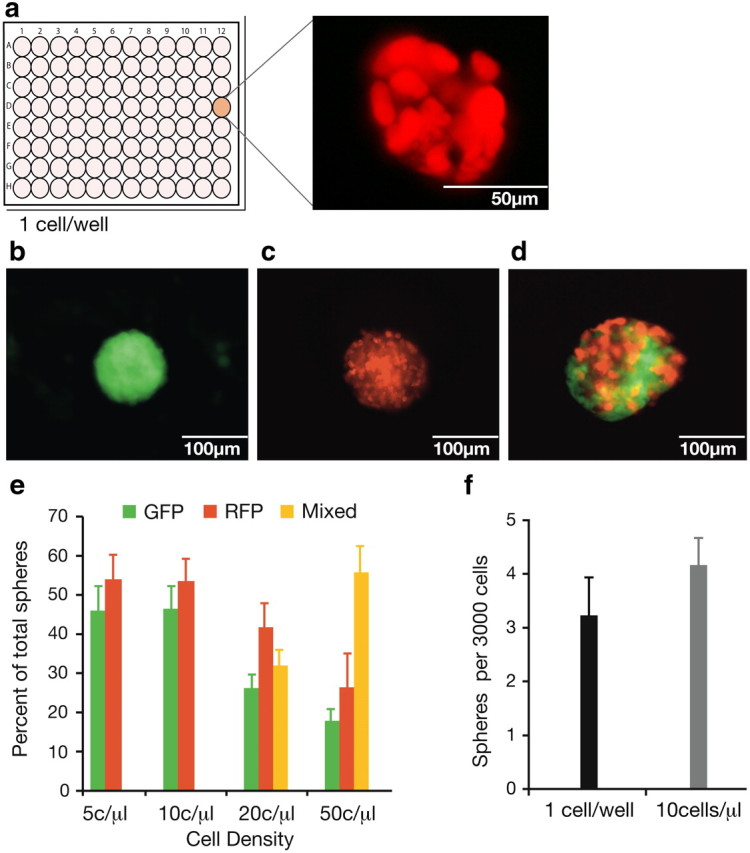
Single-cell-per-well experiments illustrate that NSC colonies grow clonally. At densities of ≤10 cells/μl, NSC colonies also grow from single cells rather than by multicellular aggregation. a, Single-cell-per-well experiments tracked the growth of 3881 individual cells and confirmed that ∼1 in 900 cells was capable of forming a clonal neural colony in serum-free conditions. b, c, e, When equal numbers of RFP and GFP cells from hESC colonies were plated together (n = 4), spheres of one color (b, c) that indicate a clonal origin, only arose at 10 and 5 cells/μl (e). d, e, Some chimeric spheres (d) arose at densities of 20 and 50 cells/μl (e), indicating that these colonies had formed by the aggregation of ≥2 cells. f, A comparison of sphere formation from single cells per well versus 10 cells/μl (3000 cells/well) showed that the frequency of sphere formation of isolated single cells was equivalent to when cells were plated at 10 cells/μl (t(4) = 1.22, p = 0.32), n = 3. Data are represented as mean ± SEM.
For the purpose of practical large-scale expansion of hESCs to NSCs, growth of single cells at low densities is more efficient than a single cell per well. To establish the density at which single hESCs could proliferate clonally to form neurospheres rather than by aggregation with other cells, equal numbers of RFP and GFP hESCs were plated at final densities of 5, 10, 20, 50, and 100 cells/μl. The resulting neurospheres were scored for chimerism. At 5 and 10 cells/μl, only unmixed spheres were found, indicating a clonal origin (Fig. 3b,c,e), while at densities of 20–100 cells/μl, some spheres of mixed colors formed, indicating that these colonies had arisen by aggregation (Fig. 3d,e). When single cells from hESC colonies were plated at 10 cells/μl, 0.14% of the starting single cell population formed neurospheres. This frequency is similar to that of neurosphere formation when cells from hESC colonies are plated at one cell per microwell (Fig. 3e).
Single neurospheres were also serially passaged to confirm that the progeny from a single cell can produce large numbers of NSCs at each passage. The vast majority of individual single-cell-derived primary colonies passaged to form an average of 38 ± 3 secondary, tertiary, and quaternary spheres per colony. This represents substantial expansion; however, it is less than when colonies are dissociated in bulk followed by single-cell plating at clonal density because a majority of cells die during the extreme stress of single-sphere dissociation.
hESC colony-derived NSCs are FGF dependent, and ROCK inhibitor improves survival but biases cell fate
NSCs derived from embryonic and adult cells are dependent on different growth factors for their propagation in vitro. mESC-derived primary neurospheres originate from “primitive” NSCs (pNSCs) that rely on leukemia inhibitory factor (LIF) to mediate self-renewal, where passaging of these neurospheres yields “definitive” NSCs (dNSCs) dependent on FGF (Tropepe et al., 2001; Hitoshi et al., 2004; Smukler et al., 2006). Embryonic and adult mouse forebrain ventricular tissue contains NSCs that require FGF or EGF to form primary and secondary colonies (Tropepe et al., 1999). To ascertain whether there is a specific growth factor dependency of input cells that survive to proliferate as NSCs, cells from hESC colonies were plated at clonal densities in wells containing LIF, FGF, EGF, and the culture supplement B27 either singly or in combination. At low cell densities, input cells were dependent on FGF but not LIF, EGF, or B27 (Fig. 4a). Cell survival and proliferation are extremely limited in the minimal culture conditions provided by SFM. We assessed cell survival by plating 50 cells per well and then scoring live cells by trypan blue exclusion at various time points. When 50 cells/well were plated in SFM alone, 69.7 ± 7.9%, 38.6 ± 4.9%, and 15.5 ± 3.3% of cells survived to 4 h, 24 h, and 3 d, respectively (doublets and aggregates were not scored). The addition of FGF and B27 improved survival to 87.92 ± 2.8%, 65.8 ± 5.8%, and 33.5 ± 8.3% at 4 h, 24 h, and 3 d, respectively (Fig. 4b).
Figure 4.
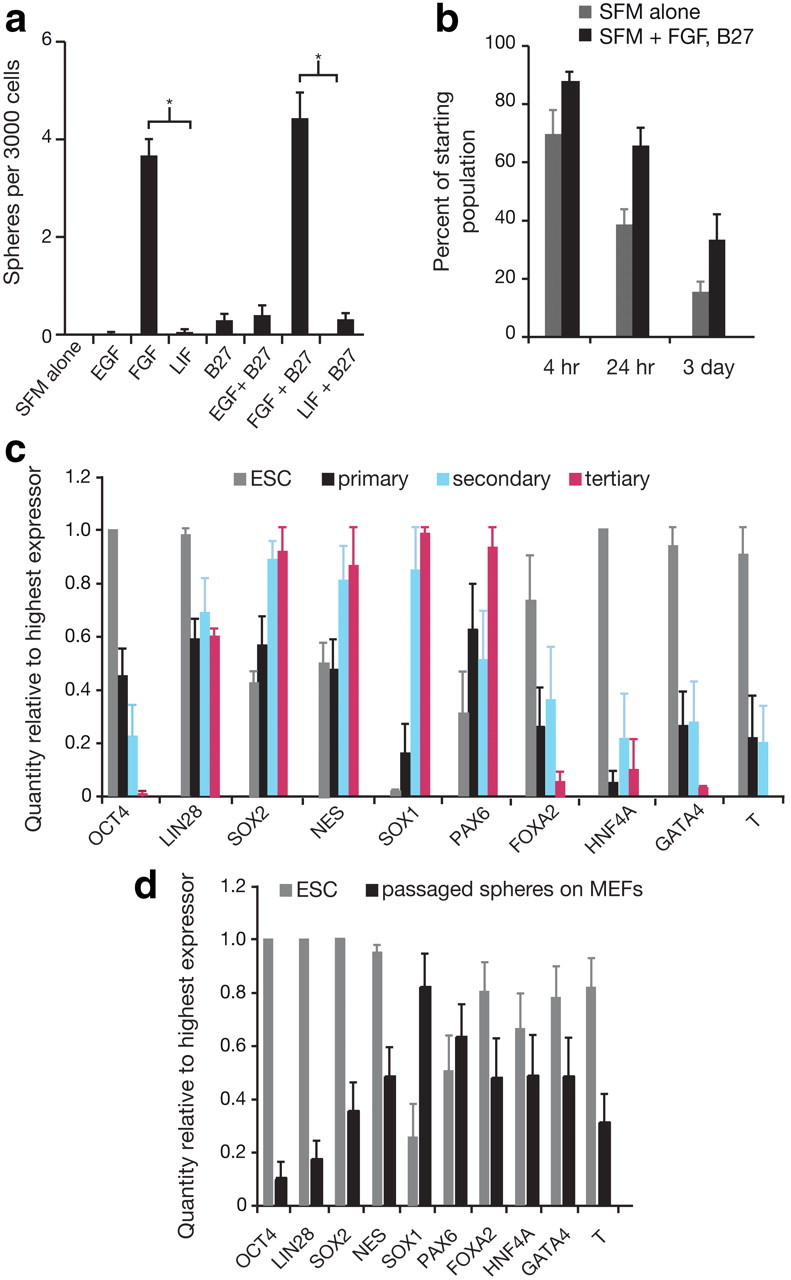
Primary neurosphere formation is dependent on FGF, and the addition of growth factors improves survival in SFM, but ROCK inhibitor influences cell fate. a, A comparison of culture conditions with addition of different growth factors reveals that the ability of single cells from hESC colonies to form neurospheres was reliant on the addition of FGF. One-way ANOVA showed a significant main effect of growth factors (F(7,31) = 56.46, p < 0.0001), while Bonferroni's multiple-comparison tests showed a significant difference between FGF and LIF (p < 0.05) and FGF+B27 compared with LIF+B27 (p < 0.05), illustrating that neurospheres are FGF dependent and not LIF dependent, n = 4. b, Single-cell survival in SFM versus SFM with growth factors represents the survivors as a percentage of starting cell populations. Two-way ANOVA showed that the survival of single cells in both conditions is diminished over time (F(2,30) = 42.40, p < 0.0001) and that the addition of growth factors to single cells in SFM improves survival (F(1,30) = 19.23, p = 0.0001), n = 3. c, Comparison of mRNA levels by Q-PCR showed that primary, secondary, and tertiary spheres cultured with ROCK inhibitor maintain expression of pluripotency (OCT4, LIN28) and non-neural genes (FOXA2, HNF4A, T and GATA4) unlike neurospheres from standard culture (Fig. 1k,l) that only express neural markers, n = 6. d, Q-PCR analysis of mRNA transcripts show that when ROCK inhibitor-cultured spheres are plated on MEFs in hESC maintenance conditions, ESC-like colonies form, and FOXA2 (p < 0.001), HNF4A (p < 0.001), and GATA4 (p < 0.001) expression is upregulated (by Bonferroni post-test comparison of primary, secondary, and tertiary spheres to passaged spheres on MEFs), n = 10. The data from c and d indicate that exposure to ROCK inhibitor prevents complete neuralization of hESCs in minimal conditions, resulting in colonies containing cells that are not stably committed to a neural lineage. Data are represented as mean ± SEM; for Q-PCR, the highest expressing sample (value of “1”) was used as the calibrator.
It has been shown that addition of ROCK inhibitor Y-27632 diminishes the dissociation-induced apoptosis that challenges hESC survival after dissociation to single cells (Watanabe et al., 2007). Survival of hESCs in SFM with FGF and B27 can be further improved by the addition of ROCK inhibitor such that the frequency of sphere generation is increased by 14-fold to 60.8 ± 15.3 spheres per 3000 cells (2% of the starting hESC population). In addition, these colonies are ready to be collected in 11 d rather than in 2 to 4 weeks. Unfortunately, primary, secondary, and tertiary neurospheres cultured with ROCK inhibitor do not completely neuralize (Fig. 4c) and also display residual pluripotency, as evidenced by their ability to form ESC-like colonies when plated on MEFs (Fig. 4d). Even when ROCK inhibitor is used only in primary neurosphere culture and then removed from subsequent culture, passaged spheres retain pluripotency (data not shown), and as such could be unsuitable for cell therapies. Q-PCR for regional marker expression also indicated differences between NSCs derived with and without ROCK inhibitor: NSCs derived clonally without ROCK inhibitor are regionally uncommitted in that they express both anterior (EMX1, OTX1) and posterior (EGR2, HOXB4) markers, while NSCs grown with ROCK inhibitor present an anterior phenotype. In ROCK spheres, there is a 16-fold increase in OTX expression, a 1.7-fold increase in EMX1 expression, a 6.5-fold decrease in EGR2 expression, and a 94.6-fold decrease in HOXB4 expression compared with neurospheres grown without ROCK inhibitor (fold difference relative to control, normalized to 18S).
Unlike mESCs, neural specification of hESCs does not occur in minimal culture but in maintenance culture conditions
During vertebrate gastrulation, the first neurally specified cells arise from the ectoderm (Spemann and Mangold, 1924) through a process of release from the inhibition of local BMPs (Hemmati-Brivanlou and Melton, 1994). BMPs are members of the TGFB superfamily that are strong inhibitors of neural differentiation (Wilson and Hemmati-Brivanlou, 1995). Noggin, Chordin, and Follistatin secreted from a specialized group of dorsal mesodermal cells (organizer/node) bind BMPs extracellularly, preventing them from binding to their receptors on ectoderm cells and promoting neuralization (Piccolo et al., 1996). The underlying assertion of this model is that in the absence of extrinsic signaling, ectodermal cells (and even earlier embryonic cells) become neural. This “default” mechanism of neural fate specification was elucidated by experiments in which single cells from early cleavage and gastrulating embryos were cultured at low density in serum-free conditions, mimicking an environment free of extrinsic signaling. In these conditions, cells autonomously adopted a neural identity (Godsave and Slack, 1989; Grunz and Tacke, 1989; Sato and Sargent, 1989). Low-density culture in minimal, serum-free conditions with mESCs yielded similar findings as these cells adopted a neural phenotype within 4 to 24 h (Tropepe et al., 2001; Smukler et al., 2006).
Based on these studies, we hypothesized that single pluripotent hESCs would directly transition to a neural identity in minimal conditions by a default mechanism. To test this, we cultured dissociated single hESC colony cells at 1 cell/μl in SFM with no growth factors (neural default conditions) and collected the cells at various time points for analysis by Q-PCR. By day 3, hESCs had begun to downregulate expression of pluripotency and mesodermal genes, but not endodermal genes (Fig. 5a). Taking into account the longer cell cycle time of hESCs (analysis at day 3), this was still a different outcome than the experiments with mESCs that showed exclusively neural gene expression by 24 h. This result made us question the assumption of our original hypothesis that NSCs would transition directly from pluripotent hESCs in minimal conditions—i.e., are hESCs becoming prespecified in maintenance conditions? To address this assumption, we sorted cells from confluent hESC colonies labeled with cell surface antigens TRA-1-60 and SSEA4. These markers are expressed on pluripotent hESCs and become downregulated during differentiation (Henderson et al., 2002). We then cultured all four fractions of TRA-1-60 and SSEA4 sorted cells in the clonal neurosphere assay to determine which fractions could give rise to NSC-containing neurospheres. Surprisingly, the pluripotent (double-positive) cells did not form neurospheres. In fact, it was the TRA-1-60−/SSEA4− fraction that contained the sphere-originating cells (Fig. 5b). In confluent colonies TRA-1-60−/SSEA4−, cells represent only 6.1% of the mixed population (Fig. 5c) and TRA-1-60−/SSEA4− cells produced clonal neurospheres at a frequency of 0.7% (adjusted to control for loss of cells in FACS). This means that in 3000 cells plated, ∼180 cells will be TRA-1-60−/SSEA4−, and with a sphere formation frequency of 0.7% should produce ∼12 neurospheres. In fact, when unsorted cells were placed in the neurosphere assay, the frequency of sphere production was 0.1% or 4.1 ± 0.5 spheres per 3000 cells, indicating that even in mixed cultures it may only be the single −/− cells that give rise to spheres. These data suggest that neural fate specification is taking place in the hESC maintenance cultures, and that cells primed for neural differentiation are those surviving to proliferate into spheres.
Figure 5.

hESCs default to a mixed ectodermal and endodermal identity in SFM alone. Neurosphere-originating cells are TRA-1-60−/SSEA4−, make up 6.14% of cells in confluent colonies, and are characterized by elevated levels of neural and endodermal gene expression. a, A comparison of gene expression by Q-PCR analysis of mRNAs from confluent cells from hESC colonies in maintenance conditions versus single cells from hESC colonies at low density in SFM for 3 d shows that when cells from hESC colonies are free of extrinsic signals their phenotype shifts from pluripotent to mixed endodermal and ectodermal. Two-way ANOVA revealed a significant interaction between gene expression and time in SFM alone (F(10,54) = 8.24, p < 0.0001). Bonferroni post-tests showed a significant decline in pluripotency genes OCT4 (p < 0.01), LIN28 (p < 0.01), and SOX2 (p < 0.05), as well as the mesoderm gene T (p < 0.001), along with a significant increase in expression of the neural gene SOX1 (p < 0.001), n = 4. b, When single cells from hES colonies were sorted on TRA-1-60 and SSEA4 expression and the resulting four fractions were plated at clonal density in SFM plus growth factors, only cells from the double-negative fraction could form neurospheres, indicating that the neurosphere-originating cell in an unsorted population is a TRA-1-60−/SSEA4− cell. One-way ANOVA showed a significant main effect of starting cell population (F(6,34) = 10.06, p < 0.0001), while Bonferroni's post-test revealed a significant difference in the sphere-forming capacity of TRA-1-60+/SSEA4+ cells vs TRA-1-60−/SSEA4− cells (p < 0.05), n = 7. c, FACS shows that a minority of cells in confluent cells from hES colonies are TRA-1-60−/SSEA4− (6.14 ± 1.35%), supporting the idea that neurospheres are arising solely from this fraction. The majority of hESCs in confluent colonies are TRA-1-60+/SSEA4+ (65.78 ± 3.23%), with the remaining 7.12 ± 2.31% being TRA-1-60+/SSEA4−, and 21.24 ± 3.84% being TRA-1-60−/SSEA4+, n = 9. d, A comparison of mRNA transcript levels by Q-PCR in TRA-1-60−/SSEA4− versus TRA-1-60+/SSEA4+ cells showed significant differences in pluripotency, and neural and endodermal gene expression (significant interaction between sorted fraction and gene expression by two-way ANOVA: F(10,58) = 14.67, p < 0.0001). The TRA-1-60−/SSEA4− fraction, which contains the neurosphere-forming cell, is characterized by the significant upregulation of neural genes SOX1 (p < 0.001) and PAX6 (p < 0.001), and endodermal genes FOXA2 (p < 0.001), HNF4A (p < 0.001), and GATA4 (p < 0.001); and downregulation of OCT4 (p < 0.001) (all by Bonferroni post-tests), n = 4. Data are represented as mean ± SEM; for Q-PCR, the highest expressing sample (value of “1”) was used as the calibrator.
To establish an expression profile of the neurosphere-forming cell, gene expression of TRA-1-60−/SSEA4− was compared with TRA-1-60+/SSEA4+ cells by Q-PCR. This analysis revealed a downregulation of pluripotency gene OCT4 along with an upregulation in the neural genes SOX1 and PAX6 and endodermal genes FOXA2, HNF4A, and GATA4 in the double-negative fraction (Fig. 5d). We predict that the TRA-1-60−/SSEA4− cells contain separate neural and endodermal populations. This is supported by studies on colony morphology demonstrating that extraembryonic endoderm-like cells tend to ring the outsides of hESC colonies, while small clusters of neuroepithelial-like cells are contained within (Peerani et al., 2007).
Noggin promotes neural fate specification only in hESC maintenance cultures
As previously described, neural fate specification in the embryo relies on the inhibition of BMPs by antagonists such as Noggin (Zimmerman et al., 1996; Muñoz-Sanjuán and Brivanlou, 2002). Neuroectodermal differentiation of hESCs is improved by culture with Noggin; however, this has been achieved by concomitant growth on transgenic MS5-Wnt1 stromal feeders that facilitated neural differentiation (Sonntag et al., 2007). In another study, Noggin treatment was administered to multicellular hESC aggregates that proliferated in minimal conditions and were highly but not exclusively enriched for neural progenitors (Itsykson et al., 2005). As previously mentioned, neural default studies suggest that Noggin promotes neural fate specification by inhibiting paracrine signaling in vivo (Zimmerman et al., 1996), while single cells grown at low cell densities will acquire a neural fate autonomously (Godsave and Slack, 1989; Grunz and Tacke, 1989; Sato and Sargent, 1989). This suggested that to use Noggin to direct differentiation in our clonal assay, it might be more effective to administer it to hESC colonies in maintenance culture conditions rather than to single cells in neurosphere-forming conditions. We added Noggin to hESC maintenance cultures each day for 6–7 d before colonies reached confluency. We then plated these Noggin-treated cells from hESC colonies in the clonal sphere-forming assay. Noggin-treated cells from hESC colonies produced significantly more neurospheres than controls (Fig. 6), indicating that inhibition of BMPs facilitates the process of neuralization of hESCs. To test the effects of Noggin on single cells from hESC colonies in low-density minimal conditions (SFM plus growth factors), we grew hESC colonies to confluency in the absence of Noggin and then added Noggin to our neurosphere culture. In this case, there was no increase in the number of spheres produced (Fig. 6), indicating that inhibition of BMPs by Noggin mediates the hESC-to-NSC transition by inhibiting paracrine signaling in high-density ESC maintenance cultures rather than inhibiting autocrine signaling in low-density, minimal culture conditions.
Figure 6.

Noggin treatment of hESC colonies enhances the generation of NSCs but administration of Noggin to low-density neurosphere culture has no effect. When Noggin was administered as a “treatment” to hESC colonies grown to confluency in maintenance conditions, these cells went on to produce significantly more neurospheres than when Noggin was added directly to single cells in hESC colonies in neurosphere-forming minimal conditions (“Noggin in SFM”). Data are represented as a fold of control (neurosphere formation from single cells grown in SFM plus growth factors in the absence of Noggin). Two-way ANOVA shows a main effect of treatment (F(1,8) = 7.17, p = 0.03), n = 3. This result illustrates that Noggin-mediated neuralization takes place when cells are grown at high densities, rather than at low (clonal) densities, where paracrine signaling has ceased. Data are represented as mean ± SEM.
Transplanted clonal NSCs integrate into host mouse striatal tissue, differentiate into neurons and glia, and do not produce tumors
To evaluate the tumorigenicity of clonal NSCs and to assess their ability to integrate and form neurons and glia in vivo, clonal NSCs were transplanted into or near the striatum of neonatal mice. After 3 weeks, clonal NSC progeny differentiated to form separate neural precursor cells positive for NES (Fig. 7a,b), neurons positive for NF (Fig. 7c,d), and glia positive for GFAP (Fig. 7e,f). Transgene shutoff during differentiation of transplanted cells was noted, as many cells positive for human specific NES did not have RFP+ nuclei (Fig. 7a). Nonetheless, only cells with RFP+ nuclei were scored, so differentiated cell counts may underestimate the percentage of human cells positive for each marker. Transplanted clonal NSCs integrated into mouse tissue (Fig. 7a,c,e) and, importantly, did not produce tumors in any of the hosts, whereas tumors arose in two of three mice transplanted with hESCs (Fig. 7g–i). Despite the residual pluripotency displayed by neurosphere cells cultured in the presence of the ROCK inhibitor, these cells did not produce tumors when transplanted in hosts (n = 4).
Figure 7.
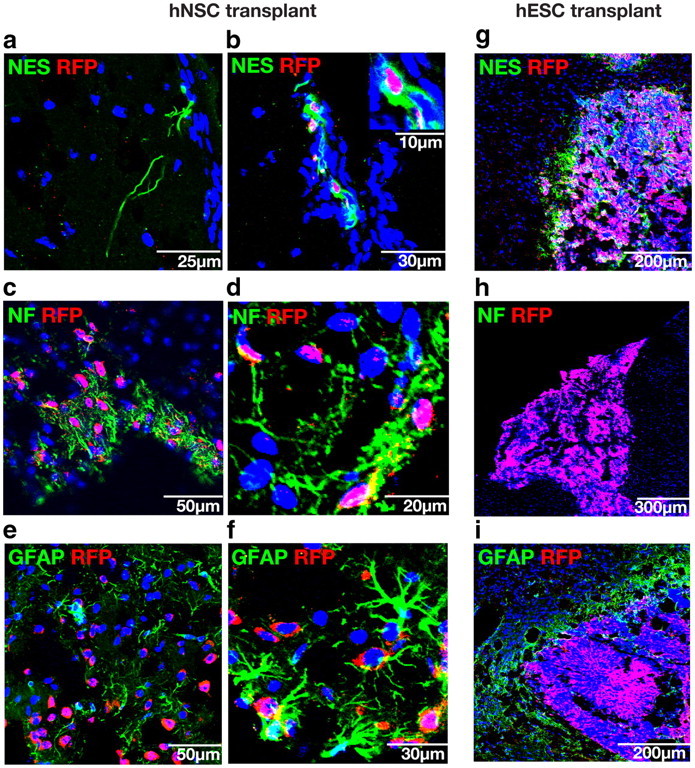
Transplanted clonal hNSCs integrate into host tissue, differentiate into neurons and glia, and do not form tumors, while transplanted hESCs do not integrate and form tumors. a, c, e, Integration and differentiation of transplanted human cells into host tissue. b, d, f, Quantification of marker proteins in transplanted hESC-derived clonal neurosphere cells grown without ROCK inhibitor shows that 16.1 ± 3.3% (n = 3) are NES+ (b); 35.25%±8.1% (n = 4) are NF+ (d); and 4.9%±0.03% (n = 3) are GFAP+ (f). a, Transgene shutoff occurred in some differentiated cells, confirmed by human specific antibody staining in the absence of RFP+ nuclei. g–i, Transplanted hESCs do not integrate and give rise to hyperproliferative masses in two of three hosts. All nuclei were counterstained with Hoechst stain (blue). Data are represented as mean ± SEM.
Discussion
This article reports the development of an assay that allows for the clonal isolation of NSCs from hESC colonies. In the absence of serum, feeder layers, or EB formation, single cells from hESC colonies differentiate into colony-forming NSCs that can be propagated, expanded long term, produce progenitor cells that differentiate into neurons and glia in vitro and in vivo, and are not tumorigenic in vivo. There have been many previous studies reporting the derivation of neural cells from hESCs; however, these protocols have relied on high-density cell culture (Nistor et al., 2005; Chambers et al., 2009), long-term differentiation (Reubinoff et al., 2001), and EB formation (Schuldiner et al., 2001; Zhang et al., 2001; Kang et al., 2007; Elkabetz et al., 2008). Under high-density culture conditions, it is impossible to assess the properties of an NSC, and there is potential carryover of teratoma-forming ES cells to transplantable populations. Some of these studies also describe the formation of neurospheres as a method of neural precursor selection (Reubinoff et al., 2001; Zhang et al., 2001; Nistor et al., 2005; Kang et al., 2007), but, again, this is done at high cell densities or with cell clusters. It must be emphasized that the protocol presented in this article produces clonal neurospheres. This enables us to distinguish multipotent NSCs from other cell types. We also observed differentiation of these clonal neurospheres into neurons and astrocytes similar to that reported by Reubinoff et al. (2001) and Zhang et al. (2001). As was found in those studies, oligodendrocyte formation was very low in our differentiation cultures, possibly because oligodendrocytes require specific growth factors and much longer periods for differentiation to mature, morphologically distinct phenotypes (Nistor et al., 2005; Kang et al., 2007).
The current study defines the clonal neurosphere-forming cell as a TRA-1-60−/SSEA4− cell that grows within hESC colonies and reveals that BMP inhibition with Noggin mediates the acquisition of this identity. It is possible that pluripotent TRA-1-60+/SSEA4+ hESCs could form neurospheres directly but that the cell death associated with dissociation of pluripotent cells to single cells prevents this. Cloning of single pluripotent hESCs is notoriously difficult. Perhaps this has less to do with the sensitivity of single cells than the culture requirements of colony-forming cells. Initial studies on the dependence of undifferentiated culture of hESCs on bFGF examined heterogeneous populations of cells for the presence of FGF receptors but assumed it was the undifferentiated fraction that possessed them (Xu et al., 2005a,b). It has since been shown that all known FGF receptors are expressed on the subfraction of hESCs that lack OCT4 expression and are incapable of forming pluripotent colonies. A signaling loop is established between differentiated cells that receive FGF and in turn secrete insulin-like growth factor (IGF) for which pluripotent, clonogenic hESCs have receptors (Bendall et al., 2007). We propose that in the minimal conditions we use to generate clonal neurospheres, cell densities are so low that paracrine signaling ceases between differentiated cells and pluripotent cells, such that there is insufficient IGF for pluripotent cells to survive. Being FGF dependent, the TRA-1-60−/SSEA4− fraction is sustained by growth factor supplementation and proliferates to form clonal NSC colonies. We observed that SOX1 was expressed in the TRA-1-60−/SSEA4− population and in neurospheres but not in hESCs. This suggests that Sox-1 may be a specific, early marker for identifying neural stem cells.
Neural fate specification in mESCs takes place in the absence of extrinsic signals, paralleling a model of BMP inhibition in vivo (Tropepe et al., 2001; Smukler et al., 2006). Although studies using hESCs have supported the concept of neural differentiation by default, the analyses were not clonal (Schulz et al., 2004; Vallier et al., 2004). In our clonal neural default assay, substantial cell death and the heterogeneity of starting cell populations confound this experimental approach. However, our finding that pluripotent hESCs do not become entirely neural when grown in serum-free conditions in the absence of growth factors does not rule out a neural default program. The ability of the BMP inhibitor Noggin to increase the frequency of sphere formation when added to hESC maintenance culture but not sphere-formation media suggests that neural specification occurs in hESC culture by the removal of an inhibitor (the in vivo demonstration of a default mechanism). The inherent heterogeneity of hESC cultures could be creating local signaling microenvironments (akin to the regional inhibitory environment that leads to neural specification in the embryo) that can influence cell fate along extraembryonic endodermal or neuroectodermal lineages (Sonntag et al., 2007). However, it is only neurally specified cells that survive and proliferate to form colonies in serum-free media, suggesting that the neural cells of the TRA-1-60−/SSEA4− fraction have a survival advantage. These minimal conditions also seem to select for dNSCs rather than the pNSCs that are selected from mESCs in neural default conditions (Smukler et al., 2006) because, first, the human cells have been specified previously in maintenance hESC culture conditions, and, second, only differentiated FGF-dependent human NSCs (hNSCs) survive in minimal neural culture conditions. Indeed, the lack of dependence of hESCs and their early progeny on exogenous LIF suggests that it is impossible to use LIF in minimal culture conditions to isolate a pNSC from hESCs as is done in mouse ESC cultures. Recently, LIF-dependent “naive” hESCs were generated from normal or “primed” hESCs by ectopic expression of OCT4, KLF2, and KLF4 (Hanna et al., 2010). The use of these naive hESCs in neural default studies may better aid the investigation of the acquisition of neural identity by pluripotent cells. The mechanism by which TRA-1-60+/SSEA4+ cells are prespecified to TRA-1-60−/SSEA4− cells in maintenance cultures has yet to be elucidated. As such we have not succeeded in completely specifying the mechanisms underlying the direct differentiation of pluripotent hESCs to hNSCs.
The influence on cell fate of certain culture agents begs further investigation in light of our findings that neurospheres cultured with ROCK inhibitor harbor cells with some pluripotent ability even after multiple passages in conditions that would normally specify cells irreversibly to a neural lineage. We observed that LIN28 expression decreased significantly in spheres grown without ROCK inhibitor but was maintained in neurospheres cultured in the presence of ROCK inhibitor. Also, though OCT4 expression has essentially been shut off in tertiary spheres that are able to re-form ES-like colonies, LIN28 expression is maintained. LIN28 is an RNA-binding protein highly expressed in undifferentiated hESCs (Balzer et al., 2010) and is known to regulate OCT4 (Qiu et al., 2010). It is possible that sustained expression of LIN28 is the mechanism by which spheres grown in ROCK inhibitor maintain an ability to revert to a pluripotent phenotype. It is unclear whether the addition of ROCK inhibitor to our sphere-formation cultures selected for a different, more pluripotent, proliferative cell, or altered the fate of the NSC. In either case, these cells were specified but not committed to the neural lineage, leaving the potential for them to form other, possibly tumorigenic, cell types. Interestingly, in vivo transplantation of NSCs grown with ROCK inhibitor did not produce tumors in hosts.
The importance of this work lies in the clonality of the method. The ability to select and expand single hESC colony-derived NSCs is crucial for a variety of reasons. hESC colonies are heterogeneous, containing cells with differing developmental potential. Dissection of neuroepithelial rosettes from hESC cultures and/or uncontrolled differentiation to neural subtypes through an EB stage has produced mixed populations of cells, some of which retain unsuitable capabilities such as teratoma formation in graft recipients (Brederlau et al., 2006; Roy et al., 2006). Commitment to the neural lineage must be stable and complete before cells are expanded for the purpose of transplantation, and this process can by aided by the selection and expansion of single neural stem cells. The propensity of hESCs maintained in vitro to undergo genetic and epigenetic alterations (Draper et al., 2004; Maitra et al., 2005), as well as the standard heterogeneity of colonies, make clonal selection of untransformed and specified cells a prudent step in the differentiation and expansion of hESCs to transplantable cell types.
Footnotes
This work was funded by the Canadian Institutes of Health Research, the Canadian Stem Cell Network, the Regenerative Medicine Development Centre, an Ontario Ministry of Economic Development and Innovation (MEDI) grant, the Krembil Foundation, and the Ontario Graduate Scholarship Program.
References
- Balzer E, Heine C, Jiang Q, Lee VM, Moss EG. LIN 28 alters cell fate succession and acts independently of the let-7 microRNA during neurogliogenesis in vitro. Development. 2010;137:891–900. doi: 10.1242/dev.042895. [DOI] [PubMed] [Google Scholar]
- Bendall SC, Stewart MH, Menendez P, George D, Vijayaragavan K, Werbowetski-Ogilvie T, Ramos-Mejia V, Rouleau A, Yang J, Bossé M, Lajoie G, Bhatia M. IGF and FGF cooperatively establish the regulatory stem cell niche of pluripotent human cells in vitro. Nature. 2007;448:1015–1021. doi: 10.1038/nature06027. [DOI] [PubMed] [Google Scholar]
- Brederlau A, Correia AS, Anisimov SV, Elmi M, Paul G, Roybon L, Morizane A, Bergquist F, Riebe I, Nannmark U, Carta M, Hanse E, Takahashi J, Sasai Y, Funa K, Brundin P, Eriksson PS, Li JY. Transplantation of human embryonic stem cell-derived cells into a rat model of Parkinson's disease: effect of in vitro differentiation on graft survival and teratoma formation. Stem Cells. 2006;24:1433–1440. doi: 10.1634/stemcells.2005-0393. [DOI] [PubMed] [Google Scholar]
- Cai J, Yang M, Poremsky E, Kidd S, Schneider JS, Iacovitti L. Host-dependent tumorigenesis of embryonic stem cell transplantation in experimental stroke. Stem Cells Dev. 2010;19:1017–1023. doi: 10.1089/scd.2009.0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti L, Pollard SM, Gorba T, Reitano E, Toselli M, Biella G, Sun Y, Sanzone S, Ying QL, Cattaneo E, Smith A. Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol. 2005;3:e283. doi: 10.1371/journal.pbio.0030283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang SM, Kyba M, Perlingeiro R, Daley GQ, Zandstra PW. Efficiency of embryoid body formation and hematopoietic development from embryonic stem cells in different culture systems. Biotechnol Bioeng. 2002;78:442–453. doi: 10.1002/bit.10220. [DOI] [PubMed] [Google Scholar]
- Dhara SK, Stice SL. Neural differentiation of human embryonic stem cells. J Cell Biochem. 2008;105:633–640. doi: 10.1002/jcb.21891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper JS, Smith K, Gokhale P, Moore HD, Maltby E, Johnson J, Meisner L, Zwaka TP, Thomson JA, Andrews PW. Recurrent gain of chromosomes 17q and 12 in cultured embryonic stem cells. Nat Biotechnol. 2004;22:53–54. doi: 10.1038/nbt922. [DOI] [PubMed] [Google Scholar]
- Elkabetz Y, Panagiotakos G, Al Shamy G, Socci ND, Tabar V, Studer L. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 2008;22:152–165. doi: 10.1101/gad.1616208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdö F, Bührle C, Blunk J, Hoehn M, Xia Y, Fleischmann B, Föcking M, Küstermann E, Kolossov E, Hescheler J, Hossmann KA, Trapp T. Host-dependent tumorigenesis of embryonic stem cell transplantation in experimental stroke. J Cereb Blood Flow Metab. 2003;23:780–785. doi: 10.1097/01.WCB.0000071886.63724.FB. [DOI] [PubMed] [Google Scholar]
- Godsave SF, Slack JM. Clonal analysis of mesoderm induction in Xenopus laevis. Dev Biol. 1989;134:486–490. doi: 10.1016/0012-1606(89)90122-x. [DOI] [PubMed] [Google Scholar]
- Grunz H, Tacke L. Neural differentiation of Xenopus Laevis ectoderm takes place after disaggregation and delayed reaggregation without inducer. Cell Differ Dev. 1989;28:211–217. doi: 10.1016/0922-3371(89)90006-3. [DOI] [PubMed] [Google Scholar]
- Hanna J, Cheng AW, Saha K, Kim J, Lengner CJ, Soldner F, Cassady JP, Muffat J, Carey BW, Jaenisch R. Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc Natl Acad Sci U S A. 2010;107:9222–9227. doi: 10.1073/pnas.1004584107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmati-Brivanlou A, Melton DA. Inhibition of activin receptor signalling promotes neuralization in Xenopus. Cell. 1994;77:273–281. doi: 10.1016/0092-8674(94)90319-0. [DOI] [PubMed] [Google Scholar]
- Henderson JK, Draper JS, Baillie HS, Fishel S, Thomson JA, Moore H, Andrews PW. Preimplantation human embryos and embryonic stem cells show comparable expression of stage-specific embryonic antigens. Stem Cells. 2002;20:329–337. doi: 10.1634/stemcells.20-4-329. [DOI] [PubMed] [Google Scholar]
- Hitoshi S, Seaberg RM, Koscik C, Alexson T, Kusunoki S, Kanazawa I, Tsuji S, van der Kooy D. Primitive neural stem cells from the mammalian epiblast differentiate to definitive neural stem cells under the control of Notch signalling. Genes Dev. 2004;18:1806–1811. doi: 10.1101/gad.1208404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itsykson P, Ilouz N, Turetsky T, Goldstein RS, Pera MF, Fishbein I, Segal M, Reubinoff BE. Derivation of neural precursors from human embryonic stem cells in the presence of noggin. Mol Cell Neurosci. 2005;30:24–36. doi: 10.1016/j.mcn.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Kang SM, Cho MS, Seo H, Yoon CJ, Oh SK, Choi YM, Kim DW. Efficient induction of oligodendrocytes from human embryonic stem cells. Stem Cells. 2007;25:419–424. doi: 10.1634/stemcells.2005-0482. [DOI] [PubMed] [Google Scholar]
- Koch P, Opitz T, Steinbeck JA, Ladewig J, Brüstle O. A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. Proc Natl Acad Sci U S A. 2009;106(9):3225–3230. doi: 10.1073/pnas.0808387106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis SA, Rietze RL, Deleyrolle L, Wagey RE, Thomas TE, Eaves AC, Reynolds BA. Enumeration of neural stem and progenitor cells in the neural colony-forming cell assay. Stem Cells. 2008;26:988–996. doi: 10.1634/stemcells.2007-0867. [DOI] [PubMed] [Google Scholar]
- Maitra A, Arking DE, Shivapurkar N, Ikeda M, Stastny V, Kassauei K, Sui G, Cutler DJ, Liu Y, Brimble SN, Noaksson K, Hyllner J, Schulz TC, Zeng X, Freed WJ, Crook J, Abraham S, Colman A, Sartipy P, Matsui S, et al. Genomic alterations in cultured human embryonic stem cells. Nat Genet. 2005;37:1099–1103. doi: 10.1038/ng1631. [DOI] [PubMed] [Google Scholar]
- Mitalipova MM, Rao RR, Hoyer DM, Johnson JA, Meisner LF, Jones KL, Dalton S, Stice SL. Preserving the genetic integrity of human embryonic stem cells. Nat Biotechnol. 2005;23:19–20. doi: 10.1038/nbt0105-19. [DOI] [PubMed] [Google Scholar]
- Muñoz-Sanjuán I, Brivanlou AH. Neural induction, the default model and embryonic stem cells. Nat Rev Neurosci. 2002;3:271–280. doi: 10.1038/nrn786. [DOI] [PubMed] [Google Scholar]
- Nistor GI, Totoiu MO, Haque N, Carpenter MK, Keirstead HS. Human embryonic stem cells differentiate into oligodendrocytes in high purity and myelinate after spinal cord transplantation. Glia. 2005;49:385–396. doi: 10.1002/glia.20127. [DOI] [PubMed] [Google Scholar]
- Peerani R, Rao BM, Bauwens C, Yin T, Wood GA, Nagy A, Kumacheva E, Zandstra PW. Niche-mediated control of human embryonic stem cell self renewal and differentiation. EMBO J. 2007;26:4744–4755. doi: 10.1038/sj.emboj.7601896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccolo S, Sasai Y, Lu B, De Robertis EM. Dorsoventral patterning in Xenopus: inhibition of ventral signals by direct binding of chordin to BMP-4. Cell. 1996;86:589–598. doi: 10.1016/s0092-8674(00)80132-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu C, Ma Y, Wang J, Peng S, Huang Y. Lin28-mediated post-transcriptional regulation of Oct4 expression in human embryonic stem cells. Nucleic Acids Res. 2010;38:1240–1248. doi: 10.1093/nar/gkp1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reubinoff BE, Itsykson P, Turetsky T, Pera MF, Reinhartz E, Itzik A, Ben-Hur T. Neural progenitors from human embryonic stem cells. Nat Biotechnol. 2001;19:1134–1140. doi: 10.1038/nbt1201-1134. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S. Clonal population analyses demonstrate than an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev Biol. 1996;175:1–13. doi: 10.1006/dbio.1996.0090. [DOI] [PubMed] [Google Scholar]
- Roy NS, Cleren C, Singh SK, Yang L, Beal MF, Goldman SA. Functional engraftment of human ES cell-derived dopaminergic neurons enriched by coculture with telomerase-immortalized mid-brain astrocytes. Nat Med. 2006;12:1259–1268. doi: 10.1038/nm1495. [DOI] [PubMed] [Google Scholar]
- Sato SM, Sargent TD. Development of neural inducing capacity in dissociated Xenopus embryos. Dev Biol. 1989;134:263–266. doi: 10.1016/0012-1606(89)90096-1. [DOI] [PubMed] [Google Scholar]
- Schuldiner M, Eiges R, Eden A, Yanuka O, Itskovitz-Eldor J, Goldstein RS, Benvenisty N. Induced neuronal differentiation of human embryonic stem cells. Brain Res. 2001;913:201–205. doi: 10.1016/s0006-8993(01)02776-7. [DOI] [PubMed] [Google Scholar]
- Schulz TC, Noggle SA, Palmarini GM, Weiler DA, Lyons IG, Pensa KA, Meedeniya AC, Davidson BP, Lambert NA, Condie BG. Differentiation of human embryonic stem cells to dopaminergic neurons in suspension culture. Stem Cells. 2004;22:1218–1238. doi: 10.1634/stemcells.2004-0114. [DOI] [PubMed] [Google Scholar]
- Seaberg RM, van der Kooy D. Adult neurogenic regions: the ventricular subependyma contains neural stem cells, but the dentate gyrus contains restricted progenitors. J Neurosci. 2002;22:1784–1793. doi: 10.1523/JNEUROSCI.22-05-01784.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smukler SR, Runciman SB, Xu S, van der Kooy D. Embryonic stem cells assume a primitive neural stem cell fate in the absence of extrinsic influences. J Cell Biol. 2006;172:79–90. doi: 10.1083/jcb.200508085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonntag KC, Pruszak J, Yoshizaki T, van Arensbergen J, Sanchez-Pernaute R, Isacson O. Enhanced yield of neuroepithelial precursors and midbrain-like dopaminergic neurons from human embryonic stem cells using the bone morphogenic protein antagonist noggin. Stem Cells. 2007;25:411–418. doi: 10.1634/stemcells.2006-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spemann H, Mangold H. Uber Induktion von embryonanlagen durch implantation artfrender organization. Arch Mikr Anat EntwMech. 1924;100:599–638. [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Tropepe V, Sibilia M, Ciruna BG, Rossant J, Wagner EF, van der Kooy D. Distinct neural stem cells proliferate in response to EGF and FGF in the developing mouse telencephalon. Dev Biol. 1999;208:166–188. doi: 10.1006/dbio.1998.9192. [DOI] [PubMed] [Google Scholar]
- Tropepe V, Hitoshi S, Sirard C, Mak TW, Rossant J, van der Kooy D. Direct neural fate specification from embryonic stem cells: a primitive mammalian neural stem cell stage acquired through a default mechanism. Neuron. 2001;30:65–78. doi: 10.1016/s0896-6273(01)00263-x. [DOI] [PubMed] [Google Scholar]
- Ungrin MD, Joshi C, Nica A, Bauwens C, Zandstra PW. Reproducible, ultra high-throughput formation of multicellular organization from single cell suspension-derived human embryonic stem cell aggregates. PLoS One. 2008;3:e1565. doi: 10.1371/journal.pone.0001565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallier L, Reynolds D, Pedersen RA. Nodal inhibits differentiation of human embryonic stem cells along the neuroectodermal default pathway. Dev Biol. 2004;275:403–421. doi: 10.1016/j.ydbio.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Vallier L, Touboul T, Chng Z, Brimpari M, Hannan N, Millan E, Smithers LE, Trotter M, Rugg-Gunn P, Weber A, Pedersen RA. Early cell fate decisions of human embryonic stem cells and mouse epiblast stem cells are controlled by the same signaling pathways. PLoS One. 2009;4:e6082. doi: 10.1371/journal.pone.0006082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T, Takahashi JB, Nishikawa S, Muguruma K, Sasai Y. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol. 2007;25:681–686. doi: 10.1038/nbt1310. [DOI] [PubMed] [Google Scholar]
- Wilson PA, Hemmati-Brivanlou A. Induction of epidermis and inhibition of neural fate by Bmp-4. Nature. 1995;376:331–333. doi: 10.1038/376331a0. [DOI] [PubMed] [Google Scholar]
- Xu C, Rosler E, Jiang J, Lebkowski JS, Gold JD, O'Sullivan C, Delavan-Boorsma K, Mok M, Bronstein A, Carpenter MK. Basic fibroblast growth factor supports undifferentiated human embryonic stem cell growth without conditioned medium. Stem Cells. 2005a;23:315–323. doi: 10.1634/stemcells.2004-0211. [DOI] [PubMed] [Google Scholar]
- Xu RH, Peck RM, Li DS, Feng X, Ludwig T, Thomson JA. Basic FGF and suppression of BMP signalling sustain undifferentiated proliferation of human ES cells. Nat Methods. 2005b;2:185–190. doi: 10.1038/nmeth744. [DOI] [PubMed] [Google Scholar]
- Zhang SC, Wernig M, Duncan ID, Brüstle O, Thomson JA. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol. 2001;19:1129–1133. doi: 10.1038/nbt1201-1129. [DOI] [PubMed] [Google Scholar]
- Zimmerman LB, De Jesús-Escobar JM, Harland RM. The Spemann organizer signal noggin binds and inactivates bone morphogenic protein 4. Cell. 1996;86:599–606. doi: 10.1016/s0092-8674(00)80133-6. [DOI] [PubMed] [Google Scholar]