

Abstract
Nicotine improves cognitive functions by modulating neuroplasticity and cortical excitability in nonsmoking subjects. As shown recently, the positive effect of nicotine on cognition might at least partially be caused by a focusing effect of nicotine on neuroplasticity in these subjects. Concordant to this, smokers under nicotine withdrawal show reduced cognitive abilities, which are at least partially restituted by nicotine consumption. We aimed to explore the neurophysiological foundation of these effects by exploring nonfocal and focal plasticity-inducing protocols in human smokers under nicotine withdrawal and exposition. Focal, synapse-specific plasticity was induced by paired associative stimulation (PAS), while nonfocal plasticity was induced by transcranial direct current stimulation (tDCS). Each subject (12) received placebo and nicotine patches combined with one of the stimulation protocols to the primary motor cortex. Corticospinal excitability was monitored by transcranial magnetic stimulation-induced motor-evoked potential amplitudes. In smokers during nicotine withdrawal, facilitatory plasticity induced by tDCS and PAS was abolished, but restituted by nicotine. In contrast, excitability-diminishing plasticity was not affected by nicotine withdrawal. Under nicotine, the inhibitory aftereffects of PAS were delayed and prolonged, while the tDCS-generated excitability reduction was abolished. Thus, absent facilitatory plasticity in smokers during nicotine withdrawal is restituted by nicotine, favoring the deficit-compensating hypothesis of nicotine consumption. These results might shed further light on the proposed mechanism of nicotine on cognition and attention, which might be connected to nicotine addiction and probability of relapse in smokers.
Introduction
Nicotine is the most commonly used drug of abuse and is responsible for the addictive properties of tobacco smoking. Beyond the addictive component, nicotine has prominent effects on cognition. Studies in animals and nonsmoking humans have shown improvements in attention, working, and episodic memories (Hahn and Stolerman, 2002; Jubelt et al., 2008; Heishman et al., 2010). In smokers, nicotine withdrawal leads to reduced verbal and working memory capacity, which could be partially replaced by nicotine replacement therapy (Jacobsen et al., 2005; Cole et al., 2010). The basis of the functional effects of nicotine is still unclear, but might be linked to its impact on cortical excitability and neuroplasticity by affecting the nicotinic acetylcholine receptors (nAChR), which are ligand-gated cation channels. The calcium permeability of nAChRs is thought to be important for the central effects of nicotine (Burnashev, 1998; Dajas-Bailador and Wonnacott, 2004), particularly for the induction and modulation of neuroplasticity (Lisman, 2001). Beside the nAChRs, nicotine also affects the dopaminergic, adrenergic, serotonergic, glutamatergic, and GABAergic systems, all known to be involved in generating and modulating plasticity (Levin et al., 2006). The impact of nicotine on plasticity has so far been mainly explored in animal experiments, especially in the hippocampus, where activation of nicotinic receptors leads to facilitation of long-term potentiation (LTP), both dependent and independent of NMDA receptor activation (Sawada et al., 1994; Huerta and Lisman, 1995; Matsuyama et al., 2000). Studies exploring the nicotinergic effects on long-term depression (LTD) are rare. In rats, reversal of stress-induced impairment of LTD has been found (Aleisa et al., 2006).
Recently, the impact of the cholinergic system and nicotinic receptors on cortical neuroplasticity has been explored in humans. For global cholinergic enhancement, it was shown that the cholinesterase inhibitor rivastigmine, as well as nicotinic receptor activation, enhanced or preserved focal, but abolished or reversed nonfocal facilitatory plasticity, thus resulting in a focusing effect (Kuo et al., 2007; Thirugnanasambandam et al., 2011). This focusing effect on facilitatory plasticity might be relevant for the cognition-enhancing effect of cholinergic and nicotinergic agents. Facilitatory and inhibitory plasticity were induced by two noninvasive brain stimulation protocols, transcranial direct current stimulation (tDCS) (Nitsche and Paulus, 2000, 2001) and paired associative stimulation (PAS) (Stefan et al., 2000). Both protocols induce NMDA- and calcium channel-dependent plasticity (Stefan et al., 2002; Nitsche et al., 2003b, 2004); however, tDCS induces a nonselective kind of plasticity affecting most neurons situated under the large stimulation electrodes (Purpura and McMurtry, 1965; Nitsche et al., 2007), while PAS-induced plasticity is thought to be restricted to synaptic connections between somatosensory and motor cortex neurons (Weise et al., 2006).
The aim of our study was to examine the effect of nicotine withdrawal and nicotine administration on tDCS- and PAS-induced LTP- and LTD-like aftereffects in otherwise healthy smoking subjects. We hypothesized that nicotine withdrawal would abolish plasticity, and exposition to the substance would restitute it, in accordance with the deficit-compensating hypothesis of nicotine consumption.
Materials and Methods
Subjects.
Altogether, 48 healthy subjects participated in the study (28 male and 20 female). Twenty-four subjects received tDCS (anodal or cathodal) and 24 subjects received 10 or 25 ms of paired-associative stimulation (PAS10 or PAS25, respectively). Table 1 displays the subject characteristics for the different groups. Subjects were smokers with a cigarette consumption of at least 10 cigarettes a day for at least 10 years and a Fagerstrom score of at least 1 point, indicating a light degree of nicotine dependence. Since nicotine has a blood serum half-life of 2–3 h with an interindividual range between 1 and 4 h (Benowitz, 1996), participants were not allowed to smoke 10 h (2 to 3 half-lives) before starting the experiments or during the experimental course to avoid nicotinic overdose with regard to the nicotine patch administration and heterogeneous nicotine levels due to uncontrolled cigarette smoking. Chronic and acute medical diseases or any history of neurological/psychiatric disease were excluded before entering the study, as was intake of chronic and acute medication. Pregnancy, family histories of epilepsy, presence of any metallic implant or cardiac pacemaker were ruled out. All subjects gave written informed consent before participating in the study. The experiments were approved by the local ethics committee and conformed to the principles laid down in the Declaration of Helsinki. Allocation of the subjects to the respective experimental conditions as well as order of sessions was randomized.
Table 1.
Comparison of subjects participating in the four different experiments
| Stimulation parameter | Anodal tDCS | Cathodal tDCS | PAS10 | PAS25 |
|---|---|---|---|---|
| Number of subjects | 12 | 12 | 12 | 12 |
| Number of females (%) | 6 (50) | 4 (33.33) | 5 (42.66) | 5 (42.66) |
| Age of subjects (mean ± SD; in years) | 24.3 ± 2.8 | 26.9 ± 2.9 | 25.75 ± 2.9 | 25.83 ± 2.9 |
| Fagerstrom scale (mean ± SD) | 3.92 ± 0.67 | 3.42 ± 1.9 | 3.17 ± 1.6 | 3.25 ± 0.75 |
Transcranial direct current stimulation.
Twenty-four subjects participated in the tDCS experiments. We used a battery-driven constant current stimulator (Schneider Electronics) with a maximum output of 2 mA. tDCS was administered via rubber electrodes covered by saline-soaked sponges (35 cm2). One electrode was positioned over the motor cortex representational area of the right abductor digiti minimi muscle (ADM), the other electrode above the right orbit. All subjects received 1 mA of either anodal or cathodal stimulation for 13 min (anodal tDCS) or 9 min (cathodal tDCS), which had been demonstrated to induce cortical excitability enhancement or inhibition lasting for ∼1 h after the end of stimulation (Nitsche and Paulus, 2001; Nitsche et al., 2003a), combined with nicotine or placebo medication in different experimental sessions. Twelve subjects received anodal tDCS and 12 subjects received cathodal tDCS under both placebo and nicotine patch conditions.
Paired associative stimulation.
Twenty-four subjects participated in the PAS experiment. Peripheral nerve stimulation was delivered to the right ulnar nerve at the wrist level by a D185 multipulse stimulator (Digitimer) at an intensity of 300% of the sensory perceptual threshold, followed by single-pulse transcranial magnetic stimulation (TMS) applied with a stimulator output resulting in motor-evoked potentials (MEPs) of ∼1 mV amplitude (baseline intensity; see Monitoring of cortical excitability, below). The paired pulses were repeated 90 times at a frequency of 0.05 Hz. This protocol induces long-lasting excitability changes in the motor cortex depending on the interstimulus interval (ISI). An ISI of 10 ms (PAS10) induces excitability diminution, whereas an ISI of 25 ms (PAS25) induces facilitation (Stefan et al., 2000; Wolters et al., 2003). Twelve subjects received PAS10 and 12 subjects received PAS25 under placebo and nicotine conditions.
Monitoring of motor cortex excitability.
TMS-elicited MEPs were recorded to measure excitability changes of the representational motor cortical area of the right ADM. Single-pulse TMS was conducted by a Magstim 200 magnetic stimulator at a frequency of 0.25 Hz with a figure-of-eight coil (diameter of one winding, 70 mm; peak magnetic field, 2.2 T). The coil was held tangentially to the scalp at an angle of 45° to the sagittal plane with the coil handle pointing laterally and posterior. The optimal position was defined as the site where stimulation resulted consistently in the largest MEPs. Surface EMG was recorded from the right ADM with Ag–AgCl electrodes in a belly–tendon montage. The signals were amplified and filtered with a time constant of 10 ms and a low-pass filter of 2.5 kHz, then digitized at an analog-to-digital rate of 5 kHz and further relayed into a laboratory computer using the Signal software and CED 1401 hardware (Cambridge Electronic Design). The intensity was adjusted to elicit, on average, baseline MEPs of 1 mV peak-to-peak amplitude and was kept constant for the poststimulation excitability measures.
Pharmacological intervention.
Each subject participated in two sessions in randomized order. Nicotine transdermal patches (30 cm2, containing 0.83 mg/cm2 nicotine, releasing 15 mg nicotine over 16 h) or placebo patches were administered to all subjects in combination with one of the stimulation protocols (anodal tDCS, cathodal tDCS, PAS10, or PAS25). By this dosage of nicotine, physiologically and behaviorally relevant plasma levels are accomplished (Tønnesen et al., 1991; Thirugnanasambandam et al., 2011). The patch was administered to the subjects 6 h before the start of the stimulation. This is the approximate time for the plasma level to reach its maximum following application of the patch (Nørregaard et al., 1992). To keep plasma levels stable, the patch was retained until the end of the last measurements of the experiment on the evening of the second day.
Course of the experiment.
Nicotine patch or placebo were adhered to the left upper arm and remained there until the end of the last measurement the following evening. Six hours after patch application, subjects were seated comfortably in a reclined chair with a headrest and armrests and asked to relax completely. EMG electrodes were placed at the right ADM as described above. Their exact position was marked with a pen. Then TMS was applied over the left representational area of the right ADM to determine the spot with the consistently highest MEPs in the resting ADM (optimal site), which was then marked with a pen. The TMS-intensity was adjusted to elicit MEP amplitudes of 1 mV (S1 mV). Twenty MEPs were recorded at this stimulus intensity and the mean MEP amplitude was calculated at baseline. One of the stimulation protocols, either tDCS or PAS, was administered, followed by immediate recording of at least 20 MEPs at the time points 0, 5, 10, 15, 20, 25, 30, 60, 90, and 120 min. For the nicotine patch condition, measurements were also conducted the evening of the stimulation day and in the morning and evening of the day following the plasticity induction procedure. Sessions were conducted in randomized order, and an intersession interval of at least 1 week was obligatory to avoid interferences (Fig. 1).
Figure 1.

Experimental design. Each subject took part in two experimental sessions. At the beginning of the session, the subject received either placebo or nicotine patch (randomized). After 6 h baseline, motor cortical excitability was determined via TMS-induced MEP with an amplitude of ∼1 mV over the cortical representational area of the right musculus ADM. One of the four excitability-inducing protocols (anodal atDCS, cathodal ctDCS, PAS10, PAS25) was then applied. Measurements started immediately after the application of the protocols and were conducted every 5 min for the first 30 min, then every 30 min up to minute 120 for both sessions. MEP amplitudes were also recorded the next morning and next evening for the nicotine patch sessions.
Data analysis and statistics.
The individual means of the 20 MEP amplitudes recorded at each time point were calculated for all subjects. The postintervention mean MEP amplitudes from each subject were then normalized to the respective individual mean baseline MEP amplitude (quotient of postintervention vs preintervention MEP amplitudes). A repeated-measures ANOVA was performed on the normalized data. MEP amplitude was the dependent variable, including all time points up to 120 min after stimulation. Drug (nicotine vs placebo) and time points were included as within-subjects factors. Stimulation (anodal tDCS or cathodal tDCS; PAS10 or PAS25) served as between-subject factors. Mauchly's sphericity test was performed and Greenhouse–Geisser correction was applied when necessary. Conditional on significant results for the ANOVA, Student's t tests (paired samples, two-tailed, p < 0.05, not adjusted for multiple comparisons) were performed to compare the MEP amplitudes before and after the interventional brain stimulations in each condition and between drug conditions (nicotine/placebo) for each time point. A p value of <0.05 was considered significant for all statistical analyses. All results are given as mean and SEM.
Results
All subjects tolerated the experimental procedure well. Three subjects complained of mild side effects such as dizziness and transient nausea with nicotine patch, but symptoms subsided before the beginning of the neuroplasticity-inducing experiments. There were no significant differences concerning gender (χ2 = 2.083, p = 0.149), age (Student's unpaired t tests; p > 0.05 for all cases), or Fagerstrom scale values (Student's unpaired t tests; p > 0.05 for all cases) between stimulation groups. Absolute TMS stimulator intensities and MEP amplitudes at baseline did not differ significantly within or between stimulation groups (Table 2) and medication conditions (Student's t test, two-tailed, unpaired/paired, p > 0.05 for all cases). The ANOVA revealed a significant main effect of the between-subjects factor stimulation (F(3,44) = 18.691, p < 0.001) and the main effects of nicotine (F(1,44) = 19.579, p < 0.001). The interactions between nicotine × stimulation (F(3,44) = 3.628, p < 0.05), time point × stimulation (F(30,440) = 2.473; p < 0.001) and nicotine × time point (F(10,440) = 2.382; p < 0.05) were also significant.
Table 2.
TMS stimulator intensities and baseline MEP amplitudes of different medication and stimulation conditions
| Stimulation group | Anodal tDCS | Cathodal tDCS | PAS10 | PAS25 |
|---|---|---|---|---|
| TMS stimulator intensity (placebo) (%) | 45.67 | 45.42 | 43.08 | 45.17 |
| TMS stimulator intensity (patch) (%) | 45.17 | 46.25 | 44.08 | 45.83 |
| p value | 0.531 | 0.573 | 0.400 | 0.803 |
| MEP amplitude (placebo) (mV) | 1.00 | 1.15 | 1.11 | 1.02 |
| MEP amplitude (patch) (mV) | 0.95 | 1.08 | 1.08 | 0.98 |
| p value | 0.132 | 0.105 | 0.348 | 0.469 |
Nicotinergic impact on PAS-induced plasticity
As shown by the t tests, without nicotine, PAS25 elicited nearly no excitability alterations, whereas under nicotine, MEP amplitudes were significantly enhanced for >120 min after intervention (Fig. 2). PAS10 without nicotine induced a significant excitability diminution lasting for 20 min after stimulation. Under nicotine, the inhibitory effects of PAS10 were trendwise reduced versus placebo medication, but then prolonged until the morning after plasticity induction.
Figure 2.
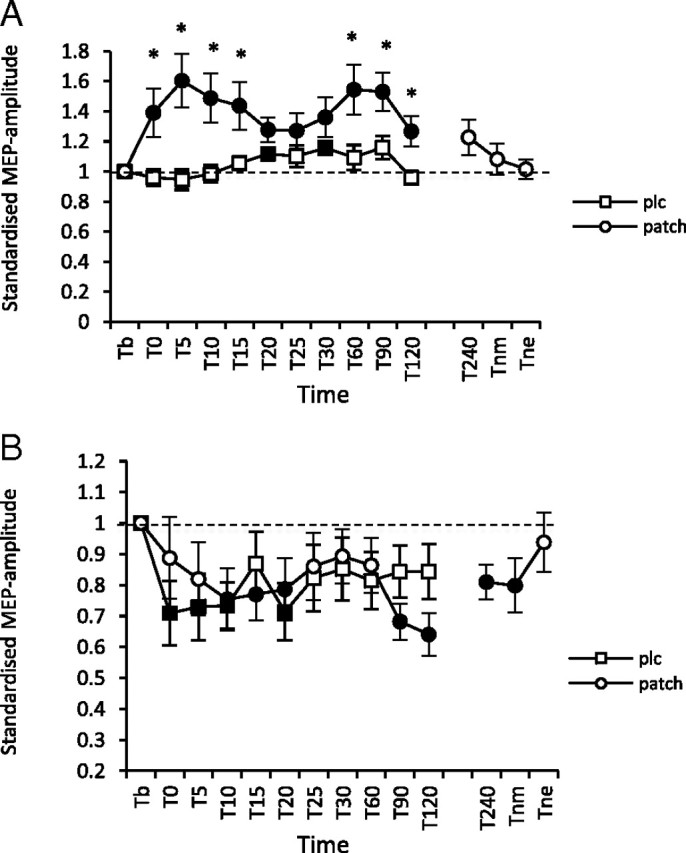
A, B, Nicotinergic impact on PAS-induced neuroplasticity. Shown are graphs with MEP standardized to the baseline on the y-axis plotted against different time points poststimulation on the x-axis. A, In smokers with placebo medication, PAS25-induced excitability enhancements were reduced, while nicotine led to a prominent facilitation lasting for 120 min after plasticity induction. B, In the placebo condition (squares), PAS10 was followed by a significant excitability diminution of 20 min. Nicotine administration (circles) delayed, but then prolonged the inhibitory effects of PAS10 until the morning after plasticity induction. Filled symbols indicate statistically significant deviations from baseline and asterisks indicate significant differences between the placebo medication and nicotine conditions (Student's t test, paired, two-tailed, p < 0.05). T, Time; b, baseline; nm, next morning; ne, next evening; plc, placebo. Baseline was MEP amplitude before begin of the stimulation protocols (standardized). Error bars indicate SEM.
Effect of nicotine on tDCS-induced plasticity
In the control condition without nicotine, anodal tDCS did not enhance MEP amplitudes significantly, while under nicotine, the stimulation-induced excitability enhancement was significant for up to 240 min post-tDCS. Cathodal tDCS stimulation under placebo medication induced a significant excitability diminution present for 60 min, whereas nicotine administration nearly abolished these aftereffects (Fig. 3).
Figure 3.
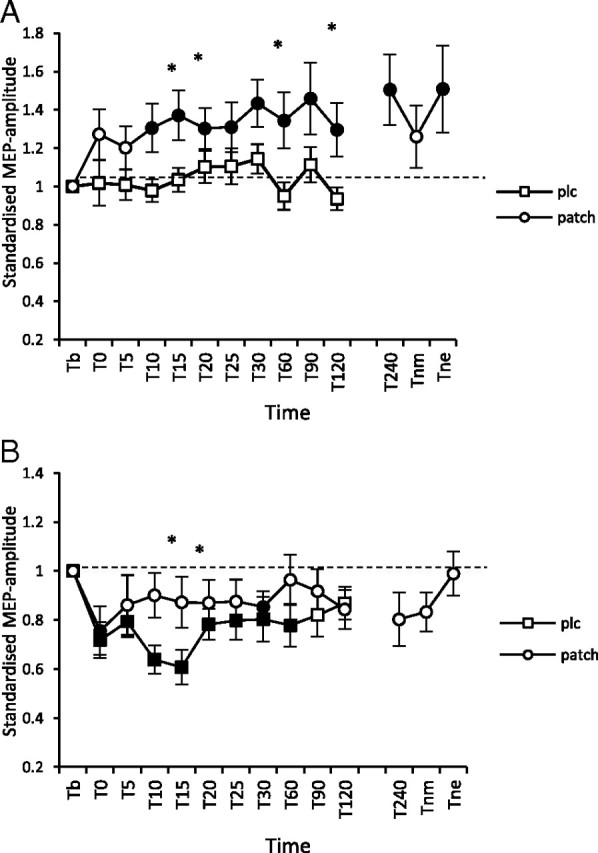
A, B, Nicotinergic impact on tDCS-induced excitability changes. Shown are graphs with MEP standardized to the baseline on the y-axis plotted against different time points postintervention on the x-axis. A, Anodal tDCS in smokers under placebo medication does not lead to an enhancement of the size of MEP amplitudes. Nicotine restitutes the tDCS-generated facilitatory aftereffects that last for up to 240 min after tDCS. B, Under placebo medication, cathodal tDCS induced an excitability reduction lasting for 60 min after plasticity induction, while nicotine abolished the tDCS-derived excitability diminution. Filled symbols indicate statistically significant deviations from baseline and asterisks indicate significant differences between the control and nicotine conditions (Student's t test, paired, two-tailed, p < 0.05). T, Time; b, baseline; nm, next morning; ne, next evening; plc, placebo. Baseline was MEP amplitude before begin of the stimulation protocols (standardized). Error bars indicate SEM.
Comparison of neuroplastic effects of nicotine in smoking and nonsmoking subjects
To draw a comparison between smokers and nonsmokers, we conducted an overall ANOVA with the results of our smoking group and the result of a nonsmoking group whose data was previously published (Thirugnanasambandam et al., 2011). The experimental procedure of the age- and gender-matched nonsmoking group was identical to our protocol. The overall ANOVA included all combinations of conditions for the smokers and nonsmokers groups and showed significant results for the MEP time points, stimulation, condition, and the respective interactions of group × condition and group × stimulation × condition, the latter indicating significant differences between the smoking and nonsmoking groups for placebo and patch conditions. Consecutively, we conducted separate ANOVAs for all medication/stimulation conditions to evaluate intergroup differences. These ANOVAs revealed significant group differences for specific placebo/nicotine medication conditions and facilitatory plasticity-eliciting stimulation protocols (Table 3). While in nonsmoking individuals, anodal tDCS and PAS25 induced a prominent motor cortex excitability enhancement without nicotine, in smokers, no facilitatory aftereffects were exhibited after stimulation. The respective intergroup differences were significant. Another significant intergroup difference exists for anodal tDCS under nicotine. Nicotine abolished anodal tDCS-induced nonfocal plasticity in nonsmokers, but reestablished compromised facilitatory plasticity in smokers. For PAS25, the nicotinic effects on excitatory aftereffects were similar in both groups, with reestablished facilitatory plasticity in smokers and prolonged focal plasticity in nonsmokers. For inhibitory plasticity, the intergroup differences are less prominent. Inhibitory plasticity was not largely affected by nicotine withdrawal in smokers, and was reduced or delayed by nicotine. Likewise, nicotine reduced or abolished inhibitory plasticity in nonsmokers. In conclusion, nicotine patch in nonsmokers leads to a focusing effect on facilitatory plasticity, because it abolished nonfocal tDCS-induced plasticity, but prolonged PAS-induced focal plasticity, whereas in smokers, it reestablishes tDCS and PAS-induced facilitatory plasticity.
Table 3.
Results of repeated-measures ANOVA
| Parameters | df | F value | p value |
|---|---|---|---|
| PAS10 placebo | |||
| Time point | 9 | 1.120 | 0.350 |
| Group | 1 | 0.497 | 0.488 |
| Time point × group | 9 | 0.771 | 0.644 |
| PAS10 patch | |||
| Time point | 9 | 2.077 | 0.033* |
| Group | 1 | 1.869 | 0.185 |
| Time point × group | 9 | 0.463 | 0.898 |
| PAS25 placebo | |||
| Time point | 9 | 2.258 | 0.019* |
| Group | 1 | 11.120 | 0.003* |
| Time point × group | 9 | 1.137 | 0.338 |
| PAS25 patch | |||
| Time point | 9 | 0.401 | 0.943 |
| Group | 1 | 0.000 | 0.992 |
| Time point × group | 9 | 1.298 | 0.240 |
| Cathodal tDCS placebo | |||
| Time point | 9 | 1.287 | 0.246 |
| Group | 1 | 0.627 | 0.437 |
| Time point × group | 9 | 1.591 | 0.120 |
| Cathodal tDCS patch | |||
| Time point | 9 | 0.322 | 0.945 |
| Group | 1 | 0.207 | 0.654 |
| Time point × group | 9 | 0.991 | 0.449 |
| Anodal tDCS placebo | |||
| Time point | 9 | 2.719 | 0.005* |
| Group | 1 | 9.898 | 0.005* |
| Time point × group | 9 | 1.094 | 0.369 |
| Anodal tDCS patch | |||
| Time point | 9 | 1.881 | 0.057 |
| Group | 1 | 7.113 | 0.014* |
| Time point × group | 9 | 0.493 | 0.879 |
*Significant p values.
Discussion
The results of the present study show that under nicotine withdrawal, facilitatory plasticity is absent in smokers regardless of the plasticity induction procedure (tDCS and PAS25), whereas inhibitory plasticity is largely unaltered. Nicotine exposition reestablishes compromised facilitatory plasticity in this subject group, again independent of the focality of the induction procedure. In contrast, nicotine results in a trendwise reduction of the amplitude of inhibitory plasticity compared with the nicotine withdrawal condition. However, for PAS-generated inhibitory plasticity, the duration of the effects seems to be prolonged.
For facilitatory plasticity, the results under nicotine withdrawal and exposition differ strikingly from those obtained in a foregoing study of our group, where the effects of identical plasticity stimulation protocols were explored in a matched group of nonsmoking subjects (Thirugnanasambandam et al., 2011). In nonsmokers, nicotine induced a focusing effect with prolongation of PAS25-induced excitatory aftereffects and abolishment of tDCS-induced excitatory aftereffects, while in smokers it reestablished lacking facilitatory plasticity of anodal tDCS and PAS25.
Interestingly, the results of the present study with regard to facilitatory plasticity are in principal accordance with those of another study, where cortical excitability of smokers under (somewhat shorter) nicotine withdrawal was compared with excitability in nonsmokers: smokers displayed deficient intracortical facilitation (Lang et al., 2008).
Proposed mechanism of action
nAChRs are widely distributed throughout the brain (Albuquerque et al., 2009). Depending on subunit composition, the receptors exhibit different pharmacological and physiological properties. In the brain, the two major types of functional nAChRs are formed by the heteromeric assembly of α4β2 and the homomeric α7 subunits (Jones et al., 1999). Glutamatergic plasticity, as accomplished by tDCS and PAS, is thought to depend on enhancement of the intracellular calcium levels. Both the α7 subtype and α4β2 receptor increase intracellular calcium levels and transmitter release by serving as presynaptic and postsynaptic ligand-gated calcium channels (Radcliffe and Dani, 1998; Fujii et al., 1999). Nicotinic activation of these channels has been shown to induce LTP (Welsby et al., 2006) in rat dentate gyrus. In humans, studies probing the induction of LTP-like long-lasting plasticity have so far been restricted to the neocortex. Facilitatory plasticity-inducing protocols, namely PAS25 and anodal tDCS, result both in LTP-like plasticity that is dependent on NMDA-receptor and calcium channel activity (Stefan et al., 2002; Nitsche et al., 2003b, 2004), and thus resembles the induction of hippocampal LTP, at least to some degree. Administration of nicotine in smokers might result in an increased calcium level via activation of nAChRs and in this way cause an enhancement of PAS25 and anodal tDCS-induced LTP-like aftereffects. A main difference in nonsmoking subjects is the fact that smokers under nicotine withdrawal did not develop any LTP-like plasticity at all after tDCS and PAS. Here, the desensitization of nAChRs, especially the α7 and β2*, might play a major role (Picciotto et al., 2008). It has been shown that chronic nicotine consumption desensitizes nAChRs in different areas of the brain (Wooltorton et al., 2003) and that the duration of desensitization depends critically on the duration of nicotine exposure (Lester and Dani, 1994). Therefore, it might be speculated that chronic smokers during nicotine withdrawal are not able to develop LTP-like plasticity due to deficient calcium influx, and that an enhancement of nicotine via nicotine patch administration overrides receptor desensitization, thus enabling a sufficiently large intracellular calcium increase to induce LTP-like effects by both stimulation techniques. This might also explain why the effects of nicotine on facilitatory plasticity differ in nonsmoking humans. The nonfocal tonic neuronal membrane depolarization accomplished by anodal tDCS, which should result in a relatively large calcium influx, might, together with the further enhancement of calcium influx via nicotinic receptor activation, result in intracellular calcium overflow. This will lead to a counter-regulation accomplished by activation of hyperpolarizing potassium channels, thus abolishing plasticity (Misonou et al., 2004). In contrast, the synapse-specific effects of the PAS plasticity induction procedure might result in a relatively lower intraneuronal calcium concentration not sufficient to induce such a counter-regulation in the presence of nicotine in nonsmokers, and thus here nicotinic receptor activation will enhance the efficacy of plasticity induction. A similar calcium-dependent mechanism of action might apply for the effects of nicotine on excitability-diminishing plasticity in smokers and nonsmoking individuals. As compared with LTP, a minor enhancement of intracellular calcium suffices to induce LTD in animal experiments (Malenka and Bear, 2004). Between these low-level calcium enhancements needed to generate LTD, and high-level calcium increases sufficient to induce LTP, a no man's land exists, which induces no plasticity (Lisman, 2001). It can be speculated that the calcium alterations accomplished by excitability-diminishing tDCS and PAS in smokers still suffice to induce inhibitory plasticity despite reduced calcium influx similar to the effects accomplished in nonsmokers. Intracellular calcium influx will then be enhanced by nicotine, and might drive the system into the no man's land of plasticity induction. This effect of nicotine might be somewhat weaker in smokers because of the nicotinic receptor desensitization.
Although attractive, these proposed mechanisms of action are speculative at present and need to be experimentally tested in future.
General remarks
The results of our study demonstrate that nicotine withdrawal and nicotine itself influence neuroplasticity in smoking subjects prominently. Nicotine withdrawal of 10 h prevents the induction of LTP-like plasticity, and nicotine administration reestablishes absent plasticity, thus favoring the deficit-compensating hypothesis of nicotine consumption. Nicotine abstinence in nicotine-dependent smokers is often associated with reduced working and verbal memory capacity compared with nonsmokers, while nicotine has been shown to improve attention, working, and long-term memories in animals and humans. From the results of the present study, it might be speculated that these effects of nicotine on cognition might at least partially be based on the respective alterations of plasticity, especially with regard to learning and memory formation. This potential connection should be explored directly in future studies. Studies exploring the impact of nicotinic receptor subtypes by pharmacological intervention or exploration of genetic differences between smoking and nonsmoking subjects might help to shed more light on the specific receptor mechanisms underlying these effects. Moreover, further work needs to be done concerning probable dose-dependent effects of nicotine, which have been shown previously to be of special importance for other neuromodulators, e.g., dopaminergic agents (Monte-Silva et al., 2010).
Conclusion
The results of this study show that nicotine and nicotine withdrawal have a prominent impact on the formation of neuroplasticity in smoking subjects, the likely physiological basis of learning and memory formation. During nicotine withdrawal, facilitatory plasticity is abolished in tobacco smokers, but restituted by nicotine administration. These neurophysiological results support the deficit-compensating hypothesis of nicotine consumption. Moreover, this effect on neuroplasticity might also be relevant for starting nicotine consumption, addiction, and the high probability of relapse in heavy smokers. Further studies are needed to explore whether a priori differences in neuroplasticity between smokers and nonsmokers result in nicotine addiction, like has been postulated for schizophrenic patients, or are the result of nicotine addiction. With regard to the application of brain stimulation protocols, the results of our study show clearly that the activity of the nicotinergic system might affect the efficacy of plasticity-inducing procedures and thus needs to be critically taken into account in these kinds of studies.
Limiting conditions
Some limitation should be taken into account. One limitation of our study is the fact that we did not obtain blood levels or breath CO concentration to verify compliance of smoking subjects. However, since experiments began in the morning and smoking subjects were only moderately dependent on the Fagerstrom scale, it was realistic to rely on the statements of the subjects. Only smokers with a light nicotine dependency participated in the current study; heavily dependent smokers may show different results. Another limitation lies in the fact that we did not connect neurophysiologic results to cognitive functions directly. Because the effects of nicotine withdrawal and readministration in cognitive functions [improved working and verbal memory function after nicotine readministration (Atzori et al., 2008)] imply an association between both outcome measurements, further studies need to verify this association directly.
Footnotes
The work was supported by the Deutsche Forschungsgemeinschaft (DFG Grants NI683/4-1 and NI 683/4-2).
References
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleisa AM, Alzoubi KH, Alkadhi KA. Nicotine prevents stress-induced enhancement of long-term depression in hippocampal area CA1: electrophysiological and molecular studies. J Neurosci Res. 2006;83:309–317. doi: 10.1002/jnr.20716. [DOI] [PubMed] [Google Scholar]
- Atzori G, Lemmonds CA, Kotler ML, Durcan MJ, Boyle J. Efficacy of a nicotine (4 mg)-containing lozenge on the cognitive impairment of nicotine withdrawal. J Clin Psychopharmacol. 2008;28:667–674. doi: 10.1097/JCP.0b013e31818c9bb8. [DOI] [PubMed] [Google Scholar]
- Benowitz NL. Pharmacology of nicotine: addiction and therapeutics. Annu Rev Pharmacol Toxicol. 1996;36:597–613. doi: 10.1146/annurev.pa.36.040196.003121. [DOI] [PubMed] [Google Scholar]
- Burnashev N. Calcium permeability of ligand-gated channels. Cell Calcium. 1998;24:325–332. doi: 10.1016/s0143-4160(98)90056-2. [DOI] [PubMed] [Google Scholar]
- Cole DM, Beckmann CF, Long CJ, Matthews PM, Durcan MJ, Beaver JD. Nicotine replacement in abstinent smokers improves cognitive withdrawal symptoms with modulation of resting brain network dynamics. Neuroimage. 2010;52:590–599. doi: 10.1016/j.neuroimage.2010.04.251. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador F, Wonnacott S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci. 2004;25:317–324. doi: 10.1016/j.tips.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Fujii S, Ji Z, Morita N, Sumikawa K. Acute and chronic nicotine exposure differentially facilitate the induction of LTP. Brain Res. 1999;846:137–143. doi: 10.1016/s0006-8993(99)01982-4. [DOI] [PubMed] [Google Scholar]
- Hahn B, Stolerman IP. Nicotine-induced attentional enhancement in rats: effects of chronic exposure to nicotine. Neuropsychopharmacology. 2002;27:712–722. doi: 10.1016/S0893-133X(02)00348-2. [DOI] [PubMed] [Google Scholar]
- Heishman SJ, Kleykamp BA, Singleton EG. Meta-analysis of the acute effects of nicotine and smoking on human performance. Psychopharmacology. 2010;210:453–469. doi: 10.1007/s00213-010-1848-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta PT, Lisman JE. Bidirectional synaptic plasticity induced by a single burst during cholinergic theta oscillation in CA1 in vitro. Neuron. 1995;15:1053–1063. doi: 10.1016/0896-6273(95)90094-2. [DOI] [PubMed] [Google Scholar]
- Jacobsen LK, Krystal JH, Mencl WE, Westerveld M, Frost SJ, Pugh KR. Effects of smoking and smoking abstinence on cognition in adolescent tobacco smokers. Biol Psychiatry. 2005;57:56–66. doi: 10.1016/j.biopsych.2004.10.022. [DOI] [PubMed] [Google Scholar]
- Jones HE, Garrett BE, Griffiths RR. Subjective and physiological effects of intravenous nicotine and cocaine in cigarette smoking cocaine abusers. J Pharmacol Exp Ther. 1999;288:188–197. [PubMed] [Google Scholar]
- Jubelt LE, Barr RS, Goff DC, Logvinenko T, Weiss AP, Evins AE. Effects of transdermal nicotine on episodic memory in non-smokers with and without schizophrenia. Psychopharmacology. 2008;199:89–98. doi: 10.1007/s00213-008-1133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo MF, Grosch J, Fregni F, Paulus W, Nitsche MA. Focusing effect of acetylcholine on neuroplasticity in the human motor cortex. J Neurosci. 2007;27:14442–14447. doi: 10.1523/JNEUROSCI.4104-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang N, Hasan A, Sueske E, Paulus W, Nitsche MA. Cortical hypoexcitability in chronic smokers? A transcranial magnetic stimulation study. Neuropsychopharmacology. 2008;33:2517–2523. doi: 10.1038/sj.npp.1301645. [DOI] [PubMed] [Google Scholar]
- Lester RA, Dani JA. Time-dependant changes in central nicotinic acetylcholine channel kinetics in outside-out patches. Neuropharmacology. 1994;33:27–34. doi: 10.1016/0028-3908(94)90093-0. [DOI] [PubMed] [Google Scholar]
- Levin ED, McClernon FJ, Rezvani AH. Nicotinic effects on cognitive function: behavioral characterization, pharmacological specification, and anatomic localization. Psychopharmacology (Berl) 2006;184:523–539. doi: 10.1007/s00213-005-0164-7. [DOI] [PubMed] [Google Scholar]
- Lisman JE. Three Ca2+ levels affect plasticity differently: the LTP zone, the LTD zone and no man's land. J Physiol. 2001;532:285. doi: 10.1111/j.1469-7793.2001.0285f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Matsuyama S, Matsumoto A, Enomoto T, Nishizaki T. Activation of nicotinic acetylcholine receptors induces long-term potentiation in vivo in the intact mouse dentate gyrus. Eur J Neurosci. 2000;12:3741–3747. doi: 10.1046/j.1460-9568.2000.00259.x. [DOI] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE, Trimmer JS. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci. 2004;7:711–718. doi: 10.1038/nn1260. [DOI] [PubMed] [Google Scholar]
- Monte-Silva K, Liebetanz D, Grundey J, Paulus W, Nitsche MA. Dosage-dependent non-linear effect of l-dopa on human motor cortex plasticity. J Physiol. 2010;588:3415–3424. doi: 10.1113/jphysiol.2010.190181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsche MA, Paulus W. Excitability changes induced in the human motor cortex by weak transcranial direct current stimulation. J Physiol. 2000;527:633–919. doi: 10.1111/j.1469-7793.2000.t01-1-00633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsche MA, Paulus W. Sustained excitability elevations induced by transcranial DC motor cortex stimulation in humans. Neurology. 2001;57:1899–1901. doi: 10.1212/wnl.57.10.1899. [DOI] [PubMed] [Google Scholar]
- Nitsche MA, Nitsche MS, Klein CC, Tergau F, Rothwell JC, Paulus W. Level of action of cathodal DC polarisation induced inhibition of the human motor cortex. Clin Neurophysiol. 2003a;114:600–604. doi: 10.1016/s1388-2457(02)00412-1. [DOI] [PubMed] [Google Scholar]
- Nitsche MA, Fricke K, Henschke U, Schlitterlau A, Liebetanz D, Lang N, Henning S, Tergau F, Paulus W. Pharmacological modulation of cortical excitability shifts induced by transcranial direct current stimulation in humans. J Physiol. 2003b;553:293–301. doi: 10.1113/jphysiol.2003.049916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsche MA, Jaussi W, Liebetanz D, Lang N, Tergau F, Paulus W. Consolidation of human motor cortical neuroplasticity by d-cycloserine. Neuropsychopharmacology. 2004;29:1573–1578. doi: 10.1038/sj.npp.1300517. [DOI] [PubMed] [Google Scholar]
- Nitsche MA, Roth A, Kuo MF, Fischer AK, Liebetanz D, Lang N, Tergau F, Paulus W. Timing-dependent modulation of associative plasticity by general network excitability in the human motor cortex. J Neurosci. 2007;27:3807–3812. doi: 10.1523/JNEUROSCI.5348-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nørregaard J, Tønnesen P, Simonsen K, Säwe U. Long-term nicotine substitution after application of a 16-h nicotine patch in smoking cessation. Eur J Clin Pharmacol. 1992;43:57–60. doi: 10.1007/BF02280755. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Addy NA, Mineur YS, Brunzell DH. It is not “either/or”: activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog Neurobiol. 2008;84:329–342. doi: 10.1016/j.pneurobio.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purpura DP, McMurtry JG. Intracellular activities and evoked potential changes during polarization of motor cortex. J Neurophysiol. 1965;28:166–185. doi: 10.1152/jn.1965.28.1.166. [DOI] [PubMed] [Google Scholar]
- Radcliffe KA, Dani JA. Nicotinic stimulation produces multiple forms of increased glutamatergic synaptic transmission. J Neurosci. 1998;18:7075–7083. doi: 10.1523/JNEUROSCI.18-18-07075.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada S, Yamamoto C, Ohno-Shosaku T. Long-term potentiation and depression in the dentate gyrus, and effects of nicotine. Neurosci Res. 1994;20:323–329. doi: 10.1016/0168-0102(94)90054-x. [DOI] [PubMed] [Google Scholar]
- Stefan K, Kunesch E, Cohen LG, Benecke R, Classen J. Induction of plasticity in the human motor cortex by paired associative stimulation. Brain. 2000;123:572–584. doi: 10.1093/brain/123.3.572. [DOI] [PubMed] [Google Scholar]
- Stefan K, Kunesch E, Benecke R, Cohen LG, Classen J. Mechanisms of enhancement of human motor cortex excitability induced by interventional paired associative stimulation. J Physiol. 2002;543:699–708. doi: 10.1113/jphysiol.2002.023317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirugnanasambandam N, Grundey J, Adam K, Drees A, Skwirba AC, Lang N, Paulus W, Nitsche MA. Nicotinergic impact on focal and non-focal neuroplasticity induced by non-invasive brain stimulation in non-smoking humans. Neuropsychopharmacology. 2011;36:879–886. doi: 10.1038/npp.2010.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tønnesen P, Nørregaard J, Simonsen K, Säwe U. A double-blind trial of a 16-h transdermal nicotine patch in smoking cessation. N Engl J Med. 1991;325:311–315. doi: 10.1056/NEJM199108013250503. [DOI] [PubMed] [Google Scholar]
- Weise D, Schramm A, Stefan K, Wolters A, Reiners K, Naumann M, Classen J. The two sides of associative plasticity in writer's cramp. Brain. 2006;129:2709–2721. doi: 10.1093/brain/awl221. [DOI] [PubMed] [Google Scholar]
- Welsby P, Rowan M, Anwyl R. Nicotinic receptor-mediated enhancement of long-term potentiation involves activation of metabotropic glutamate receptors and ryanodine sensitive calcium stores in the dentate gyrus. Eur J Neurosci. 2006;24:3109–3118. doi: 10.1111/j.1460-9568.2006.05187.x. [DOI] [PubMed] [Google Scholar]
- Wolters A, Sandbrink F, Schlottmann A, Kunesch E, Stefan K, Cohen LG, Benecke R, Classen J. A temporally asymmetric Hebbian rule governing plasticity in the human motor cortex. J Neurophysiol. 2003;89:2339–2345. doi: 10.1152/jn.00900.2002. [DOI] [PubMed] [Google Scholar]
- Wooltorton JR, Pidoplichko VI, Broide RS, Dani JA. Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J Neurosci. 2003;23:3176–3185. doi: 10.1523/JNEUROSCI.23-08-03176.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]