

Abstract
Neuronal migration is a critical feature to ensure proper location and wiring of neurons during cortical development. Postmitotic neurons migrate from the ventricular zone into the cortical plate to establish neuronal lamina in an “inside-out” gradient of maturation. Here, we report that the mitotic kinase Aurora-A is critical for the regulation of microtubule organization during neuronal migration via an Aurora-A–NDEL1 pathway in the mouse. Suppression of Aurora-A activity by inhibitors or siRNA resulted in severe impairment of neuronal migration of granular neurons. In addition, in utero injection of the Aurora-A kinase-dead mutant provoked defective migration of cortical neurons. Furthermore, we demonstrated that suppression of Aurora-A impaired microtubule modulation in migrating neurons. Interestingly, suppression of CDK5 by an inhibitor or siRNA reduced Aurora-A activity and NDEL1 phosphorylation by Aurora-A, which led to defective neuronal migration. We found that CDK5RAP2 is a key molecule that mediates functional interaction and is essential for centrosomal targeting of Aurora-A. Our observations demonstrated novel and surprising cross talk between Aurora-A and CDK5 during neuronal migration.
Introduction
Widespread cell migrations are the hallmark of vertebrate brain development (Sidman and Rakic, 1973). After primary neurogenesis in compact ventricular zones has commenced, postmitotic neurons migrate away from the germinal ventricular zone to take their position in the developing cortex, guided by the processes of radial glial cells (Hatten, 1999). The remarkable morphological transformations of neurons as they migrate, extend axons and dendrites, and establish synaptic connections, imply a strictly regulated process of structural organization and dynamic remodeling of the cytoskeleton. A central component of the cytoskeletal structure in cells is the microtubule array, which has the microtubule-organizing center, also called centrosome, at its base. The centrosome provides a structural foundation for the microtubule array, and it also is the major microtubule nucleation site that is involved in many different cell processes, particularly during cell division, cell migration, and differentiation (Doxsey et al., 2005; Azimzadeh and Bornens, 2007; Bettencourt-Dias and Glover, 2007; Nigg and Raff, 2009). The position of the centrosome is precisely regulated during neurogenesis, migration, and differentiation, leading to the segregation of cell fate factors, efficient nucleokinesis, and directed neurite outgrowth, respectively. In particular, the positioning of the centrosome and the dynamic regulation that couples and uncouples the nucleus underlies directed migration of neurons (Tsai and Gleeson, 2005; Higginbotham and Gleeson, 2007).
The centrosome provides numerous kinases a platform to encounter their target molecules. Among them, the many mitotic kinases including Aurora (Aur) (A, B, C), Polo-like (Plk1–4), and Nek (NIMA1-11) regulate the centrosome cycle, spindle checkpoint and microtubule-kinetochore attachment, spindle assembly, and chromosome condensation (Nigg, 2001; Nigg and Raff, 2009). For example, Aurora kinase was discovered in a screen to identify genes involved in mitotic spindle function (Glover et al., 1995). In particular, Aurora-A, a centrosome-localized mitotic kinase involved in spindle assembly and stability, phosphorylates proteins such as Eg5, TACC, and TPX2 (Giet et al., 1999, 2002; Kufer et al., 2002). We previously reported that NDEL1 is a substrate of the mitotic kinase Aurora-A (Mori et al., 2007), by which NDEL1 connects Aurora-A to other target molecules for the regulation of microtubule organization. Furthermore, we uncovered that an atypical protein kinase C (aPKC)–Aurora-A–NDEL1 pathway is critical for the regulation of microtubule organization during neurite extension (Mori et al., 2009), suggesting that mitotic machineries in postmitotic cells will provide unique insights into the role of such genes in development. Here, we report that an Aurora-A–NDEL1 pathway is essential for neuronal migration via regulation of microtubule remodeling. In addition, we demonstrated that cyclin-dependent kinase 5 (CDK5) partially shares the common pathway with Aurora-A during neuronal migration via CDK5RAP2. Our observations provide a surprising mechanistic relationship between mitotic and migration kinases.
Materials and Methods
siRNA and inhibitors.
siRNAs to target the mouse Aurora-A and Cdk5 were purchased from Sigma-Aldrich (MISSION siRNA): siRNA against Aurora-A, SASI_Mm01_00194885; siRNA I against CDK5, SASI_Mm01_00052852; siRNA II against CDK5, SASI_Mm01_00052853. siRNAs were transfected using Neon Transfection System (Invitrogen). After transfection, granular neurons of either sex were subjected to incubation for 24 h in noncoated 24-well dish (Falcon; BD Biosciences Discovery Labware) before start of cell culture on the coated dish. Cerebellar granule cells of either sex were isolated as described previously (Hatten, 1985; Sasaki et al., 2005; Toyo-Oka et al., 2005) and were subjected to transfection at 105 based on standard protocols. Isolated granular neurons were treated with a CDK5 inhibitor, Roscivitine (Selleck; 20 μm), or an Aurora-A inhibitor, Aurora-A Inhibitor I (Selleck; 0.17 μm) or VX-680 (Selleck; 0.03 μm). To prevent proteasomal degradation, granular neurons were applied Proteasome Inhibitor II (Merck) with 0.5 μm at the moment of transfection of siRNA. For developmental experiments of mouse cortigogenesis, we injected Roscivitine (14 μg/g dam weight), Aurora-A Inhibitor I (0.6 μg/g dam weight), or VX-680 (90 ng/g dam weight) at E15.5 and E17.5.
In utero transfection.
Expression vectors were introduced into fetal brains of either sex by an in utero electroporation-mediated gene transfer method (Saito and Nakatsuji, 2001; Tabata and Nakajima, 2001). Briefly, pregnant mice were deeply anesthetized on E16.5, and the uterine horns were exposed. pCAGGS-GFP control plasmid, pCAGGS-Aurora [kinase-dead (KD)] control plasmid, or pCAGGS-mRFP-Cre were dissolved in HBS (21 mm HEPES, pH 7.0, 137 mm NaCl, 5 mm KCl, 0.7 mm Na2HPO4, 1 mg/L glucose) at a final concentration of 10 mg/ml together with Fast Green (final concentration, 0.01%). For cotransfection, a molar ratio of 1 [pCAGGS-Aurora (KD)] to 3–6 [pCMV-Ndel1 (S251A or S251E)] was used. Approximately 2 μl of plasmid solution was injected into the lateral ventricle from outside the uteri with a glass micropipette (GD-1.5; Narishige). Each embryo of either sex in the uterus was then placed between the tweezers-type electrodes described above, and electronic pulses (45 V; 50 ms duration) were applied five times at intervals of 950 ms (GUY21; Bexco). The uterine horns were then placed back into the abdominal cavity to allow the embryos to continue normal development. Histological examination was performed 5 d after in utero injection (P2–P3).
Reaggregate neuronal migration assay, immunocytochemistry, and immunoblotting.
Cerebellar granule of either sex cells were isolated as described previously (Hatten, 1985; Sasaki et al., 2005; Toyo-Oka et al., 2005) and cultured at 105 cells/ml for 12 h, resulting in uniform-sized reaggregates (100–150 μm in diameter), which were then transferred to poly-l-lysine (Sigma-Aldrich)- and laminin (Sigma-Aldrich)-treated slides and incubated for 8 h. Inhibitors or control DMSO was added and the cultures were further incubated for 16 h. Images were captured using a 20× objective lens and analyzed using a confocal microscope (TCS-SP5; Leica). Cells were fixed in 4% PFA in PBS followed by permeabilization with 0.2% Triton X-100 in PBS. Coverslips were blocked for 1 h in PBS supplemented with 5% BSA, and were incubated for 1 h in primary antibody, washed, and incubated for 1 h using Alexa 488-conjugated secondary antibodies, Alexa 546-conjugated secondary, and DAPI (Invitrogen). Primary antibodies were an anti-Aurora-A (phospho-Thr288) antibody (Cell Signaling), an anti-Aurora-A antibody (Santa Cruz), an anti-CDK5 antibody (Santa Cruz), an anti-CDK5RAP2 antibody (Santa Cruz), and an anti-Centrin-2 antibody (Santa Cruz). For detection of NDEL1 and p-NDEL1, we used our original antibodies (Mori et al., 2007; Yamada et al., 2008). For immunoblotting, total protein from granular neurons was extracted by lysis buffer (20 mm HEPES, pH 7.5, 150 mm NaCl, 1 mm EDTA, 50 mm NaF, 1 mm Na3VO4, 10 μg/ml leupeptin, 1 mm PMSF, 1.8 μg/ml aprotinin, 0.2% Nonidet P-40) for 15 min on ice. Granular neurons were scraped, and lysates were collected. Extracts were cleared by centrifugation at 15,000 × g for 5 min at 4°C. Protein extracts were resuspended in SDS-PAGE sample buffer, separated on 10% SDS polyacrylamide gels, and subjected to immunoblotting. For rescue experiments, pCAG-Ndel1 (S251A or S251E) or pCAG-Ndel1 (3A or 3E) (Mori et al., 2007, 2009) was transfected into granular neurons using Neon Transfection System (Invitrogen).
Bromodeoxyuridine birth-dating study.
For bromodeoxyuridine (BrdU) experiments, pregnant dams (E15.5) were injected with BrdU (50 μg/g, i.p.). Subsequently, the distribution of BrdU-positive cells was determined at P5. For pulse labeling to trace proliferation of neuroepithelial stem cell, pregnant dams (E14.5) were injected with BrdU (150 μg/g, i.p.). Subsequently, the distribution of BrdU-positive cells was determined 1 h after injection. The incorporation of BrdU in cells was detected with a mouse anti-BrdU monoclonal primary antibody (Roche) followed by an alkaline phosphatase-conjugated secondary antibody (Roche Diagnostics). We analyzed three independent mice for each genotype.
Histological examination and immunohistochemistry.
After perfusion with 4% PFA fixative, tissues from wild-type and various mutant mice of either sex were subsequently embedded in paraffin and sectioned at 5 μm thickness. After deparaffination, endogenous peroxidase activity was blocked by incubating the sections in 1.5% peroxide in methanol for 20 min. The sections were then boiled in 0.01 m/L citrate buffer, pH 6.0, for 20 min and cooled slowly. Before staining, the sections were blocked with rodent block (Lab Vision) for 60 min. The sections were washed in PBS and incubated with an anti-Brn-1 antibody (Santa Cruz).
Analysis of MT dynamics.
Images were obtained, exported as tiff files, and processed with ACUACOSMOS (Hamamatsu). Black-and-white raw data were converted to color images to visualize EB3-mCherry and the end of MTs easily, and processed with the bandpass filter function of ImageJ software to remove haze. EB3-mCherry and the end of MTs were traced by a mouse-driven cursor under ImageJ software plugged in the manual track function (developed and programmed by Fabrice Cordelières, Institut Curie, Orsay, France). Changes of >0.5 μm between two points were considered growth or shortening events. Changes of <0.5 μm were considered as a pause. More than 100 microtubules from 10 cells were analyzed. The results are indicated as the mean and SEM, and at least three independent experiments were performed.
Results
Aurora-A is expressed and activated in migrating neurons
We previously demonstrated that activation of Aurora-A is essential for neurite extension (Mori et al., 2009). We postulated that activation of Aurora-A may be more generally important during neurogenesis. To address this issue, we first examined the expression and activation of Aurora-A using granular neurons isolated from the cerebellum. Immunocytochemistry demonstrated that Aurora-A was expressed and activated in granular neurons. We next examined the subcellular localization of Aurora-A. The position of the centrosome is precisely regulated during neurogenesis, migration, and differentiation, leading to the segregation of cell fate factors and efficient nucleokinesis (Tsai and Gleeson, 2005; Higginbotham and Gleeson, 2007). However, migration of rhombic lip-derived neurons of zebrafish is characterized by a centrosome that does not persistently lead the nucleus, but that is instead regularly overtaken by the nucleus (Distel et al., 2010). Aurora-A in granular neurons was localized and activated ahead of the nucleus in the direction of migration (Fig. 1a). We previously demonstrated that NDEL1 is substrate of Aurora-A, which is essential for proper modulation of microtubule organization (Mori et al., 2007). We examined whether NDEL1 is phosphorylated by Aurora-A. We found that NDEL1 was phosphorylated by Aurora-A (Fig. 1b), and phosphorylated NDEL1 was colocalized with activated Aurora-A at the centrosome (Fig. 1c,d). Next, we quantitated proteins or phosphorylated proteins by the Western blotting during neuronal migration. While the amounts of Aurora-A and NDEL1 were relatively unchanged, phosphorylated forms of both Aurora-A and NDEL1 were detected immediately after the start of migration and gradually increased during neuronal migration (Fig. 1e). These observations suggest that Aurora-A plays an essential role for neuronal migration, presumably via regulation of microtubule organization.
Figure 1.
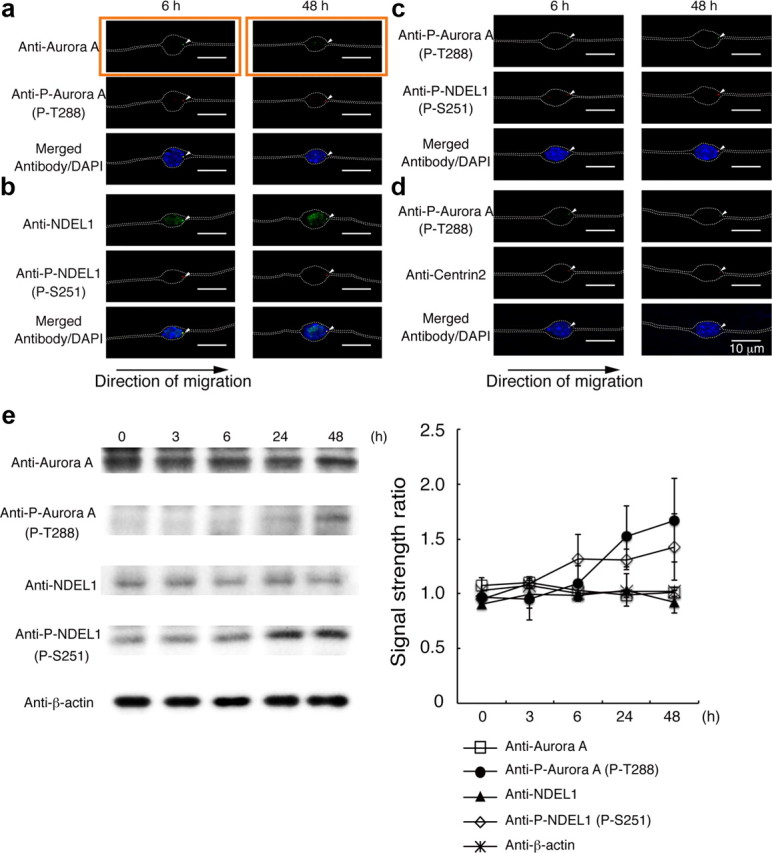
Aurora-A is expressed and phosphorylated NDEL1 in granular neurons. a, Examination of subcellular localization of Aurora-A (top), phospho-T288 Aurora-A (middle), and DAPI (bottom). Granular neurons displayed condensed expression (arrowhead) of Aurora-A (top) and phospho-T288 Aurora-A (middle). Time (in hours) after the start of culture is indicated above the panels. The white arrowheads indicate antibody signal. Phosphorylation of Aurora begins early after the start of migration. The white dotted lines indicate the outline of granular neurons. b, Examination of subcellular localization of NDEL1 (top), phospho-S251 NDEL1 (middle), and DAPI (bottom) is shown, and the white arrowheads indicate accumulation of these proteins. c, Comparison of phospho-T288 Aurora-A (top), phospho-S251 NDEL1 (middle), and DAPI (bottom). d, Comparison of phospho-T288 Aurora-A (top), Centrin-2 (middle), and DAPI (bottom). At early stage and later stage of neuronal migration, phosphorylated Aurora-A overlapped with Centrin-2, and the centrosomes were located at the front of the migrating nucleus. e, Western blotting analysis of each protein or phosphorylated protein. Either Aurora-A or NDEL1 displayed similar expression levels during culture, whereas phosphorylated Aurora-A or NDEL1 were increased during culture. Intensity was normalized to β-actin. Statistical analysis of three independent experiments is shown.
Phosphorylation of NDEL1 by Aurora-A is essential for neuronal migration
We previously reported that phosphorylation of S251 NDEL1 is essential for neurite extension (Mori et al., 2007, 2009). NDEL1 is also essential for the proper distribution of LIS1 and cytoplasmic dynein (Yamada et al., 2008). To delineate the developmental role of phosphorylation of S251 NDEL1 by Aurora-A, we first examined the motility of granular neurons isolated from Ndel1 conditional KO mice (Sasaki et al., 2005). Isolated granular neurons from Ndel1cko/cko mice displayed reduction of extension of neurites and outward movement without Ndel1 disruption by Cre-mediated recombinaiton (Fig. 2a–c). A neo gene inserted into intron III of Ndel1 for selection during gene targeting reduced NDEL1 expression, and the resulting Ndel1cko/cko mice are a hypomorphic mutant (Sasaki et al., 2005). Remarkably, defective nuclear movement was disproportionally more severely impaired than neurite extension, suggesting that nuclear movement is more sensitive to levels of NDEL1 expression. This impaired extension of neurites and defective nuclear migration were not rescued by the expression of GFP-NDEL1(S251A) (Fig. 2a–c), whereas the expression of a phosphomimic form, GFP-NDEL1(S251E), clearly rescued extension of neurites and nuclear migration (Fig. 2a–c). To address a developmental role in embryos, we injected mRFP-CRE into Ndel1cko/cko embryos to delete the Ndel1 gene at E16.5. Histological examination was performed 5 d after transfection (P2–P3). Although granular neurons displayed partial reduction of migration, corticogenesis of Ndel1cko/cko mice was intact, suggesting that the lower levels of NDEL1 in these mice were sufficient for neuronal migration in vivo. In clear contrast, transfection of mRFP-CRE in utero with the GFP marker led to severe aberrations of the positioning of mRFP-CRE/GFP-expressing cells, in which large numbers remained in deeper ventral region (Fig. 2d,e). This aberrant positioning is attributable to impaired proliferation of neuronal progenitors resulting in mitotic arrest and/or defective migration of postmitotic neurons. To dissect the underlying mechanism, we examined the distribution of mRFP-CRE/GFP-expressing cells within the brain. Interkinetic nuclear migration results in the movement of neuroepithelial and radial glial cell nuclei along the apical–basal axis in relation to the cell cycle. Mitosis of neuroepithelial cells occurs at (or very close to) the apical surface of the neuroepithelium, whereas S phase usually takes place at a more basal location, with apical-to-basal nuclear migration occurring in G1 and basal-to-apical nuclear migration in G2 (Sauer and Walker, 1959). If disruption of Ndel1 selectively affects cell proliferation, mRFP-CRE/GFP-expressing cells should remain within the ventricular zone. By contrast, if disruption of Ndel1 affects cell migration, mRFP-CRE/GFP-expressing cells should move beyond the ventricular zone. The characterization of their distributions revealed that a fraction of Ndel1-disrupted cells remained within the ventricular zone, attributable to impaired proliferation (Fig. 2e). We also found that Ndel1-disrupted cells were located beyond the ventricular zone, which must result from defective migration (Fig. 3e). We next performed rescue experiments. The reduction of cell number and aberrant positioning after deletion of Ndel1 gene were not significantly rescued by the expression of GFP-NDEL1(S251A) (Fig. 2d,e), whereas the expression of GFP-NDEL1(S251E) clearly rescued these phenotypes (Fig. 2d,e). Complete loss of NDEL1 was not compatible with proliferation of neuronal progenitors (Sasaki et al., 2005). Presumably, early and complete loss of NDEL1 causes mitotic arrest before neurogenesis, whereas partial or late inactivation of Ndel1 allows cell division and generation of postmitotic neurons, which is revealed by defective migration.
Figure 2.
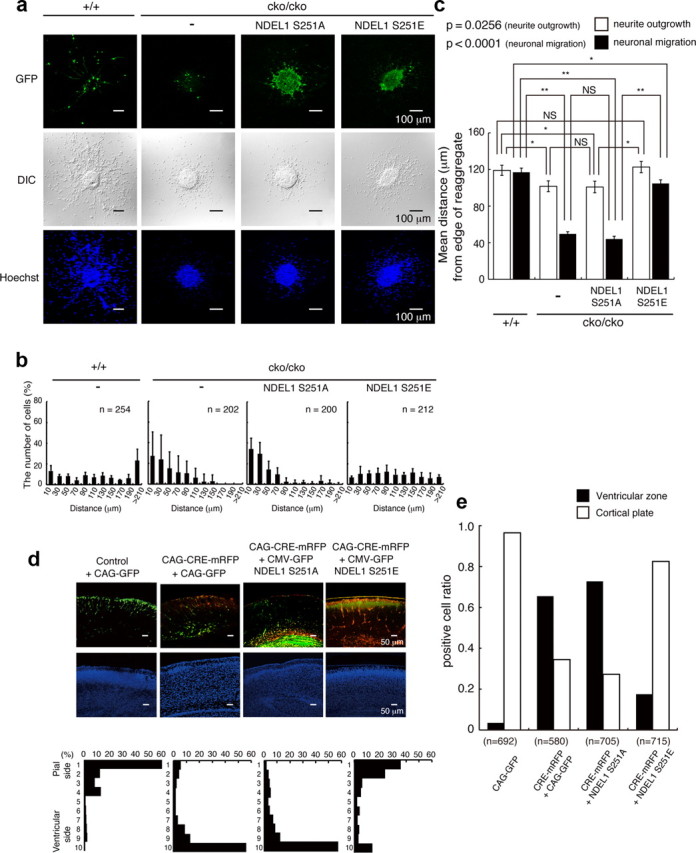
Defective migration of Ndel1 hypomorphic mutant and rescue experiments. a, Migration assay using cerebellar granule neurons after transfection of Control GFP, GFP-Ndel1 (S251A), or GFP-Ndel1 (S251E). GFP images (top), differential interference contrast (DIC) images (middle), and Hoechst staining images (bottom) are shown. Ndel1cko/cko neurons displayed defective migration. b, The migration distance of each neuron 48 h after the start of culture was binned. n is the number of neurons measured for each examination. Wild-type neurons displayed normal migration distances, whereas Ndel1cko/cko neurons displayed a shift in the distribution of bins toward the left. Expression of GFP-Ndel1 (S251A) had no effect on migration of Ndel1cko/cko neurons, whereas expression of GFP-Ndel1 (S251E) significantly improved migration of these neurons. c, Mean length of neurites (open bar) and nuclear position (solid bar) from the edge of central aggregation. Note: Granular neurons from Ndel1cko/cko mice displayed reduced neurite extension and nuclear migration. Reduction of nuclear migration appeared to be disproportionally more severe than impaired neurite extension. NDEL1 (S251E) efficiently rescued impaired extension of the leading processes and defective neural migration. Statistical analysis was performed for neurite outgrowth and nuclear migration by an ANOVA followed by t test with correction: *p < 0.05 and **p < 0.01. Error bars in graphs represent mean ± SEM. d, In utero injection of mRFP-Cre, control GFP, GFP-Ndel1 (S251A), or GFP-Ndel1 (S251E) into the E16.5 mouse neocortex of Ndel1cko/cko embryos by electroporation. GFP or mRFP/GFP images are shown in the top panels. Counterstains of a neighboring section by DAPI are displayed in middle panels. The distribution of migrated neurons are shown in the bottom panels. The cortex was divided into 10 bins, and the numbers of neurons were counted in each bin. GFP or mRFP/GFP images of cryosections are representative of six different pups from two independent in utero injections. In the control, GFP-positive neurons moved into surface area of the cortex (first lane). By contrast, disruption of Ndel1 by CRE recombinase severely impaired upward localization of cortical neurons (second lane), and this defective localization was efficiently rescued by cotransfection of GFP-Ndel1 (S251E: fourth lane) but not by GFP-Ndel1 (S251A: third lane). e, Distribution of mRFP-CRE/GFP-expressing cells within the brain. A fraction of mRFP-CRE/GFP-expressing cells remained within the ventricular zone after disruption of Ndel1. Expression of GFP-NDEL1(S251E) rescued these phenotypes. The total numbers of counted cells are indicated.
Figure 3.
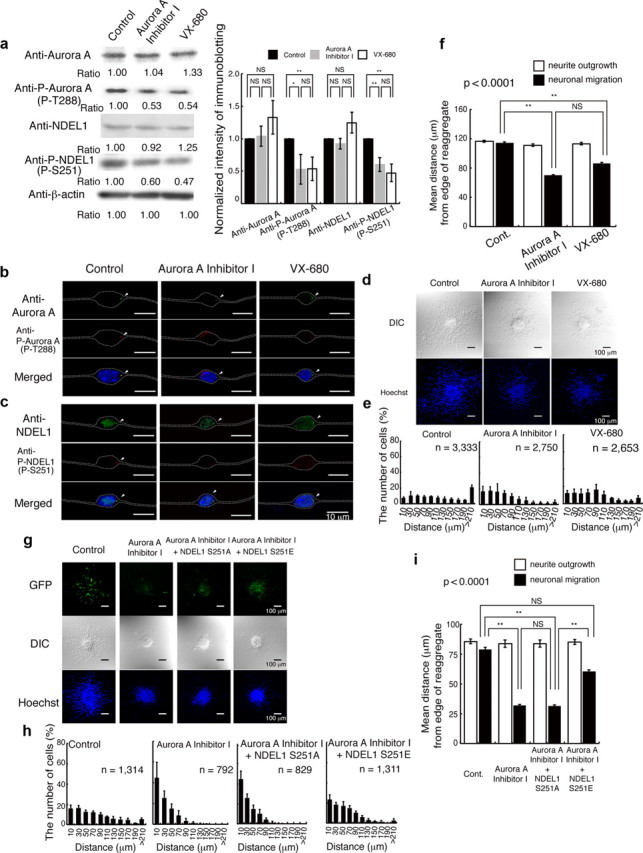
Aurora-A inhibitors severely impair neuronal migration. Migration of granular neurons after treatment of Aurora-A inhibitors was examined. a, Western blotting analysis of proteins or phosphorylated proteins. Aurora-A and NDEL1 displayed similar expression levels, whereas phosphorylated Aurora-A and NDEL1 proteins were decreased during treatment with Aurora-A inhibitors. Relative intensities of the bands of Western blotting are displayed at the bottom. Intensity was normalized with β-actin. Statistical examination was performed by unpaired Student's t test, with *p < 0.05 and **p < 0.01 (bottom panel). b, Immunocytochemistry with antibodies to Aurora-A (b) or NDEL1 (c) revealed similar expression levels, whereas phosphorylated proteins were decreased during treatment with Aurora-A inhibitors. The white dotted lines indicate the outline of granular neurons. d, Migration assays using cerebellar granule neurons. DIC images (top) and Hoechst staining images (bottom) are shown. e, The migration distance of each neuron 24 h after start of culture was binned. n is the number of neurons measured for each examination. Wild-type neurons displayed normal migration distances, whereas neurons treated with inhibitors display a shift in the distribution of bins toward the left. f, Mean length of neurites (open bar) and nuclear position (solid bar) from the edge of central aggregation. Note: Extension of neurites was not affected by inhibitors, whereas nuclear migration was significantly reduced. Statistical examination was performed to nuclear migration by an ANOVA followed by t test with correction: **p < 0.01. g, Rescue experiments of Aurora-A Inhibitor I after transfection of control GFP, GFP-Ndel1 (S251A), or GFP-Ndel1 (S251E). GFP images (top), DIC images (middle), and Hoechst staining images (bottom) are shown. Expression of GFP-Ndel1 (S251A) had no effect on migration after Aurora-A Inhibitor I treatment, whereas expression of GFP-Ndel1 (S251E) significantly improved reduced migration. h, The migration distance of each neuron 24 h after start of culture was binned. n is the number of neurons measured for each examination. Expression of GFP-Ndel1 (S251E) significantly improved migration after Aurora-A Inhibitor I treatment, which was characterized by a rightward shift of migration. i, Mean length of neurites (open bar) and nuclear position (solid bar) from the edge of central aggregation. Statistical examination was performed to nuclear migration by an ANOVA followed by t test with correction: **p < 0.01.
Inhibition of Aurora-A impairs neuronal migration, which is rescued by the expression of S251E NDEL1
To address the developmental role of Aurora-A, which is phosphorylated and activated during neuronal migration, we applied Aurora-A inhibitors to granular neurons in the migration assay. We selected two inhibitors that have relatively high specificity against Aurora-A, including Aurora-A Inhibitor I (Aliagas-Martin et al., 2009) and VX-680 (Harrington et al., 2004). We first quantitated proteins or phosphorylated proteins by the Western blotting after administration of Aurora-A inhibitors. While the amounts of Aurora-A and NDEL1 were relatively unchanged, levels of phosphorylated Aurora-A and NDEL1 were significantly suppressed by Aurora-A inhibitors (Fig. 3a). Immunocytochemistry also revealed significant reduction of T288 phosphorylation of Aurora-A accompanied by reduction of S251 phosphorylation of NDEL1 (Fig. 3b,c). In the control experiment, granular neurons displayed robust outward movement from central aggregation (Fig. 3d–f). In contrast, application of Aurora-A Inhibitor I and structurally unrelated inhibitor VX-680 resulted in significant suppression of neuronal migration, whereas extension of neurites remained intact (Fig. 3d–f,i). These observations suggest that phosphorylation of NDEL1 by Aurora-A is involved specifically in neuronal migration. To rule out off-target effects, we performed rescue experiments for the Aurora-A inhibitor phenotype by ectopic expression of a drug-resistant transgene. Expression of GFP-NDEL1(S251A) did not have any obvious influence on granular cell migration (Fig. 3g–i). By contrast, expression of GFP-NDEL1(S251E) significantly increased granular cell migration (Fig. 3g–i). Similar results were found after rescue of VX-680 inhibition of migration with these NDEL1 expression constructs (Fig. 4a–c). These observations support a pivotal role for the Aurora-A–NDEL1 pathway in neuronal migration.
Figure 4.
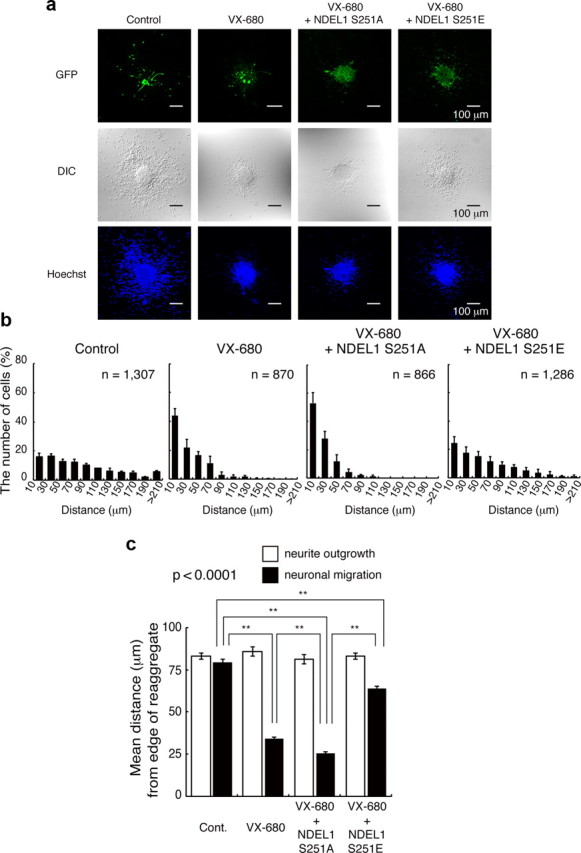
Rescue experiments of VX-680. a, Control GFP, GFP-Ndel1 (S251A), or GFP-Ndel1 (S251E) was transfected. GFP images (top), DIC images (middle), and Hoechst staining images (bottom) are shown. b, The migration distance of each neuron 24 h after start of culture was binned. n is the number of neurons measured for each examination. Expression of GFP-Ndel1 (S251A) had no effect on migration inhibited by VX-680, whereas expression of GFP-Ndel1 (S251E) significantly improved defective migration (a, b), which was characterized by rightward shift. c, Mean length of neurites (open bar) and nuclear position (solid bar) from the edge of central aggregation. Statistical examination was performed by an ANOVA followed by t test with correction: **p < 0.01.
We further investigated whether administration of these Aurora-A inhibitors affected neuronal migration during corticogenesis by BrdU birth dating. To address this issue, we first injected BrdU into the intraperitonial space of pregnant dams at E14.5 to label newly generated neurons, followed by injection of Aurora-A inhibitors twice at E15.5 and E17.5. We examined positioning of BrdU-positive cells and neurons at P21 to discriminate migration deficit from mitotic arrest by chemical inhibitors. In the control, BrdU-positive cells localized at superficial positions (Fig. 5a). By contrast, administration of Aurora-A inhibitors clearly suppressed the upward movement of BrdU-positive cells (Fig. 5a). We next confirmed these results using a layer II–III-specific marker, Brn-1 (Yamada et al., 2009). Brn-1-stained neurons were localized at superficial positions in controls (Fig. 5b). After administration of Aurora-A inhibitors, Brn-1-stained neurons were distributed over a deeper, broader region (Fig. 5b). These in vivo data indicate that the Aurora-A–NDEL1 pathway is essential for cortical neuronal migration. In contrast, the structure of hippocampus was relatively intact (data not shown).
Figure 5.
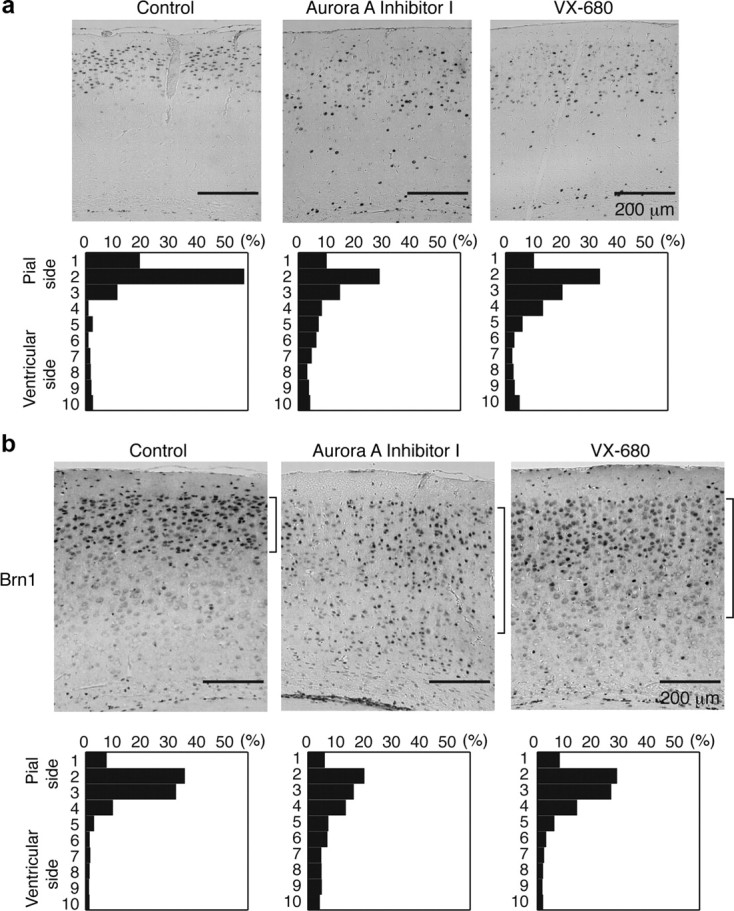
Defective neuronal migration by intraperitoneal injection of Aurora-A inhibitors. a, BrdU birth-dating analysis revealed neuronal migration defects after intraperitoneal injection of Aurora-A inhibitors. Mice were injected with BrdU at E14.5 and killed at P21. Aurora-A inhibitors were injected twice at E15.5 and E17.5. Quantitative analysis was performed by measuring the distribution of BrdU-labeled cells in each of 10 bins that equally divided the cortex from the molecular layer (ML) to the subplate (SP) (bottom panels). The staining patterns are representative of 10 different experiments. Note the shift downward toward the ventricular surface after intraperitoneal injection of Aurora-A inhibitors. b, The distribution of Brn-1-positive cells is indicated at the right side of each panel. Brn-1-positive cells were more dispersed after intraperitoneal injection of Aurora-A inhibitors. Quantitative analysis was performed by measuring the distribution of Brn-1-positive cells in each of 10 bins that equally divided the cortex from ML to SP (bottom panels). The staining patterns are representative of 10 different experiments.
Depletion of Aurora-A impairs neuronal migration, which is rescued by the expression of S251E NDEL1
Chemical inhibitors against Aurora-A clearly inhibited neuronal migration, but such inhibitory effects might occur through Aurora-A-independent or indirect mechanisms. To confirm this possibility, we applied siRNA against Aurora-A. siRNA depleted endogenous Aurora-A in granular neurons (Fig. 6a). Depletion of Aurora-A also reduced S251 phosphorylation of NDEL1 (Fig. 6a). Immunocytochemistry confirmed depletion of endogenous Aurora-A (Fig. 6b,c), which led to reduction of S251 phosphorylation of NDEL1 (Fig. 6b,c). Depletion of Aurora-A in granular neurons impaired outward movement from the central aggregates, whereas neurite extension remained intact (Fig. 6d–f). These observations support the interpretation that impairment of neuronal migration is attributable to the loss of function of Aurora-A rather than off-target effects. We next performed rescue experiments for the siRNA-induced migration phenotype by ectopic expression of NDEL1 transgenes. Expression of GFP-NDEL1(S251A) did not have any obvious rescue effect on granular cell migration after Aurora-A siRNA (Fig. 6d–f). In clear contrast, expression of GFP-NDEL1(S251E) rescued reduced migration after siRNA (Fig. 6d–f). We next examined whether suppression of Aurora-A affected radial migration in the embryo using an in utero gene transfer method. GFP-expressing cells migrated into superficial layers after in utero transfection of GFP at E16.5 (Fig. 7a). Interestingly, exogenous expression of wild-type GFP-Aurora-A led to facilitation of neuronal migration (Fig. 7a). In contrast, expression of GFP-Aurora-A (D145N) (Krystyniak et al., 2006), a KD mutant, suppressed outward migration (Fig. 7a). We examined the distribution of GFP-Aurora (KD)-expressing cells within the brain (Fig. 8c). Their distribution revealed that a fraction of GFP-Aurora (KD)-expressing cells remained within the ventricular zone (Fig. 7c). We also found that GFP-Aurora (KD)-expressing cells were located beyond the ventricular zone (Fig. 7a,c). The phenotypes that were revealed by GFP-Aurora (KD)-expressing cells were milder than those due to disruption of Ndel1. We next performed rescue experiments. The impaired migration after suppression of Aurora-A was not rescued by the expression of GFP-NDEL1(S251A), which instead exacerbated defective migration (Fig. 7b,c), whereas the expression of GFP-NDEL1(S251E) rescued granular cell migration (Fig. 7b,c). Our interpretation is that NDEL1 is essential for both cell proliferation and migration, and that S251 phosphorylation of NDEL1 by Aurora-A plays a pivotal role in both processes. Presumably, complete inactivation of Aurora-A resulted in more drastic phenotypes, similar to Ndel1 disruption. We assume that the difference in severity of phenotypes is the result of methodological difference rather than functional importance.
Figure 6.
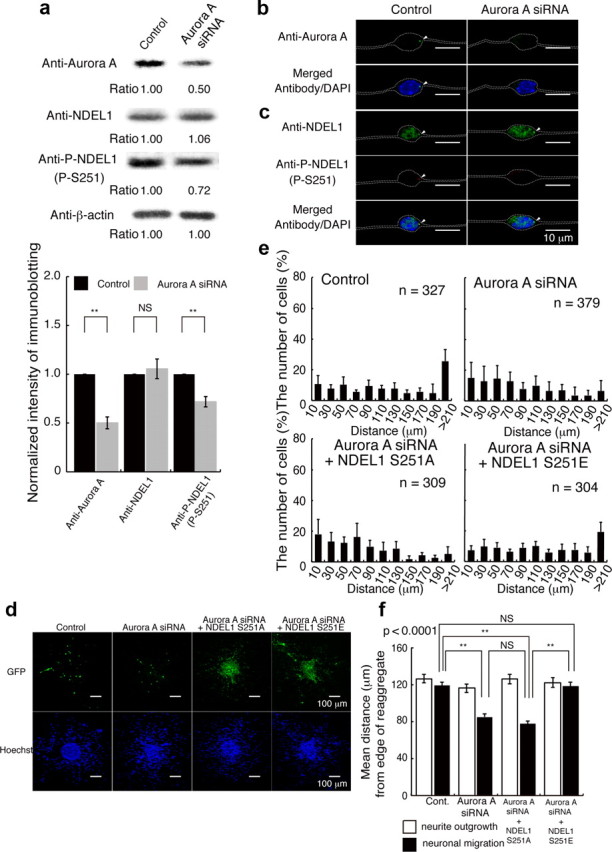
Depletion of Aurora-A by siRNA impairs neuronal migration. a, Granular neurons were transfected with siRNA against Aurora-A. Western blotting was performed 48 h after start of culture of the siRNA. Aurora-A was clearly reduced after siRNA, whereas NDEL1 displayed similar expression levels. Phosphorylated NDEL1 was decreased after knockdown of Aurora-A. Relative intensities of the bands of Western blotting is shown at the bottom. Intensities were normalized against β-actin. Statistical examination was performed by unpaired Student's t test, with **p < 0.01 (bottom panel). b, c, Immunocytochemistry revealed that the Aurora-A signal (b) was nearly invisible. NDEL1 expression (c) was unchanged, whereas phosphorylated NDEL1 was decreased after treatment of siRNA to knockdown Aurora-A. The white dotted lines indicate the outline of granular neurons. d, Rescue experiments after siRNA treatment to knockdown Aurora-A using control GFP, GFP-Ndel1 (S251A), or GFP-Ndel1 (S251E) transfections. GFP images are shown in the top panels. Hoechst images are shown in the bottom panels. Expression of GFP-Ndel1 (S251A) had no effect on reduced migration after siRNA knockdown of Aurora-A, whereas expression of GFP-Ndel1 (S251E) significantly improved this reduced migration. e, The migration distance of each neuron 48 h after the start of culture was binned. n is the number of neurons measured for each examination. Expression of GFP-Ndel1 (S251E) significantly improved migration, which was characterized by a rightward shift. f, Mean length of neurites (open bar) and nuclear position (solid bar) from the edge of central aggregation. Statistical examination was performed for nuclear migration by an ANOVA followed by t test with correction: **p < 0.01.
Figure 7.
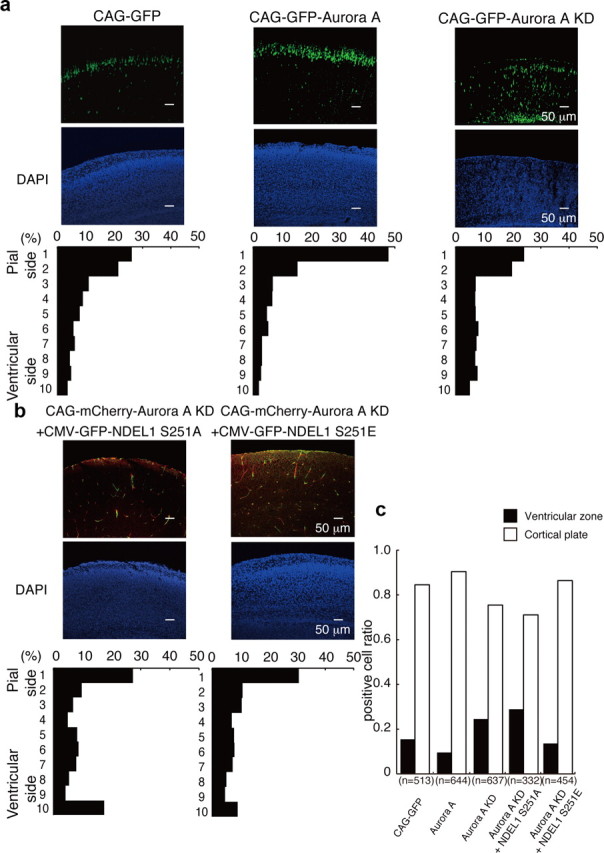
In utero injection of Aurora-A KD resulted in defective neuronal migration. a, In utero injection of control GFP, GFP-Aurora-A, or GFP-Aurora-A KD into the E16.5 wild-type mouse neocortex by electroporation. GFP images are shown in the top panels. Counterstains of a neighboring section by DAPI are shown in the middle panels. The distribution of migrated neurons is shown in the bottom panels. Interestingly, expression of GFP-Aurora-A facilitated upward localization of GFP-positive neurons compared with control GFP, whereas expression of GFP-Aurora (KD) reduced upward localization of GFP-positive neurons. b, Rescue experiments with coexpression of either GFP-Ndel1 (S251A) or GFP-Ndel1 (S251E). Expression of GFP-Ndel1 (S251A) had no effect on defective migration after expression of mCherry-Aurora-A (KD), whereas expression of GFP-Ndel1 (S251E) significantly improved this defective migration. c, Distribution of GFP-Aurora (KD)-expressing cells within the brain. A fraction of GFP-Aurora (KD)-expressing cells remained within the ventricular zone. Expression of GFP-NDEL1(S251E) rescued the phenotype.
Figure 8.
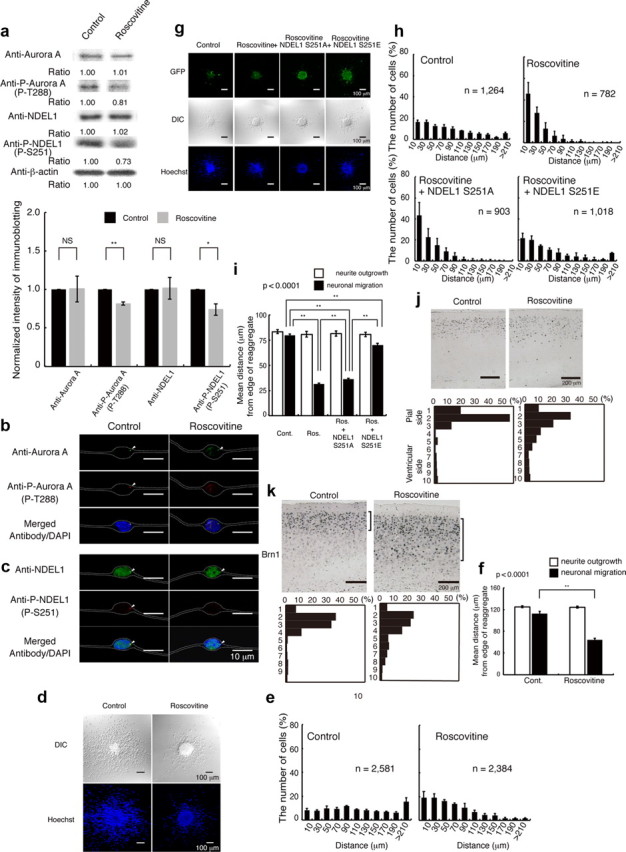
Inhibition of CDK5 by roscovitine resulted in defective neuronal migration, which was rescued by expression of GFP-Ndel1 (S251E). a, Granular neurons were treated with roscovitine. Western blotting was performed 24 h after start of culture. Aurora-A and NDEL1 displayed similar expression levels with untreated neurons, whereas the levels of phosphorylated Aurora-A and NDEL1 proteins were decreased after treatment with roscovitine. Relative intensities of the bands of Western blotting are shown at the bottom. Intensity was normalized to β-actin. Statistical examination was performed by unpaired Student's t test, with *p < 0.05 and **p < 0.01 (bottom panel). b, Immunocytochemistry against either Aurora-A (b) or NDEL1 (c) revealed similar expression levels after treatment, whereas phosphorylated proteins were decreased after treatment with roscovitine. The white dotted lines indicate the outline of granular neurons. d, Migration assay using cerebellar granule neurons. DIC images (top) and Hoechst staining images (bottom) are shown. e, The migration distance of each neuron 24 h after the start of culture was binned. n is the number of neurons measured for each examination. Wild-type neurons displayed normal migration distances, whereas neurons treated with roscovitine displayed a shift in the distribution of bins toward the left. f, Mean length of neurites (open bar) and nuclear position (solid bar) from the edge of central aggregation. Statistical examination was performed by an ANOVA followed by t test with correction: **p < 0.01. g, Rescue experiments after roscovitine treatment with control GFP, GFP-Ndel1 (S251A), or GFP-Ndel1 (S251E) transfection. GFP images are shown. Expression of GFP-Ndel1 (S251A) had no effect on defective migration, whereas expression of GFP-Ndel1 (S251E) significantly improved migration after roscovitine treatment. h, The migration distance of each neuron 24 h after the start of culture was binned. n is the number of neurons measured for each examination. Expression of GFP-Ndel1 (S251E) significantly improved migration, which was characterized by a rightward shift. i, Mean length of neurites (open bar) and nuclear position (solid bar) from the edge of central aggregation. Statistical examination was performed by an ANOVA followed by t test with correction: **p < 0.01. j, BrdU birth-dating analysis revealed neuronal migration defects after intraperitoneal injection of roscovitine. Mice were injected with BrdU at E14.5 and killed at P21. Roscovitine was injected twice at E15.5 and E17.5. Quantitative analysis was performed by measuring the distribution of BrdU-labeled cells in 10 bins that equally divided the cortex from ML to SP (bottom panels). The staining patterns are representative of 10 different experiments. Note the shift downward of neuron localization toward the ventricular side after intraperitoneal injection of roscovitine. k, The distribution of Brn-1-positive cells is indicated at the right side of each panel. Brn-1-positive cells were more dispersed after intraperitoneal injection of roscovitine. Quantitative analysis was performed by measuring the distribution of Brn-1-positive cells in each of 10 bins that equally divided the cortex from ML to SP (bottom panels). The staining patterns are representative of 10 different experiments.
CDK5 potentially shares a common pathway with Aurora-A
CDKs are a group of serine/threonine protein kinases activated by binding to a regulatory subunit cyclin. These kinases are key regulators of eukaryotic cell cycle progression. CDK5 is a unique member of the CDK family in that it does not act as a checkpoint kinase to regulate cell cycle progression, but as a regulatory kinase involved in other postmitotic processes such as neuronal activity. Recent evidence also suggests a pivotal role for CDK5 in synaptic plasticity, behavior, and cognition (Dhavan and Tsai, 2001; Gupta and Tsai, 2003; Su and Tsai, 2011). Interestingly, NDEL1 is differentially phosphorylated by CDK1 (Yan et al., 2003) and CDK5 (Niethammer et al., 2000). Upon entry into mitosis, Aurora-A becomes fully activated downstream of CDK1 activation (Van Horn et al., 2010). The functional relevance prompted us to examine whether Aurora-A might share this common pathway with CDK5. We first examined the effect of a specific CDK inhibitor, roscovitine (Patrick et al., 1998). Application of roscovitine significantly suppressed T288 phosphorylation of Aurora-A associated with reduction of S251 phosphorylation of NDEL1, which was confirmed by Western blotting and immunocytochemistry (Fig. 8a–c). Roscovitine also suppressed the outward movement of granular neurons, whereas neurite extension was not influenced (Fig. 8d–f). We performed rescue experiments. Expression of GFP-NDEL1(S251A) did not have any obvious influences on roscovitine-inhibited granular cell migration (Fig. 8g,h). By contrast, expression of GFP-NDEL1(S251E) significantly facilitated roscovitine-inhibited granular cell migration (Fig. 8g–i). We further determined whether administration of roscovitine affected neuronal migration during corticogenesis. In the control, BrdU-positive cells localized to more superficial cortical regions (Fig. 8j), while administration of roscovitine clearly suppressed upward localization of those BrdU-positive cells, suggesting reduced migration (Fig. 8j). We confirmed this interpretation using the layer II–III-specific marker Brn-1 (Yamada et al., 2009). Brn-1-stained neurons were localized in more superficial cortical layers (Fig. 8k), but after administration of roscovitine, Brn-1 stained neurons were distributed deeper and more broadly in the cortex (Fig. 8k). These observations suggest that CDK5 may share a common pathway with Aurora-A in neuronal migration.
CDK5 is required for centrosomal targeting of Aurora-A via CDK5RAP2
We next applied siRNA depletion against CDK5. siRNA I depleted endogenous CDK5 in granular neurons 0, 24, and 48 h after the start of cell culture (Fig. 9a). Surprisingly, depletion of CDK5 also reduced endogenous Aurora-A at 24 h after treatment (Fig. 9a). To confirm this, we applied a second siRNA, siRNA II, to knock down CDK5. siRNA II also depleted CDK5 and Aurora-A, and reduction of Aurora-A continued 48 h after start of cell culture (Fig. 9c). To address the mechanism of Aurora-A reduction, we applied inhibitors against calpain or the proteasome, and found that the Proteasome Inhibitor II clearly augmented Aurora-A (Fig. 9d), suggesting that depletion of CDK5 facilitated proteasome-mediated Aurora-A degradation. Next, we examined the distribution of Aurora-A in the presence of the Proteasome Inhibitor II. Interestingly, depletion of CDK5 led to loss of centrosomal targeting of Aurora-A and displayed broader localization (Fig. 9e). These findings suggest the presence of a factor that mediates cross talk between CDK5 and Aurora-A. CDK5RAP2 is a centrosomin family protein. Centrosomin orthologs have been extensively characterized in the fruit fly Drosophila melanogaster and the fission yeast Schizosaccharomyces pombe, where these proteins are essential for microtubule organizing center (MTOC) activity, and the MTOC is an important target of Aurora kinase (Megraw et al., 2011). In particular, Aurora-A and centrosomin are mutually dependent for proper localization at the spindle poles and together are required for proper targeting of γ-tubulin and other centrosomal components to the centrosome (Terada et al., 2003). Homozygous mutations in the genes CDK5RAP2 results in autosomal recessive primary microcephaly (Bond et al., 2005; Lizarraga et al., 2010). However, CDK5RAP2 is a potential regulator of CDK5 activity via its interaction with CDK5R1 (Wang et al., 2000). Therefore, we examined whether CDK5RAP2 was able to interact with either CDK5 and Aurora-A by immunoprecipitation and found that CDK5RAP2 interact with both kinases (Fig. 9f). In addition, the depletion of CDK5 led to loss of centrosomal targeting of CDK5RAP2 (Fig. 9g). Thus, we concluded that centrosomal targeting of Aurora-A is regulated by CDK5 via CDK5RAP2 in granular neurons.
Figure 9.
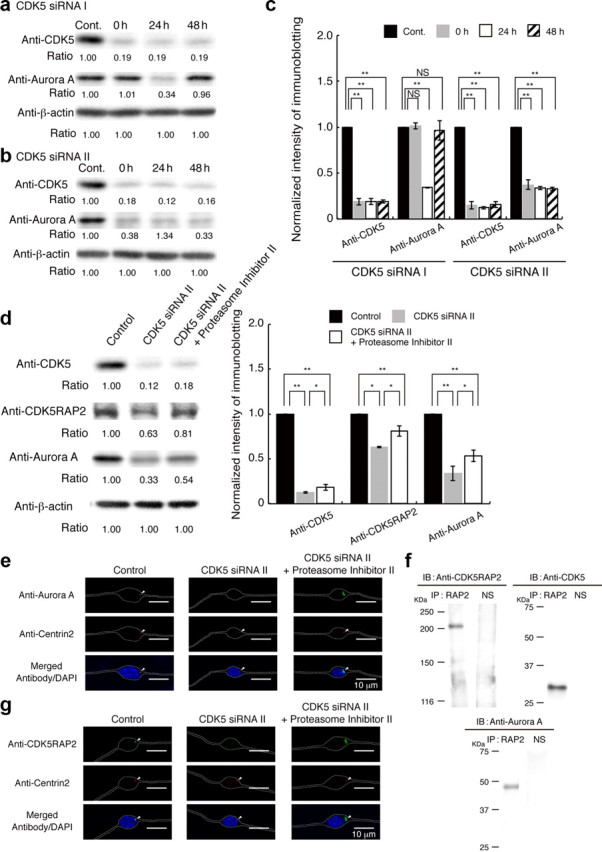
CDK5 regulates centrosomal targeting of Aurora-A via CDK5RAP2. a, Granular neurons were transfected with siRNA I against CDK5, followed by incubation for 24 h to make cell aggregations. Cell aggregations of granular neurons were plated onto coated dishes (see Materials and Methods). Western blotting was performed at 0, 24, and 48 h after the start of culture. CDK5 was clearly reduced at the start of culture. Aurora-A was also reduced at 24 h after the start of culture, followed by a recovery 48 h after start of culture. Control indicates granular neurons before transfection. b, Granular neurons were transfected with siRNA II against CDK5. Western blotting was performed at 0, 24, and 48 h after start of culture. CDK5 and Aurora-A were reduced at the start of culture. Note: Aurora-A reduction continued 48 h after the start of culture. c, Relative intensity of the bands of Western blotting is displayed. Intensity was normalized with β-actin. Statistical examination was performed by unpaired Student's t test, with *p < 0.05 and **p < 0.01. d, Aurora-A reduction was prevented by administration of Proteasome Inhibitor II at a concentration of 0.5 μm. Proteasome Inhibitor II was applied at the time of siRNA transfection. Note: Proteasome Inhibitor II had no influence on the depletion of CDK5, whereas Proteasome Inhibitor II partially prevented degradation of Aurora-A. We also used other proteasome inhibitors. However, given the toxicity of these compounds to granular neurons, we were not able to continue the experiments in culture. Relative intensity of the bands of Western blotting is displayed at the bottom. Intensity was normalized with β-actin. Statistical examination was performed by unpaired Student's t test, with *p < 0.05 and **p < 0.01. e, Subcellular distribution of Aurora-A transfected with siRNA II in the presence of Proteasome Inhibitor II. Twenty-four hours after the start of culture, there was loss of centrosomal targeting and broader localization of Aurora-A. The white dotted lines indicate the outline of granular neurons. Centrin-2 was used as a centriole marker. f, Immunoprecipitation assay using an anti-CDK5RAP2 antibody. After coimmunoprecipitation, endogenous CDK5RAP2 was detected (right). CDK5 (middle) and Aurora-A (left) were also coprecipitated with CDK5RAP2. N.S. indicates nonimmune serum. g, Subcellular distribution of CDK5RAP2 24 h after the start of culture in the presence of Proteasome Inhibitor II. There was a loss of centrosomal targeting and broader localization of CDK5RAP2. The white dotted lines indicate the outline of granular neurons.
CDK5 and Aurora-A functionally converge onto NDEL1 during neuronal migration
To uncover the functional relevance of CDK5 and Aurora-A, we performed immunocytochemistry and found that the depletion of endogenous CDK5 resulted in reduction of T288 phosphorylation and S251 phosphorylation of Aurora-A and NDEL1, respectively (Fig. 10a,b). Depletion of CDK5 in granular neurons impaired outward movement from the center of aggregates (Fig. 10c,d). We next performed rescue experiments for this CDK5 siRNA-induced phenotype by ectopic expression of a transgenes of NDEL1. Expression of GFP-NDEL1(S251A) did not rescue CDK5-siRNA inhibition of granular cell migration (Fig. 10c,d), while expression of GFP-NDEL1(S251E) clearly rescued granular cell migration (Fig. 10c,d). Similarly, expression of a phosphomimic mutant of NDEL1 for CDK5 sites, mCherry-NDEL1–3E, rescued defective granular cell migration, while the nonphosphorylated form, mCherry-NDEL1–3A, did not rescue (Fig. 10e,f). Our observations suggest that defective migration by inactivation of CDK5 is partially attributable to suppression of Aurora-A pathway and their functions converge onto NDEL1.
Figure 10.
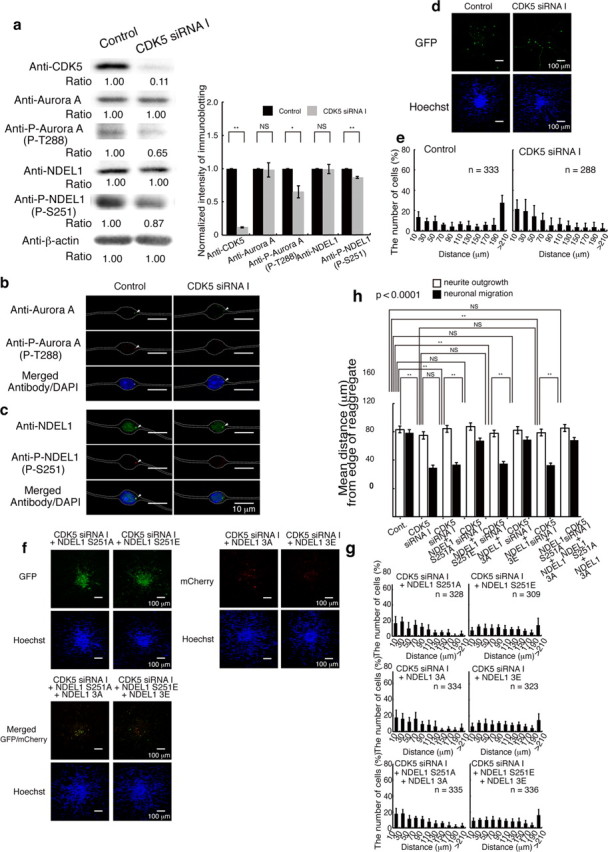
Depletion of CDK5 resulted in defective neuronal migration, which was rescued by expression of GFP-Ndel1 (S251E) and GFP-Ndel1 (3E). a, Granular neurons were transfected with siRNA I against CDK5. Western blotting was performed 24 h after start of culture. CDK5 was clearly reduced by siRNA. Aurora-A and NDEL1 displayed similar expression levels after CDK5 knockdown, whereas Aurora-A and NDEL1 phosphorylated proteins were decreased after treatment of siRNA. Relative intensity of the bands of Western blotting is displayed at the bottom. Intensity was normalized with β-actin. Statistical examination was performed by unpaired Student's t test, with *p < 0.05 and **p < 0.01 (bottom panel). b, Immunocytochemistry using antibodies Aurora-A (b) or NDEL1 (c) revealed similar expression levels of these proteins, whereas levels of Aurora-A and NDEL1 phosphorylated proteins were decreased after treatment with siRNA I. d, Migration assay using granular neurons after depletion of CDK5. Hoechst images are shown at the bottom. e, The migration distance of each neuron 48 h after start of culture was binned. n is the number of neurons measured for each examination. f, Rescue experiments of siRNA knockdown of CDK5 after transfection of control GFP, GFP-Ndel1 (S251A), GFP-Ndel1 (S251E), mCherry-Ndel1 (3A), mCherry-Ndel1 (3E), and double transfection of GFP-Ndel1 (S251A), mCherry-Ndel1 (3A) and GFP-Ndel1 (S251E), mCherry-Ndel1 (3E). GFP images are shown in the top panels and Hoechst images are shown at the bottom. Expression of GFP-Ndel1 (S251A) or mCherry-Ndel1 (3A) had no effect on defective migration, whereas expression of GFP-Ndel1 (S251E) or mCherry-Ndel1 (3E) significantly improved defective migration due to siRNA knockdown of CDK5. g, The migration distance of each neuron 48 h after the start of culture was binned. n is the number of neurons measured for each examination. Expression of GFP-Ndel1 (S251E) or mCherry-Ndel1 (3E) significantly improved migration, which was characterized by a rightward shift. h, Statistical examination was performed to nuclear migration by an ANOVA followed by t test with correction: **p < 0.01. Note: Either GFP-Ndel1 (S251E) or mCherry-Ndel1 (3E) efficiently rescued defective migration, whereas double transfection of GFP-Ndel1 (S251E) and mCherry-Ndel1 (3E) did not result in further improvement.
Phosphorylation of S251 of Aurora-A is essential for microtubule remodeling
We previously demonstrated that the Aurora-A–NDEL1 pathway plays an important role in microtubule remodeling during mitosis (Mori et al., 2007), prompting us to analyze the role of the Aurora-A–NDEL1 pathway on microtubule dynamics during neuronal migration. During neuronal migration, visualization of individual microtubules is technically challenging due to their high density, so we used EB3-mCherry as a marker of growing distal tips of microtubules (Mimori-Kiyosue et al., 2000) to trace the tracks of microtubules. During neuronal migration, EB3-mCherry was directed into the leading process of granular neurons from the leading side of the nucleus, where the centrosome was located. We examined whether suppression of Aurora-A by siRNA or a hypomorphic mutant of Ndel1 affected microtubule dynamics using EB3-mCherry. A hypomorphic mutant of Ndel1 reduced emanation frequency and speed of EB3-mCherry (Fig. 11a,b). Similar to the Aurora-A-depleted experiments, expression of GFP-NDEL1 (S251A) failed to rescue this reduction (frequency, 4.5/min; speed, 16.6 μm/min) (Fig. 11a,b), whereas GFP-NDEL1 (S251E) rescued the defective microtubule emanation due to hypomorphic Ndel1 (frequency, 5.8/min; speed, 38.2 μm/min) (Fig. 11a,b). The depletion of Aurora-A clearly suppressed the emanation frequency and speed of EB3-mCherry (Fig. 11c,d). Importantly, expression of GFP-NDEL1 (S251A) failed to rescue this phenotype (frequency, 4.4/min; speed, 14.8 μm/min) (Fig. 11c,d), whereas GFP-NDEL1 (S251E) rescued the defective microtubule emanation in Aurora-A-depleted neurons (frequency, 8.5/min; speed, 53.8 μm/min) (Fig. 11c,d). Although movement of EB3 provides only the dynamics at the plus end of microtubules, our findings suggest that emanation frequency and emanation speed were disrupted by perturbations of the Aurora-A–NDEL1 pathway.
Figure 11.

Microtubule projections from MTOC was reduced by depletion of Aurora-A or the hypomorphic mutant of Ndel1. a, The projection speed of individual EB3-mCherry spots, and the calculated average extension length of microtubules per minute in the hypomorphic mutant. Experimental conditions are indicated at the bottom. The hypomorphic mutant of Ndel1 resulted in a reduction of EB3-mCherry emanation frequency. b, Extension frequency of EB3-mCherry spots from the MTOC, and the calculated average extension frequency of microtubules per minutes. Disruption of Ndel1 resulted in a reduction of EB3-mCherry extension speed. Reductions of emanation frequency and emanation speed were rescued by expression of GFP-Ndel1 (S251E), but not by GFP-Ndel1 (S251A). c, The projection speed of individual EB3-mCherry spots, and the average extension length of microtubules per minute was calculated. Experimental conditions are indicated at the bottom. Depletion of Aurora-A resulted in reduction of EB3-mCherry emanation frequency. d, Extension frequency of EB3-mCherry spots from the MTOC, and the calculated average extension frequency of microtubules per minute. Depletion of Aurora-A resulted in the reduction of EB3-mCherry extension speed. Reductions of emanation frequency and emanation speed were rescued by expression of GFP-Ndel1 (S251E), but not by GFP-Ndel1 (S251A). Statistical examination was performed by an ANOVA followed by t test with correction: **p < 0.01.
Discussion
Here, we present molecular and cell biological data that support a unique role of Aurora-A–NDEL1 pathway on microtubule dynamics during neuronal migration. We demonstrated that Aurora-A is highly activated at the centrosome during migration accompanied by phosphorylation of NDEL1 S251. In addition, we demonstrated that suppression or depletion of Aurora-A affected the proliferation of progenitor cells as well as neuronal migration and microtubule dynamics, suggesting that the Aurora-A–NDEL1 pathway is an important regulator of microtubule organization during neuronal migration as well as progenitor proliferation. We also demonstrated that a phosphomimetic form of Aurora-A phosphorylation sites in NDEL1 efficiently rescued defective migration due to inhibition of Aurora-A and CDK5 depletion. Examination of the dynamics of microtubules using EB3 provided a plus end motion of microtubules, which suggested that the Aurora-A–NDEL1 pathway regulates the organization of microtubules in granular neurons, including their emanation frequency and emanation speed.
Novel role of mitotic kinase in neuronal migration
Mitotic kinases are a major regulator of mitosis and cell division. A growing body of evidence indicates that many of these proteins play previously unexpected roles in metabolism, cardiovascular function, or neuron biology (Malumbres, 2011). Recently, the interactions between the prometastatic scaffolding protein HEF1/Cas-L/NEDD9 and the oncogenic Aurora-A kinase at the basal body of cilia had been reported (Pugacheva et al., 2007). We previously reported that Aurora-A phosphorylates NDEL1 at S251 (Mori et al., 2007). We also reported an essential role of the aPKC–Aurora-A–NDEL1 pathway on neurite elongation (Mori et al., 2009). We now implicate these pathways in microtubule organization during neuronal migration. Expression of NDEL1 (S251E) rescued the defective neuronal migration (this report) and the mitotic entry defect by Aurora-A depletion (Mori et al., 2007). However, we previously reported that the N terminus of NDEL1 restored LIS1-dependent suppression of dynein motility (Yamada et al., 2008; Torisawa et al., 2011), whereas the C terminus of NDEL1 had no effect on LIS1 suppression (Torisawa et al., 2011). In addition, Zylkiewicz et al. (2011) reported that the N terminus of NDEL1 lacking the dynein-binding site (Sasaki et al., 2000) was fully able to rescue aster formation (Zylkiewicz et al., 2011). One plausible explanation is that phosphorylation at S251 of NDEL1 regulates the interaction with dynein or LIS1. NDEL1 (S251A), a single site mutant deficient in its susceptibility to Aurora-A pulled down almost no dynein (Zylkiewicz et al., 2011). Presumably, the C terminus of NDEL1 provides a regulatory mechanism for dynein binding, so that the unphosphorylated protein is autoinhibited (Zylkiewicz et al., 2011).
Aurora-A and CDK5 share a common pathway
CDK5 is a unique member of the CDK family that does not act as a checkpoint kinase to regulate cell cycle progression, but acts as a regulatory kinase involved in other postmitotic processes such as neuronal activity, neuronal migration during development, and neurite outgrowth. Over 20 functionally diverse proteins involved in cytoskeleton dynamics, cell adhesion, transport, and membrane trafficking act as CDK5 substrates elucidating the molecular mechanisms of CDK5 function (Wang et al., 2006), including phosphorylated NDEL1 (Niethammer et al., 2000). The inhibition of CDK5 activity alters NDEL1 localization and its association with the dynein complex, resulting in the alteration of neuronal migration (Dhavan and Tsai, 2001). In this paper, we demonstrated that the suppression of CDK5 reduced Aurora-A activity and NDEL1 phosphorylation by Aurora-A. Surprisingly, we found that depletion of CDK5 resulted in loss of centrosomal targeting of Aurora-A and the reduction of Aurora-A. Proteasome-mediated protein degradation was apparently responsible for this Aurora-A reduction, and the aberrant distribution of Aurora-A facilitated this degradation. We also found that CDK5RAP2 mediated the functional interaction of CDK5 and Aurora-A. CDK5RAP2 was originally identified as a neuronal CDK5 activator (Wang et al., 2000). Further characterization revealed a variety of activities that have been attributed to CDK5RAP2, including γ-tubulin recruitment, attachment of centrosomes to spindle poles, Chk1 kinase recruitment, microtubule plus-end dynamics, and spindle orientation in neural progenitors (Megraw et al., 2011). CDK5RAP2 interacts with pericentrin to maintain the neural progenitor pool in the developing neocortex (Buchman et al., 2010) and stimulates microtubule nucleation by the γ-tubulin ring complex (Choi et al., 2010). In addition, homozygous mutations in the genes CDK5RAP2 and CENPJ cause microcephaly, indicating essential role in corticogenesis (Bond et al., 2005). The D. melanogaster homolog of CDK5RAP2, centrosomin, maintains the pericentriolar matrix around centrioles during mitosis, and Aurora-A and centrosomin are mutually dependent for localization (Terada et al., 2003). We propose the following working model: CDK5 has a direct effect on NDEL1 by phosphorylation of CDK5-specific sites for remodeling of microtubule organization. CDK5 participates in another pathway via regulation of Aurora-A through CDK5RAP2. Rescue experiments by phosphorylated forms of NDEL1 support our model.
NDEL1 is differentially phosphorylated by CDK1 (Yan et al., 2003) and CDK5 (Niethammer et al., 2000). Interestingly, NDEL1 double phosphorylation by CDK1 and CDK5 reduced the amount of NDEL1 and LIS1 that coimmunoprecipitated with dynein, suggesting that phosphorylation not only strengthens NDEL1 binding to LIS1 but promotes the release of LIS1–pNDEL1 complexes from dynein (Hebbar et al., 2008). In contrast, phosphorylation by either kinase alone was insufficient to reduce dynein binding. Rather, CDK5 phosphorylation augments interaction with dynein (Hebbar et al., 2008). However, Zylkiewicz et al. (2011) reported that double-mutant NDEL1 lacking two sites phosphorylated by CDK1 and CDK5 pulled down almost no dynein. This discrepancy may be attributable to the difference of experimental design. Further elucidation for physiological roles and regulation of each phosphorylation site is required to address this discrepancy.
In our study, the importance of motor protein regulation has not been addressed directly. Although a role for motors in regulating microtubule length has been established, the mechanisms used by motors to promote microtubule growth or shrinkage are still unclear. For example, inhibition of dynein function by overexpression of dynamitin resulted in normal radial microtubule arrays (Burkhardt et al., 1997). However, disruption of Lis1 or Ndel1 leads to devastating effects on mitotic cells or neurons, presumably due to massive remodeling of microtubules and/or transport defect. These context-dependent requirements govern sensitivity to the loss of motor function. We previously reported that nud family genes are required for proper distribution of cytoplasmic dynein (Yamada et al., 2008, 2010). We must consider that mitotic kinases may also act as postmitotic kinases to regulate diverse functions in nondividing cells through regulation of microtubule organization and motor protein function.
Footnotes
This work was supported by Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan from the Ministry of Education, Science, Sports and Culture of Japan (S.H.) and Knowledge Cluster Initiative (Stage 2) Research Foundation (S.H.). This work was also supported by Takeda Science Foundation and NOVARTIS Foundation (Japan) for the Promotion of Science (S.H.) and NIH Grants NS41030 and HD47380 (A.W.-B.). We thank Chizumi Momura and Masami Yamada for technical support. We also thank Mika Egami, Yukimi Kira, and Yoriko Yabunaka for technical support, and Hiromichi Nishimura and Keiko Fujimoto for mouse breeding. We thank Dr. Fumio Matsuzaki for critical comments and encouragement.
References
- Aliagas-Martin I, Burdick D, Corson L, Dotson J, Drummond J, Fields C, Huang OW, Hunsaker T, Kleinheinz T, Krueger E, Liang J, Moffat J, Phillips G, Pulk R, Rawson TE, Ultsch M, Walker L, Wiesmann C, Zhang B, Zhu BY, et al. A class of 2,4-bisanilinopyrimidine Aurora A inhibitors with unusually high selectivity against Aurora B. J Med Chem. 2009;52:3300–3307. doi: 10.1021/jm9000314. [DOI] [PubMed] [Google Scholar]
- Azimzadeh J, Bornens M. Structure and duplication of the centrosome. J Cell Sci. 2007;120:2139–2142. doi: 10.1242/jcs.005231. [DOI] [PubMed] [Google Scholar]
- Bettencourt-Dias M, Glover DM. Centrosome biogenesis and function: centrosomics brings new understanding. Nat Rev Mol Cell Biol. 2007;8:451–463. doi: 10.1038/nrm2180. [DOI] [PubMed] [Google Scholar]
- Bond J, Roberts E, Springell K, Lizarraga SB, Scott S, Higgins J, Hampshire DJ, Morrison EE, Leal GF, Silva EO, Costa SM, Baralle D, Raponi M, Karbani G, Rashid Y, Jafri H, Bennett C, Corry P, Walsh CA, Woods CG. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet. 2005;37:353–355. doi: 10.1038/ng1539. [DOI] [PubMed] [Google Scholar]
- Buchman JJ, Tseng HC, Zhou Y, Frank CL, Xie Z, Tsai LH. Cdk5rap2 interacts with pericentrin to maintain the neural progenitor pool in the developing neocortex. Neuron. 2010;66:386–402. doi: 10.1016/j.neuron.2010.03.036. [DOI] [PubMed] [Google Scholar]
- Burkhardt JK, Echeverri CJ, Nilsson T, Vallee RB. Overexpression of the dynamitin (p50) subunit of the dynactin complex disrupts dynein-dependent maintenance of membrane organelle distribution. J Cell Biol. 1997;139:469–484. doi: 10.1083/jcb.139.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YK, Liu P, Sze SK, Dai C, Qi RZ. CDK5RAP2 stimulates microtubule nucleation by the gamma-tubulin ring complex. J Cell Biol. 2010;191:1089–1095. doi: 10.1083/jcb.201007030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–759. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- Distel M, Hocking JC, Volkmann K, Köster RW. The centrosome neither persistently leads migration nor determines the site of axonogenesis in migrating neurons in vivo. J Cell Biol. 2010;191:875–890. doi: 10.1083/jcb.201004154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxsey S, Zimmerman W, Mikule K. Centrosome control of the cell cycle. Trends Cell Biol. 2005;15:303–311. doi: 10.1016/j.tcb.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Giet R, Uzbekov R, Cubizolles F, Le Guellec K, Prigent C. The Xenopus laevis Aurora-related protein kinase pEg2 associates with and phosphorylates the kinesin-related protein XlEg5. J Biol Chem. 1999;274:15005–15013. doi: 10.1074/jbc.274.21.15005. [DOI] [PubMed] [Google Scholar]
- Giet R, McLean D, Descamps S, Lee MJ, Raff JW, Prigent C, Glover DM. Drosophila Aurora A kinase is required to localize D-TACC to centrosomes and to regulate astral microtubules. J Cell Biol. 2002;156:437–451. doi: 10.1083/jcb.200108135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover DM, Leibowitz MH, McLean DA, Parry H. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell. 1995;81:95–105. doi: 10.1016/0092-8674(95)90374-7. [DOI] [PubMed] [Google Scholar]
- Gupta A, Tsai LH. Cyclin-dependent kinase 5 and neuronal migration in the neocortex. Neurosignals. 2003;12:173–179. doi: 10.1159/000074618. [DOI] [PubMed] [Google Scholar]
- Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, Demur C, Hercend T, Diu-Hercend A, Su M, Golec JM, Miller KM. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med. 2004;10:262–267. doi: 10.1038/nm1003. [DOI] [PubMed] [Google Scholar]
- Hatten ME. Neuronal regulation of astroglial morphology and proliferation in vitro. J Cell Biol. 1985;100:384–396. doi: 10.1083/jcb.100.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatten ME. Central nervous system neuronal migration. Annu Rev Neurosci. 1999;22:511–539. doi: 10.1146/annurev.neuro.22.1.511. [DOI] [PubMed] [Google Scholar]
- Hebbar S, Mesngon MT, Guillotte AM, Desai B, Ayala R, Smith DS. Lis1 and Ndel1 influence the timing of nuclear envelope breakdown in neural stem cells. J Cell Biol. 2008;182:1063–1071. doi: 10.1083/jcb.200803071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higginbotham HR, Gleeson JG. The centrosome in neuronal development. Trends Neurosci. 2007;30:276–283. doi: 10.1016/j.tins.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Krystyniak A, Garcia-Echeverria C, Prigent C, Ferrari S. Inhibition of Aurora A in response to DNA damage. Oncogene. 2006;25:338–348. doi: 10.1038/sj.onc.1209056. [DOI] [PubMed] [Google Scholar]
- Kufer TA, Silljé HH, Körner R, Gruss OJ, Meraldi P, Nigg EA. Human TPX2 is required for targeting Aurora-A kinase to the spindle. J Cell Biol. 2002;158:617–623. doi: 10.1083/jcb.200204155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizarraga SB, Margossian SP, Harris MH, Campagna DR, Han AP, Blevins S, Mudbhary R, Barker JE, Walsh CA, Fleming MD. Cdk5rap2 regulates centrosome function and chromosome segregation in neuronal progenitors. Development. 2010;137:1907–1917. doi: 10.1242/dev.040410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M. Physiological relevance of cell cycle kinases. Physiol Rev. 2011;91:973–1007. doi: 10.1152/physrev.00025.2010. [DOI] [PubMed] [Google Scholar]
- Megraw TL, Sharkey JT, Nowakowski RS. Cdk5rap2 exposes the centrosomal root of microcephaly syndromes. Trends Cell Biol. 2011;21:470–480. doi: 10.1016/j.tcb.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimori-Kiyosue Y, Shiina N, Tsukita S. The dynamic behavior of the APC-binding protein EB1 on the distal ends of microtubules. Curr Biol. 2000;10:865–868. doi: 10.1016/s0960-9822(00)00600-x. [DOI] [PubMed] [Google Scholar]
- Mori D, Yano Y, Toyo-oka K, Yoshida N, Yamada M, Muramatsu M, Zhang D, Saya H, Toyoshima YY, Kinoshita K, Wynshaw-Boris A, Hirotsune S. NDEL1 phosphorylation by Aurora-A kinase is essential for centrosomal maturation, separation, and TACC3 recruitment. Mol Cell Biol. 2007;27:352–367. doi: 10.1128/MCB.00878-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori D, Yamada M, Mimori-Kiyosue Y, Shirai Y, Suzuki A, Ohno S, Saya H, Wynshaw-Boris A, Hirotsune S. An essential role of the aPKC-Aurora A-NDEL1 pathway in neurite elongation by modulation of microtubule dynamics. Nat Cell Biol. 2009;11:1057–1068. doi: 10.1038/ncb1919. [DOI] [PubMed] [Google Scholar]
- Niethammer M, Smith DS, Ayala R, Peng J, Ko J, Lee MS, Morabito M, Tsai LH. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron. 2000;28:697–711. doi: 10.1016/s0896-6273(00)00147-1. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2:21–32. doi: 10.1038/35048096. [DOI] [PubMed] [Google Scholar]
- Nigg EA, Raff JW. Centrioles, centrosomes, and cilia in health and disease. Cell. 2009;139:663–678. doi: 10.1016/j.cell.2009.10.036. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Zhou P, Kwon YT, Howley PM, Tsai LH. p35, the neuronal-specific activator of cyclin-dependent kinase 5 (Cdk5) is degraded by the ubiquitin-proteasome pathway. J Biol Chem. 1998;273:24057–24064. doi: 10.1074/jbc.273.37.24057. [DOI] [PubMed] [Google Scholar]
- Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell. 2007;129:1351–1363. doi: 10.1016/j.cell.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Nakatsuji N. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev Biol. 2001;240:237–246. doi: 10.1006/dbio.2001.0439. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Shionoya A, Ishida M, Gambello MJ, Yingling J, Wynshaw-Boris A, Hirotsune S. A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron. 2000;28:681–696. doi: 10.1016/s0896-6273(00)00146-x. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Mori D, Toyo-oka K, Chen A, Garrett-Beal L, Muramatsu M, Miyagawa S, Hiraiwa N, Yoshiki A, Wynshaw-Boris A, Hirotsune S. Complete loss of Ndel1 results in neuronal migration defects and early embryonic lethality. Mol Cell Biol. 2005;25:7812–7827. doi: 10.1128/MCB.25.17.7812-7827.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer ME, Walker BE. Radioautographic study of interkinetic nuclear migration in the neural tube. Proc Soc Exp Biol Med. 1959;101:557–560. doi: 10.3181/00379727-101-25014. [DOI] [PubMed] [Google Scholar]
- Sidman RL, Rakic P. Neuronal migration, with special reference to developing human brain: a review. Brain Res. 1973;62:1–35. doi: 10.1016/0006-8993(73)90617-3. [DOI] [PubMed] [Google Scholar]
- Su SC, Tsai LH. Cyclin-dependent kinases in brain development and disease. Annu Rev Cell Dev Biol. 2011;27:465–491. doi: 10.1146/annurev-cellbio-092910-154023. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Efficient in utero gene transfer system to the developing mouse brain using electroporation: visualization of neuronal migration in the developing cortex. Neuroscience. 2001;103:865–872. doi: 10.1016/s0306-4522(01)00016-1. [DOI] [PubMed] [Google Scholar]
- Terada Y, Uetake Y, Kuriyama R. Interaction of Aurora-A and centrosomin at the microtubule-nucleating site in Drosophila and mammalian cells. J Cell Biol. 2003;162:757–763. doi: 10.1083/jcb.200305048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torisawa T, Nakayama A, Furuta K, Yamada M, Hirotsune S, Toyoshima YY. Functional dissection of LIS1 and NDEL1 towards understanding the molecular mechanisms of cytoplasmic dynein regulation. J Biol Chem. 2011;286:1959–1965. doi: 10.1074/jbc.M110.169847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyo-Oka K, Sasaki S, Yano Y, Mori D, Kobayashi T, Toyoshima YY, Tokuoka SM, Ishii S, Shimizu T, Muramatsu M, Hiraiwa N, Yoshiki A, Wynshaw-Boris A, Hirotsune S. Recruitment of katanin p60 by phosphorylated NDEL1, an LIS1 interacting protein, is essential for mitotic cell division and neuronal migration. Hum Mol Genet. 2005;14:3113–3128. doi: 10.1093/hmg/ddi339. [DOI] [PubMed] [Google Scholar]
- Tsai LH, Gleeson JG. Nucleokinesis in neuronal migration. Neuron. 2005;46:383–388. doi: 10.1016/j.neuron.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Van Horn RD, Chu S, Fan L, Yin T, Du J, Beckmann R, Mader M, Zhu G, Toth J, Blanchard K, Ye XS. Cdk1 activity is required for mitotic activation of Aurora A during G2/M transition of human cells. J Biol Chem. 2010;285:21849–21857. doi: 10.1074/jbc.M110.141010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CX, Song JH, Song DK, Yong VW, Shuaib A, Hao C. Cyclin-dependent kinase-5 prevents neuronal apoptosis through ERK-mediated upregulation of Bcl-2. Cell Death Differ. 2006;13:1203–1212. doi: 10.1038/sj.cdd.4401804. [DOI] [PubMed] [Google Scholar]
- Wang X, Ching YP, Lam WH, Qi Z, Zhang M, Wang JH. Identification of a common protein association region in the neuronal Cdk5 activator. J Biol Chem. 2000;275:31763–31769. doi: 10.1074/jbc.M004358200. [DOI] [PubMed] [Google Scholar]
- Yamada M, Toba S, Yoshida Y, Haratani K, Mori D, Yano Y, Mimori-Kiyosue Y, Nakamura T, Itoh K, Fushiki S, Setou M, Wynshaw-Boris A, Torisawa T, Toyoshima YY, Hirotsune S. LIS1 and NDEL1 coordinate the plus-end-directed transport of cytoplasmic dynein. EMBO J. 2008;27:2471–2483. doi: 10.1038/emboj.2008.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Yoshida Y, Mori D, Takitoh T, Kengaku M, Umeshima H, Takao K, Miyakawa T, Sato M, Sorimachi H, Wynshaw-Boris A, Hirotsune S. Inhibition of calpain increases LIS1 expression and partially rescues in vivo phenotypes in a mouse model of lissencephaly. Nat Med. 2009;15:1202–1207. doi: 10.1038/nm.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Toba S, Takitoh T, Yoshida Y, Mori D, Nakamura T, Iwane AH, Yanagida T, Imai H, Yu-Lee LY, Schroer T, Wynshaw-Boris A, Hirotsune S. mNUDC is required for plus-end-directed transport of cytoplasmic dynein and dynactins by kinesin-1. EMBO J. 2010;29:517–531. doi: 10.1038/emboj.2009.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Li F, Liang Y, Shen Y, Zhao X, Huang Q, Zhu X. Human Nudel and NudE as regulators of cytoplasmic dynein in poleward protein transport along the mitotic spindle. Mol Cell Biol. 2003;23:1239–1250. doi: 10.1128/MCB.23.4.1239-1250.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylkiewicz E, Kijanska M, Choi WC, Derewenda U, Derewenda ZS, Stukenberg PT. The N-terminal coiled-coil of Ndel1 is a regulated scaffold that recruits LIS1 to dynein. J Cell Biol. 2011;192:433–445. doi: 10.1083/jcb.201011142. [DOI] [PMC free article] [PubMed] [Google Scholar]