

Abstract
Neuronal and synaptic degeneration are the best pathological correlates for memory decline in Alzheimer's disease (AD). Although the accumulation of soluble low-molecular-weight amyloid-β (Aβ) oligomers has been suggested to trigger neurodegeneration in AD, animal models overexpressing or infused with Aβ lack neuronal loss at the onset of memory deficits. Using a novel in vivo approach, we found that repeated hippocampal injections of small soluble Aβ1–42 oligomers in awake, freely moving mice were able to induce marked neuronal loss, tau hyperphosphorylation, and deficits in hippocampus-dependent memory. The neurotoxicity of small Aβ1–42 species was observed in vivo as well as in vitro in association with increased caspase-3 activity and reduced levels of the NMDA receptor subunit NR2B. We found that the sequestering agent transthyretin is able to bind the toxic Aβ1–42 species and attenuated the loss of neurons and memory deficits. Our novel mouse model provides evidence that small, soluble Aβ1–42 oligomers are able to induce extensive neuronal loss in vivo and initiate a cascade of events that mimic the key neuropathological hallmarks of AD.
Introduction
Substantial synaptic and neuronal losses are observed in the early stages of Alzheimer's disease (AD) when the loss of hippocampus-dependent memory becomes clinically detectable (Gómez-Isla et al., 1996; Scheff et al., 2006; Crews and Masliah, 2010). The perforant pathway provides input from the entorhinal cortex (EC) to the dentate gyrus (DG), and is one of the earliest and most severely affected pathways in AD (Gómez-Isla et al., 1996; Scheff et al., 2006). Individuals with mild cognitive impairment (MCI), a prodromal state of AD, have 32% fewer neurons in the EC and show synaptic loss in the DG, which correlates with cognitive deficits (Gómez-Isla et al., 1996; Scheff et al., 2006). This cellular disconnection suggests that the DG may be one of the earliest sites to display synaptic defects, and that attenuating degeneration in this area could prove useful in preventing memory deficits in AD.
Recent studies have emphasized the toxic role of soluble low-molecular-weight amyloid-β (Aβ) oligomers such as dimers, trimers, tetramers, nonamers, and dodecamers, which have all been individually identified as the main neurotoxic culprit (Lambert et al., 1998; Lesné et al., 2006; Shankar et al., 2008; Ono et al., 2009). Given the rapid oligomerization of Aβ, some authors have suggested that toxicity could be due to Aβ species ranging from 10 to 100 kDa present in the AD brain at the same time, rather than to just one particular type of oligomer (McLean et al., 1999; Hepler et al., 2006; Martins et al., 2008).
Thus far, the neurotoxic effect of low-molecular-weight Aβ oligomers was tested exclusively on primary neuronal cultures or organotypic brain slices since no appropriate animal model is currently available to determine the neurodegenerative effect of Aβ species in vivo (Lambert et al., 1998; Lesné et al., 2006; Hung et al., 2008; Shankar et al., 2008). Some of these studies have observed memory impairment following single brain infusion (Lesné et al., 2006; Shankar et al., 2008), but the toxicity of small Aβ1–42 oligomers in vivo still needs to be established. The Aβ toxicity could not be determined using an osmotic pump system for prolonged administration of more mature Aβ species, which dynamically continues to oligomerize during this time and may stick into the pump (Malm et al., 2006; Miller et al., 2009; Ji et al., 2011). Other reports have shown that transgenic models overexpressing Aβ develop early synaptic alterations but lack the extensive cell death seen at the onset of memory decline in AD (Irizarry et al., 1997; Chishti et al., 2001; Oddo et al., 2003).
In this study, we developed a new in vivo approach to analyze the toxic effects of small, soluble Aβ1–42 oligomers. To avoid any confounding effects of anesthetic agents on intracellular pathways and tau metabolism (Planel et al., 2007), Aβ1–42 injections were performed in awake, freely moving mice. We found that repeated intrahippocampal injections led to Aβ deposition, marked neuronal loss, and tau hyperphosphorylation and that the sequestering agent transthyretin (TTR) attenuated the effects of Aβ1–42 oligomers.
Materials and Methods
Mice.
Twelve-month-old female C57BL/6 mice were used in this study. All experiments were performed with the approval of the local Animal Resources Committee, following standards for the care and use of laboratory animals and French and European Community rules.
Surgical procedures and injection.
Bilateral cannulae (328OPD-2.8/Spc with a removable dummy wire; Plastics One) were stereotaxically implanted into the DG of the hippocampus (coordinates with respect to bregma: −2.2 mm anteroposterior [AP], ± 1.4 mm mediolateral [ML], −2.1 mm dorsoventral [DV], according to the Paxinos and Franklin mouse brain atlas) (Paxinos and Watson, 2005) in anesthetized mice (100 mg/kg of ketamine and 10 mg/kg of xylazine, i.p.). After 1 week of recovery, awake and freely moving mice were injected with 0.2 μg/μl of soluble Aβ1–42 oligomers (rPeptide) coincubated or not with full-length native human TTR (Abcam), 0.2 μg/μl of scrambled Aβ1–42 (rPeptide), and an equal volume (2 μl) of vehicle buffer (50 mm Tris, 1 mm EDTA, pH 7.5) at a rate of 0.4 μl/min via cannulae PE50 tubing (Plastics One) connected to a 10 μl Hamilton syringe pump system (KDS310; KD Scientific). The tubing was left in place for another 3 min at the end of each injection, and the cannulae capped to prevent reflux of the injected solution.
Aβ preparation and TTR-Aβ mixture.
Aβ1–42 peptide solution was prepared as described previously (Kuperstein et al., 2010; Broersen et al., 2011). Briefly, Aβ1–42 peptide and scrambled Aβ1–42 were dissolved at a concentration of 1 mg/ml in 99% hexafluoroisopropanol (HFIP) (Sigma-Aldrich). The HFIP was evaporated using a gentle stream of nitrogen gas and the peptide film was dissolved at a final concentration of 1 mg/ml in dimethylsulfoxide (DMSO; Sigma-Aldrich). Complete removal of DMSO was achieved by eluting Aβ1–42 peptide from a 5 ml HiTrap desalting column (GE Healthcare) with 50 mm Tris, 1 mm EDTA buffer at pH 7.5. Aβ1–42 concentration was measured using a BCA protein assay kit (Pierce). Aβ1–42 was kept on ice until the experiments were started with a maximum lag time of 30 min. Samples were kept on ice until the experiments were started, with a maximum lag time of 30 min. The biophysical and biological properties of this Aβ preparation had been previously characterized by others (Kuperstein et al., 2010; Broersen et al., 2011). The preparation was checked by transmission electron microscopy (TEM): aliquots (5 μl) of the Aβ1–42 preparation were adsorbed to carbon-coated grids (Euromedex) for 1 min. The grids were blotted, washed twice in droplets of Milli-Q water, and stained with 1% (w/v) uranyl acetate. Samples were studied with a Zeiss EM902 microscope. Full-length native human TTR was freshly diluted in PBS 10 mm before adding Aβ at the molar ratio indicated in the text. The neutral pH mixture was kept at 37°C during 24 h before administration in vitro and in vivo.
Primary hippocampal culture.
Primary cultures of hippocampal neurons were prepared from C57BL/6 mouse fetuses at embryonic day 16. Hippocampal tissues were treated with 0.25% trypsin for 20 min at 37°C and dissociated by trituration. Cells were plated onto poly-d-lysine-coated (0.1 mg/ml) wells in Minimum Essential Medium supplemented with 10% horse serum, 25 mm d-glucose, 2 mm l-glutamine, 3 mm sodium pyruvate, and 1% penicillin/streptomycin/ amphotericin (Sigma-Aldrich), and incubated at 37°C in a humidified 5% CO2 atmosphere. After 3 h all media were replaced with Neurobasal medium supplemented with 2% B27, 0.5 mm l-glutamine, and 0.5% penicillin/streptomycin/amphotericin, and primary neurons were cultured for 7 d.
MTS and resazurin cell viability assays.
The percentage of living neurons in culture was evaluated by the measurement of absorbance following MTS or resazurin bioreduction, according to the manufacturer's protocol (Promega). Briefly, MTS or resazurin was added to the Neurobasal medium containing the cells (v/v ratio of 1:5). After 1.5 h of incubation at 37°C, absorbance was recorded at 490 nm for the MTS assay and at 570 nm for the resazurin assay.
Western blot analysis.
Primary hippocampal cultures were washed with cold 10 mm PBS and scraped into cold radioimmunoprecipitation assay (RIPA) buffer (Pierce) supplemented with 0.5% CHAPS (GE Healthcare), protease inhibitors (Roche), 0.125 mm okadaic acid, and 1 mm sodium orthovanadate. For immunoblot analyses following in vivo experiments, hippocampus sections extending 1 mm on either side of the injection site were quickly dissected on ice using a coronal brain matrix (EMS). The tissue was immediately frozen on dry ice before being stored at −80°C until use, and homogenized into cold RIPA solution. In vitro and in vivo samples were sonicated, gently agitated for 45 min at 4°C, and centrifuged at 11500 × g for 20 min at 4°C. The total protein content of the resulting supernatant was determined using a BCA protein assay kit.
Equal amounts of total protein (30 μg/lane; 0.4 μg/lane for Aβ1–42 preparations) were separated by SDS-NuPAGE Novex 4–12% Bis-Tris gel electrophoresis (Invitrogen) and transferred to Hybond-ECL membranes (GE Healthcare) by electroblotting. Membranes were incubated with blotting buffer containing 5% nonfat milk for 1 h at room temperature and incubated with the appropriate primary antibody at 4°C overnight. Tau phosphorylation was detected using AT8 (S202/T205), AT180 (T231), and AT270 (T181) antibodies, all purchased from Pierce; Tau pS422 (Invitrogen); and AD2 against pS396/404 (Bio-Rad). Total tau was investigated with an antibody which recognizes 19 phospho-independent amino acids in the tau N-terminal region. The other antibodies used for immunoblotting were as follows: anti-Aβ 6E10 (Covance), anti-spinophilin (Millipore), anti-procaspase-3 and cleaved caspase-3 (Cell Signaling Technology), anti-NR2B (Cell Signaling Technology), and anti-GAPDH (Santa Cruz Biotechnology).
Following primary antibody incubation, the membranes were washed, treated with an horseradish peroxidase-labeled anti-rabbit or anti-mouse secondary antibody (Vector Laboratories) for 1 h, washed again, and chemiluminescence visualized by an ECL detection kit (GE Healthcare) using Image Reader LAS-3000 (Fujifilm). Quantitative densitometric analysis was performed using Multi-Gauge V3.0 software (Fujifilm). Results are all expressed normalized to GAPDH.
Formic acid treatment for insoluble Aβ1-42.
To retrieve insoluble Aβ1–42 stuck on the side of low-adhesion tubes, the solution containing soluble Aβ1–42 was removed and 75 μl of 100% formic acid was added to the tube and vortexed for 1 min. The formic acid was evaporated using a gentle stream of nitrogen gas and loading buffer containing 2 mm PBS, 60% NuPAGE LDS buffer (Invitrogen), and 20% NuPAGE reducing agent (Invitrogen) was added for immunoblot analysis.
Immunohistochemistry and staining with cresyl violet and Fluoro-Jade B.
Mice were anesthetized and transcardially perfused with 0.9% NaCl followed by 4% paraformaldehyde (PFA) in 0.1 m PBS (pH 7.4). Brains were dissected, immersed in 4% PFA for 48 h, incubated in 0.1 m PBS containing 15% sucrose for 24 h and finally frozen and stored until use. Immunohistochemical staining was performed in free-floating coronal sections (40 μm) obtained using a cryostat (Leica). Sections were treated with 0.01 m PBS containing 0.3% H2O2 for 30 min, blocked in 1% goat serum for 1 h, and incubated overnight at 4°C with 6E10 or anti-TTR antibody (Abcam) diluted in 0.01 m PBS containing 0.2% Triton X-100. Sections were treated with 80% formic acid for 3 min before applying the 6E10 antibody. Biotinylated antibodies were detected by using the ABC complex (Vector Laboratories) at room temperature for 2 h and labeling was visualized by incubating sections with 0.1 m Tris buffer containing 0.05% 3,3′-diaminobenzidine (Sigma-Aldrich) and 0.05% H2O2. Sections were rinsed three times for 10 min in 0.01 m PBS with 0.2% Triton X-100 after each step. Sections were mounted on gelatin-coated glass slides, air-dried for 2 h, dehydrated in an ascending alcohol series (30, 70, 95, and 100%; 5 min each), incubated three times in toluene for 5 min, and coverslipped with VectaMount Permanent (Vector Laboratories).
For cresyl violet staining, mounted cryosections were rinsed for 1 min in 0.01 m PBS, incubated in a 0.5% cresyl violet solution (Sigma-Aldrich), transferred for 1 min to a 0.5% acetic acid solution, decolored in an alcohol series (70, 95, and 100%; 20 s each), and finally immersed in toluene (three times for 5 min each) before being coverslipped. Staining with Fluoro-Jade B (Millipore), a marker for damaged neurons, has previously been described in detail (Schmued and Hopkins, 2000). Positive Fluoro-Jade B objects around the site of injection for each animal were quantified on five hippocampal slices that were 200 μm apart. Quantifications were made by two independent researchers (N.Z. and R.C.), and both observers were blinded to treatment group membership.
Images for immunohistochemical and cresyl violet staining were captured using a Leica DMRB microscope equipped with a Leica DC 300FX digital camera and a Leica 2.5× objective. Fluoro-Jade B staining was analyzed using an Axio Imager.Z1 ApoTome microscope (Zeiss) equipped with an AxioCam MRm Zeiss camera and visualized with a green filter.
Passive avoidance conditioning.
Passive avoidance training was performed 24 h after the last Aβ injection as described previously (Martins et al., 2008). Briefly, mice bilaterally injected in the DG with Aβ1–42 oligomers, TTR-Aβ, or vehicle were trained at this memory task 24 h after the end of the six daily injections. During training, we recorded latency to enter the dark chamber where a slight footshock (0.3 mA, 2 s) was delivered. Retention was tested 24 h later with a criterion time of 300 s.
Y-maze test.
Five days after the last injection, mice were tested for hippocampus-dependent spatial memory using a two-trial Y-maze task as described previously (Troquier et al., 2012). Briefly, the arms of the maze were 22 cm long, 6.4 cm wide, and 15 cm deep. The floor of the maze was covered with sawdust that was mixed after each trial to eliminate olfactory cues. Various extra-maze cues were placed on the surrounding walls. Experiments were conducted with an ambient light level of 6 lux. During the exposure phase, mice were assigned to two arms (the “start arm” and the “other arm”) that they were allowed to explore ad libitum for 5 min, without access to the third arm of the maze (the “novel arm”) blocked by an opaque door. The assignment of arms was counterbalanced within each experimental group. Mice were then removed from the maze and returned to their home cage for 2 min. During the test phase, mice were placed at the end of the same start arm and allowed to explore ad libitum all three arms for 2 min. The amount of time spent in each of the arms was recorded using EthovisionXT (Noldus).
Data analysis and statistics.
Statistical analysis of multiple samples was performed using the appropriate ANOVA, and simple main effects were analyzed using the F test when the interaction was significant. Tukey's HSD test was used for post hoc pairwise comparisons when necessary. The statistical significance between two samples was analyzed using an unpaired Student's t test. The α level used was p < 0.05.
Results
Profile of soluble Aβ1–42 oligomers
Given the complexity of the biophysical environment of Aβ aggregation in vivo, we analyzed the dynamic features of this process under simplified and controlled conditions in vitro using synthetic Aβ preparations. To identify toxic Aβ species, we used a standardized assay to generate oligomers from synthetic Aβ that were previously assessed under denaturing and nondenaturing conditions using TEM, atomic force microscopy, Fourier transform infrared spectroscopy, electrospray-ionization mass spectrometry, and nuclear magnetic resonance spectroscopy (Kuperstein et al., 2010; Broersen et al., 2011). As previously shown by others at both the biophysical and biological levels (Kuperstein et al., 2010; Broersen et al., 2011; Soura et al., 2012), the preparation of soluble Aβ1–42 oligomers used here is almost exclusively composed of low-molecular-weight Aβ1–42 species, including monomers, dimers, trimers, and tetramers (Fig. 1A).
Figure 1.
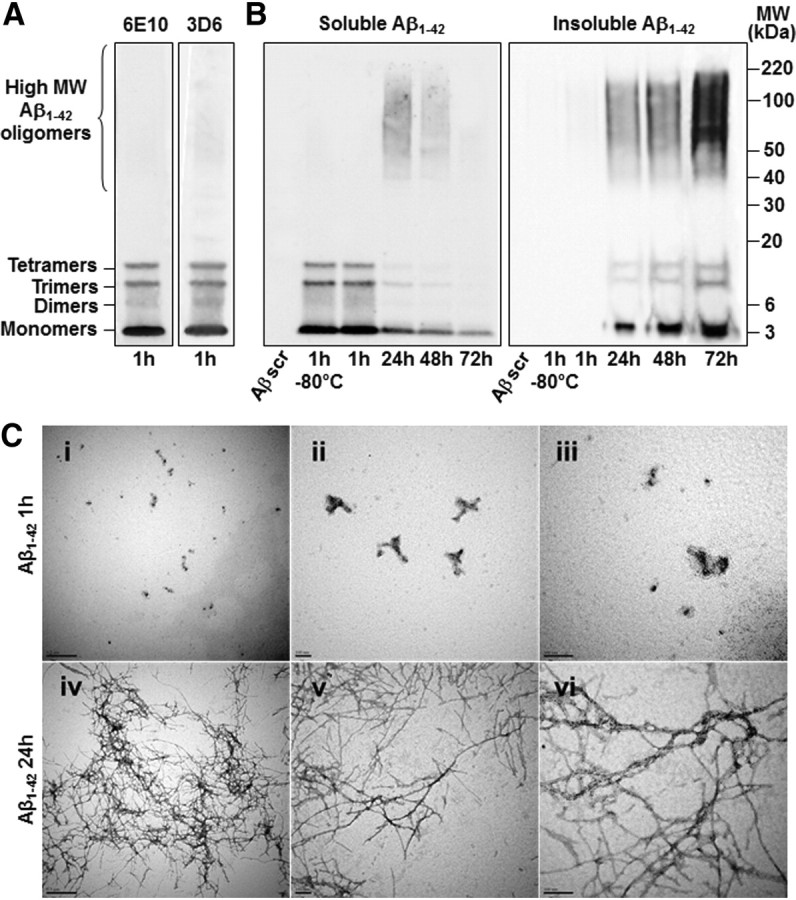
General profiles of Aβ1–42 oligomers injected in vivo and oligomerization of Aβ1–42 over time. A, Representative immunoblot of the Aβ1–42 solution used in our mouse model. The Aβ1–42 preparation is kept at 25°C for 1 h before administration and is almost exclusively composed of low-molecular-weight Aβ1–42 oligomers as revealed by electrophoresis on an SDS-NuPAGE Novex 4–12% Bis-Tris gel system using the Aβ antibodies 6E10 and 3D6. B, Representative 6E10-immunoblots of Aβ1–42 oligomerization. Soluble Aβ1–42 forms higher molecular weight (MW) oligomers over time and its concentration decreases in favor of insoluble Aβ1–42 species, which were recovered from the side of low-adhesion tubes with formic acid treatment. Scrambled Aβ1–42 (Aβscr) is not recognized by the 6E10 antibody (n = 3 for each gel in 3 independent experiments). C, The Aβ1–42 solution was verified in nondenaturing condition by TEM 1 h (i–iii) and 24 h (iv–vi) after preparation (i and iv, ×30000, scale bars 0, 5 μm; ii and v, ×85000, scale bars 0, 1 μm; iii and vi, ×140000, scale bars 0, 1 μm).
In the in vivo approach that we describe here, the Aβ1–42 solution was freshly prepared on the first and fourth days of injection, and the remainder of the solution was aliquoted and conserved at −80°C for use in the next 2 d. As described previously, there was no change in the profile of Aβ1–42 oligomers between freshly prepared Aβ1–42 solution and that frozen at −80°C for 3 d (Fig. 1B) (Broersen et al., 2011). Small Aβ1–42 species obtained 1 h following preparation had dynamically oligomerized over time to form larger oligomers (Fig. 1B). The level of soluble Aβ1–42 oligomers was inversely proportional to that of insoluble oligomers, which were found to stick to the sides of low-adhesion tubes over time. These results were confirmed in nondenaturing conditions by TEM, where Aβ fibril formations were only apparent following incubation of Aβ1–42 solution during 24 h (Fig. 1C).
Repeated injections of soluble Aβ1–42 oligomers in mice
To examine the neurotoxic effect of Aβ in vivo, bilateral cannulae were implanted in the DG and collateral injection of vehicle and soluble Aβ1–42 oligomers was done once a day for 6 consecutive days (Fig. 2A). Twenty-four hours following the last injection, we observed that soluble Aβ1–42 oligomers had consistently accumulated in the DG, extending up to ∼1 mm on either side of the injection site. Pronounced neuronal death was seen in the vicinity of oligomer deposition and was accompanied by decreased levels of the NMDA receptor subunit NR2B and increased levels of cleaved caspase-3, the active form of the enzyme (Fig. 2B,C). Hippocampal protein extraction was performed to determine the profile of toxic Aβ1–42 oligomers that had accumulated for 3 d or 6 d (Fig. 2D). Similar proportions of the various Aβ1–42 oligomers were obtained following 3 and 6 d of injection, with more pronounced Aβ1–42 deposits after 6 d. These results suggest that low-molecular-weight Aβ oligomers are able to accumulate at the level of the cell bodies of the granule neurons in a time-dependent manner.
Figure 2.

Neuronal deaths induced by Aβ1–42-oligomer deposition and tau phosphorylation following repeated intrahippocampal injections. A, Bilateral cannulae were stereotaxically implanted in the DG. One week following surgery, Aβ1–42 oligomers (Aβ; 0.2 μg/μl; 2 μl) and vehicle (Ctl) were simultaneously injected collaterally in awake, freely moving mice once a day for 6 consecutive days and killed 24 h following the last injection. B, Representative accumulation of Aβ1–42 oligomers in the DG on a section immediately next to the cannulae insertion site. Bottom, Representative staining with Fluoro-Jade B, cresyl violet, cleaved caspase-3 antibody, and NR2B antibody. C, Quantification of Fluoro-Jade B staining within the DG 24 h following the last injection. Marked surface (MS) versus total surface (TS) counted (n = 4, ***p < 0.001). D, Representative immunoblots of Aβ1–42 oligomer profiles 24 h following the last injection. E, Representative 6E10-immunostained section showing cannulae insertion. F, Representative accumulation of Aβ1–42 oligomers and cell death when the Aβ1–42 preparation is injected in the more ventral part of the hippocampus (−3.4 mm AP, ±2.0 mm ML, −2.4 mm DV from bregma) in sections stained with the 6E10 antibody and cresyl violet. G, Representative DG sections stained with 6E10 antibody and cresyl violet following collateral injections (1 per day for 6 d) of vehicle (Ctl) and scrambled Aβ1–42 (Aβscr; 0.2 μg/μl; 2 μl). H, Representative immunoblots of tau phosphorylation in the dorsal hippocampus of mice injected collaterally with vehicle (Ctl) and Aβ1–42 oligomers for 6 d. Bottom, Densitometric quantification of changes expressed as the mean ratio of phospho-tau to total tau antibody (Tau N-ter) staining. Error bars indicate ± SEM. **p < 0.01. I, Representative AT8-immunostained section 24 h and 5 d after the last injection (n = 4, two independent experiments). Scale bars: 50 μm.
Cell death was not restricted to the dorsal hippocampus, but was also observed in the ventral part of the hippocampus where Aβ1–42 oligomers accumulated following modification of the injection site (Fig. 2F). As an additional control, scrambled Aβ1–42 peptide was injected into the hippocampus and had no neurotoxic effect when compared with vehicle (Fig. 2G). Immunolabeling with AT8 (S202/T205) revealed an increase in tau phosphorylation whereas AT180 (T231), AD2 (S396/404), AT270 (T181), Tau pS422, or the phosphorylation-independent total tau antibody Tau N-ter showed no signal difference between vehicle- and Aβ-injected hippocampus (Fig. 2H,I). Interestingly, no more AT8 staining was observed 5 d following the last Aβ injection (Fig. 2I), suggesting that tau phosphorylation returns to basal levels as Aβ is gradually washed out of the brain over time, as shown below (Fig. 3).
Figure 3.
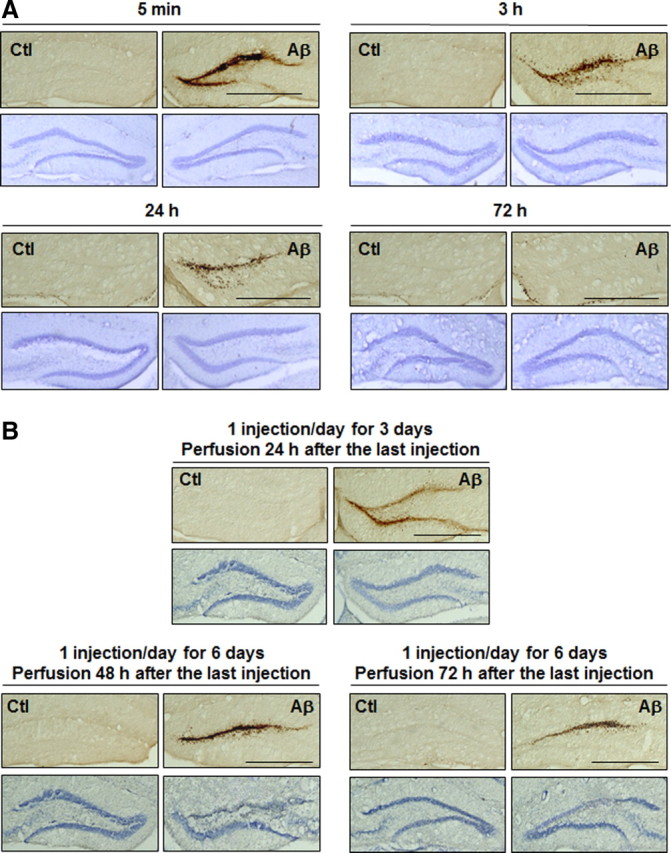
Time-dependent deposition of Aβ1–42 oligomers and neuronal death. Representative DG sections following single (A) or multiple (B) injections of Aβ1–42 oligomers (Aβ) and vehicle (Ctl) at the time points indicated and stained with the 6E10 antibody and cresyl violet. Scale bars: 50 μm (n = 4 for each time point, two independent experiments).
Time-dependent deposition of Aβ1–42 oligomers
Single and multiple injections were done to characterize the dynamic deposition of Aβ1–42 oligomers and neuronal death in the DG. Five minutes following a single injection, a large amount of low-molecular-weight Aβ1–42 oligomers was found in proximity of the injection site (Fig. 3A). Aβ1–42 oligomers were cleared gradually and no cell loss was observed at any time point following a single injection. In mice injected once a day for 3 d, a small but apparent neuronal death was seen in areas where higher levels of Aβ had accumulated (Fig. 3B). Aβ1–42 oligomers were also washed out over time following six injections and extensive neuronal loss was detected 48 and 72 h following the last injection.
Effects of Aβ1–42 oligomers in vitro
Soluble Aβ1–42 oligomers were added to primary hippocampal cultures to delineate the cellular mechanisms that could underlie their toxicity. Neuronal viability was decreased following treatment for 72 h with 2 μm of soluble Aβ1–42 oligomers as compared with vehicle alone or 2 μm of scrambled Aβ1–42 (Fig. 4A). Aβ1–42 species administered to cultured neurons induced tau hyperphosphorylation on the same epitope as in our in vivo model, namely Ser202 and Thr205 (Fig. 4B). Neuronal death induced by Aβ1–42 was accompanied by a decrease in the synaptic marker spinophilin at 24, 48, and 72 h, whereas the increased levels of spinophilin in vehicle-treated wells over time reflected the formation of new synaptic connections under control conditions (Fig. 4C). We observed that cleaved caspase-3, the active form of the enzyme, is increased during the process of neuronal death. Decreased levels of the NMDA receptor subunit NR2B were also found at all time points following Aβ1–42 treatment.
Figure 4.

Neurotoxicity induced by soluble Aβ1–42 oligomers in primary hippocampal culture is accompanied by tau phosphorylation, increased caspase-3 activity, and decreased levels of spinophilin and NR2B subunit. A, MTS and resazurin assays following 72 h treatment with vehicle (Veh), 2 μm of scrambled Aβ1–42 oligomers (Aβscr), or 2 μm of soluble Aβ1–42 oligomers (Aβ) kept at 25°C for 1 h before administration. The histograms are mean values expressed as a percentage of control (cultures without any treatment). B, C, Representative immunoblots of tau phosphorylation (B) as well as spinophilin, procaspase-3 and cleaved caspase-3, NR2B, and GAPDH (C) from neurons treated with vehicle (Ctl) or soluble Aβ1–42 oligomers (2 μm). For tau phosphorylation, densitometric quantification of changes is expressed as the mean ratio of phospho-tau to total tau antibody (Tau N-ter) staining. Results are all expressed normalized to GAPDH. Error bars indicate SEM. *p < 0.05, **p < 0.01, ***p < 0.001 (3 independent experiments performed in duplicate).
Neurotoxicity of Aβ1–42 oligomers 1 and 24 h after preparation
One strategy to prevent the neurotoxicity of soluble Aβ1–42 oligomers would be to find ligands that can bind to various Aβ1–42 species and prevent their deleterious effects. TTR is an interesting candidate because it has been found to be the major Aβ-binding protein in the CSF and to bind Aβ1–42 species with high affinity (Schwarzman et al., 1994; Carro et al., 2002). Since a coincubation time of 24 h has been found to be necessary for the formation of the TTR-Aβ complex in vitro at 37°C and neutral pH (Du and Murphy, 2010), we first assessed the toxicity of Aβ1–42 in these conditions. As reported previously, TTR tetramer assembly is strongly favored at neutral pH but can also dissociate slowly into monomers (Du and Murphy, 2010).
A clear self-association of smaller Aβ1–42 oligomers to form higher molecular weight Aβ1–42 oligomers was observed within 24 h at 37°C when compared with Aβ1–42 solution kept at 25°C for 1 h (Fig. 5A). Both preparations induced similar Aβ1–42 deposition and neuronal death following daily injection over 6 d (Fig. 5B). Cell death was also observed in neuronal cultures following application of Aβ1–42 preincubated for 1 h at 25°C or for 24 h at 37°C, although the solution containing smaller Aβ1–42 oligomers was more toxic (p < 0.05). The difference in profiles between Aβ1–42 solutions preincubated for 1 or 24 h was conserved following in vivo injections, with decreased amounts of low-molecular-weight species after 24 h of preincubation (Fig. 5A,C). The ∼25 kDa band observed in vehicle- and Aβ-treated hippocampi results from the nonspecific interaction of the anti-mouse secondary antibody used with the 6E10 antibody (Fig. 5C).
Figure 5.

Neurotoxicity induced by soluble Aβ1–42 oligomers prepared 1 or 24 h before administration. A, Profiles of Aβ1–42 oligomers kept at 25°C for 1 h (Aβ 1 h) or 37°C for 24 h (Aβ 24 h) before administration. B, Representative DG sections 24 h following the last injection (6 injections, 1 per day) of Aβ1–42 oligomers (1 and 24 h; 0.2 μg/μl; 2 μl) or vehicle (Ctl) stained with the 6E10 antibody and cresyl violet. Scale bars: 50 μm (n = 4, two independent experiments). Bottom, Resazurin assays on neuronal cultures treated with vehicle (Veh) or 2 μm of Aβ 1 h or Aβ 24 h. Histograms show mean values expressed as a percentage of controls (cultures without any treatment) ± SEM. *p < 0.05, **p < 0.01 (3 independent experiments performed in duplicate). C, Hippocampal Aβ1–42 oligomer profiles 24 h following the last injection (6 injections, 1 per day) using the 6E10 antibody. Bottom, The membrane was incubated with the anti-mouse secondary antibody (AB) without the 6E10 primary mouse monoclonal antibody (n = 4, two independent experiments).
Sequestration of Aβ1–42 oligomers by TTR
The sequestration of Aβ by TTR has been suggested to regulate the transport and clearance of Aβ from the CNS to prevent its deposition and toxicity (Schwarzman et al., 1994; Link, 1995; Du and Murphy, 2010). Using in vitro conditions, we evaluated which potential toxic Aβ species could complex with TTR during the oligomerization process. Aβ1–42 peptides and TTR were coincubated at 25°C or 37°C for 24 h at a molar TTR-Aβ ratio of 2:1 (Fig. 6A). As indicated above, leaving Aβ peptides in low-adhesion polystyrene tubes leads to their aggregation and deposition on the plastic walls over time. Sequestration with TTR permitted higher levels of soluble Aβ1–42 oligomers to be retrieved than with Aβ1–42 alone, especially at 37°C. The amount of soluble Aβ1–42 oligomers retrieved depended on the concentration of TTR in the mixture and was inversely proportional to the amount of insoluble Aβ1–42 oligomers retrieved with formic acid treatment (Fig. 6B). As shown previously (Du and Murphy, 2010), these results suggest that TTR is capable of binding various soluble Aβ1–42 species and preventing their oligomerization into longer insoluble amyloid fibrils.
Figure 6.

Sequestration of soluble Aβ1–42 oligomers with TTR. A, Representative 6E10-immunoblot of soluble Aβ1–42 oligomers 1 or 24 h following Aβ1–42 preparation with TTR (TTR-Aβ, molar (M) ratio of 2:1) or Aβ1–42 alone (Aβ) at the temperature indicated. B, Increasing doses of TTR mixed with Aβ1–42 at 37°C during 24 h promoted the sequestration and retrieval of soluble Aβ1–42 oligomers on an SDS-NuPAGE gel system using the 6E10 antibody. Lower levels of insoluble Aβ1–42 stuck to the side of low-adhesion tubes were recovered with formic acid treatment while increasing the TTR concentration in Aβ1–42 solutions (right blot) (n = 3 for each gel, 3 independent experiments).
TTR attenuation of the effects of Aβ1–42 oligomers
Even if Aβ1–42 oligomers continued to accumulate in the brain when mixed with TTR, the sequestration of Aβ1–42 with TTR was sufficient to reduce the toxicity induced by Aβ1–42 oligomers (Fig. 7A). In addition, tau epitopes Ser202 and Thr205 were found to be unphosphorylated following the sequestration of Aβ1–42 by TTR (Fig. 7B). Next, we determined whether the neurodegeneration induced by small Aβ1–42 species promoted hippocampus-dependent memory deficits using the passive avoidance task and the Y-maze test (Fig. 7C,D). Decreased memory capacity in the Aβ-oligomer-injected group was attenuated following TTR sequestration in both behavioral paradigms.
Figure 7.

Neuronal death, tau phosphorylation, and memory deficits induced by Aβ1–42 oligomers are attenuated by TTR. A, Representative DG section 24 h after the last injection (1 per day during 6 d) with Aβ1–42 oligomers alone (0.2 μg/μl, 2 μl) or with TTR (TTR-Aβ, molar ratio of 2:1), TTR alone, or vehicle (Ctl), kept at 37°C for 24 h before administration. Scale bars: 50 μm. B, Representative immunoblots of hippocampal tau phosphorylation in mice injected collaterally with Aβ1–42 oligomers and TTR-Aβ. Densitometric quantification of changes is expressed as the mean ratio of AT8 antibody to total Tau N-ter. Error bars indicate ± SEM. *p < 0.05 (n = 4, 2 independent experiments). C, D, Mice bilaterally injected in the DG with control vehicle, Aβ1–42 oligomers, or TTR-Aβ were tested in the passive avoidance memory task (C) and the Y maze (D). During the passive avoidance conditioning, latency of entry into the dark compartment was evaluated during the training phase and the testing phase 24 h later. For the Y maze, we analyzed the percentage of time spent in the arm (other) already explored during the training phase and the new arm available during the testing phase. Values are mean latency ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 (n = 8 for each experimental group for the passive avoidance task, n = 4 for each experimental group for the Y maze).
Using primary hippocampal cultures, decreased neuronal survival following a 72 h treatment with 2 μm of soluble Aβ1–42 oligomers was reversed by coincubation of Aβ1–42 peptides with TTR (Fig. 8A). As observed in vivo, the sequestration of Aβ1–42 species by TTR reduced the level of tau hyperphosphorylation at epitopes Ser202 and Thr205 seen after treatment with Aβ1–42 alone (Fig. 8B). The increased caspase-3 activity and the reduced levels of spinophilin and NR2B subunit seen following Aβ1–42 treatment were also significantly reversed by TTR sequestration (Fig. 8C).
Figure 8.

TTR prevented the deleterious effects of soluble Aβ1–42 oligomers in primary hippocampal cultures. A, Resazurin assays following a 72 h treatment with vehicle (Veh), 2 μm of scramble Aβ1–42 oligomers (Aβscr), or 2 μm of soluble Aβ1–42 oligomers alone (Aβ) or with TTR (TTR-Aβ; molar ratio of 2:1) kept at 37°C for 24 h before administration. B, C, Representative immunoblots of tau phosphorylation (B) as well as spinophilin, caspase-3, NR2B, and GAPDH (C) for neurons treated with vehicle (Ctl), Aβ, or TTR-Aβ (molar ratio of 2:1). For tau phosphorylation, densitometric quantification of changes is expressed as the mean ratio of AT8 to Tau N-ter antibody. The histograms are mean values expressed as a percentage of control ± SEM. * or ** indicates a statistical significant variation between Ctl and Aβ, and † or ‡ indicates a difference between Aβ and TTR-Aβ. *,†p < 0.05, **,‡p < 0.01, ***p < 0.001 (3 independent experiments performed in duplicate).
Discussion
Marked synaptic and neuronal losses are detected in individuals with MCI and early AD (Gómez-Isla et al., 1996; Scheff et al., 2006; Arendt, 2009; Crews and Masliah, 2010). Despite recent evidence suggesting that soluble Aβ oligomers may initiate these early pathophysiological events (Hardy and Selkoe, 2002; Benilova et al., 2012), the neurotoxic effect of small Aβ oligomers in vivo and the resulting impact on memory performance needed to be established.
Our results indicate that a single daily intrahippocampal injection of soluble Aβ1–42 species for 6 d is able to induce oligomer accumulation and extended neuronal death. Although the causal link between the effects of anesthesia and AD still needs to be firmly established, we performed Aβ injections in awake, freely moving mice to avoid any confounding effects of anesthetic agents on intracellular pathways related to Aβ and tau metabolism. Indeed, several studies have shown that anesthesia is able to affect tau phosphorylation, induce memory impairments, enhance fibril formation and the cytotoxicity of Aβ, increase plaque deposition in an AD mouse model, and affect many other molecules that were reported to be implicated in the evolution of AD pathology, such as GSK3β, Erk1/2, Akt, JNK, PP2A, and caspase-3 (Eckenhoff et al., 2004; Planel et al., 2007; Bianchi et al., 2008; Whittington et al., 2011; Le Freche et al., 2012). A relatively high concentration of Aβ was injected to accelerate amyloid deposition, a process that normally takes decades in AD patients. The neuronal damage was associated with tau hyperphosphorylation and memory deficits. Our in vivo approach has thus proven to be useful in mimicking the key neuropathological phenomena associated with AD and in providing evidence that small soluble Aβ oligomers play a major role in the etiology of the disease.
Consistent with our in vivo data, decreases in neuronal survival and synaptic marker as well as tau hyperphosphorylation were also observed following the application of small soluble Aβ1–42 oligomers to primary hippocampal cultures. Several lines of evidence indicate that caspase-3 activation may participate in apoptotic cell death and synaptic dysfunction in AD (Gervais et al., 1999; D'Amelio et al., 2011). The progressive increase in cleaved caspase-3 that we observed suggests that the activation of this enzyme could be part of the molecular mechanism by which soluble Aβ1–42 oligomers induce neuronal damage. Previous studies have shown that synaptic loss following treatment with high concentrations or prolonged exposure of Aβ was associated with rapid decrease in membrane expression of NMDA receptors (Snyder et al., 2005; Lacor et al., 2007; Shankar et al., 2007). In line with these reports, our results indicate that the NMDA receptor subunit NR2B diminishes in a sustained fashion during the neurodegenerative process initiated by soluble Aβ1–42 oligomers in vitro as well as in vivo.
Many recent studies have tried to identify one particular soluble Aβ species as the toxic culprit in AD and several interesting candidates have been proposed (Lambert et al., 1998; Lesné et al., 2006; Hung et al., 2008; Shankar et al., 2008; Ono et al., 2009). Unifying all of these findings, one could argue that different types of soluble Aβ oligomers are deleterious and that we need to consider their toxicity as a whole since they occur simultaneously in the brain due to the rapid oligomerization process. Recently, several small Aβ species have been detected in the AD brain and have been proposed to induce the multiple biological effects attributed to these oligomers in the disease (McLean et al., 1999).
Consistent with this hypothesis, we observed that our Aβ preparation was mainly composed of low-molecular-weight oligomers, which rapidly self-associated to form larger oligomers. We observed that the Aβ solution was more toxic when it contained an elevated concentration of small Aβ species. Nevertheless, our Aβ1–42 preparation containing larger oligomers when kept at 37°C for 24 h was also able to induce neuronal death in vitro and in vivo. Thus our data seem more compatible with a dynamic interpretation of Aβ toxicity rather than any specific toxic Aβ species with a defined structure that is stable over time. Further investigation requires the determination of whether the toxicity that we observed in vivo with Aβ can be extended to other amyloidogenic proteins, but our model could be used to answer this question. Further investigations are also needed to determine the long-term effects of Aβ oligomers both at the molecular and behavioral levels.
If the neurotoxic effect of Aβ is attributable to a variety of species acting together instead of one particular type of oligomer, how can the accumulation of several soluble forms of Aβ disturb the normal functioning of neurons? Although there is no clear response to this question at present, it is likely that part of the answer lies with the “sticky properties” of Aβ as observed here by their adhesion to the walls of low-adhesion tubes. Indeed, there is compelling evidence to suggest that small Aβ oligomers are highly hydrophobic and could induce neurotoxicity by interacting with lipid membranes, which in turn could alter membrane trafficking and neuronal transmission (Tickler et al., 2005; Hung et al., 2008; Benilova et al., 2012).
Various Aβ species could also exert toxic effects by binding different membrane proteins such as the cellular prion proteins (PrPc) (Laurén et al., 2009), the metabotropic glutamate receptors (mGLuR5) (Renner et al., 2010), the NMDA receptors (De Felice et al., 2007), the acetylcholine receptors (Dougherty et al., 2003), and the angiotensin II receptors (AbdAlla et al., 2009). We observed that Aβ species accumulated mostly in the granule cell layer where these receptors are mainly located in the DG. The retrieval of low-molecular-weight Aβ1–42 oligomers following 6 d of injection (Fig. 2C) suggests that the interaction with receptors and lipid membranes stabilized Aβ conformation, as observed when soluble Aβ1–42 species bind to TTR (Fig. 6).
The sequestering agent TTR has been found to be the major Aβ-binding protein in the CSF (Schwarzman et al., 1994; Carro et al., 2002) and represents a potential biomarker for AD (Schultz et al., 2010). The interaction of Aβ with TTR strongly depends on the quaternary structure of TTR (Du and Murphy, 2010). TTR monomers assemble into dimers that in turn self-associate to form homotetramers. TTR monomers have previously been shown to bind more extensively to Aβ monomers, impeding the further growth of Aβ aggregates (Du and Murphy, 2010). On the other hand, TTR tetramers interact more with Aβ aggregates than with Aβ monomers and have been observed to disrupt fibril formation (Du and Murphy, 2010). Thus, the sequestration of discrete toxic species and the arrest of Aβ monomer growth into multimers represent two possible mechanisms for the TTR-mediated protection against Aβ toxicity. Our data are in line with previous studies indicating that the overexpression of TTR reduces Aβ toxicity whereas its neutralization increases the AD-like phenotype in transgenic mouse models (Stein and Johnson, 2002; Stein et al., 2004; Choi et al., 2007; Buxbaum et al., 2008), although other reports have shown no beneficial effects of TTR in AD mouse models (Wati et al., 2009; Doggui et al., 2010). Moreover, decreased hippocampal TTR expression is associated with memory deficits in aged rats and the complete deletion of TTR leads to memory deficits during aging in mice (Brouillette and Quirion, 2008).
Because of inappropriate animal models, the study of toxic effect of small, soluble Aβ oligomers was restricted to neuronal cultures or organotypic brain slice cultures (Lambert et al., 1998; Lesné et al., 2006; Hung et al., 2008; Shankar et al., 2008; Ono et al., 2009). In this study, we described a novel in vivo approach to determine the toxicity of Aβ1–42 preparations as a function of their temporal profile. Since the intrahippocampal injections are done in awake, freely moving mice there are no confounding interference effects between any anesthetic agents and the Aβ solution on intracellular pathways. The effects of soluble Aβ1–42 oligomers were determined during the process of aging in 12-month-old mice, and the collateral injection of soluble Aβ1–42 oligomers and vehicle permitted the control of any alteration within the same mouse. We observed that one injection per day of low-molecular-weight Aβ1–42 species for 6 d triggers key AD features such as Aβ accumulation in a time-dependent manner, marked neurodegeneration, abnormal tau phosphorylation, and memory dysfunction.
Since substantial synaptic and neuronal losses are observed in the early stages of AD when memory deficits become clinically detectable, our animal model can now be used for further pathway analysis involved in Aβ neurodegeneration.
Footnotes
This work has been developed and supported in part through the LABEX DISTALZ (Laboratory of Excellence, Program for Investing in the Future and Development of Innovative Strategies for a Transdisciplinary Approach to Alzheimer's Disease), and by grants from the French National Research Agency (ANR MNP: AMYTOXTAU), the FP7 European Consortium MEMOSAD to L.B. and B.D.S., a Methusalem Grant (KULeuven-Flanders Government) to B.D.S., and a postdoctoral fellowship to J.B. from the Canadian Institutes of Health Research. We thank Ingrid Brion and Delphine Tailleu from the animal facility for the mouse work. We are grateful to K. Broersen (University of Twente, MIRA Institute for Biomedical Technology and Technical Medicine, Enschede, the Netherlands) for the Aβ preparation technique and A. Loyens (IMPRT, Lille, France) for transmission electron microscopy experiments.
The authors declare no competing financial interests.
References
- AbdAlla S, Lother H, el Missiry A, Langer A, Sergeev P, el Faramawy Y, Quitterer U. Angiotensin II AT2 receptor oligomers mediate G-protein dysfunction in an animal model of Alzheimer disease. J Biol Chem. 2009;284:6554–6565. doi: 10.1074/jbc.M807746200. [DOI] [PubMed] [Google Scholar]
- Arendt T. Synaptic degeneration in Alzheimer's disease. Acta Neuropathol. 2009;118:167–179. doi: 10.1007/s00401-009-0536-x. [DOI] [PubMed] [Google Scholar]
- Benilova I, Karran E, De Strooper B. The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- Bianchi SL, Tran T, Liu C, Lin S, Li Y, Keller JM, Eckenhoff RG, Eckenhoff MF. Brain and behavior changes in 12-month-old Tg2576 and nontransgenic mice exposed to anesthetics. Neurobiol Aging. 2008;29:1002–1010. doi: 10.1016/j.neurobiolaging.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broersen K, Jonckheere W, Rozenski J, Vandersteen A, Pauwels K, Pastore A, Rousseau F, Schymkowitz J. A standardized and biocompatible preparation of aggregate-free amyloid beta peptide for biophysical and biological studies of Alzheimer's disease. Protein Eng Des Sel. 2011;24:743–750. doi: 10.1093/protein/gzr020. [DOI] [PubMed] [Google Scholar]
- Brouillette J, Quirion R. Transthyretin: a key gene involved in the maintenance of memory capacities during aging. Neurobiol Aging. 2008;29:1721–1732. doi: 10.1016/j.neurobiolaging.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Buxbaum JN, Ye Z, Reixach N, Friske L, Levy C, Das P, Golde T, Masliah E, Roberts AR, Bartfai T. Transthyretin protects Alzheimer's mice from the behavioral and biochemical effects of Abeta toxicity. Proc Natl Acad Sci U S A. 2008;105:2681–2686. doi: 10.1073/pnas.0712197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro E, Trejo JL, Gomez-Isla T, LeRoith D, Torres-Aleman I. Serum insulin-like growth factor I regulates brain amyloid-beta levels. Nat Med. 2002;8:1390–1397. doi: 10.1038/nm1202-793. [DOI] [PubMed] [Google Scholar]
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- Choi SH, Leight SN, Lee VM, Li T, Wong PC, Johnson JA, Saraiva MJ, Sisodia SS. Accelerated Abeta deposition in APPswe/PS1deltaE9 mice with hemizygous deletions of TTR (transthyretin) J Neurosci. 2007;27:7006–7010. doi: 10.1523/JNEUROSCI.1919-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews L, Masliah E. Molecular mechanisms of neurodegeneration in Alzheimer's disease. Hum Mol Genet. 2010;19:R12–R20. doi: 10.1093/hmg/ddq160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amelio M, Cavallucci V, Middei S, Marchetti C, Pacioni S, Ferri A, Diamantini A, De Zio D, Carrara P, Battistini L, Moreno S, Bacci A, Ammassari-Teule M, Marie H, Cecconi F. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer's disease. Nat Neurosci. 2011;14:69–76. doi: 10.1038/nn.2709. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- Doggui S, Brouillette J, Chabot JG, Farso M, Quirion R. Possible involvement of transthyretin in hippocampal beta-amyloid burden and learning behaviors in a mouse model of Alzheimer's disease (TgCRND8) Neurodegener Dis. 2010;7:88–95. doi: 10.1159/000285513. [DOI] [PubMed] [Google Scholar]
- Dougherty JJ, Wu J, Nichols RA. Beta-amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J Neurosci. 2003;23:6740–6747. doi: 10.1523/JNEUROSCI.23-17-06740.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Murphy RM. Characterization of the interaction of beta-amyloid with transthyretin monomers and tetramers. Biochemistry. 2010;49:8276–8289. doi: 10.1021/bi101280t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckenhoff RG, Johansson JS, Wei H, Carnini A, Kang B, Wei W, Pidikiti R, Keller JM, Eckenhoff MF. Inhaled anesthetic enhancement of amyloid-beta oligomerization and cytotoxicity. Anesthesiology. 2004;101:703–709. doi: 10.1097/00000542-200409000-00019. [DOI] [PubMed] [Google Scholar]
- Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW. Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- Gómez-Isla T, Price JL, McKeel DW, Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci. 1996;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hepler RW, Grimm KM, Nahas DD, Breese R, Dodson EC, Acton P, Keller PM, Yeager M, Wang H, Shughrue P, Kinney G, Joyce JG. Solution state characterization of amyloid beta-derived diffusible ligands. Biochemistry. 2006;45:15157–15167. doi: 10.1021/bi061850f. [DOI] [PubMed] [Google Scholar]
- Hung LW, Ciccotosto GD, Giannakis E, Tew DJ, Perez K, Masters CL, Cappai R, Wade JD, Barnham KJ. Amyloid-beta peptide (Abeta) neurotoxicity is modulated by the rate of peptide aggregation: Abeta dimers and trimers correlate with neurotoxicity. J Neurosci. 2008;28:11950–11958. doi: 10.1523/JNEUROSCI.3916-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT. Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci. 1997;17:7053–7059. doi: 10.1523/JNEUROSCI.17-18-07053.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C, Song C, Zuo P. The mechanism of memory impairment induced by Abeta chronic administration involves imbalance between cytokines and neurotrophins in the rat hippocampus. Curr Alzheimer Res. 2011;8:410–420. doi: 10.2174/156720511795745366. [DOI] [PubMed] [Google Scholar]
- Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers-Nolten I, Van Der Werf K, Subramaniam V, Braeken D, Callewaert G, Bartic C, D'Hooge R, Martins IC, Rousseau F, Schymkowitz J, De Strooper B. Neurotoxicity of Alzheimer's disease Abeta peptides is induced by small changes in the Abeta42 to Abeta40 ratio. EMBO J. 2010;29:3408–3420. doi: 10.1038/emboj.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Freche H, Brouillette J, Fernandez-Gomez FJ, Patin P, Caillierez R, Zommer N, Sergeant N, Buée-Scherrer V, Lebuffe G, Blum D, Buée L. Tau phosphorylation and sevoflurane anesthesia: an association to postoperative cognitive impairment. Anesthesiology. 2012;116:779–787. doi: 10.1097/ALN.0b013e31824be8c7. [DOI] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Link CD. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1995;92:9368–9372. doi: 10.1073/pnas.92.20.9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm T, Ort M, Tähtivaara L, Jukarainen N, Goldsteins G, Puoliväli J, Nurmi A, Pussinen R, Ahtoniemi T, Miettinen TK, Kanninen K, Leskinen S, Vartiainen N, Yrjänheikki J, Laatikainen R, Harris-White ME, Koistinaho M, Frautschy SA, Bures J, Koistinaho J. beta-Amyloid infusion results in delayed and age-dependent learning deficits without role of inflammation or beta-amyloid deposits. Proc Natl Acad Sci U S A. 2006;103:8852–8857. doi: 10.1073/pnas.0602896103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins IC, Kuperstein I, Wilkinson H, Maes E, Vanbrabant M, Jonckheere W, Van Gelder P, Hartmann D, D'Hooge R, De Strooper B, Schymkowitz J, Rousseau F. Lipids revert inert Abeta amyloid fibrils to neurotoxic protofibrils that affect learning in mice. EMBO J. 2008;27:224–233. doi: 10.1038/sj.emboj.7601953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Miller AM, Piazza A, Martin DS, Walsh M, Mandel A, Bolton AE, Lynch MA. The deficit in long-term potentiation induced by chronic administration of amyloid-beta is attenuated by treatment of rats with a novel phospholipid-based drug formulation, VP025. Exp Gerontol. 2009;44:300–304. doi: 10.1016/j.exger.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Ono K, Condron MM, Teplow DB. Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc Natl Acad Sci U S A. 2009;106:14745–14750. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 5. Burlington, MA: Elsevier Academic; 2005. [Google Scholar]
- Planel E, Richter KE, Nolan CE, Finley JE, Liu L, Wen Y, Krishnamurthy P, Herman M, Wang L, Schachter JB, Nelson RB, Lau LF, Duff KE. Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J Neurosci. 2007;27:3090–3097. doi: 10.1523/JNEUROSCI.4854-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, Triller A. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron. 2010;66:739–754. doi: 10.1016/j.neuron.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol Aging. 2006;27:1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Schultz K, Nilsson K, Nielsen JE, Lindquist SG, Hjermind LE, Andersen BB, Wallin A, Nilsson C, Petersén A. Transthyretin as a potential CSF biomarker for Alzheimer's disease and dementia with Lewy bodies: effects of treatment with cholinesterase inhibitors. Eur J Neurol. 2010;17:456–460. doi: 10.1111/j.1468-1331.2009.02841.x. [DOI] [PubMed] [Google Scholar]
- Schwarzman AL, Gregori L, Vitek MP, Lyubski S, Strittmatter WJ, Enghilde JJ, Bhasin R, Silverman J, Weisgraber KH, Coyle PK. Transthyretin sequesters amyloid beta protein and prevents amyloid formation. Proc Natl Acad Sci U S A. 1994;91:8368–8372. doi: 10.1073/pnas.91.18.8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Soura V, Stewart-Parker M, Williams TL, Ratnayaka A, Atherton J, Gorringe K, Tuffin J, Darwent E, Rambaran R, Klein W, Lacor P, Staras K, Thorpe J, Serpell LC. Visualization of co-localization in Abeta42-administered neuroblastoma cells reveals lysosome damage and autophagosome accumulation related to cell death. Biochem J. 2012;441:579–590. doi: 10.1042/BJ20110749. [DOI] [PubMed] [Google Scholar]
- Stein TD, Johnson JA. Lack of neurodegeneration in transgenic mice overexpressing mutant amyloid precursor protein is associated with increased levels of transthyretin and the activation of cell survival pathways. J Neurosci. 2002;22:7380–7388. doi: 10.1523/JNEUROSCI.22-17-07380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein TD, Anders NJ, DeCarli C, Chan SL, Mattson MP, Johnson JA. Neutralization of transthyretin reverses the neuroprotective effects of secreted amyloid precursor protein (APP) in APPSW mice resulting in tau phosphorylation and loss of hippocampal neurons: support for the amyloid hypothesis. J Neurosci. 2004;24:7707–7717. doi: 10.1523/JNEUROSCI.2211-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tickler AK, Smith DG, Ciccotosto GD, Tew DJ, Curtain CC, Carrington D, Masters CL, Bush AI, Cherny RA, Cappai R, Wade JD, Barnham KJ. Methylation of the imidazole side chains of the Alzheimer disease amyloid-beta peptide results in abolition of superoxide dismutase-like structures and inhibition of neurotoxicity. J Biol Chem. 2005;280:13355–13363. doi: 10.1074/jbc.M414178200. [DOI] [PubMed] [Google Scholar]
- Troquier L, Caillierez M, Burnouf S, Fernandez-Gomez FJ, Grosjean MJ, Zommer N, Sergeant N, Schraen-Maschke S, Blum D, Buee L. Targeting phospho-Ser422 by active Tau immunotherapy in the THY-Tau22 mouse model: a suitable therapeutic approach. Curr Alzheimer Res. 2012;9:397–405. doi: 10.2174/156720512800492503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wati H, Kawarabayashi T, Matsubara E, Kasai A, Hirasawa T, Kubota T, Harigaya Y, Shoji M, Maeda S. Transthyretin accelerates vascular Abeta deposition in a mouse model of Alzheimer's disease. Brain Pathol. 2009;19:48–57. doi: 10.1111/j.1750-3639.2008.00166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington RA, Virág L, Marcouiller F, Papon MA, El Khoury NB, Julien C, Morin F, Emala CW, Planel E. Propofol directly increases tau phosphorylation. PLoS One. 2011;6:e16648. doi: 10.1371/journal.pone.0016648. [DOI] [PMC free article] [PubMed] [Google Scholar]