

Abstract
Accumulation of β-amyloid (Aβ) inside brain neurons is an early and crucial event in Alzheimer's disease (AD). Studies in brains of AD patients and mice models of AD suggested that cholesterol homeostasis is altered in neurons that accumulate Aβ. Here we directly investigated the role of intracellular oligomeric Aβ42 (oAβ42) in neuronal cholesterol homeostasis. We report that oAβ42 induces cholesterol sequestration without increasing cellular cholesterol mass. Several features of AD, such as endosomal abnormalities, brain accumulation of Aβ and neurofibrillary tangles, and influence of apolipoprotein E genotype, are also present in Niemann-Pick type C, a disease characterized by impairment of intracellular cholesterol trafficking. These common features and data presented here suggest that a pathological mechanism involving abnormal cholesterol trafficking could take place in AD. Cholesterol sequestration in Aβ-treated neurons results from impairment of intracellular cholesterol trafficking secondary to inhibition of protein prenylation. oAβ42 reduces sterol regulatory element-binding protein-2 (SREBP-2) cleavage, causing decrease of protein prenylation. Inhibition of protein prenylation represents a mechanism of oAβ42-induced neuronal death. Supply of the isoprenoid geranylgeranyl pyrophosphate to oAβ42-treated neurons recovers normal protein prenylation, reduces cholesterol sequestration, and prevents Aβ-induced neurotoxicity. Significant to AD, reduced levels of protein prenylation are present in the cerebral cortex of the TgCRND8 mouse model. In conclusion, we demonstrate a significant inhibitory effect of Aβ on protein prenylation and identify SREBP-2 as a target of oAβ42, directly linking Aβ to cholesterol homeostasis impairment.
Introduction
Alzheimer's disease (AD), the most common form of age-related dementia, is characterized by accumulation of β-amyloid peptide (Aβ) and neurofibrillary tangles in the brain (Selkoe, 2004). Intraneuronal accumulation of Aβ precedes neurofibrillary tangles and Aβ plaques deposition (D'Andrea et al., 2001; Wirths et al., 2002) and has been causally linked to cell death (LaFerla et al., 1995; Chui et al., 2001; Zhang et al., 2002; Blanchard et al., 2003; Casas et al., 2004; Christensen et al., 2008, 2010; Tomiyama et al., 2010), synaptic dysfunction (Takahashi et al., 2002; Oddo et al., 2003), and cognitive impairment (Billings et al., 2005). Aβ42 is degraded poorly inside neurons and accumulates in vesicles identified as lysosomes or late endosomes/multivesicular bodies (MVBs) (D'Andrea et al., 2001; Ditaranto et al., 2001; Takahashi et al., 2002; Langui et al., 2004). Aβ accumulation in the endocytic pathway causes endosomal alterations characteristic of AD (Nixon, 2005). These endosomal abnormalities are similar to those observed in patients with Niemann-Pick type C disease (NPC), a disorder characterized by impairment of intracellular cholesterol trafficking (Yamazaki et al., 2001; Runz et al., 2002; Saito et al., 2002; Jin et al., 2004; Nixon, 2005; Koh and Cheung, 2006). NPC and AD share other features. Inhibition of intracellular cholesterol transport increases amyloidogeneic cleavage of amyloid precursor protein (APP) and Aβ production in animal models of NPC and in neurons treated with U18666A (3-β-[2-(diethylamino)ethoxy]androst-5-en-17-one), a chemical agent that induces cholesterol sequestration and mimics some aspects of the NPC phenotype (Runz et al., 2002). Conversely, studies in human AD brains have shown cholesterol or cholesterol esters accumulation specifically in Aβ-immunopositive neurons (Ohm et al., 2003; Gómez-Ramos and Asunción Morán, 2007; Xiong et al., 2008). In the AD mouse model APP/PS1, brain cholesterol accumulation was preceded by Aβ accumulation, suggesting that Aβ could regulate cholesterol homeostasis and/or trafficking (Fernández et al., 2009). We have undertaken this study to examine the effects of Aβ accumulation on neuronal cholesterol homeostasis. Here we report that oligomeric Aβ42 (oAβ42) causes cholesterol sequestration within primary neurons. Cholesterol accumulation is secondary to decreased protein prenylation and reduced intracellular trafficking. The inhibition of protein prenylation is also a mechanism of oAβ42 neurotoxicity and can be prevented by replenishing the neuronal pool of isoprenoids.
Materials and Methods
Reagents.
Aβ42 was purchased from American Peptide. The same lot number was used throughout this study. Reverse Aβ42 (revAβ42) was obtained from Alpha Diagnostic. Leibovitz L-15–CO2 culture medium was from Invitrogen. Mouse NGF (2.5 S) was purchased from Alomone Labs. EZ-Link NHS-S-S-biotin and ImmunoPure immobilized Streptavidin were purchased from Pierce. Amplex Red cholesterol assay kit was from Invitrogen. Immobilon polyvinylidene difluoride (PVDF) was from Bio-Rad. Enhanced chemiluminescence reagents were from GE Healthcare. U18666A was purchased Biomol. Geranylgeranyl pyrophosphate (GGPP) was from Sigma-Aldrich and [3H]GGPP from PerkinElmer Life and Analytical Sciences. Filipin, methyl β cyclodextrin, heptane, and diisopropylether were purchased from Sigma. GGTI-2133 (4-[[N-(Imidazol-4-yl)methyleneamino]-2-(1-naphthyl)benzoyl]leucine) was obtained from Calbiochem. All other reagents were from Thermo Fisher Scientific.
Culture of neurons.
Rat basal forebrain and sympathetic neurons were prepared and cultured as described previously (Saavedra et al., 2007). Basal forebrain cholinergic neurons were isolated from brains of E17–E18 Sprague Dawley rats of either sex (Health Science Lab Animal Services, University of Alberta, Edmonton, AB, Canada) by enzymatic dissociation with 0.1% w/v of trypsin for 15 min and plated in poly-d-lysine-coated plates in Neurobasal medium supplemented with MEM, HEPES, penicillin/streptomycin/l-glutamine, and B27. Sympathetic neurons isolated from superior cervical ganglia of newborn Harlan Sprague Dawley rats of either sex (Health Science Lab Animal Services, University of Alberta) were prepared by enzymatic dissociation with 1% (w/v) of collagenase and 0.1% (w/v) of trypsin followed by mechanical dissociation using a flame-polished Pasteur pipette. After filtration of the cell suspension through cell strainer (40 μm nylon; Falcon), neurons were plated. Standard culture medium was L-15–CO2 supplemented with 0.4% methylcellulose. Non-neuronal cells were eliminated with 10–15 μm cytosine arabinoside during the first 5–6 d in culture. Two types of neuronal cultures were used: mass cultures and compartmented cultures. For mass cultures, neurons were plated in 24-well dishes at a density of 1–2 ganglia per well in medium supplemented with 2.5% rat serum, 1 mg/ml ascorbic acid, and 50 ng/ml NGF. Alternatively, sympathetic neurons were cultured in three-compartment culture dishes as described previously (Saavedra et al., 2007). Briefly, dissociated neurons were plated in the center compartment at a density of 0.2 ganglia per dish in medium containing 2.5% rat serum, 1 mg/ml ascorbic acid, 10–15 μm cytosine arabinoside, and 10 ng/ml NGF. After 5–6 d, cytosine arabinoside treatment was discontinued, and NGF (50 ng/ml) was confined to the side compartments. All experiments started at day 7–8 in culture. Rat cortical neurons were prepared according to Brewer and Torricelli (2007).
oAβ42 preparation.
oAβ42 was prepared following a published protocol (Dahlgren et al., 2002). Aβ peptide was initially dissolved to 1 mm in hexafluoroisopropanol and separated into aliquots in sterile microcentrifuge tubes. Hexafluoroisopropanol was dried under a stream of N2, and the peptide film was desiccated at −20°C. The peptide was resuspended in Me2SO at a concentration of 5 mm. L-15–CO2 medium (phenol red-free, antibiotic-free, and serum-free) was added to bring the peptide to a final concentration of 100 μm and incubated at 4°C for 24 h. All oAβ42 preparations used were similar to those characterized previously (Dahlgren et al., 2002; Saavedra et al., 2007) in that they contain monomers and oligomers of Aβ but do not contain fibrils. The neurotoxic ability of these Aβ preparations toward primary neurons has been characterized previously (Song et al., 2006).
Immunofluorescence and confocal microscopy.
For confocal microscopy studies, neurons were fixed with 4% paraformaldehyde for 20 min at room temperature, permeabilized with 0.3% saponin for 15 min at room temperature, and blocked by incubation with 2% bovine serum albumin for 1 h on ice. Primary antibodies were prepared in the same blocking solution. Primary antibodies used include the following: anti-lamp1 (sc-8098; Santa Cruz Biotechnology) at 1:100, anti-EEA1 (sc-33585; Santa Cruz Biotechnology) at 1:100, anti-COX-IV (ab14744; Abcam) at 1:400, and anti-giantin (a generous gift from Dr. Hobman, University of Alberta) at 1:300. Incubation was performed overnight at 4°C. Secondary antibodies (Alexa Fluor 594-labeled or Alexa Fluor 488-labeled anti-mouse or anti-rabbit) prepared in blocking buffer (1:1000) were incubated for 2 h at room temperature. For filipin staining, fixed neurons were incubated with 100 μg/ml filipin (Sigma) at room temperature for 2 h. Nuclei were stained with Draq5 (Biostatus Limited) at 1:4000. Pictures were taken using a laser scanning confocal microscope Carl Zeiss LSM 710 equipped with an S-Fluor 40×/1.3 NA oil-immersion objective using appropriate filter sets and excitation wavelengths. Within an experiment, all pictures were taken with the same setting for a particular fluorophore. If required for printing purposes, brightness and contrast were adjusted using Photoshop software (Adobe Systems). All images were adjusted with the same parameters. The pictures selected are representative of at least three separate experiments performed under identical conditions.
Cholesterol mass.
Cholesterol mass from cultured neurons was measured using the enzymatic Amplex red cholesterol assay kit or by gas chromatography (Lipid and Lipid Metabolite Analysis Core Facility, University of Alberta). Results obtained by the two methods were identical.
Cholesterol synthesis.
Cholesterol synthesis was determined by measuring incorporation of [3H]acetate using methods described by de Chaves et al. (1997) and modified as described herein. Neurons received the treatment indicated in each case with the addition of the radioactive cholesterol precursor [1-3H]acetic acid (100 μCi/ml) for the last 2 h. The radioactive medium was removed, neurons were washed twice with cold PBS, and cellular material was harvested and sonicated. An aliquot was separated for protein determination. A second aliquot was used for lipid isolation and quantification. Lipids were isolated from the cell lysate according to the method of Folch (Folch et al., 1959) with chloroform/methanol/water ratio of 2:1:1 (v/v). Individual lipid separation was accomplished by thin-layer chromatography (TLC). TLC plates were developed in the solvent system heptane/isopropyl ether/acetic acid (60:40:4, v/v) using unlabeled cholesterol as carrier. The band corresponding to authentic unesterified cholesterol was scraped from the plate, and radioactivity was measured. Radioactivity was normalized to protein mass and expressed in disintegration per minute (dpm) per micrograms protein. At least four cultures per treatment were used. To combine three or more experiments, the results were expressed compared with the untreated neurons, which were given a value of 100%.
Cholesterol trafficking.
Neurons were cultured in compartmented dishes. [3H]Acetate at 150 μCi/ml was added to the cell-body-containing compartment for the last 24 h of treatment. After rinsing, the cellular material of the cell-body-containing compartment and the distal-axon-containing compartment was harvested separately. Lipid were extracted and separated by TLC as indicated above. Radioactivity of [3H]cholesterol was expressed as % (total) = dpm in cell bodies or distal axons/(dpm in cell bodies + dpm in distal axons).
Immunoblot analysis.
Proteins were separated by SDS-PAGE using gels at 10 or 12% containing 0.1% SDS. Transfer of proteins to PVDF membranes was performed overnight at 4°C in 25 mm Tris, 192 mm glycine, and 16% methanol buffer, pH 8.3. Membranes were blocked for 1 h in Tris-buffered saline, 0.1% Tween 20 (TTBS) containing 5% nonfat milk (blocking buffer) and incubated overnight in the primary antibody solution prepared in TTBS containing 5% nonfat milk. Primary antibodies for β-actin (catalog #4967, 1:1000; Cell Signaling Technology), sterol regulatory element-binding protein-2 (SREBP-2) (ab-30682,1:500; Abcam), β-tubulin (T4026, 1:200; Sigma), Ras (catalog #05-516, 1:500; Millipore), Flotillin-1 (catalog #610821, 1:1000; BD Transduction Labs), Rab7 (catalog #R8779, 1:1000; Sigma), and Rab9 (ab2810, 1:500; Abcam) were used. Membranes were washed two times with Tris-buffered saline, two times with TTBS, and two times with Tris-buffered saline and then incubated for 1 h with the secondary antibody (1:2000) in blocking buffer at room temperature with gentle agitation. Immunoreactivity was detected by ECL Plus Western Blotting detection system (GE Healthcare). UN-SCAN-IT gel 5.3 software was used for semiquantification of the bands on immunoblots.
Biotinylation and retrograde transport of NGF receptor TrkA.
Surface axonal proteins were biotinylated by incubation with 1.5 mg/ml of the membrane-impermeable derivative of biotin (sulfo-NHS-S-S-biotin) (Pierce) in Leibovitz L-15–CO2 medium (free of NGF, phenol red, antibiotics, and serum) for 20 min at 4°C, in the side compartments of neurons cultured in compartmented dishes. Remaining reactive biotin reagent was quenched by incubating cells with 50 mm glycine in modified PBS twice each for 10 min at 4°C. Immediately after, fresh medium containing NGF without or with 20 μm oAβ1–42 was added to the axons, and neurons were incubated under regular conditions for 6 and 18 h. NGF induces TrkA activation, endocytosis, and retrograde transport. The cellular material from cell-body-containing compartment and distal-axon-containing compartments was collected separately (two dishes were pooled) and lysed in modified RIPA buffer (1% Triton X-100, 0.1% SDS, 0.5% deoxycholic acid, 50 mm Tris-HCl, 100 mm NaCl, 1 mm EDTA, 2 mm EGTA, 50 mm NaF, 10 mm sodium pyrophosphate, and 1 mm sodium orthovanadate, pH 7.4, with protease inhibitor mixture) at 4°C for 30 min. The lysate was centrifuged at 16,000 × g for 15 min at 4°C, and the resulting supernatants were incubated with 40 μl of 50% Streptavidin agarose beads (Pierce) overnight at 4°C. The beads were then washed three times with modified RIPA buffer, and the bound proteins were eluted with SDS sample buffer containing dithiothreitol (50 mm final concentration) boiling for 5 min. Biotinylated proteins were separated by SDS-PAGE using 8% gels. Immunoblot analysis of TrkA was performed as described previously (Song et al., 2006). In all cases, the supernatants of incubation with Streptavidin agarose beads were examined by SDS-PAGE, protein transfer to PDVF membrane, and biotinylated protein detection with Streptavidin linked to horseradish peroxidase and ECL to ensure the complete binding of biotinylated proteins to the beads.
Preparation of membrane and cytosol fractions from cultured neurons.
Neurons were harvested in 10 mm HEPES, pH 7.6, containing 250 mm sucrose, 10 mm KCl, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA and protease inhibitor cocktail, 1 mm NaF, and 2 mm NaVO4. Cellular material was homogenized by passing 20 times through a 25 gauge needle. After resting 20 min on ice, the cell homogenate was centrifuged at 1000 × g for 10 min at 4°C to eliminate unbroken cells and cellular debris. Protein content of the supernatant was determined (BCA; Pierce) and equal amounts of protein from each treatment were centrifuged at 105,000 × g for 1 h at 4°C in a Beckman Coulter Optima MAX-XP Utracentrifuge using the MLA-130 fixed-angle rotor. Supernatants (cytosol fractions) and the pellets (membrane fractions) were separated.
Preparation of membrane and cytosol fractions from mice brains.
Frontal cortices from 10-week-old male wild-type and TgCRND8 mice were cut in small pieces and homogenized in the same buffer used for membrane/cytosol separation from cultured neurons by passing through a 22 gauge needle 15 times. After 20 min on ice, the homogenate was cleared by centrifugation at 1000 × g for 10 min at 4°C. The supernatant was centrifuged at 105,000 × g for 1 h at 4°C in a Beckman Coulter Optima MAX-XP Utracentrifuge using the MLA-130 fixed-angle rotor. The supernatant of this is the cytosol fraction, and the pellet represents the membrane fraction.
Protein prenylation.
Protein prenylation was examined by extraction of prenylated proteins with Triton X-114 or recombinant GDP dissociating inhibitors fused to glutathione S-transferase (GST–GDI).
Separation of prenylated and unprenylated Rab using Triton X-114.
The proportion of prenylated and unprenylated Rabs were examined using Triton X-114 according to Coxon et al. (2005). Two-phase mixtures of Triton X-114 were formed at 37°C so that lipophilic, prenylated Rabs partition into the detergent-rich phase, whereas unprenylated Rabs remain in the aqueous phase. For experiments with cultured neurons, neurons were harvested at the end of the corresponding treatments in Tris buffer containing Triton X-114 (20 mm Tris-HCl, pH 7.5, containing 150 mm NaCl, 1% Triton X-114, and protease inhibitor cocktail) and incubated for 15–20 min at 4°C. Lysates were cleared by centrifugation at 13,000 × g for 15 min at 4°C. Protein content was determined in the clear supernatants. For experiments in mice brains, frontal cortices from 10-week-old wild-type and APP-Tg (TgCRND8) mice were lysed separately with Triton X-114 as for cultured neurons. In all cases, equal amounts of proteins were adjusted to the same volume with Tris buffer containing Triton X-114, were loaded on top of a cushion solution (20 mm Tris-HCl, pH 7.4, containing 150 mm NaCl, 6% sucrose, 0.06% Triton X-114, and protease inhibitor cocktail) and were incubated at 37°C for 10 min. After centrifugation at 16,000 × g for 5 min, the top aqueous clear layer was separated from the detergent-rich, oil-like droplet. Both layers were boiled with SDS sample buffer, and proteins were resolved by 12% SDS-PAGE. Immunoblot analysis was performed with anti-Rab9 (ab-2810, 1:500; Abcam) and/or anti-Rab7 (R8779, 1:1000; Sigma). Notice that prenylated Rabs always run faster than unprenylated Rabs under our experimental SDS-PAGE conditions. The lower band is present in the detergent phase.
Rab–GDI capture.
GDI capture was performed according to Narita et al. (2005). Neuronal lysates were prepared by homogenization of cultured neurons in buffer containing 250 mm sucrose, 3 mm imidazole, pH 7.4, with 1 mm DTT and protease inhibitors by passing through a 22 gauge needle 10 times on ice, followed by centrifugation at 300 × g for 5 min to eliminate unbroken cells and cellular debris. Alternatively cytosol and membranes fractions from neuronal cultures or mice brains prepared as indicated above were used. Protein content was measured, and equal amounts of proteins from all samples were diluted fivefold with extraction buffer (30 mm HEPES, 75 mm potassium acetate, 5 mm MgCl2, 100 μm ATP, and 500 μm GDP) containing 10 μm GDI–GST and incubated for 20 min at 30°C. Rabs bound to GDI were recovered by incubating with glutathione-Sepharose 4B beads (GE Healthcare), eluted by boiling with SDS-sample buffer for 10 min, and analyzed by SDS-PAGE and immunoblotting with anti Rab7 or anti Rab9 antibodies. As indicated for Triton X-114 extraction, prenylated Rabs ran faster than unprenylated Rabs. The lower band is the only band present in membrane preparations, as is the band that binds to GDI.
Incorporation of exogenous GGPP in prenylated proteins.
Neurons were treated with 20 μm oAβ42 or 50 μm pravastatin. [3H]GGPP (22.4 Ci/mmol) was added at a concentration of 200 μCi/ml (9.1 μm). After 24 h, neurons were rinsed several times, harvested, and lysed. Radioactivity was counted in an aliquot of the neuronal lysate and was used to calculate GGPP uptake. Proteins in neuronal lysates were separated and identified by SDS-PAGE and immunoblotting for Rab7 and actin. PDVF membranes were dyed with Ponceau Red, and bands corresponding to the molecular weight of Rab7 were cut for radioactivity counting. Radioactivity of the rest of the lane was counted separately. The results were referred to actin content and expressed in disintegration per minute in Rabs/actin pixels or total protein disintegration per minute/actin pixels.
Evaluation of metabolic activity.
Metabolic activity was evaluated using the cell 3-(4,5-dimethylthiozolyl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay (Promega). This assay is based on the conversion of MTT to a blue/purple formazan crystal by reduced or phosphorylated nicotinamide adenine dinucleotide (NADH or NADPH) produced by dehydrogenase enzymes in metabolically active cells (Berridge and Tan, 1993). After neuron treatment, MTT was added to the medium, and incubation continued for 1 h in CO2 incubator at 37°C. The blue formazan product of MTT reduction was visualized under the microscope. Direct observation of decreased color formation in bright-field images is indicative of decreased metabolic activity.
Statistical analysis.
Prism 4 computer software (GraphPad Software) was used. Data were from three to five experiments with three to five determinations each. Values were expressed as means ± SE. Statistical significance was analyzed using one-way ANOVA Kruskal–Wallis statistic with Dunn's multiple comparison test and is indicated (*p < 0.05, **p < 0.01, or ***p < 0.001). In experiments with only two groups, t test was used.
Results
Aβ42 causes intracellular accumulation of endogenous cholesterol
Because of poor degradation, Aβ42 accumulates inside neurons with dramatic consequences. We observed that, in neurons exposed to toxic concentrations of oAβ42, there was extensive accumulation of unesterified cholesterol as detected by filipin staining (Fig. 1A). This effect was specific to oAβ42 because revAβ42 did not induce cholesterol accumulation (Fig. 1A). The pattern of cholesterol staining with filipin in oAβ42-treated neurons resembled, but was not identical to that displayed by neurons treated with the amphiphilic amine U18666A, conventionally used to mimic the accumulation of cholesterol characteristic of NPC phenotype (Karten et al., 2002). Cholesterol significantly colocalized with LAMP1, a marker for MVBs/lysosomes, and partially colocalized with giantin a marker for the Golgi (Fig. 1B). No evidence of colocalization of cholesterol with EEA1, a marker for early endosomes, or COX-IV, a marker for mitochondria, was observed.
Figure 1.
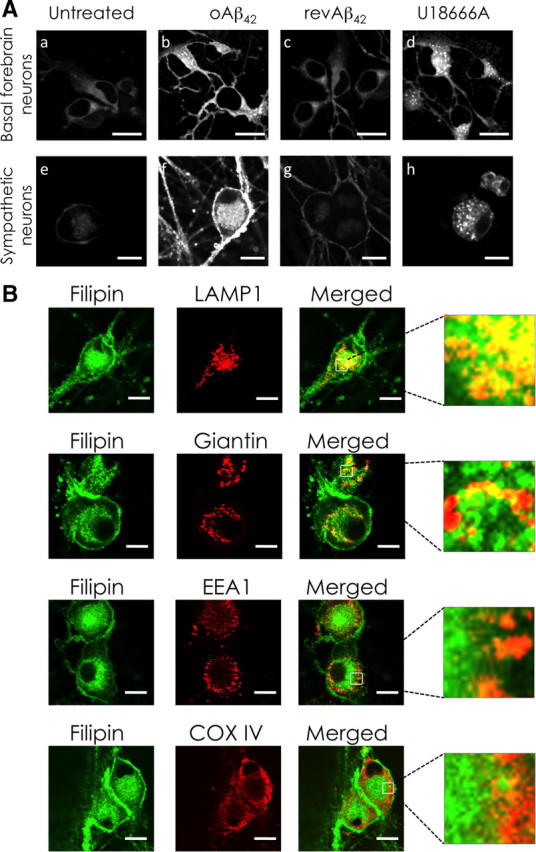
oAβ42 causes intracellular accumulation of endogenous cholesterol. A, Cholesterol accumulation: basal forebrain and sympathetic neurons were treated with 20 μm oAβ42, 20 μm revAβ42, or 3 μm U18666A for 24 h in serum-free media. Neuronal cholesterol was detected with filipin staining and confocal microscopy. B, Intracellular cholesterol distribution: sympathetic neurons were treated with 20 μm oAβ42 for 24 h. Intracellular localization of cholesterol accumulation was examined by immunofluorescence confocal microscopy using LAMP1 as a marker of late endosome/MVBs, giantin as a Golgi marker, EEA1 as a marker of early endosomes, and COX-IV as mitochondria marker. Filipin was used to detect cholesterol (green pseudo color). Scale bars, 10 μm.
To determine whether the increased filipin fluorescence displayed by oAβ42-treated neurons represented a bona fide increase in cholesterol content, we measured cholesterol mass but we did not detect any significant change induced by oAβ42 (Fig. 2A). This finding was not surprising because cholesterol mass is not elevated in NPC neurons despite that they sequester cholesterol intracellularly (Karten et al., 2002). The simultaneous occurrence of increased cellular filipin staining with normal cellular cholesterol levels in NPC neurons has been explained by mislocalization of cellular cholesterol attributable to intracellular trafficking dysfunction. Notably, our previous work using neurons cultured in compartmented dishes suggested that oAβ42-induced apoptosis was attributable in part to decrease in trafficking of survival signals to cell bodies (Song et al., 2006). Therefore, we directly tested the effect of oAβ42 on trafficking in neurons in compartmented cultures (Fig. 2B). We found that intracellular trafficking of cholesterol was significantly reduced in neurons challenged with oAβ42 as it was in U18666A-treated neurons (Fig. 2C). The inhibitory effect of U18666A on axonal transport of endogenous cholesterol had been reported previously (Karten et al., 2003). We also investigated the trafficking of proteins by assessing the retrograde transport of the NGF receptor TrkA, which reaches the cell bodies by vesicular transport. We observed significant decrease in TrkA retrograde trafficking in neurons challenged with oAβ42 (Fig. 2D).
Figure 2.

oAβ42 does not increase cholesterol mass but impairs intracellular trafficking. A, Basal forebrain neurons and sympathetic neurons were treated with 20 μm oAβ42 for 24 h in the absence of serum. Cholesterol mass was measured using Amplex red. B, The three-compartment model for culture neurons (a) illustrates an entire culture and (b) is an enlargement of a single track of a culture in which neurons are plated in the center compartment. Neurites extend under silicone grease barriers and into complete separate fluid environments of left and right compartments. Cellular material from the center (cell bodies/proximal axons) and side (distal axons) compartments can be harvested and processed separately. C, Cholesterol trafficking. Sympathetic neurons cultured in compartmented dishes were treated with 20 μm oAβ42 in all compartments or 1.5 μm U18666A in cell bodies/proximal axons for 48 h. During the final 24 h of incubation, 150 μCi/ml [3H]acetate was included in the medium of the center compartment. At the end of the incubation, cell bodies/proximal neurites and distal axons were harvested separately. Three dishes were combined for each sample. Cellular lipids were extracted and separated by TLC, and radioactivity was measured in unesterified cholesterol. Data are expressed as radioactivity in unesterified cholesterol in distal axons as percentages of total radioactivity present in unesterified cholesterol in cell bodies/proximal neurites + distal axons. Data represent means ± SE of three independent experiments performed in quintuplicate. *p < 0.05, **p < 0.01, one-way ANOVA. D, Retrograde transport of NGF receptor TrkA. Sympathetic neurons were cultured in three-compartment dishes. Surface axonal proteins were biotinylated in the absence of NGF at 4°C for 30 min just before the addition of 20 μm oAβ1–42 in the axons. Treatment with oAβ1–42 was performed in the presence of 50 ng/ml NGF. After 6 and 18 h, the material from the cell-body-containing compartment and distal-axon-containing compartment was independently harvested (2 dishes were pooled). Biotinylated proteins were pooled down from the cell lysate with Streptavidin agarose, and the proteins in the pellet were resolved by SDS-PAGE. Total TrkA and activated TrkA (pTrkA) were detected by immunoblot analysis. Non-biotinylated proteins in the supernatant were analyzed by SDS-PAGE and immunoblot analysis for tubulin as loading control. The experiment was repeated twice.
Together, our findings indicate that oAβ42 inhibits intracellular trafficking and causes sequestration of cholesterol in late endosomes/MVBs and partially at the Golgi.
Aβ inhibits cholesterol synthesis by interfering with SREBP-2 processing
Under our experimental conditions, cholesterol sequestered in neurons challenged with oAβ42 derives from endogenous synthesis exclusively, because neurons were cultured in the absence of serum during the treatments with oAβ42. Thus, we attempted to prevent cholesterol accumulation by inhibiting cholesterol synthesis with pravastatin (de Chaves et al., 1997; Saavedra et al., 2007). Contrary to our expectations, cholesterol still accumulated in neurons that received pravastatin with oAβ42 (Fig. 3A), despite the effectiveness of pravastatin in reducing cholesterol synthesis (Fig. 3B).
Figure 3.

oAβ inhibits the mevalonate pathway. A, Inhibition of cholesterol synthesis does not prevent cholesterol sequestration. Sympathetic neurons were treated with 20 μm oAβ42, 50 μm pravastatin or 20 μm oAβ42 and 50 μm pravastatin for 24 h in serum-free media. Cholesterol was detected with filipin and examined by confocal microscopy. Scale bars, 10 μm. B, oAβ inhibits cholesterol synthesis. Basal forebrain, sympathetic, and cortical neurons were treated with oAβ42 (20 μm), pravastatin (50 μm), or revAβ42 (20 μm) for 24 h (sympathetic and basal forebrain neurons) or 48 h (cortical neurons). [3H]Acetate (100 μCi/ml) was added for the last 2 h of treatment. Lipids were extracted and separated by TLC. Incorporation of [3H]acetate into unesterified cholesterol was calculated as disintegration per minute and per microgram of protein and expressed as percentage of values obtained for untreated neurons. The results are the means ± SE of three experiments performed in three to five replicates. C, Dose–response of oAβ42 effect on cholesterol synthesis. The same protocol as in B was used. The results are the means ± SE of three experiments performed in three to five replicates. D, Dose–response of oAβ42-induced apoptosis. Neurons were incubated with different concentrations of oAβ42 or with 20 μm revAβ42. After 36 h, the percentage of apoptotic nuclei was evaluated by Hoechst 33258 staining. Data are expressed as means ± SE of three experiments. Each experiment was performed in quintuplicate, and 500–1000 neurons per treatment were counted. E, Effect of pravastatin on neuronal survival. Neurons were incubated with different concentrations of pravastatin. After 36 h, the percentage of apoptotic nuclei was evaluated by Hoechst 33258 staining. Data are expressed as means ± SE of three experiments. Each experiment was performed in quintuplicate, and 500–1000 neurons per treatment were counted. F, Analysis of SREBP-2 processing. Neurons were incubated as indicated for 24 h, at which time they were harvested and lysed. Proteins were separated by SDS-PAGE, and SREBP-2 was detected by immunoblot analysis of total neuronal lysates. G, Densitometric analysis of SREBP-2 cleavage combining five experiments. Data are means ± SE. For all experiments, *p < 0.05), **p < 0.01, and ***p < 0.001, one-way ANOVA.
Cellular cholesterol levels are sensed by the pool of cholesterol in the endoplasmic reticulum (ER) (Lange et al., 1999). We rationalized that, because oAβ42 impairs cholesterol trafficking, oAβ42 could prevent cholesterol transport to the ER, thus increasing cholesterol synthesis. However, we found that oAβ42 significantly reduced cholesterol synthesis in basal forebrain, cortical, and sympathetic neurons (Fig. 3B). oAβ42 inhibited cholesterol synthesis only at concentrations at which it caused neuronal apoptosis (Fig. 3C,D), suggesting a link between these two phenomena. However, the lack of significant apoptosis in neurons that received pravastatin (Fig. 3E) indicates that cholesterol synthesis inhibition is not sufficient to induce apoptosis under our experimental conditions.
Regulation of cholesterol synthesis at the molecular level is mediated by the transcription factor SREBP-2 (Goldstein et al., 2006). For activation, SREBP-2 requires to be transported from the ER to the Golgi, in which it undergoes proteolytic cleavage, and the mature form of SREBP-2 translocates into the nucleus (Goldstein et al., 2006). Finding that oAβ42 inhibited cholesterol synthesis and impaired intracellular trafficking prompted us to examine whether oAβ42 regulates proteolytic activation of SREBP-2. As in other cells, in neurons SREBP-2 cleavage was increased in response to sterol depletion (serum deprivation); however, in the presence of oAβ42, proteolytic activation of SREBP-2 was essentially hindered (Fig. 3F,G). These results clearly demonstrate that oAβ42 inhibited SREBP-2 proteolytic cleavage.
Aβ inhibits protein prenylation
SREBP-2 controls transcription of all genes of the mevalonate pathway, a biosynthetic pathway for cholesterol and non-steroid isoprenoids farnesyl pyrophosphate and GGPP among others (Horton et al., 2002). Isoprenoids are essential for posttranslational modification of >100 proteins (Hooff et al., 2010). Hence, we hypothesized that, by inhibiting the mevalonate pathway, oAβ42 would affect protein prenylation. We examined prenylation of Rab proteins that regulate vesicular trafficking (Stenmark, 2009). Rab9 and Rab7 reside in late endosomes and participate in vesicular trafficking to late endosome and trans-Golgi (Lombardi et al., 1993; Feng et al., 1995). Rabs require prenylation for membrane targeting and for binding to Rab GDIs. GDIs keep prenylated Rabs in a soluble state in the cytosol and deliver them to proper membrane locations (Pfeffer et al., 1995). To assess the membrane and cytosolic pools of prenylated Rabs, we used two alternative approaches. First, we performed extraction of prenylated proteins with Triton X-114 and found reduced prenylation of Rab9 in oAβ42-treated neurons to an extent similar to prenylation reduction by treatment with geranylgeranyl transferase inhibitor (GGTI-2133) (Fig. 4A). Next, we performed extraction of prenylated proteins with recombinant Rab GDI. The level of prenylated Rab7 extracted from oAβ42-treated neurons was significantly lower than in untreated neurons but was unchanged when neurons were given GGPP together with oAβ42 (Fig. 4B). GDI capture of prenylated Rabs from cellular membranes may be physically hindered by cholesterol accumulation within membranes (Lebrand et al., 2002; Choudhury et al., 2004; Ganley and Pfeffer, 2006). Thus, to avoid confounding physical effects attributable to the accumulation of cholesterol in neurons treated with oAβ42, we repeated the GDI Rab-capturing experiments in cytosolic and membrane fractions separately and confirmed that protein prenylation is reduced in oAβ42-treated neurons (Fig. 4C,D). To exclude that the inhibition of prenylation by oAβ42 was specific to Rabs, we examined the level of membrane-associated Ras, which is an indication of Ras prenylation. oAβ42 caused significant decrease of Ras in neuronal membranes, but when neurons received GGPP with oAβ42, membrane Ras levels were unaffected (Fig. 4E). Although Ras proteins are preferentially modified by farnesylation, geranylgeranylation of KRAS and to a lesser extent NRAS becomes important when farnesylation is blocked (Downward, 2003). Together, these results indicate that oAβ42 effectively inhibits protein prenylation. The finding that GGPP prevented Aβ inhibition of protein prenylation suggests that the reduction in protein prenylation results from decrease in isoprenoid production. The use of GGPP can be limited by its relative membrane impermeability, but there is documented evidence of GGPP ability to enter cultured cells (Kukar et al., 2005) and to counteract some effects of inhibitors of the mevalonate pathway (Meske et al., 2003; Schulz et al., 2004; Ostrowski et al., 2007). We confirmed GGPP uptake by measuring the incorporation of [3H]GGPP in cellular proteins. Neurons were able to take up 0.2 ± 0.015% of the total [3H]GGPP added to the medium in 24 h, and [3H]GGPP uptake was not affected by Aβ. However, neurons treated with oAβ42 incorporated twofold to threefold more GGPP in proteins than untreated neurons or neurons treated with pravastatin (156.94 dpm/actin pixel in oAβ-treated neurons vs 48.51 dpm/actin pixel in untreated neurons and 56.35 dpm/actin pixel in pravastatin-treated neurons). This experiment has two main implications: (1) oAβ42 significantly reduces the endogenous pool of GGPP; and (2) GGPP does not neutralize oAβ42 effects but truly serves as a substrate for protein prenylation. In addition, these data also show that, under our experimental conditions, pravastatin does not significantly affect isoprenoids levels, which agrees with the lack of effect of pravastatin on protein prenylation (Fig. 4F) and cell survival (Fig. 3E).
Figure 4.

oAβ1–42 inhibits protein prenylation. Neurons were treated with 20 μm oAβ42 with or without the isoprenoid GGPP (10 μm) or with 5 μm GGTI for 24 h. A, Analysis of Rab prenylation by extraction with Triton X-114: neuronal lysates (equal amount of protein) were extracted with Triton X-114. Proteins in detergent (D, prenylated proteins) and aqueous (Aq, unprenylated proteins) phases were analyzed by SDS-PAGE and immunoblotting. Ratios between total pixels of Rab9 in detergent/total pixels of Rab 9 in detergent plus aqueous were calculated. The data are presented as percentage control (untreated neurons). Data are means ± SE of three experiments. B, Analysis of Rab prenylation by Rab–GDI capture. Neuronal lysates were prepared. Half sample was used to assess total protein input (I). The other half was used to extract prenylated proteins with recombinant GST–GDI (E). Proteins were analyzed by immunoblot analysis. Ratios between total pixels of Rab7 in GDI-bound (E)/total pixels of Rab7 in lysate before extraction (I) were calculated. Densitometric analysis of I included both bands. Data are presented as percentage control (untreated neurons). C, D, GDI capture from cytosol and membranes. Neuronal lysates were fractionated in membranes and cytosol before GST–GDI extraction. Extraction followed the same protocol and considerations as in B. E, Ras delivery to membranes. Membrane fractions were prepared from treated neurons. Equal amounts of membrane proteins were separated by SDS-PAGE and analyzed by immunoblotting. Densitometric analysis of Ras/flotillin combined four experiments. Values expressed as percentage of untreated neurons. F, Pravastatin does not impair protein prenylation. Sympathetic neurons were treated with oAβ1–42 (20 μm), revAβ42–1 (20 μm), or pravastatin 50 μm for 24 h. Protein prenylation was examined by extraction with Triton X-114 or GST–GDI. G, Rab–GDI capture from brains of TgCRND8 mice: brain cortex cytosol fractions were prepared, and prenylated Rabs were extracted with GST–GDI as for cultured neurons. Results obtained from six individual brains for each group have been combined in the quantification plot. H, Separation of prenylated and unprenylated Rabs using Triton X-114: proteins were separated with Triton X-114 from mouse brain cortex homogenates. Densitometric analysis of prenylated (detergent)/prenylated + unprenylated (aqueous) combined results from four brains in each group. In all experiments, data are means ± SE of values. *p < 0.05 and **p < 0.01, one-way ANOVA.
We next investigated whether the decrease in protein prenylation induced by oAβ42 in cultured neurons was recapitulated in the brain of the TgCRND8 mice overexpressing APP. We found that prenylated pools of Rab7 and Rab9 were reduced in cortex of TgCRND8 mice compared with aged-matched wild-type mice (Fig. 4G,H).
Isoprenoid supply prevents Aβ-induced cholesterol sequestration and protects against Aβ-induced neurotoxicity
Based on the evidence presented above, we hypothesized that cholesterol sequestration in oAβ42-treated neurons results from protein prenylation inhibition and could be prevented by GGPP. Supporting this hypothesis, the prenylation inhibitor GGTI caused intracellular cholesterol accumulation similar to treatment with oAβ42 (Fig. 5A). Administration of GGPP prevented oAβ42-induced, but not GGTI-induced, cholesterol sequestration (Fig. 5A). Furthermore, GGPP blocked the detrimental effect of oAβ42 on neuronal metabolic activity (Fig. 5B) and significantly reduced oAβ42-induced but not GGTI-induced apoptosis (Fig. 5C).
Figure 5.

GGPP reduces oAβ42-induced cholesterol sequestration and oAβ42-induced neurotoxicity. Neurons were treated with 20 μm oAβ42 or 5 μm GGTI with or without 10 μm GGPP for 36 h. A, Cholesterol sequestration was detected by filipin staining. B, Metabolic activity was examined by MTT assay in situ. White arrows point to healthy cell bodies and neurites containing the formazan compound. Black arrows indicate cell bodies or neurites devoid of the MTT reduction product attributable to reduced metabolic activity. C, Apoptosis was quantified by counting nuclei stained with Hoechst 33258. Data are expressed as means ± SE of three experiments. Each experiment was performed in quintuplicate, and 500–1000 neurons per treatment were counted. ***p < 0.001, one-way ANOVA.
Discussion
Intracellular cholesterol accumulation
Studies in AD brains reported cholesterol accumulation specifically in Aβ-immunopositive neurons (Ohm et al., 2003; Gómez-Ramos and Asunción Morän, 2007; Xiong et al., 2008), and studies in APP/PS1 mouse brains showed that Aβ accumulation preceded cholesterol accumulation (Fernández et al., 2009). In line with these findings and with the report of increase of free cholesterol caused by Aβ40 in hippocampal neurons (Liu et al., 1998), we show that Aβ can directly induce intracellular cholesterol sequestration. This is important because a causal relationship between cholesterol sequestration and cell death was identified in NPC (Aqul et al., 2011). NPC and AD present similar endosomal abnormalities, brain accumulation of Aβ and neurofibrillary tangles, and influence of apolipoprotein E genotype (Yamazaki et al., 2001; Jin et al., 2004; Koh and Cheung, 2006). These common features and our data suggest that a pathological mechanism involving abnormal cholesterol trafficking could take place in AD. Alternatively, intracellular cholesterol accumulation may be an epiphenomenon of Aβ effects on intracellular trafficking but not a direct cause of neuronal toxicity. Supporting this view, several studies indicated that, depending on the death insult, cells that accumulate cholesterol are not more susceptible to die or they might even survive better (Karten et al., 2002; Appelqvist et al., 2011; Reiners et al., 2011). However, simultaneous accumulation of cholesterol and Aβ42 in AD would have more severe consequences to lysosomes than accumulation of cholesterol alone because Aβ42 causes lysosome disruption (Ditaranto et al., 2001; Liu et al., 2010). With respect to the Golgi, the presence of cholesterol in the Golgi of Aβ-treated neurons coincides with the intracellular localization of Aβ in brains of APPxPS1 mouse (Langui et al., 2004) and in astrocytes treated with Aβ42 (Igbavboa et al., 2003, 2009). Aβ accumulation in the Golgi of astrocytes disrupts cholesterol homeostasis (Igbavboa et al., 2003). We are currently investigating whether Aβ localization to the Golgi plays a direct role in inhibition of SREBP-2 cleavage in this organelle.
Aβ inhibits the mevalonate pathway
oAβ42-treated neurons do not contain more cholesterol, similar to other cells that sequester cholesterol, such as NPC neurons (Karten et al., 2002) and U18666A-treated neurons (Tashiro et al., 2004). Moreover, we found that oAβ42 inhibits cholesterol synthesis, which at first seemed paradoxical to the intensive filipin staining. Nevertheless, U18666A, a potent inhibitor of cholesterol synthesis (Runz et al., 2002; Jin et al., 2004), also causes endosomal cholesterol accumulation (Karten et al., 2002; Cenedella, 2009). As for U18666A, the intense filipin staining in Aβ-treated neurons represents impairment in cholesterol trafficking and not net increase in cholesterol.
Inhibition of cholesterol synthesis by Aβ40 but not Aβ42 has been reported previously (Gong et al., 2002; Grimm et al., 2005). The consequences of cholesterol synthesis inhibition in neurons are unclear. Neurons have lower rate of cholesterol synthesis than glia (Nieweg et al., 2009), and in adulthood, neurons might not synthesize cholesterol but outsource cholesterol from astrocytes (Fünfschilling et al., 2007; Aqul et al., 2011), although some studies showed that neurons might maintain some level of cholesterol synthesis (Valdez et al., 2010). Our data suggest that inhibition of cholesterol synthesis per se is not a direct cause of neuronal death because cholesterol synthesis inhibition by pravastatin did not cause neuronal death (de Chaves et al., 1997; Saavedra et al., 2007).
In addition to cholesterol, the mevalonate pathway produces other intermediates, such as isoprenoids, which are required for protein prenylation. We show that Rab and Ras prenylation were significantly reduced in Aβ-treated neurons, and protein prenylation in brain cortex from the TgCRND8 mouse model of AD was significantly lower than in cortex of age-matched wild-type animals. Decreased prenylation in neurons challenged with Aβ was attributable to shortage of isoprenoids because supply of GGPP prevented prenylation inhibition and cholesterol sequestration, suggesting that normal intracellular trafficking was restored. Inhibition of protein prenylation appears as a pivotal mechanism of neuronal death. GGTI inhibited protein prenylation, caused cholesterol accumulation, and induced neuronal death. GGPP was readily incorporated into prenylated proteins and significantly reduced Aβ-induced neuronal death but, as expected, it could not rescue neurons treated with GGTI.
Isoprenoids and protein prenylation play important roles in the nervous system, especially in AD (Hooff et al., 2010). Reduced isoprenoid production explained the decrease of long-term potentiation in hippocampal slices (Matthies et al., 1997; Kotti et al., 2006, 2008), the increase in Tau hyperphosphorylation (Meske et al., 2003), and the prevention of statin-induced neurotoxicity by isoprenoids (Tanaka et al., 2000; Meske et al., 2003; Schulz et al., 2004). Protein prenylation is important for APP trafficking and cleavage. Reduction of protein prenylation results in increased or decreased Aβ production, depending on the cell type (Cole et al., 2005; Ostrowski et al., 2007). Moreover, Ras prenylation reduction represents a mechanism of neurodegeneration (Zipp et al., 2007). In our studies, the decrease in protein prenylation is accompanied by cholesterol accumulation within intracellular membranes, which would favor APP cleavage and Aβ production.
Recent studies showed that GGPP, farnesyl pyrophosphate, and the mRNA of their respective synthases are elevated in AD brains (Eckert et al., 2009). The significance of this elevation is still unknown because protein prenylation was not examined in this study and elevation of isoprenoids does not warrant an increase in protein prenylation. Our findings suggest that a decrease in isoprenoid synthesis will take place specifically in cells that accumulate Aβ, most likely neurons. Depending on the size of the cell population that contains intracellular Aβ, this might or might not impact the overall content of isoprenoids in the brain. An interesting model has been proposed in which SREBPs in astrocytes are involved in lipid synthesis for supply to neurons for neurite outgrowth, synaptogenesis, and synaptic plasticity (Camargo et al., 2009). According to this model, glia SREBPs may work as control points of neuronal function, providing neurons with appropriate lipids when neurons cannot make their own. Whether astrocytes can supply isoprenoids has not been investigated to our knowledge, but the increase of isoprenoids in AD brains could represent an astrocytic attempt to compensate a decrease in SREBP-dependent metabolic pathways in neurons.
In addition to the direct effect of Aβ on prenylation, Aβ could also affect indirectly the function of remaining prenylated Rabs by means of cholesterol accumulation within membranes. The level of cholesterol in endocytic compartments plays a role in Rabs fate and function (Lebrand et al., 2002; Choudhury et al., 2004; Ganley and Pfeffer, 2006). GDI capture of prenylated proteins from membranes of oAβ42-treated neurons is reduced, suggesting that in Aβ-treated neurons there are membrane structural changes similar to those present in NPC cells (Choudhury et al., 2004; Ganley and Pfeffer, 2006). These changes could result from a combination of cholesterol accumulation and the direct interaction of Aβ with cholesterol and other lipids, such as GM1 (Kimura and Yanagisawa, 2007).
Our results indicate that one mechanism of oAβ42 inhibition of the mevalonate pathway is a decrease in SREBP-2 processing. This is different from what has been shown for Aβ40. Gong and collaborators found that Aβ40 did not affect SREBP-2 proteolysis (Gong et al., 2002), and Hartmann's group found no inhibition of cholesterol synthesis when mevalonate was used as a precursor, indicating that hydroxy-methyl-glutaryl coenzyme A reductase was the target of Aβ40 (Grimm et al., 2005) but also suggesting that Aβ40 does not affect SREBP-2. Importantly, in Gong's and Hartmann's studies, Aβ was used at non-toxic concentrations, and our data indicate that only toxic concentrations of oAβ42 inhibit cholesterol synthesis. We are currently investigating the mechanism(s) of inhibition of SREBP-2 cleavage by oAβ42. We are testing whether Aβ alters the interaction between SCAP and Insig in the ER, inhibits transport of the complex SREBP-2/SCAP to the Golgi (possibly by inhibition of Akt activation), or directly affects SREBP-2 cleavage by SP1 and SP2 in the Golgi.
Conclusions
The implications of the identification of SREBP-2 as a target of oAβ42 could extend beyond the mevalonate pathway and protein prenylation. SREBP-2 is a positive regulator of the transcription factor liver X receptor (LXR), through generation of oxysterol ligands for LXR (Wong et al., 2006). LXR activation positively regulates transcription of the cholesterol efflux gene, ATP-binding cassette transporter A1 (ABCA1), among other genes. Our work suggests that Aβ would reduce LXR activation and ABCA1 transcription. In line with this idea, Aβ42 reduces expression of ABCA1 in an astrocytic cell line, and ABCA1 protein expression is reduced in brain of APP/PS1 mice (Canepa et al., 2011). More importantly, LXR agonists are being regarded as possible therapeutic agents in AD based on studies in which these agents showed beneficial effects with respect to reduced APP processing into Aβ, reduced levels of Aβ peptides, and improved memory function (Hirsch-Reinshagen et al., 2009). Another important gene that would be downregulated by Aβ inhibition of SREBP-2 cleavage is Dhcr24. The Dhcr24 gene product Seladin-1/DHCR24 is the enzyme that converts desmosterol into cholesterol. Dhcr24 is downregulated in AD brains (Greeve et al., 2000). Seladin-1 is considered neuroprotective because of its anti-apoptotic activity and its involvement in hormone-mediated neuroprotection in AD (Peri et al., 2011). Interestingly, Seladin-1 is an LXR target (Wang et al., 2008). Altogether, the available data indicate that reduction of LXR and Seladin-1 are important in AD, but the causes for their downregulation have not been elucidated yet. Our work opens the possibility that Aβ could regulate Seladin-1 levels and LXR activation by inhibiting SREBP-2 cleavage.
Footnotes
This work was supported by grants from the Canadian Institutes of Health Research and the Alzheimer Society of Canada. We thank Dr. S. Kar (University of Alberta, Edmonton, AB, Canada) for providing mice brains, Dr. J. Gruenberg (University of Geneva, Geneva, Switzerland) for providing the GDI–GST plasmid, and Dr. K. Ko (University of Alberta) for helping in expression and purification of GST–GDI. We also thank B. Khaniya and A. Leung for technical support.
The authors declare no competing financial interests.
References
- Appelqvist H, Nilsson C, Garner B, Brown AJ, Kågedal K, Ollinger K. Attenuation of the lysosomal death pathway by lysosomal cholesterol accumulation. Am J Pathol. 2011;178:629–639. doi: 10.1016/j.ajpath.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aqul A, Liu B, Ramirez CM, Pieper AA, Estill SJ, Burns DK, Liu B, Repa JJ, Turley SD, Dietschy JM. Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment. J Neurosci. 2011;31:9404–9413. doi: 10.1523/JNEUROSCI.1317-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MV, Tan AS. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch Biochem Biophys. 1993;303:474–482. doi: 10.1006/abbi.1993.1311. [DOI] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Blanchard V, Moussaoui S, Czech C, Touchet N, Bonici B, Planche M, Canton T, Jedidi I, Gohin M, Wirths O, Bayer TA, Langui D, Duyckaerts C, Tremp G, Pradier L. Time sequence of maturation of dystrophic neurites associated with Abeta deposits in APP/PS1 transgenic mice. Exp Neurol. 2003;184:247–263. doi: 10.1016/s0014-4886(03)00252-8. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR. Isolation and culture of adult neurons and neurospheres. Nat Protoc. 2007;2:1490–1498. doi: 10.1038/nprot.2007.207. [DOI] [PubMed] [Google Scholar]
- Camargo N, Smit AB, Verheijen MH. SREBPs: SREBP function in glia-neuron interactions. FEBS J. 2009;276:628–636. doi: 10.1111/j.1742-4658.2008.06808.x. [DOI] [PubMed] [Google Scholar]
- Canepa E, Borghi R, Viña J, Traverso N, Gambini J, Domenicotti C, Marinari UM, Poli G, Pronzato MA, Ricciarelli R. Cholesterol and amyloid-beta: evidence for a cross-talk between astrocytes and neuronal cells. J Alzheimers Dis. 2011;25:645–653. doi: 10.3233/JAD-2011-110053. [DOI] [PubMed] [Google Scholar]
- Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol. 2004;165:1289–1300. doi: 10.1016/s0002-9440(10)63388-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenedella RJ. Cholesterol synthesis inhibitor U18666A and the role of sterol metabolism and trafficking in numerous pathophysiological processes. Lipids. 2009;44:477–487. doi: 10.1007/s11745-009-3305-7. [DOI] [PubMed] [Google Scholar]
- Choudhury A, Sharma DK, Marks DL, Pagano RE. Elevated endosomal cholesterol levels in Niemann-Pick cells inhibit rab4 and perturb membrane recycling. Mol Biol Cell. 2004;15:4500–4511. doi: 10.1091/mbc.E04-05-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen DZ, Kraus SL, Flohr A, Cotel MC, Wirths O, Bayer TA. Transient intraneuronal A beta rather than extracellular plaque pathology correlates with neuron loss in the frontal cortex of APP/PS1KI mice. Acta Neuropathol. 2008;116:647–655. doi: 10.1007/s00401-008-0451-6. [DOI] [PubMed] [Google Scholar]
- Christensen DZ, Bayer TA, Wirths O. Intracellular Abeta triggers neuron loss in the cholinergic system of the APP/PS1KI mouse model of Alzheimer's disease. Neurobiol Aging. 2010;31:1153–1163. doi: 10.1016/j.neurobiolaging.2008.07.022. [DOI] [PubMed] [Google Scholar]
- Chui DH, Dobo E, Makifuchi T, Akiyama H, Kawakatsu S, Petit A, Checler F, Araki W, Takahashi K, Tabira T. Apoptotic neurons in Alzheimer's disease frequently show intracellular Abeta42 labeling. J Alzheimers Dis. 2001;3:231–239. doi: 10.3233/jad-2001-3208. [DOI] [PubMed] [Google Scholar]
- Cole SL, Grudzien A, Manhart IO, Kelly BL, Oakley H, Vassar R. Statins cause intracellular accumulation of amyloid precursor protein, beta-secretase-cleaved fragments, and amyloid beta-peptide via an isoprenoid-dependent mechanism. J Biol Chem. 2005;280:18755–18770. doi: 10.1074/jbc.M413895200. [DOI] [PubMed] [Google Scholar]
- Coxon FP, Ebetino FH, Mules EH, Seabra MC, McKenna CE, Rogers MJ. Phosphonocarboxylate inhibitors of Rab geranylgeranyl transferase disrupt the prenylation and membrane localization of Rab proteins in osteoclasts in vitro and in vivo. Bone. 2005;37:349–358. doi: 10.1016/j.bone.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Dahlgren KN, Manelli AM, Stine WB, Jr, Baker LK, Krafft GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- D'Andrea MR, Nagele RG, Wang HY, Peterson PA, Lee DH. Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer's disease. Histopathology. 2001;38:120–134. doi: 10.1046/j.1365-2559.2001.01082.x. [DOI] [PubMed] [Google Scholar]
- de Chaves EI, Rusiñol AE, Vance DE, Campenot RB, Vance JE. Role of lipoproteins in the delivery of lipids to axons during axonal regeneration. J Biol Chem. 1997;272:30766–30773. doi: 10.1074/jbc.272.49.30766. [DOI] [PubMed] [Google Scholar]
- Ditaranto K, Tekirian TL, Yang AJ. Lysosomal membrane damage in soluble Abeta-mediated cell death in Alzheimer's disease. Neurobiol Dis. 2001;8:19–31. doi: 10.1006/nbdi.2000.0364. [DOI] [PubMed] [Google Scholar]
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- Eckert GP, Hooff GP, Strandjord DM, Igbavboa U, Volmer DA, Müller WE, Wood WG. Regulation of the brain isoprenoids farnesyl- and geranylgeranylpyrophosphate is altered in male Alzheimer patients. Neurobiol Dis. 2009;35:251–257. doi: 10.1016/j.nbd.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Press B, Wandinger-Ness A. Rab 7: an important regulator of late endocytic membrane traffic. J Cell Biol. 1995;131:1435–1452. doi: 10.1083/jcb.131.6.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández A, Llacuna L, Fernández-Checa JC, Colell A. Mitochondrial cholesterol loading exacerbates amyloid β peptide-induced inflammation and neurotoxicity. J Neurosci. 2009;29:6394–6405. doi: 10.1523/JNEUROSCI.4909-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane-Stanley GH. A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem. 1959;226:495–509. [PubMed] [Google Scholar]
- Fünfschilling U, Saher G, Xiao L, Möbius W, Nave KA. Survival of adult neurons lacking cholesterol synthesis in vivo. BMC Neurosci. 2007;8:1. doi: 10.1186/1471-2202-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley IG, Pfeffer SR. Cholesterol accumulation sequesters Rab9 and disrupts late endosome function in NPC1-deficient cells. J Biol Chem. 2006;281:17890–17899. doi: 10.1074/jbc.M601679200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Gómez-Ramos P, Asunción Morán M. Ultrastructural localization of intraneuronal Abeta-peptide in Alzheimer disease brains. J Alzheimers Dis. 2007;11:53–59. doi: 10.3233/jad-2007-11109. [DOI] [PubMed] [Google Scholar]
- Gong JS, Sawamura N, Zou K, Sakai J, Yanagisawa K, Michikawa M. Amyloid beta-protein affects cholesterol metabolism in cultured neurons: implications for pivotal role of cholesterol in the amyloid cascade. J Neurosci Res. 2002;70:438–446. doi: 10.1002/jnr.10347. [DOI] [PubMed] [Google Scholar]
- Greeve I, Hermans-Borgmeyer I, Brellinger C, Kasper D, Gomez-Isla T, Behl C, Levkau B, Nitsch RM. The human DIMINUTO/DWARF1 homolog seladin-1 confers resistance to Alzheimer's disease-associated neurodegeneration and oxidative stress. J Neurosci. 2000;20:7345–7352. doi: 10.1523/JNEUROSCI.20-19-07345.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm MO, Grimm HS, Pätzold AJ, Zinser EG, Halonen R, Duering M, Tschäpe JA, De Strooper B, Müller U, Shen J, Hartmann T. Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nat Cell Biol. 2005;7:1118–1123. doi: 10.1038/ncb1313. [DOI] [PubMed] [Google Scholar]
- Hirsch-Reinshagen V, Burgess BL, Wellington CL. Why lipids are important for Alzheimer disease? Mol Cell Biochem. 2009;326:121–129. doi: 10.1007/s11010-008-0012-2. [DOI] [PubMed] [Google Scholar]
- Hooff GP, Wood WG, Müller WE, Eckert GP. Isoprenoids, small GTPases and Alzheimer's disease. Biochim Biophys Acta. 2010;1801:896–905. doi: 10.1016/j.bbalip.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igbavboa U, Pidcock JM, Johnson LN, Malo TM, Studniski AE, Yu S, Sun GY, Wood WG. Cholesterol distribution in the Golgi complex of DITNC1 astrocytes is differentially altered by fresh and aged amyloid beta-peptide-(1-42) J Biol Chem. 2003;278:17150–17157. doi: 10.1074/jbc.M301150200. [DOI] [PubMed] [Google Scholar]
- Igbavboa U, Sun GY, Weisman GA, He Y, Wood WG. Amyloid beta-protein stimulates trafficking of cholesterol and caveolin-1 from the plasma membrane to the Golgi complex in mouse primary astrocytes. Neuroscience. 2009;162:328–338. doi: 10.1016/j.neuroscience.2009.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin LW, Shie FS, Maezawa I, Vincent I, Bird T. Intracellular accumulation of amyloidogenic fragments of amyloid-beta precursor protein in neurons with Niemann-Pick type C defects is associated with endosomal abnormalities. Am J Pathol. 2004;164:975–985. doi: 10.1016/s0002-9440(10)63185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karten B, Vance DE, Campenot RB, Vance JE. Cholesterol accumulates in cell bodies, but is decreased in distal axons, of Niemann-Pick C1-deficient neurons. J Neurochem. 2002;83:1154–1163. doi: 10.1046/j.1471-4159.2002.01220.x. [DOI] [PubMed] [Google Scholar]
- Karten B, Vance DE, Campenot RB, Vance JE. Trafficking of cholesterol from cell bodies to distal axons in Niemann Pick C1-deficient neurons. J Biol Chem. 2003;278:4168–4175. doi: 10.1074/jbc.M205406200. [DOI] [PubMed] [Google Scholar]
- Kimura N, Yanagisawa K. Endosomal accumulation of GM1 ganglioside-bound amyloid beta-protein in neurons of aged monkey brains. Neuroreport. 2007;18:1669–1673. doi: 10.1097/WNR.0b013e3282f0d2ab. [DOI] [PubMed] [Google Scholar]
- Koh CH, Cheung NS. Cellular mechanism of U18666A-mediated apoptosis in cultured murine cortical neurons: bridging Niemann-Pick disease type C and Alzheimer's disease. Cell Signal. 2006;18:1844–1853. doi: 10.1016/j.cellsig.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Kotti TJ, Ramirez DM, Pfeiffer BE, Huber KM, Russell DW. Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc Natl Acad Sci U S A. 2006;103:3869–3874. doi: 10.1073/pnas.0600316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotti T, Head DD, McKenna CE, Russell DW. Biphasic requirement for geranylgeraniol in hippocampal long-term potentiation. Proc Natl Acad Sci U S A. 2008;105:11394–11399. doi: 10.1073/pnas.0805556105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukar T, Murphy MP, Eriksen JL, Sagi SA, Weggen S, Smith TE, Ladd T, Khan MA, Kache R, Beard J, Dodson M, Merit S, Ozols VV, Anastasiadis PZ, Das P, Fauq A, Koo EH, Golde TE. Diverse compounds mimic Alzheimer disease-causing mutations by augmenting Abeta42 production. Nat Med. 2005;11:545–550. doi: 10.1038/nm1235. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Tinkle BT, Bieberich CJ, Haudenschild CC, Jay G. The Alzheimer's A beta peptide induces neurodegeneration and apoptotic cell death in transgenic mice. Nat Genet. 1995;9:21–30. doi: 10.1038/ng0195-21. [DOI] [PubMed] [Google Scholar]
- Lange Y, Ye J, Rigney M, Steck TL. Regulation of endoplasmic reticulum cholesterol by plasma membrane cholesterol. J Lipid Res. 1999;40:2264–2270. [PubMed] [Google Scholar]
- Langui D, Girardot N, El Hachimi KH, Allinquant B, Blanchard V, Pradier L, Duyckaerts C. Subcellular topography of neuronal Abeta peptide in APPxPS1 transgenic mice. Am J Pathol. 2004;165:1465–1477. doi: 10.1016/s0002-9440(10)63405-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrand C, Corti M, Goodson H, Cosson P, Cavalli V, Mayran N, Faur é J, Gruenberg J. Late endosome motility depends on lipids via the small GTPase Rab7. EMBO J. 2002;21:1289–1300. doi: 10.1093/emboj/21.6.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JP, Tang Y, Zhou S, Toh BH, McLean C, Li H. Cholesterol involvement in the pathogenesis of neurodegenerative diseases. Mol Cell Neurosci. 2010;43:33–42. doi: 10.1016/j.mcn.2009.07.013. [DOI] [PubMed] [Google Scholar]
- Liu Y, Peterson DA, Schubert D. Amyloid beta peptide alters intracellular vesicle trafficking and cholesterol homeostasis. Proc Natl Acad Sci U S A. 1998;95:13266–13271. doi: 10.1073/pnas.95.22.13266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi D, Soldati T, Riederer MA, Goda Y, Zerial M, Pfeffer SR. Rab9 functions in transport between late endosomes and the trans Golgi network. EMBO J. 1993;12:677–682. doi: 10.1002/j.1460-2075.1993.tb05701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthies H, Jr, Schulz S, Hollt V, Krug M. Inhibition by compactin demonstrates a requirement of isoprenoid metabolism for long-term potentiation in rat hippocampal slices. Neuroscience. 1997;79:341–346. doi: 10.1016/s0306-4522(96)00703-8. [DOI] [PubMed] [Google Scholar]
- Meske V, Albert F, Richter D, Schwarze J, Ohm TG. Blockade of HMG-CoA reductase activity causes changes in microtubule-stabilizing protein tau via suppression of geranylgeranylpyrophosphate formation: implications for Alzheimer's disease. Eur J Neurosci. 2003;17:93–102. doi: 10.1046/j.1460-9568.2003.02433.x. [DOI] [PubMed] [Google Scholar]
- Narita K, Choudhury A, Dobrenis K, Sharma DK, Holicky EL, Marks DL, Walkley SU, Pagano RE. Protein transduction of Rab9 in Niemann-Pick C cells reduces cholesterol storage. FASEB J. 2005;19:1558–1560. doi: 10.1096/fj.04-2714fje. [DOI] [PubMed] [Google Scholar]
- Nieweg K, Schaller H, Pfrieger FW. Marked differences in cholesterol synthesis between neurons and glial cells from postnatal rats. J Neurochem. 2009;109:125–134. doi: 10.1111/j.1471-4159.2009.05917.x. [DOI] [PubMed] [Google Scholar]
- Nixon RA. Endosome function and dysfunction in Alzheimer's disease and other neurodegenerative diseases. Neurobiol Aging. 2005;26:373–382. doi: 10.1016/j.neurobiolaging.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Ohm TG, Treiber-Held S, Distl R, Glockner F, Schonheit B, Tamanai M, Meske V. Cholesterol and tau protein–findings in Alzheimer's and Niemann Pick C's disease. Pharmacopsychiatry. 2003;36(Suppl 2):S120–S126. doi: 10.1055/s-2003-43060. [DOI] [PubMed] [Google Scholar]
- Ostrowski SM, Wilkinson BL, Golde TE, Landreth G. Statins reduce amyloid-beta production through inhibition of protein isoprenylation. J Biol Chem. 2007;282:26832–26844. doi: 10.1074/jbc.M702640200. [DOI] [PubMed] [Google Scholar]
- Peri A, Benvenuti S, Luciani P, Deledda C, Cellai I. Membrane cholesterol as a mediator of the neuroprotective effects of estrogens. Neuroscience. 2011;191:107–117. doi: 10.1016/j.neuroscience.2011.03.011. [DOI] [PubMed] [Google Scholar]
- Pfeffer SR, Dirac-Svejstrup AB, Soldati T. Rab GDP dissociation inhibitor: putting rab GTPases in the right place. J Biol Chem. 1995;270:17057–17059. doi: 10.1074/jbc.270.29.17057. [DOI] [PubMed] [Google Scholar]
- Reiners JJ, Jr, Kleinman M, Kessel D, Mathieu PA, Caruso JA. Nonesterified cholesterol content of lysosomes modulates susceptibility to oxidant-induced permeabilization. Free Radic Biol Med. 2011;50:281–294. doi: 10.1016/j.freeradbiomed.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runz H, Rietdorf J, Tomic I, de Bernard M, Beyreuther K, Pepperkok R, Hartmann T. Inhibition of intracellular cholesterol transport alters presenilin localization and amyloid precursor protein processing in neuronal cells. J Neurosci. 2002;22:1679–1689. doi: 10.1523/JNEUROSCI.22-05-01679.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra L, Mohamed A, Ma V, Kar S, de Chaves EP. Internalization of beta-amyloid peptide by primary neurons in the absence of apolipoprotein E. J Biol Chem. 2007;282:35722–35732. doi: 10.1074/jbc.M701823200. [DOI] [PubMed] [Google Scholar]
- Saito Y, Suzuki K, Nanba E, Yamamoto T, Ohno K, Murayama S. Niemann-Pick type C disease: accelerated neurofibrillary tangle formation and amyloid beta deposition associated with apolipoprotein E epsilon 4 homozygosity. Ann Neurol. 2002;52:351–355. doi: 10.1002/ana.10266. [DOI] [PubMed] [Google Scholar]
- Schulz JG, Bösel J, Stoeckel M, Megow D, Dirnagl U, Endres M. HMG-CoA reductase inhibition causes neurite loss by interfering with geranylgeranylpyrophosphate synthesis. J Neurochem. 2004;89:24–32. doi: 10.1046/j.1471-4159.2003.02305.x. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer disease: mechanistic understanding predicts novel therapies. Ann Intern Med. 2004;140:627–638. doi: 10.7326/0003-4819-140-8-200404200-00047. [DOI] [PubMed] [Google Scholar]
- Song MS, Saavedra L, de Chaves EI. Apoptosis is secondary to non-apoptotic axonal degeneration in neurons exposed to Abeta in distal axons. Neurobiol Aging. 2006;27:1224–1238. doi: 10.1016/j.neurobiolaging.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Tatsuno I, Uchida D, Moroo I, Morio H, Nakamura S, Noguchi Y, Yasuda T, Kitagawa M, Saito Y, Hirai A. Geranylgeranyl-pyrophosphate, an isoprenoid of mevalonate cascade, is a critical compound for rat primary cultured cortical neurons to protect the cell death induced by 3-hydroxy-3-methylglutaryl-CoA reductase inhibition. J Neurosci. 2000;20:2852–2859. doi: 10.1523/JNEUROSCI.20-08-02852.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro Y, Yamazaki T, Shimada Y, Ohno-Iwashita Y, Okamoto K. Axon-dominant localization of cell-surface cholesterol in cultured hippocampal neurons and its disappearance in Niemann-Pick type C model cells. Eur J Neurosci. 2004;20:2015–2021. doi: 10.1111/j.1460-9568.2004.03677.x. [DOI] [PubMed] [Google Scholar]
- Tomiyama T, Matsuyama S, Iso H, Umeda T, Takuma H, Ohnishi K, Ishibashi K, Teraoka R, Sakama N, Yamashita T, Nishitsuji K, Ito K, Shimada H, Lambert MP, Klein WL, Mori H. A mouse model of amyloid β oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J Neurosci. 2010;30:4845–4856. doi: 10.1523/JNEUROSCI.5825-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez CM, Smith MA, Perry G, Phelix CF, Santamaria F. Cholesterol homeostasis markers are localized to mouse hippocampal pyramidal and granule layers. Hippocampus. 2010;20:902–905. doi: 10.1002/hipo.20743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Rogers PM, Stayrook KR, Su C, Varga G, Shen Q, Nagpal S, Burris TP. The selective Alzheimer's disease indicator-1 gene (Seladin-1/DHCR24) is a liver X receptor target gene. Mol Pharmacol. 2008;74:1716–1721. doi: 10.1124/mol.108.048538. [DOI] [PubMed] [Google Scholar]
- Wirths O, Multhaup G, Czech C, Feldmann N, Blanchard V, Tremp G, Beyreuther K, Pradier L, Bayer TA. Intraneuronal APP/A beta trafficking and plaque formation in beta-amyloid precursor protein and presenilin-1 transgenic mice. Brain Pathol. 2002;12:275–286. doi: 10.1111/j.1750-3639.2002.tb00442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J, Quinn CM, Brown AJ. SREBP-2 positively regulates transcription of the cholesterol efflux gene, ABCA1, by generating oxysterol ligands for LXR. Biochem J. 2006;400:485–491. doi: 10.1042/BJ20060914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H, Callaghan D, Jones A, Walker DG, Lue LF, Beach TG, Sue LI, Woulfe J, Xu H, Stanimirovic DB, Zhang W. Cholesterol retention in Alzheimer's brain is responsible for high beta- and gamma-secretase activities and Abeta production. Neurobiol Dis. 2008;29:422–437. doi: 10.1016/j.nbd.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Chang TY, Haass C, Ihara Y. Accumulation and aggregation of amyloid beta-protein in late endosomes of Niemann-pick type C cells. J Biol Chem. 2001;276:4454–4460. doi: 10.1074/jbc.M009598200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, McLaughlin R, Goodyer C, LeBlanc A. Selective cytotoxicity of intracellular amyloid beta peptide1–42 through p53 and Bax in cultured primary human neurons. J Cell Biol. 2002;156:519–529. doi: 10.1083/jcb.200110119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipp F, Waiczies S, Aktas O, Neuhaus O, Hemmer B, Schraven B, Nitsch R, Hartung HP. Impact of HMG-CoA reductase inhibition on brain pathology. Trends Pharmacol Sci. 2007;28:342–349. doi: 10.1016/j.tips.2007.05.001. [DOI] [PubMed] [Google Scholar]