

Abstract
Although sleep is defined as a behavioral state, at the cortical level sleep has local and use-dependent features suggesting that it is a property of neuronal assemblies requiring sleep in function of the activation experienced during prior wakefulness. Here we show that mature cortical cultured neurons display a default state characterized by synchronized burst–pause firing activity reminiscent of sleep. This default sleep-like state can be changed to transient tonic firing reminiscent of wakefulness when cultures are stimulated with a mixture of waking neurotransmitters and spontaneously returns to sleep-like state. In addition to electrophysiological similarities, the transcriptome of stimulated cultures strikingly resembles the cortical transcriptome of sleep-deprived mice, and plastic changes as reflected by AMPA receptors phosphorylation are also similar. We used our in vitro model and sleep-deprived animals to map the metabolic pathways activated by waking. Only a few metabolic pathways were identified, including glycolysis, aminoacid, and lipids. Unexpectedly large increases in lysolipids were found both in vivo after sleep deprivation and in vitro after stimulation, strongly suggesting that sleep might play a major role in reestablishing the neuronal membrane homeostasis. With our in vitro model, the cellular and molecular consequences of sleep and wakefulness can now be investigated in a dish.
Introduction
Sleep is believed to be a period during which restorative processes occur. However, the nature and mechanisms underlying restoration during sleep are completely unknown. Evidence for sleep's restorative function comes from the observation that sleep is homeostatically regulated, with longer periods of wakefulness being followed by deeper and/or longer sleep (Daan et al., 1984).
In birds and mammals, the gold standard to determine sleep is polysomnography, which combines measures of the EEG, EMG, and EOG. Based on these signals, two states can be determined [rapid eye movement sleep (REMS) and non-REMS (NREMS)] that alternate in cyclic fashion. Obviously, in species lacking a fully developed cortex, eye movements, or changes in muscle tone, alternative methods have been developed to quantify sleep (Campbell and Tobler, 1984) and sleep-like states have also been observed in organisms such as worms and fruit flies (Hendricks et al., 2000; Shaw et al., 2000; Raizen et al., 2008), challenging the classic view that sleep is an emerging property of a complex brain such as that of birds and mammals.
EEG analyses in mammals revealed that recovery occurs locally and in a use-dependent manner (Krueger and Obál, 1993; Krueger et al., 2008). This local aspect of sleep was, however, already suggested after the observation of sleep- and wake-like states in isolated cortical islands (Kristiansen and Courtois, 1949). Stimulation of specific brain regions (e.g., vibration of one hand in humans or unilateral stimulation of vibrissae in rodents) results in an increased intensity of sleep in the corresponding cortical area (Kattler et al., 1994; Vyazovskiy et al., 2000). It has also been shown that local sleep can occur in waking animals (Pigarev et al., 1997; Vyazovskiy et al., 2011). It has been suggested that cortical columns represent a basic neural unit capable of oscillating between the states of sleep and wakefulness (Rector et al., 2005).
Other measurable correlates of sleep and wakefulness are the consistent changes in gene expression and their effects on metabolism (Cirelli et al., 2004; Rector et al., 2005; Mackiewicz et al., 2007; Maret et al., 2007). Many genes induced during wakefulness are involved in neuronal plasticity, while genes upregulated during sleep are involved in macromolecule biosynthesis and metabolism (Mackiewicz et al., 2007). Although changes in metabolic activities were reported in gene expression studies, no systematic metabolomic analysis is yet available.
Sleep, as most other behaviors and physiological processes, undergoes a circadian rhythmicity. Similarly to the circadian process, which is a self-sustained mechanism present in all cells, sleep might also be a self-sustained recovery process that occurs at the cellular level. This basic property cannot be investigated in vivo because of the complexity of the mammalian brain, in which neurons are interconnected and receive both local and global chemical and electrical inputs. Here we demonstrate for the first time that sleep is a property of simple neuronal networks in vitro, paving the way to investigate this complex behavior at cellular and molecular levels.
Materials and Methods
Primary cortical culture.
Dissociated cortical primary cultures were prepared from C57BL/6J mouse brains either at birth or at embryonic day 16–18 (E16–E18; of either sex). Briefly, cortical tissues from pups or embryos were dissected in a solution of phosphate buffer containing HEPES, 33 mm glucose, and 40 mm sucrose. The isolated cortices were digested with a solution containing 50 U of Papain for 30 min at 37°C. Digestion was stopped by the addition of trypsin inhibitor for 10 min. Cells were dissociated mechanically through a fire-polished glass pipette several times in neurobasal medium supplemented with 2% B-27, 0.5 mm Glutamax, and penicillin/streptomycin. Cells were plated either on 35 mm dishes (at 1.5 × 106 cells; density = 1560 cells/mm2) precoated with 0.1 mg/ml poly-L-lysine or on 60-channel microelectrode arrays (MEAs; Multichannel Systems) precoated with 0.1% polyethyleneimine plus 20 μg/ml laminin (same number of cells, resulting in a density of 4780 cells/mm2). Cultures were maintained in a humidified CO2 incubator (5% CO2, 37°C) and half of the medium was changed once a week. All experimental procedure were conducted in accordance with regulatory standards and approved by the Vaud Veterinary Office, Switzerland.
Quantitative PCR, microarrays expression profiling, and data analysis.
Total RNA from male mice cortex for the microarray experiment was isolated with the RNeasy Lipid Tissue Midi kit (Quiagen). Total RNA from cell cultures and from mice cortex for the time course experiment were extracted using RNA easy mini kit (Quiagen). All RNA samples were DNase-treated and quantified on a NanoDrop ND-1000 spectrophotometer. To quantify the RNA expression level, 500 ng of RNA were reverse-transcribed in 20 μl using random hexamers and Superscript II reverse transcriptase (Invitrogen) according to standard procedures. The cDNA was diluted 10 times and 2 μl were amplified in a 10 μl TaqMan reaction on ABI PRISM HT 7900 detection system in technical triplicate. Cycler conditions were 50°C 2 min, 95°C 10 min, and 45 cycles at 95°C 15 s and 60°C 1 min. Forward primer, reverse primer, and probe sequences are available on request. The gene expression level was normalized to three reference genes (Rsp9, TBP, and EEF1a1) using Qbase software (Hellemans et al., 2007). The fold changes indicative of the relative gene expression are based on the mean of three biological replicates in relation to control samples.
For microarray analysis, RNA was extracted from the cortex of 27 male C57BL/6J mice: nine sleep-deprived for 6 h, nine home-cage controls, and nine after 24 h of recovery from a 6 h sleep deprivation. RNA was also extracted from three dishes of three different cultures 3 h after stimulation with an excitatory cocktail, 3 h after sham stimulation with water, and 24 h after stimulation. The RNA quality was assessed on the Agilent 2100 bioanalyzer chips. Three pools of three samples were prepared for analysis. RNA (100 ng) was used to perform target preparation using WT Expression Kit (Ambion). Next, 5.5 μg of each fragmented cDNA was end-labeled with biotin and hybridized to a Mouse Exon 1.0 ST array as per manufacturer's instructions, then processed and scanned according to standard procedures. Expression signals were normalized separately for in vitro and in vivo datasets at the genes level from Affymetrix CEL files using RMA normalization method implemented in the Affymetrix Expression Console software. Data have been deposited in NCBIs Gene Expression Omnibus (GEO; www.ncbi.nlm.nih.gov/geo) and are accessible through GEO Series accession number GSE33491.
All subsequent statistical analyses were performed using R (http://www.R-project.org) (R Development Core Team, 2004) and Bioconductor packages (http://www.Bioconductor.org). To identify sleep responsive genes, we used limma (Smyth, 2004) and applied a linear model with three coefficients: an intercept, the stimulated (or sleep-deprived) condition versus the control condition (SvsC), and the stimulated (or sleep-deprived) condition versus the recovery condition (SvsR). We extracted a contrast that averages SvsC and SvsR to obtain one statistic for the sleep effect. The top 200 most significant probe sets in each dataset were used as sleep signature. The signatures were split in upregulated and downregulated probe sets. To test whether the sleep signature probe sets were also affected in the other dataset, a two-tailed Mann–Whitney U test was performed to check whether the upregulated (downregulated) sleep signature probe sets from one dataset were also upregulated (downregulated) in the ranked ordered limma moderated t statistics of the other dataset. To identify genes affected in both datasets, a meta-analytical approach was used in which the p values were transformed into Z scores, taking into account the sign of the t statistics. Z scores from the two different datasets were combined as described previously (Wirapati et al., 2008). p values calculated from the meta-analysis were adjusted using Bonferroni corrections, and adjusted p values <0.1 were considered significant.
MEA recording.
Electrophysiological signals were acquired using the complete MEA60-BC system (Multi Channel Systems). The set-up consists of a MEA 1060-Inv-BC amplifier integrating 60-channel and filter amplifiers with a bandwidth of 0.1 Hz–10 KHz and a gain of 1100×. The set-up was connected to a computer equipped with a PCI data acquisition board and with the commercial software MCRack (Multichannel Systems) to record and analyze raw data. Primary cultures were seeded on standard MEA60 biosensors composed of 59 planar TiN/SiN microelectrodes (30 μm diameter, 200 μm interelectrode distances) plus one internal reference electrode. Recordings from MEA60 were performed in a humidified CO2 incubator immediately after transferring each culture into the recording device.
MEA data analysis.
Each channel was sampled at a frequency of 25 kHz and raw data were high-pass filtered at 200 Hz and low-pass filtered at 2 kHz. Spikes were sorted from the noise (10 μV) using the threshold tool of the MCRack software calculated as a multiple of the standard deviation (7*SD) of the biological noise for each recording channel (Chiappalone et al., 2006). Sorted spikes were processed by using a personal developed software tool in Matlab environment calculating the array-wide synchronized electrodes as the sum of the electrodes that fired together into 100 ms bins over each 5 min recordings.
To quantify the bursting activity, the recordings were divided in 1 s bins and the rates of spikes were counted to determine the fraction of spikes containing the 15% most active bins (f15). We systematically varied the epoch duration and the 1 s bin was found to best capture bursting activity, as previously reported (Wagenaar et al., 2005). The burstiness index (BI) was defined as BI = (f15 − 0.15)/0.85, as previously described (Wagenaar et al., 2005). Bursts were sorted using the burst detection tool from the MCRack software with parameters fixed at 60 ms for the maximal interval between spikes in a burst, 20 ms for the minimal duration of a burst, and a minimum of three consecutive spikes belonging to a burst.
Synaptoneurosome preparation.
Frozen cortical mouse samples (half hemisphere or 8–10 millions of cultured cells) were homogenized in a tissue homogenizer (Pyrex) with 500 μl of ice-cold homogenization buffer [10 mm HEPES, 1 mm EDTA, 2 mm EGTA, 0.5 mm DTT, 0.1 mm PMSF, 1 pellet of proteases inhibitor (Roche), 100 mm NaF, 100 mm Na3VO4]. Homogenates were passed through two 100-μm-pore nylon mesh (Small Parts) and an aliquot of the flow-through was preserved as the total fraction and stored at −20°C. The rest was passed through a 5-μm-pore filter (Durapore; Millipore) twice before centrifugation at 10,000 g for 10 min at 4°C. Pellets were resuspended in homogenization buffer with 1% SDS and stored at −20°C. Protein concentration was determined by BCA assay (Pierce).
Quantitative immunoblotting.
Twenty or 50 μg of synaptoneurosomes from each animal or from each culture sample, respectively, were spiked with nonimmune chicken IgY (40 ng/lane) before separation by SDS-PAGE in Tris/glycine/SDS buffer, transferred on PVDF membranes in Tris base/glycine/methanol blotting buffer, and blocked 1 h in Tween/Tris-buffered saline supplemented with 5% nonfat milk and 1 mm NaF (to preserve serine-threonine phosphorylation). Membranes were immunoblotted with rabbit polyclonal anti-GluR1-phospho S845 antibody (Abcam) followed by horseradish peroxidase-conjugated goat anti-rabbit antibody (Dako). Blots were also reprobed with horseradish peroxidase-conjugated anti-chicken IgY antibody as loading control. The secondary antibodies were revealed using ECL-prime reagents and Hyperfilm MP (GE Healthcare) before scanning and analysis by ImageJ (Gel Analysis tool).
Real-time PER2::luciferase expression.
Bioluminescence was continuously recorded in a light-tight incubator (Actimetrics) using photomultiplier tube detector assemblies. Total photons emitted by PER2::Luciferase (Luc) protein were integrated over 10 min intervals. Background from empty dishes was subtracted and bioluminescence for each condition was counted in triplicates before being averaged. Periods and amplitude of the signals were measured using Lumicycle Analysis software (Actimetrics). Period calculation was determined for every well between the first two minima in bioluminescence after adding the cocktail. Effects on circadian phase after the second stimulation were assessed by determining the time of the first maximum reached in stimulated cultures versus control cultures (water). Phase shifts were expressed in circadian hours.
In vivo calorimetry.
Metabolic measurement was performed using an Oxymax indirect calorimetry system (Columbus Instruments). Twelve C57BL/6J mice (10 weeks old) were maintained for 4 nights and 3 d in individual calorimetric chambers with a 12 h light/12 h dark cycle in an ambient temperature of 22–24°C. VO2 and VCO2 rates were determined under Oxymax system settings as follows: air flow, 0.4 L/min; sample flow, 0.6 L/min; settling time, 90 s; and measuring time, 1 min (with zirconia O2 probe). The system was calibrated against a standard gas mixture to measure O2 consumed (VO2, ml/kg/h) and metabolic rate (VO2) was evaluated over the entire period.
Seahorse experiment.
Primary cortical neurons were seeded at 200,000 cells/well in XF24 cell culture microplates (Seahorse Bioscience) and maintained for 12 d before analysis. Oxygen consumption was measured using the Seahorse XF24 equipment under basal conditions, followed by injection of neurotransmitter mixture in neurobasal medium or of neurobasal medium alone as control and continued measurement for 2 h.
Metabolome.
Eight C57BL/6J were sleep deprived for 6 h while eight others were left undisturbed in their home cage. Mice were killed by cervical elongation and brains quickly removed and stored at −80°. The cortex was dissected from frozen brains for metabolome analysis. Cell cultures were stimulated with our excitatory cocktail or sham stimulated with water and harvested 3 h later. Three culture dishes from three different cultures were used in each condition, resulting in nine by nine comparisons. Metabolomic profiling analysis was performed by Metabolon as previously described (Reitman et al., 2011). All samples were maintained at −80°C until processed. Samples were prepared using the automated MicroLab STAR system from Hamilton. A recovery standard was added before the first step in the extraction process for quality control (QC) purposes. Sample preparation was conducted using aqueous methanol extraction process to remove the protein fraction while allowing maximum recovery of small molecules. The resulting extract was divided into four fractions: one for analysis by ultra-performance liquid chromatography (UPLC)/mass spectrometry (MS)/MS (positive mode), one for UPLC/MS/MS (negative mode), one for gas chromatography (GC)/MS, and one for backup. Samples were placed briefly on a TurboVap (Zymark) to remove the organic solvent. Each sample was then frozen and dried under vacuum. Samples were then prepared for the appropriate instrument, either UPLC/MS/MS or GC/MS. The liquid chromatography (LC)/MS portion of the platform was based on a Waters ACQUITY UPLC and a Thermo-Finnigan linear trap quadrupole mass spectrometer, which consisted of an electrospray ionization source and linear ion-trap mass analyzer. The sample extract was dried, then reconstituted in acidic or basic LC-compatible solvents, each of which contained eight or more injection standards at fixed concentrations to ensure injection and chromatographic consistency. One aliquot was analyzed using acidic positive ion optimized conditions and the other using basic negative ion optimized conditions in two independent injections using separate dedicated columns. Extracts reconstituted in acidic conditions were gradient eluted using water and methanol containing 0.1% formic acid, while the basic extracts, which also used water/methanol, contained 6.5 mm ammonium bicarbonate. The MS analysis alternated between MS and data-dependent MS2 scans using dynamic exclusion. Raw data files were archived and extracted as described below.
The samples destined for GC/MS analysis were redried under vacuum desiccation for a minimum of 24 h before being derivatized under dried nitrogen using bistrimethyl-silyl-triflouroacetamide. The GC column was 5% phenyl and the temperature ramp was from 40° to 300°C in a 16 min period. Samples were analyzed on a Thermo-Finnigan Trace DSQ fast-scanning single-quadrupole mass spectrometer using electron impact ionization. The instrument was tuned and calibrated for mass resolution and mass accuracy on a daily basis. The information output from the raw data files was automatically extracted, as discussed below.
For QA/QC purposes, additional samples were included with each day's analysis. These samples included extracts of a pool of well characterized human plasma, extracts of a pool created from a small aliquot of the experimental samples, and process blanks. QC samples were spaced evenly among the injections and all experimental samples were randomly distributed throughout the run. A selection of QC compounds was added to every sample for chromatographic alignment, including those under test. These compounds were carefully chosen so as not to interfere with the measurement of the endogenous compounds.
Raw data were extracted, peak-identified, and QC processed using Metabolon's hardware and software. These systems are built on a web service platform using Microsoft's .NET technologies, which run on high-performance application servers and fiber-channel storage arrays in clusters to provide active failover and load balancing. Compounds were identified by comparison to library entries of purified standards or recurrent unknown entities. More than 2400 commercially available purified standard compounds have been acquired and registered into the Laboratory Information System for distribution to both the LC and GC platforms for determination of their analytical characteristics. Raw data from cortex extracts were normalized to the median value for each biochemical and analyzed by a linear model with two factors: intercept and condition (sleep-deprived and non-sleep-deprived). Raw data from cell extracts were normalized first to the amount of proteins, then to the mean value of control samples for each of the three cultures and analyzed by a linear model with three factors: intercept, condition (cocktail vs sham stimulation), and series (cultures).
Results
Sleep as a default state of mature neuronal assemblies in vitro
Intracellular recordings from cortical neurons in cats identified a slow oscillation, typically within 0.16–0.3 Hz, in addition to delta oscillations (1–4 Hz) (Steriade et al., 1993), a finding replicated in humans at the EEG level (Achermann and Borbély, 1997). Cortical cultures have been used previously to study firing properties of cortical networks and MEA recordings revealed spontaneous synchronized bursting across culture resembling slow-wave sleep (Van Pelt et al., 2004; Corner, 2008; Corner et al., 2008). To verify the activity of cortical neurons in vitro, embryonic dissociated cortical neurons were cultured on MEAs and their firing activity was recorded over the course of maturation (Fig. 1). At 7 d in vitro (DIV), cortical cells started firing randomly. This pattern progressively changed into an array-wide synchronized burst–pause firing pattern from 10 DIV on (Fig. 1D). This bursting activity, quantified as a BI (Wagenaar et al., 2005), increased progressively from 6 to 10 DIV and remained stable thereafter (Fig. 1E). This synchronous, low-frequency (0.07–0.2 Hz) firing pattern is highly similar to the in vivo slow cortical oscillations recorded in sleeping animals (Steriade et al., 1993), suggesting that by 10 DIV, cultured cortical neurons are spontaneously in a sleep-like state. In the absence of excitatory input, this state seems to be the default state of simple neuronal assemblies in vitro.
Figure 1.
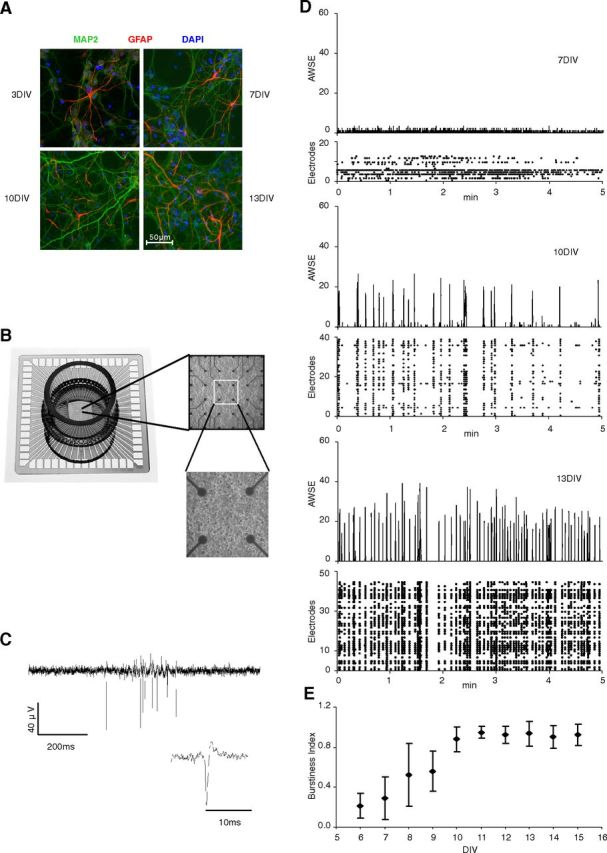
The sleep-like state emerges after 10 DIV. A, The structural organization of cortical cultures at different time points shows a progressive establishment of complex interconnected neuronal networks. Neurons are stained with anti-MAP2 (red), astrocytes with anti-GFAP (green), and cell nuclei with DAPI (blue). Note that cultures are enriched in neurons (86–94% neurons and 1–14% astrocytes from 3–13 DIV). B, The MEA used for recording firing activity is composed of 60 electrodes arranged on a culture surface. C, Typical bursting activity recorded from one electrode with an expanded illustration of a single spike (amplitude scale bar applies to both panels). D, Five-minute MEA recordings performed from 6 DIV onwards at daily intervals (7, 10, and 13 DIV shown) show initial random activity (7 DIV), which becomes progressively synchronized throughout the array by 10 DIV. For each DIV, the bottom panel (Electrodes) depicts timing of firing at individual electrodes. The top panel shows the number of electrodes for which simultaneous firing activity was observed. AWSE, Array-wide synchronous electrodes. E, Synchronized burst–pause activity, quantified with the burstiness index (with an index of 1 indicating synchronized firing over all recorded electrodes; see Materials and Methods), increases until 10 DIV, after which a plateau is reached, representing the prevailing default firing mode in mature cortical cultures. Values for each DIV represent means ±1 SD of at least three different cultures. Note that not all 60 electrodes of each MEA detect firing activity, but that their number increases with the maturation of cortical cultures.
Stimulation of dissociated cortical cultures dose-dependently induces a transient wake-like state
To change the synchronized default sleep-like state into an active wake-like state, cortical cultures were stimulated with various excitatory neurotransmitters or their agonists: NMDA, kainate, AMPA, ibotenic acid (glutamatergic), carbachol (mixed cholinergic), norepinephrine, serotonin, histamine, dopamine, and orexin A (hypocretin-1), either at literature-reported concentrations or based on dose–response curves generated by ourselves (data not shown). We used changes in expression of the candidate genes Homer1a, Arc, and Dbp as initial read-outs because these transcripts reliably change in function of extended wakefulness in vivo (Maret et al., 2007). None of the neurotransmitters alone reproduced a pattern of gene expression similar to that observed in vivo (data not shown). After testing several mixtures, we decided on one containing all major excitatory neurotransmitters known to be upregulated with wakefulness (1 μm NMDA, AMPA, kainate, ibotenic acid, serotonin, histamine, dopamine and noradrenaline; 10 μm carbachol; and 0.01 μm orexin). Since we noticed that washing the cultures to stop stimulation is invariably accompanied by a strong stimulation, the mixture was added and the cultures left undisturbed for the rest of the experiment.
Embryonic cultures between 12 and 14 DIV were sham-stimulated (H2O) or stimulated with our mixture and recorded for 5 min before stimulation; 5 min after stimulation; and 1-, 3-, 6-, or 24 h after stimulation. Sham stimulation did not change the burst–pause firing pattern of the culture over the experiment, whereas mixture stimulation induced a tonic firing that progressively replaced the burst–pause firing pattern. Six hours after stimulation, no or only a few bursts could be detected; 24 h after stimulation (i.e., at the same time of day time as the onset of stimulation), all cultures reverted to the default burst–pause synchronized firing (Fig. 2A). To characterize the stimulation-induced changes in neuronal firing, detailed analyses were performed on recorded spiking and bursting activities. Mean number, percentage, and frequency of spikes per burst were found to be the most affected measures in our stimulation. These results indicate that bursting activity, quantified as BI, best characterizes changes in firing induced by stimulation. Mature cultures showed the highest BI levels, which almost completely disappeared 6 h after stimulation, then reappeared 24 h poststimulation (Fig. 2B). Nevertheless, the synchronous burst–pause activity after recovery was not identical to the resting activity; e.g., bursting activity was faster, suggesting either that plastic changes occurred during and/or following neuronal stimulation or that the sleep-like state increased in intensity (see below).
Figure 2.
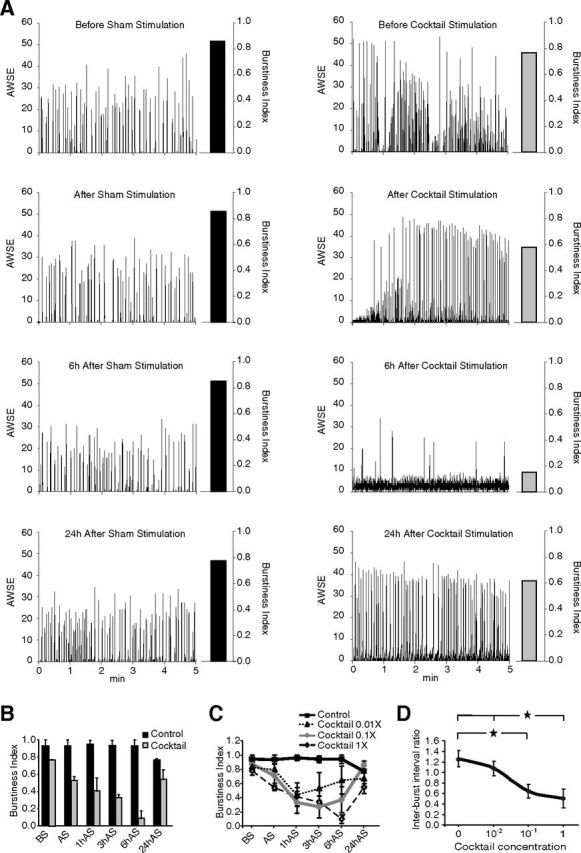
The sleep-like state can be changed into active wake-like state. A, Mature cortical cultures (11–13 DIV) were stimulated with either distilled water (Sham, left) or the neurotransmitter cocktail (right). Typical 5 min MEA recordings before (BS), immediately after (AS), 6 h after (6hAS), and 24 h after (24hAS) the stimulation are shown. Stimulation completely abolished the typical burst–pause activity 6 h after stimulation and reappeared 24 h later. Within each panel, the array-wide synchronous electrodes (AWSE) and corresponding burstiness index (vertical bar) for the 5 min recordings are indicated (see Fig. 1 for details). B, Mean burstiness index (+1 SD; n = 3+) in the sham and cocktail-stimulated conditions was calculated in the 5 min before and 5 min and 1, 3, 6, and 24 h after the stimulation. The stimulation significantly changed bursting pattern (two-way ANOVA with factors: condition, F(1,27) = 69.95, p < 10−8; time, F(5,27) = 3.56, p < 0.02; and interaction, F(5,27) = 5.04, p < 0.003). C, Cortical cultures stimulated with different concentrations of the neurotransmitter cocktail (0.01×, 0.1×, and 1×; n = 3, mean ± 1 SD) show changes in bursting activity in a dose-dependent manner (two-way ANOVA with factors: dose, F(3,45) = 29.20, p < 10−10; time, F(5,45) = 9.58, p < 3 × 10−6; and interaction, F(15,45) = 3.12, p < 0.002). Note that minimum bursting activity is reached later (i.e., 1, 3, and 6 h after stimulation) with increasing concentration and that values revert to control levels thereafter. D, Interburst intervals are shorter after stimulation with different concentrations of the neurotransmitter cocktail (0×, 0.01×, 0.1×, and 1×; n = 3, mean ± 1 SD) compared with baseline (before stimulation) in a dose-dependent manner (ANOVA: F(3,8) = 12.4, p < 0.003; *p < 0.05, post hoc Tukey test).
Sleep need is proportional to the preceding duration and/or intensity of wakefulness (sleep homeostasis). To mimic the time course of changes in sleep need, cortical cultures were stimulated with 0.01×, 0.1×, and 1× concentration of our neurotransmitter mixture and recorded 1, 3, 6, and 24 h later. MEA recordings showed that burst–pause firing activity (sleep-like state) changed proportionally to the intensity of stimulation. The BI after the lowest concentration showed a later decrease and faster recovery while that of higher concentrations showed faster decrease and later recovery after stimulation (Fig. 2C). Carbachol-induced prolonged tonic firing in cortical cultures, similar to our stimulation with the waking cocktail, was followed by enhanced frequency of spontaneous burst–pause firing (Corner, 2010). This observation was interpreted as an intensification of synchronized firing (Corner, 2010). Similarly, prolonged wakefulness was followed by a more frequent alternation between up- and down-states (burst–pause) in the slow oscillations in the human NREMS EEG, again indicating higher synchrony (Bersagliere and Achermann, 2010). The frequency of up- and down-state alternation could thus index a homeostatically regulated process. To verify whether periods of tonic firing were also followed by increased synchrony, we quantified and compared interburst interval (i.e., down-state) and burst frequency before and after stimulation. The ratio between interburst intervals after and before sham, 0.01×, 0.1×, and 1× stimulations dose-dependently decreased (Fig. 2D), strongly suggesting that more intense stimulation results in more intense synchronization. This was also evidenced in changes in burst frequency (0.07 ± 0.01 vs 0.07 ± 0.01 Hz before and after sham stimulation and 0.06 ± 0.03 vs 0.17 ± 0.08 Hz before and after 1× stimulation, p = 0.065).
Stimulation of cortical cultures induces similar changes in gene expression as sleep deprivation in living animals
Brain gene expression experiments in living animals indicate that distinct classes of genes are differentially expressed during sleep and wakefulness (Pompeiano et al., 1994, 1997; Cirelli et al., 1996, 2004, 2006; Cirelli and Tononi, 1999, 2000a,b; Wisor et al., 2002; Terao et al., 2003; Mackiewicz et al., 2007; Maret et al., 2007; Mongrain et al., 2010). Many of the genes, overexpressed by spontaneous or enforced wakefulness relative to sleep, are immediate early genes (IEG). We have shown that among these, Homer1a expression is the most reliable marker for sleep need (Maret et al., 2007). We thus aimed at verifying whether the sleep- and wake-like states identified by MEA recordings in culture are associated with similar transcriptome changes as observed in vivo.
For the in vivo transcriptome analyses, mice were sleep deprived for 6 h and brains collected immediately afterward. For the in vitro analyses, cortical cultures were stimulated with the 1× cocktail and RNAs extracted 3 h afterward. Control mice were kept undisturbed in their home cage and control cultures were sham-stimulated with water. Another set of sleep-deprived mice and stimulated cultures were allowed to recover before being sampled the next day at the time of day at which sleep deprivation or stimulation ended the day before. Gene expression profiling was performed on the cortex of sleep-deprived, control, and recovered mice and on stimulated, control, and recovered cortical cultures. The effect of loss of sleep on gene expression was calculated as the average effect of two comparisons: stimulation (or sleep deprivation) versus control and stimulation (or sleep deprivation) versus recovery. As previously reported (Maret et al., 2007), two-thirds of changes concerned upregulated transcripts and one-third concerned downregulated genes after sleep deprivation or cell stimulation. The in vitro and in vivo expression signatures for genes upregulated and downregulated were cross-validated in the other dataset to determine whether a significant overlap between the two was present (Fig. 3A). Genes similarly affected in vitro and in vivo were then identified using meta-analysis (Fig. 3B). As expected, Homer1a, Arc, and Dbp were among the most significantly affected genes, but other IEGs such as Per1, Per2, Egr2, Fosl2, and Fos were all overexpressed by sleep deprivation, as previously reported (see above) (Maret et al., 2007), as well as by stimulation in cell cultures. Additionally, overexpressed probe sets encompass genes involved in glucose metabolism such as Slc2a1 and -3 (facilitated glucose transporters 1 and 3), and Ppapdc2 (a phosphatidic acid phosphatase) and in lipid metabolism such as Chka (choline kinase A). Increased expression of cell cycle inhibitors Cdkn1a, Gadd45b, and apoptosis regulator Lats2 suggests a negative effect of enforced waking and cell stimulation on cell survival. Although many IEGs were found overexpressed, several serine-threonine protein kinases (Dusp1 and 4) that inhibit MAPKs in ERK pathways were also overexpressed, probably buffering the extensive increase in IEG expression. Additionally, Pak7 (a serine threonine protein kinase involved in proliferation and cell survival) was significantly downregulated, as was Oxsm (3-oxoacyl-acyl-carrier-protein synthase), which is involved in lipoic and long-chain fatty acids biosynthesis in mitochondria.
Figure 3.
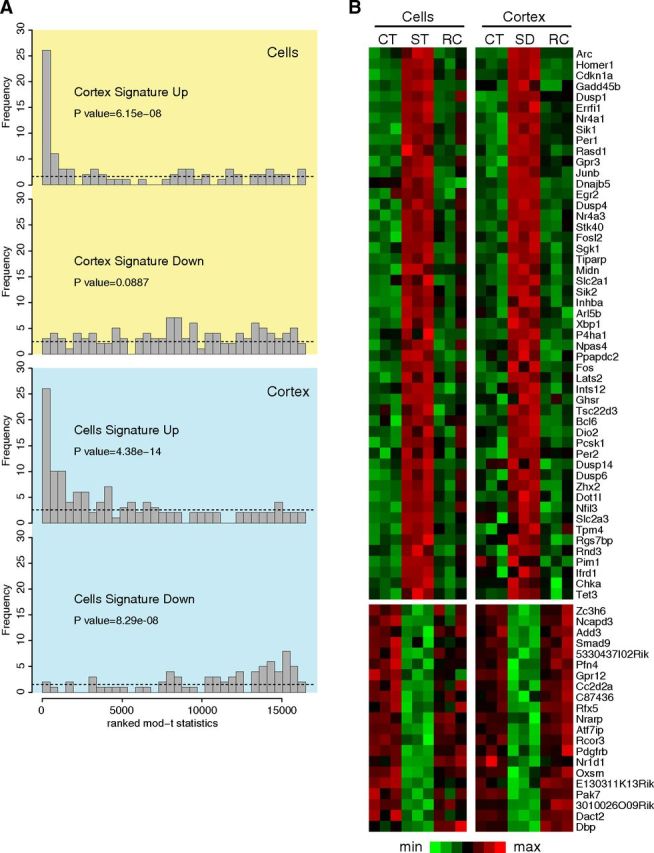
Gene expression signatures in vitro and in vivo. A, Probe sets were ranked by their moderated t statistic and binned into 34 bins. The bins were ordered from the highest t statistic (left) to the lowest t statistic (right); the yellow box is from the in vitro dataset and in the blue box is from the in vivo dataset. The number of upregulated (or downregulated) probe sets from the sleep signature of the other condition dataset is displayed in each bin (the height of the bar). Under the assumption that the transcription signature of one dataset is not differentially regulated in the other dataset, probe sets from the signature should be uniformly distributed among bins, as indicated by the dashed line. The peaks on the left show that the upregulated sleep signature of one dataset tend to be upregulated in the other dataset. p values from the Mann–Whitney U test are indicated. B, Heat map with genes identified as sleep-responsive genes both in cortex and in cell cultures. Expression data were scaled (mean-centered and variance normalized) for each dataset separately. CT, Control; ST, stimulated; SD, sleep-deprived; RC, recovery. Genes are ordered by Z scores.
To verify that the pattern of gene expression associated with the in vitro-induced wake-like state varies in function of stimulation strength, cortical cultures were stimulated with 0.001×, 0.01×, 0.1×, and 1× concentrations of the cocktail. In addition, cultures stimulated with the 1× cocktail for 3 h were allowed 2, 4, or 6 h of recovery. In parallel, C57BL/6J mice were sleep deprived for 2, 4, or 6 h and mice sleep deprived for 6 h were allowed 2, 4, or 6 h of recovery sleep. Additional C57BL/6J mice were killed 4 h after lights off as a reference for spontaneous waking [C57BL/6J mice stay almost completely awake during this period (Franken et al., 1998, 1999)]. Both in vivo and in vitro samples showed increasing levels of Homer1a and Arc and decreasing levels of Dbp expression proportional to the duration of prior waking or to the concentration of the neurotransmitter cocktail (Fig. 4). Nevertheless, Dbp expression did not revert to baseline during the 6 h recovery period. Indeed, the expression of Dbp recovers between 12 and 24 h after stimulation. We also measured the expression of circadian genes Per2, Bmal1, and Cry1 and none returned to the baseline level before 12 h after stimulation (data not shown). Spontaneous wakefulness of 4 h duration resulted in a level of expression of Homer1a, Arc, and Dbp near the 4 h sleep-deprivation condition (Fig. 4), suggesting that spontaneous and enforced wakefulness are accompanied by similar changes in gene expression, as previously reported (Cirelli et al., 2004; Franken et al., 2007; Maret et al., 2007; Mongrain et al., 2010).
Figure 4.
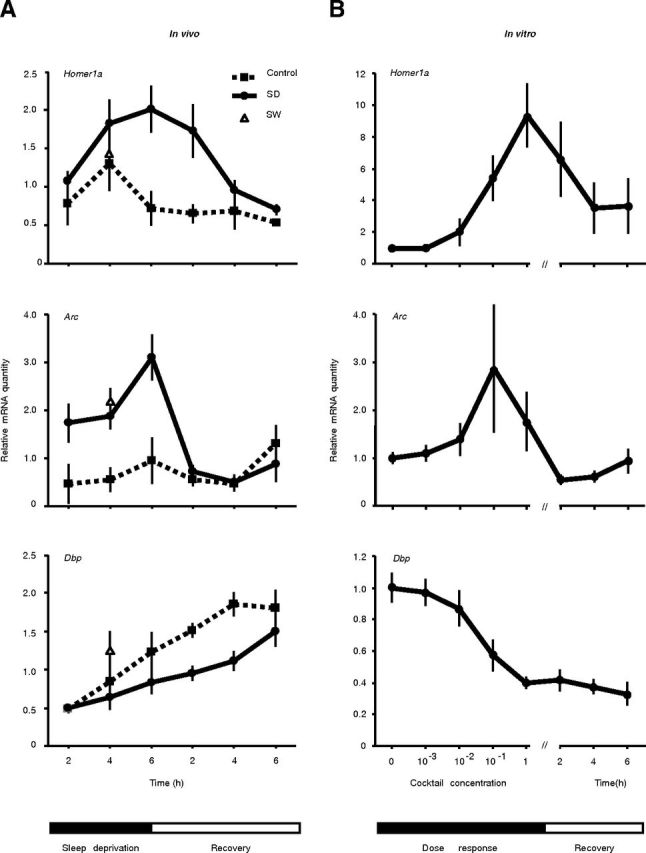
Similarities in the time course of gene expression in vivo (A) and in vitro (B). The time course of gene expression was followed in vivo by sleep depriving (SD) mice (n = 4 for each time point) for 2, 4, and 6 h; 6-h-sleep-deprived mice were allowed 2, 4, or 6 h of recovery (left). Undisturbed home-cage controls were also assessed at the same time points (Control). A reference level of expression corresponding to 4 h of spontaneous wakefulness (SW) is indicated by a triangle (note that this point is 4 h into the dark period and not at the same circadian time as the 4 h of sleep deprivation after lights-on). Homer1a is overexpressed proportional to the time kept awake and decreases slowly during the recovery period, while Arc shows a rapid recovery after 6 h of sleep deprivation. Dbp is decreased by sleep deprivation and the recovery is slow. To show the similarity in the time course of gene expression, primary cortical cultures (n = 3 measured in triplicates) were stimulated at indicated concentrations of our waking cocktail; those stimulated for 3 h with 1× cocktail were allowed 2, 4, or 6 h of recovery (right).
The changes in circadian gene expression after stimulation of the cortical neurons may result from direct effects of the stimulation on gene expression or from phase shifts induced by stimulation. To verify this possibility, primary cortical cultures were prepared from PER2::Luciferase knock-in embryos (Yoo et al., 2004). At 8 DIV, after changing half of the medium, a low-amplitude circadian expression of PER2::Luc protein (period between 23 and 25.8 h) was observed that disappeared after the third day, indicating that the change of the medium synchronized circadian rhythms of cortical neurons in vitro (data not shown). Four days later, cultures stimulated with the cocktail displayed a strong and high-amplitude circadian oscillation (Fig. 5). These results suggest that the delay in reaching the basal expression levels observed in the above experiments might have resulted from the long-term circadian modulation of gene expression in addition to the acute, activity-induced changes after the stimulation. To determine whether the stimulation can affect the phase of PER2::Luc expression in cortical neurons, we stimulated a second time at two different circadian times. When stimulated after the minimum of bioluminescence on day 2, the subsequent phase of the rhythm was clearly advanced and delayed, respectively, by ∼4 h (Fig. 5).
Figure 5.

Circadian rhythms in vitro. Recording of circadian changes in PER2-bioluminescence in primary cortical cultures prepared from PerLuc knock-in embryos. A, Stimulation at 0 h (blue triangle) with 1× neurotransmitter cocktail after 11 d in vitro induced a circadian oscillation in PER2-bioluminescence with a mean period length of 23.5 h (n = 15; including recordings in B). Period length was calculated over the first cycle as the difference between the time of the second and first trough. B, Cocktail-induced phase shifts depend on circadian phase. A second stimulation (blue triangle) with cocktail (or water) given at circadian time (CT)3.5 (red) or CT11.7 (green line; n = 3/condition) resulted in a phase advance or delay, respectively. Phase shifts were calculated according to differences in the time PER2-bioluminescence peaked during the cycle after the stimulation between stimulated (cocktail) and control (water) conditions. Values represent circadian hours calculated according to individual period length in the cycle before stimulation. Note that CT0, denoting the time trough values are reached in culture, corresponds to approximately Zeitgeber time (ZT)6 in vivo in the cortex of adult PerLuc mice under entrained conditions. Waveforms represent 100 min moving averages at 10 min intervals (n = 3/condition). C, The mean (±1 SEM) phase delay was significant (green bar; *p < 0.05), while the phase advance only showed a trend (red bar; Xp < 0.09; t tests; n = 3/condition). Black bars show the change in phase in the two control conditions (H2O) relative to the recording in A (p > 0.10; n = 3/condition).
Synaptic changes between sleep-like and wake-like states in cultured neurons are similar to sleeping and waking brains
Waking is associated with synaptic plastic changes believed to be crucial for the acquisition and consolidation of memory (Tononi and Cirelli, 2006). While synaptic stimulation during wakefulness induces long-term potentiation accompanied by an increase in the expression of plasticity-related genes (see above) and receptor trafficking and phosphorylation, sleep is supposed to restore the overall synaptic homeostasis (Tononi and Cirelli, 2006; Vyazovskiy et al., 2008b). A key correlate of these plastic changes is the trafficking and phosphorylation of postsynaptic glutamatergic AMPA receptors containing GluR1 subunit (Collingridge et al., 2004). To show that similar plastic changes occur in vitro, we prepared synaptic-enriched protein samples (synaptoneurosomes) before and after stimulation of our primary cortical cultures. For comparisons, synaptoneurosomes were also prepared from cortex of mice after variable durations of sleep deprivation and recovery after 6 h sleep deprivation. Western blot analysis of phosphorylated GluR1s at Ser845 clearly indicated that 2 and 6 h sleep deprivations increased GluR1s phosphorylation that decreased after 2–4 h recovery after 6 h sleep deprivation in the cortex of mice (Fig. 6A). Similarly, stimulation of primary cultures rapidly increased GluR1s phosphorylation levels that decreased after 6 h and returned to the control level 24 h after stimulation (Fig. 6B), strongly suggesting similar synaptic changes in sleep-like and wake-like states as the ones identified in vitro.
Figure 6.
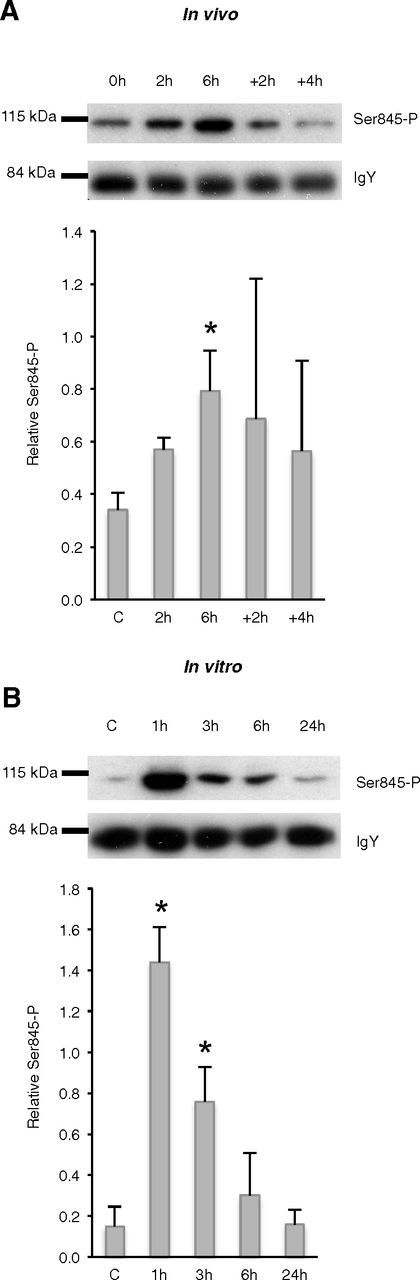
Similarities in the Ser845 phosphorylation of GluR1 subunit in vivo and in vitro. A, Western blot of synaptoneurosomes from half cortical hemisphere of C57BL/6J mice sleep deprived for 0, 2, or 6 h and sleep deprived for 6 h and let recovered for 2 or 4 h. A representative blot of Ser845-phosphorylated GluR1 and IgY loading control is shown. Quantitative analysis were performed with ImageJ and presented as the phosphorylated Ser845-GluR1 amount relative to IgY internal control (mean + SD of three mice per condition). B, Western blot of synaptoneurosomes from mature cortical cultures (11–13 DIV) stimulated with either distilled water (C, control) or the neurotransmitter cocktail and sampled after 1, 3, 6, or 24 h. A representative blot of Ser845-phosphorylated GluR1 and IgY loading control is shown. Quantitative analysis is presented as the phosphorylated Ser845-GluR1 amount relative to IgY internal control (mean + SD of three independent cultures). *Significant (p < 0.01) differences with the control condition (post hoc Tukey test).
Metabolic changes between sleep-like and wake-like states in cultured neurons are similar to sleeping and waking animals
Several hypotheses on sleep function suggest a role for sleep in correcting a metabolic imbalance incurred by the preceding wakefulness (Benington and Heller, 1995; Mackiewicz et al., 2007; Porkka-Heiskanen and Kalinchuk, 2011). How the changes in brain and body metabolism that occur with changes in behavioral states relate to sleep function remains, however, unknown. So far, the evidence is based on glucose metabolism or gene expression related to metabolism of lipids and proteins (Kennedy et al., 1982; Everson et al., 1994; Cirelli and Tononi, 2000b; Netchiporouk et al., 2001; Franken et al., 2003; Brown, 2004; Mackiewicz et al., 2007; Vyazovskiy et al., 2008a). Changes in metabolism in living animals are regulated both locally (activity-dependent) and globally through circulating humoral factors. In the brain, changes in oxygen and glucose consumption are major hallmarks of changes in neuronal activity and these changes are finely tuned by local and many global regulatory mechanisms, including changes in blood flow, that are mostly missing in primary cultures. Given that an in vitro model is readily amenable to biochemical analysis, we next analyzed the metabolic changes occurring by cell stimulation as a first demonstration of the utility of our model in finding the cellular function(s) of sleep. First, mice were monitored in calorimetric chambers to measure changes in basal metabolism over 24 h. Oxygen consumption of mice in vivo was 40% higher during the active (nighttime, wakefulness) compared with the inactive (daytime, sleep) period (Fig. 7A). We then validated our findings in vitro. We recorded changes in oxygen consumption at baseline and after stimulation of primary cortical cultures. Cultures were either sham-stimulated or stimulated with our cocktail, and oxygen consumption rate (OCR) was measured for 2 h with Seahorse XF extracellular flux analyzer. A rapid increase in OCR was observed and remained 30–40% higher than sham stimulation (Fig. 7B), indicating a sustained metabolic increase.
Figure 7.

Oxygen consumption increases in stimulated cortical cultures. A, In vivo oxygen consumption of C57BL/6J mice increased by 40% during the dark active period compared with the light rest period. The VO2 at each time point is expressed relative to the mean of the VO2 of the first 4 h of the first light period (black bar). Data indicate means ± SEM. B, Stimulation of cortical neurons increased by 40% oxygen consumption within 5 min, and increased oxygen consumption is maintained during 2 h. Data are from 10 wells per condition (mean ± SD).
We next analyzed the changes in cortical metabolites between sleep-deprived and non-sleep-deprived mice by liquid and gas chromatography-mass spectrometry (Evans et al., 2009). Two hundred twenty-six biochemicals were reliably detected, but only a few were significantly changed by sleep deprivation. Basically, two major pathways were identified. The first concerns the glycolytic pathway with increased alanine and lactate levels and a tendency for increase in glucose and acetyl-CoA, while pyruvate concentration was unchanged. The second pathway was unexpected and concerns lipid metabolism and, more specifically, points to a large increase in membrane breakdown of phospholipids (releasing lysolipids) following sleep deprivation. Long-chain fatty acids were carnitine-conjugated, suggesting that they may undergo β-oxidation. Decreased glutamine might result from increased uptake and recycling of glutamate (which was unchanged). Also, medium- and short-chain fatty acids were significantly decreased, corroborating the hypothesis that part of the fatty acids generated during wakefulness are being used by β-oxidation. We also noticed that two metabolites (aspartate and 1-methylimidazoleacetate) in the histidine pathway, probably from catabolism of the waking neurotransmitter histamine, were also increased. Note that aspartate also participates in the glycolysis pathway. Interestingly, 4-hydroxybuytrate, an endogenous neuromodulator acting on GABA-B receptors, was also significantly increased after sleep deprivation and might contribute to the increase in EEG delta power observed both after sleep deprivation and after stimulation of the GABAB receptors (Vienne et al., 2010).
The same analysis was also performed with cortical cultures that were sham-stimulated or stimulated with our waking cocktail. Two hundred twenty-two biochemicals were detected. One hundred forty-four biochemicals were common between the two models and included metabolites showing the largest changes in both conditions. Again, a large increase in lysolipids was observed. To identify common metabolic pathways, the data from the two models were meta-analyzed; very few changes were observed. As expected, increased lactate, a by-product of glycolysis, was among the most significantly changed metabolites in both in vivo sleep deprivation and in vitro stimulation. Several amino acids and their derivatives, including lysine, threonine, N-acetyl-aspartyl-glutamate, and N-methylthioadenosine, were decreased, while N-acetylmethionine was increased by both sleep deprivation and cell stimulation (Table 1). Pantothenic acid, essential for coenzyme A synthesis and acetylation, was also consistently decreased after sleep deprivation and cell stimulation. Surprisingly, the major pathway with substantial upregulation was lipid metabolism. More specifically, many lysolipids were increased after sleep deprivation or cell stimulation (Table 1), although individual lysolipids were not identical between the two models. Searching our transcriptome results after sleep deprivation indicated that mRNAs of the secreted phospholipase Pla2g3 and the membrane-associated Pla2g4c were increased, while the cytosolic phospholipases Pla2g4e and Pla2g4a were decreased. This unexpected finding suggests a strong impact of wakefulness and cortical stimulation on membrane homeostasis.
Table 1.
Metabolic changes induced by sleep deprivation and cell stimulation
| Metabolite | Pathway | Change | META_Pval | META_Padj |
|---|---|---|---|---|
| Lactate | Glycolysis, gluconeogenesis, pyruvate metabolism | Up | 0.0014 | 0.0437 |
| Pantothenate | Pantothenate and CoA metabolism | Down | 0.0002 | 0.0264 |
| 5-Methylthioadenosine | Polyamine metabolism | Down | 0.0022 | 0.0437 |
| N-acetyl-aspartyl-glutamate | Glutamate metabolism | Down | 0.0044 | 0.0515 |
| N-acetylmethionine | Cysteine, methionine, S-adenosyl methionine, taurine metabolism | Up | 0.0045 | 0.0515 |
| Laurate (12:0) | Medium-chain fatty acid | Down | 0.0026 | 0.0437 |
| 1-Stearoylglycerol (1-monostearin) | Monoacylglycerol | Up | 0.0020 | 0.0437 |
| 1-Palmitoylglycerophosphocholine | Lysolipid | Up | 0.0025 | 0.0437 |
| 1-Palmitoylglycerophosphoinositol | Lysolipid | Up | 0.0026 | 0.0437 |
| 1-Linoleoylglycerophosphocholine | Lysolipid | Up | 0.0027 | 0.0437 |
| 1-Oleoylglycerophosphocholine | Lysolipid | Up | 0.0032 | 0.0463 |
| 1-Stearoylglycerophosphocholine | Lysolipid | Up | 0.0046 | 0.0515 |
| 1-Stearoylglycerophosphoethanolamine | Lysolipid | Up | 0.0075 | 0.0725 |
| 1-Palmitoleoylglycerophosphocholine | Lysolipid | Up | 0.0091 | 0.0757 |
| 1-Stearoylglycerophosphoinositol | Lysolipid | Up | 0.0092 | 0.0757 |
| 1-Myristoylglycerophosphocholine | Lysolipid | Up | 0.0094 | 0.0757 |
| 2-Palmitoleoylglycerophosphoethanolamine | Lysolipid | Up | 0.0135 | 0.0991 |
| 2-Oleoylglycerophosphocholine | Lysolipid | Up | 0.0137 | 0.0991 |
| γ-Glutamylleucine | γ-Glutamyl | Down | 0.0024 | 0.0437 |
| γ-Glutamylglutamine | γ-Glutamyl | Down | 0.0050 | 0.0515 |
Upregulated and downregulated metabolites identified by meta-analysis of global metabolomics of the cortex and primary cortical cultures. For detailed analysis, see Materials and Methods. META_Pval indicates p value after meta-analysis; META_Padj indicates the adjusted p value for multiple testing.
Discussion
On the basis of electrophysiological, transcriptional, and metabolic criteria, we have identified a sleep-like state that is the default state in vitro. A similar observation was also recently reported by another laboratory (Sengupta et al., 2011). This state can be actively changed into a wake-like state that returns spontaneously back to sleep-like state. We showed here that wake-like and sleep-like states in vitro are accompanied by similar changes in gene expression as in the cerebral cortex of living animals. The induction of many activity-induced genes is interpreted as signs of plastic changes that occur during sleep and wakefulness. Their implication in sleep–wake-dependent neuronal plasticity can now be analyzed in vitro.
The pattern of electrical activity of cultured neuronal assemblies is highly similar to in vivo activity of cortical neurons during slow-wave sleep. The appearance of a culture-wide synchronous activity parallels the maturation of simple neuronal networks. The default spontaneous synchronized firing of mature cortical cultures is proposed to result from deafferentation (lack of structured stimulatory input) (Wagenaar et al., 2005). Single cortical neurons in vivo show a burst–pause synchronous firing <1 Hz (Steriade et al., 1993). This slow oscillation represents the intrinsic discharge mode of sleeping cortical neurons. We have shown that matured cortical cultures also fire with a typical burst–pause low-frequency pattern. Undisturbed cultures show this unique electrical activity (default mode) that can only be altered by electrical or chemical stimulation (Van Pelt et al., 2004; Corner, 2008; Corner et al., 2008). However, as opposed to the cortex of living animals, no activity in delta frequency range (1–4 Hz) was observed in cortical cultures, although during recovery after stimulation, the frequency of bursting activity reached the lower (0.5–0.6 Hz) delta frequency range. Delta oscillations result from thalamocortical feedback loops lost in dissociated cortical cultures. Whether dissociated thalamic or corticothalamic cocultures may show delta oscillations can now be investigated in vitro. Note that cortical cultures not only lack the complex connectivity with other brain regions but also have a much simpler monolayer architectural organization compared with even simple cortical columns.
Finally, wakefulness is accompanied by metabolic changes that are believed to necessitate recovery that is most efficiently accomplished during sleep. Gene expression analysis suggests that metabolic changes induced by sleep deprivation are tissue-specific (Maret et al., 2007). In the living cortex, extended wakefulness resulted in few changes in specific pathways (amino acid and lysolipid metabolism). Importantly, we show here that the same metabolic pathways undergo similar changes in vitro as in living animals. Increased aerobic glycolysis and fatty acid oxidation during wakefulness or cell stimulation is consistently accompanied by increased oxygen consumption and lactate production. Also, we discovered that lipid metabolism is compromised during wakefulness and cell stimulation with a specific impact on membrane phospholipids.
Our findings demonstrate that key signatures of sleep and wakefulness can be found in vitro in simple neuronal networks. Thus, the developmental changes, plasticity properties, single gene, or metabolite effects can readily and reliably be investigated. Moreover, in vitro changes can be studied in cultures from loss- or gain-of-function mutations as well as systematic gene knockdown by simple transfection. Finally, the in vitro model might represent an ideal approach in the pharmacology of sleep–wake states.
Notes
Supplemental material for this article is available at http://www.genomyx.ch/cig/mt/files/Index.html. Supplemental tables and figures. This material has not been peer reviewed.
Footnotes
This work was supported by the Swiss National Science Foundation (Grants 130487 to M.T. and 124713 to J.A.), the Novartis Foundation, the University of Lausanne, the Ecole Polytechnique Fédérale de Lausanne, NIH (Grant DK59820), the EU Ideas program (Grant ERC-2008-AdG-23118), and the Velux Stiftung. R.H.H. is supported by a Rubicon fellowship of the Netherlands Organization for Scientific Research. J.A. is the Nestlé Chair in Energy Metabolism. We thank James Krueger, Stéphanie Maret, Howard Riezman, Ursula Loizides, and Ueli Schibler for critical reading and comments of our earlier drafts. We also thank Angélique Vaucher for primary cultures and Sonia Jimenez, Yann Emmeneger, Gianina Luca, David Gatfield, members of the Genomic Technology Facility, and the Mouse Metabolic Evaluation Facility of the University of Lausanne for technical assistance.
The authors declare no financial conflicts of interest.
References
- Achermann P, Borbély AA. Low-frequency (<1 Hz) oscillations in the human sleep electroencephalogram. Neuroscience. 1997;81:213–222. doi: 10.1016/s0306-4522(97)00186-3. [DOI] [PubMed] [Google Scholar]
- Benington JH, Heller HC. Restoration of brain energy metabolism as the function of sleep. Prog Neurobiol. 1995;45:347–360. doi: 10.1016/0301-0082(94)00057-o. [DOI] [PubMed] [Google Scholar]
- Bersagliere A, Achermann P. Slow oscillations in human non-rapid eye movement sleep electroencephalogram: effects of increased sleep pressure. J Sleep Res. 2010;19:228–237. doi: 10.1111/j.1365-2869.2009.00775.x. [DOI] [PubMed] [Google Scholar]
- Brown AM. Brain glycogen re-awakened. J Neurochem. 2004;89:537–552. doi: 10.1111/j.1471-4159.2004.02421.x. [DOI] [PubMed] [Google Scholar]
- Campbell SS, Tobler I. Animal sleep: a review of sleep duration across phylogeny. Neurosci Biobehav Rev. 1984;8:269–300. doi: 10.1016/0149-7634(84)90054-x. [DOI] [PubMed] [Google Scholar]
- Chiappalone M, Bove M, Vato A, Tedesco M, Martinoia S. Dissociated cortical networks show spontaneously correlated activity patterns during in vitro development. Brain Res. 2006;1093:41–53. doi: 10.1016/j.brainres.2006.03.049. [DOI] [PubMed] [Google Scholar]
- Cirelli C, Tononi G. Differences in brain gene expression between sleep and waking as revealed by mRNA differential display and cDNA microarray technology. J Sleep Res. 1999;8(Suppl 1):44–52. doi: 10.1046/j.1365-2869.1999.00008.x. [DOI] [PubMed] [Google Scholar]
- Cirelli C, Tononi G. Differential expression of plasticity-related genes in waking and sleep and their regulation by the noradrenergic system. J Neurosci. 2000a;20:9187–9194. doi: 10.1523/JNEUROSCI.20-24-09187.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirelli C, Tononi G. Gene expression in the brain across the sleep-waking cycle. Brain Res. 2000b;885:303–321. doi: 10.1016/s0006-8993(00)03008-0. [DOI] [PubMed] [Google Scholar]
- Cirelli C, Pompeiano M, Tononi G. Neuronal gene expression in the waking state: a role for the locus coeruleus. Science. 1996;274:1211–1215. doi: 10.1126/science.274.5290.1211. [DOI] [PubMed] [Google Scholar]
- Cirelli C, Gutierrez CM, Tononi G. Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron. 2004;41:35–43. doi: 10.1016/s0896-6273(03)00814-6. [DOI] [PubMed] [Google Scholar]
- Cirelli C, Faraguna U, Tononi G. Changes in brain gene expression after long-term sleep deprivation. J Neurochem. 2006;98:1632–1645. doi: 10.1111/j.1471-4159.2006.04058.x. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5:952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- Corner MA. Spontaneous neuronal burst discharges as dependent and independent variables in the maturation of cerebral cortex tissue cultured in vitro: a review of activity-dependent studies in live ‘model’ systems for the development of intrinsically generated bioelectric slow-wave sleep patterns. Brain Res Rev. 2008;59:221–244. doi: 10.1016/j.brainresrev.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Corner MA. The sleep-like nature of early mammalian behavioral rhythms: theoretical comment on Todd et al. (2010) Behav Neurosci. 2010;124:175–178. doi: 10.1037/a0018483. [DOI] [PubMed] [Google Scholar]
- Corner MA, Baker RE, van Pelt J. Physiological consequences of selective suppression of synaptic transmission in developing cerebral cortical networks in vitro: differential effects on intrinsically generated bioelectric discharges in a living ‘model’ system for slow-wave sleep activity. Neurosci Biobehav Rev. 2008;32:1569–1600. doi: 10.1016/j.neubiorev.2008.06.008. [DOI] [PubMed] [Google Scholar]
- Daan S, Beersma DG, Borbély AA. Timing of human sleep: recovery process gated by a circadian pacemaker. Am J Physiol. 1984;246:R161–R183. doi: 10.1152/ajpregu.1984.246.2.R161. [DOI] [PubMed] [Google Scholar]
- Evans AM, DeHaven CD, Barrett T, Mitchell M, Milgram E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem. 2009;81:6656–6667. doi: 10.1021/ac901536h. [DOI] [PubMed] [Google Scholar]
- Everson CA, Smith CB, Sokoloff L. Effects of prolonged sleep deprivation on local rates of cerebral energy metabolism in freely moving rats. J Neurosci. 1994;14:6769–6778. doi: 10.1523/JNEUROSCI.14-11-06769.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franken P, Malafosse A, Tafti M. Genetic variation in EEG activity during sleep in inbred mice. Am J Physiol. 1998;275:R1127–R1137. doi: 10.1152/ajpregu.1998.275.4.R1127. [DOI] [PubMed] [Google Scholar]
- Franken P, Malafosse A, Tafti M. Genetic determinants of sleep regulation in inbred mice. Sleep. 1999;22:155–169. [PubMed] [Google Scholar]
- Franken P, Gip P, Hagiwara G, Ruby NF, Heller HC. Changes in brain glycogen after sleep deprivation vary with genotype. Am J Physiol Regul Integr Comp Physiol. 2003;285:R413–R419. doi: 10.1152/ajpregu.00668.2002. [DOI] [PubMed] [Google Scholar]
- Franken P, Thomason R, Heller HC, O'Hara BF. A non-circadian role for clock-genes in sleep homeostasis: a strain comparison. BMC Neurosci. 2007;8:87. doi: 10.1186/1471-2202-8-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8:R19. doi: 10.1186/gb-2007-8-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks JC, Finn SM, Panckeri KA, Chavkin J, Williams JA, Sehgal A, Pack AI. Rest in Drosophila is a sleep-like state. Neuron. 2000;25:129–138. doi: 10.1016/s0896-6273(00)80877-6. [DOI] [PubMed] [Google Scholar]
- Kattler H, Dijk DJ, Borbély AA. Effect of unilateral somatosensory stimulation prior to sleep on the sleep EEG in humans. J Sleep Res. 1994;3:159–164. doi: 10.1111/j.1365-2869.1994.tb00123.x. [DOI] [PubMed] [Google Scholar]
- Kennedy C, Gillin JC, Mendelson W, Suda S, Miyaoka M, Ito M, Nakamura RK, Storch FI, Pettigrew K, Mishkin M, Sokoloff L. Local cerebral glucose utilization in non-rapid eye movement sleep. Nature. 1982;297:325–327. doi: 10.1038/297325a0. [DOI] [PubMed] [Google Scholar]
- Kristiansen K, Courtois G. Rhythmic electrical activity from isolated cerebral cortex. Electroencephalogr Clin Neurophysiol. 1949;1:265–272. [PubMed] [Google Scholar]
- Krueger JM, Obál F. A neuronal group theory of sleep function. J Sleep Res. 1993;2:63–69. doi: 10.1111/j.1365-2869.1993.tb00064.x. [DOI] [PubMed] [Google Scholar]
- Krueger JM, Rector DM, Roy S, Van Dongen HP, Belenky G, Panksepp J. Sleep as a fundamental property of neuronal assemblies. Nat Rev Neurosci. 2008;9:910–919. doi: 10.1038/nrn2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackiewicz M, Shockley KR, Romer MA, Galante RJ, Zimmerman JE, Naidoo N, Baldwin DA, Jensen ST, Churchill GA, Pack AI. Macromolecule biosynthesis: a key function of sleep. Physiol Genomics. 2007;31:441–457. doi: 10.1152/physiolgenomics.00275.2006. [DOI] [PubMed] [Google Scholar]
- Maret S, Dorsaz S, Gurcel L, Pradervand S, Petit B, Pfister C, Hagenbuchle O, O'Hara BF, Franken P, Tafti M. Homer1a is a core brain molecular correlate of sleep loss. Proc Natl Acad Sci U S A. 2007;104:20090–20095. doi: 10.1073/pnas.0710131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongrain V, Hernandez SA, Pradervand S, Dorsaz S, Curie T, Hagiwara G, Gip P, Heller HC, Franken P. Separating the contribution of glucocorticoids and wakefulness to the molecular and electrophysiological correlates of sleep homeostasis. Sleep. 2010;33:1147–1157. doi: 10.1093/sleep/33.9.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netchiporouk L, Shram N, Salvert D, Cespuglio R. Brain extracellular glucose assessed by voltammetry throughout the rat sleep-wake cycle. Eur J Neurosci. 2001;13:1429–1434. doi: 10.1046/j.0953-816x.2001.01503.x. [DOI] [PubMed] [Google Scholar]
- Pigarev IN, Nothdurft HC, Kastner S. Evidence for asynchronous development of sleep in cortical areas. Neuroreport. 1997;8:2557–2560. doi: 10.1097/00001756-199707280-00027. [DOI] [PubMed] [Google Scholar]
- Pompeiano M, Cirelli C, Tononi G. Immediate-early genes in spontaneous wakefulness and sleep: expression of c-fos and NGFI-A mRNA and protein. J Sleep Res. 1994;3:80–96. doi: 10.1111/j.1365-2869.1994.tb00111.x. [DOI] [PubMed] [Google Scholar]
- Pompeiano M, Cirelli C, Ronca-Testoni S, Tononi G. NGFI-A expression in the rat brain after sleep deprivation. Brain Res Mol Brain Res. 1997;46:143–153. doi: 10.1016/s0169-328x(96)00295-1. [DOI] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Kalinchuk AV. Adenosine, energy metabolism and sleep homeostasis. Sleep Med Rev. 2011;15:123–135. doi: 10.1016/j.smrv.2010.06.005. [DOI] [PubMed] [Google Scholar]
- Raizen DM, Zimmerman JE, Maycock MH, Ta UD, You YJ, Sundaram MV, Pack AI. Lethargus is a Caenorhabditis elegans sleep-like state. Nature. 2008;451:569–572. doi: 10.1038/nature06535. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2004. [Google Scholar]
- Rector DM, Topchiy IA, Carter KM, Rojas MJ. Local functional state differences between rat cortical columns. Brain Res. 2005;1047:45–55. doi: 10.1016/j.brainres.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Reitman ZJ, Jin G, Karoly ED, Spasojevic I, Yang J, Kinzler KW, He Y, Bigner DD, Vogelstein B, Yan H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc Natl Acad Sci U S A. 2011;108:3270–3275. doi: 10.1073/pnas.1019393108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta P, Davis CJ, Clinton JM, Roy S, Krueger JM. Networks of cultured neurons show oscillatory dynamics. Sleep. 2011;34:A36–A37. [Google Scholar]
- Shaw PJ, Cirelli C, Greenspan RJ, Tononi G. Correlates of sleep and waking in Drosophila melanogaster. Science. 2000;287:1834–1837. doi: 10.1126/science.287.5459.1834. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- Steriade M, Contreras D, Curró Dossi R, Nuñez A. The slow (< 1 Hz) oscillation in reticular thalamic and thalamocortical neurons: scenario of sleep rhythm generation in interacting thalamic and neocortical networks. J Neurosci. 1993;13:3284–3299. doi: 10.1523/JNEUROSCI.13-08-03284.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terao A, Steininger TL, Hyder K, Apte-Deshpande A, Ding J, Rishipathak D, Davis RW, Heller HC, Kilduff TS. Differential increase in the expression of heat shock protein family members during sleep deprivation and during sleep. Neuroscience. 2003;116:187–200. doi: 10.1016/s0306-4522(02)00695-4. [DOI] [PubMed] [Google Scholar]
- Tononi G, Cirelli C. Sleep function and synaptic homeostasis. Sleep Med Rev. 2006;10:49–62. doi: 10.1016/j.smrv.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Van Pelt J, Corner MA, Wolters PS, Rutten WL, Ramakers GJ. Longterm stability and developmental changes in spontaneous network burst firing patterns in dissociated rat cerebral cortex cell cultures on multielectrode arrays. Neurosci Lett. 2004;361:86–89. doi: 10.1016/j.neulet.2003.12.062. [DOI] [PubMed] [Google Scholar]
- Vienne J, Bettler B, Franken P, Tafti M. Differential effects of GABAB receptor subtypes, {gamma}-hydroxybutyric acid, and baclofen on EEG activity and sleep regulation. J Neurosci. 2010;30:14194–14204. doi: 10.1523/JNEUROSCI.3145-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyazovskiy VV, Cirelli C, Tononi G, Tobler I. Cortical metabolic rates as measured by 2-deoxyglucose-uptake are increased after waking and decreased after sleep in mice. Brain Res Bull. 2008a;75:591–597. doi: 10.1016/j.brainresbull.2007.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyazovskiy VV, Cirelli C, Pfister-Genskow M, Faraguna U, Tononi G. Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat Neurosci. 2008b;11:200–208. doi: 10.1038/nn2035. [DOI] [PubMed] [Google Scholar]
- Vyazovskiy V, Borbély AA, Tobler I. Unilateral vibrissae stimulation during waking induces interhemispheric EEG asymmetry during subsequent sleep in the rat. J Sleep Res. 2000;9:367–371. doi: 10.1046/j.1365-2869.2000.00230.x. [DOI] [PubMed] [Google Scholar]
- Vyazovskiy VV, Olcese U, Hanlon EC, Nir Y, Cirelli C, Tononi G. Local sleep in awake rats. Nature. 2011;472:443–447. doi: 10.1038/nature10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenaar DA, Madhavan R, Pine J, Potter SM. Controlling bursting in cortical cultures with closed-loop multi-electrode stimulation. J Neurosci. 2005;25:680–688. doi: 10.1523/JNEUROSCI.4209-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirapati P, Sotiriou C, Kunkel S, Farmer P, Pradervand S, Haibe-Kains B, Desmedt C, Ignatiadis M, Sengstag T, Schütz F, Goldstein DR, Piccart M, Delorenzi M. Meta-analysis of gene expression profiles in breast cancer: toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res. 2008;10:R65. doi: 10.1186/bcr2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisor JP, O'Hara BF, Terao A, Selby CP, Kilduff TS, Sancar A, Edgar DM, Franken P. A role for cryptochromes in sleep regulation. BMC Neurosci. 2002;3:20. doi: 10.1186/1471-2202-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong HK, Oh WJ, Yoo OJ, Menaker M, Takahashi JS. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci U S A. 2004;101:5339–5346. doi: 10.1073/pnas.0308709101. [DOI] [PMC free article] [PubMed] [Google Scholar]