

Abstract
Axonal outgrowth is a coordinated process of cytoskeletal dynamics and membrane trafficking; however, little is known about proteins responsible for regulating the membrane supply. LMTK1 (lemur kinase 1)/AATYK1 (apoptosis-associated tyrosine kinase 1) is a serine/threonine kinase that is highly expressed in neurons. We recently reported that LMTK1 plays a role in recycling endosomal trafficking in CHO-K1 cells. Here we explore the role of LMTK1 in axonal outgrowth and its regulation by Cdk5 using mouse brain cortical neurons. LMTK1 was expressed and was phosphorylated at Ser34, the Cdk5 phosphorylation site, at the time of axonal outgrowth in culture and colocalized with Rab11A, the small GTPase that regulates recycling endosome traffic, at the perinuclear region and in the axon. Overexpression of the unphosphorylated mutant LMTK1-S34A dramatically promoted axonal outgrowth in cultured neurons. Enhanced axonal outgrowth was diminished by the inactivation of Rab11A, placing LMTK1 upstream of Rab11A. Unexpectedly, the downregulation of LMTK1 by knockdown or gene targeting also significantly enhanced axonal elongation. Rab11A-positive vesicles were transported anterogradely more quickly in the axons of LMTK1-deficient neurons than in those of wild-type neurons. The enhanced axonal outgrowth was reversed by LMTK1-WT or the LMTK1-S34D mutant, which mimics the phosphorylated state, but not by LMTK1-S34A. Thus, LMTK1 can negatively control axonal outgrowth by regulating Rab11A activity in a Cdk5-dependent manner, and Cdk5–LMTK1–Rab11 is a novel signaling pathway involved in axonal outgrowth.
Introduction
Neurons are polarized cells with two functionally and structurally different processes, an axon and one or more dendrites. Axons and dendrites are formed sequentially after neurons differentiate. The formation of axons and dendrites can be reproduced in isolated cultured neurons. A single neurite among several unspecified neurites first elongates and differentiates into the axon, and the remaining neurites then become dendrites (Dotti et al., 1988). Axonal elongation includes the coordinated actions of cytoskeletal rearrangements and directed membrane delivery. Although the cytoskeletal components involved in axonal elongation have been studied extensively (Arimura and Kaibuchi, 2007; Polleux and Snider, 2010), little is known about the molecular mechanism of membrane supply to the axons (Sann et al., 2009; Bloom and Morgan, 2011).
The membrane components required for axonal outgrowth are provided by recycling endosomes in the cell soma (Ng and Tang, 2008). Recycling endosome trafficking is regulated by Rab11 small GTPase. Rab11 has been studied mostly in nonpolarized cultured cells (Ullrich et al., 1996; Sonnichsen et al., 2000), and only a few reports are available regarding Rab11 in neurons (Ng and Tang, 2008). For example, it has not been determined whether the GTP or the GDP form of Rab11 is involved in neurite outgrowth. We recently reported that LMTK1A (lemur kinase 1A)/AATYK1A (apoptosis-associated tyrosine kinase 1A) promotes the formation of Rab11A-positive recycling endosomes in CHO-K1 cells (Takano et al., 2010; Tsutsumi et al., 2010). LMTK1 is an uncharacterized serine/threonine (Ser/Thr) kinase that is highly expressed in the nervous system (Gaozza et al., 1997; Baker et al., 2001; Tomomura et al., 2007). We hypothesized that LMTK1 participates in axonal outgrowth by regulating Rab11 activity for recycling endosomes.
We also reported that the activity of LMTK1A is regulated by phosphorylation at Ser34 by Cdk5-p35 (Takano et al., 2010). Cdk5 is a member of the family of cyclin-dependent, proline-directed Ser/Thr kinases, which are activated predominantly in postmitotic neurons when bound to the regulatory subunit p35 or p39 (Dhavan and Tsai, 2001; Hisanaga and Endo, 2010). Cdk5 regulates neurite outgrowth (Nikolic et al., 1996; Paglini et al., 1998; Cheung et al., 2007) and phosphorylates cytoskeletal proteins, such as MAP1B and CRMP-2, that are involved in neurite outgrowth (Inagaki et al., 2001; Brown et al., 2004; Kawauchi et al., 2005; Uchida et al., 2005; Cole et al., 2008). Thus, Cdk5 may play a role in axonal outgrowth by phosphorylating cytoskeletal proteins. Cdk5 is, however, a membrane-associated kinase (Patrick et al., 1999; Asada et al., 2008). Cdk5-p35 is localized to recycling endosomes in neurons (Tsutsumi et al., 2010) and thus is likely to regulate axonal outgrowth by regulating membrane transport. However, a role for Cdk5 in trafficking of membrane components during neurite outgrowth has not yet been investigated.
Here we explored the function of LMTK1 in recycling endosomes during axon formation in primary cultured neurons. We show that LMTK1 is a novel regulator of axonal outgrowth, acting negatively through Rab11A, which is dependent on phosphorylation by Cdk5.
Materials and Methods
Antibodies.
Anti-p35 (C-19), anti-Cdk5 (DC17), anti-Trk (B-3), and monoclonal anti-myc (9E10) were purchased from Santa Cruz Biotechnology. Anti-Rab11 was from Invitrogen. Monoclonal anti-GFP was from Roche Diagnostics. Polyclonal rabbit anti-GFP was from MBL. Monoclonal anti-neuron-specific class III β-tubulin was from Techne. Anti-actin was from Sigma-Aldrich. Anti-mouse/rat β1-integrin (CD29) conjugated with Alexa Fluor 647 was from BioLegend. Monoclonal anti-T7 was from Merck Biosciences and Novagen. Anti-LMTK1 and anti-phospho-Ser34 of LMTK1 (pS34) have been described previously (Tsutsumi et al., 2008, 2010), and anti-neurofilament has been described previously (Komuta et al., 2007).
Plasmid construction.
Double-stranded oligonucleotides encoding siRNA sequences were cloned into a pcDNA6.2-GW/EmGFP-miR vector (Invitrogen). The siRNA sequences used were as follows: LMTK1-miRNA1, 5′-TGCTGATCAGATAGAGCCAGCTGCAGGTTTTGGCCACTGACTGACCTGCAGCTCTCTATCTGAT-3′; LMTK1-miRNA2, 5′-TGCTGTGCTTCTGCAGACTGCGCAAGGTTTTGGCCACTGACTGACCTTGCGCACTGCAGAAGCA-3′; LMTK1-mRNA3, 5′-TGCTGTGAAGCTGCTAACATAGCCAGGTTTTGGCCACTGACTGACCTGGCTATTAGCAGCTTCA-3′; LMTK1-miRNA4, 5′-TGCTGATCTGAGTCAGATATGTGGGTGTTTTGGCCACTGACTGACACCCACATCTGACTCAGAT-3′; the control scrambled (miRNA-SC) sequence, 5′-GAAATGTACTGCGCGTGGAGACGTTTTGGCCACTGACTGACGTCTCCACGCAGTACATTT-3′, contains the same percentage of C and G nucleotides but has no sequence homology. cDNA encoding a small hairpin RNA (shRNA) against Rab11A (target sequence: 5′-TCTGGAAAGCAAGAGTACC-3′) was inserted into pSilencer 2.1-U6 neo vector (Applied Biosystems). shRNA against p35 (target sequence; 5′-CAGCTACCAGAGCAACATCGC-3′) has been described previously (Endo et al., 2009). The LMTK1 alanine mutant and the phosphorylation-mimic aspartic acid mutant at Ser34 have been described (Takano et al., 2010; Tsutsumi et al., 2010). The LMTK1 kinase-deficient mutant (D206V) has been described (Tomomura et al., 2001). pEGFP-Rab5A, pEGFP-Rab7, pEGFP-Rab11A, and pEF-T7-FIP2C have been described (Fukuda, 2003; Fukuda et al., 2008).
Primary cortical neuron cultures and transfection.
ICR mice were obtained from Sankyo Laboratory Service. Mice were housed in a temperature-controlled room under a 12 h light/dark cycle with ad libitum access to food and water. All experiments were performed according to the guidelines for animal experimentation of Tokyo Metropolitan University (Tokyo, Japan). Brain cortices from embryonic day 17 or 18 mice were dissected and plated in polyethyleneimine-coated dishes or on polyethyleneimine-coated glass coverslips at a density of 5.2 × 105 neurons/cm2 (Saito et al., 2007). Plasmid vectors were introduced into neurons by electroporation with an Amaxa Nucleofector apparatus according to the manufacturer's instructions. Briefly, 1 × 107 neurons were resuspended in 100 μl of Nucleofector solution. After the addition of 4 μg of plasmid DNA, the neurons were subjected to electroporation using the mouse hippocampal neurons program (O-005) supplied with the Amaxa Nucleofector apparatus and were then cultured with 1 × 107 untransfected neurons.
Targeting construct and generation of LMTK1-KO mice.
Homologous recombination was used to replace the genomic region of LMTK1 that contains exons 4–7 and encodes the kinase domain with the neomycin resistance (Neo) cassette. LMTK1-targeted ES clones were injected into C57B6/J blastocysts. Male chimeras were mated with C57B6/J females to obtain heterozygous (LMTK1+/−) F1 offspring. We intercrossed the F1 progeny to produce homozygous LMTK1−/− mice. Genotyping was performed by PCR with specific primers to amplify either the mutant or the wild-type allele.
Immunofluorescence staining and statistical analysis.
Neurons were fixed with 4% paraformaldehyde in PBS for 20 min at room temperature and permeabilized in PBS containing 0.1% Triton X-100 and 5% normal goat serum for 30 min. The cells were probed with indicated primary antibodies followed by secondary antibodies conjugated to Alexa-488, Alexa-546, or Alexa-647 (Invitrogen). Fluorescence images were acquired with an LSM 5 EXCITER confocal microscope (Carl Zeiss). When the fluorescence intensity of EGFP-Rab11A was measured, Z-stack images of the entire axon thickness were acquired along a 20 μm length from the middle region of each axon. The EGFP intensity was estimated using Zen software (Zeiss) and normalized against the intensity in the cell soma of each neuron. For cell quantification, ≥90 neurons were counted in three independent experiments for each assay. The statistical significance of the data was evaluated with a Student's t test.
Live-cell imaging.
Mouse brain cortical neurons were prepared on a glass-bottom dish (CELLview; Greiner) as described above. Three days after seeding, the culture medium was replaced with Neurobasal medium (Invitrogen) without phenol red and was supplemented with B-27 (Invitrogen). The culture dish was mounted on a temperature-controlled stage on a Zeiss confocal microscope (LSM 710) at 37°C in 5% CO2. The images of vesicle transport were captured in the middle region of axons for at least 5 min at 2 s intervals using a 63× oil-immersion lens.
Preparation of cell extracts and immunoblotting.
Neurons were disrupted in RIPA buffer [20 mm Tris-HCl, pH 7.5; 150 mm NaCl; 1 mm EGTA; 1 mm EDTA; 1% Nonidet P-40; 0.1% SDS; 0.1% sodium deoxycholate; 0.4 mm Pefabloc (Roche Applied Science; Sigma-Aldrich); 10 μg/ml leupeptin; 10 mm NaF; 10 mm β-glycerophosphate; 1 mm Na3VO4]. After brief centrifugation at 2500 × g for 5 min at 4°C, the lysate was suspended in SDS sample buffer and boiled for 5 min. SDS-PAGE and immunoblotting were performed as described previously (Takano et al., 2010).
GTP-Rab11A pull-down assay.
T7-FIP2C was transfected into COS-7 cells using PolyFect (Qiagen) transfection reagent. COS-7 cells were lysed in 10 mm MOPS, pH 7.5; 150 mm NaCl; 1 mm MgCl2; 1 mm EGTA; 0.2% Nonidet P-40; 0.4 mm Pefabloc; and 10 μg/ml leupeptin. The cell lysate, which was obtained by centrifugation at 15,000 × g for 10 min at 4°C, was incubated with protein G-Sepharose beads bound to anti-T7. After centrifugation at 2500 × g for 5 min, T7-FIP2C–protein G beads were incubated for 1 h at 4°C with extracts of mouse brains obtained by centrifuging at 15,000 × g for 10 min at 4°C. SDS-PAGE and immunoblotting were performed as described previously (Takano et al., 2010). Rab11A band intensities were quantified with Image J software.
Tissue preparation and immunohistochemical staining.
The dissected brains of mice were immersed in ice-cold PBS containing 4% paraformaldehyde overnight and then transferred to 20% sucrose in PBS until they sank. The brains were frozen in crushed dry ice, and consecutive 10-μm-thick horizontal sections were cut on a cryostat. The sections were kept at −80°C for processing for immunohistochemistry as described previously (Yoshioka et al., 2011). Briefly, the sections were incubated overnight at 4°C with either anti-LMTK1 (1:1000) or anti-neurofilament (1:10,000). After incubation with biotinylated anti-rabbit IgG (1:200; Vector Laboratories) for 30 min at 37°C, immunoreactivity was visualized with 50 mm Tris-HCl at pH 7.4 containing 0.01% diaminobenzidine tetrahydrochloride and 0.01% hydrogen peroxide at 37°C for 5–10 min. Sections were embedded in Entellan Neu (Merck) after dehydration through ethanol and xylene.
Results
Localization of LMTK1 in Rab11A-positive recycling endosomes in neurons
We examined the expression of total LMTK1 and LMTK1 phosphorylated by Cdk5 in cultured cortical neurons derived from embryonic day 17 or 18 mice, which were used throughout this study. Neurons began to express LMTK1 as early as the second day in vitro (DIV2), and expression increased thereafter (Fig. 1A, top). Levels of LMTK1 phosphorylated at Ser34, the functionally important Cdk5 phosphorylation site, increased from DIV2 in parallel with the expression of the Cdk5 activator p35 (Fig. 1A, second and fifth panels). Rab11A, a recycling endosomal protein, showed constant expression during this time (Fig. 1A, third panel).
Figure 1.

Expression and localization of LMTK1 in neurons. A, Immunoblots show expression of endogenous LMTK1, Rab11A, Cdk5, and p35 in a culture of mouse cortical neurons from DIV1 to DIV6. Cell lysates were collected at DIV1–6 and immunoblotted with antibodies against LMTK1, phospho-Ser34 of LMTK1 (pS34), Rab11A, Cdk5, and p35. Actin was used as the loading control. B, Immunofluorescence staining of primary cortical neurons with anti-pS34. Cortical neurons transfected with LMTK1-myc were doubly labeled with anti-myc (top) and anti-pS34 (middle). Merged images are shown in the bottom. The cell body and growth cone are indicated by the arrow and arrowhead, respectively. Higher-magnification views of the boxed areas are (left, right). C, Localization of LMTK1 to Rab11-positive recycling endosomes in neurons. LMTK1 was cotransfected into primary cortical neurons with EGFP-Rab5A, EGFP-Rab7, or EGFP-Rab11A. LMTK1 was visualized by immunostaining with anti-LMTK1 (left) 3 d after transfection. Rabs (middle). Merged images (right). Cell body (top). Axon (bottom). Arrows indicate colocalization of LMTK1 with Rabs in axons. N, Nucleus. Scale bars: B, 40 μm; C, 5 μm.
We reported that phospho-Ser34 LMTK1 is present in the growth cone of PC12D cells (Tsutsumi et al., 2010). We sought to determine whether this is also the case in cultured neurons. Because anti-pS34 does not detect endogenous phospho-LMTK1 by immunofluorescence staining (data not shown), we expressed LMTK1 exogenously in cortical neurons. Phospho-LMTK1 was found in the growth cone as well as in the perinuclear region of the cell soma (Fig. 1B).
Next, we observed the localization of LMTK1 in recycling endosomes in neurons. Cortical neurons were cotransfected with expression vectors encoding LMTK1 and EGFP-Rab5A, EGFP-Rab7, or EGFP-Rab11A, markers of early, late, and recycling endosomes, respectively. Fluorescence images of cell bodies and axons at DIV3 are shown in Figure 1C. The longest neurite at this stage is the axon (Dotti et al., 1988). Hereafter, we refer to this process as the axon, and we use the term “neurite” to indicate all processes, including the axon. LMTK1 accumulated mainly in perinuclear regions of the cell soma and as small particles in axons (Fig. 1C, left). Rab5A, Rab7, and Rab11A were detected mainly in perinuclear regions and axons (Fig. 1C, middle). Merged images indicate that LMTK1 colocalized extensively with Rab11A and partly with Rab5A, but not with Rab7 (Fig. 1C, right). This colocalization with Rab11A was clearly observed in axons (Fig. 1C, arrows, bottom). Thus, LMTK1 colocalizes with Rab11A-positive recycling endosomes in both perinuclear regions and axons of cortical neurons.
Effect of phosphorylation of LMTK1 at Ser34 on axonal outgrowth
The effect of LMTK1 on the accumulation of recycling endosomes is inhibited by the phosphorylation of LMTK1 at Ser34 by Cdk5-p35 (Takano et al., 2010). We examined the effect of overexpression of two LMTK1 mutants on axonal outgrowth: S34A, which is constitutively unphosphorylated, and S34D, which mimics the phosphorylated form. LMTK1-WT, LMTK1-S34A, and LMTK1-S34D were individually transfected into primary cortical neurons at DIV0, and axonal outgrowth was observed at DIV3. Neurons were immunostained with anti-myc to examine the cellular distribution of overexpressed LMTK1-myc proteins. LMTK1 was distributed in the cell body and neurites (Fig. 2A) similarly to cotransfected EGFP. Axons of neurons expressing LMTK1-S34A were remarkably longer than those of neurons that expressed LMTK1-S34D or -WT (Fig. 2A). We measured the axon length, the number of neurites (>20 μm), and total neurite length in LMTK1-overexpressing neurons. Overexpression of LMTK1-S34A increased axon length (307 ± 18 μm) by ∼2.5-fold compared with axons that expressed LMTK1-WT or LMTK1-S34D (124 ± 13 and 125 ± 16 μm, respectively; Fig. 2B). In contrast, the number of neurites per neuron was decreased to a mean of 1.8 in neurons expressing LMTK1-S34A compared with means of 2.2 and 2.3 in those expressing LMTK1-S34D and LMTK1-WT, respectively (Fig. 2C). The total length of neurites was not different with the overexpression of any LMTK1 constructs (Fig. 2D).
Figure 2.

Effect of LMTK1 overexpression on axonal outgrowth. A, LMTK1-WT, LMTK1–S34A, and LMTK1–S34D tagged with myc at the C terminus were individually transfected into cortical neurons at DIV0. Control neurons (Cont) were transfected with EGFP. The neurons were immunostained with anti-myc at DIV3. B, Axon length in LMTK1-expressing neurons (mean ± SEM of 90 neurons from three independent experiments; *p < 0.01, Student's t test). C, The number of neurites (>20 μm) extending from LMTK1-expressing neurons (90 neurons from three independent experiments). D, Total length of neurites in LMTK1-expressing neurons (mean ± SEM of 90 neurons from three independent experiments; n.s., not significant; Student's t test). E, The effect of downregulation of p35 expression on LMTK1-induced axonal outgrowth. Cortical neurons were cotransfected with shRNA for p35 and LMTK1-WT, LMTK1–S34A, LMTK1–S34D, or empty vector (Cont). Axons were observed with EGFP at DIV3. F, Axon length of EGFP-expressing neurons (mean ± SEM of 90 neurons from three independent experiments; *p < 0.01, Student's t test). Scale bars: A, E, 20 μm.
LMTK1-WT, like LMTK1-S34D, did not show axonal outgrowth activity. This result is in contrast to previous findings in CHO-K1 cells (Takano et al., 2010), in which LMTK1-WT promotes the recycling of endosomes to the pericentrosomal compartment, as does LMTK1-S34A. The difference may be explained by the difference in Cdk5 activity between these cell types; CHO-K1 cells do not show Cdk5 activity. If this were the case, then the downregulation of Cdk5 activity should change the axonal outgrowth activity of LMTK1-WT to that of LMTK1-S34A in neurons. We performed this experiment by the knockdown of p35, a major Cdk5 activator (Endo et al., 2009). Decreased Cdk5 activity by p35 knockdown indeed converted the axonal outgrowth activity of LMTK1-WT from the LMTK1-S34D type to the LMTK1-S34A type (Fig. 2E, compare B and F), whereas the activities of LMTK1-S34A and -S34D were not affected (Fig. 2F). These results indicate that the axonal outgrowth effect of LMTK1 is regulated by Cdk5 activity in neurons.
An LMTK1 kinase-deficient mutant enhances axonal outgrowth of neurons
LMTK1 is a Ser/Thr kinase, although its physiological substrates are not yet known. We next examined whether LMTK1 regulates axonal outgrowth through its kinase activity using the kinase-negative mutant LMTK1-D206V (Tomomura et al., 2001). The overexpression of LMTK1-D206V increased axon length (265 ± 17 μm) by 1.9-fold compared with axons from neurons transfected with LMTK1-WT (142 ± 8 μm) (Fig. 3A,B). The number of neurites per neuron was concomitantly decreased to 2.1 in LMTK1-D206V-expressing neurons compared with 2.8 in neurons that expressed LMTK1-WT (Fig. 3C), but the total length of neurites was not different between them (Fig. 3D). Thus, the kinase activity of LMTK1 was required for the suppression of axonal outgrowth.
Figure 3.

A kinase-negative mutant of LMTK1 promotes axonal outgrowth in cortical neurons. A, LMTK1-WT and the kinase-negative mutant LMTK1–D206V were individually cotransfected into cortical neurons with EGFP at DIV0, and axonal outgrowth was observed at DIV3. The control (Cont) image represents neurons transfected with EGFP alone. B, Axon length of EGFP-expressing neurons (mean ± SEM of 90 neurons from three independent experiments; *p < 0.01, Student's t test). C, The percentage of EGFP-expressing neurons with the indicated number of neurites (90 neurons from three independent experiments). D, Total length of neurites in LMTK1-expressing neurons (mean ± SEM of 90 neurons from three independent experiments; n.s., not significant; Student's t test). Scale bar, 20 μm.
LMTK1 that is unphosphorylated at Ser34 localizes to Rab11A-positive recycling endosomes in axons
We investigated the effects of the phosphorylation of LMTK1 at Ser34 on the subcellular localization of Rab11A in neurons. We transfected cultured cortical neurons with constructs encoding EGFP-Rab11A and LMTK1-S34A or LMTK1-S34D. Both LMTK1-S34A and LMTK1-S34D localized with Rab11A in perinuclear regions, and Rab11A distribution did not change significantly after cotransfection with LMTK1-S34A or LMTK1-S34D (compare Figs. 4A, 1C). In axons, however, LMTK1-S34A showed stronger colocalization with Rab11A than did LMTK1-S34D (Fig. 4A). The amount of Rab11A was also increased in axons by coexpression with LMTK1-S34A. To confirm this, we measured the fluorescence intensity of EGFP-Rab11A in the axons of neurons in which LMTK1-WT, LMTK1-S34D, LMTK1-S34A, or empty vector (Cont) was cotransfected. Coexpression of LMTK1-S34A increased axonal Rab11A-positive recycling endosomes ∼1.4-fold with respect to control, whereas LMTK1-WT and LMTK1-S34D did not (Fig. 4B). We also tested whether LMTK1-WT colocalizes with constitutively active Rab11A-Q70L or dominant-negative Rab11A-S25N. LMTK1 appeared to colocalize with both Rab11A-Q70L and Rab11A-S25N in perinuclear regions in the cell soma, but colocalization of LMTK1-WT with Rab11A-Q70L was also clearly seen in axons and to a greater extent than that with Rab11-S25N (Fig. 4C). Expression of active Rab11A-Q70L increased the extent of Rab11A localization in the axon. This was confirmed quantitatively by measuring EGFP-Rab11A in axons and cell bodies (Fig. 4D). Thus, LMTK1 colocalizes preferentially with the active GTP-bound form of Rab11A in axons, and LMTK1-S34A increases the number of axonal Rab11A-positive recycling endosomes.
Figure 4.
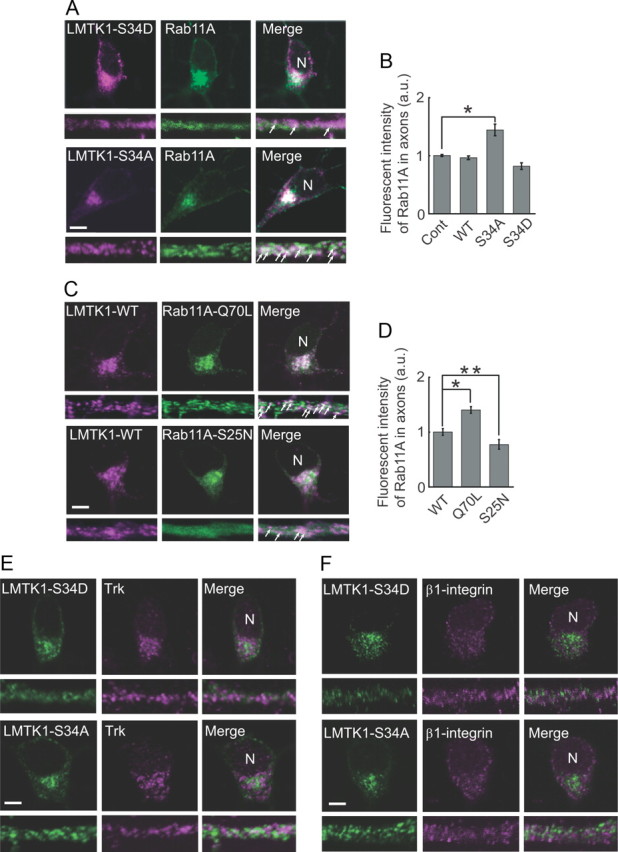
Colocalization of LMTK1 and Rab11A in cortical neurons. A, Localization of LMTK1–S34D and LMTK1–S34A in Rab11-positive recycling endosomes. Cortical neurons were cotransfected with EGFP-Rab11A and LMTK1–S34D or LMTK1–S34A, and the cellular localization of LMTK1 mutants was detected by immunofluorescence staining with anti-LMTK1 (left). Rab11A (middle). Cell body (top). Axon (bottom). Arrows indicate colocalization of LMTK1 with Rab11A in axons. B, Quantification of Rab11A in axons. Cortical neurons were cotransfected with EGFP-Rab11A and LMTK1-WT, LMTK1–S34D, LMTK1–S34A, or empty vector (Cont). Fluorescence intensity of EGFP-Rab11A was measured along a 20 μm length in the middle region of the axons. After normalization against EGFP intensity in the cell soma, the relative intensity was expressed with respect to that of the control empty vector (mean ± SEM of 90 neurons from three independent experiments; *p < 0.05, Student's t test). C, Colocalization of LMTK1-WT with the active or dominant-negative form of Rab11A. Cortical neurons expressing LMTK1 and constitutively active Rab11A-Q70L or dominant-negative Rab11A-S25N were stained with anti-LMTK1. Rab11A (middle). Cell body (top). Axon (bottom). Arrows indicate colocalization of LMTK1 with Rab11A. D, The active form of Rab11A increases its axonal localization. Cortical neurons were cotransfected with LMTK1 and EGFP-Rab11A-WT, EGFP-Rab11A-Q70L, or EGFP-Rab11A-S25N. Fluorescence intensity of EGFP-Rab11A in the axon and cell soma was measured as in B, and the abundance of Rab11A-Q70L and Rab11A-S25N in the axon was expressed relative to that of Rab11A-WT (mean ± SEM of 90 neurons from three independent experiments; *p < 0.01, **p < 0.05, Student's t test). E, Trk receptors do not colocalize with LMTK1 in primary neurons. Cortical neurons were transfected with LMTK1–S34D or LMTK1–S34A, and the cellular localization of LMTK1 mutants was detected by immunofluorescence staining with anti-LMTK1 (left). Trk receptors were stained fluorescently with anti-pan Trk (middle). Merged images (right). Cell body (top). Axon (bottom). F, β1-Integrin does not colocalize with LMTK1 in primary neurons. Cortical neurons were transfected with LMTK1–S34D or LMTK1–S34A, and the cellular localization of LMTK1 mutants was detected by immunofluorescence staining with anti-LMTK1 (left). β1-Integrin was stained fluorescently with anti-β1-integrin (middle). Cell body (top). Axon (bottom). N, Nucleus. Scale bars: A, C, E, F, 5 μm.
We examined whether Trks and β1-integrin, as typical examples of trophic factor receptors and extracellular matrix receptors, respectively, are transported by LMTK1-dependent recycling endosomes. Trks and β1-integrin showed punctate staining in the cell soma and axon, but their distribution did not coincide with that of LMTK1-WT, LMTK1-S34D, or LMTK1-S34A (Fig. 4E,F; and data not shown). Furthermore, LMTK1 overexpression did not markedly change the distribution of Trks and β1-integrin in the distal regions of the axon. These results suggest that LMTK1 regulates the trafficking of recycling endosomes that are distinct from those transporting Trks and β1-integrin.
Role of Rab11A in axonal outgrowth
There are only a few reports describing how Rab11 regulates neurite elongation. In addition, there is disagreement about whether the active or inactive form of Rab11 promotes neurite outgrowth (Shirane and Nakayama, 2006; Ascano et al., 2009). Therefore, we examined how Rab11A is involved in axonal outgrowth in our experimental paradigm. We first examined the effect of knocking down Rab11A on axonal outgrowth. Decreased expression of endogenous Rab11A was shown with immunoblotting (Fig. 5A) and immunostaining (Fig. 5B) after the introduction of shRNA-Rab11A compared with control scrambled shRNA (shRNA-SC). The downregulation of Rab11A decreased axon length (116 ± 7 μm) by 26% compared with the length (146 ± 7 μm) in control shRNA-SC–treated neurons (Fig. 5C). Next, we examined whether constitutively active or dominant-negative Rab11A is involved in axonal outgrowth. When Rab11A-WT, -Q70L, and -S25N were each expressed in neurons, all Rab11 proteins were distributed throughout neurites (data not shown). Therefore, we measured axon length using EGFP tagged to Rab11. Rab11A-WT significantly enhanced axonal outgrowth compared with EGFP alone (Fig. 5D,E). Rab11A-Q70L stimulated axonal outgrowth more than Rab11A-WT, whereas the axonal outgrowth activity of Rab11A-S25N was considerably low compared with that of Rab11A-WT and Rab11A-Q70L (Fig. 5D,E). As shown in Figure 5E, however, the axon length of neurons expressing Rab11A-S25N was not shorter than that of control neurons expressing EGFP, although Rab11A-S25N is a dominant-negative mutant. This result may suggest the abundant expression of Rab11A guanine nucleotide exchange factor, whose activity Rab11A-S25N would inhibit. In contrast, neither Rab11A-Q70L nor Rab11A-S25N affected the number of neurites, which was similar to the number in neurons transfected with EGFP alone or Rab11A-WT (Fig. 5F). Thus, the GTP-bound active form of Rab11A promotes the axonal outgrowth of cortical neurons.
Figure 5.
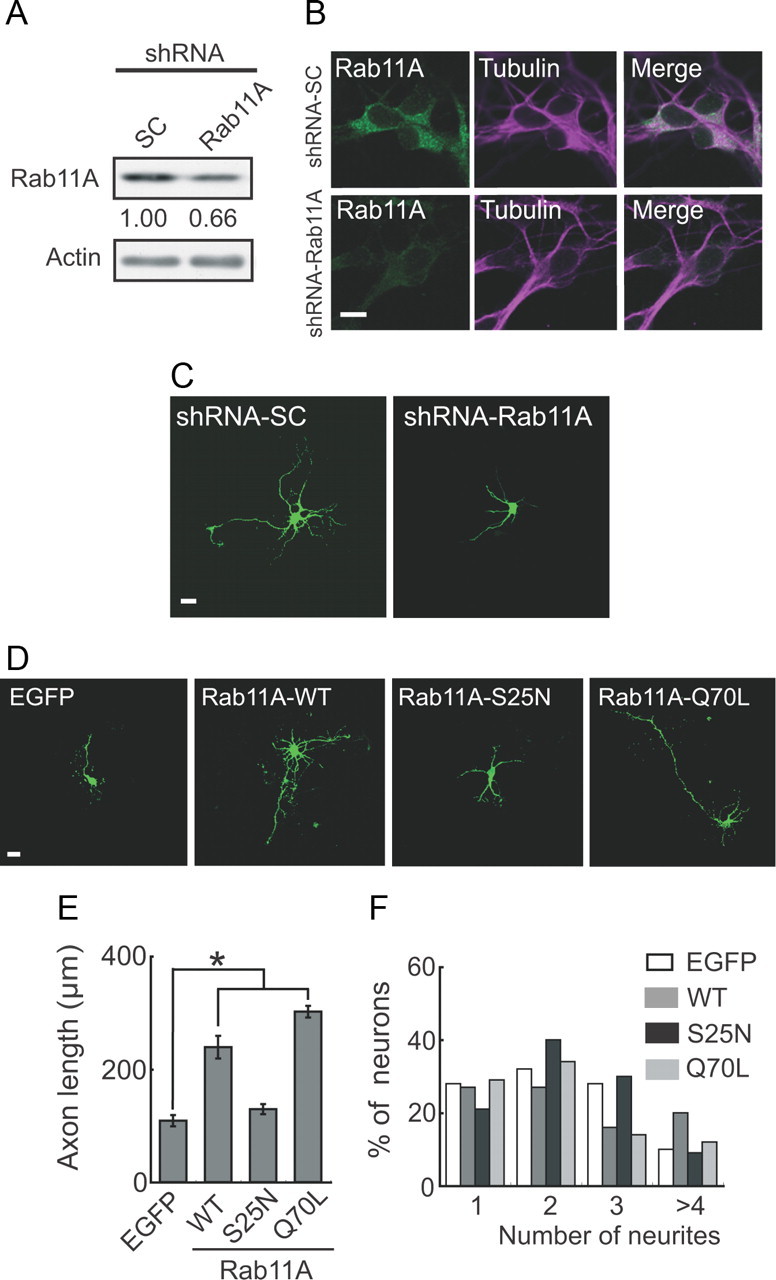
Effect of knockdown and overexpression of Rab11A on axonal outgrowth. A, Immunoblots showing knockdown of Rab11A with RNA interference. Cortical neurons were transfected with pSilencer-Rab11A (shRNA-Rab11A) or shRNA-SC at DIV0. Three days after transfection, they were immunoblotted with anti-Rab11. Knockdown efficiency is indicated below the blot. Actin was used as the loading control. B, Immunostaining showing knockdown of Rab11A by RNAi. Cortical neurons were transfected with shRNA-SC (top) or shRNA-Rab11A (bottom) at DIV0. Three days after transfection, they were immunostained with anti-Rab11 (left) and anti-tubulin (middle). Merged images (right). C, Effect of downregulation of Rab11A on axonal outgrowth. Cortical neurons were cotransfected with EGFP and either shRNA-Rab11A or shRNA-SC at DIV0, and EGFP images were obtained at DIV3. D, EGFP, EGFP-Rab11A-WT, EGFP-Rab11A-S25N, and EGFP-Rab11A-Q70L were individually transfected into cortical neurons at DIV0, and axonal outgrowth was observed at DIV3. E, Axon length in EGFP-expressing neurons (mean ± SEM of 180 neurons from three independent experiments; *p < 0.01, Student's t test). F, Number of neurites extending from EGFP-expressing neurons (90 neurons from three independent experiments). Scale bars: B, 10 μm; C, D, 20 μm.
LMTK1 regulates axonal outgrowth through Rab11 activity
To determine the signaling hierarchy of LMTK1 and Rab11A, we asked whether the axonal outgrowth activity of LMTK1 was affected by Rab11A expression. Enhancement of axonal outgrowth by LMTK1-S34A was strongly abrogated, from 294 ± 30 to 185 ± 21 μm, with cotransfection of shRNA-Rab11A (Fig. 6A), suggesting that LMTK1 regulates axonal outgrowth through Rab11A. We further tested the order of LMTK1 and Rab11A using combinatory expression of Rab11A-Q70L or Rab11A-S25N and LMTK1-S34A or LMTK1-S34D. Expression of Rab11A-S25N inhibited the axonal outgrowth-promoting activity of LMTK1-S34A, whereas coexpression with Rab11A-Q70L did not affect the length of axons (Fig. 6B,C). Reduction in the number of neurites induced by LMTK1-S34A was reversed by Rab11A-S25N but not by Rab11-Q70L (Fig. 6B,D). In contrast, the expression of Rab11A-Q70L, but not Rab11A-S25N, enhanced the axonal elongation in LMTK1-S34D-expressing neurons by more than twofold (Fig. 6E,F). The number of neurites did not, however, change significantly with the coexpression of Rab11A-S25N or Rab11A-Q70L (Fig. 6E,G). Together, these results show that Rab11A is a stronger determinant of both the axon length and the number of neurites than LMTK1, indicating that LMTK1 is upstream of Rab11A.
Figure 6.
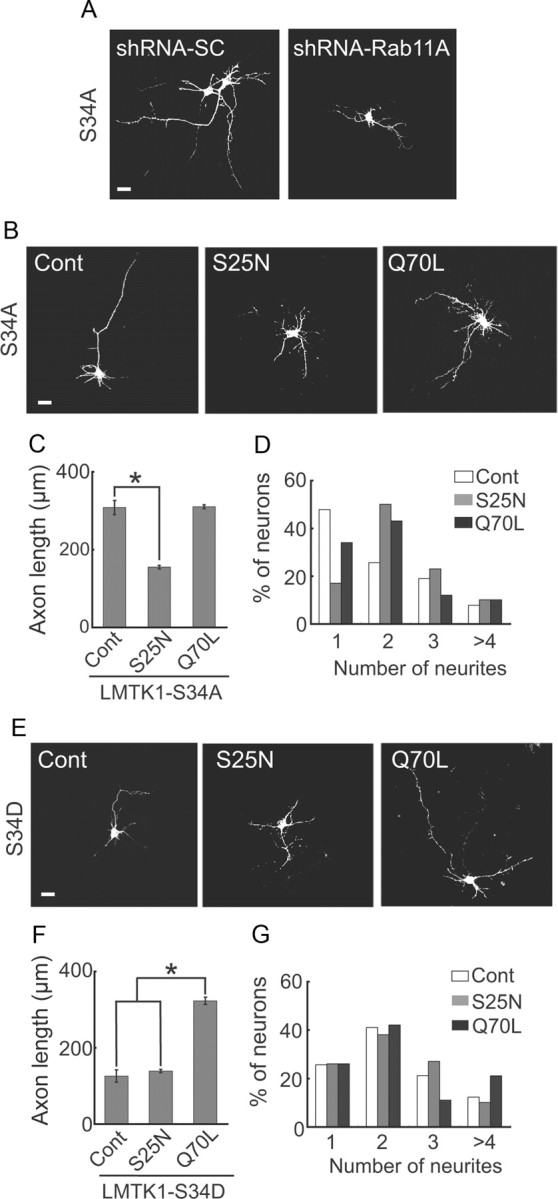
LMTK1 regulates axonal outgrowth through Rab11 activity. A, Axonal outgrowth induced by LMTK1–S34A was suppressed by the downregulation of Rab11A. LMTK1–S34A was coexpressed with either shRNA-Rab11A or shRNA-SC in cortical neurons at DIV0. Neurons were immunostained with anti-myc at DIV3. B–D, Axonal outgrowth induced by LMTK1–S34A was suppressed by the inactive form of Rab11A. LMTK1–S34A was coexpressed with either EGFP-Rab11A-S25N or EGFP-Rab11A-Q70L in cortical neurons. B, LMTK1 was immunostained with anti-myc. C, Axon length in LMTK1-expressing neurons (mean ± SEM of 90 neurons from three independent experiments; *p < 0.01, Student's t test). D, Percentage of LMTK1-expressing neurons with the indicated number of neurites (90 neurons from three independent experiments). E–G, The active form of Rab11A stimulated axonal outgrowth even in the presence of LMTK1–S34D. LMTK1–S34D was coexpressed with either EGFP-Rab11A-S25N or EGFP-Rab11A-Q70L in cortical neurons. E, LMTK1 was immunostained with anti-myc. F, Axon length in LMTK1-expressing neurons (mean ± SEM of 90 neurons from three independent experiments; *p < 0.01, Student's t test). G, Percentage of LMTK1-expressing neurons with the indicated number of neurites (90 neurons from three independent experiments). Scale bars: A, B, E, 20 μm.
LMTK1 knockdown enhances axonal outgrowth of neurons
We next used miRNA to examine the effect of knocking down LMTK1 on axonal outgrowth. If the phosphorylated form of LMTK1 is predominantly expressed in neurons, the knockdown of LMTK1 should result in increased axonal outgrowth. EmGFP-miRNA vectors encoding four different LMTK1 sequences, LMTK1 miRNA1–miRNA4, were transfected into cortical neurons. Immunoblotting showed that LMTK1 miRNA3 and miRNA4 decreased the expression level of LMTK1 to 38% and 49%, respectively, of control miRNA-SC (Fig. 7A). Staining with anti-LMTK1 was also greatly reduced in neurons transfected with miRNA3 or miRNA4, whereas neurons transfected with miRNA-SC showed no reduction in anti-LMTK1 staining (Fig. 7B). Unexpectedly, we found that cortical neurons expressing LMTK1 miRNA3 or miRNA4 displayed longer axons than did those expressing miRNA-SC at DIV3 (Fig. 7C). Knockdown of LMTK1 by miRNA3 or miRNA4 increased axon length by almost threefold, from 114 ± 15 μm to 351 ± 10 and 312 ± 18 μm, respectively (Fig. 7D). LMTK1 miRNAs also decreased the number of neurites per neuron. The number of neurons with a single neurite increased markedly, and the number of neurons with three or more neurites decreased markedly after the transfection of miRNA3 or miRNA4 (Fig. 7E). The total length of neurites was not, however, different between neurons expressing LMTK1 miRNAs and those expressing miRNA-SC (Fig. 7F). Thus, LMTK1 can also suppress axonal outgrowth.
Figure 7.
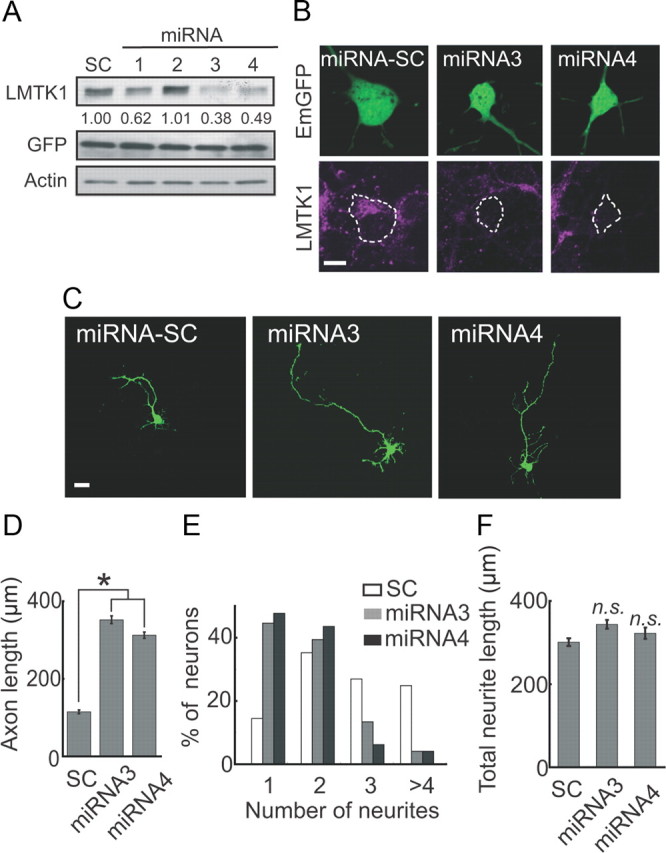
Knockdown of LMTK1 promotes axonal outgrowth in cortical neurons. A, Knockdown of LMTK1 by miRNA. Cortical neurons were transfected with EmGFP-miRNA1–4 or scrambled sequence (miRNA-SC) at DIV0. Three days after transfection, cell lysates were immunoblotted with anti-LMTK1 and anti-GFP. Actin was used as the loading control. Knockdown efficiencies are indicated below the blot. B, Cortical neurons were transfected with EmGFP-miRNA-SC, EmGFP-miRNA3, or EmGFP-miRNA4 at DIV0. Three days after transfection, they were immunostained with anti-LMTK1 (bottom). C, Effect of downregulating LMTK1 on axonal outgrowth. Cortical neurons were transfected with EmGFP-miRNA-SC, EmGFP-miRNA3, or EmGFP-miRNA4 at DIV0, and EmGFP images were obtained at DIV3. D, Axon length of EmGFP-expressing neurons (mean ± SEM of 90 neurons from three independent experiments; *p < 0.01, Student's t test). E, Percentage of EGFP-expressing neurons with the indicated number of neurites (90 neurons from three independent experiments). F, Total length of neurites in LMTK1-expressing neurons (mean ± SEM of 90 neurons from three independent experiments; n.s., not significant; Student's t test). Scale bars: B, 10 μm; C, 20 μm.
LMTK1−/− mouse neurons exhibit enhanced axonal outgrowth
We confirmed the results of the LMTK1 knockdown using neurons prepared from the brains of LMTK1−/− mice. LMTK1−/− mice were produced by homologous recombination, in which the genomic region of LMTK1 that contains exons 4–7 and that encodes the kinase domain was replaced with the Neo cassette (Fig. 8A). The brains of LMTK1−/− mice were slightly smaller than those of wild-type mice (data not shown), but histological abnormalities were not observed in the cerebral cortex (Fig. 8B, middle) or in the axonal pathway between the cerebral cortex and the thalamus (Fig. 8B, right). Neurofilament-immunoreactive axons invaded the cerebral cortex through the internal capsule in both LMTK1+/+ and LMTK1−/− mouse brains (Fig. 8B, IC).
Figure 8.
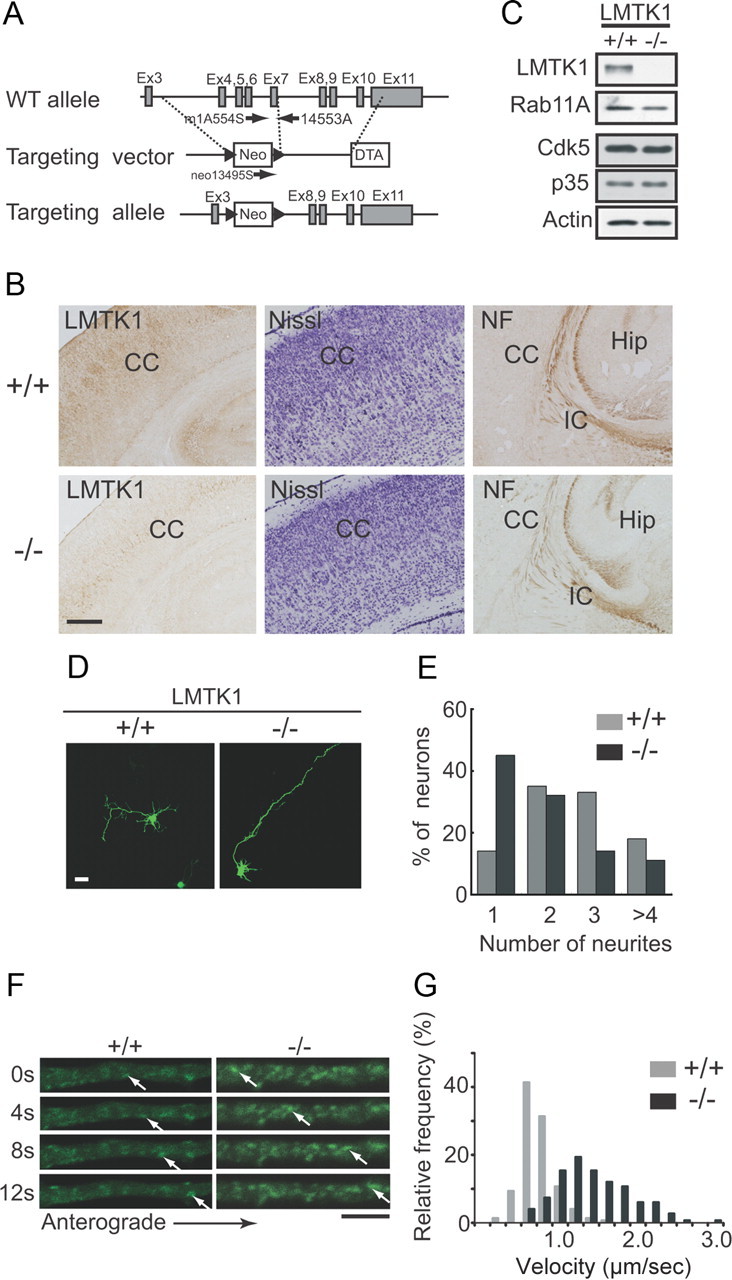
LMTK1−/− neurons have longer axons. A, Schematic diagram of the WT allele of mouse LMTK1 gene, targeting vector, and mutant allele resulting from homologous recombination. Gray and white boxes represent exons and selection marker genes, respectively. Arrows indicate primers used for genotyping by PCR. B, Immunostaining of cortical sections of LMTK1+/+ (top) and LMTK1−/− (bottom) mice with anti-LMTK1. Cortical structures of LMTK1+/+ and LMTK1−/− mouse brains at P5. Sections of frontal cortex were stained with cresyl violet (Nissl) (middle). Sections of thalamocortical tract at P5 were stained with anti-NF. C, Immunoblots showing LMTK1 deficiency. Cortical neurons isolated from brains of littermate LMTK1+/+ (left) or LMTK1−/− (right) mice were immunoblotted with antibodies to LMTK1, Rab11A, Cdk5, and p35. Actin was used as the loading control. D, Axons of LMTK1−/− mouse neurons. Cortical neurons from LMTK1+/+ (left) or LMTK1−/− (right) brains were transfected with EGFP at DIV0 and observed at DIV3. E, Percentage of EGFP-expressing neurons with the indicated number of neurites (120 neurons from three independent experiments). F, Dynamics of Rab11A-positive recycling endosomes in axons of LMTK1−/− neurons. Neurons prepared from LMTK1+/+ or LMTK1−/− mouse brains were transfected with EGFP-Rab11A. Movement of Rab11A-positive recycling endosomes was analyzed by time-lapse imaging. Sequential frames >12 s with a 4 s interval are shown for LMTK1+/+ (left) and LMTK1−/− (right) axons. Cell body is outside the frame on the left side. The movement of a typical Rab11A vesicle is indicated by arrows in each series of images. G, Histogram showing the instantaneous velocity of Rab11A-positive vesicles during 6 s period (three frames, 2 s interval). CC, Cerebral cortex; DTA, diphtheria toxin A-chain gene; Hip, hippocampus; IC, internal capsule; NF, neurofilament; Neo, neomycin resistance gene. Scale bars: B, 100 μm; D, 20 μm; F, 5 μm.
We examined the effect of LMTK1 deficiency on axon formation with cultured cortical neurons. The deletion of LMTK1 was confirmed by immunoblotting with anti-LMTK1 (Fig. 8C, top). Expression of Rab11A, Cdk5, and p35 was not altered in cultured neurons prepared from LMTK1−/− mouse brains (Fig. 8C). When LMTK1−/− neurons were stained with anti-tubulin, however, neurites appeared to be thinner than were those of neurons prepared from the brains of wild-type littermates (data not shown). We observed axonal outgrowth based on EGFP expression. Axons were remarkably longer in LMTK1−/− neurons than in wild-type neurons (Fig. 8D). Quantification indicated that LMTK1−/− neurons had axons (380 ± 15 μm) that were ∼1.6-fold longer than were those (234 ± 13 μm) from LMTK1+/+ control neurons. Of note, cortical neurons from wild-type C57B6/J mice (genetic background of LMTK1−/− mice) showed longer axons (∼200 μm at DIV3) than did those of wild-type ICR mice (∼100 μm at DIV3). The latter is the strain of mice used in experiments shown in Figures 1–7. Therefore, the relative increase was lower in LMTK1−/− neurons (∼1.6-fold) than the relative increase obtained with the knockdown experiment (3-fold; Fig. 7D). Concomitantly, the mean number of neurites in LMTK1−/− neurons was reduced to 1.9 from a mean of 2.6 in LMTK1+/+ neurons (Fig. 8D,E). The total neurite length at 410 ± 16 μm of LMTK1−/− neurons was not, however, different from 384 ± 12 μm of LMTK1+/+ neurons. These results clearly show that LMTK1 negatively regulates axon length, resulting in an increased number of neurites in developing neurons.
To analyze how axonal outgrowth is enhanced in LMTK1−/− neurons, we visualized the movement of EGFP-Rab11A-positive recycling endosomes in the axons of LMTK1−/− neurons and compared it with the movement in LMTK1+/+ axons (Fig. 8F). Rab11A-positive vesicles were increased in the axons of LMTK1−/− neurons (∼1.7-fold of LMTK1+/+ neurons) (Fig. 8F). Recycling endosomes were also more dynamic in LMTK1−/− axons than in LMTK1+/+ axons (Fig. 8F). Rab11A-positive recycling endosomes in LMTK1−/− neurons moved longer distances anterogradely with a mean velocity of 1.3 μm/s, which was faster than that of endosomes from LMTK1+/+ neurons at 0.6 μm/s (Fig. 8F,G). Thus, the absence of LMTK1 enhanced both the amount of recycling endosomes and their moving velocity in axons.
Expression of LMTK1 rescues the LMTK1−/− phenotype
Both LMTK1 downregulation and overexpression of LMTK1-S34A promoted axonal outgrowth. To confirm that these opposite effects on LMTK1 activity indeed result in the same apparent phenotype, we introduced LMTK1-WT, LMTK1-S34A, and LMTK1-S34D into LMTK1−/− neurons. Expression of LMTK1-WT or LMTK1-S34D in LMTK1−/− neurons reversed the knock-out effects by shortening the axon length and increasing the number of neurites to levels observed in WT mouse neurons (Fig. 9A–C). In contrast, LMTK1-S34A did not rescue the effect on axon length seen in the LMTK1−/− neurons (Fig. 9A–C). Thus, LMTK1 negatively regulates axonal outgrowth, depending on its phosphorylation state.
Figure 9.
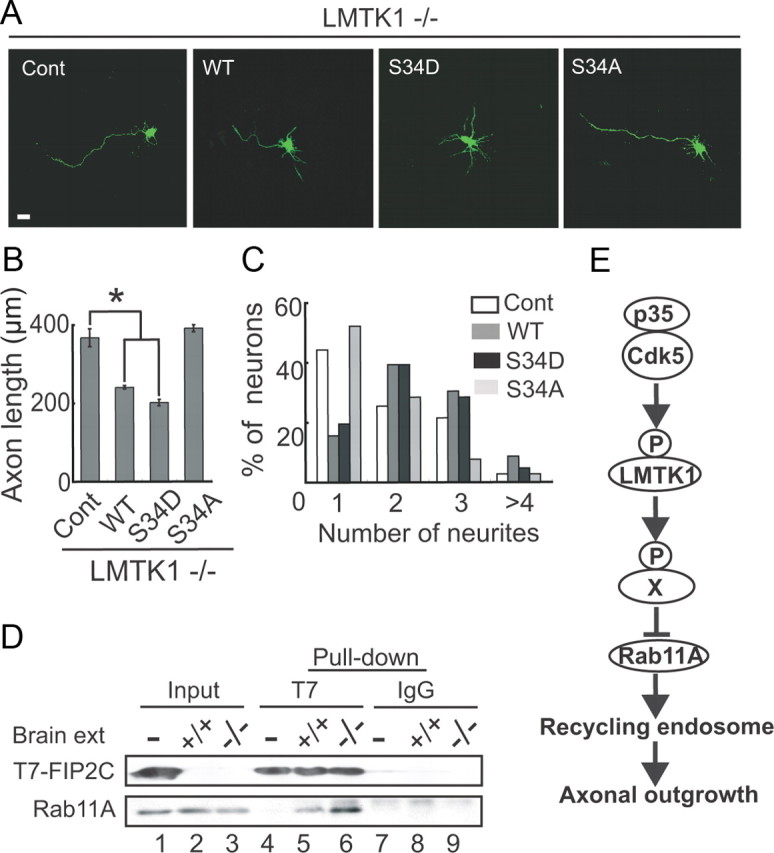
Expression of LMTK1 reverses the effect of LMTK1 knock-out on axonal outgrowth. A, Overexpression of LMTK1-WT or S34D reverses the LMTK1−/− phenotype. Empty vector (Cont), LMTK1-WT, LMTK1-S34D, and LMTK1-S34A were individually cotransfected with EGFP into LMTK1−/− neurons at DIV0, and axonal outgrowth was observed at DIV3. Scale bar, 20 μm. B, Axon length in EGFP-expressing neurons (mean ± SEM of 90 neurons from three independent experiments; *p < 0.01, Student's t test). C, Number of neurites extending from EGFP-expressing neurons (90 neurons from three independent experiments). D, Rab11A-GTP is increased in LMTK1−/− brains. T7-FIP2C was expressed in COS-7 cells (lane 1) and bound to protein G-Sepharose beads linked to anti-T7. T7-FIP2C-bound beads were incubated with brain extracts (ext) from LMTK1+/+ (lane 2) and LMTK1−/− (lane 3) mice. Rab11A was pulled down and detected by immunoblotting with anti-Rab11A (bottom). T7-FIP2C was detected by immunoblotting with anti-T7 (top). Lane 4, Control T7-FIP2C beads; lane 5, Rab11A pulled down from LMTK1+/+ mouse brain extract; lane 6, Rab11A from LMTK1−/− mouse brain extract. Control experiments using control IgG are shown in lanes 7–9. E, Scheme representing the role of LMTK1 in axonal outgrowth. Cdk5 phosphorylates LMTK1 at Ser34. Phosphorylated LMTK1 may phosphorylate an unknown substrate (X), which would inhibit Rab11A activity, leading to reduced transport of recycling endosomes to the growing tip of the axon, resulting in suppression of its overelongation. The Cdk5-LMTK1-Rab11A pathway is a novel regulatory mechanism for axonal outgrowth of mammalian neurons.
If the axonal outgrowth stimulation induced by LMTK1 depletion is mediated through Rab11A, the active GTP-bound form of Rab11A should be increased in LMTK1-deficient mouse neurons. To quantify the amount of active Rab11A, we pulled down Rab11A-GTP from LMTK1+/+ and LMTK1−/− brain extracts with T7-FIP2C, which is a Rab11A effector that is able to bind only the GTP-bound form of Rab11A (Lindsay and McCaffrey, 2002). The amount of Rab11A bound to FIP2C was increased ∼2.2-fold in LMTK1−/− brains compared with LMTK1+/+ brains (Fig. 9D), indicating that more Rab11A is activated in LMTK1−/− brains than in LMTK1+/+ brains.
Discussion
Axonal outgrowth is a coordinated process of cytoskeletal reorganization and membrane transport (Tang, 2001; Arimura and Kaibuchi, 2007; Polleux and Snider, 2010). This process has been extensively investigated, and a number of molecules involved in neurite outgrowth have been delineated in recent years. Most are axonal cytoskeletal proteins and their regulators, and a few are involved in membrane supply. Although Rab11 is the most well known component involved in membrane supply (Ng and Tang, 2008), its role in neurite outgrowth was only recently shown experimentally. For example, the expression of Rab11 in dorsal root ganglion neurons increases neurite length, and the knockdown of Rab11 in PC12 cells with siRNA decreases neurite outgrowth (Eva et al., 2010). Our results with Rab11A RNA interference and Rab11A overexpression are consistent with this line of evidence. In contrast, whether the GTP- or GDP-bound form of Rab11 promotes neurite outgrowth is still controversial. Although the GTP-bound active form of Rab11A (Rab11A-Q70L) enhances nerve growth factor-induced axon extension in superior cervical ganglion neurons (Ascano et al., 2009), the GDP-bound form of Rab11 stimulates neurite outgrowth in PC12 cells (Shirane and Nakayama, 2006). Using cortical neurons, we have shown that constitutively active Rab11A-Q70L, but not dominant-negative Rab11A-S25N, promotes axonal outgrowth. Considering the principal role of active Rab11 in promoting the trafficking of recycling endosomes, it is conceivable that the active GTP-bound form stimulates axonal outgrowth by providing axons with membranes. Rab11 may, however, operate differently, depending on the type of neuron or process.
We have previously reported that LMTK1 regulates Rab11A-dependent trafficking of recycling endosomes in CHO-K1 cells (Takano et al., 2010) and have shown here that LMTK1 is involved in axonal outgrowth by regulating the dynamics of Rab11A-dependent recycling endosomes. Together, these findings indicate that LMTK1 is a new player in the regulation of recycling endosome dynamics and axonal outgrowth. We might, however, have underestimated the cell-specific nature of LMTK1 signaling in our previous article (Takano et al., 2010). In CHO-K1 cells, in which Cdk5 activity is absent and LMTK1 expression is low, the overexpression of LMTK1-S34A as well as LMTK1-WT promoted the transport of Rab11A-positive recycling endosomes to the pericentrosomal compartment from the perinuclear region, whereas LMTK1-S34D suppressed pericentrosomal recycling endosome formation (Takano et al., 2010). Based on those results, we thought the unphosphorylated form of LMTK1 was responsible for promoting recycling endosome trafficking and that the phosphorylation of LMTK1 inactivated this activity. In addition, the overexpression of unphosphorylated LMTK1-S34A dramatically stimulated axonal outgrowth. Based on those results in CHO-K1 cells, we thought initially that LMTK1 was a positive regulator of axonal outgrowth. Unexpectedly, however, we subsequently found that axonal outgrowth was dramatically enhanced when LMTK1 was knocked down or knocked out in neurons. These results indicate that LMTK1 plays an inhibitory role in axonal elongation. Considering that LMTK1-S34D, but not LMTK1-S34A, reversed the LMTK1 knock-out phenotype, the phosphorylated form of LMTK1 at Ser34, which is represented by LMTK1-S34D, would be the functional form that suppresses axonal outgrowth, and the unphosphorylated form of LMTK1, which is represented by LMTK1-S34A, would have worked as a dominant-negative factor. If this is the case, in CHO-K1 cells, LMTK1-S34A may promote the trafficking of recycling endosomes by inhibiting (in a dominant-negative fashion) a protein that is functionally homologous to LMTK1 such as other LMTK1 family members (Tomomura et al., 2007).
Overexpression of LMTK1-WT led to conflicting effects in neurons and CHO-K1 cells; whereas LMTK1-WT promoted recycling endosome transport, as did LMTK1-S34A, in CHO-K1 cells (Takano et al., 2010), it did not affect axon length, which was similar to the effect of LMTK1-S34D in neurons (Fig. 2A). This difference is due to the difference in phosphorylation states of LMTK1 between the two cell types; LMTK1 is phosphorylated by Cdk5 in neurons but is not phosphorylated in CHO-K1 cells, which do not have Cdk5 activity. LMTK1-WT would thus acquire its inhibitory activity in neurons after phosphorylation with Cdk5. This was clearly shown in neurons in which Cdk5 activity was decreased by the knockdown of p35. The reduction of Cdk5 activity converted LMTK1-WT from a S34D-type effect to an S34A-type effect with respect to axonal outgrowth activity. To our knowledge, there are no other molecules that function in this manner, at least with regard to axonal outgrowth. This type of regulation is likely to work more efficiently than the simple on/off regulation of molecules and would allow LMTK1 to regulate the transport of membrane components to axons with respect to both the required amount and the required time. In particular, in the perinuclear region and growth cone, where Cdk5 activity targeting LMTK1 may be high, LMTK1 would be phosphorylated and would suppress Rab11A activity to maintain the proper elongation rate of the axon.
Thus, Cdk5 plays an important role in the regulation of LMTK1 activity and then axonal outgrowth. According to the above scenario, Cdk5 would be a negative factor for axonal outgrowth. There is, however, disagreement on this point. Cdk5 phosphorylates the axonal cytoskeletal proteins CRMP-2 and MAP1B to reduce their interactions with microtubules (Brown et al., 2004; Uchida et al., 2005). Activation of Cdk5 also induces the shrinkage of growth cones (Sasaki et al., 2002). These results suggest that Cdk5 plays a negative role in axon formation. In contrast, previous studies have demonstrated a positive role for Cdk5 in neurite outgrowth: neurite length is reduced by dominant-negative Cdk5 and by the inhibition of Cdk5 by antisense oligonucleotides in cerebellar and cortical neurons (Nikolic et al., 1996; Paglini et al., 1998) and by the Cdk5 inhibitor roscovitine in retinal ganglion cells (Hahn et al., 2005). Overexpression of Mib1 ubiquitin ligase inhibits neurite outgrowth, and this inhibitory effect is blocked by p35 coexpression (Choe et al., 2007). These results indicate that Cdk5-p35 has a positive effect on axon formation, prompting us to examine the role of Cdk5 in axonal outgrowth using our experimental paradigm. When p35 was knocked down with shRNA, axon length was decreased slightly in cortical neurons (data not shown), consistent with the positive role of Cdk5-p35 in axonal outgrowth (Nikolic et al., 1996; Paglini et al., 1998) rather than our earlier suggestion to the contrary. However, when we conducted similar experiments with hippocampal neurons, p35 knockdown increased axonal outgrowth slightly (data not shown). Cdk5 is a multifunctional protein kinase that phosphorylates many cytoskeletal and membrane proteins. The knockdown effect of p35 would thus appear to be the overall outcome of various effects on the cytoskeleton, membrane trafficking and so on, instead of a simple output from the Cdk5–LMTK1 pathway. To understand the role of Cdk5 in axonal outgrowth, it would be necessary to investigate more axonal substrates for Cdk5 in detail and to determine how they are coordinated and integrated in the processes that lead to axonal outgrowth.
Axon length in LMTK1-null neurons was much longer than that in wild-type neurons. What is the mechanism? Neither axon formation nor polarity was affected by knockdown or knockout of LMTK1. Several molecules with axonal outgrowth activity determine polarity rather than regulate axon length itself (Polleux and Snider, 2010), but LMTK1 is not a polarity-establishing protein. Because total neurite length did not change after the overexpression of LMTK1-S34A or the knockdown of LMTK1, axons were lengthened at the expense of the elongation of other neurites. LMTK1 may modulate the direction of membrane sorting between the axon and dendrites. Alternatively, LMTK1 may activate the dynamics of Rab11A-dependent recycling endosomes only in the axon. LMTK1 may promote a specific subtype of recycling endosomes that contains neither neurotrophic factor receptors nor β-integrin adhesion protein. It would be interesting to determine what kind of recycling endosomes are conveyed under LMTK1 regulation. Depletion of the inhibitory LMTK1 activity should release the suppression of axonal outgrowth. In axons from LMTK1 knock-out neurons, Rab11A-positive vesicles were abundant, dynamic, and rapidly transported anterogradely. These results suggest that membrane supply is a rate-limiting step in axonal elongation. The length of axons in LMTK1 knock-out neurons was similar to that observed in LMTK1-S34A-overexpressing neurons, in which the membrane supply might have been excessive as a result of the activation of the Rab11A-dependent recycling endosome pathway. The maximum elongation rate may also be determined by the ability of the cytoskeleton to lengthen the axon.
The axonal outgrowth activity of LMTK1-S34A was compromised by dominant-negative Rab11A-S25N, and LMTK1-S34D did not display negative activity in the presence of constitutively active Rab11A-Q70L, indicating that LMTK1 functions upstream of Rab11A. LMTK1 is the first and only reported factor upstream of Rab11A to act on axonal outgrowth. Regarding downstream effectors or Rab11-binding proteins, only two proteins, Rab coupling protein and protrudin, have been reported to play a role in neurite extension (Shirane and Nakayama, 2006; Eva et al., 2010). Because LMTK1 does not interact with Rab11A directly (Takano et al., 2010), there should be an unknown component that functions between LMTK1 and Rab11A (Fig. 9E, component X). Our previous results suggest that this component affects the exchange rate of guanine nucleotide bound to Rab11A (Takano et al., 2010). Because the kinase-negative mutant of LMTK1 lost axonal outgrowth inhibitory activity, as did LMTK1-S34A, it may be that LMTK1 functions by phosphorylating component X to suppress Rab11A activity (Fig. 9E). Although component X remains to be identified, we would like to propose that the Cdk5–LMTK1–Rab11A pathway is a novel signaling pathway that regulates axonal outgrowth during early neuronal development.
Footnotes
This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (S.H.).
References
- Arimura N, Kaibuchi K. Neuronal polarity: from extracellular signals to intracellular mechanisms. Nat Rev Neurosci. 2007;8:194–205. doi: 10.1038/nrn2056. [DOI] [PubMed] [Google Scholar]
- Asada A, Yamamoto N, Gohda M, Saito T, Hayashi N, Hisanaga S. Myristoylation of p39 and p35 is a determinant of cytoplasmic or nuclear localization of active cyclin-dependent kinase 5 complexes. J Neurochem. 2008;106:1325–1336. doi: 10.1111/j.1471-4159.2008.05500.x. [DOI] [PubMed] [Google Scholar]
- Ascaño M, Richmond A, Borden P, Kuruvilla R. Axonal targeting of Trk receptors via transcytosis regulates sensitivity to neurotrophin responses. J Neurosci. 2009;29:11674–11685. doi: 10.1523/JNEUROSCI.1542-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SJ, Sumerson R, Reddy CD, Berrebi AS, Flynn DC, Reddy EP. Characterization of an alternatively spliced AATYK mRNA: expression pattern of AATYK in the brain and neuronal cells. Oncogene. 2001;20:1015–1021. doi: 10.1038/sj.onc.1204209. [DOI] [PubMed] [Google Scholar]
- Bloom OE, Morgan JR. Membrane trafficking events underlying axon repair, growth, and regeneration. Mol Cell Neurosci. 2011;48:339–348. doi: 10.1016/j.mcn.2011.04.003. [DOI] [PubMed] [Google Scholar]
- Brown M, Jacobs T, Eickholt B, Ferrari G, Teo M, Monfries C, Qi RZ, Leung T, Lim L, Hall C. α2-Chimaerin, cyclin-dependent kinase 5/p35, and its target collapsin response mediator protein-2 are essential components in semaphorin 3A-induced growth-cone collapse. J Neurosci. 2004;24:8994–9004. doi: 10.1523/JNEUROSCI.3184-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung ZH, Chin WH, Chen Y, Ng YP, Ip NY. Cdk5 is involved in BDNF-stimulated dendritic growth in hippocampal neurons. PLoS Biol. 2007;5:e63. doi: 10.1371/journal.pbio.0050063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe EA, Liao L, Zhou JY, Cheng D, Duong DM, Jin P, Tsai LH, Peng J. Neuronal morphogenesis is regulated by the interplay between cyclin-dependent kinase 5 and the ubiquitin ligase mind bomb 1. J Neurosci. 2007;27:9503–9512. doi: 10.1523/JNEUROSCI.1408-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole AR, Soutar MP, Rembutsu M, van Aalten L, Hastie CJ, McLauchlan H, Peggie M, Balastik M, Lu KP, Sutherland C. Relative resistance of Cdk5-phosphorylated CRMP2 to dephosphorylation. J Biol Chem. 2008;283:18227–18237. doi: 10.1074/jbc.M801645200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–759. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8:1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo R, Saito T, Asada A, Kawahara H, Ohshima T, Hisanaga S. Commitment of 1-methyl-4-phenylprinidinium ion-induced neuronal cell death by proteasome-mediated degradation of p35 cyclin-dependent kinase 5 activator. J Biol Chem. 2009;284:26029–26039. doi: 10.1074/jbc.M109.026443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eva R, Dassie E, Caswell PT, Dick G, ffrench-Constant C, Norman JC, Fawcett JW. Rab11 and its effector Rab coupling protein contribute to the trafficking of β1 integrins during axon growth in adult dorsal root ganglion neurons and PC12 cells. J Neurosci. 2010;30:11654–11669. doi: 10.1523/JNEUROSCI.2425-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M. Distinct Rab binding specificity of Rim1, Rim2, rabphilin, and Noc2: identification of a critical determinant of Rab3A/Rab27A recognition by Rim2. J Biol Chem. 2003;278:15373–15380. doi: 10.1074/jbc.M212341200. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Kanno E, Ishibashi K, Itoh T. Large scale screening for novel rab effectors reveals unexpected broad Rab binding specificity. Mol Cell Proteomics. 2008;7:1031–1042. doi: 10.1074/mcp.M700569-MCP200. [DOI] [PubMed] [Google Scholar]
- Gaozza E, Baker SJ, Vora RK, Reddy EP. AATYK: a novel tyrosine kinase induced during growth arrest and apoptosis of myeloid cells. Oncogene. 1997;15:3127–3135. doi: 10.1038/sj.onc.1201575. [DOI] [PubMed] [Google Scholar]
- Hahn CM, Kleinholz H, Koester MP, Grieser S, Thelen K, Pollerberg GE. Role of cyclin-dependent kinase 5 and its activator P35 in local axon and growth cone stabilization. Neuroscience. 2005;134:449–465. doi: 10.1016/j.neuroscience.2005.04.020. [DOI] [PubMed] [Google Scholar]
- Hisanaga S, Endo R. Regulation and role of cyclin-dependent kinase activity in neuronal survival and death. J Neurochem. 2010;115:1309–1321. doi: 10.1111/j.1471-4159.2010.07050.x. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Chihara K, Arimura N, Ménager C, Kawano Y, Matsuo N, Nishimura T, Amano M, Kaibuchi K. CRMP-2 induces axons in cultured hippocampal neurons. Nat Neurosci. 2001;4:781–782. doi: 10.1038/90476. [DOI] [PubMed] [Google Scholar]
- Kawauchi T, Chihama K, Nishimura YV, Nabeshima Y, Hoshino M. MAP1B phosphorylation is differentially regulated by Cdk5/p35, Cdk5/p25, and JNK. Biochem Biophys Res Commun. 2005;331:50–55. doi: 10.1016/j.bbrc.2005.03.132. [DOI] [PubMed] [Google Scholar]
- Kesavapany S, Lau KF, Ackerley S, Banner SJ, Shemilt SJ, Cooper JD, Leigh PN, Shaw CE, McLoughlin DM, Miller CC. Identification of a novel, membrane-associated neuronal kinase, cyclin-dependent kinase 5/p35-regulated kinase. J Neurosci. 2003;23:4975–4983. doi: 10.1523/JNEUROSCI.23-12-04975.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuta Y, Hibi M, Arai T, Nakamura S, Kawano H. Defects in reciprocal projections between the thalamus and cerebral cortex in the early development of Fezl-deficient mice. J Comp Neurol. 2007;503:454–465. doi: 10.1002/cne.21401. [DOI] [PubMed] [Google Scholar]
- Lindsay AJ, McCaffrey MW. Rab11-FIP2 functions in transferrin recycling and associates with endosomal membranes via its COOH-terminal domain. J Biol Chem. 2002;277:27193–27199. doi: 10.1074/jbc.M200757200. [DOI] [PubMed] [Google Scholar]
- Ng EL, Tang BL. Rab GTPases and their roles in brain neurons and glia. Brain Res Rev. 2008;58:236–246. doi: 10.1016/j.brainresrev.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Nikolic M, Dudek H, Kwon YT, Ramos YF, Tsai LH. The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 1996;10:816–825. doi: 10.1101/gad.10.7.816. [DOI] [PubMed] [Google Scholar]
- Paglini G, Pigino G, Kunda P, Morfini G, Maccioni R, Quiroga S, Ferreira A, Cáceres A. Evidence for the participation of the neuron-specific CDK5 activator P35 during laminin-enhanced axonal growth. J Neurosci. 1998;18:9858–9869. doi: 10.1523/JNEUROSCI.18-23-09858.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- Polleux F, Snider W. Initiating and growing an axon. Cold Spring Harb Perspect Biol. 2010;2:a001925. doi: 10.1101/cshperspect.a001925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Konno T, Hosokawa T, Asada A, Ishiguro K, Hisanaga S. p25/cyclin-dependent kinase 5 promotes the progression of cell death in nucleus of endoplasmic reticulum-stressed neurons. J Neurochem. 2007;102:133–140. doi: 10.1111/j.1471-4159.2007.04540.x. [DOI] [PubMed] [Google Scholar]
- Sann S, Wang Z, Brown H, Jin Y. Roles of endosomal trafficking in neurite outgrowth and guidance. Trends Cell Biol. 2009;19:317–324. doi: 10.1016/j.tcb.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Cheng C, Uchida Y, Nakajima O, Ohshima T, Yagi T, Taniguchi M, Nakayama T, Kishida R, Kudo Y, Ohno S, Nakamura F, Goshima Y. Fyn and Cdk5 mediate semaphorin-3A signaling, which is involved in regulation of dendrite orientation in cerebral cortex. Neuron. 2002;35:907–920. doi: 10.1016/s0896-6273(02)00857-7. [DOI] [PubMed] [Google Scholar]
- Shirane M, Nakayama KI. Protrudin induces neurite formation by directional membrane trafficking. Science. 2006;314:818–821. doi: 10.1126/science.1134027. [DOI] [PubMed] [Google Scholar]
- Sönnichsen B, De Renzis S, Nielsen E, Rietdorf J, Zerial M. Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J Cell Biol. 2000;149:901–914. doi: 10.1083/jcb.149.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Tsutsumi K, Saito T, Asada A, Tomomura M, Fukuda M, Hisanaga S. AATYK1A phosphorylation by Cdk5 regulates the recycling endosome pathway. Genes Cells. 2010;15:783–797. doi: 10.1111/j.1365-2443.2010.01419.x. [DOI] [PubMed] [Google Scholar]
- Tang BL. Protein trafficking mechanisms associated with neurite outgrowth and polarized sorting in neurons. J Neurochem. 2001;79:923–930. doi: 10.1046/j.1471-4159.2001.00674.x. [DOI] [PubMed] [Google Scholar]
- Tomomura M, Fernandez-Gonzales A, Yano R, Yuzaki M. Characterization of the apoptosis-associated tyrosine kinase (AATYK) expressed in the CNS. Oncogene. 2001;20:1022–1032. doi: 10.1038/sj.onc.1204210. [DOI] [PubMed] [Google Scholar]
- Tomomura M, Morita N, Yoshikawa F, Konishi A, Akiyama H, Furuichi T, Kamiguchi H. Structural and functional analysis of the apoptosis-associated tyrosine kinase (AATYK) family. Neuroscience. 2007;148:510–521. doi: 10.1016/j.neuroscience.2007.05.048. [DOI] [PubMed] [Google Scholar]
- Tsutsumi K, Tomomura M, Furuichi T, Hisanaga S. Palmitoylation-dependent endosomal localization of AATYK1A and its interaction with Src. Genes Cells. 2008;13:949–964. doi: 10.1111/j.1365-2443.2008.01219.x. [DOI] [PubMed] [Google Scholar]
- Tsutsumi K, Takano T, Endo R, Fukuda M, Ohshima T, Tomomura M, Hisanaga S. Phosphorylation of AATYK1 by Cdk5 suppresses its tyrosine phosphorylation. PLoS One. 2010;5:e10260. doi: 10.1371/journal.pone.0010260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida Y, Ohshima T, Sasaki Y, Suzuki H, Yanai S, Yamashita N, Nakamura F, Takei K, Ihara Y, Mikoshiba K, Kolattukudy P, Honnorat J, Goshima Y. Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer's disease. Genes Cells. 2005;10:165–179. doi: 10.1111/j.1365-2443.2005.00827.x. [DOI] [PubMed] [Google Scholar]
- Ullrich O, Reinsch S, Urbé S, Zerial M, Parton RG. Rab11 regulates recycling through the pericentriolar recycling endosome. J Cell Biol. 1996;135:913–924. doi: 10.1083/jcb.135.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka N, Kimura-Kuroda J, Saito T, Kawamura K, Hisanaga S, Kawano H. Small molecule inhibitor of type I transforming growth factor-beta receptor kinase ameliorates the inhibitory milieu in injured brain and promotes regeneration of nigrostriatal dopaminergic axons. J Neurosci Res. 2011;89:381–393. doi: 10.1002/jnr.22552. [DOI] [PubMed] [Google Scholar]