

Abstract
Canonical transient receptor potential (TRPC3) nonselective cation channels are effectors of G-protein-coupled receptors (GPCRs), activated via phospholipase C–diacylglycerol signaling. In cerebellar Purkinje cells, TRPC3 channels cause the metabotropic glutamate receptor (mGluR)-mediated slow EPSC (sEPSC). TRPC3 channels also provide negative feedback regulation of cytosolic Ca2+, mediated by a C terminus “calmodulin and inositol trisphosphate receptor binding” (CIRB) domain. Here we report the alternative splicing of the TRPC3 mRNA transcript (designated TRPC3c), resulting in omission of exon 9 (approximately half of the CIRB domain) in mice, rats, and guinea pigs. TRPC3c expression is brain region specific, with prevalence in the cerebellum and brainstem. The TRPC3c channels expressed in HEK293 cells exhibit increased basal and GPCR-activated channel currents, and increased Ca2+ fluorescence responses, compared with the previously characterized (TRPC3b) isoform when activated via either the endogenous M3 muscarinic acetylcholine receptor, or via coexpressed mGluR1. GPCR-induced TRPC3c channel opening rate (cell-attached patch) matched the maximum activation achieved with inside-out patches with zero cytosolic Ca2+, whereas the GPCR-induced TRPC3b activation frequency was significantly less. Both TRPC3 channel isoforms were blocked with 2 mm Ca2+, attributable to CIRB domain regulation. In addition, genistein blocked Purkinje cell (S)-2-amino-2-(3,5-dihydroxyphenyl) acetic acid (mGluR1)-activated TPRC3 current as for recombinant TRPC3c current. This novel TRPC3c ion channel therefore has enhanced efficacy as a neuronal GPCR-Ca2+ signaling effector, and is associated with sensorimotor coordination, neuronal development, and brain injury.
Introduction
Canonical transient receptor potential (TRPC3) nonselective cation channels have broad representation in the brain (Riccio et al., 2002; Chung et al., 2006). The channels are activated by Gq-protein-coupled receptor–phospholipase Cβ (PLCβ)–diacylglycerol (DAG) and tyrosine protein kinase receptor–phospholipase Cγ–DAG pathways (Birnbaumer, 2009), and are modulated by Ca2+-calmodulin (CaM) and IP3 receptor binding.
TRPC3 channels have broad physiological significance in neural tissues. For example, in the cochlea TRPC3 channels are expressed in spiral ganglion neurons and sensory hair cells (Phan et al., 2010; Tadros et al., 2010). In the hair cells, the channels regulate resting Ca2+ against a high level of plasma membrane Ca2+ ATPase 2 (PMCA2) Ca2+ extrusion (Raybould et al., 2007). In the brain, TRPC3 is the predominant subtype of TRPC subunit (Dietrich et al., 2005). TRPC3 has been implicated in promoting neurite outgrowth and survival of cerebellar neurons (Li et al., 2005). TRPC3 is necessary for brain-derived neurotrophic factor (BDNF) to confer a neuroprotective effect in neonatal cerebellar granule cells by allowing BDNF to activate cAMP/Ca2+ response element binding (CREB) protein that leads to neuronal survival following serum deprivation (Jia et al., 2007). In substantia nigra, TRPC3 channels regulate GABAergic neuron firing that is integral to basal ganglia motor output (Zhou et al., 2008). A key study by Hartmann et al. (2008) used the TRPC3 knock-out mouse model to determine that the slow EPSC (sEPSC) in the cerebellar Purkinje neurons, which arises from activation of the metabotropic glutamate receptor (mGluR1), during normal synaptic transmission, is solely attributable to the TRPC3 channel. The TRPC3-null mice exhibited ataxia, consistent with the altered regulation of motor coordination by impaired Purkinje neuron function. It has recently been reported that these cerebellar Purkinje cell TRPC3 channels have different phosphorylation properties to the recombinant protein (TRPC3b isoform), which has implications for long-term depression in these cells that underpin motor control (Nelson and Glitsch, 2012).
A calmodulin/IP3R-binding site (CIRB domain) coded within exons 9 and 10 of TRPC3 (Tang et al., 2001; Zhang et al., 2001; Wedel et al., 2003) contributes to the modulation of TRPC3 channel activation in association with juxtaposed IP3R-gated Ca2+ release. IP3 can activate the TRPC3 channel by promoting binding of the IP3R to the CIRB domain; however, this is not obligatory for TRPC3 activation (Vazquez et al., 2003). TRPC3 channel activation is inhibited by membrane-bound Ca2+-calmodulin via the CIRB domain. Thus extrusion of Ca2+, for example, by PMCA, can be balanced by Ca2+ entering alongside Na+ through TRPC3 cation channels when cytosolic Ca2+ levels fall (Zhang et al., 2001). A fall in cytosolic [Ca2+] leads to dissociation of the Ca2+-CaM complex and hence reduction of the block of the TRPC3 channels by this complex; enabling restoration of resting Ca2+.
In the present study, we characterize the TRPC3c ion channel that arises from splicing out exon 9 (approximately half of the CIRB domain coding region). We demonstrate that this channel is the dominant TRPC3 isoform in the cerebellum, and it exhibits significant enhancement of cation flux and Ca2+ entry, which likely impacts on receptor-coupled neuronal signaling.
Materials and Methods
Reverse transcription and PCR amplification of the TRPC3b and TRPC3c mRNA.
RNA was extracted from mouse, rat, and guinea pig brain tissues (cerebellum, midbrain, medulla, and cerebrum) of either sex. For mouse (C57BL/6J strain) and rat (Wistar) brain tissues, the total RNA was extracted using Trizol (Invitrogen) according to the manufacturer's instruction. For guinea pig (colored) tissues, total RNA was extracted using Purelink total RNA isolation kit (Invitrogen). The RNA was then reverse transcribed using Superscript III Reverse Transcription System (Invitrogen), with random hexamer priming, to produce first-strand cDNA template. The TRPC3 cDNA was amplified using primers that span exon 9 to investigate the relative expression of each isoform. The PCR amplification (40 cycles) used forward and reverse primers that targeted the coding regions of exon 8 and 10 of TRPC3 mRNA, respectively: denaturation at 98°C for 10 s, annealing at 58°C for 15 s, and extension at 72°C for 30 s. The sequence of the primers (and size of the amplicons of both isoforms in base pairs) were as follows: mouse: forward 5′-CTAACTTTTCCAAATGCAGGAGGAGAAG-3′; reverse 5′-TCGCATGATAAAGGTAGGGAACACTAGA-3′ (TRPC3b: 501, TRPC3c: 417), rat: forward 5′-CAGTGATGTAGAGTGGAAGTTTGC-3′; reverse 5′-CTCCCTCATTCACACCTCAGC-3′ (TRPC3b: 408, TRPC3c: 324), guinea pig: forward 5′-GGATCATTAACTTTTCCAAATGTAGAAGG-3′; reverse 5′-TCTCAGCACGCTGGGATTCAGTTTCT-3′ (TRPC3b: 374, TRPC3c: 290). The PCR produced amplicons for TRPC3b isoform and TRPC3c isoform (84 bp shorter than TRPC3b). The amplicons were then separated using electrophoresis on 1% agarose gel. The cDNA of each TRPC3 isoform was quantified via measurement of the optical density of the respective bands (SYBR-safe stained; Invitrogen) using semiquantitative analysis software Genesnap (v6.08; PerkinElmer). The number of animals for each brain region in these experiments were as follows: mouse: n = 9 cerebellum, n = 5 midbrain, medulla, n = 4 cerebrum; rat n = 6 cerebellum, midbrain, medulla, n = 5 cerebrum; guinea pig n = 5 cerebellum, n = 6 midbrain, medulla, cerebrum. The cDNAs for both TRPC3 isoforms were sequenced. The TRPC3 cDNA sequences were verified via alignment with TRPC3b mRNA sequences in the National Center for Biotechnology Information database (GenBank accession numbers: mouse AK080619, rat NM021771, and guinea pig FJ207473). Full-length mouse TRPC3 transcripts were also obtained by PCR using similar thermal cycling parameters. Full-length TRPC3b and TRPC3c transcripts were detected by reverse transcription (RT)-PCR from cerebrum and cerebellum of mouse, respectively, using 5′ sense and 3′ antisense primers that targeted the regions of the start and stop codons (primer sequences: forward 5′-ACAGAATTCCTGCGGGGATGCGTGACA-3′, reverse 5′-AGCGGATCCCCTCACTCACATCTCAGCA-3′). The restriction sites for EcoR1 and BamH1 were incorporated into the 5′ end of the forward and reverse primers, respectively, to facilitate cloning into the pIRES-DsRed2 mammalian expression vector (Clontech). All PCRs used Finnzyme high-fidelity TaqDNA polymerase and supplied reaction mix (ThermoFisher Scientific). Animal experiments were undertaken with protocol approval of the University of New South Wales (Australia) and University of Auckland (New Zealand) animal ethics committees.
Cell culture and transfection with TRPC3-pIRES-DsRed2 construct and mGluR1-eYFP.
Human embryonic kidney (HEK) 293 cells (Invitrogen) were cultured to 90% confluence in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS) in a humidified atmosphere of 95% O2 and 5% CO2 at 37°C. Cells were transfected with either mouse full-length TRPC3b or TRPC3c cDNA cloned into a pIRES-DsRed2 vector construct (Clontech). The transfection of the HEK293 cells with the vectors was made using Lipofectamine 2000 (Invitrogen) according to manufacturer's instructions. Cells were then treated with 1.1 mg/ml G418 antibiotic (Invitrogen) for 4 weeks to select for stably transfected cells. The expression of the mouse TRPC3 gene construct in the G418-resistant cells was verified by RT-PCR amplification and sequencing of the TRPC3 cDNA. The stably transfected cell lines were sorted using fluorescence-activated cell sorting (FACS) for high DsRed2 fluorescence. To do this, cells were trypsinized and sorted using FACSaria (BD Bioscience) cell sorting apparatus, which selected for DsRed2 signal using a PE-A filter. Cells with top 5% level of DsRed2 fluorescence were selected for experiments. Coexpression of mGluR1 with the TRPC3 isoforms used a mouse mGluR1-eYFP fusion protein encoding cDNA construct downstream of the cytomegalovirus promoter (Masu et al., 1991). The HEK293 cells stably expressing either TRPC3 isoform were then transformed with the mGluR1-eYFP fusion protein cDNA using Lipofectamine 2000 (Invitrogen).
Western blotting—expression of TRPC3b and TRPC3c isoforms in HEK293 cells.
Expression of TRPC3b and TRPC3c proteins was quantified in transfected HEK293 cells by Western blotting. HEK293 cells stably expressing TRPC3b or TRPC3c, and untransfected cells (control), were grown in minimum essential medium containing 10% FBS, streptomycin, and penicillin and before collection and were plated out overnight at a density of 1.5 × 105 cells/well in poly-d-lysine-coated 6-well (6.9 cm2 area) culture dishes at 37°C with 5% CO2. Whole-cell lysates were prepared by incubating cells in lysis buffer containing (in mm): 137 NaCl; 20 Tris; 1 EDTA; 1% Triton X-100; 1% sodium deoxycholate; 0.1% SDS; protease inhibitors (Complete Mini protease inhibitor mixture; Roche Applied Sciences), adjusted to pH 7.5 with HCl, for 30 min at room temperature with agitation; and insoluble content was removed by centrifugation. Cell lysates were separated by 10% SDS-PAGE at 10 μg/lane in 2× Laemmeli sample buffer (125 mm Tris, 4% SDS, 20% glycerol, and 10% β-mercaptoethanol). TRPC3b and TRPC3c proteins were detected by Western blotting using polyclonal rabbit antibody directed to amino acids 822–835 of mouse TRPC3 at 2 μg/ml (ACC-016, lot no. AN-07; Alomone Labs) with a goat anti-rabbit IgG-horseradish peroxidase conjugate secondary antibody (1:20,000, lot no. L9704446 RevA; Bio-Rad). Chemiluminescence was detected using enhanced chemiluminescence reagent (Bio-Rad) and a ChemiDoc digital imaging system (Bio-Rad). To confirm equal protein loading of the whole-cell lysate samples, blots were stripped of TRPC3 antibodies and actin expression level was detected with a rabbit anti-actin affinity isolated antibody (1:1000, lot no. 048k4861; Sigma-Aldrich).
Integration of TRPC3 channel subunits into the plasma membrane was determined by Western blotting following extraction of the plasma membrane fraction using membrane-impermeant biotinylation reagent (EZ-Link Sulfo-NHS-SS-Biotin; Pierce Biotechnology). Cells were washed three times with 2 ml ice-cold PBS, incubated with 0.5 ml of 1.5 mg/ml sulfo-NHS-SS-Biotin in PBS for 20 min at 4°C, removed by aspiration, and repeated with a fresh aliquot of sulfo-NHS-SS biotin. Unbound biotin was removed by aspiration and cells thoroughly washed and quenched with ice-cold PBS with 100 mm glycine on ice before being solubilized in lysis buffer by gentle agitation on ice for 30 min. Cell lysates were collected by centrifugation. To isolate biotinylated cell-surface protein, NeutrAvidin beads (Pierce Biotechnology) prepared as 50% suspension in lysis buffer were added at 50 μl to 0.19 ml of each lysate supernatant and incubated for 1 h at 4°C with occasional mixing. NeutrAvidin beads–biotinylated membrane protein complex was pelleted by centrifugation at 4°C. The unbound supernatant fraction was removed, the pellet washed three times with 0.5 ml lysis buffer, and the biotinylated proteins were extracted from the beads by adding 50 μl 2× Laemmeli sample buffer and incubated for 30 min at room temperature for 1 h followed by centrifugation. Biotinylated proteins were analyzed by SDS-PAGE and Western blotting. Purity of the isolated cell surface protein sample was confirmed by nil expression of actin (data not shown).
TRPC3 immunolocalization.
TRPC3 protein expression was localized in the mouse cerebellum and in HEK293 cells expressing the recombinant mouse TRPC3b and TRPC3c variants by immunofluorescence and confocal microscopy. For fixation and collection of the cerebellar tissue, the mice (C129 SvEv background strain) were killed with sodium pentobarbital solution (100 mg/ml; 100 mg/kg body weight) and intracardially perfused with 10 ml of 0.5% sodium nitroprusside in 0.9% saline followed by perfusion with 20 ml of 4% paraformaldehyde (PFA) in 0.1 m phosphate buffer. Cerebellum was then dissected and postfixed in the PFA solution overnight. The tissue was then cryoprotected (10, 20, and 30% sucrose in PBS), embedded in Tissue-Tek OCT compound (Sakura Finetek), and sectioned at 50 μm using a cryostat (floating sections). HEK293 cells grown on poly-d-lysine-coated (Sigma-Aldrich) coverslips, were fixed in situ using 4% PFA in PBS for 10 min, then the cells were washed with PBS. The cells or brain sections were then permeabilized with 1% Triton X-100 in PBS with 10% normal goat serum (NGS; Vector Laboratories) (2 h for cerebellum sections, 10 min for HEK293 cells) at room temperature, followed by overnight incubation with TRPC3 antibody (1:1000; lot no. AN-03 or AN-07; Alomone Labs) in PBS with 5% NGS and 0.1% Triton X-100, at 4°C. After washing in PBS (3 × 30 min), Alexa Fluor 488 goat anti-rabbit IgG secondary antibody (Invitrogen) (1:500, 5% NGS, 0.1% Triton X-100, PBS) was applied for 4 h at room temperature, followed by 2 h at 4°C, during which the tissue was protected from light. The coverslips with the HEK293 cells or floating cerebellar sections were then washed in PBS several times and mounted using Vectashield (Vector Laboratories). The immunolabeling was then visualized using a Zeiss Z1 AxioExaminer NLO710 confocal microscope with 40× objective, and 488 nm excitation laser (495–550 nm emission). Controls included incubation without the primary antibody (to assess nonspecific secondary binding) and use of cerebellar sections from TRPC3 knock-out mice or untransfected HEK293 cells (data not shown).
Whole-cell electrophysiology.
For patch-clamp recordings, HEK293 cells stably expressing recombinant TRPC3 ion channels were grown to 85–95% confluence on a coverslip coated with poly-d-lysine and collagen (both from Sigma-Aldrich). Recording pipettes were made from borosilicate glass (GC120TF-10; Harvard Apparatus). The pipette resistance was at 3–6 MΩ (PC-10; Narishige). The internal solution had the composition containing (in mm): 130 CsCl, 2.0 MgCl2, 10 EGTA, 0.3 ATP, 0.03 GTP, pH at 7.3, adjusted with CsOH. Cells on the coverslip were placed in a microchamber on an inverted microscope (Leica DMIL) and superfused with HEPES-buffered physiological salt solution (HPSS) containing 120 mm NaCl, 5.4 mm KCl, 2 mm CaCl2, 1.13 mm MgCl2, 10 mm glucose, and 20 mm HEPES, pH 7.4, at room temperature. Whole-cell patch-clamp recordings were made following a gigaseal, using an Axopatch 200 patch-clamp amplifier (Molecular Devices) controlled by software (pClamp 10.2; Molecular Devices). Cell capacitance was canceled and series resistance was compensated by ∼90%. Holding voltage was −40 mV, with a voltage ramp (−100 to +50 mV) over 1 s in every 5 s of recording to determine basal and TRPC3 channel-mediated membrane conductance. Carbachol [CCh; used to activate TRPC3 channels via the endogenous M3 acetylcholine receptor (AChR)] was purchased from Sigma-Aldrich. (S)-2-Amino-2-(3,5-dihydroxyphenyl) acetic acid (DHPG), a group 1 mGluR agonist (Schoepp et al., 1994), was purchased from Tocris Bioscience. All experiments were undertaken at room temperature.
Single-channel electrophysiology.
Single-channel activity was recorded using cell-attached and inside-out patch-clamp configurations in HEK293 cells stably expressing the recombinant mouse TRPC3 channels. The bath solution consisted of HPSS solution. Ca2+-free HPSS was identical to the normal HPSS except it consisted of 10 mm EGTA with an additional 2 mm MgCl2 substituting for Ca2+ (reducing free [Ca2+] to <10 nm). Membrane patch recordings were made using a pipette potential of +100 mV (i.e., a holding potential of −100 mV in an excised patch). The single-channel recording was made using an Axopatch 200 patch-clamp amplifier (Molecular Devices) controlled by software pClamp 10.2 (Molecular Devices). Sampling rate was at 125 kHz and the lowpass filter frequency was 5 kHz. Single-channel data were analyzed using Clampfit 10.2 (Molecular Devices). The frequency of the channel opening was analyzed using a threshold crossing function. The value for threshold was set as 7 × SD of the stable baseline (>15 s in continuous length). Channel-opening frequency was quantified for 30 s epochs ∼1 min after the CCh (100 μm) application. Each transient with an amplitude greater than the set threshold was counted as a single-channel opening event. To estimate single-channel conductance (from current amplitude), ∼100 opening events were analyzed from each of a series of voltage-clamp recordings in inside-out patches in Ca2+-free HPSS solution. These data were parsed using a 50 Hz band filter and then detected using the threshold search function of Clampfit. Peak amplitude was measured for opening events that exceeded a threshold set 4.5 pA above the noise floor and showed no evidence of multichannel activation.
Indo-1 and Fluo-4 microfluorometric Ca2+ imaging.
Cells were grown in DMEM media on 18 mm circular coverslips coated with poly-d-lysine (25 μg/ml) and collagen (25 μg/ml) at 1:1 ratio. The cells were washed with HPSS. The coverslips were then incubated with HPSS and 0.1% pluronic acid, along with either 1 μm Indo-1 AM or Fluo-4AM Ca2+ indicator (Invitrogen) for an hour before the experiment. The cells were then placed into HPSS with 5 μm GdCl3 to block endogenous Ca2+ entry (added to all superfusions) (Zhu et al., 1998). For Indo-1 experiments, the cells were mounted on a Nikon TMD inverted microscope fitted with an Indo-1 filter set (Nikon Indo-1 filter cube, 485 nm/DM455 nm/405 nm) and illuminated with a mercury lamp. Cells were excited at 350 nm, with dual emission of the field was detected at 410 and 480 nm using two photomultiplier tubes, and the ratioed emission was determined in real time using in-house software. Calibration was performed using a calibration kit and Indo-1 K+ salt (Invitrogen). Ratios were converted to free Ca2+ concentrations using the formula from Grynkiewicz et al. (1985): [Ca2+] = Kd.Q.(R − Rmin)/(Rmax − R), where Kd is the estimated dissociation constant of the Indo-1 and Ca2+ with a value of 250 nm, Q is the ratio of Fmin and Fmax at λ2 (480 nm), and R represents the fluorescence intensity ratio Fλ1/Fλ2, in which λ1 is at 410 nm. For Fluo-4 experiments, the cells were mounted on an inverted microscope (Leica DMIL) with a 20× 0.4 NA objective (Leica HCX PL Fluotar), and illuminated with a mercury lamp using a green fluorescent protein filter (part no. 11504164; excitation 470/40 nm, dichroic 500 nm, emission 525/50 nm) every 5 s (Andor iXon+ 885 EMCCD Camera) with a gated shutter (Ludl Electronic Products), controlled via Andor IQ (v1.8.1) software. The images were then analyzed using ImageJ software (National Institutes of Health) with individual regions of interest identified for HEK293 cells responding to M3 or mGluR1 agonists with increased Ca2+ signal. Change in fluorescence was then presented as a ratio of the basal fluorescence (F0) at the nominal intracellular [Ca2+] before changing to Ca2+-free solution in the bath.
Purkinje cell TRPC3 current block.
Parasagittal cerebellar brain slices (400 μm) were prepared using standard techniques (Power and Sah, 2007). Mice (C129-SvEv strain, 4–8 weeks old) were anesthetized with pentobarbital and decapitated. The brain was removed and submerged in an ice-cold modified artificial CSF (ACSF) solution containing (in mm): 119 NaCl, 2.5 KCl, 3.3 MgCl2, 0.5 CaCl2, 1.0 Na2PO4, 26.2 NaHCO3, and 11 glucose, equilibrated with 95% CO2, 5% O2. Slices were cut with a VT1200 vibratome (Leica) and were allowed to recover for at least 1 h at room temperature in a standard ACSF solution containing (in mm): 119 NaCl, 2.5 KCl, 1.3 MgCl2, 2.5 CaCl2, 1.0 Na2PO4, 26.2 NaHCO3, and 11 glucose, equilibrated with 95% CO2, 5% O2. For recording, slices were transferred to the stage of a Zeiss Examiner D1 microscope and continuously perfused with ACSF heated to 30°C. Whole-cell patch-clamp recordings were made from the soma of Purkinje neurons identified using infrared differential interference contrast videomicroscopy. Patch pipettes (3–5 MΩ) were filled with an internal solution containing (in mm): 135 cesium methanesulfonate, 8 NaCl, 10 HEPES, 2 Mg2ATP, 0.3 Na3GTP, and 0.1 spermine (pH 7.3 with KOH, osmolarity 290–300 mosmol l−1). Alexa 594 (30 μm; Invitrogen) was added to the internal solution to visualize the dendritic tree. Whole-cell currents at a holding potential of −70 mV were amplified with an Axopatch 2B amplifier (Molecular Devices), filtered at 5 kHz and digitized at 20 kHz with a Digidata1440 (Molecular Devices), and controlled using PClamp 10.2 Software (Molecular Devices). Whole-field fluorescence measurements were made with a 40× water-immersion objective (NA 1.0; Zeiss) using an F43 filter block: excitation 545/25 nm, dichroic 570 nm, and emission 605/70 nm. Images were acquired with a cooled CCD camera (ProgRes MF-Cool; Jenoptik). DHPG (50 μm) was applied onto the dendritic field by focal pressure application (in ACSF) through a patch pipette.
Results
Alternative splicing of TRPC3 varies with brain region
TRPC3 gene transcription in brain tissue from mouse, rat, and guinea pig was determined by RT-PCR. PCR amplicons arising from primers that spanned exons 8–10 of TRPC3 indicated brain region-dependent alternative splicing of TRPC3 mRNA (Fig. 1A). In all three species, the cerebellum showed dominant expression of a novel isoform designated TRPC3c (GenBank accession numbers FJ207476, JN160741, and FJ207474 for mouse, rat, and guinea pig, respectively), which was smaller than the TRPC3b (unspliced) isoform by 84 bp as determined by sequencing of cloned cDNA. The proportion of TRPC3c relative to TRPC3b in different brain regions (cerebellum, midbrain, medulla, and cerebral cortex) was compared by optical density measurement of agarose gel electrophoresis images (Fig. 1B). ANOVA for these data for each species indicated that there were significant differences (mouse, p < 0.001; rat, p < 0.001; guinea pig, p < 0.001) in the proportion of TRPC3c:TRPC3b transcript across brain regions. In both mouse and rat cerebellum, the TRPC3c isoform comprised >80% of the cDNA amplicon; with guinea pig cerebellar tissue exhibiting approximately equivalent levels of TRPC3c and TRPC3b expression (Fig. 1B). The TRPC3c isoform was also detectable in the other brain regions, with the lowest relative level in each species found in the cerebral cortex (Fig. 1A,B). For the mouse cDNA template, primers spanning the full coding region were used to exclude additional alternative splicing. As previously described, using immunofluorescence (Huang et al., 2007; Hartmann et al., 2008), the localization of TRPC3 in the mouse cerebellum was largely confined to the Purkinje neurons (Fig. 1C). Based on our semiquantitative mRNA expression data we conclude that the TRPC3c isoform contributes the majority of the TRPC3 immunoreactivity present in Purkinje cells.
Figure 1.
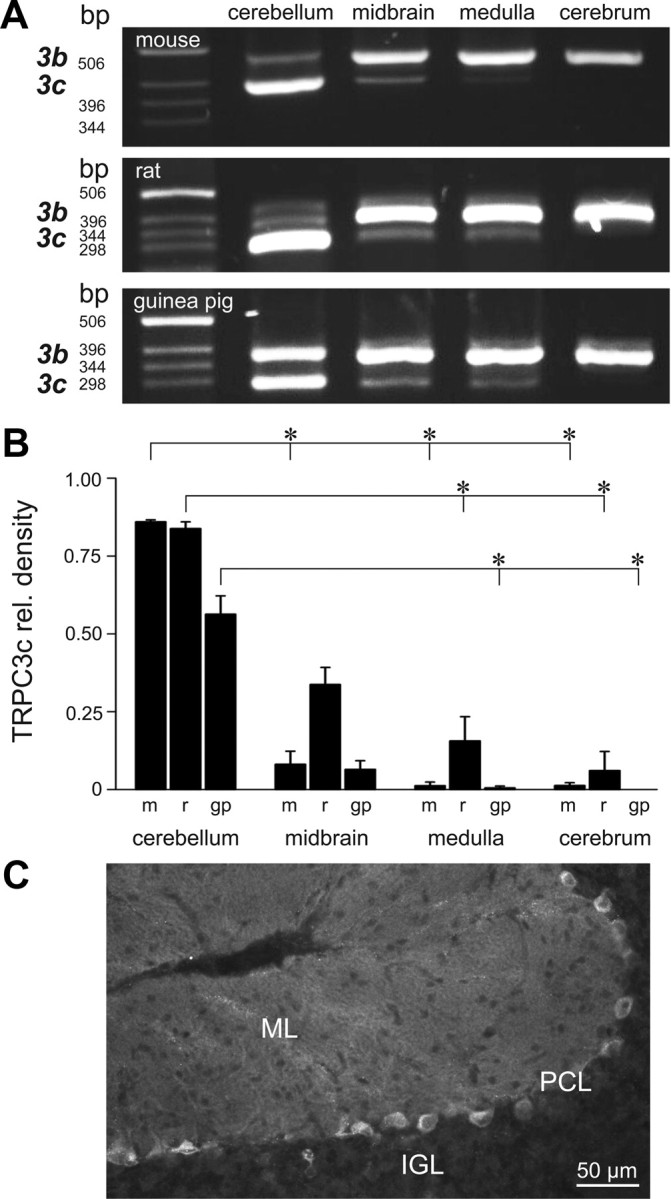
TRPC3 isoform expression in the brain. A, Agarose gel electrophoresis showing TRPC3b (upper) and TRPC3c (lower) RT-PCR amplicons from different brain regions of mouse, rat, and guinea pig. Note the predominance of the TRPC3c isoform in the cerebellum. B, Semiquantification of the expression of TRPC3c cDNA amplicon fluorescence intensity on the agarose gel, as a proportion of the combined TRPC3c + TRPC3b signals, as shown in A (expressed as mean ± SEM). Regional differences in TRPC3c expression are apparent (*p < 0.05, Dunn's pairwise post hoc comparison of ranked data from ANOVA) with highest relative levels in cerebellum followed by midbrain. m, mouse; r, rat; g, guinea pig. C, Immunolabeling of the TRPC3 protein in the mouse cerebellum, showing the high level of staining in the Purkinje neurons including their neurite projections into the molecular layer. PCL, Purkinje cell layer; IGL, internal granule cell layer; ML, molecular layer.
The TRPC3c isoform arose from omission of the complete exon 9 coding region (84 bp, corresponding to 28 aa) (Fig. 2), which, in mouse TRPC3 protein, is equivalent to 749–783 aa. Exon 9 encodes a significant portion of the CIRB domain. There is 100% conservancy of this coding region at the protein level in all three species and also in the human. Alternative splicing of the TRPC3c isoform in the human is predicted based on conservation of the intron–exon boundaries.
Figure 2.

Predicted amino acid sequences in the CIRB domain for the TRPC3b and TRPC3c splice variants (mouse; GenBank accession numbers FJ207475 and FJ207476, respectively). Alternative splicing in the TRPC3c isoform results in loss of exon 9, which codes for ∼50% of the CIRB domain (Tang et al., 2001; Zhang et al., 2001). The truncated CIRB region retained in the TRPC3c variant corresponds to the first 19/21 aa of the TRPC3 C8 fusion protein of Zhang et al. (2001), which had limited CaM binding. Further, the retained CIRB region in the TRPC3c variant also corresponds to last 19/21 aa of a fusion protein TRPC3 CΔ78 sequence necessary for trafficking of the TRPC3 protein to the plasma membrane (Wedel et al., 2003).
Expression of recombinant mouse TRPC3b and TRPC3c channels in HEK293 cells
Protein expression of recombinant TRPC3b and TRPC3c channels in stably transfected HEK293 cells was confirmed by Western blotting with the specific rabbit anti-rat/mouse TRPC3 antibody (Tadros et al., 2010), which recognizes both TRPC3 splice variants. Channel-specific protein species were detected at ∼75 kDa. The TRPC3c isoform migrated slightly further, consistent with the small size predicted from the loss of the 28 aa encoded by exon 9. TRPC3 expression was not detected in whole-cell lysates from untransfected HEK293 cells (Fig. 3A). HEK293-TRPC3c protein levels appeared somewhat less than TRPC3b, most likely due to differences in copy numbers. Equal protein loading was confirmed after stripping and reprobing for β-actin (Fig. 3B). Treatment of transfected and untransfected HEK293 cells with sulfo-HNS-SS-biotin followed by purification of biotinylated proteins on NeutrAvidin beads confirmed expression of TRPC3 channels in the plasma membrane of transfected cells only. Consistent with the whole-cell lysates, the biotinylated (cell surface) TRPC3c protein migrated further, given its slightly smaller molecular weight; with equivalent expression level to TRPC3b (Fig. 3C). Trafficking of both TRPC3 isoforms to the plasma membrane was evident with confocal immunofluorescence (Fig. 3D).
Figure 3.
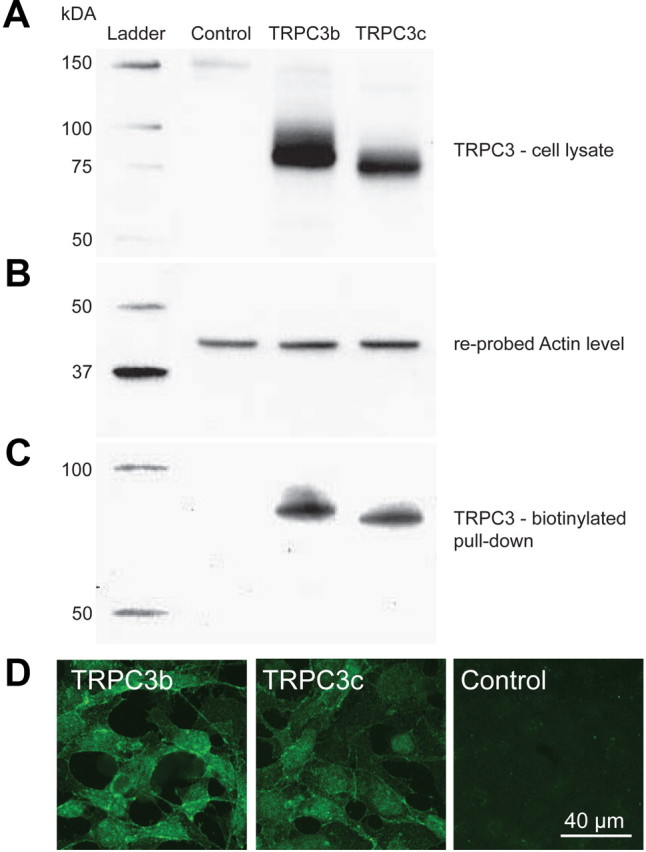
Expression of recombinant TRPC3b and TRPC3c proteins in transfected HEK293 cells detected by Western blotting and immunohistochemistry. A, Whole-cell lysate samples of transfected and untransfected HEK293 cells separated by 10% SDS-PAGE gel, blotted onto polyvinylidene difluoride membrane, and probed for TRPC3 protein with rabbit anti-TRPC3 antibody shown as ∼75 kDa protein species. Note that the TRPC3c isoform has a slightly smaller size, which is predicted based on the loss of 28 aa, encoded by exon 9 (equivalent to ∼3.1 kDa). B, Detection of actin in the same blot after anti-TRPC3 strip-off provides a control for protein loading (43 kDa). C, TRPC3 immunodetection of the membrane-bound fraction labeled with NHS-biotin and purified by adsorption onto NeutrAvidin beads, separated by a 10% SDS-PAGE gel followed by Western blotting with anti-TRPC3 antibody, ∼75 kDa. D, TRPC3b and TRPC3c expression in transfected HEK293 cells detected with anti-TRPC3 antibody by immunofluorescence confocal microscopy. The images are consistent with lower expression of TRPC3c as indicated the Western blot above (A). TRPC3-specific immunolabeling was localized to the plasma membrane and cytoplasm in the transfected cells; untransfected cells (control) were unlabeled.
TRPC3c variant exhibits increased membrane conductance due to enhanced channel opening frequency
Recombinant mouse TRPC3 ion channels expressed in HEK293 cells were activated by bath application of CCh (100 μm in HPSS), which activates the endogenously expressed mAChR (Zhu et al., 1998). Whole-cell recordings demonstrated slowly activating sustained inward currents with CCh, which were significantly greater in the TRPC3c-expressing cells (Fig. 4). The mean peak CCh-activated inward current for TRPC3b-expressing versus TRPC3c-expressing cells was −235.0 ± 28.7 pA and −809.4 ± 36.9 pA, respectively, at the holding potential Vh = −40 mV (SEM; n = 25 and n = 29; p < 0.001; unpaired t test). Voltage-ramps confirmed an increased slope conductance with CCh activation that was significantly greater in the TRPC3c-expressing cells (CCh increased TRPC3b from 2.6 ± 0.6 nS to 9.9 ± 1.2 nS, n = 8; TRPC3c slope conductance increased from 2.8 ± 0.5 nS to 20.4 ± 1.7 nS, n = 14; measured ∼ −40 mV; p < 0.001 t test). The corresponding right shifts in zero-current potential (Vz) evident in the current/voltage relationships (I/V; Fig. 4B) changed from −16.9 ± 3.5 mV to −5.6 ± 1.1 mV and −13.9 ± 3.6 mV to −0.4 ± 1.0 mV for CCh-treated HEK293 cells expressing TRPC3b and TRPC3c, respectively. There were no significant differences in the reversal potentials (Erev) of the isolated TRPC3 conductances determined by subtracting the I/Vs obtained during activation by CCh, with the respective I/Vs at rest (Erev(TRPC3b) = −1.9 ± 1.9 mV, n = 8; Erev(TRPC3c) = 1.1 ± 1.5 mV, n = 14; p = 0.241, t test) (Fig. 4B), indicating that the ion selectivities of the two isoforms were similar. Despite the larger TRPC3c currents, TRPC3c expression by the HEK293 cells was weaker than the TRPC3b level, as determined directly by Western blot (Fig. 3) and also by FACS of isolated cells using the DsRed2 reporter fluorescence, where the mean fluorescence for TRPC3b was 20.7 ± 0.2, n = 17024, and for TRPC3c was 11.6 ± 0.2 (n = 9149 (p < 0.001, t test).
Figure 4.
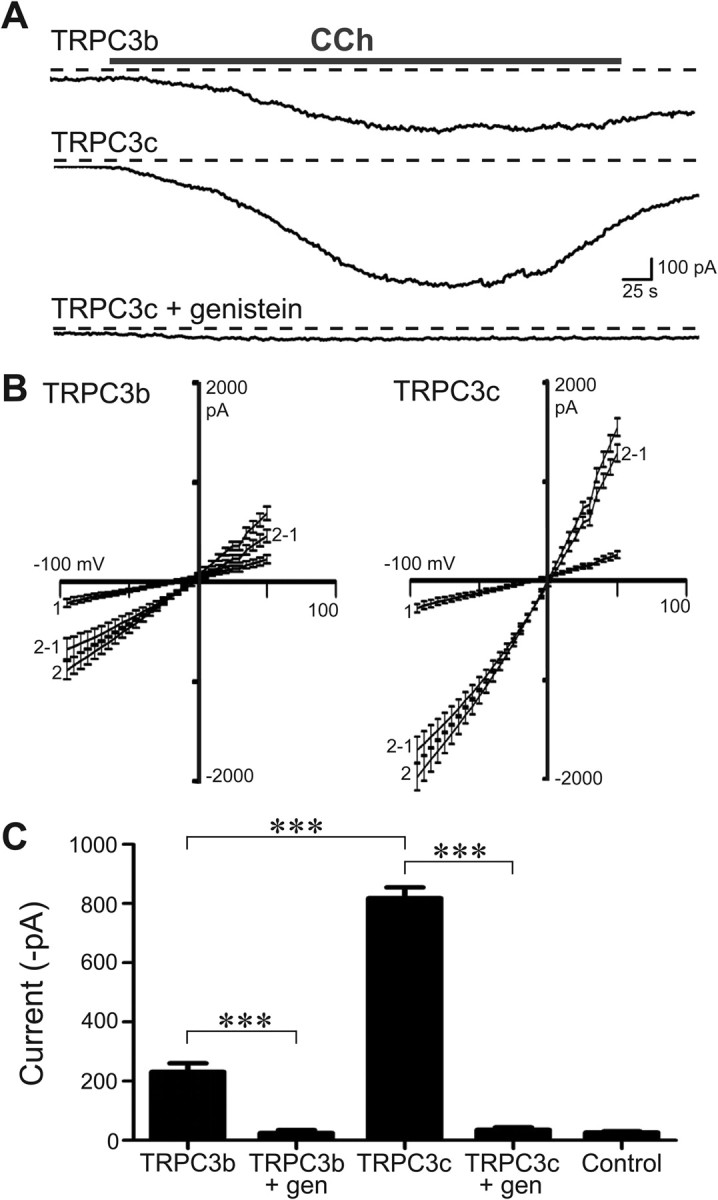
Whole-cell voltage-clamp of HEK293 cells expressing recombinant TRPC3b or TRPC3c channels. A, Example of the larger currents produced by the TRPC3c transfected cells (currents activated by bath application of CCh (100 μm). The currents were blocked by pre-incubation with genistein (10 min; 200 μm). Example shows block of TRPC3c current; Vh = −40 mV; dashed lines indicate zero-current. B, Current/voltage relationships (I/Vs; mean ± SEM) for TRPC3b and TRPC3c (1 = control ramp before CCh; 2 = ramp during CCh response; 2–1 represents the isolated ITRPC3 I/V (trace 2-trace 1). The reversal potential (Erev) of ITRPC3 was close to 0 mV for both isoforms, indicating that the ion selectivity of the two channel isoforms was similarly nonselective. C, Mean peak whole-cell current responses for both isoforms of TRPC3 channels, genistein block for each, and control data (untransfected cells). ***p < 0.001; two-way ranked ANOVA, Holm–Sidak multiple pairwise comparisons).
We did not observe any CCh-activated current in untransfected cells (mean = −25.4 ± 4.2 pA; n = 7; Vh = −40 mV). In addition, both TRPC3 isoform current responses could be reliably inhibited by genistein, which is a tyrosine kinase inhibitor (200 μm; TRPC3b + CCh + genistein = −24.8 ± 9.7 pA, n = 6; TRPC3c + CCh + genistein = −35.4 ± 8.1 pA, n = 7; Fig. 4C). Tyrosine kinases, such as Src kinase, are believed to have obligatory roles in activation pathways of TRPC3 channels, and its blockers such as genistein have been shown to inhibit TRPC3 channel activity (Vazquez et al., 2003).
TRPC3b and TRPC3c single-channel currents were recorded at a pipette voltage of −100 mV (Vh = +100 mV). The single-channel recordings consisted of four patch-clamp conditions as shown in Figure 5Ai–iv). Each recording started with measurement of the baseline (Ai), then activation by CCh (Aii) in cell-attached configuration. The patch was then excised, to form an inside-out patch, with the intracellular side of the patch exposed to Ca2+-free solution (Aiii), and finally the patch was exposed to 2 mm Ca2+ (Aiv). The channel activity featured very brief transients (often <50 μs), that limited the analysis of channel kinetics. Figure 5B provides detail of the transient TRPC3 channel-opening events. Single-channel opening events from inside-out patches in Ca2+-free solution of >100 μs duration were analyzed using threshold-crossing discrimination to estimate current amplitude [TRPC3b and TRPC3c; 7.7 ± 0.21 pA (n = 9); and 8.0 ± 0.36 pA (n = 5), respectively, p = 0.93, t test]. Given the +100 mV holding potential, this provides an estimate of single-channel conductance of ∼80 pS for both isoforms. These features are consistent with previous characterization of TRPC3 channels (Zitt et al., 1997; Zhang et al., 2001).
Figure 5.
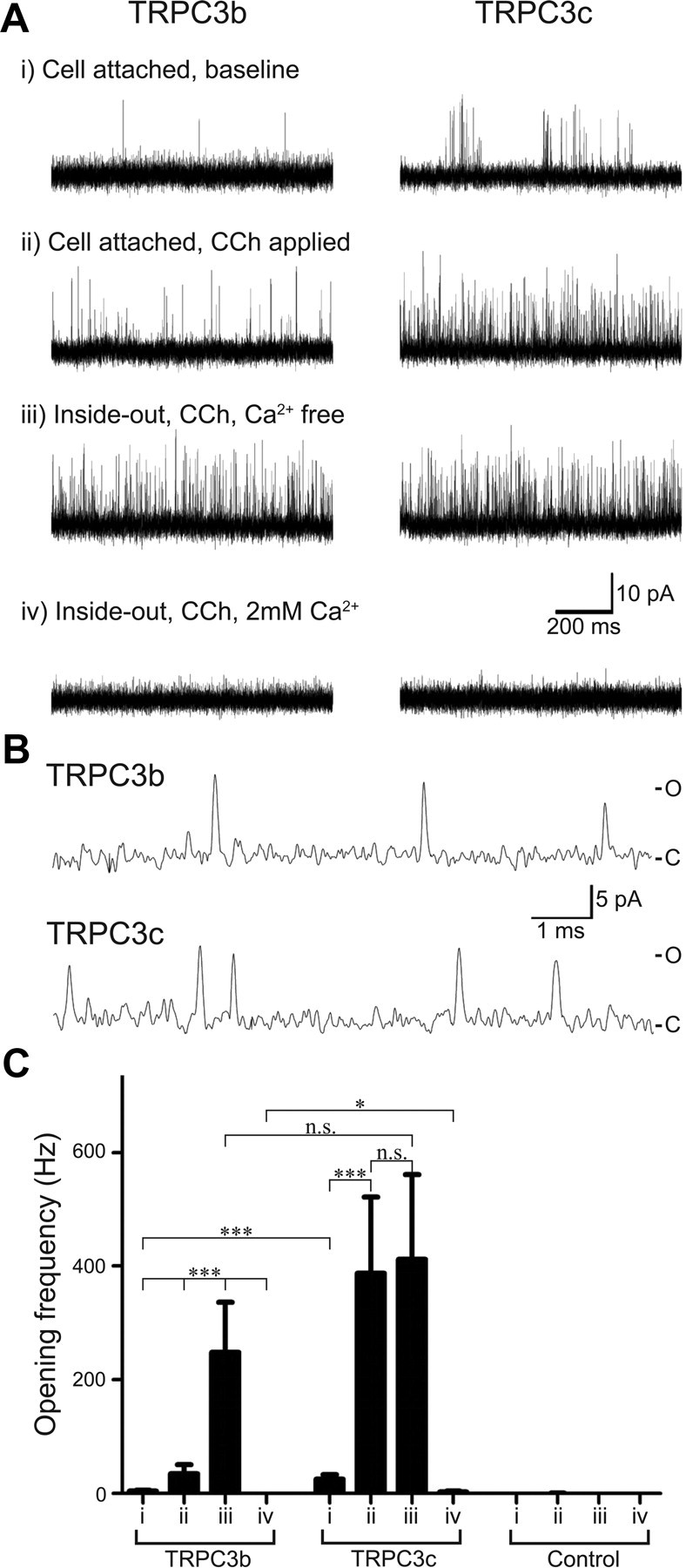
Single-channel patch-clamp recording of HEK293 cells expressing recombinant TRPC3 channels. A, Current traces of HEK293 cells expressing TRPC3b and TRPC3c channels. Each cell group is from the same patch recording and contains four experimental modes as shown (i, ii, iii, and iv). B, Representative single-channel current transients in cell-attached mode shown at high temporal resolution, with CCh (100 μm), as for Aii. C, closed state; O, open state. C, Mean channel opening frequency of membrane patches containing TRPC3b and TRPC3c channels, as well as control patches (no recombinant TRPC3 channel). n.s. indicates that the differences were not significant (p > 0.05).
Opening frequency was compared for the baseline condition and following activation by CCh (Fig. 5C), with statistical analysis by nonparametric ranked two-way ANOVA with Holm–Sidak post hoc multiple pairwise comparisons (α = 0.05). In the cell-attached patch configuration, baseline channel-opening frequency was greater for the TRPC3c isoform (25.2 ± 8.3 Hz, n = 16) than that of the TRPC3b isoform (4.4 ± 1.5 Hz, n = 19; p < 0.001; Fig. 5Ai,Ci). Addition of CCh to the bath caused an increase in channel-opening activity that was ∼10-fold greater in the TRPC3c isoform compared with TRPC3b (318.3 ± 116.9 Hz, n = 16; 34.6 ± 16.3, n = 19, respectively; p < 0.001) (Fig. 5Aii,B).
In both TRPC3 isoforms, the subsequent excision and exposure of the intracellular side of the patch to the Ca2+-free solution elicited a high frequency of channel opening (Fig. 5Aiii,Ciii). In TRPC3b channels, this was a significant increase (to 248.0 ± 88.0 Hz, n = 9) from the cell-attached, CCh-activated state (p < 0.001). In contrast, for TRPC3c channels there was no significant difference above the already high opening rate after CCh activation (Ca2+-free inside-out patch, 411.9 ± 149.1 Hz,; n = 5; p = 0.43). The apparent maximum activation via the Ca2+-free inside-out patch recordings between the TRPC3b and TRPC3c were not significantly different (p = 0.238). Finally, exposure of the intracellular side of the inside-out patch to 2 mm Ca2+ rapidly reduced channel opening in both TRPC3b isoforms, although the TRPC3c isoform retained a small residual opening rate (TRPC3b, 0.1 ± 0.9 Hz, n = 10; TRPC3c, 2.9 ± 1.2 Hz, n = 10; p = 0.019).
The mean opening frequency of untransfected cells was 0.2 ± 0.2 Hz for baseline and 0.3 ± 0.1 Hz for CCh activation (cell-attached patch) (n = 6). There was no significant difference between baseline and CCh treatment (p = 0.694; paired t test).
TRPC3c variant exhibits increased Ca2+ entry
Ca2+ entry was compared between the two TRPC3 channel isoforms with Ca2+ microfluorimetry. We investigated the TRPC3-mediated Ca2+ entry through two distinct activation pathways: (1) via the M3 receptor–PLCβ–DAG pathway endogenous to HEK293 cells, using Indo-1 as a Ca2+ indicator and (2) via mGluR1–PLCβ–DAG through coexpression of the mGluR1, using Fluo-4 as a Ca2+ indicator.
The M3 AChR-mediated TRPC3 activation was achieved by bath application of CCh. The initial rise in intracellular [Ca2+] in Ca2+-free solution reflected release from Ca2+ stores. CCh presentation was maintained and extracellular Ca2+ restored to nominal levels. This enabled Ca2+ entry via the TRPC3 channels (Fig. 6). The resultant peak [Ca2+] in HEK293 expressing TRPC3c (575.6 ± 44.2 nm, n = 25) was significantly greater than that in TRPC3b-expressing cells (182.7 ± 20.8 nm, n = 24; p < 0.001). The baseline [Ca2+] TRPC3c, TRPC3b, and untransfected cells before CCh treatment were not significantly different (70.1 ± 6.6 nm; 56.2 ± 6.5 nm; 67.4 ± 6.3 nm, respectively, p = 0.723, one-way ANOVA). The average peak baseline [Ca2+] with return of extracellular Ca2+ in untransfected cells was 62.5 ± 5.3 nm, n = 6, reflecting an absence of endogenous Ca2+ entry under these conditions. CCh-activated Ca2+ entry via both TRPC3 channel isoforms was completely blocked by pre-incubation of the cells with genistein, a tyrosine kinase inhibitor (200 μm) (Fig. 6A, typical result for TRPC3c) (TRPC3c, 81.9 ± 19.8 nm, n = 5; TRPC3b, 86.7 ± 8.7 nm, n = 5). These values did not differ significantly from untransfected control cells with CCh (p = 0.341, one-way ANOVA).
Figure 6.
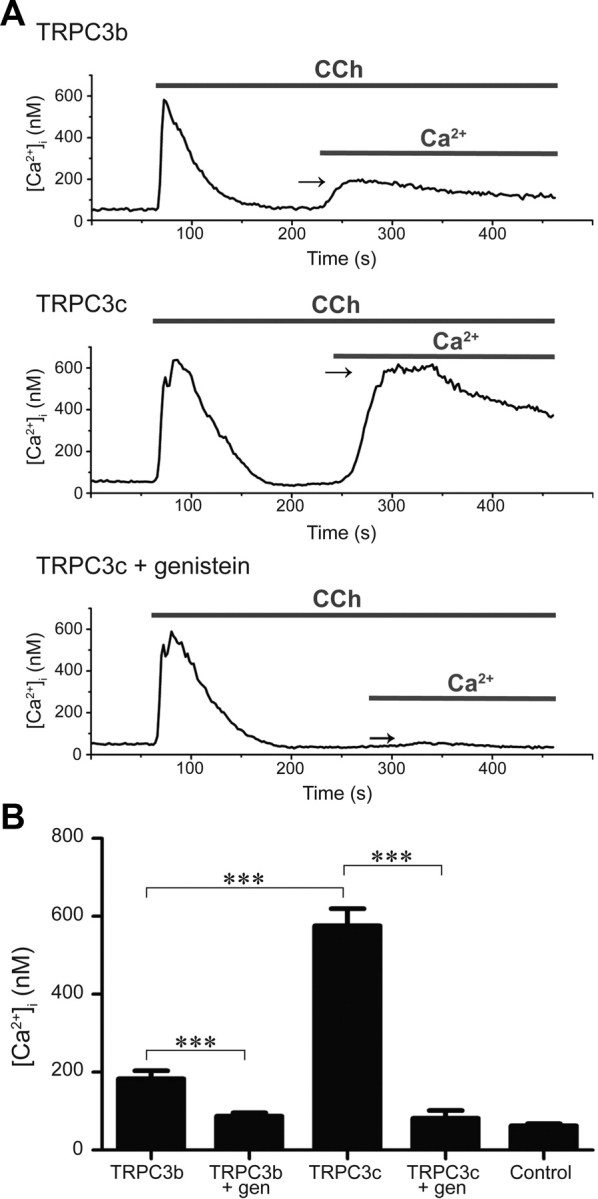
Ratiometric Ca2+ imaging of a field of HEK293 cells expressing recombinant TRPC3 channels using Indo-1 Ca2+ indicator dye. Cells were superfused with nominal Ca2+-free solution followed by application of CCh (100 μm), which causes release of stored Ca2+ via IP3R activation. Once released Ca2+ has been eliminated from the cell, the extracellular Ca2+ is returned to the bath, enabling TRPC3 channel-mediated Ca2+ entry (arrows). A, Greater Ca2+ entry in TRPC3c-expressing cells compared with TRPC3b-expressing cells or genistein block (200 μm; throughout the experiment). B, Mean peak [Ca2+]i arising from TRPC3b- and TRPC3c-mediated Ca2+ entry, genistein block and control (untransfected cell) data. ***p < 0.001; two-way ranked ANOVA, Holm–Sidak multiple pairwise comparison, and Mann–Whitney rank sum test.
The mGluR1-mediated TRPC3 activation was achieved by bath application of DHPG. Application of DHPG to HEK293 cells coexpressing TRPC3 and mGluR1 caused an initial rise in Fluo-4 fluorescence in Ca2+-free bath solution (expressed as F/F0), reflecting IP3R-gated Ca2+ store activation, as for the CCh experiments. This was followed, with return of Ca2+-containing external solution, by TRPC3-mediated Ca2+ entry (Fig. 7). The Fluo-4 fluorescence (average of 5–10 cells per experiment) was significantly greater in cells expressing TRPC3c (TRPC3c, 2.71 ± 0.217, n = 12; TRPC3b, 1.56 ± 0.0713, n = 10, p < 0.001, one-way ANOVA). Pre-application of 200 μm genistein abolished the TRPC3-mediated Ca2+ entry (TRPC3c, 0.876 ± 0.0621, n = 6; TRPC3b, 1.031 ± 0.0144, n = 6, p < 0.001, one-way ANOVA).
Figure 7.
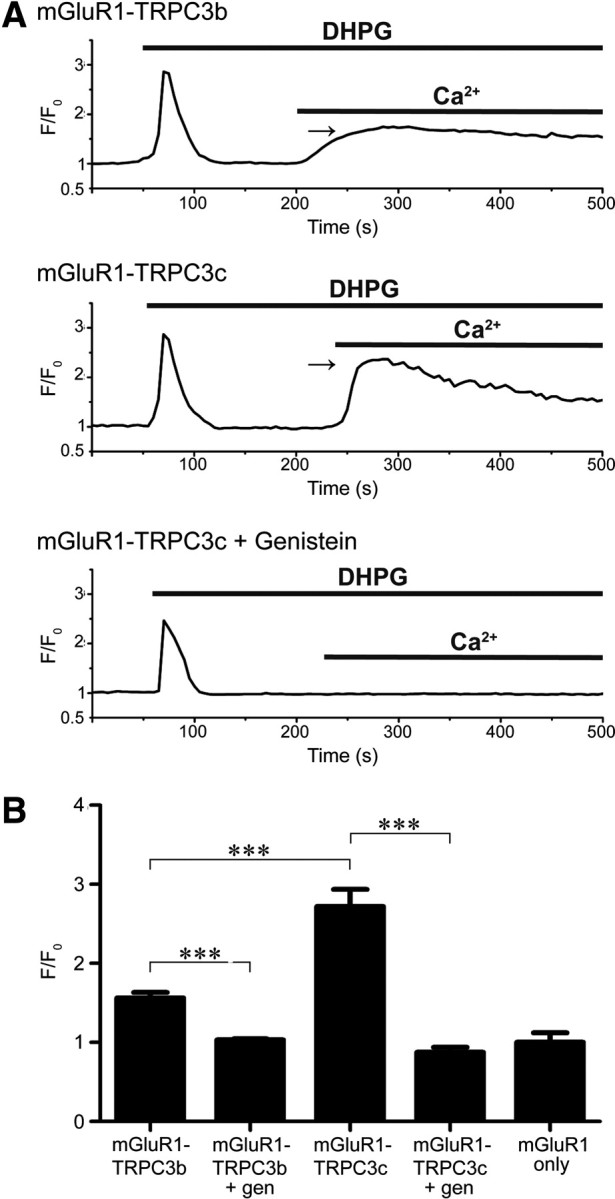
Fluo-4AM Ca2+ imaging of HEK293 cells coexpressing recombinant TRPC3 channels and mGluR1. A, Rise in [Ca2+]i from Ca2+ store release is shown for a single cell with application of the mGluR1 agonist DHPG (200 μm) in Ca2+-free solution. Fluorescence signal declines as the Ca2+ is extruded from the cell, and then rises again with TRPC3-mediated Ca2+ entry upon return of Ca2+ to the bath (arrows). Greater Ca2+ entry in TRPC3c-expressing cells compared with TRPC3b-expressing cells, or genistein block (200 μm; throughout the experiment). F0 represents the Ca2+ signal just before DHPG application. B, Relative mean peak [Ca2+]i (F/Fo) arising from TRPC3b- and TRPC3c-mediated Ca2+ entry, genistein block, and control (expression of mGluR1 only, no TRPC3). ***p < 0.001; one-way ranked ANOVA, Holm–Sidak multiple pairwise comparison.
Block of mGluR1-activated TRPC3c current in cerebellar Purkinje cells
The TRPC3 blocker genistein (100 μm) reduced the DHPG (50 μm)-evoked Purkinje cell inward current by 90 ± 12% (n = 3; paired t test; p = 0.036) in mouse cerebellar brain slices (Fig. 8). DHPG-evoked responses could be repeatedly generated at 3 min intervals before addition of genistein to the bath. Genistein produced a block of the current over ∼10–15 min. The sensitivity of the DHPG-activated current to genistein confirms the coupling of the mGluR to the Purkinje cell TRPC3 channels as reported by Hartmann et al. (2008) in mouse, and by Nelson and Glitsch (2012) for rat and mouse. The peak of the evoked current before genistein was −400 ± 55 pA, and −77 ± 50 pA 15 min after genistein (p = 0.06) The integrated area of the current (or net charge) was −878 ± 11 pC and −74 ± 105 pC, before and after genistein, respectively.
Figure 8.

Whole-cell voltage-clamp recordings of DHPG-evoked inward currents in Purkinje cells. The mGluR agonist DHPG (50 μm) was applied onto the Purkinje cell's dendrites by pressure application (50 ms, 70 kPa) through a patch pipette. A, Bright-field image of the cerebellar slice shows the recording pipette (r) on the Purkinje cell (PC) soma and drug pipette (d) containing DHPG (50 μm) positioned over the dendritic field (see B). B, Fluorescence image of the Purkinje cell loaded with Alexa 594 via the patch-clamp pipette (r). C, The current (Vm = −70 mV) evoked by DHPG before and after bath application of the TRPC3 channel blocker genistein (100 μm). Arrowhead indicates the timing of DHPG application. D, Time-course plot showing the normalized integrated area of repeated (3 min intervals) DHPG-evoked currents before and during application of genistein (indicated by bar). Mean ± SEM (n = 3) responses.
Discussion
The current study demonstrates region-specific expression of alternative splicing of the novel mRNA transcript encoding the TRPC3c channel in the mammalian hindbrain across three species (mouse, rat, and guinea pig). The splicing results in omission of exon 9, encoding a substantial part of the cytosolic C-terminal region CIRB domain. In cerebellar tissue, the TRPC3c isoform was dominant, whereas in other brainstem samples TRPC3c expression was lower than the previously described (TRPC3b) isoform. TRPC3c mRNA levels were minimal in the cerebral cortex compared with TRPC3b. Immunohistochemistry of the adult mouse cerebellum confirmed previous studies (Huang et al., 2007; Hartmann et al., 2008) showing principally Purkinje neuron labeling. Utilizing heterologous expression in HEK293 cells, we found that the novel TRPC3c isoform was translocated to the plasma membrane and assembled into ion channels with enhanced spontaneous and GPCR-activated opening frequency. The channel activity was modulated by cytosolic Ca2+, where removal of Ca2+ provided maximum activation of the channels, and addition of Ca2+ rapidly inhibited channel opening. The enhanced Ca2+ entry arising from the increased opening frequency of the TRPC3c channels was associated with a fivefold increase in cytosolic Ca2+ concentration following channel activation, compared with the TRPC3b-expressing cells. Homomeric TRPC3 channels are known to be the sole effector of the mGluR-mediated sEPSC in Purkinje neurons (Hartmann et al., 2008). In the present study the mouse cerebellar Purkinje cell mGluR1-activated TRPC3 current was blocked by genistein, as was the mGluR1-mediated activation of recombinant TRPC3c channels expressed in HEK293 cells. The TRPC3c isoform-mediated Ca2+ entry described here is likely to contribute to regulation of this major output pathway for motor coordination and learning.
Exon 9 codes for a major part of the CIRB domain, which has a critical role in regulating TRPC3 channel function by cytosolic [Ca2+], via competitive binding of the IP3 receptor and CaM (Zhang et al., 2001). Hence, the omission of exon 9 may lead to significant alteration of either, or both, of these regulatory elements. Earlier studies have attempted to determine the role of the CIRB domain, by expressing the TRPC3b protein with targeted deletions. Zhang et al. (2001) expressed a synthetic human TRPC3b protein, designated Trp3ΔC8 with a deletion within exon 10, equivalent to 765–783 aa in mouse TRPC3b. This Trp3ΔC8 mutant was shown to assemble into a functional ion channel that lacked CaM binding; rather than enabling channel activation, spontaneous activation was suppressed. Subsequently, Wedel et al. (2003) undertook a study with a similar mutant known as T3T-CΔ78, (equivalent to deletion of 763–782 aa in mouse TRPC3b, including two amino acids from exon 9). The channel protein failed to be transported to the HEK293 cell membrane, suggesting that this region of the CIRB domain is involved in membrane trafficking (note that this was a fusion protein with YFP reporter on the C terminus, which may have influenced this finding). In the present study, Western blotting of the biotinylated membrane TRPC3c protein and immunohistochemistry of the HEK293-TRPC3c cells, as well as the electrophysiology and Ca2+ imaging, clearly validate the membrane trafficking and functional integrity of this naturally occurring variant.
Our electrophysiological characterization has shown that the TRPC3c isoform confers increased cation flux. This was in the face of TRPC3c protein expression levels that were less than that of the TRPC3b-expressing cells. The single-channel electrophysiology study shows that this larger cation flux in TRPC3c-expressing cells is due to increased channel-opening frequency, while there appears to be no difference in channel conductance or selectivity, as shown by the similarity in the amplitude of the channel openings, and in the reversal potentials of the isolated TRPC3c versus TRPC3b currents (Fig. 4B). Thus exon 9 is unlikely to influence these ion channel properties, consistent with the separation from the pore-forming region (Zhu et al., 1996). We also noticed from the single-channel study that both TRPC3 channel types were able to be activated by the DAG produced via PLCβ activation following CCh-induced stimulation of the M3 receptors outside the recording patch. This is likely due to the membrane-bound DAG translocating under the rim of the patch pipette, to the TRPC3 channels within the patch.
The increased opening rate of the TRPC3c channel is likely to be instigated by altered regulation at the CIRB domain. Based on the findings from Zhang et al. (2001), the most likely explanation is that TRPC3c channels have reduced affinity for CaM binding to the CIRB domain, thereby reducing its inhibitory effect on the channel overall. Our single-channel data show that the two TRPC3 isoforms show different sensitivity to [Ca2+]. TRPC3c channels exposed to nominal intracellular [Ca2+] (∼70 nm; cell-attached patch, Fig. 5Ai) had a greater spontaneous opening rate. In addition, when the intracellular side of the patch was exposed to zero [Ca2+], the TRPC3c channels did not show further increased opening frequency, while the TRPC3b channels did, approaching that of the TRPC3c (Fig. 5B).
Alternative splicing of TRPC3 has been reported in the past. An isoform designated TRPC3a was identified in a cDNA library of human olfactory neurons (Yildirim et al., 2005). This isoform consists of an additional exon designated as exon 0 at the N terminus. This has not been reported in other tissues. Another N-terminal alternatively spliced isoform of TRPC3 was discovered in rat myocytes (GenBank accession no. AB022331) (Ohki et al., 2000). While these spliced isoforms have not been functionally compared with the native TRPC3b isoform, it is possible that they have an altered phenotype that adds to the functional diversity of the TRPC3 ion channels. Analysis of the splice donor and receptor sites within the intron regions spanning exon 9 of TRPC3c indicates full conservation across the three species studied and in human, making it highly likely that this isoform is broadly prevalent in homomeric configuration in the Purkinje neurons, and in heteromeric configurations with the TRPC3b isoform and other compatible TRPC subunits in other brain regions.
As described, TRPC3 ion channels can be activated by both DAG, via PLC, and by cytosolic allosteric protein–protein regulation, e.g., CaM. PLC is engaged by a broad range of Gα/q-protein-coupled receptors (GPCR-PLCβ). Thus in neurons, mGluR, P2Y receptors, muscarinic AChR, for example, can result in multiplexing of Ca2+ signaling via direct Ca2+ entry through a common TRPC3 channel effector (Abramowitz and Birnbaumer, 2009). In addition, the substantial Na+ entry depolarizes the cells, enabling parallel Ca2+ entry through other pathways, such as NMDA receptors and voltage-dependent Ca2+ channels (Abramowitz and Birnbaumer, 2009). In the case of the TRPC3c ion channel described here, the membrane depolarization and Ca2+ entry are comparable to that of the NMDA receptor (MacDermott et al., 1986). This raised the specter that the sustained depolarization and Ca2+ entry associated with activation of the mGluR, physiologically represented as the sEPSC during normal neurotransmission, may contribute to ischemic brain injury as a pathophysiological response to unregulated glutamate release during hindbrain stroke. Indeed, cerebellar Purkinje cells are particularly susceptible to ischemic injury (Hausmann et al., 2007), despite the absence of NMDA receptor expression in the mature neurons (Monyer et al., 1994), and brainstem stroke has the poorest prognosis (Sairanen et al., 2011).
TRPC channels are also a principal downstream effector of neurotrophin-Trk receptor-PLCγ-signaling, and they feature prominently in neural development. There is broad TRPC expression in the cerebellum, where as shown in the rat, shortly after birth, a developmental switch upregulates TRPC3 compared with the other TRPC isoforms (Huang et al., 2007). In this animal model, expression of TRPC3 and the TrkB receptor for the BDNF neurotrophin show almost complete overlap during early postnatal development of the olfactory bulb, cerebral cortex, amygdala, pons, and cerebellar Purkinje neurons (Li et al., 1999). In pontine neurons, BDNF-activated nonselective cation currents were attributable to TrkB receptor-PLCγ-DAG-mediated activation of TRPC3 ion channels (Li et al., 1999). TRPC3 expression has been associated with the development of the dendritic arbor of Purkinje neurons (Becker et al., 2009); however this is independent of TrkB signaling (Bosman et al., 2006). TRPC3 and TRPC6 have been shown to contribute to BDNF-mediated protection of cerebellar granule cells from apoptosis by Ca2+-signal-dependent CREB activation (Jia et al., 2007). Downregulating TRPC3 or TRPC6 in neonatal rat cerebellar granule cells induced apoptosis, and this could be rescued by overexpressing these TRPC channel subunits. In cerebellar Purkinje neurons, where TRPC3c expression is dominant, constitutive activation of TRPC3 channels has been shown to underlie cerebellar ataxias. The moonwalker mouse (Mwk), which provides a principal model of cerebellar ataxia, has a gain-of-function point mutation in the TRPC3 gene that alters channel gating. This mouse model exhibits profound impairment of Purkinje neuron dendrite development and loss of these neurons (Becker et al., 2009). Thus we can hypothesize that the TRPC3c ion channel we describe has a “ying-yang” capability, where, depending upon the site and severity of injury, it may provide neuroprotection, or with unregulated activation of the mGluR1 in the cerebellum in stroke injury, the potent Ca2+ entry and membrane depolarization likely stimulate apoptosis and necrosis. Study of the contribution of TRPC3 channels to brain pathophysiology is clearly warranted given the physiological properties of this alternatively spliced TRPC3c channel.
Footnotes
We acknowledge funding support from the Australian Research Council (ARC DP1097202) and thank Mr. Edward Crawford for support of the Ca2+ imaging.
The authors declare no competing financial interests.
References
- Abramowitz J, Birnbaumer L. Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J. 2009;23:297–328. doi: 10.1096/fj.08-119495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker EB, Oliver PL, Glitsch MD, Banks GT, Achilli F, Hardy A, Nolan PM, Fisher EM, Davies KE. A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc Natl Acad Sci U S A. 2009;106:6706–6711. doi: 10.1073/pnas.0810599106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaumer L. The TRPC class of ion channels: a critical review of their roles in slow, sustained increases in intracellular Ca2+ concentrations. Annu Rev Pharmacol Toxicol. 2009;49:395–426. doi: 10.1146/annurev.pharmtox.48.113006.094928. [DOI] [PubMed] [Google Scholar]
- Bosman LW, Hartmann J, Barski JJ, Lepier A, Noll-Hussong M, Reichardt LF, Konnerth A. Requirement of TrkB for synapse elimination in developing cerebellar Purkinje cells. Brain Cell Biol. 2006;35:87–101. doi: 10.1007/s11068-006-9002-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung YH, Sun Ahn H, Kim D, Hoon Shin D, Su Kim S, Yong Kim K, Bok Lee W, Ik Cha C. Immunohistochemical study on the distribution of TRPC channels in the rat hippocampus. Brain Res. 2006;1085:132–137. doi: 10.1016/j.brainres.2006.02.087. [DOI] [PubMed] [Google Scholar]
- Dietrich A, Kalwa H, Rost BR, Gudermann T. The diacylgylcerol-sensitive TRPC3/6/7 subfamily of cation channels: Functional characterization and physiological relevance. Pflugers Arch. 2005;451:72–80. doi: 10.1007/s00424-005-1460-0. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hartmann J, Dragicevic E, Adelsberger H, Henning HA, Sumser M, Abramowitz J, Blum R, Dietrich A, Freichel M, Flockerzi V, Birnbaumer L, Konnerth A. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron. 2008;59:392–398. doi: 10.1016/j.neuron.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausmann R, Seidl S, Betz P. Hypoxic changes in Purkinje cells of the human cerebellum. Int J Legal Med. 2007;121:175–183. doi: 10.1007/s00414-006-0122-x. [DOI] [PubMed] [Google Scholar]
- Huang WC, Young JS, Glitsch MD. Changes in TRPC channel expression during postnatal development of cerebellar neurons. Cell Calcium. 2007;42:1–10. doi: 10.1016/j.ceca.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Jia Y, Zhou J, Tai Y, Wang Y. TRPC channels promote cerebellar granule neuron survival. Nat Neurosci. 2007;10:559–567. doi: 10.1038/nn1870. [DOI] [PubMed] [Google Scholar]
- Li HS, Xu XZ, Montell C. Activation of a TRPC3-dependent cation current through the neurotrophin BDNF. Neuron. 1999;24:261–273. doi: 10.1016/s0896-6273(00)80838-7. [DOI] [PubMed] [Google Scholar]
- Li Y, Jia YC, Cui K, Li N, Zheng ZY, Wang YZ, Yuan XB. Essential role of TRPC channels in the guidance of nerve growth cones by brain-derived neurotrophic factor. Nature. 2005;434:894–898. doi: 10.1038/nature03477. [DOI] [PubMed] [Google Scholar]
- MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature. 1986;321:519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- Masu M, Tanabe Y, Tsuchida K, Shigemoto R, Nakanishi S. Sequence and expression of a metabotropic glutamate receptor. Nature. 1991;349:760–765. doi: 10.1038/349760a0. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Nelson C, Glitsch MD. Lack of kinase regulation of canonical transient receptor potential 3 (TRPC3) channel-dependent currents in cerebellar Purkinje cells. J Biol Chem. 2012;287:6326–6335. doi: 10.1074/jbc.M111.246553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohki G, Miyoshi T, Murata M, Ishibashi K, Imai M, Suzuki M. A calcium-activated cation current by an alternatively spliced form of Trp3 in the heart. J Biol Chem. 2000;275:39055–39060. doi: 10.1074/jbc.M003606200. [DOI] [PubMed] [Google Scholar]
- Phan PA, Tadros SF, Kim Y, Birnbaumer L, Housley GD. Developmental regulation of TRPC3 ion channel expression in the mouse cochlea. Histochem Cell Biol. 2010;133:437–448. doi: 10.1007/s00418-010-0686-x. [DOI] [PubMed] [Google Scholar]
- Power JM, Sah P. Compartmentalisation of IP3 mediated calcium responses in basolateral amygdala neurons. J Physiol. 2007;580:835–857. doi: 10.1113/jphysiol.2006.125062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybould NP, Jagger DJ, Kanjhan R, Greenwood D, Laslo P, Hoya N, Soeller C, Cannell MB, Housley GD. TRPC-like conductance mediates restoration of intracellular Ca2+ in cochlear outer hair cells in the guinea pig and rat. J Physiol. 2007;579:101–113. doi: 10.1113/jphysiol.2006.122929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio A, Medhurst AD, Mattei C, Kelsell RE, Calver AR, Randall AD, Benham CD, Pangalos MN. mRNA distribution analysis of human TRPC family in CNS and peripheral tissues. Brain Res Mol Brain Res. 2002;109:95–104. doi: 10.1016/s0169-328x(02)00527-2. [DOI] [PubMed] [Google Scholar]
- Sairanen T, Strbian D, Soinne L, Silvennoinen H, Salonen O, Artto V, Koskela I, Häppölä O, Kaste M, Lindsberg PJ. Intravenous thrombolysis of basilar artery occlusion: predictors of recanalization and outcome. Stroke. 2011;42:2175–2179. doi: 10.1161/STROKEAHA.110.605584. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Goldsworthy J, Johnson BG, Salhoff CR, Baker SR. 3,5-dihydroxyphenylglycine is a highly selective agonist for phosphoinositide-linked metabotropic glutamate receptors in the rat hippocampus. J Neurochem. 1994;63:769–772. doi: 10.1046/j.1471-4159.1994.63020769.x. [DOI] [PubMed] [Google Scholar]
- Tadros SF, Kim Y, Phan PA, Birnbaumer L, Housley GD. TRPC3 ion channel subunit immunolocalization in the cochlea. Histochem Cell Biol. 2010;133:137–147. doi: 10.1007/s00418-009-0653-6. [DOI] [PubMed] [Google Scholar]
- Tang J, Lin Y, Zhang Z, Tikunova S, Birnbaumer L, Zhu MX. Identification of common binding sites for calmodulin and inositol 1,4,5-trisphosphate receptors on the carboxyl termini of trp channels. J Biol Chem. 2001;276:21303–21310. doi: 10.1074/jbc.M102316200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez G, Wedel BJ, Trebak M, St John Bird G, Putney JW., Jr Expression level of the canonical transient receptor potential 3 (TRPC3) channel determines its mechanism of activation. J Biol Chem. 2003;278:21649–21654. doi: 10.1074/jbc.M302162200. [DOI] [PubMed] [Google Scholar]
- Wedel BJ, Vazquez G, McKay RR, St J Bird G, Putney JW., Jr A calmodulin/inositol 1,4,5-trisphosphate (IP3) receptor-binding region targets TRPC3 to the plasma membrane in a calmodulin/IP3 receptor-independent process. J Biol Chem. 2003;278:25758–25765. doi: 10.1074/jbc.M303890200. [DOI] [PubMed] [Google Scholar]
- Yildirim E, Kawasaki BT, Birnbaumer L. Molecular cloning of TRPC3a, an N-terminally extended, store-operated variant of the human C3 transient receptor potential channel. Proc Natl Acad Sci U S A. 2005;102:3307–3311. doi: 10.1073/pnas.0409908102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Tang J, Tikunova S, Johnson JD, Chen Z, Qin N, Dietrich A, Stefani E, Birnbaumer L, Zhu MX. Activation of Trp3 by inositol 1,4,5-trisphosphate receptors through displacement of inhibitory calmodulin from a common binding domain. Proc Natl Acad Sci U S A. 2001;98:3168–3173. doi: 10.1073/pnas.051632698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FW, Matta SG, Zhou FM. Constitutively active TRPC3 channels regulate basal ganglia output neurons. J Neurosci. 2008;28:473–482. doi: 10.1523/JNEUROSCI.3978-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Jiang M, Peyton M, Boulay G, Hurst R, Stefani E, Birnbaumer L. Trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell. 1996;85:661–671. doi: 10.1016/s0092-8674(00)81233-7. [DOI] [PubMed] [Google Scholar]
- Zhu X, Jiang M, Birnbaumer L. Receptor-activated Ca2+ influx via human Trp3 stably expressed in human embryonic kidney (HEK)293 cells. Evidence for a non-capacitative Ca2+ entry. J Biol Chem. 1998;273:133–142. doi: 10.1074/jbc.273.1.133. [DOI] [PubMed] [Google Scholar]
- Zitt C, Obukhov AG, Strübing C, Zobel A, Kalkbrenner F, Lückhoff A, Schultz G. Expression of TRPC3 in Chinese hamster ovary cells results in calcium-activated cation currents not related to store depletion. J Cell Biol. 1997;138:1333–1341. doi: 10.1083/jcb.138.6.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]