

Abstract
The precision of sound information transmitted to the brain depends on the transfer characteristics of the inner hair cell (IHC) ribbon synapse and its multiple contacting auditory fibers. We found that brain derived neurotrophic factor (BDNF) differentially influences IHC characteristics in the intact and injured cochlea. Using conditional knock-out mice (BDNFPax2 KO) we found that resting membrane potentials, membrane capacitance and resting linear leak conductance of adult BDNFPax2 KO IHCs showed a normal maturation. Likewise, in BDNFPax2 KO membrane capacitance (ΔCm) as a function of inward calcium current (ICa) follows the linear relationship typical for normal adult IHCs. In contrast the maximal ΔCm, but not the maximal size of the calcium current, was significantly reduced by 45% in basal but not in apical cochlear turns in BDNFPax2 KO IHCs. Maximal ΔCm correlated with a loss of IHC ribbons in these cochlear turns and a reduced activity of the auditory nerve (auditory brainstem response wave I). Remarkably, a noise-induced loss of IHC ribbons, followed by reduced activity of the auditory nerve and reduced centrally generated wave II and III observed in control mice, was prevented in equally noise-exposed BDNFPax2 KO mice. Data suggest that BDNF expressed in the cochlea is essential for maintenance of adult IHC transmitter release sites and that BDNF upholds opposing afferents in high-frequency turns and scales them down following noise exposure.
Introduction
The precision of processing sound information in the mammalian cochlea depends on the release of neurotransmitters from ribbon synapses in inner hair cells (IHCs), which drive postsynaptic auditory fibers at high rates of spikes (Liberman et al., 1990). Synaptic ribbons are electron-dense presynaptic specializations that tether synaptic vesicles for exocytosis at the active zone (for review: Fuchs, 2005; Moser et al., 2006; Schmitz, 2009). Ribbon synapses support an extremely high rate of exocytosis through maintenance of a large ready releasable pool, thereby enabling a fine intensity discrimination over a wide dynamic range (Johnson et al., 2005; Khimich et al., 2005). When ribbons are lost, as, for example, in Bassoon mouse mutants, the reliability of spikes is disturbed and temporal resolving power of the auditory system is reduced (Buran et al., 2010). Thus, disturbance of ribbon integrity is expected to influence the temporal resolution of processing sound information. Recent studies showed that acoustic overexposure leads to a rapid and irreversible loss of peripheral cochlear nerve terminals on IHCs, and to a reduction of up to 50% of synaptic ribbons, leading to slow degeneration of spiral ganglia neurons (SG) (Kujawa and Liberman, 2009; Lin et al., 2011). Under prevailing circumstances this may even endanger the typical capacity of central target neurons to increase discharge rate and compensate with increased central circuitry responsiveness for auditory nerve (AN) degradation (Knipper et al., 2010). Recent studies suggested that the trigger for SG loss in the cochlea does not originate from IHCs but rather from IHCs supporting cells such as inner border and inner phalangeal cells, suggested to be essential for neuronal survival after IHC damage (Zilberstein et al., 2012).
Brain-derived neurotrophic factor (BDNF) is expressed from birth onwards below IHCs, in cells most likely corresponding to supporting cells (Sobkowicz et al., 2002). Moreover BDNF is a crucial survival and growth factor for the developing CNS (for review: Barde, 1990; Poo, 2001). In the adult CNS it displays an important role for memory and cognitive functions (Thoenen, 2000; Bramham and Messaoudi, 2005; Minichiello, 2009), as well as for the development and maintenance of mature dendritic complexity and spine density in selected brain centers (Rauskolb et al., 2010). On the other hand, unbalanced regulation of BDNF can also be harmful as discussed in the context of neuropathic pain (Biggs et al., 2010) or in psychiatric and neurodegenerative disorders (Hu and Russek, 2008; Lu and Martinowich, 2008; Krishnan and Nestler, 2010; Pardon, 2010; Aznar and Knudsen, 2011). So far it is elusive whether either of these predicted roles of BDNF is relevant for the mature auditory system because constitutive BDNF KO mice, in which a role of BDNF for survival of neonatal vestibular neurons has been described (Ernfors et al., 1995; Fritzsch et al., 2004), die too early to assess the role of BDNF in the mature auditory system. We therefore generated a BDNF conditional mouse line (BDNFPax2 KO) with the aim to clarify the function of BDNF in the mature cochlea in the intact organ as well as in the injured organ. By analyzing the BDNFPax2 KO mice, we found that BDNF is essential to maintain mature exocytosis, ribbon number and afferents in high-frequency cochlear regions in the mature organ and is responsible for IHCs synapse degradation during noise exposure.
Materials and Methods
Generation of conditional knock-out mice.
Animals in which the Cre gene is controlled by the Pax2 promoter and mutants carrying a floxed BDNF allele (both obtained by the Mutant Mouse Regional Research Center, MMRRC) have been described (Rios et al., 2001; Ohyama and Groves, 2004). Genotyping of mutant alleles were performed as described previously (Rios et al., 2001). For hearing measurements, noise exposure and molecular analysis, mice of either sex aged 9–12 weeks were used. The care and use of the animals and the experimental protocol were reviewed and approved by the animal welfare commissioner and the regional board for scientific animal experiments in Tübingen.
Hearing measurements: auditory brainstem response and distortion product otoacoustic emission.
Auditory brainstem responses (ABRs) to click and pure tone auditory stimuli and the cubic 2*f1-f2 distortion product otoacoustic emission (DPOAE) for f2 = 1.24*f1 and L2 = L1 + 10 dB were recorded in anesthetized mice aged 9–12 weeks in a soundproof chamber as described previously (Engel et al., 2006). In short, ABR thresholds were determined with click (100 μs), noise burst (1 ms), or pure tone stimuli (3 ms, including 1 ms cosine squared rise and fall envelope, 2–45 kHz). OHC function was assessed by the threshold and growth function of the DPOAE.
Noise exposure.
For acoustic trauma, animals were exposed under anesthesia to intense pure tone noise (10 kHz, 116 dB SPL for 40 min) in a reverberating chamber, binaurally in open field as previously described (Engel et al., 2006). Sham-exposed animals were anesthetized and placed in the reverberating chamber without acoustic stimulus (i.e., the speaker remained turned off). These animals have completely normal hearing.
Electrophysiology.
Electrophysiological recordings were made from IHCs of BDNFPax2 KO and control mice maintained at body temperature, using physiological 1.3 mm extracellular Ca2+ as previously described (Johnson et al., 2008, 2010).
Apical and basal turn IHCs (n = 37) from control and BDNFPax2 KO mice of either sex were investigated in acutely dissected organs of Corti from postnatal day 15 (P15) to P19. Animals were killed by cervical dislocation in accordance with U.K. Home Office regulations. Cochleae were dissected as previously described (Johnson et al., 2008) in normal extracellular solution containing (in mm): 135 NaCl, 5.8 KCl, 1.3 CaCl2, 0.9 MgCl2, 0.7 NaH2PO4, 5.6 d-glucose, 10 HEPES-NaOH, 2 sodium pyruvate, amino acids and vitamins (pH 7.48; osmolality ∼308 mmol kg−1). All recordings were performed near body temperature (35–37°C) using 1.3 mm Ca2+ in the extracellular solution.
The general biophysical properties were obtained using the following intracellular solution (in mm): 131 KCl, 3 MgCl2, 1 EGTA-KOH, 5 Na2ATP, 5 HEPES-KOH, 10 sodium phosphocreatine, pH 7.3. The pipette intracellular solution for exocytosis measurements contained the following (in mm): 106 Cs-glutamate, 20 CsCl, 3 MgCl2, 1 EGTA-CsOH, 5 Na2ATP, 0.3 Na2GTP, 5 HEPES-CsOH, 10 Na2-phosphocreatine, pH 7.3. Patch pipettes were coated with surf wax (Mr. Zoggs SexWax) to minimize the fast patch pipette capacitance transient. Electrophysiological recordings were made using the Optopatch (Cairn Research) amplifier. Data acquisition was controlled by pClamp software using a Digidata (Molecular Devices). Current and voltage recordings were low-pass filtered at 2.5 kHz (8-pole Bessel), sampled at 5 kHz and stored on computer for off-line analysis (Origin, OriginLab). Membrane potentials were corrected for the Rs (3.4 ± 0.3 MΩ, n = 27) and liquid junction potential (Cl−- and glutamate-based intracellular solution: −4 mV and −11 mV, respectively). Real-time changes in membrane capacitance (ΔCm) were measured using the Optopatch as previously described (Johnson et al., 2008, 2010). Briefly, a 4 kHz sine wave of 13 mV RMS was applied to IHCs from −81 mV and was interrupted for the duration of the voltage step. The capacitance signal from the Optopatch was amplified (×50), filtered at 250 Hz, and sampled at 5 kHz. Capacitance changes were measured by averaging the Cm traces after the voltage step (∼200 ms) and subtracting the prepulse baseline. ΔCm was recorded while applying 30 mm TEA and 15 mm 4AP (Fluka) and 80 μm linopirdine (Tocris Bioscience) to reduce K+ currents.
Tissue preparation.
For immunohistochemistry, cochleae were isolated, prepared, cryosectioned at 10 μm, and mounted on SuperFrost*/plus microscope slides at −20°C as previously described (Knipper et al., 2000). For RNA isolation, cochleae and different brain regions were dissected with small forceps, immediately frozen in liquid nitrogen, and stored at −80°C before use. For whole-mount preparation, cochlear turns were dissected and mounted on slides with the tissue adhesive Cell-Tak (BD Biosciences) in PBS and performed immunohistochemistry.
Riboprobe synthesis.
Riboprobes targeting regions of BDNF exon IX and cyclophilin were amplified using specific primers (BDNF: for 5′-GAGGACCAGAAGGTTCG-3′; rev 5′-TTTATCTGCCGCTGTGAC-3′; cyclophilin: for 5′-CGTGCTCTGAGCACTGGGGAGAAA-3′; rev 5′-CATGCCTTCTTTCACCTTCCCAAAGAC-3′). Riboprobes were synthesized as previously described (Tan et al., 2007).
Immunohistochemistry.
For immunohistochemistry, mouse cochlear sections were stained as described previously (Tan et al., 2007). Antibodies directed against CtBP2/RIBEYE (mouse, BD Transduction Laboratories), BDNF (rabbit, Santa Cruz Biotechnology), and NF200 (mouse, Sigma-Aldrich) were used and detected using appropriate Cy3-conjugated (Jackson Immunoresearch) or Alexa488-conjugated (Invitrogen) secondary antibodies. For double labeling studies, both antibodies were simultaneously incubated for identical time periods. Sections were viewed by an AX70 Olympus microscope as previously described (Zampini et al., 2010). For whole mount preparations hair cell region was imaged by z-stacking, 3 dimensionally deconvoluted (ADVMLE, cellSens, Olympus) and displayed as maximum intensity projection over z. Figure 4C (bottom) displays isoprojections for the different labelings of the whole-mount preparation derived from the deconvoluted z-stacks (top; Voxel Viewer, cellSens).
Figure 4.
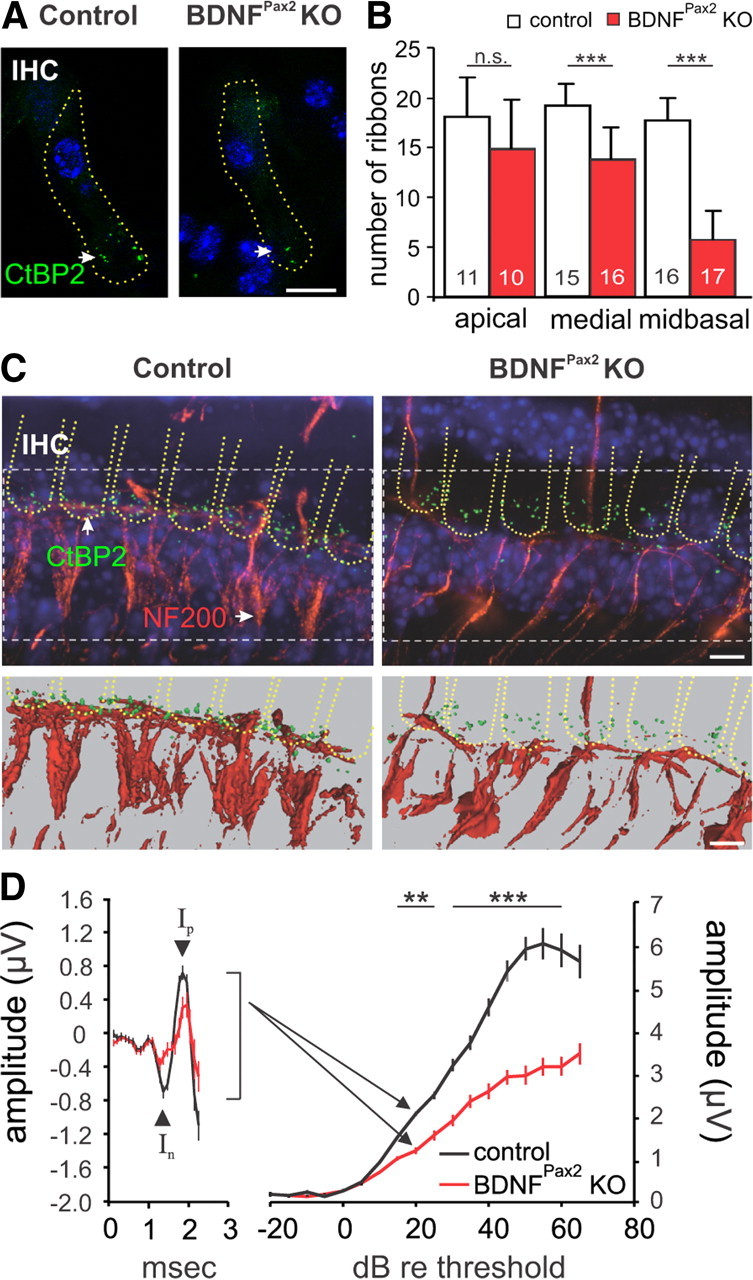
Reduced synaptic ribbons and fibers in BDNFPax2 KO mice. A, Immunohistochemistry for CtBP2/RIBEYE (green) in controls and BDNFPax2 KO mice, shown for the midbasal turn. Cell nuclei were counterstained with DAPI; scale bar, 10 μm. B, Ribbon counts ± SD in different cochlear turns (t test: p < 0.001). Numbers of IHCs counted are given in the bars; n = 3 mice. C, Whole mount preparation of the medial turn in controls and BDNFPax2 KO mice labeled with antibodies against CtBP2/RIBEYE (green) and NF200 (red; top). Cell nuclei were counterstained with DAPI; scale bar, 5 μm. The lower panel displays isoprojections for the different labelings of the upper panel. D, Reduction of ABR wave I amplitude at 20 dB above hearing threshold (left) and growth function (right) in BDNFPax2 KO mice compared with control, mean ± SEM. Controls: n = 17/34 mice/ears; BDNFPax2 KO: n = 17/34 mice/ears; two-way ANOVA: p < 0.001.
β-Galactosidase staining.
For detection of β-galactosidase activity in the Pax2 Cre/ROSA26 mouse line, mice (between P12 and P18) were perfused transcardially with 4% PFA. Cochleae were additionally postfixed by immersion in 4% PFA for 30 min and stained 24–48 h at 37°C in X-gal solution. Brains were postfixed in 4% PFA overnight, cryosectioned at 60 μm, and stained in free floating conditions. Slices were additionally postfixed in 4% PFA and 2.5% glutaraldehyde and stained in X-gal solution for 24–48 h at 37°C. Preparations were viewed with an AX70 Olympus microscope.
Northern blot.
The mRNA isolation was performed using the Oligotex mRNADirect Mini Kit (QIAGEN). The mRNA was loaded onto a denaturing 0.8% agarose formaldehyde gel and transferred onto a nylon membrane (Roche). The membrane was blocked and hybridized overnight at 65°C with riboprobes for BDNF and cyclophilin. The membrane was incubated with anti-Dig-AP (Roche; 1:20,000). mRNA was detected with CDP-Star ready to use (Roche) and exposed to x-ray films.
Western blot.
Proteins were extracted using the NucleoSpin RNA/protein kit (Macherey-Nagel) following the manufacturer's instructions. SDS-PAGE and Western blotting were performed using the “XCell II SureLock Mini-Cell and XCell II Blot Module” (Invitrogen), as previously described (Heidrych et al., 2008). The blotted proteins were incubated with either rabbit polyclonal or mouse monoclonal antibodies: BDNF (Santa Cruz Biotechnology); GAPDH (Abcam).
ABR waveform amplitude analysis.
ABR functions were analyzed for consecutive amplitude deflections (waves), each wave consisting of a starting negative (n) deflection and the following positive (p) deflection. Amplitudes of ABR waves I–V were defined as follows: wave I: Ip–In (latency, 1.2–1.9 ms), wave II: IIp–IIn (latency, 2.2–2.8 ms), wave III: IIIp–IIIn (latency, 3.2–3.7 ms), wave IV: IVp–IVn (latency, 4.1–4.7 ms), wave V: Vp–Vn (latency, 5.5–6.3 ms). An algorithm for an automated determination of ABR amplitudes based on the definitions given above was programmed in MATLAB (MathWorks). A threshold-corrected analysis of ABR wave I–V amplitudes was obtained by comparing the amplitude levels 20 dB above hearing threshold for the individual groups. For ABR growth functions, ABR amplitudes for wave I were derived from individual ears' responses for increasing stimulus levels from 0 to 70 dB above threshold.
Statistical analysis.
Data are presented as mean ± SD or ± SEM. Differences of the mean were compared for statistical significance either by Student's t test, one-way or two-way ANOVA test and t test (with α level adjusted for multiple testing), or Bonferroni test as post hoc test. Statistical significance was tested at α = 0.05, and resulting p values are reported in the figure legends. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant.
Results
Mild hearing loss in BDNFPax2 KO mice
Constitutive BDNF KO mice (Ernfors et al., 1994; Jones et al., 1994) do not survive long enough to assess the role of BDNF in mature SG (Schimmang et al., 2003). Thus BDNF was conditionally inactivated in the auditory system by mating Pax2Cre mice (Ohyama and Groves, 2004) with BDNF lox/lox mice (Rios et al., 2001), which resulted in viable BDNFPax2 KO mice. By breeding Pax2Cre transgenic mice with the ROSA26 reporter mouse line (Soriano, 1999), β-galactosidase reporter expression was confirmed in the mature cochlea (Fig. 1A). BDNF expression was also observed in the dorsal cochlear nucleus (DCN) and inferior colliculus (IC, Fig. 1B,D), but not in the ventral cochlear nucleus (VCN, Fig. 1B), olivary complex (MSO/LSO, Fig. 1C) or auditory cortex (AC, Fig. 1D, inset). In line with previous findings using electron microscopy (Sobkowicz et al., 2002), immunohistochemistry revealed BDNF protein expression in control mice below IHCs, most likely corresponding to phalangeal cells and also in SG (Fig. 1E). Very weak staining was observed in apical cochlear turns (data not shown). BDNF staining was absent in BDNFPax2 KO mice (Fig. 1F), confirming antibody specificity. Northern blot analysis showed that in BDNFPax2 KO mice, BDNF mRNA was absent in the mature cochlea and IC but present in the AC (Fig. 1G). Loss of BDNF in BDNFPax2 KO mice was also confirmed at protein level using Western blot analysis (Fig. 1H).
Figure 1.

Inactivation of BDNF in the auditory system. A–D, X-Gal staining of mice carrying the Pax2-Cre transgene on a ROSA26R background (Soriano, 1999), revealed Cre activity in the mature cochlea (A, organ of Corti, OC, and spiral ganglion neurons, SG), the dorsal (but not ventral) cochlear nucleus (B, DCN, VCN) and inferior colliculus (D, IC). No staining was observed in the olivary complex (C, MSO/LSO) and in all layers of the auditory cortex (D, inset, AC). Scale bar, 200 μm; n = 4. E, F, Immunohistochemistry for BDNF (red) on cochlear sections from control (E), BDNFPax2 KO (F) mice at the level of SG (left) and IHCs (right). Cell nuclei were counterstained with DAPI, n = 3 mice, done in triplicate. Scale bar, 10 μm. G, Northern blot of mRNA from cochlea, IC and AC tissue of control and BDNFPax2 KO mice, hybridized with a probe for BDNF exon IX recognizing BDNF mRNA isoforms (1.8 kb and 4 kb; Timmusk et al., 1993). Cyclophilin (CP) was used as reference gene, n = 4 mice, done in duplicate. H, Western blot detection of BDNF in IC and AC from control and BDNFPax2 mice. GAPDH was used as loading control, n = 4 mice, done in duplicate.
To test whether the lack of BDNF affects hearing function in adult mice, acoustically evoked ABRs were recorded. ABRs, evoked by short sound stimuli, represent the summed activity of neurons in the ascending auditory pathway and are measured by averaging the evoked electrical response recorded via subcutaneous electrodes. Compared with control mice (20.8 ± 6.04 dB SPL, n = 27 mice) ABR thresholds in mutants (32 ± 9.27 dB SPL, n = 26 mice) were significantly elevated 10–15 decibels (dB) for click stimuli (t test: p < 0.001) and for most pure tone stimulus frequencies (two-way ANOVA: p < 0.001; Fig. 2A,B). Since OHC function contributes to the thresholds for sound-evoked neural potentials (El-Badry and McFadden, 2007), their function was analyzed by measuring the DPOAE, an objective indicator for OHC electromotility. Best emission amplitudes of the DPOAE input/output (I/O) function (Fig. 2C) and best thresholds (Fig. 2D) were found to be similar at most frequencies between control (n = 7/12 mice/ears) and BDNFPax2 KO mice (n = 7/12 mice/ears), indicating that threshold loss in the absence of BDNF did not originate from altered motility of OHCs.
Figure 2.

Mild impairment of hearing threshold but similar OHC function in BDNFPax2 KO mice. A, Average ABR thresholds (horizontal dashes) and single ear thresholds (circles and squares) for control (black dash and open circles) and BDNFPax2 KO mice (red dash and squares). Average ABR thresholds ± SD for click stimuli were 11.2 dB higher in ears from BDNFPax2 KO mice (32 dB SPL ± 9.27 SD, n = 26/52 mice/ears) than in ears from control mice (20.8 dB SPL ± 6.04 SD, n = 27/54 mice/ears, t test: p < 0.001). B, Average frequency-specific ABR thresholds ± SD confirmed a significant increase in thresholds in BDNFPax2 KO mice over most frequencies measured (controls: n = 27/27 mice/ear; BDNFPax2 KO: n = 26/26 mice/ear; two-way ANOVA: p < 0.001). C, D, DPOAE growth function ± 95% confidence interval at f2 = 11.3 kHz (C) and 2f1–f2 DPOAE thresholds ± SD (dB SPL f1) (D) in control (n = 7/12 mice/ears) and BDNFPax2 KO mice (n = 7/12 mice/ears), revealed mostly similar OHC thresholds and signal amplitudes in both mouse lines.
IHC function and ABR wave I amplitude are affected in BDNFPax2 KO mice
The increased ABR thresholds observed in BDNFPax2 KO mice may result from abnormalities in sensory IHCs. Therefore we investigated some of the general biophysical properties of mature IHCs (postnatal day 15–19) from control and BDNFPax2 KO mice. We found that the resting membrane potentials (control: −70.8 ± 1.6 mV, n = 6; BDNFPax2 KO: −68.0 ± 1.6 mV, n = 5), the membrane capacitance (control: 8.5 ± 0.3 pF; BDNFPax2 KO: 7.9 ± 0.3 pF) and resting linear leak conductance (control: 0.48 ± 0.09 nS; BDNFPax2 KO: 0.38 ± 0.12 nS) were similar in basal IHCs recorded from both genotypes. We next investigated the presynaptic function of BDNFPax2 KO IHCs by measuring exocytosis. Exocytosis was estimated by monitoring increases in cell membrane capacitance (ΔCm) following depolarizing voltage steps, a measure of indication of neurotransmitter release from IHC presynaptic sites. At around the onset of hearing (postnatal day 12 in most rodents) the synaptic machinery of IHCs becomes more sensitive to Ca2+, causing docked vesicles to be released linearly with increases in intracellular Ca2+ concentration (Johnson et al., 2008, 2010). We found that in basal turn IHCs from adult BDNFPax2 KO mice the maximal ΔCm, but not the maximal size of the Ca2+ current, was significantly reduced by 45% (t test: p < 0.01) compared with that of control cells (Fig. 3A,B,D). Despite this, the exocytotic Ca2+ dependence, defined as the variation in ΔCm as a function of ICa and displayed as a synaptic transfer function, was similar between the two genotypes (Fig. 3C) and showed the normal linear relation of adult IHCs (Johnson et al., 2008, 2010). The ΔCm responses were not affected in apical turn IHCs, again showing a normal linear relation in both genotypes, but ICa was slightly larger in BDNFPax2 KO mice (Fig. 3D and data not shown).
Figure 3.

Reduced exocytosis in the basal IHCs of BDNFPax2 KO mice. A, B, ICa and ΔCm responses from adult basal turn control and BDNFPax2 KO IHCs. Recordings were obtained in response to 100 ms voltage steps, in 10 mV increments, from the holding potential of −81 mV. For clarity, only maximal responses are shown in A. C, The relation between Ca2+ entry and exocytosis in IHCs, estimated using a synaptic transfer function, was obtained by plotting ΔCm against the peak ICa for voltage steps from −71 mV to that where the maximal ICa occurred from the I-V curves shown in B. Data were approximated using a power function: ΔCm ∝ ICaN, where N is the power. N was 1.3 ± 0.1 (n = 5) in control and 1.1 ± 0.1 (n = 4) in BDNFPax2 KO mice. D, Ca2+ currents and changes in cell membrane capacitance from apical (ap) and basal (ba) IHCs.
We next investigated whether the altered exocytosis in BDNFPax2 KO mice may be due to a reduced number of synaptic ribbons by counting them in three cochlear regions (apical: 2–7 kHz, medial: 7–16 kHz and midbasal: > 17 kHz; (Engel et al., 2006)) following immunostaining for a component of the presynaptic ribbon (CtBP2/RIBEYE, Fig. 4A). Ribbon numbers in control IHCs from mice with a mature hearing were found to be similar to those measured in previous studies (Kujawa and Liberman, 2009), with the highest number present in the medial turn (Fig. 4B). However, IHCs from age-matched BDNFPax2 KO mice revealed a significantly reduced number of ribbon synapses that was most prominent toward the basal high-frequency cochlear region (Fig. 4B, Table 1, controls: 17.69 ± 2.27 SD, n = 16 IHC; BDNFPax2 KO: 5.81 ± 2.79 SD, n = 17 IHC; 68% reduction, t test: p < 0.001), thus explaining the reduced ΔCm in these basal IHCs (Fig. 3D). Noteworthy, ribbon numbers in IHCs were unsuspicious in BDNFPax2 KO until P12 between (data not shown).
Table 1.
Number of inner hair cell ribbons in control and BDNFPax2 KO mice
| Apical | Medial | Midbasal | |
|---|---|---|---|
| Control, mean ± SD | 17.82 ± 3.97 | 19.20 ± 2.21 | 17.69 ± 2.27 |
| BDNFPax2 KO, mean ± SD | 14.8 ± 4.98 | 13.73 ± 3.22 | 5.81 ± 2.79 |
| Significance | n.s. | *** | *** |
Average number of ribbons counted in IHCs of indicated cochlear turns from three animals in three independent experiments. n.s., not significant;
***t test, p < 0.001.
When afferent fibers were analyzed using cochlear whole mount preparation, we observed a strong reduction in afferent fibers labeled with neurofilament NF200 in BDNFPax2 KO mice compared with controls, in cochlear turns with reduced IHCs ribbons (Fig. 4C). In agreement with this finding, the amplitude of the ABR wave I, which reflects the summed activity of the AN (Johnson and Kiang, 1976), was also reduced by ∼ 40% in BDNFPax2 KO mice at 15 dB threshold and above (Fig. 4D, controls: n = 17/34 mice/ears; BDNFPax2 KO: n = 17/34 mice/ears; two-way ANOVA: p < 0.001).
We can conclude from these findings that after hearing onset BDNF is essential for maintaining a normal maximal exocytosis and mature number of ribbons and afferent fibers in high-frequency IHCs.
Lack of BDNF in the cochlea protects from noise-induced hearing loss
Acoustic overexposure is known to induce neural degeneration at the IHC synapse (Kujawa and Liberman, 2009; Lin et al., 2011). Therefore, we studied a possible role of BDNF during noise damage by comparing ABR thresholds between control and BDNFPax2 KO mice after exposing them for 40 min to a 10 kHz tone of 116 dB SPL under anesthesia. Sham-exposed animals were anesthetized and placed in the reverberating chamber without acoustic stimulus (i.e., the speaker remained turned off). These animals showed completely normal hearing. In noise-exposed mice, the degree of the ABR threshold shift was measured seven days after noise exposure, when noise-induced permanent threshold shifts (PTS, Liberman et al., 1986) have settled and a recovery from damage is no longer expected. At this time point auditory fibers show a recovery from excitotoxic swelling and normal-appearing synapses in the IHC area are observed by electron microscopy (Lin et al., 2011). Hearing thresholds to click stimuli and frequency-specific pure tone ABR thresholds were significantly increased (click stimuli, one-way ANOVA: p < 0.001, frequency-specific stimuli, two-way ANOVA: p < 0.001) in noise-exposed control mice (n = 8/16 mice/ears; 64.5 dB SPL ± 16.64 SD) compared with nonexposed controls (n = 6/12 mice/ears; 18.6 dB SPL ± 3.92 SD), indicating a PTS (Fig. 5A,B). On the other hand, no significant ABR threshold shifts were observed between noise-exposed BDNFPax2KO mice (n = 8/16 mice/ears; 41.9 dB SPL ± 16.71 SD) and sham-exposed KO mice (n = 6/12 mice/ears; 30.3 dB SPL ± 5.3 SD, Fig. 5A,B). Moreover hearing thresholds to click stimuli and frequency-specific pure tone ABR thresholds were significantly lower in noise-exposed BDNFPax2 KO mice compared with noise-exposed controls (Fig. 5A,B, 5.6–22.6 kHz, two-way ANOVA: p < 0.001).
Figure 5.

BDNFPax2 KO mice are less vulnerable to noise trauma. A, B, Mean ABR thresholds ± SD for click stimuli (A) and mean frequency-specific ABR thresholds ± SD (B), seven days after noise exposure (AT, 116 dB SPL, 10 kHz, for 40 min) in control and BDNFPax2 KO mice (nonexposed control: n = 6/12 mice/ears; control +AT: n = 8/16; nonexposed BDNFPax2 KO: n = 6/12, BDNFPax2 KO +AT: n = 8/16; A, one-way ANOVA: p < 0.001; B, two-way ANOVA: p < 0.001). C, Ribbon counts from three different frequency cochlear regions in sham- or noise-exposed controls and BDNFPax2 KO mice (one-way ANOVA: p < 0.001); n = 3 mice. Numbers of IHCs counted are given in the bars. D, E, Immunohistochemistry for CtBP2/RIBEYE (green) in controls (D) and BDNFPax2 KO mice (E) before and 14 d after noise exposure (+AT), shown for the midbasal turn. Cell nuclei were counterstained with DAPI; scale bar, 10 μm. n = 3 mice, done in triplicate.
We next analyzed ribbon synapses from control mice 2 weeks after noise exposure and observed a significant loss of ribbons in basal/midbasal high-frequency regions representing the cochlear turns most damaged by the 10 kHz tone (Fig. 5C–E, Table 2, one-way ANOVA: p < 0.001). In contrast, ribbon counts from equally noise-exposed BDNFPax2 KO mice were rather similar to unexposed controls (Fig. 5C, Table 2), indicating a partial recovery of ribbon number 2 weeks after noise exposure. To determine whether these ribbons were able to make functional contacts with afferents, we analyzed click-evoked ABR waveform amplitudes that are expected to change proportionally to the size of discharge rates and number of synchronously firing auditory fibers (Johnson and Kiang, 1976). Click-evoked ABR waveform amplitudes were compared before and after noise exposure at 20 dB above threshold for latencies corresponding to the AN (wave I), cochlear nucleus (II), superior olivary complex (SOC, III), lateral lemniscus and IC (IV) and IC output activity (V) (Melcher and Kiang, 1996). As expected from the higher PTS observed in control mice after acoustic overexposure, an overall reduction of the amplitudes was observed compared with the pre-exposure situation (Fig. 6A). In contrast, BDNFPax2 KO mice showed no significant alteration in ABR waveform amplitudes upon noise exposure (Fig. 6B). When the main peaks of the ABR waves (Fig. 6A, arrowheads) were analyzed in detail, all amplitudes were significantly reduced in control mice after noise exposure (two-way ANOVA: p < 0.001), whereas, despite reduced ABR wave I, the peak amplitudes of unexposed and noise-exposed BDNFPax2 KO mice were not significantly different from pre-exposure amplitudes (Fig. 6C). As later ABR waves arise from synchronous neural activity in the auditory brainstem that are dominated by the response of globular bushy cells in the ventral cochlear nucleus (VCN, wave II) and superior olivocochlear complex (SOC, wave III) (Melcher and Kiang, 1996) where no deletion of BDNF occurs in BDNFPax2 KO, variations in the ABR wave in higher brain areas observed between BDNFPax2 KO and WT animals must have their origin in different levels of BDNF in the cochlea. Finally, the fact that despite elevated ribbon numbers in IHCs (Fig. 6C–E), ABR wave I amplitude did not recover in noise-exposed BDNFPax2 KO mice, indicates that in the absence of BDNF, an overstimulation can trigger an increase in IHCs ribbons that however is not translated in functional afferent contacts.
Table 2.
Number of inner hair cell ribbons in control and BDNFPax2 KO mice before and after noise exposure
| Apical | Medial | Midbasal | |
|---|---|---|---|
| Control, mean ± SD | 16.33 ± 3.26 | 19.17 ± 1.8 | 15.86 ± 2.51 |
| BDNFPax2 KO, mean ± SD | 12.08 ± 2.56 | 10.31 ± 2.63 | 5.77 ± 2.24 |
| Control + AT, mean ± SD | 14.25 ± 2.82 | 11.67 ± 1.32 | 7.18 ± 1.83 |
| BDNFPax2 KO + AT, mean ± SD | 17.33 ± 1.94 | 17.67 ± 2.74 | 12.7 ± 2.31 |
Average number of ribbons counted in IHCs of indicated cochlear turns from three animals in three independent experiments before and after noise overexposure (+AT).
Figure 6.

Peak-to-peak amplitudes of ABR waves are not reduced in BDNFPax2 KO mice following noise exposure. ABR waves illustrate the difference in signal amplitude in control (A) and BDNFPax2 KO mice (B) before and 7 d after trauma (AT; ±SEM), shown for stimulation with clicks presented at 20 dB above the hearing threshold. C, Peak to peak amplitudes at 20 dB above hearing threshold for five selected peak-to-peak amplitudes (wave I-V, arrows in D, two-way ANOVA: p < 0.001). After exposure the amplitude of control mice was significantly reduced for waves II-V compared with pre-exposure (two-way ANOVA: p < 0.05). Amplitudes of BDNFPax2 KO mice were not significantly decreased after exposure. Control, n = 8/16 mice/ears; BDNFPax2 KO, n = 8/16.
Discussion
Using a conditional BDNF mouse line with specific deletion of BDNF in the auditory periphery and some ascending nuclei, the present study suggests that BDNF is required to upgrade mature maximal exocytosis rates and normal amplitudes of sound-induced potentials in the peripheral and central auditory pathway. In contrast during injury BDNF in the cochlea can downscale the AN response to a degree that is insufficient to enable successful central compensation upon degradation of AN response.
BDNF is crucial for normal exocytosis and maintenance of ribbon number in high-frequency cochlear turns
Before onset of hearing BDNF is expressed in both IHCs and OHCs (Wiechers et al., 1999; Fritzsch et al., 2004). According to constitutive BDNF KO mouse models, BDNF is necessary for the recruitment of afferent type II fibers to OHCs in the high-frequency region of the cochlea during early postnatal development (Schimmang et al., 2003), and for the survival of vestibular neurons (Ernfors et al., 1994). From birth onwards BDNF is downregulated in hair cells and upregulated in SG where it is maintained throughout adult stages in a tonotopic gradient, increasing toward higher frequency cochlear turns (Adamson et al., 2002; Sobkowicz et al., 2002; Schimmang et al., 2003). As shown by electron microscopy (Sobkowicz et al., 2002), BDNF is also expressed in inner border and phalangeal cells that ensheath IHCs. However the role of BDNF in the mature cochlea is still elusive, because BDNF constitutive KO mice die before onset of hearing (Ernfors et al., 1995; Fritzsch et al., 2004).
By generating a conditional BDNF mouse line (BDNFPax2 KO), the present study documents a reduction in the maximal rate of exocytosis and reduced ribbon number in IHCs positioned in high-frequency cochlear turns of BDNFPax2 KO mice with mature hearing. The reduction in the maximal ΔCm in IHCs of BDNFPax2 KO mice occurred despite mature biophysics of the Ca2+ current (Johnson et al., 2008), and regardless of the linearization of the exocytotic Ca2+ dependence in IHCs (Marcotti et al., 2003). Since IHC ribbon numbers were normal in BDNFPax2 KO mice at the onset of hearing, it is unlikely that we observe a developmental aberration. We rather suggest a requirement of BDNF for maintaining the mature number of ribbon-containing active zones in IHCs.
The intracellular mechanism controlled by BDNF that leads to altered ribbon numbers is elusive, but it is challenging to consider this response from the perspective of BDNF expression in supporting phalangeal cells (Sobkowicz et al., 2002). BDNF may influence IHC synapses through a TrkB-mediated control of synaptic proteins as also described for synapses in the CNS (Takei et al., 1997; Tartaglia et al., 2001; Yamada et al., 2002; Matsumoto et al., 2006). The main component of ribbons, RIBEYE, was shown to be essential for stabilization of afferent contacts (Sheets et al., 2011). Thus, a requirement of BDNF to preserve ribbon numbers could also explain its role on afferent fiber density and auditory nerve activity both shown to be reduced in BDNFPax2 KO mice. A role of BDNF in supporting cells to maintain IHC ribbons would be in line with previous findings that describe a ribbon loss in vestibular sensory cells following BDNF deletion in supporting cells (Gómez-Casati et al., 2010). Also, recently the long-standing dogma that cochlear nerve degeneration is a consequence of IHC death was overturned, proposing that supporting cells that surround IHCs are crucial for auditory nerve survival (Zilberstein et al., 2012). Thus, our data indicate that BDNF in the cochlea, and perhaps specifically in supporting cells of IHCs, is crucial for the maintenance of IHC synapse integrity and summed activity of the auditory nerve in high-frequency cochlear regions. This is expected to reduce the temporal acuity of sound processing as the ∼20 ribbons that tether synaptic vesicles in active zones of ∼3000 IHCs (Glowatzki and Fuchs, 2002) drive a single postsynaptic AN fiber by a large releasable transmitter pool (Matthews and Fuchs, 2010). When ribbons are declined the temporal resolution of sound processing will worsen, as the instantaneous spikes of AN fibers particular at stimulus onset are crucial for reliability and precision of timing (Buran et al., 2010). Thus, a reduced cochlear BDNF level, as described to occur over age (Rüttiger et al., 2007), would likely reduce temporal resolution of sound processing and thereby likely speech recognition or sound localization (Wang et al., 2011).
ABR responses are differentially altered in BDNFPax2 KO mice
The click-evoked ABR waveform amplitudes measured in the present study in nontraumatized and traumatized BDNFPax2 KO and control mice give us an information about discharge rates and/or number of synchronously firing auditory fibers (Johnson and Kiang, 1976). According to Salvi et al. (1990), the central auditory system compensates for diminished input by upregulating its responsiveness in central circuitries, a process assumed to be critically dependent on response characteristics of maintained afferent fibers (Schaette and McAlpine, 2011). Central compensation that followed reduced auditory nerve activity may earliest occur at the level of the auditory brainstem. From here late ABR waves arise from synchronous neural activity in the VCN (Melcher and Kiang, 1996) through the response of globular bushy cells (wave II) toward the SOC (wave III) and from there to higher brain regions such as the lateral lemniscus (wave IV) or IC output activity (wave V). Following auditory trauma, hyperactivity spreads through the same pathway (Cai et al., 2009; Vogler et al., 2011). As conditional inactivation of BDNF, through Cre expression under the Pax2 promoter does not lead to BDNF deletion in the VCN or SOC neurons, shown by X-Gal staining of brain tissue from the Pax2 Cre/ROSA26 reporter mouse line, the differences in ABR waves II and III observed in BDNFPax2 KO and noise-exposed BDNFPax2 KO and control mice, are unlikely to be caused by changes of BDNF expression in these centers but are rather due to the difference in BDNF levels in the cochlea. Additionally, so far no descending projections from the IC have been described to terminate in the SOC (Malmierca and Merchan, 2004). During acoustic trauma, a reduction of ribbons or loss of IHC afferent fibers occurs. Depending on the absence or presence of BDNF in the cochlea this can lead to less or more central compensation. Previous studies described a loss of afferent neurodegeneration and IHCs ribbon loss in high-frequency cochlear turns in mice and guinea pig (Kujawa and Liberman, 2009; Lin et al., 2011). Using moderate trauma condition this rapid permanent primary neural degeneration was followed by a slow degeneration of spiral ganglion neurons over the next months (Lin et al., 2011), and similar as described in the present study did occur without loss of OHCs or IHCs. Our data suggest cochlear BDNF as a candidate trigger for the afferent neurodegeneration following noise trauma, and that the brainstem responsiveness following intense acoustic trauma is rescued by deletion of BDNF in the cochlea.
While further studies are essential to verify the consequences of this presumptive “non-adaptive” response, the role of BDNF in the cochlea for this process is remarkable. A crucial function of BDNF in the periphery of sensory organs may be revisited in the context of contradictory findings about the role of BDNF for brain diseases with involved disturbed homeostatic adaptation mechanism (Krishnan and Nestler, 2010). The findings may also be considered in the context of harmful effects of BDNF described to occur following peripheral nerve injury of dorsal root ganglia, when microglial-derived BDNF triggers hyperexcitability and altered sensitivity of the sciatic nerve through a TrkB-mediated alteration of the transmembrane chloride gradient (Biggs et al., 2010). This BDNF upregulation in microglial cells was shown to take place following an increase in excitability in an ATP-dependent manner (Coull et al., 2005). Thus, either excess glutamate release from IHCs (Wang et al., 2002, 2003), or a possible harmful role of purinergic signaling after cochlear damage (Housley et al., 2009), may be discussed as a trigger for detrimental BDNF activities in futures studies.
Moreover, alternative splicing events generate truncated TrkB receptors and the differential interaction of TrkB receptor isoforms with p75 neurotrophin receptor (p75 NTR) may be considered as a cause of the observed harmful effects of BDNF. Accordingly in hippocampal neurons the in vivo overexpression of truncated TrkB receptor led to deficits in long-term potentiation and depression, correlated with reduced dendritic complexity and decline of spine density. Interestingly p75 NTR overexpression rescued this phenotype, suggesting the presence of a cross-linked system (Michaelsen et al., 2010). In the cochlea p75 NTR is expressed in cochlear Schwann Cells (Provenzano et al., 2011) and full-length and truncated TrkB isoforms are expressed at the level of IHCs as well as SG (Knipper et al., 1996). In p75 KO mice however, SG survival is compromised after noise, suggesting p75 NTR mediates a rather protecting role (Tan et al., 2010). As we observed the BDNF effect on IHCs synapse and auditory fiber physiology restrictively in the high-frequency regions, a presumptive participation of a change in relative expression of TrkB FL and its isoforms may occur in a topologically specific region. Therefore a quantitative analysis of TrkB receptor isoform expression in cochlear compartments is currently not feasible.
In conclusion, cochlear BDNF seems to have an ambiguous effect on IHC physiology and auditory sound processing: a crucial role to upgrade complexity of the IHC synapse and afferents in the intact system but a harmful role when acoustic overstimulation damages the system. Changes in BDNF expression in the inner ear that may have a genetic, epigenetic, or environmental origin may thus have to be reconsidered in the context of these findings for their consequences for hearing. Accordingly, a gradual age-related loss of BDNF in the cochlea, as previously shown to occur in the third life span in rats and gerbils (Rüttiger et al., 2007), or altered BDNF levels in psychiatric and neurodegenerative disorders (Lu and Martinowich, 2008; Krishnan and Nestler, 2010; Pardon, 2010), may be discussed in the context of altered loudness perception that has been described in these diseases (Frisina, 2009; Aznar and Knudsen, 2011). Finally, the current ideas to propose BDNF as a promising future therapeutic for hearing deficits (Pettingill et al., 2011) may be revisited in the context of the present study.
Footnotes
This work was supported by the Marie Curie Research Training Network CavNET MRTN-CT-2006-035367, the Deutsche Forschungsgemeinschaft DFG-Kni-316-4-1 and Hahn Stiftung (Index AG), and by the Wellcome Trust, 088719 and 091895 (to W.M.). W.M. and S.L.J. are Royal Society University Research Fellows.
References
- Adamson CL, Reid MA, Davis RL. Opposite actions of brain-derived neurotrophic factor and neurotrophin-3 on firing features and ion channel composition of murine spiral ganglion neurons. J Neurosci. 2002;22:1385–1396. doi: 10.1523/JNEUROSCI.22-04-01385.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aznar S, Knudsen GM. Depression and Alzheimer's disease: is stress the initiating factor in a common neuropathological cascade? J Alzheimers Dis. 2011;23:177–193. doi: 10.3233/JAD-2010-100390. [DOI] [PubMed] [Google Scholar]
- Barde YA. The nerve growth factor family. Prog Growth Factor Res. 1990;2:237–248. doi: 10.1016/0955-2235(90)90021-b. [DOI] [PubMed] [Google Scholar]
- Biggs JE, Lu VB, Stebbing MJ, Balasubramanyan S, Smith PA. Is BDNF sufficient for information transfer between microglia and dorsal horn neurons during the onset of central sensitization? Mol Pain. 2010;6:44. doi: 10.1186/1744-8069-6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR, Messaoudi E. BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol. 2005;76:99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Buran BN, Strenzke N, Neef A, Gundelfinger ED, Moser T, Liberman MC. Onset coding is degraded in auditory nerve fibers from mutant mice lacking synaptic ribbons. J Neurosci. 2010;30:7587–7597. doi: 10.1523/JNEUROSCI.0389-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai S, Ma WL, Young ED. Encoding intensity in ventral cochlear nucleus following acoustic trauma: implications for loudness recruitment. J Assoc Res Otolaryngol. 2009;10:5–22. doi: 10.1007/s10162-008-0142-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- El-Badry MM, McFadden SL. Electrophysiological correlates of progressive sensorineural pathology in carboplatin-treated chinchillas. Brain Res. 2007;1134:122–130. doi: 10.1016/j.brainres.2006.11.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel J, Braig C, Rüttiger L, Kuhn S, Zimmermann U, Blin N, Sausbier M, Kalbacher H, Münkner S, Rohbock K, Ruth P, Winter H, Knipper M. Two classes of outer hair cells along the tonotopic axis of the cochlea. Neuroscience. 2006;143:837–849. doi: 10.1016/j.neuroscience.2006.08.060. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Kucera J, Lee KF, Loring J, Jaenisch R. Studies on the physiological role of brain-derived neurotrophic factor and neurotrophin-3 in knockout mice. Int J Dev Biol. 1995;39:799–807. [PubMed] [Google Scholar]
- Frisina RD. Age-related hearing loss: ear and brain mechanisms. Ann N Y Acad Sci. 2009;1170:708–717. doi: 10.1111/j.1749-6632.2009.03931.x. [DOI] [PubMed] [Google Scholar]
- Fritzsch B, Tessarollo L, Coppola E, Reichardt LF. Neurotrophins in the ear: their roles in sensory neuron survival and fiber guidance. Prog Brain Res. 2004;146:265–278. doi: 10.1016/S0079-6123(03)46017-2. [DOI] [PubMed] [Google Scholar]
- Fuchs PA. Time and intensity coding at the hair cell's ribbon synapse. J Physiol. 2005;566:7–12. doi: 10.1113/jphysiol.2004.082214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glowatzki E, Fuchs PA. Transmitter release at the hair cell ribbon synapse. Nat Neurosci. 2002;5:147–154. doi: 10.1038/nn796. [DOI] [PubMed] [Google Scholar]
- Gómez-Casati ME, Murtie JC, Rio C, Stankovic K, Liberman MC, Corfas G. Nonneuronal cells regulate synapse formation in the vestibular sensory epithelium via erbB-dependent BDNF expression. Proc Natl Acad Sci U S A. 2010;107:17005–17010. doi: 10.1073/pnas.1008938107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidrych P, Zimmermann U, Bress A, Pusch CM, Ruth P, Pfister M, Knipper M, Blin N. Rab8b GTPase, a protein transport regulator, is an interacting partner of otoferlin, defective in a human autosomal recessive deafness form. Hum Mol Genet. 2008;17:3814–3821. doi: 10.1093/hmg/ddn279. [DOI] [PubMed] [Google Scholar]
- Housley GD, Bringmann A, Reichenbach A. Purinergic signaling in special senses. Trends Neurosci. 2009;32:128–141. doi: 10.1016/j.tins.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Hu Y, Russek SJ. BDNF and the diseased nervous system: a delicate balance between adaptive and pathological processes of gene regulation. J Neurochem. 2008;105:1–17. doi: 10.1111/j.1471-4159.2008.05237.x. [DOI] [PubMed] [Google Scholar]
- Johnson DH, Kiang NY. Analysis of discharges recorded simultaneously from pairs of auditory nerve fibers. Biophys J. 1976;16:719–734. doi: 10.1016/S0006-3495(76)85724-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Marcotti W, Kros CJ. Increase in efficiency and reduction in Ca2+ dependence of exocytosis during development of mouse inner hair cells. J Physiol. 2005;563:177–191. doi: 10.1113/jphysiol.2004.074740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Forge A, Knipper M, Münkner S, Marcotti W. Tonotopic variation in the calcium dependence of neurotransmitter release and vesicle pool replenishment at mammalian auditory ribbon synapses. J Neurosci. 2008;28:7670–7678. doi: 10.1523/JNEUROSCI.0785-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Franz C, Kuhn S, Furness DN, Rüttiger L, Münkner S, Rivolta MN, Seward EP, Herschman HR, Engel J, Knipper M, Marcotti W. Synaptotagmin IV determines the linear Ca2+ dependence of vesicle fusion at auditory ribbon synapses. Nat Neurosci. 2010;13:45–52. doi: 10.1038/nn.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KR, Fariñas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khimich D, Nouvian R, Pujol R, Tom Dieck S, Egner A, Gundelfinger ED, Moser T. Hair cell synaptic ribbons are essential for synchronous auditory signalling. Nature. 2005;434:889–894. doi: 10.1038/nature03418. [DOI] [PubMed] [Google Scholar]
- Knipper M, Zimmermann U, Rohbock K, Köpschall I, Zenner HP. Expression of neurotrophin receptor trkB in rat cochlear hair cells at time of rearrangement of innervation. Cell Tissue Res. 1996;283:339–353. doi: 10.1007/s004410050545. [DOI] [PubMed] [Google Scholar]
- Knipper M, Zinn C, Maier H, Praetorius M, Rohbock K, Köpschall I, Zimmermann U. Thyroid hormone deficiency before the onset of hearing causes irreversible damage to peripheral and central auditory systems. J Neurophysiol. 2000;83:3101–3112. doi: 10.1152/jn.2000.83.5.3101. [DOI] [PubMed] [Google Scholar]
- Knipper M, Zimmermann U, Müller M. Molecular aspects of tinnitus. Hear Res. 2010;266:60–69. doi: 10.1016/j.heares.2009.07.013. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Nestler EJ. Linking molecules to mood: new insight into the biology of depression. Am J Psychiatry. 2010;167:1305–1320. doi: 10.1176/appi.ajp.2009.10030434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujawa SG, Liberman MC. Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J Neurosci. 2009;29:14077–14085. doi: 10.1523/JNEUROSCI.2845-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman MC, Dodds LW, Learson DA. Structure–function and correlation in noise-damaged ears: a light and electronmicroscopic study. In: Salvi RJ, Henderson D, Hamernik RP, Colletti V, editors. Basic and applied aspects of noise-induced hearing loss. New York: Plenum; 1986. pp. 163–177. [Google Scholar]
- Liberman MC, Dodds LW, Pierce S. Afferent and efferent innervation of the cat cochlea: quantitative analysis with light and electron microscopy. J Comp Neurol. 1990;301:443–460. doi: 10.1002/cne.903010309. [DOI] [PubMed] [Google Scholar]
- Lin HW, Furman AC, Kujawa SG, Liberman MC. Primary neural degeneration in the Guinea pig cochlea after reversible noise-induced threshold shift. J Assoc Res Otolaryngol. 2011;12:605–616. doi: 10.1007/s10162-011-0277-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Martinowich K. Cell biology of BDNF and its relevance to schizophrenia. Novartis Found Symp. 2008;289:119–129. doi: 10.1002/9780470751251.ch10. discussion 129–135, 193–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmierca M, Merchan M. The auditory system. In: Paxinos G, editor. The rat nervous system. San Diego: Academic; 2004. pp. 997–1082. [Google Scholar]
- Marcotti W, Johnson SL, Holley MC, Kros CJ. Developmental changes in the expression of potassium currents of embryonic, neonatal and mature mouse inner hair cells. J Physiol. 2003;548:383–400. doi: 10.1113/jphysiol.2002.034801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T, Numakawa T, Yokomaku D, Adachi N, Yamagishi S, Numakawa Y, Kunugi H, Taguchi T. Brain-derived neurotrophic factor-induced potentiation of glutamate and GABA release: different dependency on signaling pathways and neuronal activity. Mol Cell Neurosci. 2006;31:70–84. doi: 10.1016/j.mcn.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Matthews G, Fuchs P. The diverse roles of ribbon synapses in sensory neurotransmission. Nat Rev Neurosci. 2010;11:812–822. doi: 10.1038/nrn2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melcher JR, Kiang NY. Generators of the brainstem auditory evoked potential in cat. III: Identified cell populations. Hear Res. 1996;93:52–71. doi: 10.1016/0378-5955(95)00200-6. [DOI] [PubMed] [Google Scholar]
- Michaelsen K, Zagrebelsky M, Berndt-Huch J, Polack M, Buschler A, Sendtner M, Korte M. Neurotrophin receptors TrkB.T1 and p75NTR cooperate in modulating both functional and structural plasticity in mature hippocampal neurons. Eur J Neurosci. 2010;32:1854–1865. doi: 10.1111/j.1460-9568.2010.07460.x. [DOI] [PubMed] [Google Scholar]
- Minichiello L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci. 2009;10:850–860. doi: 10.1038/nrn2738. [DOI] [PubMed] [Google Scholar]
- Moser T, Brandt A, Lysakowski A. Hair cell ribbon synapses. Cell Tissue Res. 2006;326:347–359. doi: 10.1007/s00441-006-0276-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyama T, Groves AK. Generation of Pax2-Cre mice by modification of a Pax2 bacterial artificial chromosome. Genesis. 2004;38:195–199. doi: 10.1002/gene.20017. [DOI] [PubMed] [Google Scholar]
- Pardon MC. Role of neurotrophic factors in behavioral processes: implications for the treatment of psychiatric and neurodegenerative disorders. Vitam Horm. 2010;82:185–200. doi: 10.1016/S0083-6729(10)82010-2. [DOI] [PubMed] [Google Scholar]
- Pettingill LN, Wise AK, Geaney MS, Shepherd RK. Enhanced auditory neuron survival following cell-based BDNF treatment in the deaf guinea pig. PLoS One. 2011;6:e18733. doi: 10.1371/journal.pone.0018733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Provenzano MJ, Minner SA, Zander K, Clark JJ, Kane CJ, Green SH, Hansen MR. p75(NTR) expression and nuclear localization of p75(NTR) intracellular domain in spiral ganglion Schwann cells following deafness correlate with cell proliferation. Mol Cell Neurosci. 2011;47:306–315. doi: 10.1016/j.mcn.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauskolb S, Zagrebelsky M, Dreznjak A, Deogracias R, Matsumoto T, Wiese S, Erne B, Sendtner M, Schaeren-Wiemers N, Korte M, Barde YA. Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J Neurosci. 2010;30:1739–1749. doi: 10.1523/JNEUROSCI.5100-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, Lechan RM, Jaenisch R. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol. 2001;15:1748–1757. doi: 10.1210/mend.15.10.0706. [DOI] [PubMed] [Google Scholar]
- Rüttiger L, Panford-Walsh R, Schimmang T, Tan J, Zimmermann U, Rohbock K, Köpschall I, Limberger A, Müller M, Fraenzer JT, Cimerman J, Knipper M. BDNF mRNA expression and protein localization are changed in age-related hearing loss. Neurobiol Aging. 2007;28:586–601. doi: 10.1016/j.neurobiolaging.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Salvi RJ, Saunders SS, Gratton MA, Arehole S, Powers N. Enhanced evoked response amplitudes in the inferior colliculus of the chinchilla following acoustic trauma. Hear Res. 1990;50:245–257. doi: 10.1016/0378-5955(90)90049-u. [DOI] [PubMed] [Google Scholar]
- Schaette R, McAlpine D. Tinnitus with a normal audiogram: physiological evidence for hidden hearing loss and computational model. J Neurosci. 2011;31:13452–13457. doi: 10.1523/JNEUROSCI.2156-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmang T, Tan J, Müller M, Zimmermann U, Rohbock K, Köpschall I, Limberger A, Minichiello L, Knipper M. Lack of Bdnf and TrkB signalling in the postnatal cochlea leads to a spatial reshaping of innervation along the tonotopic axis and hearing loss. Development. 2003;130:4741–4750. doi: 10.1242/dev.00676. [DOI] [PubMed] [Google Scholar]
- Schmitz F. The making of synaptic ribbons: how they are built and what they do. Neuroscientist. 2009;15:611–624. doi: 10.1177/1073858409340253. [DOI] [PubMed] [Google Scholar]
- Sheets L, Trapani JG, Mo W, Obholzer N, Nicolson T. Ribeye is required for presynaptic Ca(V)1.3a channel localization and afferent innervation of sensory hair cells. Development. 2011;138:1309–1319. doi: 10.1242/dev.059451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobkowicz HM, August BK, Slapnick SM. Influence of neurotrophins on the synaptogenesis of inner hair cells in the deaf Bronx waltzer (bv) mouse organ of Corti in culture. Int J Dev Neurosci. 2002;20:537–554. doi: 10.1016/s0736-5748(02)00084-9. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Takei N, Sasaoka K, Inoue K, Takahashi M, Endo Y, Hatanaka H. Brain-derived neurotrophic factor increases the stimulation-evoked release of glutamate and the levels of exocytosis-associated proteins in cultured cortical neurons from embryonic rats. J Neurochem. 1997;68:370–375. doi: 10.1046/j.1471-4159.1997.68010370.x. [DOI] [PubMed] [Google Scholar]
- Tan J, Rüttiger L, Panford-Walsh R, Singer W, Schulze H, Kilian SB, Hadjab S, Zimmermann U, Köpschall I, Rohbock K, Knipper M. Tinnitus behavior and hearing function correlate with the reciprocal expression patterns of BDNF and Arg3.1/arc in auditory neurons following acoustic trauma. Neuroscience. 2007;145:715–726. doi: 10.1016/j.neuroscience.2006.11.067. [DOI] [PubMed] [Google Scholar]
- Tan J, Clarke M, Barrett G, Millard R. The p75 neurotrophin receptor protects primary auditory neurons against acoustic trauma in mice. Hear Res. 2010;268:46–59. doi: 10.1016/j.heares.2010.04.013. [DOI] [PubMed] [Google Scholar]
- Tartaglia N, Du J, Tyler WJ, Neale E, Pozzo-Miller L, Lu B. Protein synthesis-dependent and -independent regulation of hippocampal synapses by brain-derived neurotrophic factor. J Biol Chem. 2001;276:37585–37593. doi: 10.1074/jbc.M101683200. [DOI] [PubMed] [Google Scholar]
- Thoenen H. Neurotrophins and activity-dependent plasticity. Prog Brain Res. 2000;128:183–191. doi: 10.1016/S0079-6123(00)28016-3. [DOI] [PubMed] [Google Scholar]
- Timmusk T, Palm K, Metsis M, Reintam T, Paalme V, Saarma M, Persson H. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron. 1993;10:475–489. doi: 10.1016/0896-6273(93)90335-o. [DOI] [PubMed] [Google Scholar]
- Vogler DP, Robertson D, Mulders WH. Hyperactivity in the ventral cochlear nucleus after cochlear trauma. J Neurosci. 2011;31:6639–6645. doi: 10.1523/JNEUROSCI.6538-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Isik M, Borst A, Hemmert W. Auditory information coding by modeled cochlear nucleus neurons. J Comput Neurosci. 2011;30:529–542. doi: 10.1007/s10827-010-0276-x. [DOI] [PubMed] [Google Scholar]
- Wang J, Ding D, Salvi RJ. Carboplatin-induced early cochlear lesion in chinchillas. Hear Res. 2003;181:65–72. doi: 10.1016/s0378-5955(03)00176-x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Hirose K, Liberman MC. Dynamics of noise-induced cellular injury and repair in the mouse cochlea. J Assoc Res Otolaryngol. 2002;3:248–268. doi: 10.1007/s101620020028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiechers B, Gestwa G, Mack A, Carroll P, Zenner HP, Knipper M. A changing pattern of brain-derived neurotrophic factor expression correlates with the rearrangement of fibers during cochlear development of rats and mice. J Neurosci. 1999;19:3033–3042. doi: 10.1523/JNEUROSCI.19-08-03033.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada MK, Nakanishi K, Ohba S, Nakamura T, Ikegaya Y, Nishiyama N, Matsuki N. Brain-derived neurotrophic factor promotes the maturation of GABAergic mechanisms in cultured hippocampal neurons. J Neurosci. 2002;22:7580–7585. doi: 10.1523/JNEUROSCI.22-17-07580.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampini V, Johnson SL, Franz C, Lawrence ND, Münkner S, Engel J, Knipper M, Magistretti J, Masetto S, Marcotti W. Elementary properties of CaV1.3 Ca(2+) channels expressed in mouse cochlear inner hair cells. J Physiol. 2010;588:187–199. doi: 10.1113/jphysiol.2009.181917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberstein Y, Liberman MC, Corfas G. Inner hair cells are not required for survival of spiral ganglion neurons in the adult cochlea. J Neurosci. 2012;32:405–410. doi: 10.1523/JNEUROSCI.4678-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]