

Abstract
The mechanisms through which estradiol (E2) regulates gonadotropin-releasing hormone (GnRH) neurons to control fertility are unclear. Previous studies have demonstrated that E2 rapidly phosphorylates cAMP response element-binding protein (CREB) in GnRH neurons in vivo. In the present study, we used GnRH neuron-specific CREB-deleted mutant mice [GnRH-CREB knock-outs (KOs)] with and without global cAMP response element modulator (CREM) deletion (global-CREM KOs) to investigate the role of CREB in estrogen negative feedback on GnRH neurons. Evaluation of GnRH-CREB KO mice with and without global CREM deletion revealed normal puberty onset. Although estrus cycle length in adults was the same in controls and knock-out mice, cycles in mutant mice consisted of significantly longer periods of diestrus and less estrus. In GnRH-CREB KO mice, basal levels of luteinizing hormone (LH) and the postovariectomy increment in LH were normal, but the ability of E2 to rapidly suppress LH was significantly blunted. In contrast, basal and postovariectomy LH levels were abnormal in GnRH-CREB KO/global-CREM KO mice. Fecundity studies showed that GnRH-CREB KO with and without global CREM deletion were normal up to ∼9 months of age, at which time they became prematurely reproductively senescent. Morphological analysis of GnRH neurons revealed a significant reduction (p < 0.01) in GnRH somatic spine density of GnRH-CREB KO mice compared to control females. These observations implicate CREB within the GnRH neuron as an important target for E2's negative feedback actions. They also indicate that the rapid modulation of CREB by E2 is of physiological significance in the CNS.
Introduction
In addition to mediating its effects through direct nuclear estrogen receptors (ERs), estradiol (E2) has rapid, nonclassical actions via the activation of second messenger molecules such as extracellular signal regulated kinase 1/2, protein kinase A, and transcription factors such as cAMP response element-binding protein (CREB) (Nilsson et al., 2001; Abrahám and Herbison, 2005; Vasudevan and Pfaff, 2008; Micevych and Dominguez, 2009). Although recent studies have demonstrated the possible role of rapid nonclassical estrogen signaling in reproduction, sexual behavior, and nociception (Dewing et al., 2007; McDevitt et al., 2008; Boulware and Mermelstein, 2009; Herbison, 2009; Kelly and Rønnekleiv, 2009), the physiological significance of rapid estrogen actions through transcription factors such as CREB remain unknown.
The gonadotropin-releasing hormone (GnRH) neurons represent the key output cells of the neural neuronal network regulating fertility. Gonadal steroid hormones such as E2 provide one of the most important homeostatic modulators of GnRH neuronal function in mammalian species (Herbison, 2006; Moenter et al., 2009). In addition to direct genomic effects, E2 also exerts “nonclassical” actions to modulate the activity of GnRH neurons and, in particular, bring about estrogen negative feedback (Glidewell-Kenney et al., 2007). The mechanisms underlying these nonclassical actions have been probed by examining the effects of rapid E2, unlikely to involve transcriptional responses, on adult GnRH neurons (Herbison 2009; Kelly and Rønnekleiv, 2009; Moenter and Chu, 2012). Such studies have identified acute actions of E2 on GnRH neuron excitability (Kelly et al., 1976; Chu et al., 2009) and a range of effects on calcium dynamics, kinases, and transcription factors such CREB (Abrahám et al., 2003, 2004; Romano et al., 2008; Herbison, 2009; Zhao et al., 2009). The key issue that remains to be established is what importance and functional relevance these range of nonclassical E2 actions have on estrogen feedback mechanisms in vivo.
Given previous work showing that rapid E2 acutely phosphorylates CREB in GnRH neurons of adult female mice in vivo (Abrahám et al., 2003), we have generated here female mice with a GnRH neuron-specific CREB deletion. Because, cAMP response element modulator (CREM) has been found to be able to compensate for CREB deletion in other brain regions (Rudolph et al., 1998; Mantamadiotis et al., 2002), we generated GnRH neuron-specific CREB deletions on both normal and CREM global knock-out (KO) backgrounds. Together, these studies identify CREB as an important signaling molecule in GnRH neurons that is essential for normal estrogen negative feedback in vivo.
Materials and Methods
Animals.
All experiments were approved and performed in accordance with the regulations of the Australian and New Zealand Council for the Care of Animals in Research and Teaching and the University of Otago Animal Ethics Committee. All mice were bred and housed at the University of Otago Hercus Taieri Resource Unit. The animals were maintained under conditions of a 12 h light/dark cycle (lights on at 7:00 A.M.) with food and water available ad libitum. All experiments were performed on female mice. Six mouse lines were used: C57BL/6 wild-type control (C57BL/6); CREBloxP/loxP control (CREBloxP/loxP); GnRHCre;CREBloxP/loxP (GnRH-CREB KO); CREM+/+ control (CREM+/+); CREM−/− (global- CREM KO); GnRHCre;CREBloxP/loxP;CREM−/− (GnRH-CREB KO/global-CREM KO). CREM+/+ controls were the wild-type littermates from CREM heterozygous crossings.
Previous studies with the GnRH-Cre line demonstrated strong GnRH immunoreactivity in 100 of 104 randomly picked CRE-positive cells, and weak staining in the remaining four cells (Yoon et al., 2005). The cross of the GnRH-Cre line to ROSA-26 indicator mice (Srinivas et al., 2001) also verified that the Cre transgene product is functional and efficient (Yoon et al., 2005). Confirmatory studies in our laboratory have shown that Cre is expressed by 97 ± 2% of all GnRH neurons, and crossing this line with the ROSA-26 indicator mice (Soriano, 1999) revealed Cre-dependent recombination in 97 ± 1% of GnRH neurons (R. E. Campbell, R. Porteous, and A. E. Herbison, unpublished data). Unpublished data show that Cre is expressed from embryonic day 13 in GnRH neurons (J. Shin and R. Campbell, personal communication).
The CREBloxP/loxPCREM+/− mouse (provided by T. Mantamadiotis, Monash University, Victoria, Australia) (Mantamadiotis et al., 2002) has exon 10 of the CREB1 allele flanked with loxP sites. Cre-mediated recombination of the CREB1loxP allele leads to a CREB1 null allele that encodes a truncated unstable CREB protein devoid of DNA-binding and dimerization domains. The CREM-mutant line was generated by homologous recombination as described by Blendy et al. (1996). By crossing these transgenic lines, we generated mice in which CREB and/or CREM is deleted from GnRH neurons from the early embryogenesis.
Puberty and estrus cycle.
Prepubertal female mice were examined every day for vaginal opening, and once this had occurred, vaginal smears were taken each day (10:00 A.M.) until the first estrus smear was encountered. Two weeks after the first estrus, the estrus cycle was evaluated using vaginal smears for a period of 3 weeks.
Fecundity testing.
The fecundity of mice (n = 6 of each genotype) was examined by pairing them with wild-type mice for a period of 6–12 months. The day of birth, number of pups, and delivery intervals were recorded for each pair over this period.
Immunohistochemistry.
Free-floating dual-label peroxidase-based immunohistochemistry for phospho-CREB (pCREB) or CREB and GnRH was undertaken as described previously (Abrahám et al., 2003). Briefly, brain sections were incubated with primary antibodies recognizing CREB (1:1000; Cell Signaling Technology) or pCREB (1:200; Cell Signaling Technology). This was followed by biotinylated goat anti-rabbit IgGs (1:200; Vector Laboratories) and the avidin-biotin-HRP complex (1:200; Vector Elite ABC kit, Vector Laboratories) incubations. Labeling was then visualized with nickel-diaminobenzidine tetrahydrochloride (DAB) using glucose oxidase that resulted in a black precipitate within the nucleus of the labeled cell. The sections were then processed further for GnRH immunoreactivity with the LR1 antibody (1:40,000; Dr. R. Benoit, Montréal General Hospital, Montréal, Québec, Canada) followed by incubation with peroxidase-labeled anti-rabbit IgGs and revealed by DAB only to result in a brown precipitate within the cytoplasm. Specificities of the primary antibodies have been tested and reported previously (McNulty et al., 1998; Abrahám et al., 2003). The omission of the primary antibodies resulted in complete absence of the immunoreactivity.
Sections with CREB-GnRH dual-labeling immunohistochemistry were examined under an Olympus BX51 microscope (Olympus Corporation) by an investigator blinded to the experimental groupings. Analysis was undertaken by counting the numbers of single-labeled (brown cytoplasm) and dual-labeled (brown cytoplasm and black nucleus) GnRH neurons in two coronal brain sections in the regions of the medial septum (MS), rostral preoptic area (rPOA), and anterior hypothalamus (AHA), represented by plates 22–24, 25–27, and 28–31 of Paxinos and Franklin's (2000) brain atlas. The CREB and pCREB expression in GnRH neurons was calculated as a percentage of GnRH neurons at each level and also as a percentage of all of the GnRH neurons examined.
GnRH neuron spine density analysis.
GnRH immunofluorescence labeling was performed to visualize spine density of GnRH neuron somata using the GA02 antibody (1:1500; a gift from G. Anderson, University of Otago, Dunedin, New Zealand) that labels the somata and proximnal dendrites of GnRH neurons (Rizwan et al., 2012) (Anderson, unpublished results). Brain sections from intact diestrus female CREBloxP/loxP and GnRH-CREB KO mice were incubated with the GA02 primary antibody followed by incubation with biotinylated donkey anti-guinea pig IgGs (1:200; Jackson ImmunoResearch) and streptavidin-conjugated Alexa Fluor 488 (1:200; Invitrogen).
Two sections containing the largest number of GnRH neurons from the rPOA were selected from each animal, and each neuron was imaged and analyzed using a Zeiss LSM 710 upright confocal laser-scanning microscope. An argon laser exciting at 488 nm was used with a 63× Apochromate objective (numerical aperture, 1.4) and 3× zoom function. Stacks of images were captured at 400 nm intervals. For each cell, somatic spines were identified as short cytoplasmic processes emerging from the soma in a 1–5 μm range (Campbell et al., 2005; Chan et al., 2011). Thin protrusions greater in length were filopodia (Campbell et al., 2005). Spine numbers were quantified by analyzing each captured image in z-plane using the LSM 710 control software, Zen 2009. Soma circumference was measured in a stack outlining the greatest part of the soma and excluding the proximal dendrite.
Estrogen negative feedback protocol.
Adult female mice (10–12 weeks old) of all six genotypes were anesthetized with Avertin (0.1 ml/10 g body weight) and 50 μl tail blood samples collected before bilateral ovariectomy (OVX). Seven days after OVX, mice were anesthetized with Avertin and additional 50 μl tail blood samples obtained. Two days later, mice each received a subcutaneous injection of 17-β-estradiol (1 μg/20 g body weight; Sigma) in sesame oil or vehicle alone, killed 3 h later by an overdose of Avertin, a blood sample obtained from the right atrium, and the brain perfusion fixed through the heart with 4% paraformaldehyde solution to enable CREB or pCREB-GnRH dual immunohistochemistry. Blood sera were stored at −20°C until assayed for LH by RIA. Plasma LH concentrations were determined using the anti-rLH-S-11 antiserum and mouse luteinizing hormone reference preparation (mLH-RP) reference provided by A. F. Parlow (National Hormone and Peptide Program, National Institutes of Health, Bethesda, MD). Studies using the negative feedback protocol outlined here have shown that LH levels are low in the intact animals, elevated in 2 week ovariectomized mice, and suppressed at the 3 h post-E2 time point (Campbell et al., 2011).
Statistical analysis.
All data were analyzed by one-way ANOVA followed by Tukey's post hoc test with a value of p < 0.05 considered significant. Values for each animal were used to determine means, and these were used to generate ± SEM values for each group. Data in all experiments are expressed as mean ± SEM. All statistical analyses were performed using Statistica 7.0 (StarSoft) and Prism (version 5.03; GraphPad Software).
Results
GnRH neuron-specific deletion of CREB
Dual-label immunohistochemistry in control (C57BL/6, CREBloxP/loxP, and CREM+/+ mice) and global-CREM KO mice revealed that CREB was restricted to the cell nuclei of ∼30% of all GnRH neurons (Fig. 1A,B). In GnRH-CREB KO and GnRH-CREB KO/global-CREM KO mice there was a complete absence of CREB immunoreactivity in GnRH neurons (Fig. 1A,B), indicating highly efficient Cre recombination in these lines.
Figure 1.
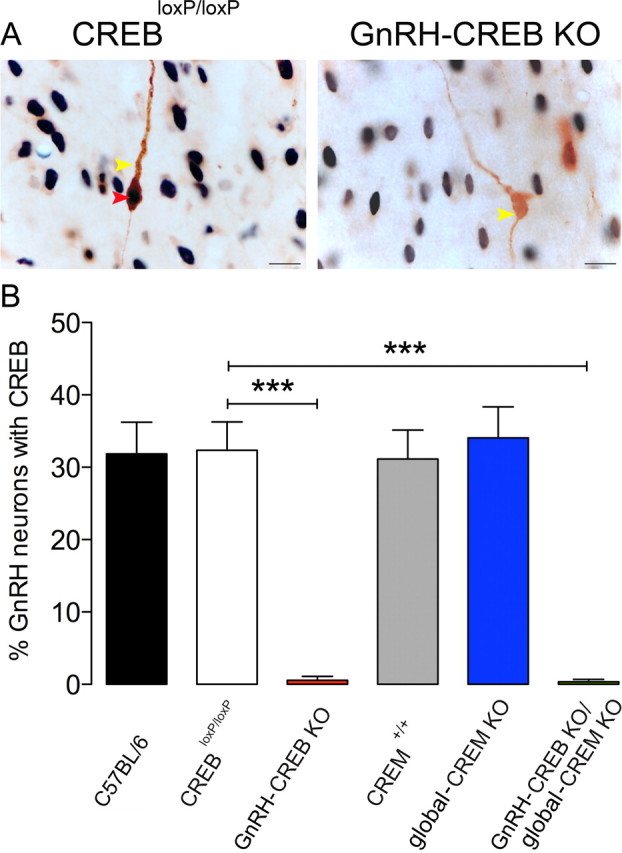
A, CREB expression in CREB mutant mouse line. The photomicrographs show nuclear CREB immunoreactivity (black nuclei, red arrowhead) in GnRH neurons (brown cytoplasm, yellow arrowhead) in CREBloxP/loxP mouse and lack of CREB expression in GnRH neurons (brown cytoplasm, yellow arrowhead) in GnRH-CREB KO mouse. Scale bars, 10 μm. B, Histogram shows the percentage of GnRH neurons expressing CREB in control (C57BL/6, CREBloxP/loxP, CREM+/+), GnRH-CREB KO, global-CREM KO, and GnRH-CREB KO/global-CREM KO mice. The histogram shows the means (+SEM); n = 7–14. ***p < 0.001 (ANOVA with Tukey's post hoc test).
The percentage of GnRH neurons expressing pCREB 3 h after E2 administration in vivo was also examined. In control (as above) and global-CREM KO mice, ∼25% of GnRH neurons (C57BL/6, 27.6 ± 2.4%; CREBloxP/loxP, 27.7 ± 1.9%; CREM+/+, 25.9 ± 3.4%; global-CREM KO, 24.5 ± 2.7%; n = 4–8) showed pCREB immunoreactivity. In contrast, there was no pCREB expression in GnRH neurons in GnRH-CREB KO and GnRH-CREB KO/global-CREM KO mice (data not shown).
Increased GnRH neuron number in global CREM KO mice
A previous report of global-CREM KO and CREB KO mice showed extensive neuronal cell loss in the brain (Mantamadiotis et al., 2002). To evaluate whether the GnRH neurons survive without CREB and/or CREM, the number of GnRH neurons was counted in the control and mutant mice. The data showed that the numbers of neurons immunoreactive for GnRH between control and GnRH-CREB KO mice were not different (Table 1). In contrast, significantly more GnRH neurons were detected within global-CREM KO (F(5,48) = 10.7, p = 0.001) and GnRH-CREB KO/global-CREM KO (F(5,48) = 10.7, p = 0.0004) animals compared with control CREM+/+ and CREBloxP/loxP mice (Table 1).
Table 1.
GnRH neuron number in CREB mutant mice
| Mouse line | MS | rPOA | AHA | Total number of GnRH neurons |
|---|---|---|---|---|
| C57BL/6 | 6 ± 0.3 | 14 ± 0.6 | 3.3 ± 0.3 | 21.8 ± 1.4 |
| CREBloxP/loxP | 6.3 ± 0.3 | 12.6 ± 0.5 | 3.4 ± 0.4 | 21.7 ± 0.6 |
| GnRH-CREB KO | 6.9 ± 0.2 | 12.6 ± 0.4 | 3.3 ± 0.3 | 21.5 ± 0.9 |
| CREM+/+ | 5 ± 0.3 | 13.3 ± 0.4 | 3.5 ± 0.3 | 18.9 ± 1 |
| Global CREM KO | 6.1 ± 0.2 | 15.1 ± 0.6 | 4.3 ± 0.4 | 25.9 ± 0.6** |
| GnRH-CREB KO/global CREM KO | 8 ± 0.4* | 15 ± 0.6 | 6.1 ± 0.2* | 28.9 ± 0.5*** |
The numbers of GnRH neurons in the MS, in the rPOA, and at the level of the AHA are shown. GnRH neurons per section per region and the total number of GnRH neurons in MS, rPOA, and AHA were examined for each of the six genotypes. The table shows the means (+SEM); n = 7–14.
*p < 0.05;
**p < 0.01;
***p < 0.001 (ANOVA with Tukey's post hoc test).
The total numbers of GnRH neurons detected in control mice varied according to anatomical location (Table 1), as reported previously in mouse (Abrahám et al., 2003; 2005). The same patterns of GnRH immunoreactivity were detected within these brain regions between the control and GnRH-CREB KO, GnRH-CREB KO/global-CREM KO, and global CREM KO brains (data not shown).
Puberty onset is normal in CREB mutant mice
An increase in GnRH neuronal activity is required for initiation of puberty during sexual development (Terasawa and Fernandez, 2001). To disclose whether the puberty was affected in GnRH-CREB mutant mice, the puberty onset was detected in those animals. Vaginal opening occurred at the same age in control and knock-out mice (Fig. 2A). Similarly, no differences were detected between the day of first estrus in control and knock-out mice (Fig. 2B). Vaginal opening, but not first estrus, was delayed by ∼3 d in control CREBloxP/loxP mice, compared with wild-type C57BL/6 mice (F(5,78) = 8.2, p = 0.03; Fig. 2A).
Figure 2.
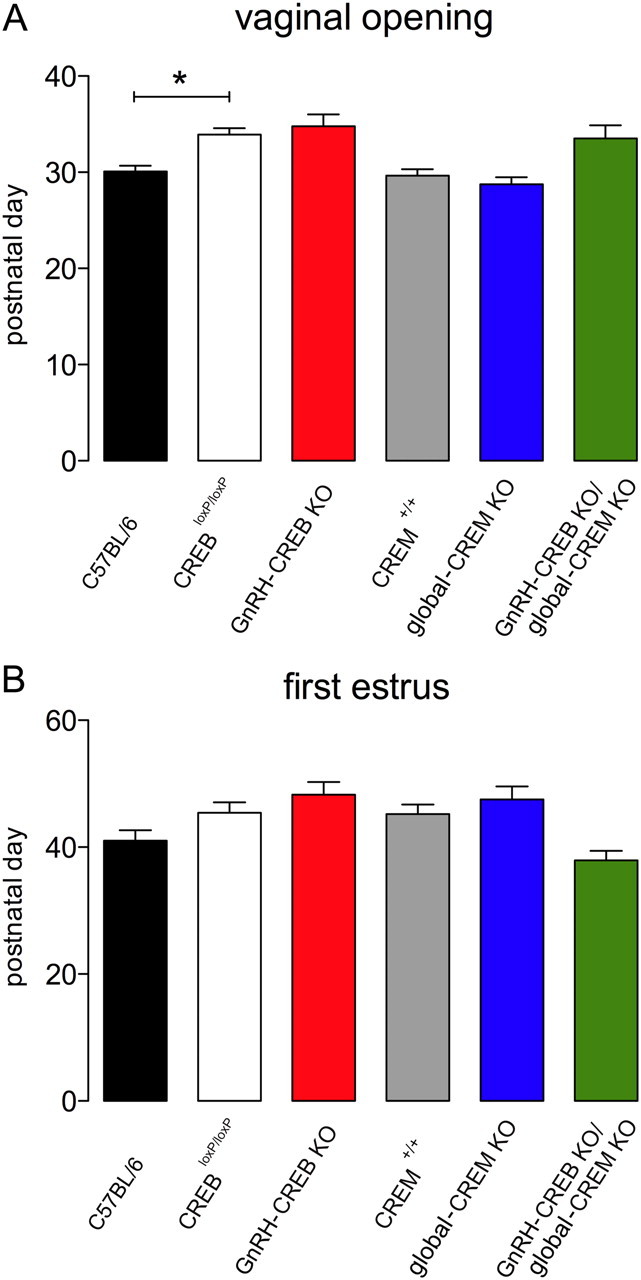
A, B, Puberty onset in CREB mutant mice. Vaginal opening (A) and first estrus (B) in transgenic and control mice. The mean (+SEM) days of vaginal opening and first estrus are given for each of the six genotypes (n = 8–14). *p < 0.05 (ANOVA with Tukey's post hoc test).
Estrus cyclicity is disrupted in CREB mutant mice
After puberty, the GnRH neurons plays pivotal role in the regulation of estrus cycle. Accordingly, estrus cyclicity was evaluated using vaginal smears for a period of 3 weeks. These data showed that the number of complete cycles and length of each cycle was variable but not significantly different between mutant mice and controls (Fig. 3A,B). However, the percentage of time spent in diestrus and estrus within these cycles was significantly different. GnRH-CREB KO and GnRH-CREB KO/global-CREM KO mice experienced longer periods of diestrus (F(5,42) = 7.6; GnRH-CREB KO, p = 0.003; GnRH-CREB KO/global-CREM KO, p = 0.01) and spent significantly less time in estrus (F(5,42) = 5.7; GnRH-CREB KO, p = 0.0004; GnRH-CREB KO/global-CREM KO, p = 0.03) than controls (Fig. 3C,D). Representative estrus cycle profiles demonstrating these alterations are presented in Figure 3E. The estrus cycles (Fig. 3C–E) of global CREM KO mice were not different from those of controls.
Figure 3.

Abnormal estrus cyclicity in CREB knock-out mice. A–D, The number of cycles (A), cycle length (B), and percentage of time spent in estrus (C) and diestrus (D) in CREB mutant mice. E, Representative estrus cycle profiles (for individual mice over a 21 d period) demonstrating the alterations in estrus cyclicity in CREB KO mice compared with controls. Both control and mutant mice show clearly distinguishable estrus cycle phases and proestrus–metestrus intermediate phases at similar frequency. The histograms show the means (+SEM) for each of the six genotypes; n = 8–16. *p < 0.05; **p < 0.01; ***p < 0.001 (ANOVA with Tukey's post hoc test). E, Estrus; P, proestrus; D, diestrus; M, metestrus.
Impaired fecundity of CREB mutant mice
To demonstrate whether the disrupted estrus cyclicity resulted in any change in fertility, a fecundity test was performed evaluating birth, number of pups, and delivery intervals. No significant differences were detected between control and GnRH-CREB KO or GnRH-CREB KO/global-CREM KO mice in terms of the number of delivered litters or pups per litter delivered during the first 6 months of the testing period (Fig. 4A,B). Fifty percent of the tested global-CREM KO mutant females were infertile (n = 6), while the fertile global CREM KO females produced a normal number of litters and a normal number of pups per litter compared to control mice. To investigate the time between the deliveries of consecutive litters, delivery intervals were calculated. During the first 6 months of testing, no significant differences were detected between control and GnRH-CREB KO, GnRH-CREB KO/global-CREM KO, or fertile global-CREM KO mice in terms of the delivery interval.
Figure 4.

Fecundity of CREB mutant female mice. A, B, The histograms show the mean (+SEM) number of litters (A) and number of pups/litter (B) born to mutant and control female mice per 6 months. C, The delivery intervals in young (Y; first 6 months of testing) and old (O; after 6 months of age) mice are given for each of the six genotypes (n = 4–6). **p < 0.01; ***p < 0.001 (ANOVA with Tukey's post hoc test).
However, after 6 months of age (6.3 ± 0.2 months; n = 3) the fertile global-CREM KO females stopped producing litters. At approximately 9 months of age, GnRH-CREB KO (8 ± 0.2 months; n = 6) and GnRH-CREB KO/global-CREM KO (9.5 ± 0.8; n = 6) mice also completely stopped producing litters, while controls remained fertile delivering litters until the experiment was discontinued. After 6 month of age, the delivery interval had doubled in GnRh-CREB-KO (F(5,30) = 10.7, p = 0.003) and GnRH-CREB KO/global-CREM KO animals (F(5,30) = 10.7, p = 0.0001) (Fig. 4C).
Abnormal estrogen negative feedback in CREB mutant female mice
Estrogen negative feedback on GnRH neurons is known to be critical for maintaining normal estrus cyclicity and fecundity. To assess the estrogen action on GnRH neurons, an estrogen negative feedback protocol was applied where the LH levels were measured at basal conditions, after ovariectomy and 3 h of E2 administration.
The basal serum LH levels of GnRH-CREB KO/global-CREM KO mice were significantly lower than basal LH levels of CREBloxP/loxP (control), GnRH-CREB KO, CREM+/+, and global-CREM KO mice in diestrus (Fig. 5).
Figure 5.

Effects of GnRH-specific deletion of CREB on estrogen negative feedback mechanisms. A, Chart summarizing the serum LH levels in mice before OVX (intact), 1 week after OVX, and 3 h after treatment with E2. B, C, Intact serum LH levels (B) and the increment from basal LH level (C) after OVX. D, LH levels decrease after treatment with E2 normalized to post-OVX LH levels. The histograms show the means (+SEM); n = 9–16. *p < 0.05; **p < 0.01; ***p < 0.001 (ANOVA with Tukey's post hoc test).
Ovariectomy induced a fourfold increase in the LH levels in C57BL/6, CREBloxP/loxP, GnRH-CREB KO, CREM+/+, and global CREM KO mice (Fig. 5). In contrast, the LH rise in GnRH-CREB KO/global-CREM KO mutants was significantly attenuated (F(5,78) = 6.6, p = 0.0001; Fig. 5A,C).
Whereas E2 treatment suppressed LH levels in C57BL/6, CREBloxP/loxP, CREM+/+, global-CREM KO, and GnRH-CREB KO/global-CREM KO mice, it was significantly less effective in GnRH-CREB KO mice (F(5,42) = 6, p = 0.002; Fig. 5A). In GnRH-CREB KO mice, E2 induced a 36 ± 13% decrease in LH levels compared to an 83 ± 7% decrease in CREBloxP/loxP controls (F(5,42) = 5, p = 0.002; Fig. 5A,D).
Decreased somatic spine density of GnRH neurons in CREB knock-out mice
Previous work has shown that the density of spines on the soma of GnRH neurons is regulated by estrogen (Chan et al., 2011). As such, we examined here whether soma spine density was altered in GnRH-CREB KO mice.
GnRH immunofluorescence labeling on brain sections from intact female CREBloxP/loxP and GnRH-CREB KO mice in diestrus revealed that deletion of CREB from GnRH neurons significantly alters somatic spine density in GnRH neurons of the rPOA (Fig. 6A–C). GnRH-CREB KO mice demonstrated a significant decrease in GnRH somatic spine density (F(2,15) = 34.95, p = 0.002, n = 112 GnRH neurons from five mice; Fig. 6C) compared with CREBloxP/loxP mice (n = 129 GnRH neurons from six mice). No significant differences were observed in GnRH soma circumference between control and GnRH-CREB KO mice (Fig. 6D). The number of spines per GnRH neuron also showed a significant decrease (F(2,15) = 39.84, p = 0.001; Fig. 6E) in GnRH-CREB KO mice compared to controls.
Figure 6.

Somatic spine density of GnRH neurons in CREB knock-out mice. A, B, Confocal stacks (400 nm optical thickness) of GnRH-immunolabeled neurons from CREBloxP/loxP (A) and GnRH-CREB KO (B) mice. Insets, Typical somatic spines are shown in confocal stacks from corresponding white boxes in GnRH neurons in A and B. Scale bar, 5 μm. C–E, Spine densities for the somata (C), soma circumferences (D), and numbers of spines on GnRH neurons (E) of control (C57BL/6, CREBloxP/loxP) and GnRH-CREB KO mice. The histograms show the means (+SEM); n = 4–6. **p < 0.01 (ANOVA with Tukey's post hoc test).
Discussion
We report here on the generation and reproductive phenotypes of mutant mouse models with different combinations of GnRH neuron-specific CREB and global CREM deletions. Our most important result is that mice with a GnRH neuron-specific CREB deletion, with or without global CREM knock-out, exhibit normal puberty, but have disordered estrous cycles and E2 negative feedback, show premature reproductive senescence, and exhibit a 50% reduction in GnRH somatic spine density. In addition, we note reproductive deficits of global-CREM KO mice and the interesting observation that the numbers of GnRH neurons in the brain are increased following global CREM deletion.
To define the role of CREB in GnRH neurons we generated a GnRH neuron-specific CREB mutant mouse line using a Cre-LoxP conditional mutagenesis strategy. We show here that crossing this GnRH-Cre mouse with the CREBloxP/loxP strain results in a complete abolition of CREB expression in all GnRH neurons. It remains possible that this strategy will also delete CREB from the population of lateral septal cells that express GnRH in the perinatal period (Skinner et al., 2009). The functions of these lateral septal cells are unknown, but they are very unlikely to be involved in control of the hypothalamic–pituitary–gonadal axis.
One unexpected phenotype noted in the global-CREM KO mouse was a small but significant ∼30% increase in the number of GnRH neurons. This may arise from either enhanced GnRH protein expression in GnRH neurons, making more cells detectable with immunohistochemistry, or from a real increase in the number of GnRH neurons in the brain. The increase in GnRH neuron number was not restricted to one part of the GnRH neuron continuum within the MS–rPOA–AHA, suggesting that it is probably not a defect in the timing or sequence of migration (Jasoni et al., 2009).
Within the nervous system, CREB is known to be critical for many processes ranging from cell survival through to the maintenance of long-term memory (Silva et al., 1998; Finkbeiner, 2000; Mantamadiotis et al., 2002). Interestingly CREB deletion from an early embryonic time point appears to have relatively little effect on GnRH neurons. While they exhibit some reproductive deficits, the GnRH neurons continue to synthesize GnRH and are capable of supporting normal levels of fertility when young. Previous studies have shown that the great majority (at least 65%) of GnRH neurons need to be deleted before a reproductive phenotype appears (Herbison et al., 2008). This indicates that at least 35% of GnRH neurons in CREB/CREM mutant female mice are viable.
We found that puberty is normal in GnRH-CREB KO mice with and without global-CREM KO. As adults, however, GnRH-CREB KO mice with and without global-CREM KO exhibited estrus cycles of normal length but with greater time spent in diestrus and less in estrus. A similar disassociation in dysfunction between the first and latter ovulations has been found in GNR23 mice that have a markedly reduced number of GnRH neurons (Herbison et al., 2008).
This disordered estrus cyclicity did not, however, impact upon fecundity as, in the first 6 months, the numbers of litters generated by mutant mice were slightly but not significantly decreased compared to their controls. These observations indicate that GnRH-CREB KO mutant mice exhibit normal ovulation despite the disturbed negative feedback during this period. However, altered GnRH/LH output in GnRH-CREB KO/global-CREM KO mutant mice may cause subtle but destabilizing changes in LH secretion able to disrupt the normal homeostasis of the estrus cyclicity.
After 6 months of age, we began to notice marked changes in fecundity. By 8 to 9 months of age, all GnRH-CREB KO mice with and without global-CREM KO had stopped producing litters. These observations demonstrate that the absence of CREB in GnRH neurons results in premature reproductive aging. Several studies indicate that changes in GnRH neuron function, in particular their ability to generate the preovulatory GnRH/LH surge, are responsible for reproductive senescence in rodents (Nass et al., 1984; Lu et al., 1985; Matt et al., 1987; Gore et al., 2000). Modulation of GnRH neurons is an initial factor in reproductive senescence with a greater driving role in rodents and a lesser role in primates, including humans (Hall et al., 2000; Gill et al., 2002; Shaw et al., 2009; Kermath and Gore, 2012).
It has been suggested that the negative feedback actions of E2 occur in a multimodal manner involving rapid and direct actions on the GnRH neuron, transsynaptic effects, and GnRH neuron–glial remodeling (Herbison 1998; Petersen et al., 2003; Prevot et al., 2010; Handa et al., 2011). Previous studies have shown that E2 injections can quickly reduce serum LH levels (in <30 min) (Yamaji et al., 1972; Negro-Vilar et al., 1973) and portal blood GnRH (Sarkar and Fink, 1980), but the rapidity of this E2 effect has been difficult to associate with a genomic mode of E2 action (Barnea and Gorski, 1970; Lagrange et al., 1995). Furthermore, recent studies found that 10 pm E2, a physiological concentration during negative feedback, rapidly inhibited GnRH neuron firing (Chu et al., 2009). Another piece of evidence for nonclassical mechanisms in estrogen negative feedback comes from ERα mutant mice with a mutated estrogen response element-binding domain where basal LH levels are intermediate between intact and ovariectomized normal LH levels (Glidewell-Kenney et al., 2007; Zhao et al., 2009), suggesting an involvement of both classical and non-estrogen response element (ERE)-dependent mechanisms. Our previous work using ER KO mice also found that rapid E2 action is unable to suppress LH secretion in ER single and double knock-out mice, demonstrating that classical ERs are essential for the rapid inhibitory E2 actions and for normal negative feedback (Abrahám et al., 2003, 2004; Couse et al., 2003; Dorling et al., 2003; Herbison 2009). Recent data suggest the predominance of ERα in the negative feedback mechanism, with a possible role of both direct and indirect ERβ-mediated actions on GnRH neurons (Handa et al., 2011).
The experiments reported here add to this complex dynamic of E2 actions by showing the physiological importance of CREB within GnRH neurons in this process. Our previous studies showed that CREB within GnRH neurons is phosphorylated rapidly by E2 (Abrahám et al., 2003), indicating that, despite CREB's established role in regulating gene transcription in a classical manner (Walton and Dragunow, 2000; Lonze and Ginty, 2002), it may also be involved in the rapid phase of E2 negative feedback. The negative feedback experiments performed in this study support this concept, as GnRH-CREB-deleted mice were unable to exhibit the full E2-evoked acute suppression of LH secretion. We interpret this as indicating that CREB in GnRH neurons is essential for one component of the acute E2 negative feedback response of GnRH neurons. It is possible that one instance where CREM may in fact be compensating for CREB loss in GnRH neurons is in the post-OVX LH rise, where the increment in LH levels was significantly suppressed in GnRH-CREB KO/global- CREM KO mice compared with GnRH-CREB only KOs. It seems clear that CREB is not the only signaling mechanism involved in E2 feedback. First, the acute suppression of LH by E2 still occurs in GnRH-CREB KO mice but is blunted in magnitude. Thus, other rapid actions of E2 through indirect transsynaptic or direct mechanisms (Lagrange et al., 1995; Romano et al., 2008; Chu et al., 2009; Zhao et al., 2009) are also likely to be in play. Second, we note that basal LH levels are within the normal range in intact GnRH-CREB KO mice, and the 1 week post-OVX increase in LH is normal. This indicates that rapid E2 actions through CREB can only represent one part of the negative feedback mechanism, although the diestrus-dominated estrous cycles of GnRH-CREB KO mice indicate some abnormality.
Prior studies have shown that CREB mediates effects of E2 on dendritic remodeling and spine formation in the hippocampus (Murphy and Segal, 1997; Segal and Murphy, 2001). As E2 is known to regulate somatic spine density of GnRH neurons (Chan et al., 2011), we reasoned that CREB deletion may impact spine density in these cells. Indeed, we show here that CREB deletion results in a substantial 50% decrease in GnRH neuron spine density. This observation suggests that one impact of deleting CREB in GnRH neurons is that of reduced spine density and likely reduced afferent input, and that this may underlie defective negative feedback in these cells.
In summary, we report here that CREB expressed by GnRH neurons is required for normal fertility. The use of cell-specific mutagenesis has been critical in allowing us to dissect the role of CREB specifically in GnRH neurons in vivo. Together with previous evidence that E2 rapidly phosphorylates CREB in GnRH neurons, we suggest that the negative feedback deficits encountered in GnRH-CREB KO mice result from defective acute E2 modulation of GnRH neurons that ultimately impact upon spine formation and altered afferent input. It is possible that the abnormal E2 feedback in these mice also contributes to their early reproductive senescence. These studies provide evidence for the physiological importance of rapid, nonclassical E2 actions within the brain and help define the mechanism of E2 negative feedback on GnRH neurons.
Footnotes
This work was supported by the Marsden Fund, the Otago School of Medical Sciences, and the Department of Physiology of the University of Otago. We acknowledge the excellent work of the members of the Hercus Taieri Resource Unit, University of Otago. We thank Dr. Theo Mantamadiotis (Victorian College of Pharmacy, Monash University, Victoria, Australia) for the generous provision of the CREB-loxP and CREM knock-out mice.
References
- Abrahám IM, Herbison AE. Major sex differences in non-genomic estrogen actions on intracellular signaling in mouse brain in vivo. Neuroscience. 2005;131:945–951. doi: 10.1016/j.neuroscience.2004.10.046. [DOI] [PubMed] [Google Scholar]
- Abrahám IM, Han SK, Todman MG, Korach KS, Herbison AE. Estrogen receptor beta mediates rapid estrogen actions on gonadotropin-releasing hormone neurons in vivo. J Neurosci. 2003;23:5771–5777. doi: 10.1523/JNEUROSCI.23-13-05771.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahám IM, Todman MG, Korach KS, Herbison AE. Critical in vivo roles for classical estrogen receptors in rapid estrogen actions on intracellular signaling in mouse brain. Endocrinology. 2004;145:3055–3061. doi: 10.1210/en.2003-1676. [DOI] [PubMed] [Google Scholar]
- Barnea A, Gorski J. Estrogen-induced protein. Time course of synthesis. Biochemistry. 1970;9:1899–1904. doi: 10.1021/bi00811a006. [DOI] [PubMed] [Google Scholar]
- Blendy JA, Kaestner KH, Weinbauer GF, Nieschlag E, Schütz G. Severe impairment of spermatogenesis in mice lacking the CREM gene. Nature. 1996;380:162–165. doi: 10.1038/380162a0. [DOI] [PubMed] [Google Scholar]
- Boulware MI, Mermelstein PG. Membrane estrogen receptors activate metabotropic glutamate receptors to influence nervous system physiology. Steroids. 2009;74:608–613. doi: 10.1016/j.steroids.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell RE, Han SK, Herbison AE. Biocytin filling of adult gonadotropin-releasing hormone neurons in situ reveals extensive, spiny, dendritic processes. Endocrinology. 2005;146:1163–1169. doi: 10.1210/en.2004-1369. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Ducret E, Porteous R, Liu X, Herde MK, Wellerhaus K, Sonntag S, Willecke K, Herbison AE. Gap junctions between neuronal inputs but not gonadotropin-releasing hormone neurons control estrous cycles in the mouse. Endocrinology. 2011;152:2290–2301. doi: 10.1210/en.2010-1311. [DOI] [PubMed] [Google Scholar]
- Chan H, Prescott M, Ong Z, Herde MK, Herbison AE, Campbell RE. Dendritic spine plasticity in gonadatropin-releasing hormone (GnRH) neurons activated at the time of the preovulatory surge. Endocrinology. 2011;152:4906–4914. doi: 10.1210/en.2011-1522. [DOI] [PubMed] [Google Scholar]
- Chu Z, Andrade J, Shupnik MA, Moenter SM. Differential regulation of gonadotropin-releasing hormone neuron activity and membrane properties by acutely applied estradiol: dependence on dose and estrogen receptor subtype. J Neurosci. 2009;29:5616–5627. doi: 10.1523/JNEUROSCI.0352-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couse JF, Yates MM, Walker VR, Korach KS. Characterization of the hypothalamic-pituitary-gonadal axis in estrogen receptor (ER) Null mice reveals hypergonadism and endocrine sex reversal in females lacking ERalpha but not ERbeta. Mol Endocrinol. 2003;17:1039–1053. doi: 10.1210/me.2002-0398. [DOI] [PubMed] [Google Scholar]
- Dewing P, Boulware MI, Sinchak K, Christensen A, Mermelstein PG, Micevych P. Membrane estrogen receptor-alpha interactions with metabotropic glutamate receptor 1a modulate female sexual receptivity in rats. J Neurosci. 2007;27:9294–9300. doi: 10.1523/JNEUROSCI.0592-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorling AA, Todman MG, Korach KS, Herbison AE. Critical role for estrogen receptor alpha in negative feedback regulation of gonadotropin-releasing hormone mRNA expression in the female mouse. Neuroendocrinology. 2003;78:204–209. doi: 10.1159/000073703. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S. CREB couples neurotrophin signals to survival messages. Neuron. 2000;25:11–14. doi: 10.1016/s0896-6273(00)80866-1. [DOI] [PubMed] [Google Scholar]
- Gill S, Sharpless JL, Rado K, Hall JE. Evidence that GnRH decreases with gonadal steroid feedback but increases with age in postmenopausal women. J Clin Endocrinol Metab. 2002;87:2290–2296. doi: 10.1210/jcem.87.5.8508. [DOI] [PubMed] [Google Scholar]
- Glidewell-Kenney C, Hurley LA, Pfaff L, Weiss J, Levine JE, Jameson JL. Nonclassical estrogen receptor alpha signaling mediates negative feedback in the female mouse reproductive axis. Proc Natl Acad Sci U S A. 2007;104:8173–8177. doi: 10.1073/pnas.0611514104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore AC, Oung T, Yung S, Flagg RA, Woller MJ. Neuroendocrine mechanisms for reproductive senescence in the female rat: gonadotropin-releasing hormone neurons. Endocrine. 2000;13:315–323. doi: 10.1385/ENDO:13:3:315. [DOI] [PubMed] [Google Scholar]
- Hall JE, Lavoie HB, Marsh EE, Martin KA. Decrease in gonadotropin-releasing hormone (GnRH) pulse frequency with aging in postmenopausal women. J Clin Endocrinol Metab. 2000;85:1794–1800. doi: 10.1210/jcem.85.5.6612. [DOI] [PubMed] [Google Scholar]
- Handa RJ, Ogawa S, Wang JM, Herbison AE. Roles for oestrogen receptor β in adult brain function. J Neuroendocrinol. 2011;24:160–173. doi: 10.1111/j.1365-2826.2011.02206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbison AE. Multimodal influence of estrogen upon gonadotropin-releasing hormone neurons. Endocr Rev. 1998;19:302–330. doi: 10.1210/edrv.19.3.0332. [DOI] [PubMed] [Google Scholar]
- Herbison AE. Physiology of the GnRH neuronal network. In: Neill Ka., editor. Physiology of reproduction. Ed 3. San Diego: Academic; 2006. pp. 1415–1482. [Google Scholar]
- Herbison AE. Rapid actions of oestrogen on gonadotropin-releasing hormone neurons; from fantasy to physiology? J Physiol. 2009;587:5025–5030. doi: 10.1113/jphysiol.2009.179838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbison AE, Porteous R, Pape JR, Mora JM, Hurst PR. Gonadotropin-releasing hormone neuron requirements for puberty, ovulation, and fertility. Endocrinology. 2008;149:597–604. doi: 10.1210/en.2007-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasoni CL, Porteous RW, Herbison AE. Anatomical location of mature GnRH neurons corresponds with their birthdate in the developing mouse. Dev Dyn. 2009;238:524–531. doi: 10.1002/dvdy.21869. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Rønnekleiv OK. Control of CNS neuronal excitability by estrogens via membrane-initiated signaling. Mol Cell Endocrinol. 2009;308:17–25. doi: 10.1016/j.mce.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MJ, Moss RL, Dudley CA. Differential sensitivity of preoptic-septal neurons to microelectrophoresed estrogen during the estrous cycle. Brain Res. 1976;114:152–157. doi: 10.1016/0006-8993(76)91017-9. [DOI] [PubMed] [Google Scholar]
- Kermath BA, Gore AC. Neuroendocrine control of the transition to reproductive senescence: lessons learned from the female rodent model. Neuroendocrinology. 2012 doi: 10.1159/000335994. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagrange AH, Rønnekleiv OK, Kelly MJ. Estradiol-17 beta and mu-opioid peptides rapidly hyperpolarize GnRH neurons: a cellular mechanism of negative feedback? Endocrinology. 1995;136:2341–2344. doi: 10.1210/endo.136.5.7720682. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Lu JK, LaPolt PS, Nass TE, Matt DW, Judd HL. Relation of circulating estradiol and progesterone to gonadotropin secretion and estrous cyclicity in aging female rats. Endocrinology. 1985;116:1953–1959. doi: 10.1210/endo-116-5-1953. [DOI] [PubMed] [Google Scholar]
- Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Martin Villalba A, Tronche F, Kellendonk C, Gau D, Kapfhammer J, Otto C, Schmid W, Schütz G. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet. 2002;31:47–54. doi: 10.1038/ng882. [DOI] [PubMed] [Google Scholar]
- Matt DW, Coquelin A, Lu JK. Neuroendocrine control of luteinizing hormone secretion and reproductive function in spontaneously persistent-estrous aging rats. Biol Reprod. 1987;37:1198–1206. doi: 10.1095/biolreprod37.5.1198. [DOI] [PubMed] [Google Scholar]
- McDevitt MA, Glidewell-Kenney C, Jimenez MA, Ahearn PC, Weiss J, Jameson JL, Levine JE. New insights into the classical and non-classical actions of estrogen: evidence from estrogen receptor knock-out and knock-in mice. Mol Cell Endocrinol. 2008;290:24–30. doi: 10.1016/j.mce.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty S, Schurov IL, Sloper PJ, Hastings MH. Stimuli which entrain the circadian clock of the neonatal Syrian hamster in vivo regulate the phosphorylation of the transcription factor CREB in the suprachiasmatic nucleus in vitro. Eur J Neurosci. 1998;10:1063–1072. doi: 10.1046/j.1460-9568.1998.00114.x. [DOI] [PubMed] [Google Scholar]
- Micevych P, Dominguez R. Membrane estradiol signaling in the brain. Front Neuroendocrinol. 2009;30:315–327. doi: 10.1016/j.yfrne.2009.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moenter SM, Chu Z. Rapid nongenomic effects of oestradiol on gonadotrophin-releasing hormone neurones. J Neuroendocrinol. 2012;24:117–121. doi: 10.1111/j.1365-2826.2011.02135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moenter SM, Chu Z, Christian CA. Neurobiological mechanisms underlying oestradiol negative and positive feedback regulation of gonadotrophin-releasing hormone neurones. J Neuroendocrinol. 2009;21:327–333. doi: 10.1111/j.1365-2826.2009.01826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy D, Segal M. Morphological plasticity of dendritic spines in central neurons is mediated by activation of cAMP response element binding protein. Proc Natl Acad Sci U S A. 1997;94:1482–1487. doi: 10.1073/pnas.94.4.1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nass TE, LaPolt PS, Judd HL, Lu JK. Alterations in ovarian steroid and gonadotrophin secretion preceding the cessation of regular oestrous cycles in ageing female rats. J Endocrinol. 1984;100:43–50. doi: 10.1677/joe.0.1000043. [DOI] [PubMed] [Google Scholar]
- Negro-Vilar A, Orias R, McCann SM. Evidence for a pituitary site of action for the acute inhibition of LH release by estrogen in the rat. Endocrinology. 1973;92:1680–1684. doi: 10.1210/endo-92-6-1680. [DOI] [PubMed] [Google Scholar]
- Nilsson S, Mäkelä S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin FK. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2000. [Google Scholar]
- Petersen SL, Ottem EN, Carpenter CD. Direct and indirect regulation of gonadotropin-releasing hormone neurons by estradiol. Biol Reprod. 2003;69:1771–1778. doi: 10.1095/biolreprod.103.019745. [DOI] [PubMed] [Google Scholar]
- Prevot V, Hanchate NK, Bellefontaine N, Sharif A, Parkash J, Estrella C, Allet C, de Seranno S, Campagne C, de Tassigny X, Baroncini M. Function-related structural plasticity of the GnRH system: a role for neuronal-glial-endothelial interactions. Front Neuroendocrinol. 2010;31:241–258. doi: 10.1016/j.yfrne.2010.05.003. [DOI] [PubMed] [Google Scholar]
- Rizwan MZ, Poling MC, Corr M, Cornes PA, Augustine RA, Quennell JH, Kauffman AS, Anderson GM. RFamide-related peptide-3 receptor gene expression in GnRH and kisspeptin 1 neurons and GnRH-dependent mechanism of action. Endocrinology. 2012 doi: 10.1210/en.2012-1133. in press. [DOI] [PubMed] [Google Scholar]
- Romanò N, Lee K, Abrahám IM, Jasoni CL, Herbison AE. Nonclassical estrogen modulation of presynaptic GABA terminals modulates calcium dynamics in gonadotropin-releasing hormone neurons. Endocrinology. 2008;149:5335–5344. doi: 10.1210/en.2008-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph D, Tafuri A, Gass P, Hämmerling GJ, Arnold B, Schütz G. Impaired fetal T cell development and perinatal lethality in mice lacking the cAMP response element binding protein. Proc Natl Acad Sci U S A. 1998;95:4481–4486. doi: 10.1073/pnas.95.8.4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar DK, Fink G. Luteinizing hormone releasing factor in pituitary stalk plasma from long-term ovariectomized rats: effects of steroids. J Endocrinol. 1980;86:511–524. doi: 10.1677/joe.0.0860511. [DOI] [PubMed] [Google Scholar]
- Segal M, Murphy D. Estradiol induces formation of dendritic spines in hippocampal neurons: functional correlates. Horm Behav. 2001;40:156–159. doi: 10.1006/hbeh.2001.1688. [DOI] [PubMed] [Google Scholar]
- Shaw ND, Srouji SS, Histed SN, McCurnin KE, Hall JE. Aging attenuates the pituitary response to gonadotropin-releasing hormone. J Clin Endocrinol Metab. 2009;94:3259–3264. doi: 10.1210/jc.2009-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- Skinner DC, Albertson AJ, Navratil A, Smith A, Mignot M, Talbott H, Scanlan-Blake N. Effects of gonadotrophin-releasing hormone outside the hypothalamic-pituitary-reproductive axis. J Neuroendocrinol. 2009;21:282–292. doi: 10.1111/j.1365-2826.2009.01842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasawa E, Fernandez DL. Neurobiological mechanisms of the onset of puberty in primates. Endocr Rev. 2001;22:111–151. doi: 10.1210/edrv.22.1.0418. [DOI] [PubMed] [Google Scholar]
- Vasudevan N, Pfaff DW. Non-genomic actions of estrogens and their interaction with genomic actions in the brain. Front Neuroendocrinol. 2008;29:238–257. doi: 10.1016/j.yfrne.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Walton MR, Dragunow I. Is CREB a key to neuronal survival? Trends Neurosci. 2000;23:48–53. doi: 10.1016/s0166-2236(99)01500-3. [DOI] [PubMed] [Google Scholar]
- Yamaji T, Dierschke DJ, Bhattacharya AN, Knobil E. The negative feedback control by estradiol and progesterone of LH secretion in the ovariectomized rhesus monkey. Endocrinology. 1972;90:771–777. doi: 10.1210/endo-90-3-771. [DOI] [PubMed] [Google Scholar]
- Yoon H, Enquist LW, Dulac C. Olfactory inputs to hypothalamic neurons controlling reproduction and fertility. Cell. 2005;123:669–682. doi: 10.1016/j.cell.2005.08.039. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Park C, McDevitt MA, Glidewell-Kenney C, Chambon P, Weiss J, Jameson JL, Levine JE. p21-Activated kinase mediates rapid estradiol-negative feedback actions in the reproductive axis. Proc Natl Acad Sci U S A. 2009;106:7221–7226. doi: 10.1073/pnas.0812597106. [DOI] [PMC free article] [PubMed] [Google Scholar]