

Abstract
Microglia are the immune cells of the nervous system, where they act as resident macrophages during inflammatory events underlying many neuropathological conditions. Microglia derive from primitive myeloid precursors that colonize the nervous system during embryonic development. In the postnatal brain, microglia are initially mitotic, rounded in shape (amoeboid), and phagocytically active. As brain development proceeds, they gradually undergo a transition to a surveillant nonphagocytic state characterized by a highly branched (ramified) morphology. This ramification process is almost recapitulated in reverse during the process of microglia activation in the adult brain, when surveillant microglia undergo a ramified-to-amoeboid morphological transformation and become phagocytic in response to injury or disease. Little is known about the mechanisms controlling amoeboid microglial cell proliferation, activation, and ramification during brain development, despite the critical role of these processes in the establishment of the adult microglia pool and their relevance to microglia activation in the adult brain. Here we show that the mouse transcription factor Runx1, a key regulator of myeloid cell proliferation and differentiation, is expressed in forebrain amoeboid microglia during the first two postnatal weeks. Runx1 expression is then downregulated in ramified microglia. Runx1 inhibits mouse amoeboid microglia proliferation and promotes progression to the ramified state. We show further that Runx1 expression is upregulated in microglia following nerve injury in the adult mouse nervous system. These findings provide insight into the regulation of postnatal microglia activation and maturation to the ramified state and have implications for microglia biology in the developing and injured brain.
Introduction
Microglia are unique mononuclear phagocytes localized in the adult CNS parenchyma, where they act as the resident inflammatory cells. The main role of microglia is to continuously survey the local environment for signs of injury or disease. Under nonpathological conditions, adult CNS microglia have a small cell body and extensively branched (“ramified”) and motile processes, which they use to monitor the CNS environment. In the presence of invading microbes or neural disease, microglia become “activated” through a morphological transformation characterized by retraction and shortening of their processes, enlargement of their soma, and acquisition of a rounded (“amoeboid”) morphology. Amoeboid microglia are endowed with phagocytic ability and perform macrophage-like functions in the brain. The activation of ramified (“surveillant”) microglia to a phagocytic state is a common event in virtually all types of neuropathologies (Hanisch and Kettenmann, 2007; Graeber and Streit, 2010; Ransohoff and Cardona, 2010; Prinz and Mildner, 2011).
Microglia activation in the adult brain is almost a recapitulation in reverse of events occurring during brain development. Adult brain parenchymal microglia derive from primitive myeloid progenitors that seed the brain during embryonic and perinatal stages, starting as early as embryonic day 9 (E9.0) (Alliot et al., 1999; Ginhoux et al., 2010; Prinz and Mildner, 2011; Schlegelmilch et al., 2011). Embryonic and postnatal microglia are initially mitotic, amoeboid, and phagocytic. They participate in important developmental functions including cell debris phagocytosis, guidance of axons in white matter tracts, and synapse elimination (Alliot et al., 1999; Prinz and Mildner, 2011; Schlegelmilch et al., 2011). Limited numbers of amoeboid microglia are present during early brain development, but their number rapidly increases soon after birth. In rodents, this expansion is the result of a controlled burst of in situ amoeboid microglial cell proliferation during the first two postnatal weeks. As postnatal brain development progresses, amoeboid microglia become gradually less abundant, with a concomitant increase in the number of ramified microglia, which go on to acquire a surveillant role (Sminia et al., 1987; Alliot et al., 1999; Ginhoux et al., 2010; Prinz and Mildner, 2011; Schlegelmilch et al., 2011).
The molecular mechanisms regulating perinatal amoeboid microglia proliferation and transition to the ramified (“deactivated”) state are key initial steps in the establishment of the adult brain microglial cell pool, because postnatal hematopoiesis does not significantly contribute to microglia homeostasis in the adult brain parenchyma (Sminia et al., 1987; Alliot et al., 1999; Ajami et al., 2007; Ginhoux et al., 2010). Moreover, understanding the events underlying microglia proliferation and deactivation during development is important for the study of adult brain microglia activation in response to injury or disease. Little is presently known, however, about the mechanisms controlling these developmental processes. Here we provide evidence for a previously unrecognized role for the Runt-related transcription factor 1 (Runx1), a key regulator of myeloid progenitor cell proliferation and differentiation (Kurokawa, 2006; Stifani and Ma, 2009), in promoting the transition of amoeboid microglia to the ramified deactivated state in the postnatal mouse brain.
Materials and Methods
Animals and histology.
All animal procedures were conducted in accordance with the guidelines of the Canadian Council for Animal Care and were approved by the Animal Care Committee of the Montreal Neurological Institute and the McGill University Animal Care Committee. For staging of mouse embryos, the day of the appearance of the vaginal plug was considered as E0.5. Runx1LacZ/+ and Runx1Flox/+ mice were generated and genotyped as described previously (North et al., 1999; Growney et al., 2005). Pups were dissected at various postnatal stages and the brains were fixed, cryopreserved in 30% sucrose, and embedded in Tissue-Tek OCT compound (Sakura Finetek) as described previously (Zagami and Stifani, 2010). Frozen tissues were cryostat-sectioned (14 μm), mounted onto SuperFrost glass slides (Fisher), and stored at −20°C until use. β-Galactosidase (β-gal) activity in Runx1LacZ/+ embryos was detected by rinsing sections from staged embryos three times in solution A (80 mm Na2HPO4, 20 mm NaH2PO4, pH 7.4, 2 mm MgCl2, 0.2% IGEPAL, 0.1% sodium deoxycholate), followed by incubation overnight at 37°C in solution A containing 5 mm potassium ferricyanide, 5 mm potassium ferrocyanide, and 1 mg/ml 5-bromo-4-chloro-3-indolyl-β-galactopyranoside (X-gal) (Invitrogen). Following this time, sections were extensively rinsed in PBS and counterstained with eosin before mounting with Fluoromount-G (SouthernBiotech).
Surgical procedures.
Female C57BL/6 mice (8–10 weeks old) were anesthetized with ketamine/xylazine/acepromazine (50/5/1 mg/kg) and the left sciatic nerve was exposed in the region of the upper thigh. The nerve was crushed with fine forceps (Dumont No. 5) for 5 s, three times. The lesion site was ∼45 mm from the tip of the third digit. After crush injury, the skin and muscle were closed using 5–0 Ethicon sutures (Johnson & Johnson). Mice were deeply anesthetized with ketamine/xylazine/acepromazine 7 d after sciatic nerve crush and then perfused through the heart with PBS. A 10 mm length of the spinal cord was removed, centered on the L4–L5 lumbar vertebral level, and followed by fixation for 1 h in 4% paraformaldehyde (PFA) in PBS and cryoprotection with 30% sucrose for 24 h. Serial 14 μm cross sections were cut on a cryostat and mounted onto SuperFrost glass slides and stored at −20°C until analysis.
Immunofluorescence.
Immunofluorescence staining involving mouse primary antibodies was performed using a “Mouse on Mouse” Kit (Vector Laboratories). All other single- and double-label immunofluorescence experiments were performed by first blocking nonspecific staining with blocking solution containing 5% normal donkey serum (v/v) and 0.1% Triton X-100 (v/v) in PBS for 1 h. Sections were then incubated sequentially with primary (2 h) and secondary (1 h) antibodies in blocking solution. The following primary antibodies were used: goat anti-β-gal (1:1000; Biogenesis), rabbit anti-β-gal (1:2000; Cappel), rabbit anti-Runx1 (1:2000; a kind gift from Dr. T.M. Jessell, Columbia University), rabbit anti-Runx1 (1:500; Epitomics), rat anti-F4/80 (1:500; Abcam), rat anti-CD11b (1:500; Abcam), rabbit anti-Iba1 (1:1000; Wako), mouse anti-Ki67 (1:200; BD Pharmingen), and mouse anti-nestin (1:10; Developmental Studies Hybridoma Bank). The fluorescent-conjugated secondary antibodies used included the Alexa Fluor 488 and 555 series (1:1000; Invitrogen). Sections were counterstained with Hoechst 33258 (Sigma) to visualize nuclei. All images were acquired using a Digital Video Camera mounted on an Axioskop 2 fluorescence microscope (Zeiss) and Northern Eclipse software (Empix Imaging).
Image analysis.
The numbers of Hoechst-stained nuclei, as well as the numbers of Ki67+, F4/80+ amoeboid and ramified microglial cells in the entire corpus callosum and external capsule of Runx1LacZ/+ and wild-type littermate pups were counted every fifth coronal section of 14 μm each (every 70 μm) derived from three different pups for each genotype (at least 15 sections for each genotype were analyzed). Pups were obtained from three different litters. Three images per hemisphere were taken and analyzed using ImageJ software (National Institutes of Health) by counting immunolabeled cells in the corpus callosum and external capsule. The percentage of cells stained for specific markers was then calculated. The numbers of Hoechst-stained nuclei and F4/80+ cells were counted in a similar way in the cerebral cortex (where virtually all F4/80+ cells had a ramified morphology) of the same pups on three randomly selected, nonoverlapping 10× fields per section, representing 1.035 mm2 tissue area and 0.014 mm3 tissue volume (15 sections for each genotype were analyzed).
Primary microglial cell culture, transduction, immunocytochemistry, and Western blot analysis.
Primary microglial cells were isolated from mixed glial cell cultures prepared from the forebrain of postnatal day 3 (P3) CD1 (Charles River Laboratory) or Runx1Flox/Flox pups as described previously (Skaper et al., 2006). Briefly, upon reaching confluence (∼2 weeks after isolation) microglia adhering to the astroglial monolayer were dislodged by shaking; resuspended in high-glucose DMEM, supplemented with 2 mm glutamine, 1% penicillin–streptomycin (v/v) and 10% fetal bovine serum (v/v); and plated on uncoated 4-well chamber slides (Nalgene Nunc) at the density of 1.25 × 105 cells/cm2. Cells were allowed to adhere for 45 min and then washed to remove nonadhering cells. Purity of these cultures was confirmed by immunocytochemistry using antibodies against F4/80, Iba1, and CD11b to visualize microglia and against glial fibrillary acidic protein (GFAP) (1:1000; Sigma) to visualize astrocytes. In gain-of-function studies, adherent microglial cells were transduced with recombinant adenovirus expressing enhanced green fluorescent protein (GFP) alone or in combination with either Runx1 or Runx1(1–250), generated as previously described (Theriault et al., 2005), at the multiplicity of infection (MOI) of 50. Two days after infection, cells were fixed (2% PFA for 15 min at room temperature) and subjected to double-label immunocytochemical analysis with the following antibodies: rabbit anti-GFP (1:5000; Invitrogen), mouse anti-GFP (1:300; Novus Biologicals), mouse anti-Ki67, rabbit anti-phosphorylated-histone H3 (PH3; 1:200; Millipore), or mouse anti-p21Cip1 (1:200; BD Pharmingen). In loss-of-function studies, microglial cells from Runx1Flox/Flox pups were transduced with lentiviral vectors expressing enhanced GFP alone or in combination with Cre recombinase (kindly provided by Dr. P.A. Barker, McGill University) at the MOI of 5 and analyzed 4 d later. Transduced cells were subjected to double-label immunocytochemical analysis of the expression of GFP, Runx1, and Ki67. For bromodeoxyuridine (BrdU) incorporation studies, transduced cells were pulsed with 3.0 μg/ml BrdU for 6 h, fixed, and then subjected to immunocytochemistry with anti-GPP and anti-BrdU (1:100; rat monoclonal from Abcam) antibodies. Cells were counterstained with Hoechst before examination by fluorescence microscopy. Three to six random fields of each condition (per experiment) were used for quantification of the percentage of GPF-positive cells coexpressing specific markers. In experiments with lipopolysaccharide (LPS), primary microglia were either not transduced or transduced with GFP- or Runx1-expressing adenovirus, as described above. Three days later, cells received LPS (10 ng/ml; Sigma) or vehicle (water) for 16 h, followed by either double-label immunofluorescence with mouse anti-GFP and rabbit anti-iNOS (inducible isoform of nitric oxide synthase, 1:500; Abcam) antibodies or preparation of whole-cell lysates and Western blot analysis with anti-iNOS, rabbit anti-amphiphysin II (1:1500; a kind gift from Dr. P. McPherson, McGill University), and rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 1:2000; Santa Cruz Biotechnology) antibodies.
Transcription assays.
HEK293 cells were transfected with the pww-p21-luc reporter plasmid (Theriault et al., 2005) and subjected to quantification of luciferase activity in the absence or presence of plasmids pCMV2-FLAG-Runx1 or pCMV2-FLAG-Runx1(1–250) (500 ng/transfection) as described previously (Theriault et al., 2005).
Chromatin immunoprecipitation.
Chromatin immunoprecipitation (ChIP) experiments were performed using the Magna ChIP G kit (Millipore) following the manufacturer's instructions. Chromatin of primary microglial cell cultures was cross-linked for 10 min using formaldehyde, sonicated to yield 200–600 bp fragments, and subjected to immunoprecipitation with either ChIP-grade rabbit anti-Runx1 antibody (Abcam) or control pre-immune rabbit antibody. Protein:DNA complexes were harvested using protein-G magnetic beads and a magnetic separator, rinsed extensively, and incubated at 62°C in the presence of “ChIP Elution Buffer” (Millipore) containing 0.1 mg/ml proteinase K to reverse the cross-linking and recover the DNA. Immunoprecipitated DNA was purified using spin columns and reagents supplied with the Magna ChIP G kit and subjected to PCR using oligonucleotide primers p21Cip1-F (5′-GAG GCA GCT CGA CGC CAA CTG CAG) and p21Cip1-R (5′-CGT CAT CCA CCT GCC GCG GAC ACA). These oligonucleotides map to the proximal region of the mouse Cdkn1a/p21Cip1 (p21Cip1) promoter (Chi et al., 2005) and yield a 301 bp PCR product.
Statistics.
Statistical comparisons were made using two-tailed Student's t test. Values were expressed as mean ± SEM. All statistical tests were performed with GraphPad Prism software (GraphPad). Significance level was set at p < 0.05. Additional details are provided in the figure legends, where appropriate.
Results
Runx1 is expressed in a restricted group of cells in the developing mouse forebrain
Runx1 has recently emerged as a precise in vivo lineage-tracing marker for early myeloid microglia progenitors that begin to populate the CNS during gestational stages, before the onset of brain development (Ginhoux et al., 2010). This finding prompted us to determine whether Runx1 was expressed in microglial cells in the embryonic and postnatal brain. Previously described Runx1LacZ/+ knock-in mice in which β-gal immunoreactivity faithfully reproduces Runx1 gene activation were examined to characterize Runx1 expression (North et al., 1999; Stifani et al., 2008; Zagami and Stifani, 2010). A small number of β-gal-expressing cells was first detected in the forebrain of Runx1LacZ/+ embryos at late embryonic stages (Fig. 1A–H). These β-gal+ cells were observed mostly around the lateral ventricles, where they were adjacent to cells expressing the neural progenitor marker, nestin (Fig. 1A–C). Although they did not express nestin, the β-gal+ cells in this region expressed the mitotic cell marker, Ki67 (Fig. 1E–H). At early postnatal stages, a more sizeable group of β-gal+ cells was observed in the germinative zone lining the lateral ventricles of the alar telencephalon, just below the corpus callosum (Figs. 1I, 2B,H). Similar to the situation observed at embryonic stages, β-gal+ cells in the forebrain of postnatal Runx1LacZ/+ mice did not express nestin, but were adjacent to nestin+ neural progenitor cells (Fig. 1I–K). Using a previously characterized anti-Runx1 antibody (Stifani et al., 2008; Zagami and Stifani, 2010), we confirmed that β-gal expression overlapped with Runx1 immunoreactivity at all stages examined, demonstrating the presence of the Runx1 protein in β-gal+ cells (Fig. 1M,O).
Figure 1.

Runx1 is expressed in a restricted group of cells in the developing forebrain. A–H, Coronal sections through the telencephalon of either E16.5 (A–C) or E18.5 (D–H) Runx1LacZ/+ embryos were subjected to double-labeling immunofluorescence analysis of β-gal and nestin expression (A–C), determination of β-gal activity using X-gal staining (D), or double-labeling immunofluorescence analysis of β-gal and Ki67 expression (E–H). E, Box indicates area shown at higher magnification in F–H. H, Arrowheads indicate cells coexpressing β-gal and Ki67. I–K, Coronal sections through the telencephalon of P1 Runx1LacZ/+ pups were subjected to double-labeling immunofluorescence analysis of β-gal and nestin expression. β-gal+ cells do not express nestin but are located adjacent to nestin+ cells. L, Schematic of the forebrain in the coronal plane indicating the location (red square) of the area examined in panels E–K. M–O, Sagittal sections of P10 Runx1LacZ/+ brains were subjected to double-labeling analysis of β-gal and Runx1 expression. All β-gal+ cells express the Runx1 protein, indicating that β-gal expression faithfully recapitulates Runx1 expressed in the postnatal forebrain. P, Schematic of the forebrain in the sagittal plane demonstrating the location (red square) of the area examined in M–O. LV, lateral ventricle. Scale bars: (in C, O) A–C, F–K, M–O, 50 μm; E, 100 μm; D, 250 μm.
Figure 2.
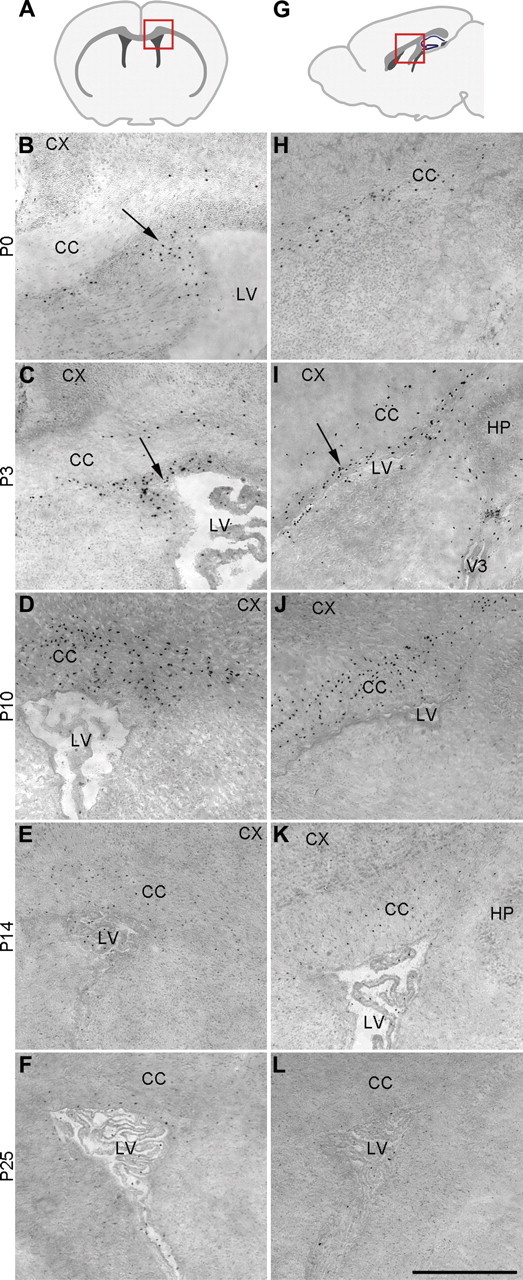
Runx1 is expressed in a restricted group of cells in the postnatal forebrain. Coronal (B–F, left column) or sagittal (H–L, right column) sections through the telencephalon of Runx1LacZ/+ mice at the indicated postnatal (P) stages were subjected to detection of the distribution of β-gal activity using the X-gal substrate, followed by counterstaining with eosin. A, G, Schematic of the forebrain in the coronal (A) or sagittal (G) plane demonstrating the location (red square) of the areas examined in the corresponding experiments shown in the columns below. B, C, I, Arrows point to groups of β-gal+ cells in the ventricular/subventricular zone. CC, corpus callosum; CX, cortex; LV, lateral ventricle; HP, hippocampus; V3, third ventricle. Scale bar, 500 μm.
The number of cells expressing β-gal in the alar telencephalon of Runx1LacZ/+ mice increased at later stages of postnatal brain development, reaching a peak at ∼P10, when many β-gal+ cells were observed within the corpus callosum (Fig. 2C,D,I,J). Forebrain β-gal expression then gradually decreased in older Runx1LacZ/+ animals and became almost undetectable by P25 (Fig. 2E–L). Together, these results demonstrate that Runx1 is expressed in a restricted number of cells in the postnatal mouse forebrain. In this region, Runx1 expression coincides spatially and temporally with the developmental appearance of periventricular amoeboid microglia, which progressively populate the germinative zone and subcortical white matter during the first 10 d after birth and then gradually decrease in number and become replaced by ramified microglia (Hristova et al., 2010; Prinz and Mildner, 2011; Schlegelmilch et al., 2011).
Runx1 is expressed in amoeboid microglia in the postnatal telencephalon
We next determined whether the β-gal+ cells in the postnatal telencephalon of Runx1LacZ/+ mice corresponded to microglia. All β-gal+ cells in this region were immunoreactive for three different antigens expressed in microglia in the developing and mature nervous system, such as F4/80, CD11b, and Iba1 (Davoust et al., 2008; Ginhoux et al., 2010) (Fig. 3B–M). In agreement with this finding, immunocytochemical analysis of primary cultures of postnatal mouse brain microglia demonstrated the expression of Runx1 in microglia, but not in occasional astrocytes present in these in vitro cultures (Fig. 3N–U).
Figure 3.
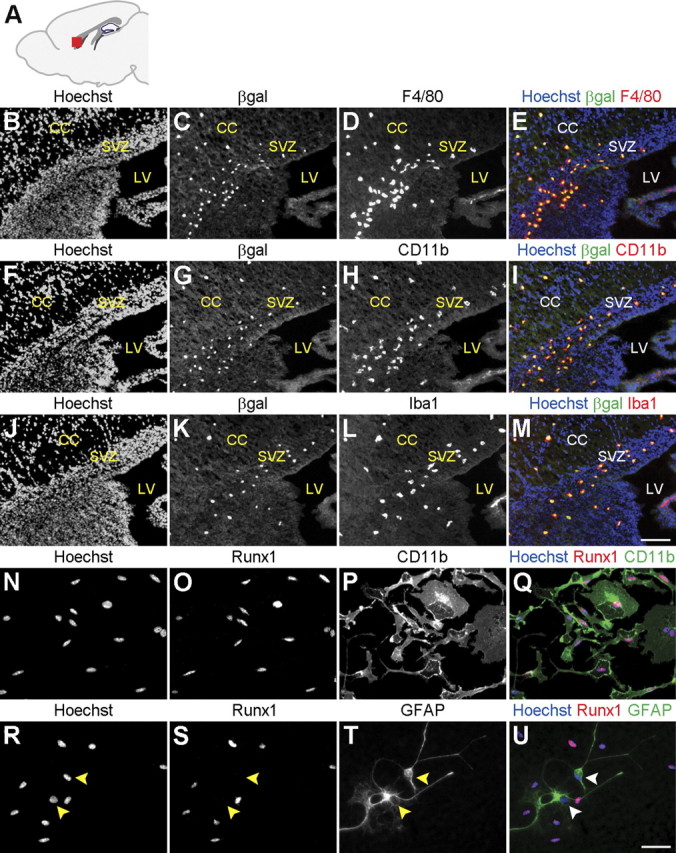
Runx1 is expressed in microglia in the postnatal forebrain. A, Schematic of the forebrain in the sagittal plane demonstrating the location (red square) of the anatomical area examined in B–M. B–M, Sagittal sections of P3 Runx1LacZ/+ brains were subjected to double-labeling analysis of β-gal and F4/80, CD11b, or Iba1 immunoreactivity, as indicated. Hoechst counterstaining is shown in the images in the left column and in the merged images in the right column. All β-gal+ cells in the ventricular zone and corpus callosum are positive for the expression of the microglial cell markers, F4/80, CD11b, and Iba1. CC, corpus callosum; LV, lateral ventricle; SVZ, subventricular zone. Scale bar, 100 μm. N–U, Primary microglial cell cultures from P3 mouse brain were subjected to double-labeling analysis of the expression of Runx1 and either the microglia marker CD11b (N–Q) or the astrocyte marker GFAP (R–U). Hoechst counterstaining is shown in N and Q and R and U. All Runx1+ cells express CD11b and do not express GFAP (R–U, arrowheads). Scale bar, 50 μm.
Further analysis showed that β-gal expression was correlated with microglial cells that exhibited a round morphology and were mainly localized in the subventricular zone and corpus callosum during the first 10 d after birth (Fig. 4A–L). In contrast, β-gal expression did not correlate with microglial cells that exhibited a ramified appearance, were located in the gray matter, and became progressively more abundant just as β-gal expression subsided at later postnatal stages (Fig. 4M–W). Together, these findings suggest that Runx1 is selectively expressed in periventricular amoeboid microglia, which are most abundant during the first 10–14 d after birth and are mainly present below the cerebral cortex (Hristova et al., 2010). They suggest further that Runx1 expression is downregulated during the transition of amoeboid microglia to the ramified state.
Figure 4.
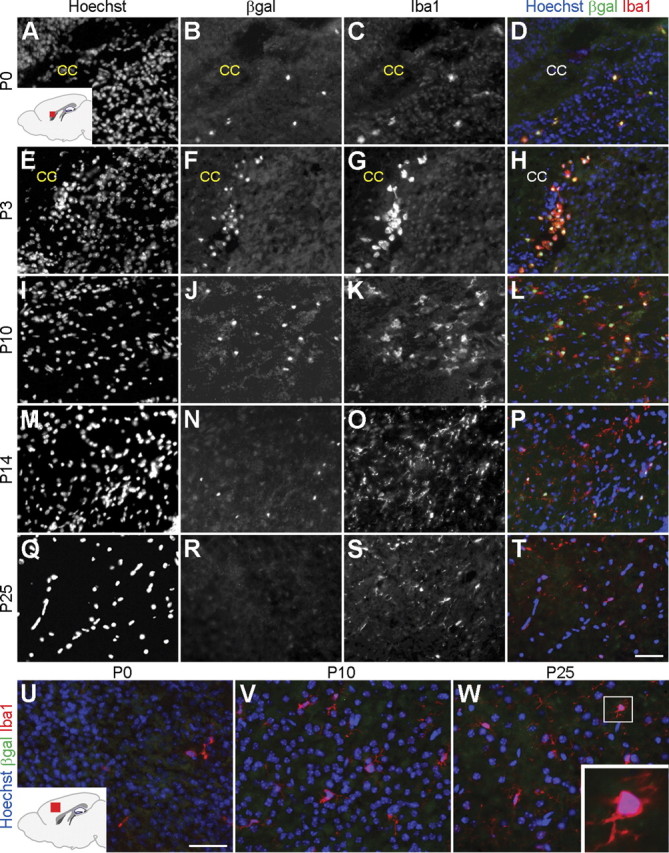
Runx1 is expressed in amoeboid microglia. Sagittal sections of the brain of Runx1LacZ/+ mice at the indicated postnatal stages were subjected to double-labeling analysis of β-gal and Iba1 expression. A–T, Analysis of the forebrain periventricular area. Inset in A shows a schematic of the forebrain in the sagittal plane demonstrating the location (red) of the area examined in A–T. Hoechst counterstaining is shown in the images in the left column and in the merged images in the right column. All Iba1+ cells express β-gal and exhibit a rounded morphology the first few days after birth (A–H). A gradual increase in the number of Iba1+ cells displaying a ramified morphology is observed during the first two postnatal weeks, just as the number of β-gal+ cells decreases (I–P). β-gal expression becomes undetectable by P25, when Iba1 expression is still observed (M–T). U–W, Analysis of the cerebral cortex parenchyma. Inset in U shows a schematic of the forebrain in the sagittal plane demonstrating the location (red) of the area examined in U–W. No β-gal expression is detected at any stage in Iba1+ cells located within the cerebral cortex. W, Inset shows higher magnification view of an Iba1+ cell with a ramified morphology. CC, corpus callosum. Scale bar, 50 μm.
Runx1 inhibits amoeboid microglial cell proliferation
Runx1 is an important regulator of cell proliferation in myeloid lineage cells (Ichikawa et al., 2004; Friedman, 2009). Postnatal amoeboid microglia are mitotic and motile during the first few days after birth but then gradually leave the cell cycle as they progress toward a ramified and deactivated state (Sminia et al., 1987; Alliot et al., 1999). To examine the involvement of Runx1 in microglia proliferation in the postnatal forebrain, we first compared the expression of β-gal and the mitotic cell marker Ki67 in Runx1LacZ/+ mice. At P0, most β-gal+ cells in the alar telencephalon of Runx1LacZ/+ mice expressed Ki67 (Fig. 5B–E). This correlation was progressively lost, however, and by P10 the majority of β-gal+ cells no longer expressed Ki67 (Fig. 5F–M). These results suggest that Runx1-expressing amoeboid microglia are initially mitotic, but they gradually leave the cell cycle before Runx1 expression is downregulated.
Figure 5.

Correlation between Runx1 expression and postnatal microglia proliferation. A, Schematic of the forebrain in the coronal plane demonstrating the location (red square) of the area examined in B–M. B–M, Coronal sections of Runx1LacZ/+ brains collected at the indicated postnatal stages were subjected to double-labeling analysis of β-gal and Ki67 expression. Boxes in D, H, and L indicate areas shown at higher magnification in E, I, and M. Arrowheads indicate cells coexpressing β-gal and Ki67. Numerous β-gal+ cells express Ki67 at P0, but this correlation is lost in older animals. CC, corpus callosum; LV, lateral ventricle. Scale bars: (in L) B–D, F–H, J–L, 100 μm; (in M) E, I, M, 50 μm.
Based on these observations, loss-of-function and gain-of-function studies were performed to characterize the involvement of Runx1 in the regulation of microglial cell proliferation. We first took advantage of the previous demonstration that Runx1 heterozygosity is haploinsufficient in the myeloid lineage (Song et al., 1999; Cai et al., 2000) to test whether reduced Runx1 activity would perturb microglial cell proliferation in vivo. A significant increase in the number of mitotic microglia (i.e., F4/80+/Ki67+ cells) was observed in the corpus callosum and external capsule of P3 Runx1LacZ/+ animals (which carry only one functional Runx1 allele) compared with Runx1+/+ littermates (Fig. 6A–I). To extend this in vivo observation, we next cultured primary microglial cells from the forebrain of P3 Runx1Flox/Flox mice, in which the Runx1 locus can be conditionally disrupted due to the insertion of a neomicin gene flanked by loxP sites (Growney et al., 2005). These primary cultures were transduced with lentiviral vectors expressing enhanced GFP alone or in combination with the Cre recombinase. Runx1 was endogenously expressed in virtually all cultured microglial cells in the absence of Cre and its expression became undetectable in cells transduced with Cre-expressing lentivirus (Fig. 6J,K). More important, the number of primary microglial cells that expressed Ki67 and incorporated BrdU was significantly increased following transduction with Cre-expressing lentivirus, compared with GFP alone (Fig. 6L–P). Together, the results of both in vivo and in vitro studies provide evidence that Runx1 is necessary for the inhibition of postnatal microglial cell proliferation.
Figure 6.
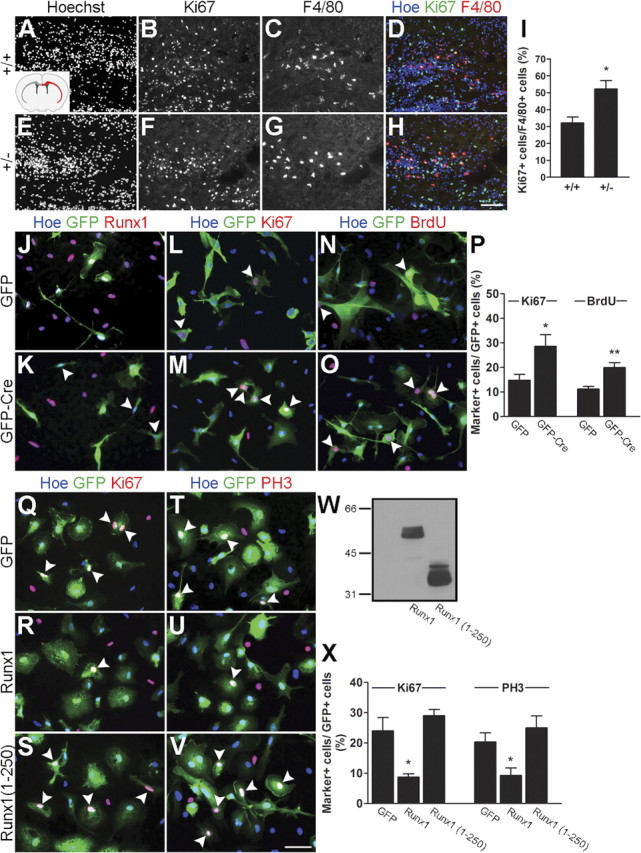
Runx1 promotes microglial cell cycle exit. A–I, Double-labeling immunofluorescence analysis of Ki67 and F4/80 expression in coronal sections of P3 Runx1+/+ and Runx1LacZ/+ brains. Inset in A shows a schematic of the forebrain in the coronal plane demonstrating the location (red) of the corpus callosum and external capsule, where cell counts were performed. I, Quantification of the percentage of microglial cells (F4/80+) expressing Ki67 in the corpus callosum and external capsule of P3 wild-type (32.13 ± 3.49%) versus Runx1Lacz+ (52.18 ± 5.08%) pups. Data are presented as mean ± SEM of 15 sections from three different pups for each genotype (n = 3). *p < 0.05, t test. Increased numbers of Ki67+ microglia are observed in Runx1LacZ/+ pups compared with control littermates. J–P, Primary cultures of mouse forebrain microglia were obtained from P3 Runx1Flox/Flox mice and transduced with lentivirus expressing GFP alone or together with the Cre recombinase, as indicated. J, K, Transduced cells were subjected to double-labeling analysis of GFP and endogenous Runx1 expression. All GFP+ cells express Runx1 in the absence of Cre (J); in contrast, Runx1 expression is not detectable in transduced cells in the presence of Cre (K, arrowheads). L–P, Transduced cells were subjected to double-labeling analysis of GFP and Ki67 (L, M) or BrdU incorporation followed by double-labeling analysis of GFP and BrdU immunoreactivity (N, O) (arrowheads, double-labeled cells). P, Quantification of the percentage of Ki67+ and BrdU+ microglia following transduction with lentivirus expressing GFP alone (14.76 ± 2.49% and 11.23 ± 1.05%, respectively) or in combination with Cre recombinase (28.60 ± 4.79% and 19.97 ± 1.96%, respectively). Data are presented as mean ± SEM from five separate experiments (n = 5). *p < 0.05 and **p < 0.005, t test. Increased numbers of Ki67+ and BrdU+ microglia are observed following expression of Cre, compared with GFP alone. Q–X, Primary cultures of mouse forebrain microglia were obtained from wild-type P3 CD1 mice and transduced with adenovirus expressing GFP alone or together with FLAG epitope-tagged forms of either full-length Runx1 or a truncated form of Runx1 containing only the first 250 aa [Runx1(1–250)], as indicated. Transduced cells were subjected to double-labeling analysis of GFP and Ki67 (Q–S) or GFP and PH3 (T–V) expression (arrowheads, examples of double-labeled cells). W, Western blotting analysis of the expression of Runx1 and Runx1(1–250) in transduced microglia using anti-FLAG antibody. X, Quantification of the percentage of Ki67+ and PH3+ microglia following transduction with adenovirus expressing GFP alone (24.05 ± 4.38% and 20.35 ± 3.04%, respectively) or in combination with either Runx1 (8.75 ± 1.05% and 9.28 ± 2.49%, respectively) or Runx1(1–250) (29.00 ± 2.12% and 25.06 ± 3.90%, respectively). Data are presented as mean ± SEM from four separated infection experiments (n = 4). *p < 0.05, t test. Decreased numbers of Ki67+ and PH3+ microglia are observed following expression of Runx1 (R, U), but not Runx1(1–250) (S, V), compared with GFP alone (Q, T). Scale bars: (in H) A–H, 100 μm; (in V) J–O and Q–V, 50 μm.
To determine whether Runx1 was sufficient to decrease microglia proliferation, converse gain-of-function experiments were performed in which an adenoviral vector was used to force exogenous expression of full-length Runx1 in primary microglial cell cultures obtained from wild-type mice. Compared with the expression of GFP alone, exogenous Runx1 caused a decrease in the number of cells expressing two mitotic cell markers, Ki67 and PH3. This effect was specific because it was not observed when we used a transcriptionally incompetent form of Runx1 containing only the first 250 aa, termed Runx1(1–250) (Theriault et al., 2005) (Fig. 6Q–X). We observed further that the decrease in the number of mitotic microglial cells caused by forced expression of exogenous Runx1 was paralleled by a converse increase in the number of cells expressing the cyclin-dependent kinase inhibitor p21Cip1 (Fig. 7A). Runx1(1–250) did not have a similar consequence, indicating that this effect required a transcriptionally competent Runx1. This observation is consistent with previous studies demonstrating that Runx1, as well as other Runx family members, regulates the expression of p21Cip1 (Lutterbach et al., 2000; Chi et al., 2005; Theriault et al., 2005; Hoi et al., 2010). Although double-labeling experiments to test for the coexpression of Runx1 and p21Cip1 in microglia in the developing forebrain proved to be technically unfeasible, chromatin immunoprecipitation studies demonstrated that endogenous Runx1 was bound to the p21Cip1 promoter in primary microglial cell cultures (Fig. 7B). Consistent with this result, we demonstrated that Runx1 was competent to activate transcription from the p21Cip1 promoter in HEK293 cells (Fig. 7C). In contrast, Runx1(1–250) did not cause promoter transactivation. Similar transcription assays proved to be technically challenging in primary microglial cells due to inadequate levels of transfection. Together, these results show that Runx1 is necessary and sufficient to inhibit amoeboid microglial cell proliferation and suggest that this function is mediated, at least in part, through activation of the expression of the cell cycle inhibitor, p21Cip1.
Figure 7.
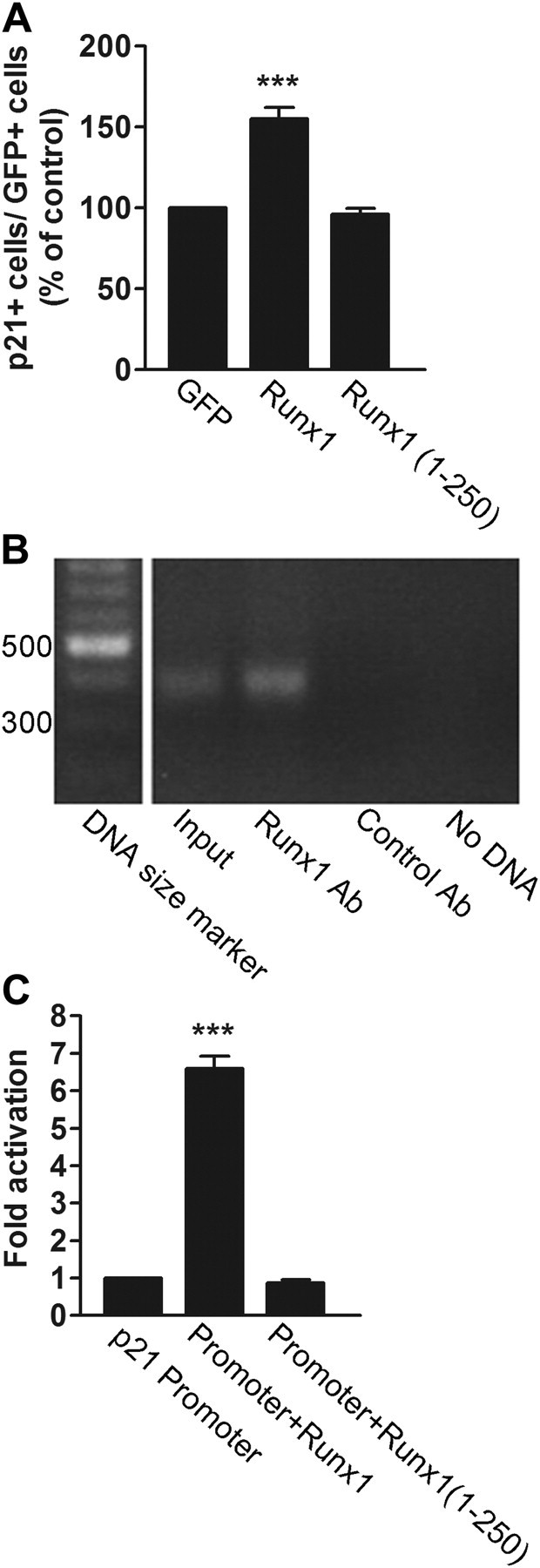
Runx1 promotes p21Cip1 expression. A, Primary cultures of forebrain microglia were obtained from wild-type P3 CD1 mice and transduced with adenovirus expressing GFP alone or together with FLAG epitope-tagged forms of either full-length Runx1 or Runx1(1–250), followed by double-labeling analysis of GFP and p21Cip1 expression and quantification of the percentage of transduced cells expressing p21Cip1. Increased numbers of p21Cip1-expressing microglia are observed following expression of Runx1 (155.09 ± 7.11%), but not Runx1(1–250) (96.20 ± 2.94%), compared with GFP alone (designated as 100%). Results are presented as mean ± SEM from five separated infection experiments (n = 5). ***p < 0.001, t test. B, Chromatin immunoprecipitation. Protein-DNA complexes from primary microglial cell cultures were subjected to immunoprecipitation with rabbit anti-Runx1 antibody or control rabbit antibody as indicated, followed by PCR with primers specific for the proximal region of the p21Cip1 promoter. Control PCRs were performed using input genomic DNA (input) or oligonucleotide primers alone (No DNA). A specific ChIP product is observed with the anti-Runx1 antibody. C, Transcription assays. HEK293 cells were transfected with a reporter plasmid encoding luciferase under the control of the p21Cip1 promoter in the absence (luciferase activity designated as one fold) or presence of plasmids encoding full-length Runx1 or Runx1(1–250). Runx1 (luciferase activity 6.60 ± 0.32) but not Runx1(1–250) (luciferase activity 0.87 ± 0.09) activates transcription from the p21Cip1 promoter (n = 4; ***p < 0.001, t test).
Runx1 inhibits microglia activation in vitro
We next examined the possibility that Runx1 might modulate microglia activation, in addition to regulating proliferation. Previous studies have shown that adenovirus infection induces microglia activation in vitro and causes increased expression of iNOS; this effects leads to the production of greater amounts of nitric oxide, which is involved in host defense functions (Bhat and Fan, 2002). Primary microglial cell cultures from P3 mouse forebrain were either untransduced or transduced with adenovirus-expressing GFP alone or together with Runx1, followed by analysis of iNOS expression. Compared with untransduced cells, infection with GFP-expressing adenovirus caused a significant increase in the expression of iNOS (Fig. 8A, compare lanes 1 and 2). This response was not observed when cells were transduced with a Runx1-expressing adenovirus, suggestive of an inhibitory effect of Runx1 on iNOS induction under these conditions (Fig. 8A, compare lanes 2 and 3). A similar result was obtained when we examined the expression of amphiphysin II, which was shown to be a component of the phagosome (Morrissette et al., 1999; Gold et al., 2000) (Fig. 8A, lanes 1–3). To confirm these observations, primary microglia were treated with LPS, which causes robust microglia activation and iNOS upregulation (Sheng et al., 2011; Wen et al., 2011). LPS treatment led to a significant increase in iNOS expression in untransduced cells and considerably enhanced the upregulation of iNOS caused by adenovirus infection (Fig. 8A, compare lanes 1, 4 and 2, 5). The latter effect was not observed, however, when the Runx1-expressing adenovirus was used (Fig. 8A, compare lanes 4–6). In agreement with these results, immunofluorescence analysis revealed that most microglia transduced with adenovirus-expressing GFP alone also coexpressed iNOS in cells treated with LPS; in contrast most of the cells transduced with GFP together with Runx1 displayed little or no expression of iNOS (Fig. 8B).Together, these results suggest that Runx1 may play a role in the regulation of both proliferation and activation in postnatal brain microglia.
Figure 8.
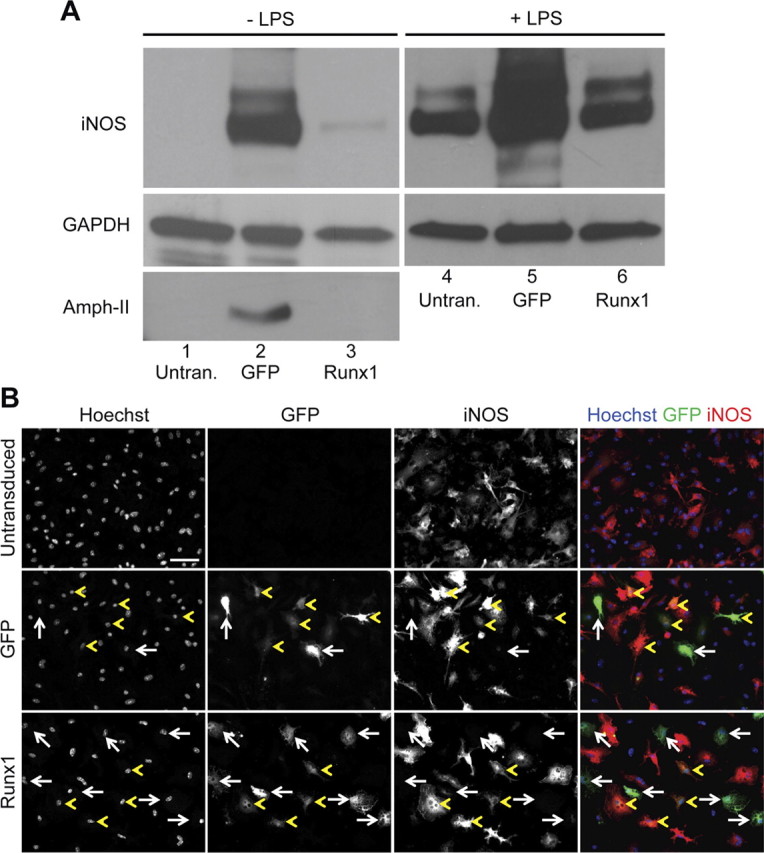
Effect of exogenous Runx1 expression on microglia activation. A, Western blot analysis of iNOS, amphiphysin II, and GAPDH expression in primary cultures of P3 mouse forebrain microglia, as indicated. Cells were not transduced (untran.) or transduced with adenovirus expressing GFP alone (GFP) or together with Runx1 (Runx1), followed by incubation in the absence or presence of LPS, as indicated. Cell lysates were collected and subjected to sequential Western blot analysis with anti-iNOS, anti-amphiphysin II (Amph-II), and anti-GAPDH antibodies. B, Double-label immunofluorescence analysis of GFP and iNOS expression in primary microglial cells that were either not transduced or transduced with adenovirus expressing GFP alone or together with Runx1, as indicated, followed by treatment with LPS. Where present, white arrows point to examples of cells expressing GFP but little or no iNOS, whereas yellow arrowheads point to examples of cells coexpressing GFP and iNOS. Notice how most cells transduced with GFP alone express iNOS, while most cells transduced with GFP + Runx1 express little or no iNOS. Scale bar, 50 μm.
Reduced Runx1 function results in delayed microglial cell ramification
The previous observations suggest that, by promoting the cell cycle exit and deactivation of postnatal amoeboid microglia, Runx1 might be important for the establishment of the ramified microglia phenotype. To examine this possibility, we compared the total number of microglial cells in the corpus callosum of Runx1+/− (i.e., Runx1LacZ/+) mice with Runx1+/+ littermates. Runx1 heterozygous mice displayed a higher number of microglia in this region consistent with a prolonged proliferation, and delayed ramification, of microglia in the presence of a reduced Runx1 gene dosage (Fig. 9A–G). However, this increase in microglia in the corpus callosum was not correlated with a parallel enlargement of the microglial cell pool in the cerebral cortex, where ramified microglia are normally found (Fig. 9H–N). These observations suggest that reduced Runx1 expression caused an expansion of the mitotic amoeboid microglia pool but did not result in the generation of supernumerary ramified microglia, possibly as a result of delayed/arrested ramification. To examine this possibility, we compared the numbers of amoeboid and ramified microglia in the forebrain of Runx1LacZ/+ heterozygous and Runx1+/+ wild-type littermates. Runx1LacZ/+ mice displayed increased numbers of amoeboid microglial cells and decreased ramified microglia compared with control littermates (Fig. 9O–Q).Together, these results provide evidence that Runx1 is important for both amoeboid microglial cell cycle exit and progression of the amoeboid-to-ramified microglia transformation.
Figure 9.
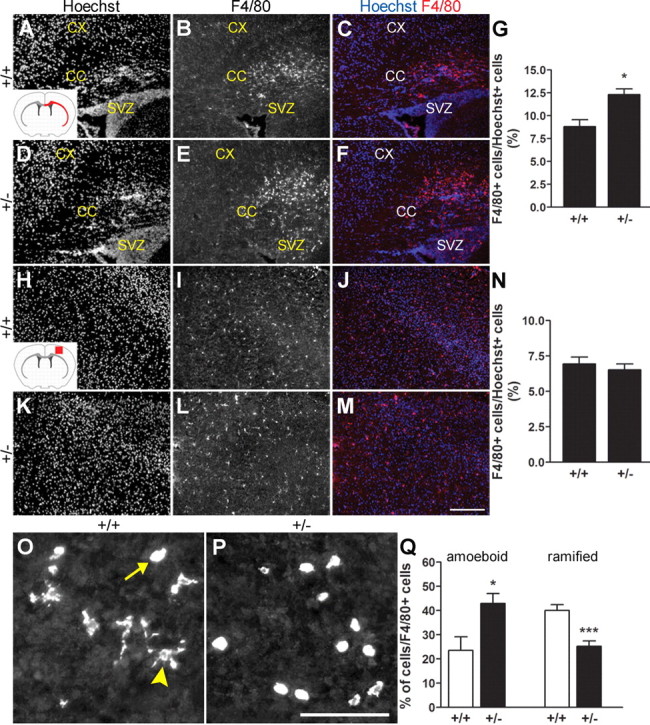
Decreased Runx1 activity impairs the amoeboid-to-ramified microglial cell transition. A–N, Analysis of F4/80 immunoreactivity in either the entire extent of the corpus callosum and external capsule (A–F) or a defined area of the cerebral cortex (H–M) of P10 Runx1+/+ or Runx1LacZ/+ mice. Red areas in the schematic of the forebrain in the coronal plane shown in the insets in A and H indicate regions subjected to cell counting analysis in either case. CC, corpus callosum; CX, cortex; SVZ, subventricular zone. G, Quantification of the number of F4/80+ cells (percentage of Hoechst+ nuclei) located in the corpus callosum and external capsule of either Runx1+/+ (8.80 ± 0.77%) or Runx1Lacz+ (12.32 ± 0.62%) littermates. Data are presented as mean ± SEM of 15 sections from three different pups for each genotype (n = 3; *p < 0.05, t test). N, Quantification of the number of F4/80+ cells (percentage of Hoechst+ nuclei) located in the cerebral cortex of either Runx1+/+ (6.94 ± 0.49%) or Runx1Lacz+ (6.53 ± 0.41%) littermates. Data are presented as mean ± SEM of 15 sections from three different pups for each genotype (n = 3). Increased numbers of microglia are detected in the subcortical white matter, but not cerebral cortex, of Runx1LacZ/+ pups compared with wild-type littermates. O–Q, Analysis of F4/80 immunoreactivity in the telencephalon of P3 Runx1+/+ or Runx1LacZ/+ mice. Arrow points to an example of cell with amoeboid morphology, while arrowhead points to an example of cell with ramified morphology. Q, Quantification of the number of amoeboid and ramified microglial cells located in the corpus callosum of either Runx1+/+ (23.50 ± 5.20% and 40.00 ± 1.44%, respectively) or Runx1Lacz/+ (42.92 ± 4.62% and 25.20 ± 0.92%, respectively) littermates. Data are presented as mean ± SEM of 15 sections from three different pups for each genotype (n = 3; *p < 0.05 and ***p < 0.001, t test). Runx1LacZ/+ pups display increased numbers of amoeboid and decreased numbers of ramified microglia compared with control littermates. Scale bars: 250 μm (A–F and H–M, 100 μm (O, P).
Runx1 expression is induced in adult nervous system microglia following nerve injury
The involvement of Runx1 in the transition from the amoeboid to the ramified microglia state in the developing CNS suggests that Runx1 expression might be induced in adult CNS microglia in response to events causing microglia activation. We therefore examined whether Runx1 expression would be upregulated in spinal cord microglia following sciatic nerve crush, a procedure that causes local microglia activation (Scholz and Woolf, 2007; Zhuang et al., 2007). No detectable Runx1 expression was observed in control adult mice ventral lumbar spinal cord, where microglia expressing Iba1, CD11b, F4/80, and exhibiting a small cell body and a ramified morphology were present (Fig. 10A–F). This finding is consistent with the absence (or expression at below detection levels) of Runx1 in ramified microglia in the developing brain. In contrast, Runx1 expression was readily detected at equivalent levels of the ventral lumbar spinal cord in mice subjected to sciatic nerve crush. All Runx1+ cells expressed CD11b and F4/80 and were part of aggregates of cells that also expressed Iba1 and displayed a microglia-like morphology (Fig. 10G–R). Within these ventral spinal cord aggregates, cells were generally less ramified than microglia in the ventral lumbar spinal cord of untreated control mice, where Runx1 was not expressed (compare Fig. 10S,T). Microglia aggregates with similar characteristics have been previously observed in the injured or diseased spinal cord and shown to contain microglia with morphological and immunological hallmarks of activation (Sanagi et al., 2010; Berger et al., 2011; David and Kroner, 2011). Interestingly, Runx1 expression was detected in only a subpopulation of microglia in these aggregates, possibly because not all cells in these clusters were equally activated. Together, these results suggest that Runx1 expression is induced in adult CNS microglia upon nerve injury. This finding in turn suggests that Runx1 might be involved in at least some of the mechanisms underlying the regulation of adult CNS microglia activation.
Figure 10.

Upregulation of Runx1 expression in adult mouse spinal cord microglia following sciatic nerve injury. A, B, Immunofluorescence analysis of Iba1 expression in the ventral spinal cord (lumbar level) of naive uninjured mice. B Represents a high-magnification view of the boxed area in A. C–F, Double-label immunofluorescence analysis of Runx1 and either CD11b (C, D) or F4/80 (E, F) expression in the spinal cord of naive uninjured mice. D, F, Represent high-magnification views of the boxed areas in C and E. G–J, Double-label immunofluorescence analysis of Iba1 and CD11b expression in the ventral spinal cord of sciatic nerve-injured mice. Dashed line in G demarcates the contour of the spinal cord. H–J, High-magnification views of the boxed area in G. K–R, Double-label immunofluorescence analysis of Runx1 and either CD11b (K–N) or F4/80 (O–R) expression in the ventral spinal cord of sciatic nerve-injured mice. L–N and P–R, High-magnification views of boxed areas in K and O, respectively. Arrowheads in panels N and R point to examples of double-labeled cells. S, T, Double-label immunofluorescence analysis of Runx1 and CD11b expression in either the ventral lumbar spinal cord of control, untreated mice, where Runx1 was not expressed (S), or in the CD11b+ cell aggregates observed in the ventral spinal cord of sciatic nerve-injured mice (T). T1–T4, Depict four different examples of Runx1+/CD11b+ cells in the ventral aggregates. Scale bars: A–R, 50 μm; or S, T, 25 μm.
Discussion
This study has demonstrated that the transcription factor Runx1, a key regulator of proliferation and differentiation along the myeloid lineage, is expressed in forebrain microglia during late embryogenesis and the first 2 weeks of postnatal development. Runx1-expressing perinatal microglia have a large rounded morphology and are mainly localized in the periventricular white matter. In contrast, little or no Runx1 expression is observed in ramified microglia in the gray matter. Together with the previous demonstration that microglia in the subcortical white matter of the postnatal forebrain are for the most part amoeboid and mitotic (Hristova et al., 2010), the present results provide evidence that Runx1 is expressed in amoeboid microglia during a critical period of perinatal development when microglia undergo a transition from the activated amoeboid state to the deactivated ramified phenotype.
We have shown further that many Runx1-expressing microglia are proliferative at P0, when mitotic microglia are also highly motile. This correlation is gradually lost during postnatal development, however, and by P10 most Runx1-expressing microglia are no longer mitotic. More important, decrease, or loss, of Runx1 function delays amoeboid microglial cell cycle exit in vivo and in vitro, resulting in an accumulation of mitotic microglia in the subcortical white matter in vivo. Conversely, Runx1 overexpression leads to decreased numbers of mitotic microglia in vitro. Runx1 regulates cell proliferation in a variety of contexts (Theriault et al., 2005; Hoi et al., 2010; Link et al., 2010). Moreover, Runx1, as well as other Runx proteins, can coordinate cell cycle exit with the progression to a more developmentally mature state in a number of cell lineages (Kagoshima et al., 2007; Appleford and Woollard, 2009).Together, these observations strongly suggest that at least one role of Runx1 in the postnatal forebrain is to inhibit the proliferative capacity of amoeboid microglia. In doing so, Runx1 might play an important function in mechanisms that ensure that amoeboid microglia undergoing the transition to the ramified state switch from a rapid proliferation mode to the slow-dividing mode typical of ramified microglia (Ajami et al., 2007).
The regulation of amoeboid microglial cell proliferation by Runx1 acquires particular importance when considered in light of the fact that it is estimated that large numbers of microglia are generated after birth, mostly due to a transient spurt in amoeboid microglia proliferation (Sminia et al., 1987; Alliot et al., 1999; Ginhoux et al., 2010). This expansionary phase is a crucial event in microglia biology because microglial cell numbers are determined in the adult brain mainly through the proliferation of resident microglia, rather than the recruitment of circulating bone marrow-derived progenitors (Ajami et al., 2007; Mildner et al., 2007; Ginhoux et al., 2010). The present results suggest that Runx1 might play a key role in the expansion of the initial contingent of mitotic microglia during postnatal development, which represents a critical first step toward achieving the required number of microglia in the adult brain.
The involvement of Runx1 in postnatal microglial cell proliferation is expected to provide significant new insight into the molecular mechanisms leading to the establishment of the correct number of resident brain microglia. Runx1 plays a key role in the regulation of cell proliferation in myeloid progenitor cells and the mechanisms underlying its function in these cells have been partly elucidated. For instance, Runx1 is directly involved in the transcriptional regulation of the expression of cell cycle progression genes, such as cyclins and cyclin-dependent kinase inhibitors, like p21Cip1 (Lutterbach et al., 2000; Bernardin-Fried et al., 2004; Friedman, 2009; Bakshi et al., 2010). With regard to the latter, Runx proteins were shown to either repress or transactivate the expression of p21Cip1 in context-dependent manners (Lutterbach et al., 2000; Theriault et al., 2005; Hoi et al., 2010). In agreement with these findings, we have demonstrated that endogenous Runx1 binds to the p21Cip1 promoter in primary microglial cells. Moreover, Runx1 can transactivate the p21Cip1 promoter and, more important, causes the appearance of supernumerary p21Cip1-positive cells when exogenously expressed in microglial cell cultures (a situation paralleled by a concomitant decrease in the number of mitotic microglia). Thus, it is reasonable to suggest that Runx1 regulates amoeboid microglial cell proliferation, at least in part, through its ability to activate expression of the cell cycle inhibitor, p21Cip1, resulting in a promotion of cell cycle exit.
This study has also provided evidence suggesting that Runx1 is sufficient to inhibit microglia activation in response to viral infection and LPS treatment in vitro. This finding suggests that Runx1 may act in postnatal amoeboid microglia to both promote cell cycle exit and modulate the activation of these cells. We have shown further that decreased Runx1 function also results in a delay/arrest of the microglia ramification process in vivo. It is therefore reasonable to suggest that Runx1 acts in amoeboid microglia to limit the length of the activated phase and guide these cells toward a deactivated phenotype. In doing so, Runx1 might play an important role in establishing the correct temporal window of microglia activation in the developing nervous system, thereby ensuring a developmental control of the activated-to-deactivated microglia transition.
Our results also strongly suggest that Runx1 expression is induced in adult spinal cord microglia following sciatic nerve crush, a procedure that causes local microglia activation. This finding raises a number of possibilities. Runx1 expression might be induced when ramified microglia receive signals to become activated, undergo a morphological transformation to a less ramified state, and mobilize to a site of injury. Under these conditions, Runx1 might promote the motility and/or morphological transformation of the activated cells. Alternatively, or in addition, circumstances causing adult CNS microglia activation might lead to Runx1 upregulation as a means of providing a negative control on the duration of microglia activation. More specifically, Runx1 might be important to limit the proliferation and/or activation of microglia in the injured/diseased CNS, thereby ensuring a transient microglia activation and a controlled and contained immune response. Through these or other possible mechanisms, Runx1 might be involved in processes that maintain a healthy balance of resting and activated microglia in the adult CNS.
In conclusion, continued analysis of the roles of Runx1, as well as its target genes and transcriptional partners, in the regulation of mononuclear phagocyte biology is expected to provide new molecular insight into the ontogeny and functions of embryonic and postnatal microglia. Characterization of genetic program(s) controlled by Runx1 in microglia has the potential to provide information on the molecular mechanisms controlling the balance between microglia activation and deactivation. This information is expected to suggest strategies to disable microglia functions experimentally at defined stages of development to clarify the key functions driving microglia infiltration into the developing brain. Moreover, it may provide new insight into the properties that distinguish microglia from other specific types of CNS mononuclear phagocytes, such as monocyte-derived cells that enter the adult CNS during disease, meningeal macrophages, or perivascular macrophages. Increased understanding of the molecular properties that distinguish microglia from these and other different types of mononuclear phagocytes of the CNS may enable researchers to develop strategies to selectively modulate microglia activation in the diseased adult CNS. Finally, the characterization of the roles of Runx1 during adult CNS microglia activation may provide important insight into the mechanisms controlling the balance between microglia activation and deactivation in the injured or diseased nervous system.
Footnotes
This work was supported by the Canadian Institutes of Health Research Operating Grants MOP-14828 (S.D.) and MOP-42479 and MOP-84577 (S.S). We thank Yeman Tang for excellent assistance in transcription assays, Estelle Llamosas and Matteo Di Piazza for important preliminary observations, Dr. Nancy Speck for Runx1Flox/+ mice, Dr. Phil Barker for Cre-expressing lentivirus, and Dr. Thomas Jessell for anti-Runx1 antibody.
The authors declare no competing financial interests.
References
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- Alliot F, Godin I, Pessac B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res. 1999;117:145–152. doi: 10.1016/s0165-3806(99)00113-3. [DOI] [PubMed] [Google Scholar]
- Appleford PJ, Woollard A. RUNX genes find a niche in stem cell biology. J Cell Biochem. 2009;108:14–21. doi: 10.1002/jcb.22249. [DOI] [PubMed] [Google Scholar]
- Bakshi R, Hassan MQ, Pratap J, Lian JB, Montecino MA, van Wijnen AJ, Stein JL, Imbalzano AN, Stein GS. The human SWI/SNF complex associates with RUNX1 to control transcription of hematopoietic target genes. J Cell Physiol. 2010;225:569–576. doi: 10.1002/jcp.22240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger JV, Deumens R, Goursaud S, Schäfer S, Lavand'homme P, Joosten EA, Hermans E. Enhanced neuroinflammation and pain hypersensitivity after peripheral nerve injury in rats expressing mutated superoxide dismutase 1. J Neuroinflammation. 2011;8:33. doi: 10.1186/1742-2094-8-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardin-Fried F, Kummalue T, Leijen S, Collector MI, Ravid K, Friedman AD. AML1/RUNX1 increases during G1 to S cell cycle progression independent of cytokine-dependent phosphorylation and induces cyclin D3 gene expression. J Biol Chem. 2004;279:15678–15687. doi: 10.1074/jbc.M310023200. [DOI] [PubMed] [Google Scholar]
- Bhat NR, Fan F. Adenovirus infection induces microglial activation: involvement of mitogen-activated protein kinase pathways. Brain Res. 2002;948:93–101. doi: 10.1016/s0006-8993(02)02953-0. [DOI] [PubMed] [Google Scholar]
- Cai Z, de Bruijn M, Ma X, Dortland B, Luteijn T, Downing RJ, Dzierzak E. Haploinsufficiency of AML1 affects the temporal and spatial generation of hematopietic stem cells in the mouse embryo. Immunity. 2000;13:423–431. doi: 10.1016/s1074-7613(00)00042-x. [DOI] [PubMed] [Google Scholar]
- Chi XZ, Yang JO, Lee KY, Ito K, Sakakura C, Li QL, Kim HR, Cha EJ, Lee YH, Kaneda A, Ushijima T, Kim WJ, Ito Y, Bae SC. RUNX3 suppresses gastric epithelial cell growth by inducing p21WAF1/Cip1 expression in cooperation with transforming growth factor β-activated SMAD. Mol Cell Biol. 2005;25:8097–8107. doi: 10.1128/MCB.25.18.8097-8107.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David S, Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci. 2011;12:388–399. doi: 10.1038/nrn3053. [DOI] [PubMed] [Google Scholar]
- Davoust N, Vuaillat C, Androdias G, Nataf S. From bone marrow to microglia: barriers and avenues. Trends Immunol. 2008;29:227–234. doi: 10.1016/j.it.2008.01.010. [DOI] [PubMed] [Google Scholar]
- Friedman AD. Cell cycle and developmental control of hematopoiesis by Runx1. J Cell Physiol. 2009;219:520–524. doi: 10.1002/jcp.21738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold ES, Morrissette NS, Underhill DM, Guo J, Bassetti M, Aderem A. Amphiphysin IIm, a novel amphiphysin II isoform, is required for macrophage phagocytosis. Immunity. 2000;12:285–292. doi: 10.1016/s1074-7613(00)80181-8. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Streit WJ. Microglia: biology and pathology. Acta Neuropathol. 2010;119:89–105. doi: 10.1007/s00401-009-0622-0. [DOI] [PubMed] [Google Scholar]
- Growney JD, Shigematsu H, Li Z, Lee BH, Adelsperger J, Rowan R, Curley DP, Kutok JL, Akashi K, Williams IR, Speck NA, Gilliland DG. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood. 2005;106:494–504. doi: 10.1182/blood-2004-08-3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Hoi CS, Lee SE, Lu SY, McDermitt DJ, Osorio KM, Piskun CM, Peters RM, Paus R, Tumbar T. Runx1 directly promotes proliferation of hair follicle stem cells and epithelial tumor formation in mouse skin. Mol Cell Biol. 2010;30:2518–2536. doi: 10.1128/MCB.01308-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hristova M, Cuthill D, Zbarsky V, Acosta-Saltos A, Wallace A, Blight K, Buckley SM, Peebles D, Heuer H, Waddington SN, Raivich G. Activation and deactivation of periventricular white matter phagocytes during postnatal mouse development. Glia. 2010;58:11–28. doi: 10.1002/glia.20896. [DOI] [PubMed] [Google Scholar]
- Ichikawa M, Asai T, Chiba S, Kurokawa M, Ogawa S. Runx1/AML-1 ranks as a master regulator of adult hematopoiesis. Cell Cycle. 2004;3:722–724. [PubMed] [Google Scholar]
- Kagoshima H, Shigesada K, Kohara Y. RUNX regulates stem cell proliferation and differentiation: insights from studies of C. elegans. J Cell Biochem. 2007;100:1119–1130. doi: 10.1002/jcb.21174. [DOI] [PubMed] [Google Scholar]
- Kurokawa M. AML1/Runx1 as a versatile regulator of hematopoiesis: regulation of its function and a role in adult hematopoiesis. Int J Hematol. 2006;84:136–142. doi: 10.1532/IJH97.06070. [DOI] [PubMed] [Google Scholar]
- Link KA, Chou FS, Mulloy JC. Core binding factor at the crossroads: determining the fate of the HSC. J Cell Physiol. 2010;222:50–56. doi: 10.1002/jcp.21950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutterbach B, Westendorf JJ, Linggi B, Isaac S, Seto E, Hiebert SW. A mechanism of repression by acute myeloid leukemia-1, the target of multiple chromosomal translocations in acute leukemia. J Biol Chem. 2000;275:651–656. doi: 10.1074/jbc.275.1.651. [DOI] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Brück W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Morrissette NS, Gold ES, Guo J, Hamerman JA, Ozinsky A, Bedian V, Aderem AA. Isolation and characterization of monoclonal antibodies directed against novel components of macrophage phagosomes. J Cell Sci. 1999;112:4705–4713. doi: 10.1242/jcs.112.24.4705. [DOI] [PubMed] [Google Scholar]
- North T, Gu TL, Stacy T, Wang Q, Howard L, Binder M, Marín-Padilla M, Speck NA. Cbfa2 is required for the formation of intra-aortic hematopoietic clusters. Development. 1999;126:2563–2575. doi: 10.1242/dev.126.11.2563. [DOI] [PubMed] [Google Scholar]
- Prinz M, Mildner A. Microglia in the CNS: immigrants from another world. Glia. 2011;59:177–187. doi: 10.1002/glia.21104. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Nature. 2010;468:253–262. doi: 10.1038/nature09615. [DOI] [PubMed] [Google Scholar]
- Sanagi T, Yuasa S, Nakamura Y, Suzuki E, Aoki M, Warita H, Itoyama Y, Uchino S, Kohsaka S, Ohsawa K. Appearance of phagocytic microglia adjacent to motoneurons in spinal cord tissue from a presymptomatic transgenic rat model of amyotrophic lateral sclerosis. J Neurosci Res. 2010;88:2736–2746. doi: 10.1002/jnr.22424. [DOI] [PubMed] [Google Scholar]
- Schlegelmilch T, Henke K, Peri F. Microglia in the developing brain: from immunity to behavior. Curr Opin Neurobiol. 2011;21:5–10. doi: 10.1016/j.conb.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- Sheng W, Zong Y, Mohammad A, Ajit D, Cui J, Han D, Hamilton JL, Simonyi A, Sun AY, Gu Z, Hong JS, Weisman GA, Sun GY. Pro-inflammatory cytokines and lipopolysaccharide induce changes in cell morphology, and upregulation of ERK1/2, iNOS and sPLA2-IIA expression in astrocytes and microglia. J Neuroinflammation. 2011;8:121. doi: 10.1186/1742-2094-8-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaper SD, Facci L, Culbert AA, Evans NA, Chessell I, Davis JB, Richardson JC. P2X(7) receptors on microglial cells mediate injury to cortical neurons in vitro. Glia. 2006;54:234–242. doi: 10.1002/glia.20379. [DOI] [PubMed] [Google Scholar]
- Sminia T, de Groot CJ, Dijkstra CD, Koetsier JC, Polman CH. Macrophages in the central nervous system of the rat. Immunobiology. 1987;174:43–50. doi: 10.1016/S0171-2985(87)80083-9. [DOI] [PubMed] [Google Scholar]
- Song WJ, Sullivan MG, Legare RD, Hutchings S, Tan X, Kufrin D, Ratajczak J, Resende IC, Haworth C, Hock R, Loh M, Felix C, Roy DC, Busque L, Kurnit D, Willman C, Gewirtz AM, Speck NA, Bushweller JH, Li FP, Gardiner K, Poncz M, Maris JM, Gilliland DG. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23:166–175. doi: 10.1038/13793. [DOI] [PubMed] [Google Scholar]
- Stifani N, Freitas AR, Liakhovitskaia A, Medvinsky A, Kania A, Stifani S. Suppression of interneuron programs and maintenance of selected spinal motor neuron fates by the transcription factor AML1/Runx1. Proc Natl Acad Sci U S A. 2008;105:6451–6456. doi: 10.1073/pnas.0711299105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stifani S, Ma Q. ‘Runxs and regulations’ of sensory and motor neuron subtype differentiation: implications for hematopoietic development. Blood Cells Mol Dis. 2009;43:20–26. doi: 10.1016/j.bcmd.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriault FM, Nuthall HN, Dong Z, Lo R, Barnabe-Heider F, Miller FD, Stifani S. Role for Runx1 in the proliferation and neuronal differentiation of selected progenitor cells in the mammalian nervous system. J Neurosci. 2005;25:2050–2061. doi: 10.1523/JNEUROSCI.5108-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, Ribeiro R, Zhang Y. Specific PKC isoforms regulate LPS-stimulated iNOS induction in murine microglial cells. J Neuroinflammation. 2011;8:38. doi: 10.1186/1742-2094-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagami CJ, Stifani S. Molecular characterization of the mouse superior lateral parabrachial nucleus through expression of the transcription factor Runx1. PLoS One. 2010;5:e13944. doi: 10.1371/journal.pone.0013944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Kawasaki Y, Tan PH, Wen YR, Huang J, Ji RR. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav Immun. 2007;21:642–651. doi: 10.1016/j.bbi.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]