

Abstract
Addictive drugs share the ability to increase dopamine (DA) levels and trigger synaptic adaptations in the mesocorticolimbic system, two cellular processes engaged in the early stages of drug seeking. Neurons located in the lateral habenula (LHb) modulate the activity of DA neurons and DA release, and adaptively tune goal-directed behaviors. Whether synaptic modifications in LHb neurons occur upon drug exposure remains, however, unknown. Here, we assessed the influence of cocaine experience on excitatory transmission onto subsets of LHb neurons using a combination of retrograde tracing and ex vivo patch-clamp recordings in mice. Recent evidence demonstrates that AMPA receptors lacking the GluA2 subunit mediate glutamatergic transmission in LHb neurons. We find that cocaine selectively potentiates AMPA receptor-mediated EPSCs in LHb neurons that send axons to the rostromedial tegmental nucleus, a GABAergic structure that modulates the activity of midbrain DA neurons. Cocaine induces a postsynaptic accumulation of AMPA receptors without modifying their subunit composition or single-channel conductance. As a consequence, a protocol pairing presynaptic glutamate release with somatic hyperpolarization, to increase the efficiency of GluA2-lacking AMPA receptors, elicited a long-term potentiation in neurons only from cocaine-treated mice. This suggests that cocaine resets the rules for the induction of synaptic long-term plasticity in the LHb. Our study unravels an early, projection-specific, cocaine-evoked synaptic potentiation in the LHb that may represent a permissive step for the functional reorganization of the mesolimbic system after drug exposure.
Introduction
Midbrain dopamine (DA) neurons are central in guiding motivated behaviors as well as drug seeking (Wise, 2004). Addictive substances with different properties modulate DA neuron activity and DA release from the ventral tegmental area (VTA) (Lüscher and Malenka, 2011). Recent evidence indicates that the lateral habenula (LHb)—a hub-like region anatomically and functionally connected to midbrain structures—bidirectionally modulates the activity of DA neurons (Matsumoto and Hikosaka, 2007). LHb neurons provide DA neurons with the signal necessary to respond to salient rewarding and aversive stimuli (Hikosaka, 2010). The LHb neurons are mostly glutamatergic, and contain GluA2-lacking AMPA receptors (AMPARs) (Li et al., 2011). They project to DA neurons through two distinct pathways (Balcita-Pedicino et al., 2011). The direct projection that targets the VTA has been recently implicated in the etiology of mood disorders (Li et al., 2011). The indirect projection tunes DA neuron activity through a GABAergic relay named rostromedial tegmental nucleus (RMTg) or tail VTA (Jhou et al., 2009; Kaufling et al., 2009). Addictive drugs have been shown to alter the physiology and the expression of immediate early genes, not only in VTA DA neurons, but also in putative RMTg and LHb neurons (Brown et al., 2010b; Kaufling et al., 2010; James et al., 2011; Lecca et al., 2011). These findings suggest that drug-induced changes in the DA circuit might be, at least in part, a consequence of adaptations taking place in structures upstream of the VTA, such as the LHb. However, the effects of drug exposure on synaptic transmission and plasticity in the LHb remain unknown.
Here, we used tract tracing in combination with ex vivo patch-clamp recordings to demonstrate that exposure to cocaine selectively potentiates AMPAR-mediated transmission in LHb neurons projecting to the RMTg (LHb→RMTg), but not to the VTA (LHb→VTA), via a postsynaptic increase in AMPAR content. Consequently, cocaine-evoked synaptic potentiation modifies the rules for long-term plasticity in LHb neurons.
Materials and Methods
Surgery.
C57BL/6J mice (20–40 d old) of either sex were used in accordance with the guidelines of the French Agriculture and Forestry Ministry for handling animals. Animals were anesthetized with ketamine (90 mg/kg)/xylazine (15 mg/kg) (Sigma-Aldrich) before bilateral injection with either Cholera-toxin-B–Alexa Fluor 488/594 or fluorobeads (Invitrogen, Lumafluor). The following coordinates were used (from bregma, in mm): RMTg: anteroposterior (A-P), −2.9; mediolateral (M-L), ±0.5; dorsoventral (D-V), −4.3; VTA: A-P, −2.4; M-L, ±0.8; D-V, −4.4. Recovery was allowed for at least 3 d.
Electrophysiology.
Animals injected with saline or cocaine (10 mg/kg), once for 2 consecutive days, were anesthetized (ketamine/xylazine) 24 h after the last injection for preparation of LHb-containing brain slices. Slicing was performed in bubbled ice-cold 95% O2/5% CO2-equilibrated solution containing the following (in mm): choline Cl 110; glucose 25; NaHCO3 25; MgCl2 7; ascorbic acid 11.6; Na+-pyruvate 3.1; KCl 2.5; NaH2PO4 1.25; and CaCl2 0.5. Sagittal slices (250 μm) were stored at room temperature in 95% O2/5% CO2-equilibrated artificial CSF containing the following (in mm): NaCl 124; NaHCO3 26.2; glucose 11; KCl 2.5; CaCl2 2.5; MgCl2 1.3; and NaH2PO4 1. Recordings (flow rate of 2.5 ml/min) were made under an Olympus BX51 microscope at 32°C. Currents were amplified, filtered at 5 kHz, and digitized at 20 kHz. Access resistance was monitored by a step of −10 mV (0.1 Hz). Experiments were discarded if the access resistance increased >20%. Miniature EPSCs (mEPSCs) were recorded in voltage-clamp mode at −60 mV in the presence of bicuculline (20 μm), TTX (1 μm), and d,l(−)-2-amino-5-phosphonovaleric acid (d,l-ap5; 100 μm). The internal solution contained the following (in mm): CsCl 130; NaCl 4; MgCl2 2; EGTA 1.1; HEPES 5; Na2ATP 2; Na+-creatine-phosphate 5; Na3GTP 0.6; and spermine 0.1 was added when indicated. The liquid junction potential was −3 mV. EPSCs were evoked through glass electrodes placed in the stria medullaris. AMPA/NMDA ratios of evoked EPSC were obtained by AMPA-EPSC +40 mV/NMDA-EPSCs at +40 mV as previously described (Ungless et al., 2001). The rectification index (RI) was calculated by the formula (EPSC −70/EPSC +40)/1.75 in the presence of d,l-ap5 and intracellular spermine. For long-term plasticity, the internal solution contained the following (in mm): K-gluconate 140; KCl 5; HEPES 10; EGTA 0.2; MgCl2 2; Na2ATP 4; Na3GTP 0.3; and creatine-phosphate 10. Recordings were corrected for liquid junction potential (12 mV). The induction protocol for long-term plasticity consisted of a 10 Hz stimulation (20 times each 5 s) paired with somatic hyperpolarization (−90 mV). Recordings were kept for at least 30–45 min.
For glutamate uncaging (4-methoxy-7-nitroindolinyl-caged l-glutamate 200 μm, Tocris-Cookson), a single-path photolysis head (Prairie Technologies) was connected to a solid-state laser (405 nm, duration 0.5 ms, diameter 3–5 μm). Photolysis was optimized by adjusting the laser beam position and power (3–10 mW) such that laser-evoked EPSCs [(L)-EPSCs] yielded amplitudes and kinetics similar to mEPSCs. AMPA/NMDA ratios of (L)-EPSCs were obtained by dividing the peak of AMPA-EPSC at −60 mV and the NMDA-EPSCs at +40 mV at 30 ms after onset as previously described (Mameli et al., 2011).
Nonstationary fluctuation analysis.
A peak-scaled nonstationary fluctuation analysis (NSFA) was performed on mEPSCs (100–200) (Synaptosoft). mEPSCs were selected by: fast rise time alignment, stable baseline holding current, and the absence of spurious fluctuations during the mEPSCs decay. The variance–amplitude relationship of mEPSC decay was plotted and fitted with the equation σ2 = iI − I2/N + σb2 (where i is the mean single-channel AMPA current, I is the mean current, N is the number of channels activated at the peak, N = mean amplitude/i; and σb2 is the baseline variance). i was estimated as the slope of the linear fit of the first portion of the parabola of the fitted mEPSC decay. The goodness-of-fit was assessed with a least-squares algorithm. The unitary current was converted in conductance based on the reversal potential of evoked EPSCs (∼0 mV). Conductance and average EPSC amplitude, mean rise time, mean decay time, access resistance, or background noise variance had no correlation (p > 0.4) (Balland et al., 2006).
Analysis and drugs.
All drugs were obtained from Abcam and Tocris Bioscience and dissolved in water, except for TTX (citric acid 1%) and cocaine HCl (NaCl 0.9%). On-line/off-line analyses were performed using IGOR-6 (Wavemetrics) and Prism (Graphpad). Compiled data are expressed as the mean ± SEM. Significance was set at p = 0.05 using Student's t test and the Kolmogorov–Smirnov (KS) test.
Results
Cocaine strengthens excitatory synaptic transmission in the LHb
LHb neurons send axons to the RMTg and the VTA (Balcita-Pedicino et al., 2011; Hong et al., 2011). To selectively identify LHb→RMTg and LHb→VTA neurons, we stereotaxically injected green and red fluorobeads in the VTA and RMTg, respectively. As previously shown in rats (Li et al., 2011), LHb→RMTg and LHb→VTA neuronal populations were segregated (Fig. 1A).
Figure 1.

Cocaine increases the efficacy of excitatory transmission onto LHb→RMTg neurons. A, LHb-containing sagittal sections illustrating the injections sites with retrobeads in the RMTg (red) and the VTA (green, left panels). Confocal images (10×/40×) illustrating retrogradely labeled LHb→RMTg and LHb→VTA neurons (right panels). sm, Stria medullaris; fr, fasciculus retroflexus. B, Sample traces of mEPSCs obtained in LHb→RMTg neurons in slices from saline-treated (black) and cocaine-treated (red) mice. C, Cumulative probability distributions (KS test, p < 0.0001) for a representative LHb→RMTg neuron, bar-graph and scatter plot for mEPSC amplitude in the saline and cocaine groups (n = 10–15; saline 14.1 ± 1.1 pA; cocaine 17.9 ± 0.9 pA; **p < 0.01; t(22) = 2.9). D, Same as C for mEPSC frequency (n = 10–15; saline 3 ± 0.9 Hz; cocaine 4.9 ± 1.3 Hz; p > 0.05; t(22) = 0.9). E, Same as B for LHb→VTA neurons. F, Same as C for LHb→VTA neurons (KS test, p > 0.05) (n = 9–14; saline 16.7 ± 2.2 pA; cocaine 14.08 ± 1.3 pA; p > 0.05; t(21) = 1.1). G, Same as F for mEPSC frequency (n = 9–14; saline 4.6 ± 1.8 Hz; cocaine 4.1 ± 0.7 Hz; p > 0.05; t(21) = 0.3). Sal, Saline; Coc, cocaine.
Addictive drugs modify excitatory synaptic transmission in the mesocorticolimbic system (Lüscher and Malenka, 2011). We examined whether cocaine affects excitatory synaptic transmission onto LHb neurons. Mice infused with retrograde tracers in the RMTg and/or VTA were injected with cocaine/saline for 2 consecutive days, a paradigm leading to a progressive rise in locomotor activity (data not shown). We prepared LHb-containing slices 24 h after the last injection, and performed whole-cell recordings from retrogradely labeled LHb neurons. We first monitored quantal release by recording pharmacologically isolated AMPAR-mediated mEPSCs. In saline-treated animals, mEPSC amplitude and frequency in LHb→RMTg and LHb→VTA neurons were comparable (Fig. 1B–G; amplitude, t(17) = 1.3; frequency, t(16) = 0.8; p > 0.05). After cocaine, mEPSCs amplitude significantly increased in LHb→RMTg but not in LHb→VTA neurons (Fig. 1B,C,E,F). In contrast, mEPSC frequency was not significantly affected regardless of the neuronal subpopulation (Fig. 1B,D,E,G). These findings suggest that cocaine exposure strengthens AMPAR-mediated synaptic transmission specifically in LHb→RMTg neurons.
Cocaine postsynaptically potentiates AMPAR-mediated transmission in LHb→RMTg neurons
We next examined the effect of cocaine on synaptically evoked EPSCs. Under our recording conditions, paired-pulse depression was detected in LHb→RMTg neurons and the paired-pulse ratio (PPR) of AMPAR-EPSCs was not affected by cocaine exposure (Fig. 2A), indicating no alterations in presynaptic release. We next compared the ratio of AMPAR and NMDAR-EPSCs recorded at +40 mV (AMPA/NMDA). Cocaine exposure increased the AMPA/NMDA ratio in LHb→RMTg neurons (Fig. 2B), indicating a postsynaptic strengthening of excitatory transmission. Next, we assessed the properties of AMPARs by recording EPSCs at −70, 0, and +40 mV, and calculating the RI in LHb→RMTg neurons. As shown in rats (Li et al., 2011), we also observed large RIs in mice, suggesting that AMPARs in LHb lack the GluA2 subunit (Fig. 2C). In the VTA, a single injection of cocaine leads to a change in AMPAR subunit composition from GluA2-containing to GluA2-lacking (Bellone and Lüscher, 2006; Mameli et al., 2011). We found that the RIs in LHb→RMTg neurons from the saline and cocaine groups were not significantly different (Fig. 2C), indicating that cocaine treatment did not change AMPAR subunit composition.
Figure 2.

Synaptic potentiation in LHb→RMTg neurons requires a larger synaptic pool of AMPARs with no change in subunit composition. A, Averaged traces and bar-graph illustrating the PPR in the saline and cocaine groups (saline 0.82 ± 0.1; cocaine 0.74 ± 0.05; p > 0.05; t(12) = 0.64; n = 6–7). B, Sample traces illustrating AMPA and NMDA-EPSCs recorded at +40 mV and AMPA/NMDA ratios in LHb→RMTg neurons in slices from saline (black) and cocaine (red) groups (n = 5–7; saline 1.1 ± 0.1; cocaine 2.6 ± 0.3; **p < 0.01; t(9) = 4.9). C, Sample traces of AMPA-EPSCs recorded at −70, 0, and +40 mV and RIs in LHb→RMTg neurons in slices from saline (black) and cocaine (red) groups (n = 6; saline 3.85 ± 1.12; cocaine 3.1 ± 0.3; p > 0.05; t(15) = 0.7). D, Example of peak-scaled NSFA of LHb→RMTg neurons in the saline- and cocaine-treated group. Insets, Overlay of 50 consecutive traces. E, F, Pooled data for conductance (γ) and number of channels (N) open at the peak (n = 5–8; γ: saline 12.9 ± 0.4 pS; cocaine 13.1 ± 0.9 pS; p > 0.05; t(11) = 0.1; N: saline 19.2 ± 4.8; cocaine 42 ± 4.2; **p < 0.01; t(11) = 3.4). Sal, Saline; Coc, cocaine.
To assess whether cocaine treatment affected AMPAR conductance or receptor number, we used peak-scaled NSFA of mEPSCs recorded from LHb→RMTg neurons. The estimated i value was not affected by cocaine treatment (Fig. 2D,E). On the other hand, the estimated number of channels opened at the peak was significantly larger (N; Fig. 2D,F). This suggests that cocaine induces an increase in the number of AMPARs without changing their biophysical properties or their subunit composition.
We next characterized the effect of cocaine exposure on AMPAR-mediated currents evoked by glutamate uncaging. We filled LHb→RMTg neurons with dye to visualize cell morphology and performed recordings in slices from saline- or cocaine-treated animals using focalized photolysis of caged glutamate in dendrites (Fig. 3A). While amplitudes of laser-evoked EPSCs at −60 mV were comparable to those of mEPSCs, these events had slower decay, suggesting activation of synaptic as well as extrasynaptic glutamate receptors by glutamate uncaging (Fig. 3B,C). (L)-EPSCs were mediated by AMPARs, as they were blocked by NBQX (Fig. 3B). We then mapped synaptic hot spots by uncaging glutamate at −60, 0, and +40 mV. (L)-EPSCs at depolarized potentials had slower decay kinetics, indicating activation of both AMPARs and NMDARs (Fig. 3D). We found that AMPA/NMDA ratios of laser-evoked currents (AMPA−60mV, peak/NMDA+40mV, 30 ms onset) in LHb→RMTg neurons of slices from cocaine-treated animals were significantly larger than those from saline-treated animals (Fig. 3E). Plotting the ratio as a function of the (L)-AMPA-EPSCs revealed that cocaine significantly shifts the distribution toward the top right (Fig. 3F), while no significant changes were detected for (L)-NMDA currents (Fig. 3G). These findings support that the increase in AMPAR-mediated component is largely responsible for the increase in AMPA/NMDA ratio. Altogether, these results suggest that cocaine triggers a synaptic potentiation by enhancing the AMPAR content at excitatory synapses onto LHb→RMTg neurons.
Figure 3.

Cocaine-evoked plasticity in LHb→RMTg neurons is postsynaptic. A, Confocal image of LHb neurons filled with Alexa Fluor Red-594 (left) and magnified dendrite (right). B, Sample mEPSCs and (L)-EPSCs. Note blockade of all events with NBQX. C, Mean values and scatter plot for mEPSC and (L)-EPSC amplitudes and decays (90–37%) in naive animals (n = 20). D, Averaged traces of (L)-EPSC at −60, 0, and +40 mV in slices from saline and cocaine-treated animals. E, Box and scatter plot for the AMPA/NMDA ratios in the saline and cocaine groups (n = 9–13 cells and 24–39 synapses; ***p < 0.001; t(61) = 5.3). F, G, (L)-AMPA-EPSC (L-AMPA) and l-NMDA-EPSC (L-NMDA) vs AMPA/NMDA ratio, scatter plot (empty circles) and mean values (full circles) (L-AMPA-EPSCs: saline 16.5 ± 1.12 pA; cocaine 20.7 ± 1; **p < 0.01; t(61) = 2.68; (L)-NMDA-EPSCs: saline 15.1 ± 1.6 pA; cocaine 12.8 ± 1.7; p > 0.05; t(61) = 0.96). Sal, Saline; Coc, cocaine.
Cocaine drives metaplasticity in the LHb
Hippocampal interneurons and DA neurons in the VTA undergo long-term synaptic changes through GluA2-lacking AMPAR-mediated calcium influx (Lamsa et al., 2007; Mameli et al., 2011). Given that cocaine increases the levels of AMPARs in LHb→RMTg neurons, we asked whether cocaine experience altered long-term synaptic plasticity rules. We tested whether synaptic activity modulated AMPAR-mediated transmission onto LHb→RMTg and LHb→VTA neurons. GluA2-lacking AMPARs conduct more efficiently at hyperpolarized potentials, therefore we used a synaptic protocol pairing presynaptic activity with postsynaptic hyperpolarization in presence of the NMDAR blocker APV (Fig. 4A,B) (Lamsa et al., 2007).
Figure 4.
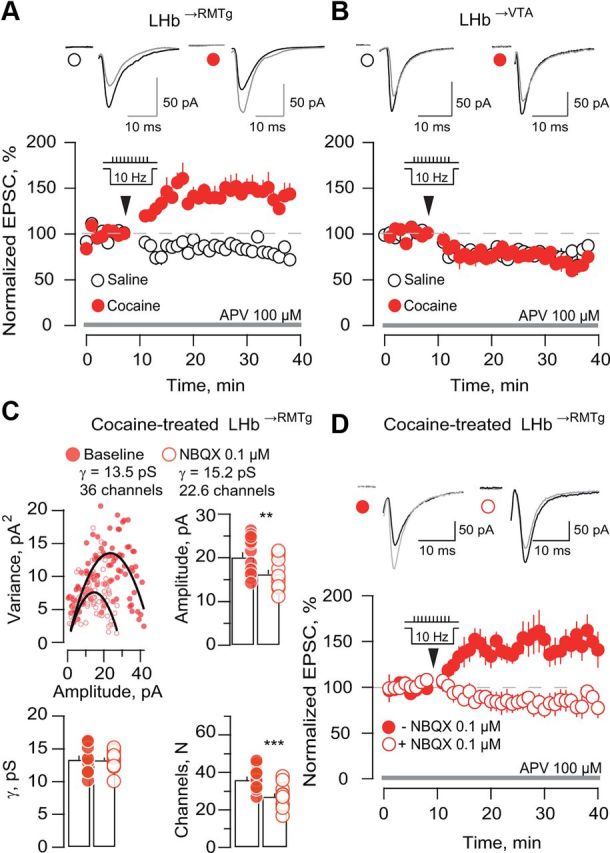
Cocaine evokes metaplasticity in LHb→RMTg neurons. A, Averaged traces and amplitudes versus time plot of AMPAR-EPSCs in LHb→RMTg neurons before and after conditioning protocol (arrow) in slices of saline and cocaine groups (n = 6–8; saline 86.6 ± 3.7%; cocaine 145.2 ± 11.8%; p < 0.01; t(12) = 4.1). B, Same as A but for LHb→VTA neurons (n = 5–6; saline 77.9 ± 4.1%; cocaine 78.2 ± 2.5%; p > 0.05; t(10) = 0.04). C, Example of peak-scaled NSFA of mEPSCs of an LHb→RMTg neuron before and after bath application of 0.1 μm NBQX in the cocaine group. Pooled data for amplitudes, conductances (γ), and number of channels (N) open at the peak of the response (n = 13; amplitude: baseline 20.1 ± 1.2 pA; NBQX 16.2 ± 1 pA; **p < 0.01; t(12) = 6.7; γ: baseline 13.3 ± 0.6 pS, NBQX 13.3 ± 0.4 pS; p > 0.5; t(12) = 0.1; N: baseline 36.1 ± 1.7; NBQX 27.2 ± 1.6; ***p < 0.001; t(12) = 6.6). D, Averaged sample traces and amplitude versus time plot of AMPAR-EPSCs from LHb→RMTg neurons before and after the conditioning protocol in slices from cocaine-exposed animals in the presence or absence of NBQX 0.1 μm (n = 7; control 149.3 ± 15.5%; NBQX 80.42 ± 9.9%; **p < 0.01; t(12) = 3.7).
In saline-treated mice, we found that a 10 Hz stimulation protocol induced a synaptic depression in both LHb→RMTg and LHb→VTA neurons (Fig. 4A,B). In contrast, after cocaine treatment, a robust LTP was observed specifically in LHb→RMTg neurons (Fig. 4A,B). To causally link the cocaine-evoked increase in quantal size (∼20%) and the metaplasticity in LHb→RMTg neurons, we used a submaximal concentration of NBQX (0.1 μm) that decreased mEPSC amplitude by ∼20%, mimicking AMPAR synaptic transmission in the control group (Fig. 4C). Peak-scaled NSFA of mEPSCs revealed that NBQX 0.1 μm decreased the number of channels opened at the peak without altering single-channel conductance (Fig. 4C). We then tested whether NBQX 0.1 μm could restore the normal pattern of synaptic plasticity in LHb→RMTg neurons. We found that the 10 Hz protocol in the presence of 0.1 μm NBQX in slices from cocaine-treated mice produced a synaptic depression similar to that observed in slices from saline-treated mice (Fig. 4D). The cocaine-mediated LTP was intact in interleaved slices in the absence of NBQX. Hence, cocaine-evoked potentiation enables metaplasticity in LHb→RMTg neurons.
Discussion
Using whole-cell recordings in conjunction with retrograde labeling techniques, we find that cocaine experience increases AMPAR-mediated transmission onto LHb→RMTg neurons by increasing the number of AMPARs at the postsynaptic level. These results indicate that cocaine profoundly alters the efficacy of excitatory transmission in LHb in a projection-specific fashion. This condition further allows presynaptic activity to trigger a form of LTP, absent in control animals, ultimately diverting the induction rules for long-term plasticity.
Our data suggest that the subunit composition of the synaptic AMPARs in LHb→RMTg neurons was not significantly changed by cocaine treatment. Recent electrophysiological evidence in rats indicates the presence of GluA2-lacking AMPARs in these neurons (Li et al., 2011). In agreement with this study, we found rectifying EPSCs in mouse slices, consistent with the expression of GluA2-lacking AMPARs. However, a residual current at positive potentials was still detected, suggesting that excitatory synapses onto LHb neurons likely contain GluA2-lacking and GluA2-containing AMPARs. Importantly, cocaine did not change the RI of LHb→RMTg neurons, indicating no alterations in AMPAR subunit composition. This differs from drug-evoked synaptic adaptations in the VTA or ventral striatum, where acute or chronic cocaine promotes the insertion of GluA2-lacking AMPARs at synapses initially devoid of these receptors (Bellone and Lüscher, 2006; Conrad et al., 2008).
NSFA provides further insight into the cellular mechanism behind cocaine potentiation, revealing a larger pool of AMPARs. Consistently, the uncaging experiments suggest that larger AMPA-EPSCs correlate with AMPA/NMDA ratios supporting the idea that synaptic strengthening relies, at least in part, on a higher number of postsynaptic AMPARs.
Abraham and Bear (1996) defined metaplasticity as the ability of presynaptic activity to induce subsequent synaptic plasticity as a consequence of synaptic or cellular adaptations (Abraham and Bear, 1996). We demonstrate that cocaine-evoked synaptic potentiation of LHb→RMTg neurons facilitates the induction of a form of metaplasticity absent in control conditions.
To demonstrate a causal relationship between cocaine-evoked synaptic strengthening and metaplasticity, we used low concentrations of NBQX to compensate for the cocaine-evoked synaptic potentiation. In cocaine-exposed mice, resetting synaptic transmission by reducing postsynaptic AMPAR availability is sufficient to restore the basal long-term synaptic plasticity. This evidence, together with larger quantal size, the increase in AMPA/NMDA ratios, and the results from NSFA after cocaine, suggest the presence of a larger fraction of AMPARs, including some lacking GluA2. This may provide a higher synaptic calcium source responsible for the induction of cocaine-induced metaplasticity onto LHb→RMTg neurons.
Future studies are nevertheless required to understand the cascade of events leading to this cocaine-evoked synaptic potentiation in LHb. In vivo evidence demonstrates that DA neuron activity and DA receptor activation modulate the firing of LHb neurons. This implies a potential role of DA signaling in triggering cocaine-evoked adaptations in the LHb (Kowski et al., 2009; Shen et al., 2012). In the VTA, cocaine triggers synaptic plasticity via its action on the DA transporter (Brown et al., 2010a). Therefore, cocaine may lead to a surge in DA in LHb, which may account for the cellular mechanism triggering the synaptic potentiation onto LHb neurons.
Anatomical studies suggest that the LHb innervation onto RMTg is the principal candidate for the net inhibition of DA neurons (Balcita-Pedicino et al., 2011; Stamatakis and Stuber, 2012). Consistently with this view, we show that cocaine-evoked synaptic potentiation is selective for LHb→RMTg neurons. From a neural circuit standpoint, the cocaine-evoked potentiation in LHb→RMTg neurons may result in a stronger inhibition of DA neurons via RMTg GABA neurons, in line with recent proposed models in which drugs increase LHb firing, adaptively dampening DA release (Baldwin et al., 2011). In this context, the modulation of the LHb-to-RMTg pathway as well of GABA inputs impinging on DA neurons is crucial for the processing of rewarding and aversive stimuli (Stamatakis and Stuber, 2012; Tan et al., 2012). Future studies are required to better probe how cocaine, reward, or aversive experiences alter specific populations of DA neurons via the LHb–RMTg–VTA path (Lammel et al., 2011).
In conclusion, we provide evidence for a synaptic potentiation in the LHb that may represent a key cellular mechanism engaged in the formation of drug-associated memories. Cellular adaptations in LHb may not only contribute to the early phases of drug exposure as reported here, but also in shaping drug-seeking behaviors (Brown et al., 2010b; James et al., 2011). We propose that cocaine may lead to behavioral adaptations by modifying synaptic efficacy and the rules for long-term plasticity in LHb, participating in the dysregulation of the DA system.
Footnotes
This work was supported by The Paris Neuroscience School (ENP), INSERM ATIP-AVENIR, and the City of Paris. We thank C. Bellone, M. Brown, M. Carta, F. J. Meye, C. Lüscher, C. F. Valenzuela, J. C. Poncer, and the whole Poncer-Levi Laboratory for helpful discussions and comments. We thank C. F. Valenzuela for assistance with English language editing, and N. Gervasi and L. Gonnard for image acquisition.
The authors declare no competing financial interests.
References
- Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- Balcita-Pedicino JJ, Omelchenko N, Bell R, Sesack SR. The inhibitory influence of the lateral habenula on midbrain dopamine cells: ultrastructural evidence for indirect mediation via the rostromedial mesopontine tegmental nucleus. J Comp Neurol. 2011;519:1143–1164. doi: 10.1002/cne.22561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin PR, Alanis R, Salas R. The role of the habenula in nicotine addiction. J Addict Res Ther. 2011;S1 doi: 10.4172/2155-6105.S1-002. 002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balland B, Lachamp P, Strube C, Kessler JP, Tell F. Glutamatergic synapses in the rat nucleus tractus solitarii develop by direct insertion of calcium-impermeable AMPA receptors and without activation of NMDA receptors. J Physiol. 2006;574:245–261. doi: 10.1113/jphysiol.2006.108738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone C, Lüscher C. Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci. 2006;9:636–641. doi: 10.1038/nn1682. [DOI] [PubMed] [Google Scholar]
- Brown MT, Bellone C, Mameli M, Labouèbe G, Bocklisch C, Balland B, Dahan L, Luján R, Deisseroth K, Lüscher C. Drug-driven AMPA receptor redistribution mimicked by selective dopamine neuron stimulation. PLoS One. 2010a;5:e15870. doi: 10.1371/journal.pone.0015870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RM, Short JL, Lawrence AJ. Identification of brain nuclei implicated in cocaine-primed reinstatement of conditioned place preference: a behaviour dissociable from sensitization. PLoS One. 2010b;5:e15889. doi: 10.1371/journal.pone.0015889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikosaka O. The habenula: from stress evasion to value-based decision-making. Nat Rev Neurosci. 2010;11:503–513. doi: 10.1038/nrn2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Jhou TC, Smith M, Saleem KS, Hikosaka O. Negative reward signals from the lateral habenula to dopamine neurons are mediated by rostromedial tegmental nucleus in primates. J Neurosci. 2011;31:11457–11471. doi: 10.1523/JNEUROSCI.1384-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James MH, Charnley JL, Flynn JR, Smith DW, Dayas CV. Propensity to “relapse” following exposure to cocaine cues is associated with the recruitment of specific thalamic and epithalamic nuclei. Neuroscience. 2011;199:235–242. doi: 10.1016/j.neuroscience.2011.09.047. [DOI] [PubMed] [Google Scholar]
- Jhou TC, Geisler S, Marinelli M, Degarmo BA, Zahm DS. The mesopontine rostromedial tegmental nucleus: a structure targeted by the lateral habenula that projects to the ventral tegmental area of Tsai and substantia nigra compacta. J Comp Neurol. 2009;513:566–596. doi: 10.1002/cne.21891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufling J, Veinante P, Pawlowski SA, Freund-Mercier MJ, Barrot M. Afferents to the GABAergic tail of the ventral tegmental area in the rat. J Comp Neurol. 2009;513:597–621. doi: 10.1002/cne.21983. [DOI] [PubMed] [Google Scholar]
- Kaufling J, Waltisperger E, Bourdy R, Valera A, Veinante P, Freund-Mercier MJ, Barrot M. Pharmacological recruitment of the GABAergic tail of the ventral tegmental area by acute drug exposure. Br J Pharmacol. 2010;161:1677–1691. doi: 10.1111/j.1476-5381.2010.00984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowski AB, Veh RW, Weiss T. Dopaminergic activation excites rat lateral habenular neurons in vivo. Neuroscience. 2009;161:1154–1165. doi: 10.1016/j.neuroscience.2009.04.026. [DOI] [PubMed] [Google Scholar]
- Lammel S, Ion DI, Roeper J, Malenka RC. Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron. 2011;70:855–862. doi: 10.1016/j.neuron.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamsa KP, Heeroma JH, Somogyi P, Rusakov DA, Kullmann DM. Anti-Hebbian long-term potentiation in the hippocampal feedback inhibitory circuit. Science. 2007;315:1262–1266. doi: 10.1126/science.1137450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecca S, Melis M, Luchicchi A, Ennas MG, Castelli MP, Muntoni AL, Pistis M. Effects of drugs of abuse on putative rostromedial tegmental neurons, inhibitory afferents to midbrain dopamine cells. Neuropsychopharmacology. 2011;36:589–602. doi: 10.1038/npp.2010.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Piriz J, Mirrione M, Chung C, Proulx CD, Schulz D, Henn F, Malinow R. Synaptic potentiation onto habenula neurons in the learned helplessness model of depression. Nature. 2011;470:535–539. doi: 10.1038/nature09742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–663. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mameli M, Bellone C, Brown MT, Lüscher C. Cocaine inverts rules for synaptic plasticity of glutamate transmission in the ventral tegmental area. Nat Neurosci. 2011;14:414–416. doi: 10.1038/nn.2763. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Hikosaka O. Lateral habenula as a source of negative reward signals in dopamine neurons. Nature. 2007;447:1111–1115. doi: 10.1038/nature05860. [DOI] [PubMed] [Google Scholar]
- Shen X, Ruan X, Zhao H. Stimulation of midbrain dopaminergic structures modifies firing rates of rat lateral habenula neurons. PLoS One. 2012;7:e34323. doi: 10.1371/journal.pone.0034323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis AM, Stuber GD. Activation of lateral habenula inputs to the ventral midbrain promotes behavioral avoidance. Nat Neurosci. 2012;15:1105–1107. doi: 10.1038/nn.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan KR, Yvon C, Turiault M, Mirzabekov JJ, Doehner J, Labouèbe G, Deisseroth K, Tye KM, Lüscher C. GABA neurons of the VTA drive conditioned place aversion. Neuron. 2012;73:1173–1183. doi: 10.1016/j.neuron.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci. 2004;5:483–494. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]