

Abstract
Prenatal exposure to infection has been linked to increased risk of neurodevelopmental brain disorders, and recent evidence implicates altered dopaminergic development in this association. However, since the relative risk size of prenatal infection appears relatively small with respect to long-term neuropsychiatric outcomes, it is likely that this prenatal insult interacts with other factors in shaping the risk of postnatal brain dysfunctions. In the present study, we show that the neuropathological consequences of prenatal viral-like immune activation are exacerbated in offspring with genetic predisposition to dopaminergic abnormalities induced by mutations in Nurr1, a transcription factor highly essential for normal dopaminergic development. We combined a mouse model of heterozygous genetic deletion of Nurr1 with a model of prenatal immune challenge by the viral mimetic poly(I:C) (polyriboinosinic polyribocytidilic acid). In our gene–environment interaction model, we demonstrate that the combination of the genetic and environmental factors not only exerts additive effects on locomotor hyperactivity and sensorimotor gating deficits, but further produces synergistic effects in the development of impaired attentional shifting and sustained attention. We further demonstrate that the combination of the two factors is necessary to trigger maldevelopment of prefrontal cortical and ventral striatal dopamine systems. Our findings provide evidence for specific gene–environment interactions in the emergence of enduring attentional impairments and neuronal abnormalities pertinent to dopamine-associated brain disorders such as schizophrenia and attention deficit/hyperactivity disorder, and further emphasize a critical role of abnormal dopaminergic development in these etiopathological processes.
Introduction
Prenatal exposure to infection is an environmental risk factor for neuropsychiatric disorders of developmental origin, especially schizophrenia and autism (Brown and Derkits, 2010; Meyer et al., 2011; Patterson, 2011). Converging evidence suggests that cytokine-associated inflammatory events (Gilmore and Jarskog, 1997; Smith et al., 2007; Meyer et al., 2008b), together with downstream pathophysiological processes such as oxidative stress (Lanté et al., 2008), hypoferremia (Aguilar-Valles et al., 2010), and zinc deficiency (Coyle et al., 2009), are critical in mediating the neurodevelopmental effects of prenatal infection. However, even though maternal infection during pregnancy is relatively common (Longman and Johnson, 2007), most offspring of infected mothers do not seem to develop severe neurodevelopmental brain disorders (Selten et al., 2010). This indicates that, if in utero exposure to infection plays a role in the etiology of developmental neuropsychiatric disorders, then it probably does so by interacting with other factors, including genetic factors (van Os et al., 2008; Ayhan et al., 2009; Abazyan et al., 2010). In support of this, the effect of prenatal infection on elevating risk of schizophrenia is substantially increased in offspring with a positive family history of psychotic disorders (Clarke et al., 2009).
Experimental work in animals shows that prenatal exposure to infection and/or inflammation has enduring consequences on dopaminergic structures and functions (Zuckerman et al., 2003; Ozawa et al., 2006; Meyer et al., 2008a; Aguilar-Valles et al., 2010; Bitanihirwe et al., 2010a,b; Romero et al., 2010; Vuillermot et al., 2010). In addition to such environmental influences, dopaminergic development is critically regulated by various genetic factors (Smidt and Burbach, 2007). Nurr1 (NR4A2) is a member of the orphan steroid hormone receptor family with pivotal functions in both the final differentiation, migration, and survival of ventral mesencephalic dopaminergic neurons (Zetterström et al., 1997; Saucedo-Cardenas et al., 1998; Kadkhodaei et al., 2009). Together with other seminal transcription factors such as Pitx3 and Lmx1b, Nurr1 also exerts a number of functions in postmitotic and mature mesencephalic dopamine neurons, including regulation of tyrosine hydroxylase (TH) and dopamine transporter (DAT) (Saucedo-Cardenas et al., 1998; Kadkhodaei et al., 2009). Nurr1 has also immunomodulatory roles by functioning as transcriptional repressor of proinflammatory genes (Saijo et al., 2009), and altered Nurr1 expression has been implicated in dopamine-associated brain disorders, including Parkinson's disease (Jankovic et al., 2005), attention deficit/hyperactivity disorder (ADHD) (Smith et al., 2005), and schizophrenia (Buervenich et al., 2000; Xing et al., 2006).
In view of the known dopaminergic effects of prenatal infection (for review, see Meyer and Feldon, 2009), one intriguing possibility is that genetic Nurr1 deficiency may interact with such early-life adversity to shape the severity and/or specificity of long-term dopaminergic brain pathology and associated behavioral/cognitive impairments. In the present study, we tested this hypothesis by combining a mouse model of heterozygous genetic deletion of Nurr1 with a model of prenatal exposure to the viral mimetic polyriboinosinic polyribocytidilic acid [poly(I:C)]. The latter is a well established model of prenatal viral-like immune activation that has been widely used to induce fetal brain inflammation and long-term brain and behavioral dysfunctions (Meyer et al., 2009).
Materials and Methods
Animals
Mice with a constitutive heterozygous deletion of the Nurr1 gene (Nurr1+/−) and their wild-type (wt) littermates were used throughout the study. In contrast to mice lacking both alleles of Nurr1 (i.e., Nurr1−/−), Nurr1+/− mice survive postnatally (Zetterström et al., 1997). Therefore, we only included Nurr1+/− and wt mice because of our interest to study the long-term effects on postnatal brain functions. The levels of Nurr1 protein in our Nurr1 heterozygous mouse colony were previously assessed (Vuillermot et al., 2011b,c), confirming that the expression of Nurr1 protein (and of downstream dopaminergic targets such as tyrosine hydroxylase) is reduced by ∼30–50% in midbrain regions of adult Nurr1+/− mice relative to wt littermates (Vuillermot et al., 2011b,c) (see also Jiang et al., 2005).
The generation of the Nurr1-deficient mouse colony with a 129/Sv genetic background has been fully described previously (Zetterström et al., 1997). Nurr1+/− mice were backcrossed for >15 generations to the C57BL/6 strain before commencement of the experiments described herein. Nurr1+/− and wt mice were kept in our in-house specific pathogen-free facility and used to generate a sufficient number of breeders necessary for the present investigations. The genotype of each animal was determined by PCR of tail DNA using specific probes for the Nurr1 gene as previously described (Zetterström et al., 1997). All animals were kept in a temperature- and humidity-controlled (21 ± 1°C; 55 ± 5%) holding facility under a reversed light/dark cycle (lights off: 8:00 A.M. to 8:00 P.M.), and they had ad libitum access to food (Kliba 3430; Kliba Mühlen) and water unless specified otherwise.
All procedures described in the present study had been previously approved by the Cantonal Veterinarian's Office of Zurich and are in agreement with the principles of laboratory animal care in the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication No. 86-23, revised 1985). All efforts were made to minimize the number of animals used and their suffering.
Maternal immune activation during pregnancy
For the purpose of the maternal immunological manipulations during pregnancy, female mice were subjected to a timed-mating procedure as described previously (Meyer et al., 2005, 2009). Pregnant wt or Nurr1+/− dams on gestation day 17 (GD17) received either a single injection of poly(I:C) (potassium salt; Sigma-Aldrich) at a dose of 2 mg/kg or vehicle (sterile pyrogen-free 0.9% NaCl) according to protocols established previously (Meyer et al., 2006, 2008a,b,c). The late gestational stage (i.e., GD17) was selected because of our previous findings showing that GD17 poly(I:C) treatment is capable of inducing long-term abnormalities in multiple behavioral and cognitive domains and of causing long-term dopaminergic abnormalities (Meyer et al., 2006, 2009; Bitanihirwe et al., 2010a,b). A low dose of poly(I:C) (i.e., 2 mg/kg, i.v.) was used to facilitate the anticipated interactions between the immunological manipulation and the genetic background (Ayhan et al., 2009; Meyer et al., 2009). Poly(I:C) was dissolved in sterile pyrogen-free 0.9% NaCl (vehicle) solution to yield a final concentration of 0.4 mg/ml and was administered via the intravenous route at the tail vein under mild physical constraint. All solutions were freshly prepared at the day of administration and injected with a volume of 5 ml/kg.
Testing of offspring
All offspring in each of the four experimental conditions [i.e., wt, saline; wt, poly(I:C); Nurr1+/−, saline; Nurr1+/−, poly(I:C)] were raised by their biological mothers (i.e., they were not cross-fostered to surrogate mothers), and they were weaned and sexed at postnatal day 21 (P21). Littermates of the same sex were caged separately and maintained in groups of two to four animals per cage as described above. Behavioral and cognitive testing was conducted when the offspring reached early adulthood (i.e., between P70 and P120) (Table 1). Only male subjects were included in all behavioral and cognitive tests to circumvent avoid bias arising from sexual dimorphism.
Table 1.
Group sizes and sequence of testing
| Experiment | wt, SAL | wt, POL | Nurr1+/−, SAL | Nurr1+/−, POL | Age | |
|---|---|---|---|---|---|---|
| Cohort I | 1. Cytokine assay | 4 f | 4 f | 5 f | 5 f | P70 |
| Cohort II | 2. Open field exploration | 14 m | 14 m | 14 m | 14 m | P70 |
| 3. Prepulse inhibition | 14 m | 14 m | 14 m | 14 m | P75 | |
| 4. Latent inhibition | ||||||
| Non-preexposed | 7 m | 7 m | 7 m | 7 m | P85 | |
| Preexposed | 7 m | 7 m | 7 m | 7 m | P85 | |
| Cohort III | 5. Visual discriminationa | 7 m | 7 m | 7 m | 7 m | P70 |
| 6. Working memory | 7 m | 7 m | 7 m | 7 m | P100 | |
| 7. Immunohistochemistry | 7 m | 7 m | 7 m | 7 m | P130 |
Animals from cohort I were used to ascertain the effects of poly(I:C) (POL) (2 mg/kg, i.v.) treatment, relative to saline (SAL) treatment, on serum proinflammatory and antiinflammatory cytokine elevations in pregnant adult (P70) wild-type (wt) and Nurr1-deficient (Nurr1+/−) female (f) mice on gestation day 17 (Experiment 1). Experimental subjects in cohorts II and III included adult offspring from SAL- or POL-exposed wt and Nurr1+/− mothers. Offspring from cohort II were tested sequentially in Experiments 2–4 assessing open-field exploration, prepulse inhibition, and latent inhibition in adulthood (P70–P85). Experiments 2–4 were separated by a intertest resting period of 3–5 d. Offspring from cohort III were first subjected to a two-choice visual discrimination test (Experiment 5) and spatial working memory test (Experiment 6). These offspring were then given a resting period of 2 weeks before being assigned to immunohistochemical analyses (Experiment 7). Only male (m) subjects were included in all behavioral, cognitive, and immunohistochemical tests performed in animals from cohorts II and III; offspring in each group derived from multiple independent litters (7–10) in each of the four experimental conditions to avoid confounds from litter effects.
aTwo animals (1 Nurr1+/−, SAL, and 1 Nurr1+/−, POL) did not reach criterion during the acquisition phase of the two-choice visual discrimination task. The final number of animals included in the statistical analyses of the two-choice visual discrimination task was therefore N(wt, SAL) = 7, N(wt, POL) = 7, N(Nurr1+/−, SAL) = 6, N(Nurr1+/−, POL) = 6.
A variety of behavioral and cognitive tests were used to assess the single and combined effects of heterozygous Nurr1 ablation and prenatal immune activation. This included tests measuring locomotor activity in a novel environment (open field), sensorimotor gating [prepulse inhibition (PPI)], sustained attention [two-choice visual discrimination test (2-CVDT)], attentional shifting [persistence of latent inhibition (LI)], and working memory (match-to-position paradigm in the dry maze). We focused on these behavioral and cognitive traits because they are highly sensitive to dopaminergic imbalances and are known to be affected by genetic Nurr1 deficiency (Rojas et al., 2007; Vuillermot et al., 2011a,c) and/or by prenatal poly(I:C) treatment (Meyer et al., 2008a; Bitanihirwe et al., 2010a,b; Vuillermot et al., 2010). The precise test procedures are fully described below, and the number of subjects in each test and the sequence of testing are given in Table 1.
Spontaneous locomotor activity in the open field
The apparatus consisted of four identical open-field arenas (40 × 40 × 35 cm high) made of wood and painted white as described previously (Meyer et al., 2005). They were located in a testing room under dim diffused lighting (∼35 lux as measured in the center of the arenas). A digital camera was mounted directly above the four arenas. Images were captured at a rate of 5 Hz and transmitted to a PC running the Ethovision tracking system (Noldus). For the purpose of data collection, the arena was conceptually partitioned into two zones: a center zone (measuring 13.5 × 13.5 cm2) in the middle of the arena and a peripheral zone occupying the remaining area. Spontaneous locomotor activity was assessed for 1 h and was indexed by the distance moved in the entire arena and center zone, expressed as a function of successive 5 min bins.
Prepulse inhibition of the acoustic startle reflex
Sensorimotor gating was assessed using the paradigm of PPI of the acoustic startle reflex. PPI of the acoustic startle reflex refers to the reduction of startle reaction in response to a startle-eliciting pulse stimulus when it is shortly preceded by a weak prepulse stimulus. The apparatus consisted of four startle chambers for mice (San Diego Instruments) as fully described previously (Meyer et al., 2005). In the demonstration of PPI of the acoustic startle reflex, subjects were presented with a series of discrete trials comprising a mixture of four trial types. These included pulse-alone trials, prepulse-plus-pulse trials, prepulse-alone trials, and no-stimulus trials in which no discrete stimulus other than the constant background noise was presented. The pulse and prepulse stimuli used were in the form of a sudden elevation in broadband white noise level (sustaining for 40 and 20 ms, respectively) from the background (65 dBA), with a rise time of 0.2–1.0 ms. In all, three different intensities of pulse (100, 110, and 120 dBA) and three intensities of prepulse (71, 77, and 83 dBA, which corresponded to 6, 12, and 18 dBA above background, respectively) were used. The stimulus-onset asynchrony of the prepulse and pulse stimuli on all prepulse-plus-pulse trials was 100 ms (onset to onset). The PPI test procedure strictly followed protocols established and fully described previously (Bitanihirwe et al., 2011). For each of the three pulse intensities (100, 110, or 120 dBA), PPI was indexed by percentage inhibition of the startle response obtained in the pulse-alone trials by the following expression: 100% × [1 − (mean reactivity on prepulse-plus-pulse trials/mean reactivity on pulse-alone trials)], for each subject, and at each of the three possible prepulse and pulse intensities.
Persistence of latent inhibition in a conditioned freezing paradigm
The LI effect is known to be a window phenomenon, in which the stimulus-no event association acquired in preexposure is only expressed under a very specific balance between the behavioral impact of preexposure and conditioning (Weiner, 2003). Organisms that tend to show impaired attentional shifting are expected to express the LI effect under parametric conditions that are insufficient for control animals to display LI (Weiner, 2003). Such attentional abnormalities are manifest in adult mice following prenatal treatment with a high dose of poly(I:C) (Bitanihirwe et al., 2010b), indicating that prenatal immune activation at high intensity is capable of inducing deficits in this cognitive domain. Here, we were interested to explore whether the combination of Nurr1 deficiency and mild prenatal immune activation may interact with each other to produce such attentional abnormalities, and therefore, we used parametric conditions with low amount of stimulus preexposure to facilitate the identification of potential LI persistence in Nurr1+/− offspring with additional prenatal poly(I:C) exposure. The apparatus comprised two sets of four test chambers as fully described previously (Meyer et al., 2005). The test procedures of the LI test consisted of three phases: preexposure, conditioning, and tone test as follows.
Preexposure phase.
The animals were placed in the appropriate test chamber: Subjects assigned to conditioned stimulus (CS) preexposure (PE) condition received 30 presentations of a 30 s tone stimulus at a variable interstimulus interval of 40 ± 30 s; non-preexposed (NPE) subjects were confined to the chamber for an equivalent period of time without any stimulus presentation.
Conditioning.
Conditioning commenced immediately at the end of the preexposure phase without removing the animals from the chambers. Conditioning comprised three discrete trials of paring between the CS and an unconditioned stimulus (US): Each trial began with the 30 s tone stimulus (identical to the one used during preexposure) followed immediately by the delivery of a 1 s footshock set at 0.3 mA (US). Each trial was preceded and followed by a 180 s interval.
Test.
The test of conditioned responding to the tone CS was conducted another 24 h later, when the animals were placed in the same test chambers again. Following an initial period of 360 s acclimatization, the tone CS was delivered and remained on for 360 s, in which the time of conditioned freezing to the tone stimulus was evaluated. The test phase was concluded with a final 360 s interval.
The data on freezing behavior collected on the three phases (preexposure, conditioning, CS test) were separately analyzed.
Sustained attention in a two-choice visual discrimination task
Sustained attention was assessed using an operant-based two-choice visual discrimination tests as established previously in mice (Bitanihirwe et al., 2011). Four operant chambers (model E10-10; Habitest System; Coulbourn Instruments) were used, each placed inside a ventilated and sound-attenuated box. The test chambers have been fully described previously (Bitanihirwe et al., 2011). The data were analyzed by the Graphic State (version 1.013) tracking system. For this experiment, the animals were food-deprived according to a gradual food deprivation regime, and at each of the eventual test days, the animals received one pellet of food 5 h after the end of each trial.
The experimental procedures consisted of four stages: habituation and magazine training, lever press shaping, visual discrimination, and test of visual discrimination with variable signal duration. Detailed information regarding the procedures used for the initial habituation and magazine training, lever press shaping, and acquisition of visual discrimination phases can be obtained elsewhere (Bitanihirwe et al., 2011). The animals were trained on the attentional task for an average of 9 d before the critical test was performed.
Once the animals had achieved high accuracy (≥80% on 2 consecutive days) in the acquisition of visual discrimination (for detailed information, see Bitanihirwe et al., 2011), the duration of the visual signal was reduced to 10, 2, 1, or 0.5 s. The animals were allowed a maximum of 30 s to perform a nose poke response following the presentation of the visual signal, before an omission would be recorded. Throughout the maximum number of 48 trials (i.e., 12 trials at each stimulus duration regardless of omission), every block of 4 trials randomly featured one of the four possible signal durations. Testing was performed in one daily session that lasted for a maximum of 50 min. Choice accuracy was taken to index sustained attentional performance and was defined by the following formula: correct responses/(correct responses + incorrect responses) × 100%.
Spatial working memory in the dry maze
The apparatus consisted of a wooden circular board (diameter, 1.1 m; thickness, 3 cm) painted in gray as fully described previously (Bitanihirwe et al., 2010a; Llano Lopez et al., 2010). It was positioned in a well lit room enriched with distal spatial cues. One surface of the board was drilled with 32 holes (diameter, 3.1 cm; depth, 1.3 cm) arranged in radial design. Each hole contained a Plexiglas bottle cap that could be equipped with a visual cue (composed of a blue flag attached to a 15 cm stick) and/or a reward in the form of 75 μl of freshly prepared condensed milk (Alicommerce SAS) used in a 1:4 (milk/water) dilution. A digital camera was mounted above the dry maze. Images were captured at a rate of 5 Hz and transmitted to a PC running the Ethovision (Noldus Information Technology) tracking system, which calculated the distance moved to reach the reward on each trial.
All animals were progressively food deprived during the initial habituation phase (days 1–5) until a minimal 90% free-feeding weight was reached. The animals were first habituated to the dry maze for two daily trials of 2 min each. To begin a trial, the animals were placed gently in the center of the dry maze. The intertrial interval (ITI) was ∼1 min. On days 6 and 7, the animals were then pretrained in the dry maze using the visual cue for two consecutive trials. One random hole was cued with the flag and rewarded with the milk solution. For both trials, each animal was left on the maze for 2 min. If it failed to reach the rewarded hole during this time, it was gently guided to the rewarded hole by the experimenter. All animals consumed the reward within the allowed maximum time of 15 s.
Following the habituation (days 1–5) and pretesting (days 6 and 7) phases, working memory testing was started and lasted for 7 testing days (days 8–14). The working memory task was based on the matching-to-position paradigm, in which the animals were required to learn the novel position of a rewarded hole revealed to them on trial 1 of each day to navigate effectively to the same location (i.e., matching) on the subsequent trial on the same day. Hence, each test day included two trials. For both trials, each animal was left on the maze for 2 min. If it failed to reach the rewarded hole during this time, it was gently guided to the rewarded hole by the experimenter as described above. The reward remained in the same position across trials on a given test day, but took a new position on each test day. The daily change of the reward location ensured that the information acquired on a given day was irrelevant for the next day, thus taxing the flexible use of working memory (Baddeley, 2003). To begin a trial, the animals were placed gently in the center of the dry maze as described previously. The allocation of the rewarded holes to a specific spatial location was counterbalanced across the four experimental conditions. An ITI of 1 min was used at each of 7 test days, corresponding to the minimal amount of time needed to clean the dry maze surface with water and dry it from the first to the second trial. Working memory was indexed by the reduction in distance moved and time needed to find the location of the reward in trial 2 relative to trial 1. The data were subjected to logarithmic transformation (base e) to better conform to the assumption of data homogeneity and normality by parametric ANOVA.
Immunohistochemistry
Adult (P130) offspring were deeply anesthetized with an overdose of Nembutal (Abbott Laboratories) and perfused transcardially with 0.9% NaCl, followed by 4% phosphate-buffered paraformaldehyde solution containing 15% picric acid. The dissected brains were postfixed in the same fixative for 6 h and processed for antigen retrieval involving overnight incubation in citric acid buffer, pH 4.5, followed by a 90 s microwave treatment at 480 W according to protocols established previously (Meyer et al., 2008a; Vuillermot et al., 2010). The brains were then cryoprotected using 30% sucrose in PBS, frozen with powdered dry ice, and stored at −80°C until further processing.
Perfused brain samples were cut coronally at 30 μm thickness from frozen blocks with a sliding microtome. Six series of sections were collected, rinsed in PBS, and stored at −20°C in antifreeze solution until further processing. For immunohistochemical staining, the slices were rinsed three times for 10 min in PBS, and blocking was done in PBS, 0.3% Triton X-100, 5% normal serum for 1 h at room temperature. The following primary antibodies were used to study dopamine-related markers in various brain areas according to protocols established previously (Meyer et al., 2008a; Bitanihirwe et al., 2010a; Vuillermot et al., 2010): rabbit anti-TH (Santa Cruz Biotechnology; diluted 1:500), rabbit anti-dopamine D1 receptor (D1R) (Millipore Bioscience Research Reagents; diluted 1:250), rabbit anti-dopamine D2 receptor (D2R) (Millipore Bioscience Research Reagents; diluted 1:500), and rabbit anti-catechol-O-methyltransferase (COMT) (SDI; diluted 1:5000). These dopaminergic markers were selected because they are known to be modulated by prenatal immune activation and/or genetic Nurr1 deficiency (Zetterström et al., 1997; Saucedo-Cardenas et al., 1998; Kadkhodaei et al., 2009; Bitanihirwe et al., 2010a; Vuillermot et al., 2010, 2011c) and because they are implicated in the dopaminergic neuropathology of schizophrenia and related disorders (Akil et al., 1999; Chen et al., 2004; Howes et al., 2007). All antibodies were diluted in PBS containing 0.3% Triton X-100 and 2% normal serum, and the sections were incubated free-floating overnight at room temperature. After three washes with PBS (10 min each), the sections were incubated for 1 h with the biotinylated secondary antibodies diluted 1:500 in PBS containing 2% NGS and 0.3% Triton X-100. Sections were washed again three times for 10 min in PBS and incubated with Vectastain kit (Vector Laboratories) diluted in PBS for 1 h. After three rinses in 0.1 m Tris-HCl, pH 7.4, the sections were stained with 1.25% 3,3-diaminobenzidine and 0.08% H2O2 for 10–15 min, rinsed again four times in PBS, dehydrated, and coverslipped with Eukitt (Kindler).
Stereological estimation of TH-positive dopaminergic midbrain neurons
The numbers of midbrain TH-positive dopamine neurons were quantified in the two relevant midbrain areas, namely in the ventral tegmental area (VTA) and substantia nigra (SN). For the latter region, TH-positive cells in both the substantia nigra pars compacta (SNc) and substantia nigra pars reticulata (SNr) were taken into account. The numbers of midbrain TH-positive dopamine neurons in either the left or right brain hemisphere were determined by unbiased stereological estimations using the optical fractionator method (Gundersen et al., 1988). With the aid of the image analysis computer software Stereo Investigator (version 6.50.1; MicroBrightField), every section of a one-in-six series was measured, resulting in an average of six sections per brain sample as previously described (Vuillermot et al., 2010). The following sampling parameters were used: (1) a fixed counting frame with a width of 40 μm and a length of 40 μm; and (2) a sampling grid size of 150 × 110 μm. The counting frames were placed randomly at the intersections of the grid within the outlined structure of interest by the software. The cells were counted following the unbiased sampling rule using the 40× oil lens [numerical aperture (NA), 1.3] and included in the measurement when they came into focus within the optical dissector (Howard and Reed, 2005).
Optical densitometry of dopaminergic markers in prefrontal cortical and striatal regions
Quantification of the immunoreactivity for TH, COMT, D1R, and D2R in prefrontal cortical and striatal regions was achieved by means of optical densitometry using NIH ImageJ software. Optical densitometry was chosen because these dopaminergic markers are highly enriched at synaptic sites in the areas of interest (Meyer et al., 2008a; Bitanihirwe et al., 2010a; Vuillermot et al., 2010). Digital images were acquired at a magnification of 2.5× (NA 0.075) using a digital camera (Axiocam MRc5; Zeiss) mounted on a Zeiss Axioplan microscope. Exposure times were set so that pixel brightness was never saturated. Pixel brightness was measured in the respective areas of one randomly selected brain hemisphere. In addition, pixel brightness was measured in the forceps minor corpus callosum (for prefrontal cortical measures) and corpus callosum (for striatal measures) as background area. The background-corrected optical densities were averaged per brain region and animal. Four to six sections per animal were analyzed in the specimens. All immunohistochemical preparations were quantified in the medial prefrontal cortex (mPFC) [including anterior cingluate (aCG), prelimbic (PrL), and infralimbic (IL) cortices], dorsal striatum [caudate–putamen (CPu)], nucleus accumbens core (NAc core), and nucleus accumbens shell (NAc shell) (see below).
Delineation of brain regions
All brain areas of interest were delineated according to The Mouse Brain in Stereotaxic Coordinates by Franklin and Paxinos (2008). The following brain areas were included in the densitometric and stereological analyses: mPFC (bregma +2.20 to +1.70 mm), CPu (bregma +1.34 to +0.14 mm), NAc core and shell (bregma +1.60 to +0.98 mm), SN (bregma −2.80 to −3.64 mm), and VTA (bregma −2.92 to −3.64 mm). Schematic coronal brain sections of the brain areas of interest with reference to bregma are provided in corresponding figure legends.
Cytokine assay
A separate cohort of pregnant female wt and Nurr1+/− mice were used to ascertain the effects of poly(I:C) treatment on serum proinflammatory and antiinflammatory cytokine elevations. For this purpose, pregnant dams were injected with either poly(I:C) (2 mg/kg, i.v.) or vehicle (sterile pyrogen-free 0.9% NaCl, i.v.) as described above, and they were killed by decapitation 2 h after treatment to collect trunk blood. The posttreatment interval was chosen to capture peak inflammatory cytokine responses to intravenous poly(I:C) treatment (Meyer et al., 2006, 2008b). Trunk blood was collected into Eppendorf tubes and was allowed to clot at room temperature for 1 h before centrifugation at 10,000 rpm for 4 min at 4°C. The resulting serum from each animal was subdivided to permit storage (−80°C) until the cytokine assays were performed. Cytokine protein levels were measured using a multiplexed particle-based flow cytometric cytokine assay as described previously (Meyer et al., 2006). Fluorokine MAP (Multi Analyte Profiling) mouse kits for interleukin-1β (IL-1β), IL-6, IL-10, and tumor necrosis factor α (TNFα) were purchased from R&D Systems. The procedures closely followed the manufacturer's instructions. The analysis was conducted using a conventional flow cytometer (LSR II; BD Biosciences). The detection limits were 0.3 pg/ml for IL-1β, 0.2 pg/ml for IL-6, 1.0 pg/ml for IL-10, and 0.2 pg/ml for TNFα.
Statistical analyses
All data were analyzed using parametric ANOVA. All cytokine data as well as TH, COMT, D1R, and D2R immunoreactivities measured in striatal and prefrontal cortical regions were first subjected to logarithmic transformation (base e) to better conform to the assumption of data homogeneity and normality by parametric ANOVA, and were then analyzed using a 2 × 2 (genotype by prenatal treatment) ANOVA. Locomotor activity in the open field was indexed by the distance moved in the entire arena and center zone and was analyzed using 2 × 2 × 12 (genotype by prenatal treatment by 5 min bin) repeated-measure ANOVAs. In the test of PPI, percentage PPI was analyzed using a 2 × 2 × 3 × 3 (genotype by prenatal treatment by pulse by prepulse) ANOVA, and the reactivity to pulse-alone trials and prepulse-alone trials was analyzed using 2 × 2 × 3 (genotype by prenatal treatment by pulse) and (genotype by prenatal treatment by prepulse) ANOVAs, respectively. In the test of LI persistence, conditioned freezing toward the tone-CS was assessed 24 h following conditioning: Percentage time freezing during the 6 min period of the tone presentation was expressed as a function of 1 min bins and subjected to a 2 × 2 × 2 × 6 (genotype by prenatal treatment by preexposure by 1 min bins) repeated-measure ANOVA. Percentage time freezing during the initial conditioning phase was analyzed using a 2 × 2 × 2 × 3 (genotype by prenatal treatment by preexposure by trials) repeated-measure ANOVA. Choice accuracy in the 2-CVDT was indexed by percentage correct responses during the test day and was analyzed using a 2 × 2 × 4 (genotype by prenatal treatment by signal duration) ANOVA. In the test of working memory in the dry maze matching-to-position paradigm, the distance moved to locate the reward served as the critical test readout and was analyzed using a 2 × 2 × 2 × 7 (genotype by prenatal treatment by trial by day) repeated-measures ANOVA. Following these initial ANOVAs, Fisher's least significant difference post hoc comparisons or restricted ANOVAS were conducted whenever appropriate. Statistical significance was set at p < 0.05. All statistical analyses were performed using the statistical software StatView (version 5.0) implemented on a PC running the Windows XP operating system.
Results
Nurr1 modulates basal and poly(I:C)-induced maternal cytokine responses
Systemic poly(I:C) treatment in pregnant mice is known to induce cytokine levels in maternal and fetal compartments (Meyer et al., 2006, 2008b; Smith et al., 2007). Here, we ascertained the efficacy of systemic treatment with a low dose (2 mg/kg, i.v.) to induce cytokine levels by measuring serum contents of prototypical proinflammatory (IL-1β, IL-6, and TNFα) and antiinflammatory (IL-10) cytokines in pregnant (GD17) wt and Nurr1+/− mice using established multiplexed particle-based flow-cytometric cytokine assays (Meyer et al., 2006). As expected (Meyer et al., 2005, 2006, 2008b), administration of poly(I:C) led to a relatively mild but significant increase in maternal serum levels of proinflammatory and antiinflammatory cytokines in wt mice compared with levels measured in vehicle-treated wt dams (Fig. 1A). Interestingly, pregnant Nurr1+/− mice displayed enhanced basal levels of the proinflammatory cytokine IL-6, as evident in the comparison between IL-6 levels in saline-treated wt and Nurr1-deficient mice (Fig. 1A). Poly(I:C) treatment in Nurr1+/− mice did not further elevate any of the cytokines of interest, but instead significantly reduced serum IL-6 levels to control levels measured in saline-treated wt mice (Fig. 1A). These patterns of changes led to the presence of significant interactions between genotype and prenatal treatment in the analysis of maternal cytokine levels (IL-6: F(1,14) = 12.86, p < 0.01; TNFα: F(1,14) = 6.50, p < 0.05; IL-10: F(1,14) = 5.57; p < 0.05). Serum IL-1β levels were below detection limit for all experimental groups.
Figure 1.

Heterozygous Nurr1 deficiency modulates peripheral maternal cytokine levels at basal conditions and following viral-like immune activation. A, The bar plots show serum levels of the prototypical proinflammatory cytokines IL-6 and TNFα and the antiinflammatory cytokine IL-10 in pregnant wt and Nurr1+/− mice treated with vehicle [saline (SAL)] or the viral mimetic poly(I:C) (POL) (2 mg/kg, i.v.) on gestation day 17. *p < 0.05 and **p < 0.01, based on post hoc group comparisons. Serum levels of IL-1β were below detection limit (0.3 pg/ml) for each experimental condition. B, The bar plots depict the balance between maternal proinflammatory versus antiinflammatory cytokines in terms of the IL-6/IL-10 and TNFα/IL-10 ratios in SAL- or POL-treated wt and Nurr1+/− dams. *p < 0.05, based on post hoc group comparisons. N(wt, SAL) = 4, N(wt, POL) = 4, N(Nurr1+/−, SAL) = 5, N(Nurr1+/−, POL) = 5. All values are means ± SEM.
Maternal serum ratios of IL-6/IL-10 and TNFα/IL-10 were then analyzed to determine the relative proinflammatory versus antiinflammatory cytokine responses to the poly(I:C) challenge or vehicle treatment. As depicted in Figure 1B, there was a significant synergistic interaction between genotype and immune treatment (F(1,14) = 4.68; p < 0.05) in the analysis of serum IL-6/IL-10 ratios, with poly(I:C)-treated Nurr1+/− mice displaying a significant reduction in the IL-6/IL-10 ratio compared with all other groups (all values of p < 0.05). No significant differences emerged in the analysis of serum TNFα/IL-10 ratios (Fig. 1B).
Additive effects between prenatal immune activation and Nurr1 deficiency in the development of spontaneous locomotor hyperactivity and prepulse inhibition deficits
In a next step, we explored whether prenatal immune activation may interact with genetic Nurr1 deficiency in the development of long-term behavioral dysfunctions. One of the abnormal behaviors previously identified in Nurr1-deficient mice is the emergence of spontaneous locomotor hyperactivity in a novel environment (Rojas et al., 2007; Vuillermot et al., 2011a). This phenotype is critically driven by dopaminergic imbalances and can be normalized by acute treatment with dopamine receptor antagonists (Rojas et al., 2007). Here, we show that this hyperactivity phenotype is significantly modulated by prenatal viral-like immune challenge: As summarized in Figure 2, we found that prenatal immune activation and Nurr1 deficiency exerted additive effects on spontaneous locomotor hyperactivity in the open field test. Hence, while the locomotor activity scores were increased by 25–30% in saline-treated Nurr1+/− and poly(I:C)-treated wt offspring relative to saline-exposed wt controls, distance moved in the open field was increased by 55–60% in poly(I:C)-exposed Nurr1+/− mice. These additive effects between genetic Nurr1 deficiency and prenatal immune activation were evident both in terms of total distance moved in the entire open field arena (main effect of genotype: F(1,52) = 21.93, p < 0.001; main effect of prenatal treatment: F(1,52) = 9.55, p < 0.01; Fig. 2A) and distance moved in the center zone (main effect of genotype: F(1,52) = 24.15, p < 0.001; main effect of prenatal treatment: F(1,52) = 8.21, p < 0.01; Fig. 2B).
Figure 2.

Additive effects between prenatal immune activation and Nurr1 deficiency in the development of spontaneous locomotor hyperactivity. Pregnant wt and Nurr1+/− mice were treated with the viral mimetic poly(I:C) (POL) (2 mg/kg, i.v.) or vehicle [saline (SAL)] on gestation day 17, and the resulting offspring were tested in early adulthood. A, The line plot depicts the total distance moved in the entire open field area as a function of 5 min bins, and the bar plot shows the mean distance moved across the 1 h test period. ***p < 0.001 and §p < 0.01, signifying the main effects of genotype and prenatal treatment, respectively. N = 14 in each experimental group; all values are means ± SEM. B, The line plot represents the distance moved in the center zone of the open field as a function of 5 min bins, and the bar plot depicts the mean center zone distance moved across the 1 h test period. ***p < 0.001 and §p < 0.01, signifying the main effects of genotype and prenatal treatment, respectively. N = 14 in each experimental group; all values are means ± SEM. C, Computer-generated path drawings of representative SAL- or POL-exposed wt and Nurr1+/− offspring in the open-field test.
We were then interested to assess the single and combined effects of prenatal immune activation and heterozygous Nurr1 ablation on sensorimotor gating using the paradigm of PPI of the acoustic startle reflex. PPI deficiency has been linked to numerous dopamine-associated neuropsychiatric disorders, including schizophrenia, obsessive-compulsive disorder, and Huntington's disease (Braff et al., 2001). Deficits in PPI have also been documented in adult Nurr1+/− mice (Vuillermot et al., 2011a,c), and this phenotype has further been correlated with presynaptic dopaminergic changes in striatal structures (Vuillermot et al., 2011c). Here, we demonstrate that prenatal immune challenge and genetic Nurr1 deficiency induce additive effects on the disruption of PPI (as indexed by percentage PPI): While PPI scores were highest in saline-exposed wt offspring and lowest in poly(I:C)-treated Nurr1+/− offspring, the two other groups [i.e., poly(I:C)-exposed wt and saline-exposed Nurr1+/− offspring] displayed intermediate levels of percentage PPI (Fig. 3A). ANOVA yielded significant main effects of genotype (F(1,52) = 4.48; p < 0.05) and prenatal treatment (F(1,52) = 4.52; p < 0.05). In addition, we found a significant (F(1,52) = 4.42; p < 0.05) interaction between genotype and prenatal treatment in the analysis of startle reactivity to pulse-alone trials, with poly(I:C)-exposed wt offspring displaying a significant increase in startle reactivity compared with all other groups (all values of p < 0.05, based on post hoc group comparison; Fig. 3B). Since differences in general startle reactivity can readily confound the interpretation of percentage PPI (Swerdlow et al., 2000), we further analyzed reactivity scores obtained on pulse-alone and prepulse-plus-pulse trials. As shown in Figure 3C, introduction of a prepulse preceding the pulse led to the typical reduction in pulse reactivity toward the succeeding pulse. This effect of PPI was markedly pronounced in saline- or poly(I:C)-exposed wt offspring, but weakened in saline or poly(I:C) Nurr1+/− offspring (Fig. 3C). This led to the presence of a significant three-way interaction between genotype, prenatal treatment, and prepulse levels (F(3,156) = 2.63, p < 0.05). Hence, the extent to which presentation of a prepulse preceding the pulse was efficient in reducing pulse reactivity toward the succeeding pulse did not seem to differ between saline-exposed and poly(I:C)-exposed wt offspring. It follows that the attenuation of percentage PPI associated with prenatal poly(I:C) treatment in wt mice (Fig. 3A) is likely confounded by the presence of increased startle reactivity, whereas the reduction of percentage PPI in saline- or poly(I:C)-exposed Nurr1+/− mice represents a genuine disruption of sensorimotor gating (Swerdlow et al., 2000). Neither heterozygous Nurr1 ablation nor prenatal poly(I:C) administration significantly affected prepulse-elicited reactivity (i.e., the reactivity to prepulse-alone trials) (data not shown).
Figure 3.

Additive effects between prenatal immune activation and Nurr1 deficiency in the development of sensorimotor gating deficiency. Pregnant wt and Nurr1+/− mice were treated with the viral mimetic poly(I:C) (POL) (2 mg/kg, i.v.) or vehicle [saline (SAL)] on gestation day 17, and the resulting offspring were tested in early adulthood. Sensorimotor gating was assessed by PPI of the acoustic startle reflex. A, The line plot shows percentage PPI as a function of three different pulse intensities (P-100, P-110, and P-120, which correspond to 100, 110, and 120 dBA) and prepulse intensities (+6, +12, and +18 dBA above background of 65 dBA); the bar plot depicts the mean percentage PPI across all prepulse and pulse stimuli used. Percentage PPI was calculated by the following expression: 100% × [1 − (mean reactivity on prepulse-plus-pulse trials/mean reactivity on pulse-alone trials)], for each subject, and at each of the three possible pulse and prepulse intensities. *p < 0.05 and §p < 0.05, reflecting the main effects of genotype and prenatal treatment, respectively. N = 14 in each experimental group; all values are means ± SEM. B, The line plot shows startle reactivity (in arbitrary units) to pulse-alone trials as a function of the three different pulse intensities (100, 110, and 120 dBA), and the bar plot depicts the mean startle reactivity across all pulse stimuli used. *p < 0.05, signifying the significant difference between POL-exposed Nurr1+/− mice and all other groups. N = 14 in each experimental group; all values are means ± SEM. C, Raw score analysis of PPI: The line plot shows the startle reactivity (in arbitrary units) obtained on pulse-alone and prepulse-plus-pulse trials averaged across all three pulse stimuli used. N = 14 in each experimental group; all values are means ± SEM.
Synergistic effects between prenatal immune activation and Nurr1 deficiency in the development of attentional impairments
In a next step, we explored the single and combined effects of Nurr1 deficiency and prenatal immune challenge on attentional shifting and sustained attention, both of which are known to be highly regulated by dopaminergic mechanisms especially within mesocortical and frontostriatal pathways (Robbins, 2002; Kehagia et al., 2010; Cools and D'Esposito, 2011). Attentional shifting was studied by assessing the presence of LI persistence in a conditioned freezing paradigm. Organisms that tend to show impaired attentional shifting are expected to express the LI effect under parametric conditions that are insufficient for control animals to display LI (Weiner, 2003). Such attentional abnormalities are manifest in adult mice following prenatal treatment with a high dose of poly(I:C) (Bitanihirwe et al., 2010b), indicating that prenatal immune activation at high intensity is capable of inducing deficits in this cognitive domain. Here, we show that mild prenatal immune challenge can synergistically interact with a genetic predisposition factor in the development of LI persistence: Whereas poly(I:C)-treated Nurr1+/− offspring displayed a marked LI effect (indexed by reduced conditioned freezing in PE subjects compared with NPE subjects), all other groups showed no similar LI effect during the critical tone-CS test phase of the LI paradigm (Fig. 4A). ANOVA yielded a significant three-way interaction between genotype, prenatal treatment, and stimulus preexposure (F(1,48) = 4.15; p < 0.05), and additional restricted analyses confirmed the significant (p < 0.01) preexposure effect in poly(I:C)-treated Nurr1+/− offspring but not in the other groups (all values of F < 1). No group differences were present during the initial acclimatization period preceding the tone-CS test phase (Fig. 4A), indicating that the presence of abnormally enhanced LI revealed in poly(I:C)-treated Nurr1+/− offspring is not confounded by possible differences in general activity and/or altered conditioned responses toward the context. Furthermore, the emergence of LI persistence in poly(I:C)-treated Nurr1+/− offspring could also not be accounted for by selective effects in the conditioning phase of the test. Indeed, even though Nurr1-deficient mice displayed a general reduction in percentage time freezing in the conditioning phase of the test (main effect of genotype: F(1,48) = 12.63, p < 0.01), this effect of heterozygous Nurr1 ablation emerged independently of the prenatal treatment and stimulus preexposure conditions. The mean ± SEM percentage time freezing scores across the three successive CS presentations during conditioning was 7.2 ± 3.7 for Nurr1+/− animals, and 18.6 ± 3.5 for wt mice. Together, our results show that the combination of prenatal immune activation and Nurr1 deficiency causes abnormally enhanced LI as manifest specifically during the critical tone-CS test phase of the LI paradigm (Fig. 4A).
Figure 4.
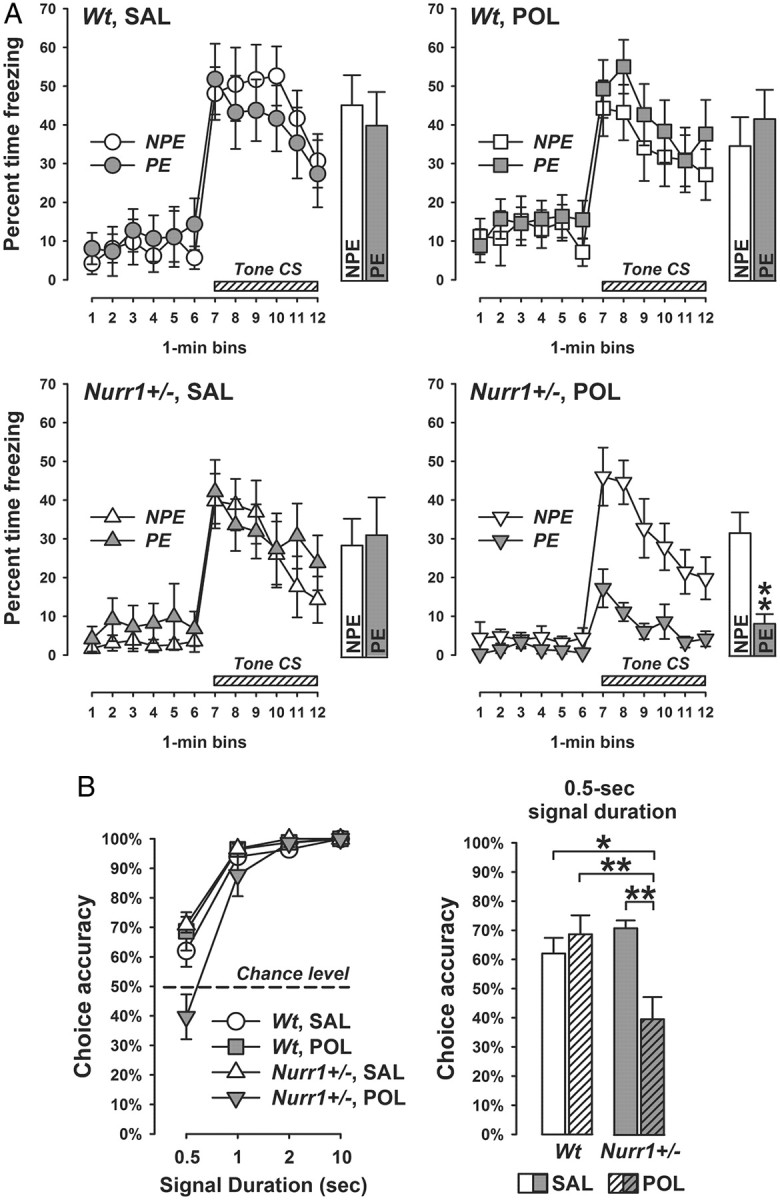
Synergistic effects between prenatal immune activation and Nurr1 deficiency in the development of attentional impairments. Pregnant wt and Nurr1+/− mice were treated with the viral mimetic poly(I:C) (POL) (2 mg/kg, i.v.) or vehicle [saline (SAL)] on gestation day 17, and the resulting offspring were tested in early adulthood. A, Persistence of the LI effect during the test of conditioned tone-freezing: The line plots show percentage time freezing as function of 1 min bins for the initial 6 min pretone period and for the subsequent 6 min tone period separately for NPE and PE subjects, and the bar plots depict the corresponding mean freezing response over the entire period of tone presentation. **p < 0.01, signifying the significant main effect of preexposure (i.e., LI effect) in POL-exposed Nurr1+/− offspring. N(NPE) = 7 and N(PE) = 7 in each experimental group; all values are means ± SEM. B, Sustained visual attention in the two-choice visual discrimination test: The line plot depicts choice accuracy (indexed by the percentage correct responses to the cued magazine) for each of the four stimulus-duration trial types (0.5, 1, 2, and 10 s), and the bar plot shows the choice accuracy at the shortest signal duration (0.5 s) condition. *p < 0.05 and **p < 0.01; N(Wt, SAL) = 7, N(Wt, POL) = 7, N(Nurr1+/−, SAL) = 6, and N(Nurr1+/−, POL) = 6. All values are means ± SEM.
We were also interested in evaluating the single and combined effects of Nurr1 deficiency and prenatal immune activation on sustained attention. Deficits in this cognitive domain are one of the most consistent findings in dopamine-associated neuropsychiatric disorders, especially schizophrenia (Nuechterlein et al., 2009) and ADHD (Del Campo et al., 2011). Here, we used an operant-based 2-CVDT that depends on effortful attention to brief visual cues. This requires the animals to learn to choose between two magazines according to a visual discriminatory signal, which can be progressively reduced to impose increasing demand on sustained visual attention (Robbins, 2002; Bitanihirwe et al., 2011). There were no group differences during the lever press shaping and acquisition of visual discrimination in training phase (Table 2), and with the exception of two animals (Table 1), all mice reached test criterion at the end of the acquisition phase. As expected, the choice accuracy significantly increased as a function of duration of the visual signal during the critical test phase (main effect of signal duration: F(3,66) = 120.57, p < 0.001; Fig. 4B). Most interestingly, choice accuracy was significantly reduced specifically in poly(I:C)-exposed Nurr1+/− offspring at the shortest signal duration condition (Fig. 4B). This led to a significant interaction between genotype, prenatal treatment, and signal duration (F(3,66) = 7.64; p < 0.001). Subsequent post hoc group comparisons conducted at the 0.5 s signal duration confirmed the significant impairment in poly(I:C)-exposed Nurr1+/− offspring relative to saline-treated wt offspring (p < 0.05), poly(I:C)-exposed wt offspring (p < 0.01), and saline-treated Nurr1+/− offspring (p < 0.01).
Table 2.
Performance during lever press shaping and acquisition of visual discrimination in the training phase of the two-choice visual discrimination test
| Phase | Criterion | Group | Mean ± SEM |
|---|---|---|---|
| Shaping | 19 of 24 trials (80%) on last day | wt, SAL | 24.0 ± 0.0 |
| wt, POL | 24.0 ± 0.0 | ||
| Nurr1+/−, SAL | 24.0 ± 0.0 | ||
| Nurr1+/−, POL | 24.0 ± 0.0 | ||
| Acquisition | 38 of 48 trials (80%) on last 2 d | wt, SAL | 47.5 ± 0.3 |
| wt, POL | 47.6 ± 0.1 | ||
| Nurr1+/−, SAL | 47.7 ± 0.2 | ||
| Nurr1+/−, POL | 47.8 ± 0.2 |
The table summarizes the mean ± SEM of correct responses during the initial lever press shaping and subsequent acquisition of visual discrimination processes for each of the four experimental groups, and further outlines the criterion to be reached in each phase. Neither genetic Nurr1 deficiency nor prenatal poly(I:C) exposure affected training performance, so that the four groups were highly comparable in the measure of correct responses during shaping and acquisition phases of the training phase. N(wt, SAL) = 7, N(wt, POL) = 7, N(Nurr1+/−, SAL) = 6, and N(Nurr1+/−, POL) = 6. SAL, Saline; POL, poly(I:C); wt, wild type; Nurr1+/−, heterozygous Nurr1 ablation.
Independent effect of prenatal immune activation on spatial working memory deficits
We also evaluated whether prenatal immune challenge would differentially affect working memory in Nurr1-deficient and wt mice. Working memory is a form of short-term memory with a limited capacity that is used to hold relevant information active for ongoing behavior, including comprehension, reasoning, and problem solving (Baddeley, 2003). Deficits in working memory are common to several neuropsychiatric conditions with presumed dopaminergic pathology, including schizophrenia, ADHD, and major depression (Goldman-Rakic, 1994; Nuechterlein et al., 2009; Del Campo et al., 2011). Here, spatial working memory was studied using a matching-to-position paradigm in the dry maze as established in mice before (Bitanihirwe et al., 2010a; Llano Lopez et al., 2010). The critical measure of working memory in this task is the reduction in distance moved and time needed (latency) to find the location of the rewarded hole from trial 1 (when the location of the reward is essentially unknown to the subjects) to trial 2.
There was an overall change in test performance as a function of successive test days, as indicated by the presence of a significant main effect of days (distance moved: F(6,144) = 4.43, p < 0.001; latency: F(6,144) = 19.61, p < 0.001), with animals generally performing faster at the end of the 7 d test procedure compared with the first days of testing. Most importantly, we found that prenatal poly(I:C) exposure disrupted working memory regardless of the Nurr1 genotype (Fig. 5): Both wt and Nurr1+/− offspring prenatally treated with saline solution displayed a clear reduction in the distance moved and time needed to find the rewarded hole in trial 2 relative to trial 1. In contrast, poly(I:C)-exposed wt or Nurr1+/− offspring displayed no similar improvement from trial 1 to 2 (Fig. 5). ANOVA yielded a significant interaction between prenatal treatment and trials (distance moved: F(1,24) = 5.08, p < 0.05; latency: F(1,24) = 4.78, p < 0.05), and subsequent analyses restricted to the two prenatal treatment conditions confirmed a significant trial effect in prenatally saline-exposed offspring (distance moved: F(1,12) = 17.41, p < 0.01; latency: F(1,12) = 24.77, p < 0.001), but not in poly(I:C)-exposed offspring (values of F < 1). Prenatally saline- and poly(I:C)-treated animals also significantly differed with respect to performance in the second trial (significant main effect of prenatal treatment in trail 2 for distance: F(1,24) = 7.97, p < 0.01; and for latency: F(1,24) = 4.81, p < 0.05).
Figure 5.

Independent effect of prenatal immune activation on spatial working memory deficits. Pregnant wt and Nurr1+/− mice were treated with the viral mimetic poly(I:C) (POL) (2 mg/kg, i.v.) or vehicle [saline (SAL)] on gestation day 17, and the resulting offspring were tested in early adulthood. A, Spatial working memory was assessed using a dry maze containing 32 holes arranged in a radial design as schematically illustrated. The working memory task was based on the matching-to-position paradigm, in which the animals were required to learn the novel position of a rewarded hole revealed to them on trial 1 of each day to navigate effectively to the same location (i.e., matching) on the subsequent trial on the same day. B, The line plots show the distance moved and latency to find the rewarded hole in trial 2 relative to trial 1. **p < 0.01 (distance) and ***p < 0.001 (latency), reflecting the significant main effects of trials in offspring subjected to prenatal SAL treatment; §p < 0.01 and #p < 0.05, reflecting the significant difference between prenatally SAL- and POL-treated offspring in trial 2 with respect to distance moved and latency, respectively. N = 7 in each experimental group; all values are means ± SEM. C, Computer-generated search path of representative SAL- or POL-exposed wt and Nurr1+/− offspring in trial 1 (T1) and trial 2 (T2) of the working memory test. S, Starting position; R, location of the rewarded hole. Note that there was a delay of ∼1 s between placing the animal in the center of the maze and starting the video tracking system, so that the seemingly discrepant starting positions are attributable to this 1 s delay, during which some animals already moved away from the center.
Independent effects of prenatal immune activation and Nurr1 deficiency on midbrain dopamine cells
Subsequent to the behavioral and cognitive phenotyping, we attempted to explore the single and combined effects of Nurr1 deficiency and prenatal immune challenge on long-term changes at the neuroanatomical level. Previous studies have shown that both the genetic (i.e., Nurr1 deficiency) and environmental (prenatal viral-like immune activation) manipulations can interfere with the normal development of the central dopamine system (Zetterström et al., 1997; Meyer et al., 2008a,c; Vuillermot et al., 2010, 2011b,c), indicating that at least parts of the detrimental neurodevelopmental effects of these manipulations may converge on the dopamine system. Here, we tested this hypothesis by analyzing the expression of various dopaminergic markers in the major dopaminergic pathways, namely the nigrostriatal, mesoaccumbal, and mesocortical pathways.
In a first step, we quantified the number of TH-positive dopamine neurons using stereological estimations on serial coronal brain sections in the two major midbrain areas containing dopamine cells, namely the VTA and SN. Consistent with previous investigations (Vuillermot et al., 2011c), we found that heterozygous constitutive deletion of Nurr1 significantly decreased TH-positive cells in the SN, and this effect emerged independently of the prenatal immune treatment history (main effect of genotype: F(1,24) = 9.97, p < 0.01; Fig. 6). However, prenatal treatment with poly(I:C) led to a small but significant reduction in the number of TH-positive dopamine cells in the VTA regardless of the genetic background (main effect of prenatal treatment: F(1,24) = 4.25, p < 0.05; Fig. 6). There were no significant interactions between the two manipulations on TH-positive cell numbers in the SN or VTA. Together, these findings highlight independent effects induced by Nurr1 deficiency and prenatal immune challenge on dopaminergic midbrain structures: The former markedly reduces SN dopamine cell numbers, whereas the latter leads to decreased TH-positive cells in the VTA (Fig. 6).
Figure 6.

Independent effects of prenatal immune activation and genetic Nurr1 deficiency on midbrain dopamine cell numbers. A, Schematic coronal brain sections delineating the ventral midbrain areas investigated with reference to bregma [adapted from The Mouse Brain in Stereotaxic Coordinates by Franklin and Paxinos (2008)]. Dopaminergic midbrain cells were quantified in sections ranging from bregma −2.92 to −3.64 mm. 1, VTA; 2a, SNc; 2b, SNr. B, Stereological estimates of TH-positive cells in the SN and VTA of adult wt and Nurr1+/− offspring subjected to prenatal poly(I:C) (POL) (2 mg/kg, i.v.) or vehicle [saline (SAL)] on gestation day 17. Stereological estimates in the SN took into account TH-positive cells in both the SNc and SNr. **p < 0.01 and §p < 0.05, signifying the main effects of genotype and prenatal treatment, respectively. N = 7 males in each experimental group; all values are means ± SEM. C, Coronal brain sections of representative SAL- or POL-exposed wt and Nurr1+/− offspring stained with anti-TH antibody. The sections highlight the SN and VTA regions as indicated by the dashed lines. Note the decrease of TH-positive cell bodies in the SN of SAL- or POL-exposed Nurr1+/− offspring (indicated by the arrowheads), and the reduction of TH-positive cells in the VTA of POL-treated wt or Nurr1+/− offspring (indicated by the white stars).
Independent and synergistic dopaminergic effects of Nurr1 deficiency and prenatal immune activation in striatal and prefrontal cortical regions
We were then interested in assessing the single and combined effects of Nurr1 deficiency and prenatal immune activation on the expression of dopaminergic markers in ventral (NAc core and shell) and dorsal (CPu) striatal regions. In mice, dopaminergic cell bodies located in the SN project primarily to dorsal parts of the striatum (CPu), forming the nigrostriatal dopaminergic pathway. However, ventral parts of the striatum (NAc shell and core) receive prominent projections from dopaminergic cell bodies of the VTA, forming parts of the mesolimbic dopamine system (Van den Heuvel and Pasterkamp, 2008).
Consistent with our previous findings (Vuillermot et al., 2011c), Nurr1+/− mice displayed a significant reduction in TH immunoreactivity specifically in the NAc shell but not other striatal areas (Fig. 7). This effect was similarly observed in saline- and poly(I:C)-exposed Nurr1+/− offspring, suggesting that prenatal immune activation did not significantly influence this aspect of accumbal dopamine pathology in Nurr1-deficient animals. ANOVA of this measure only yielded a significant main effect of genotype (F(1,24) = 17.76; p < 0.01). Most interestingly, however, we revealed a synergistic interaction between Nurr1 deficiency and prenatal immune activation in the disruption of D2R expression in the NAc core and shell subregions: As shown in Figure 7, a significant reduction of accumbal D2R immunoreactivity emerged selectively in poly(I:C)-exposed Nurr1+/− offspring. ANOVA yielded a significant interaction between prenatal treatment and genotype in the analysis of D2R density in the NAC core (F(1,24) = 4.03; p < 0.05) and NAc shell (F(1,24) = 12.75; p < 0.01), and subsequent post hoc group comparisons verified the significant difference between poly(I:C)-exposed Nurr1+/− offspring and all other groups (NAc core: all values of p < 0.05; NAc shell: all values of p < 0.01). The genetic and/or environmental manipulations did not significantly affect dorsal or ventral striatal expression of the two other markers investigated (i.e., COMT and D1R; Table 3).
Figure 7.
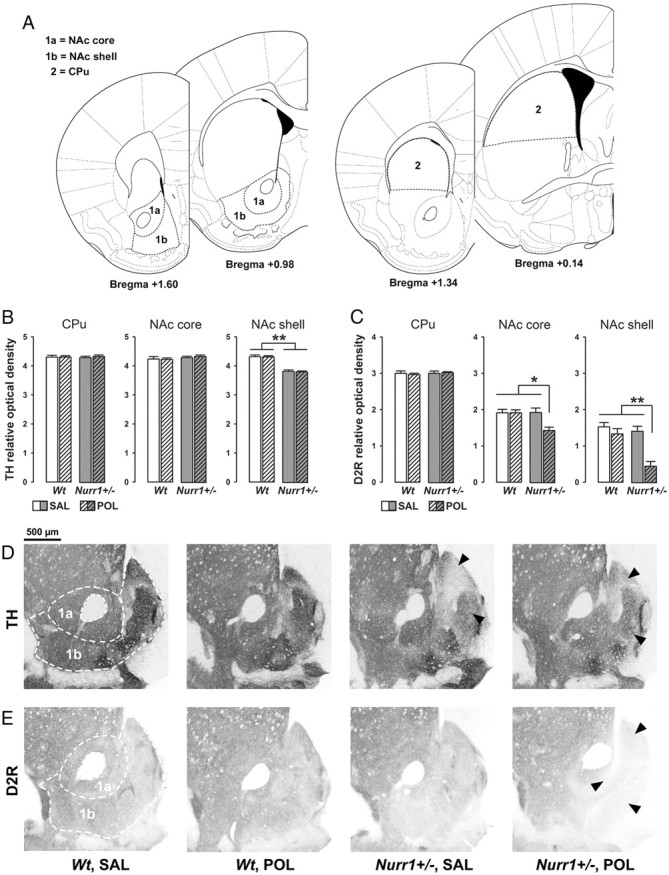
Independent and synergistic effects of prenatal immune activation and genetic Nurr1 deficiency on dopaminergic markers in the striatum. A, Schematic coronal brain sections delineating the striatal areas investigated with reference to bregma [adapted from The Mouse Brain in Stereotaxic Coordinates by Franklin and Paxinos (2008)]. Relative optical densities of dopaminergic markers in the NAc core (1a) and NAc shell (1b) were assessed in sections ranging from bregma +1.60 to +0.98 mm; relative optical densities of dopaminergic markers in the CPu (2) were assessed in sections ranging from bregma +1.34 to +0.14 mm. B, Mean + SEM relative optical density of TH in the CPu, NAc core and NAc shell of adult wt and Nurr1+/− offspring subjected to prenatal poly(I:C) (POL) (2 mg/kg, i.v.) or vehicle [saline (SAL)] on gestation day 17. **p < 0.01, signifying the main effect of genotype. N = 7 males in each experimental group. C, Mean + SEM relative optical density of D2R in the CPu, NAc core, and NAc shell of SAL- or POL-exposed wt and Nurr1+/− offspring. *p < 0.05 and **p < 0.01, reflecting the significant difference between POL-exposed Nurr1+/− offspring and all other groups. N = 7 males in each experimental group. D, Coronal brain sections of representative SAL- or POL-exposed wt and Nurr1+/− offspring stained with anti-TH antibody. The sections were taken at the level of the ventral striatum highlighting NAc core (1a) and NAc shell (1b) as indicated by the dashed lines. Note the reduction (indicated by the arrowheads) of TH immunoreactivity specifically in the NAc shell region of SAL- or POL-exposed Nurr1+/− offspring. E, Coronal brain sections of representative SAL- or POL-exposed wt and Nurr1+/− offspring stained with anti-D2R antibody. The sections were taken at the level of the ventral striatum highlighting NAc core (1a) and NAc shell (1b) as indicated by the dashed lines. Note the decrease (indicated by the arrowheads) of D2R immunoreactivity in the NAc core and shell regions emerging selectively in POL-exposed Nurr1+/− offspring.
Table 3.
Summary of the relative optical densities of COMT and D1R in the dorsal and ventral striatum
| Marker | Area | wt, SAL | wt, POL | Nurr1+/−, SAL | Nurr1+/−, POL |
|---|---|---|---|---|---|
| COMT | CPu | 2.46 ± 0.36 | 2.35 ± 0.27 | 2.32 ± 0.18 | 2.41 ± 0.32 |
| NAc core | 2.29 ± 0.21 | 2.07 ± 0.27 | 2.11 ± 0.16 | 2.23 ± 0.26 | |
| NAc shell | 2.06 ± 0.20 | 1.98 ± 0.31 | 2.01 ± 0.21 | 2.10 ± 0.34 | |
| D1R | CPu | 3.86 ± 0.10 | 3.79 ± 0.15 | 3.82 ± 0.18 | 3.78 ± 0.22 |
| NAc core | 3.34 ± 0.14 | 3.28 ± 0.23 | 3.31 ± 0.11 | 3.36 ± 0.20 | |
| NAc shell | 3.08 ± 0.17 | 2.97 ± 0.20 | 3.01 ± 0.18 | 3.10 ± 0.33 |
The table summarizes the ln-transformed measures obtained in the CPu, NAc core, and NAc shell subregions of the striatum for wt and Nurr1+/− offspring subjected to prenatal saline (SAL) or poly(I:C) (POL) treatment. All values are means ± SEM; N = 7 in each experimental group and for each measure.
Finally, we also analyzed the expression of TH, COMT, D1R, and D2R in the mPFC, which receives dense dopaminergic projections from the ventral midbrain (Van den Heuvel and Pasterkamp, 2008). Our analyses of these dopaminergic markers in the mPFC revealed a remarkable synergism between Nurr1 deficiency and prenatal immune activation in the measures of relative TH and COMT densities: The combined effects of Nurr1 ablation and prenatal poly(I:C) treatment resulted in a significant decrease and increase in mPFC TH and COMT density, respectively, compared with levels measured in all other groups (Fig. 8). ANOVA yielded a significant interaction between prenatal treatment and genotype for both markers (TH: F(1,24) = 4.30, p < 0.05; COMT: F(1,24) = 4.51, p < 0.05). Additional post hoc group comparisons confirmed the significant difference between poly(I:C)-exposed Nurr1+/− offspring and all other groups with regards to TH relative density in the mPFC (all values of p < 0.01), and between saline- and poly(I:C)-exposed Nurr1+/− offspring (p < 0.05) in the measure of COMT density. D1R and D2R IRs in the mPFC were generally low and largely indistinguishable from background staining. They were therefore not subjected to quantification and statistical analyses (data not shown).
Figure 8.
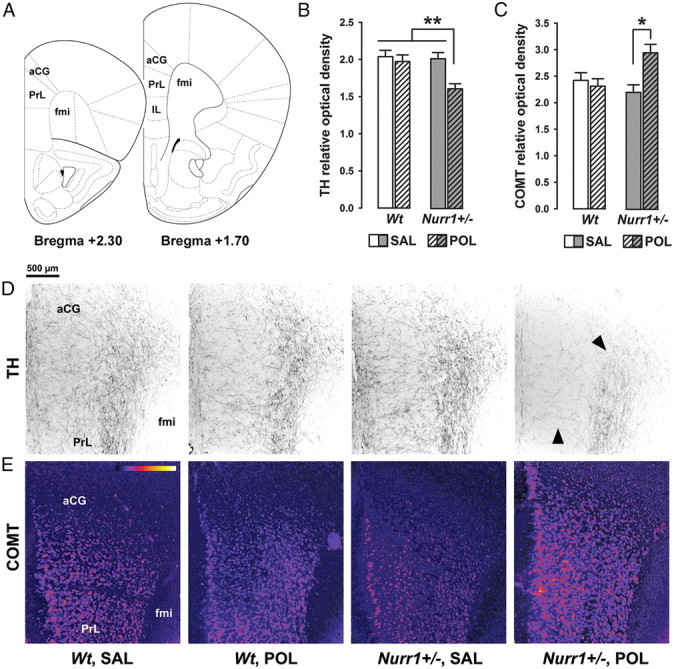
Synergistic effects of genetic Nurr1 deficiency and prenatal immune activation on the prefrontal cortical dopamine system. A, Schematic coronal brain sections delineating the prefrontal cortical areas investigated with reference to bregma [adapted from The Mouse Brain in Stereotaxic Coordinates by Franklin and Paxinos (2008)]. Assessment of the relative optical densities of dopaminergic markers in the mPFC included measurements in the aCG, PrL, and IL cortices and was performed on coronal sections ranging from bregma +2.30 to +1.70 mm. B, Mean + SEM relative optical density of TH in the mPFC of adult wt and Nurr1+/− offspring prenatally treated with poly(I:C) (POL) (2 mg/kg, i.v.) or vehicle [saline (SAL)] solution on gestation day 17. **p < 0.01; N = 7 males in each experimental group. C, Mean + SEM relative optical density of COMT in the mPFC of SAL- or POL-exposed wt and Nurr1+/− offspring. *p < 0.05; N = 7 males in each experimental group. D, Coronal brain sections of representative SAL- or POL-exposed wt and Nurr1+/− offspring stained with anti-TH antibody. The sections were taken at the level of the mPFC highlighting TH-positive fibers in the aCG and PrL cortices. Note the marked decrease (indicated by the arrowheads) of TH-positive fibers emerging selectively in POL-exposed Nurr1+/− offspring. E, Coronal brain sections of representative SAL- or POL-exposed wt and Nurr1+/− offspring stained with anti-COMT antibody. The sections were taken at the level of the mPFC highlighting COMT protein expression in the aCG and PrL cortices. The sections were color-coded for the purpose of facilitating the visualization of differential COMT protein expression; strongest staining intensities are shown in yellow, while the background is represented in dark purple (bar inset). fmi, Forceps minor corpus callosum.
Discussion
There has been increasing interested in and evidence for an important contribution of prenatal exposure to infection and/or inflammation in the etiology of developmental brain disorders (Brown and Derkits, 2010; Meyer et al., 2011; Patterson, 2011). Human epidemiological studies have further indicated that the relative effects of such prenatal adversities may be magnified by additional genetic and/or environmental risk factors (van Os et al., 2008; Clarke et al., 2009; Selten et al., 2010). The findings from the present gene–environment interaction model are consistent with these interpretations and provide a novel set of data showing that the brain and behavioral consequences of prenatal viral-like immune activation are exacerbated in offspring with genetic predisposition to dopaminergic abnormalities induced by heterozygous Nurr1 ablation. Not only did we find additive effects of Nurr1 deficiency and prenatal immune activation (as for example seen in the induction of spontaneous locomotor hyperactivity), but even more strikingly, we found numerous synergisms between the two factors at both the cognitive and neuroanatomical levels.
A major finding of the present study was the identification of synergistic effects between genetic Nurr1 deficiency and prenatal immune activation in the development of attentional impairments. First, we found that the combination of the two factors led to abnormally enhanced LI, which is also referred to as LI persistence (Weiner, 2003). LI persistence reflects retarded switching between associations and can be seen as a failure to re-deploy attention when previously irrelevant stimuli become relevant (Weiner, 2003; Weiner and Arad, 2009). Abnormally enhanced LI can therefore be taken to index impaired attentional shifting (Weiner, 2003; Weiner and Arad, 2009). Here, we went on to show that the combination of genetic Nurr1 deficiency and prenatal immune activation also caused impaired performance in the 2-CVDT, a translational paradigm that allows the assessment of sustained attention in mice (Robbins, 2002; Llano Lopez et al., 2010). The presence of impaired sustained attention has been taken as index of a failure to continuously allocate processing resources for the detection of rare events for prolonged periods of time (Robbins, 2002). Impairments in attentional shifting and sustained attention are core dysfunctions in several dopamine-associated neuropsychiatric disorders, especially schizophrenia (Floresco and Magyar, 2006; Nuechterlein et al., 2009) and ADHD (Robbins, 2002; Del Campo et al., 2011).
Imaging studies in humans and experimental work in animals have repeatedly associated abnormal dopaminergic mechanisms especially within mesocortical and frontostriatal pathways in the precipitation of impairments in attentional shifting and sustained attention (Floresco and Magyar, 2006; Kehagia et al., 2010; Cools and D'Esposito, 2011; Del Campo et al., 2011), and dopaminergic imbalances in these pathways have also long been recognized to contribute to the neuropathology of both schizophrenia (Knable and Weinberger, 1997; Howes et al., 2007) and ADHD (Del Campo et al., 2011). Here, we identified a number of neuropathological deficits that are suggestive of dopaminergic imbalances in the mesocortical and frontostriatal pathways. Although our study does not causally link distinct neuroanatomical changes with specific neurocognitive deficits, we consider it important to draw attention to some potential structural–functional relationships. In view of the known contribution of (dopaminergic) prefrontal deficits in the precipitation of attentional dysfunctions (Robbins, 2002; Kehagia et al., 2010; Cools and D'Esposito, 2011), one clear possibility would be that the neurocognitive deficits present in prenatally immune-challenged Nurr1-deficient mice may be linked to the apparent prefrontal dopaminergic abnormalities. In the context of schizophrenia, it is interesting to note that enhanced COMT activity in homozygote valine–valine (Val–Val) allele carriers is associated with impaired neurocognitive functions, including deficient executive functioning, processing speed, and sustained attention (Diaz-Asper et al., 2006; Tan et al., 2009). However, it needs to be emphasized that most studies implicating COMT in schizophrenia have focused on genetic COMT variation rather than actual mRNA or protein expression (Weinberger et al., 2001; Chen et al., 2004). One could therefore argue that our findings of enhanced COMT protein expression in poly(I:C)-exposed offspring with additional Nurr1 ablation cannot be directly compared with the clinical condition, in which enhanced COMT activity resulting from homozygosity in the valine–valine (Val–Val) allele has been correlated with schizophrenia-relevant brain and behavioral abnormalities (Weinberger et al., 2001). Even so, functional analyses of genetic COMT variation has confirmed that the Val–Val allele is linked to significant increases in prefrontal COMT protein and mRNA levels compared with Val–Met (methionine) or Met–Met carriers (Chen et al., 2004). Against this background, our findings highlight that schizophrenia-relevant alterations in prefrontal COMT protein expression can readily be induced by specific gene–environment interactions that by themselves are unrelated to genetic COMT variation.
The neuronal substrates for deficient sustained attention in prenatally immune-challenged Nurr1-deficient offspring may further include downregulation of D2R in the ventral striatum, based on previous observations that intra-NAc infusion of the D2R antagonist sulpiride impairs response accuracy in the rat five-choice serial reaction time test (Pezze et al., 2007). With respect to LI persistence, it has been shown that D2R antagonists are efficient in increasing the LI effect under parametric conditions, in which control rats do not show LI (Weiner et al., 1997). It thus seems feasible that reduced D2R-mediated signaling may further provide a relevant neurochemical substrate contributing to the development of abnormally enhanced LI in poly(I:C)-exposed Nurr1+/− offspring.
In addition to the relevance of increased prefrontal COMT density/activity to schizophrenia (Weinberger et al., 2001; Chen et al., 2004; Tan et al., 2009), our findings of reduced TH fiber density in the mPFC of poly(I:C)-exposed Nurr1+/− offspring match postmortem investigations in schizophrenic patients showing reduced densities of TH-immunoreactive axons in specific areas of the PFC (Akil et al., 1999). However, our gene–environment model falls short in mimicking postmortem and imaging findings implicating a mild but significant elevation of D2R densities in striatal structures of schizophrenic patients (Laruelle, 1998; Howes et al., 2007). Furthermore, our study does not reveal a direct relationship between prefrontal dopamine pathology (in the form of reduced and increased TH and COMT density, respectively) and disruption of working memory, the latter of which is presumably dependent on prefrontal dopamine activity (Floresco and Magyar, 2006). Indeed, whereas the prefrontal dopamine makers of interest were found to be reduced specifically in Nurr1+/− animals with additional prenatal poly(I:C) treatment, working memory was impaired by prenatal immune activation independently of the genetic background. This suggest that alterations in prefrontal TH and COMT protein densities are unlikely to be the prime mechanisms for abnormal working memory performance in our gene–environment model, so that imbalances in other neurochemical systems and/or brain areas such as the hippocampus need to be considered in this association.
In addition to its impact on normal dopaminergic development and functions (Zetterström et al., 1997; Saucedo-Cardenas et al., 1998; Kadkhodaei et al., 2009), Nurr1 has immunomodulatory roles by functioning as transcriptional repressor of proinflammatory genes (Saijo et al., 2009). Consistent with this, we found that pregnant Nurr1+/− mice displayed enhanced basal levels of the proinflammatory cytokine IL-6. However, poly(I:C) treatment in Nurr1+/− mice did not further elevate any of the proinflammatory cytokines of interest, but instead significantly reduced serum IL-6 levels to control levels measured in saline-treated wt mice. It is therefore unlikely that the development of additive and synergistic pathological long-term effects between prenatal immune activation and genetic Nurr1 deficiency simply results from exacerbated proinflammatory immune responses in Nurr1-deficient mice. Furthermore, the synergistic brain and behavioral effects between prenatal immune activation and heterozygous Nurr1 ablation cannot simply be explained by the recent hypothesis that IL-6 is most critical in altering fetal brain development and in causing long-term brain abnormalities following prenatal infection (Smith et al., 2007). If this were the case, one would have expected the IL-6 response to poly(I:C) in Nurr1+/− mice to be more pronounced compared with wt mice. Notably, this was not the case. Interestingly, however, we found a synergistic effect of maternal poly(I:C) treatment and Nurr1 deficiency in terms of shifting the relative balance between IL-6/IL-10 expression toward enhanced IL-10 signaling. Consistent with recent theoretical accounts emphasizing the importance of relative rather than absolute imbalances in maternal/fetal cytokine imbalances (Meyer et al., 2008b), our cytokine data suggest that relative shifts toward enhanced antiinflammatory cytokine signaling may be more directly relevant to the development of synergistic pathological interactions between genetic Nurr1 deficiency and prenatal immune activation.
In conclusion, the present experimental data implicate specific gene–environment interactions in the emergence of enduring attentional impairments pertinent to dopamine-associated neuropsychiatric disorders. Prenatal maternal infection and genetically driven Nurr1 deficiency have each been implicated as risk factors for neurodevelopmental brain disorders, including schizophrenia (Buervenich et al., 2000; Brown and Derkits, 2010) and ADHD (Smith et al., 2005; Pineda et al., 2007), but the relative effect size of each individual factor appears relatively small. Our experimental approach here highlights that the combination of these two risk factors can induce far more pronounced dopaminergic pathology and neurocognitive deficits pertinent to these disorders than does one factor alone, and therefore it emphasizes the importance of interacting effects between these two factors in the disruption of such brain dysfunctions. Against this background, the present model system may be helpful for future attempts to further explore how specific gene–environment interactions can shape the vulnerability for distinct neuronal and neurocognitive abnormalities pertinent to multifactorial and dopamine-associated brain disorders of developmental origin.
Footnotes
This work was supported by Swiss National Science Foundation Grants 3100AO-100309 and 3100AO-116719, and the Swedish Strategic Research Foundation for Predictive Models and Biomarkers. J.F. received additional support from a 2009 National Alliance for Research on Schizophrenia and Depression Distinguished Investigator Award, and U.M. received additional financial support from the European Union Seventh Framework Programme (FP7/2007–2011) under Grant Agreement 259679. We are extremely grateful to Liz Weber for her excellent technical assistance in the immunohistochemical studies. We also remain indebted to the laboratory animal technician team, Schwerzenbach, for animal husbandry and care.
The authors declare no competing financial interests.
References
- Abazyan B, Nomura J, Kannan G, Ishizuka K, Tamashiro KL, Nucifora F, Pogorelov V, Ladenheim B, Yang C, Krasnova IN, Cadet JL, Pardo C, Mori S, Kamiya A, Vogel MW, Sawa A, Ross CA, Pletnikov MV. Prenatal interaction of mutant DISC1 and immune activation produces adult psychopathology. Biol Psychiatry. 2010;68:1172–1181. doi: 10.1016/j.biopsych.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar-Valles A, Flores C, Luheshi GN. Prenatal inflammation-induced hypoferremia alters dopamine function in the adult offspring in rat: relevance for schizophrenia. PLoS One. 2010;5:e10967. doi: 10.1371/journal.pone.0010967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akil M, Pierri JN, Whitehead RE, Edgar CL, Mohila C, Sampson AR, Lewis DA. Lamina-specific alterations in the dopamine innervation of the prefrontal cortex in schizophrenic subjects. Am J Psychiatry. 1999;156:1580–1589. doi: 10.1176/ajp.156.10.1580. [DOI] [PubMed] [Google Scholar]
- Ayhan Y, Sawa A, Ross CA, Pletnikov MV. Animal models of gene-environment interactions in schizophrenia. Behav Brain Res. 2009;204:274–281. doi: 10.1016/j.bbr.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baddeley A. Working memory: looking back and looking forward. Nat Rev Neurosci. 2003;4:829–839. doi: 10.1038/nrn1201. [DOI] [PubMed] [Google Scholar]
- Bitanihirwe BK, Weber L, Feldon J, Meyer U. Cognitive impairment following prenatal immune challenge in mice correlates with prefrontal cortical AKT1 deficiency. Int J Neuropsychopharmacol. 2010a;13:981–996. doi: 10.1017/S1461145710000192. [DOI] [PubMed] [Google Scholar]
- Bitanihirwe BK, Peleg-Raibstein D, Mouttet F, Feldon J, Meyer U. Late prenatal immune activation in mice leads to behavioral and neurochemical abnormalities relevant to the negative symptoms of schizophrenia. Neuropsychopharmacology. 2010b;35:2462–2478. doi: 10.1038/npp.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitanihirwe BK, Dubroqua S, Singer P, Feldon J, Yee BK. Sensorimotor gating and vigilance-dependent choice accuracy: a within-subject correlative analysis in wild-type C57BL/6 mice. Behav Brain Res. 2011;217:178–187. doi: 10.1016/j.bbr.2010.10.021. [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology. 2001;156:234–258. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry. 2010;167:261–280. doi: 10.1176/appi.ajp.2009.09030361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buervenich S, Carmine A, Arvidsson M, Xiang F, Zhang Z, Sydow O, Jönsson EG, Sedvall GC, Leonard S, Ross RG, Freedman R, Chowdari KV, Nimgaonkar VL, Perlmann T, Anvret M, Olson L. Nurr1 mutations in cases of schizophrenia and manic-depressive disorder. Am J Med Genet. 2000;96:808–813. doi: 10.1002/1096-8628(20001204)96:6<808::aid-ajmg23>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Chen J, Lipska BK, Halim N, Ma QD, Matsumoto M, Melhem S, Kolachana BS, Hyde TM, Herman MM, Apud J, Egan MF, Kleinman JE, Weinberger DR. Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. Am J Hum Genet. 2004;75:807–821. doi: 10.1086/425589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke MC, Tanskanen A, Huttunen M, Whittaker JC, Cannon M. Evidence for an interaction between familial liability and prenatal exposure to infection in the causation of schizophrenia. Am J Psychiatry. 2009;166:1025–1030. doi: 10.1176/appi.ajp.2009.08010031. [DOI] [PubMed] [Google Scholar]
- Cools R, D'Esposito M. Inverted-U-shaped dopamine actions on human working memory and cognitive control. Biol Psychiatry. 2011;69:e113–125. doi: 10.1016/j.biopsych.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle P, Tran N, Fung JN, Summers BL, Rofe AM. Maternal dietary zinc supplementation prevents aberrant behaviour in an object recognition task in mice offspring exposed to LPS in early pregnancy. Behav Brain Res. 2009;197:210–218. doi: 10.1016/j.bbr.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Del Campo N, Chamberlain SR, Sahakian BJ, Robbins TW. The roles of dopamine and noradrenaline in the pathophysiology and treatment of attention-deficit/hyperactivity disorder. Biol Psychiatry. 2011;69:e145–e157. doi: 10.1016/j.biopsych.2011.02.036. [DOI] [PubMed] [Google Scholar]
- Diaz-Asper CM, Weinberger DR, Goldberg TE. Catechol-O-methyltransferase polymorphisms and some implications for cognitive therapeutics. NeuroRx. 2006;3:97–105. doi: 10.1016/j.nurx.2005.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco SB, Magyar O. Mesocortical dopamine modulation of executive functions: beyond working memory. Psychopharmacology. 2006;188:567–585. doi: 10.1007/s00213-006-0404-5. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Amsterdam: Elsevier Academic; 2008. [Google Scholar]
- Gilmore JH, Jarskog LF. Exposure to infection and brain development: cytokines in the pathogenesis of schizophrenia. Schizophr Res. 1997;24:365–367. doi: 10.1016/s0920-9964(96)00123-5. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. Working memory dysfunction in schizophrenia. J Neuropsychiatry Clin Neurosci. 1994;6:348–357. doi: 10.1176/jnp.6.4.348. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Bagger P, Bendtsen TF, Evans SM, Korbo L, Marcussen N, Møller A, Nielsen K, Nyengaard JR, Pakkenberg B, Sørensen FB, Vesterby A, West MJ. The new stereological tools: disector, fractionator, nucleator and point sampled intercepts and their use in pathological research and diagnosis. APMIS. 1988;96:857–881. doi: 10.1111/j.1699-0463.1988.tb00954.x. [DOI] [PubMed] [Google Scholar]
- Howard CV, Reed MG. Unbiased stereology. New York: Garland Science/BIOS Scientific; 2005. [Google Scholar]
- Howes OD, Montgomery AJ, Asselin MC, Murray RM, Grasby PM, McGuire PK. Molecular imaging studies of the striatal dopaminergic system in psychosis and predictions for the prodromal phase of psychosis. Br J Psychiatry. 2007;51:13–18. doi: 10.1192/bjp.191.51.s13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovic J, Chen S, Le WD. The role of Nurr1 in the development of dopaminergic neurons and Parkinson's disease. Prog Neurobiol. 2005;77:128–138. doi: 10.1016/j.pneurobio.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Jiang C, Wan X, He Y, Pan T, Jankovic J, Le W. Age-dependent dopaminergic dysfunction in Nurr1 knockout mice. Exp Neurol. 2005;191:154–162. doi: 10.1016/j.expneurol.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Kadkhodaei B, Ito T, Joodmardi E, Mattsson B, Rouillard C, Carta M, Muramatsu S, Sumi-Ichinose C, Nomura T, Metzger D, Chambon P, Lindqvist E, Larsson NG, Olson L, Björklund A, Ichinose H, Perlmann T. Nurr1 is required for maintenance of maturing and adult midbrain dopamine neurons. J Neurosci. 2009;29:15923–15932. doi: 10.1523/JNEUROSCI.3910-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehagia AA, Murray GK, Robbins TW. Learning and cognitive flexibility: frontostriatal function and monoaminergic modulation. Curr Opin Neurobiol. 2010;20:199–204. doi: 10.1016/j.conb.2010.01.007. [DOI] [PubMed] [Google Scholar]
- Knable MB, Weinberger DR. Dopamine, the prefrontal cortex and schizophrenia. J Psychopharmacol. 1997;11:123–131. doi: 10.1177/026988119701100205. [DOI] [PubMed] [Google Scholar]
- Lanté F, Meunier J, Guiramand J, De Jesus Ferreira MC, Cambonie G, Aimar R, Cohen-Solal C, Maurice T, Vignes M, Barbanel G. Late N-acetylcysteine treatment prevents the deficits induced in the offspring of dams exposed to an immune stress during gestation. Hippocampus. 2008;18:602–609. doi: 10.1002/hipo.20421. [DOI] [PubMed] [Google Scholar]
- Laruelle M. Imaging dopamine transmission in schizophrenia. A review and meta-analysis. Q J Nucl Med. 1998;42:211–221. [PubMed] [Google Scholar]
- Llano Lopez L, Hauser J, Feldon J, Gargiulo PA, Yee BK. Evaluating spatial memory function in mice: a within-subjects comparison between the water maze test and its adaptation to dry land. Behav Brain Res. 2010;209:85–92. doi: 10.1016/j.bbr.2010.01.020. [DOI] [PubMed] [Google Scholar]
- Longman RE, Johnson TR. Viral respiratory disease in pregnancy. Curr Opin Obstet Gynecol. 2007;19:120–125. doi: 10.1097/GCO.0b013e328028fdc7. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J. Prenatal exposure to infection: a primary mechanism for abnormal dopaminergic development in schizophrenia. Psychopharmacology. 2009;206:587–602. doi: 10.1007/s00213-009-1504-9. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Schedlowski M, Yee BK. Towards an immuno-precipitated neurodevelopmental animal model of schizophrenia. Neurosci Biobehav Rev. 2005;29:913–947. doi: 10.1016/j.neubiorev.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, Yee BK, Feldon J. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci. 2006;26:4752–4762. doi: 10.1523/JNEUROSCI.0099-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Schwendener S, Knuesel I, Yee BK, Feldon J. Relative prenatal and postnatal maternal contributions to schizophrenia-related neurochemical dysfunction after in utero immune challenge. Neuropsychopharmacology. 2008a;33:441–456. doi: 10.1038/sj.npp.1301413. [DOI] [PubMed] [Google Scholar]
- Meyer U, Murray PJ, Urwyler A, Yee BK, Schedlowski M, Feldon J. Adult behavioral and pharmacological dysfunctions following disruption of the fetal brain balance between pro-inflammatory and IL-10-mediated anti-inflammatory signaling. Mol Psychiatry. 2008b;13:208–221. doi: 10.1038/sj.mp.4002042. [DOI] [PubMed] [Google Scholar]
- Meyer U, Engler A, Weber L, Schedlowski M, Feldon J. Preliminary evidence for a modulation of fetal dopaminergic development by maternal immune activation during pregnancy. Neuroscience. 2008c;154:701–709. doi: 10.1016/j.neuroscience.2008.04.031. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Fatemi SH. In-vivo rodent models for the experimental investigation of prenatal immune activation effects in neurodevelopmental brain disorders. Neurosci Biobehav Rev. 2009;33:1061–1079. doi: 10.1016/j.neubiorev.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Dammann O. Schizophrenia and autism: both shared and disorder-specific pathogenesis via perinatal inflammation? Pediatr Res. 2011;69:26R–33R. doi: 10.1203/PDR.0b013e318212c196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuechterlein KH, Luck SJ, Lustig C, Sarter M. CNTRICS final task selection: control of attention. Schizophr Bull. 2009;35:182–196. doi: 10.1093/schbul/sbn158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa K, Hashimoto K, Kishimoto T, Shimizu E, Ishikura H, Iyo M. Immune activation during pregnancy in mice leads to dopaminergic hyperfunction and cognitive impairment in the offspring: a neurodevelopmental animal model of schizophrenia. Biol Psychiatry. 2006;59:546–554. doi: 10.1016/j.biopsych.2005.07.031. [DOI] [PubMed] [Google Scholar]
- Patterson PH. Maternal infection and immune involvement in autism. Trends Mol Med. 2011;17:389–394. doi: 10.1016/j.molmed.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezze MA, Dalley JW, Robbins TW. Differential roles of dopamine D1 and D2 receptors in the nucleus accumbens in attentional performance on the five-choice serial reaction time task. Neuropsychopharmacology. 2007;32:273–283. doi: 10.1038/sj.npp.1301073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda DA, Palacio LG, Puerta IC, Merchán V, Arango CP, Galvis AY, Gómez M, Aguirre DC, Lopera F, Arcos-Burgos M. Environmental influences that affect attention deficit/hyperactivity disorder: study of a genetic isolate. Eur Child Adolesc Psychiatry. 2007;16:337–346. doi: 10.1007/s00787-007-0605-4. [DOI] [PubMed] [Google Scholar]
- Robbins TW. The 5-choice serial reaction time task: behavioural pharmacology and functional neurochemistry. Psychopharmacology. 2002;163:362–380. doi: 10.1007/s00213-002-1154-7. [DOI] [PubMed] [Google Scholar]
- Rojas P, Joodmardi E, Hong Y, Perlmann T, Ogren SO. Adult mice with reduced Nurr1 expression: an animal model for schizophrenia. Mol Psychiatry. 2007;12:756–766. doi: 10.1038/sj.mp.4001993. [DOI] [PubMed] [Google Scholar]
- Romero E, Guaza C, Castellano B, Borrell J. Ontogeny of sensorimotor gating and immune impairment induced by prenatal immune challenge in rats: implications for the etiopathology of schizophrenia. Mol Psychiatry. 2010;15:372–383. doi: 10.1038/mp.2008.44. [DOI] [PubMed] [Google Scholar]
- Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, Gage FH, Glass CK. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137:47–59. doi: 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saucedo-Cardenas O, Quintana-Hau JD, Le WD, Smidt MP, Cox JJ, De Mayo F, Burbach JP, Conneely OM. Nurr1 is essential for the induction of the dopaminergic phenotype and the survival of ventral mesencephalic late dopaminergic precursor neurons. Proc Natl Acad Sci U S A. 1998;95:4013–4018. doi: 10.1073/pnas.95.7.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selten JP, Frissen A, Lensvelt-Mulders G, Morgan VA. Schizophrenia and 1957 pandemic of influenza: meta-analysis. Schizophr Bull. 2010;36:219–228. doi: 10.1093/schbul/sbp147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smidt MP, Burbach JP. How to make a mesodiencephalic dopaminergic neuron. Nat Rev Neurosci. 2007;8:21–32. doi: 10.1038/nrn2039. [DOI] [PubMed] [Google Scholar]
- Smith KM, Bauer L, Fischer M, Barkley R, Navia BA. Identification and characterization of human NR4A2 polymorphisms in attention deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2005;133B:57–63. doi: 10.1002/ajmg.b.30127. [DOI] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Braff DL, Geyer MA. Animal models of deficient sensorimotor gating: what we know, what we think we know, and what we hope to know soon. Behav Pharmacol. 2000;11:185–204. doi: 10.1097/00008877-200006000-00002. [DOI] [PubMed] [Google Scholar]
- Tan HY, Callicott JH, Weinberger DR. Prefrontal cognitive systems in schizophrenia: towards human genetic brain mechanisms. Cogn Neuropsychiatry. 2009;14:277–298. doi: 10.1080/13546800903091665. [DOI] [PubMed] [Google Scholar]
- Van den Heuvel DM, Pasterkamp RJ. Getting connected in the dopamine system. Prog Neurobiol. 2008;85:75–93. doi: 10.1016/j.pneurobio.2008.01.003. [DOI] [PubMed] [Google Scholar]
- van Os J, Rutten BP, Poulton R. Gene–environment interactions in schizophrenia: review of epidemiological findings and future directions. Schizophr Bull. 2008;34:1066–1082. doi: 10.1093/schbul/sbn117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillermot S, Weber L, Feldon J, Meyer U. A longitudinal examination of the neurodevelopmental impact of prenatal immune activation in mice reveals primary defects in dopaminergic development relevant to schizophrenia. J Neurosci. 2010;30:1270–1287. doi: 10.1523/JNEUROSCI.5408-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillermot S, Joodmardi E, Perlmann T, Ove Ögren S, Feldon J, Meyer U. Schizophrenia-relevant behaviors in a genetic mouse model of constitutive Nurr1 deficiency. Genes Brain Behav. 2011a;10:589–603. doi: 10.1111/j.1601-183X.2011.00698.x. [DOI] [PubMed] [Google Scholar]
- Vuillermot S, Feldon J, Meyer U. Nurr1 is not essential for the development of prepulse inhibition deficits induced by prenatal immune activation. Brain Behav Immun. 2011b;25:1316–1321. doi: 10.1016/j.bbi.2011.06.012. [DOI] [PubMed] [Google Scholar]
- Vuillermot S, Feldon J, Meyer U. Relationship between sensorimotor gating deficits and dopaminergic neuroanatomy in Nurr1-deficient mice. Exp Neurol. 2011c;232:22–32. doi: 10.1016/j.expneurol.2011.07.008. [DOI] [PubMed] [Google Scholar]
- Weinberger DR, Egan MF, Bertolino A, Callicott JH, Mattay VS, Lipska BK, Berman KF, Goldberg TE. Prefrontal neurons and the genetics of schizophrenia. Biol Psychiatry. 2001;50:825–844. doi: 10.1016/s0006-3223(01)01252-5. [DOI] [PubMed] [Google Scholar]
- Weiner I. The “two-headed” latent inhibition model of schizophrenia: modeling positive and negative symptoms and their treatment. Psychopharmacology. 2003;169:257–297. doi: 10.1007/s00213-002-1313-x. [DOI] [PubMed] [Google Scholar]
- Weiner I, Arad M. Using the pharmacology of latent inhibition to model domains of pathology in schizophrenia and their treatment. Behav Brain Res. 2009;204:369–386. doi: 10.1016/j.bbr.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Weiner I, Shadach E, Barkai R, Feldon J. Haloperidol- and clozapine-induced enhancement of latent inhibition with extended conditioning: implications for the mechanism of action of neuroleptic drugs. Neuropsychopharmacology. 1997;16:42–50. doi: 10.1016/S0893-133X(96)00145-5. [DOI] [PubMed] [Google Scholar]
- Xing G, Zhang L, Russell S, Post R. Reduction of dopamine-related transcription factors Nurr1 and NGFI-B in the prefrontal cortex in schizophrenia and bipolar disorders. Schizophr Res. 2006;84:36–56. doi: 10.1016/j.schres.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Zetterström RH, Solomin L, Jansson L, Hoffer BJ, Olson L, Perlmann T. Dopamine neuron agenesis in Nurr1-deficient mice. Science. 1997;276:248–250. doi: 10.1126/science.276.5310.248. [DOI] [PubMed] [Google Scholar]
- Zuckerman L, Rehavi M, Nachman R, Weiner I. Immune activation during pregnancy in rats leads to a postpubertal emergence of disrupted latent inhibition, dopaminergic hyperfunction, and altered limbic morphology in the offspring: a novel neurodevelopmental model of schizophrenia. Neuropsychopharmacology. 2003;28:1778–1789. doi: 10.1038/sj.npp.1300248. [DOI] [PubMed] [Google Scholar]