

Abstract
Systemic inflammatory response syndrome (SIRS) is a highly mortal inflammatory disease, associated with systemic inflammation and organ dysfunction. SIRS can have a sterile cause or can be initiated by an infection, called sepsis. The prevalence is high, and available treatments are ineffective and mainly supportive. Consequently, there is an urgent need for new treatments. The brain is one of the first organs affected during SIRS, and sepsis and the consequent neurological complications, such as encephalopathy, are correlated with decreased survival. The choroid plexus (CP) that forms the blood–CSF barrier (BCSFB) is thought to act as a brain “immune sensor” involved in the communication between the peripheral immune system and the CNS. Nevertheless, the involvement of BCSFB integrity in systemic inflammatory diseases is seldom investigated. We report that matrix metalloprotease-8 (MMP8) depletion or inhibition protects mice from death and hypothermia in sepsis and renal ischemia/reperfusion. This effect could be attributed to MMP8-dependent leakage of the BCSFB, caused by collagen cleavage in the extracellular matrix of CP cells, which leads to a dramatic change in cellular morphology. Disruption of the BCSFB results in increased CSF cytokine levels, brain inflammation, and downregulation of the brain glucocorticoid receptor. This receptor is necessary for dampening the inflammatory response. Consequently, MMP8+/+ mice, in contrast to MMP8−/− mice, show no anti-inflammatory response and this results in high mortality. In conclusion, we identify MMP8 as an essential mediator in SIRS and, hence, a potential drug target. We also propose that the mechanism of action of MMP8 involves disruption of the BCSFB integrity.
Introduction
The normal host response to infection or other insults is protective activation of the immune system, which efficiently eradicates the pathogen and/or resolves tissue injury. However, the host can develop an exaggerated or unbalanced immune response that is harmful and might lead to systemic inflammatory response syndrome (SIRS). This situation can aggravate into vascular dysfunction, multiple organ failure (MOF), or even death. SIRS can be caused by ischemia, inflammation, trauma, infection, or a combination of several insults. SIRS in the presence of infection is called sepsis. The prevalence of SIRS is extremely high (Stephenson et al., 2010) and is expected to increase with the aging of the population and the development of new invasive technologies. SIRS is a major cause of mortality, varying from 7% (sterile SIRS) to 46% (septic shock) (Rangel-Frausto et al., 1995). Available SIRS treatments are ineffective and mainly supportive (e.g., mechanical ventilation, hemodialysis, and fluid resuscitation), but it also includes antibiotics if infection is present. The only approved drug (recombinant human activate protein C) was recently withdrawn from the market worldwide (Barie, 2011). Consequently, there is an urgent need for more detailed analysis of processes involved in the pathogenesis of SIRS and sepsis to identify key molecules for the development of new treatments.
The brain is one of the first organs affected in SIRS patients (Young et al., 1990), and brain (Ebersoldt et al., 2007) and neuroendocrine (Chrousos, 1995) dysfunction are frequent complications of sepsis that contribute to mortality (Sprung et al., 1990). Homeostasis of the CNS is strictly regulated and dependent on three barriers that separate the CNS from the periphery: brain endothelium forming the blood–brain barrier (BBB), arachnoid epithelium forming the middle layer of the meninges, and choroid plexus (CP) epithelium, which secretes CSF and forms the blood–CSF barrier (BCSFB) (Abbott et al., 2010). However, one should realize that many different versions of the term BBB are encountered in literature and that this includes both diffusion/transport of solutes and migration of cells from the blood to CNS parenchyma (Owens et al., 2008). Septic encephalopathy, which is reported in up to 70% of septic patients (Ebersoldt et al., 2007), is associated with increased protein leakage from the circulation into the CSF (Young et al., 1992).
The BCSFB that consists of the CP cells is believed to act as a brain “immune sensor” involved in the communication of the peripheral immune system with the CNS. Nevertheless, when studying brain leakage caused by systemic inflammation, most research focuses on the BBB, whereas disruption of the BCSFB is generally neglected.
Matrix metalloproteases (MMPs) are Zn2+-dependent endopeptidases and important modulators of inflammation and innate immunity (Parks et al., 2004). Consequently, they are involved in several inflammatory diseases (Wielockx et al., 2001; Vanlaere and Libert, 2009; Dejonckheere et al., 2011b; Vandenbroucke et al., 2011). Solan et al. (2012) recently reported that MMP8 inhibition can improve outcome in experimental sepsis. Moreover, serum MMP8 levels at admission to intensive care units were high among sepsis patients with fatal outcome (Lauhio et al., 2011).
Recently, Zeni et al. (2007) published that in vitro BCSFB integrity is compromised by MMPs. CSF analysis revealed that MMP levels are elevated during systemic inflammatory disorders (Tsuge et al., 2010; Marangoni et al., 2011). Moreover, the CP epithelium was shown to be a source of MMPs (Pagenstecher et al., 2000), but so far, there are no reports explaining whether MMPs participate in BCSFB disruption in response to systemic inflammation in vivo.
Materials and Methods
Animals.
C57BL/6J MMP8−/− mice (Balbin et al., 2003) were housed in a specific pathogen-free animal facility with ad libitum access to food and water. Both male and female mice (8–12 weeks old) were used, except for ischemia/reperfusion (I/R) experiments, in which males were used for lethality experiments and females for histopathology. For batimastat (BB-94) experiments, female C57BL/6J mice (8–12 weeks old) were purchased from Janvier. All experiments were approved by the ethics committee of the Faculty of Science of Ghent University.
Endotoxemia model.
Mice were injected intraperitoneally with 17.5 mg/kg body weight lipopolysaccharide (LPS) from Salmonella enterica serotype abortus equi (Sigma), an LD100 dose for wild-type C57BL/6 mice. BB-94 (British Biotech), dissolved in PBS containing 0.01% Tween 20 and MMP8-specific inhibitor (Biasone et al., 2007) dissolved in PBS containing 5% DMSO, were injected intraperitoneally 3 and 6 h before LPS challenge, at 50 mg/kg body weight. Control animals received intraperitoneal injections of vehicle or nothing at all. Rectal temperature was measured at different times after challenge. Mice were bled by heart puncture, and serum or EDTA plasma was prepared and stored at −20°C until use.
Renal I/R model.
Renal ischemia was induced in isoflurane-anesthetized mice by occluding the left renal pedicle for 45 min with a vessel clip (Aesculap). The right kidney was removed. Sham-operated animals received identical treatment except for the clamping of the left renal pedicle. Because female mice seem to be less sensitive to renal I/R (Hutchens et al., 2008), only males were included for lethality and serum analysis, whereas females were used for the histopathology study. Rectal temperature was measured at different times after challenge. Mice were bled by heart puncture, and serum or EDTA plasma was prepared and stored at −20°C until use.
Cecal ligation and puncture model.
Severe sepsis was induced in isoflurane-anesthetized mice by ligation of the cecum, followed by twice puncturing with a 21-gauge needle as described previously (Rittirsch et al., 2009). All animals received two doses of antibiotic therapy intraperitonally (25 mg/kg ceftriaxone and 12.5 mg/kg metronidazole) at 9 and 24 h after cecal ligation and puncture (CLP).
Real-time qPCR.
Organs were stored in RNALater (Ambion), and RNA was isolated using the RNeasy Mini Kit (Qiagen). cDNA was synthesized by the iScript cDNA Synthesis Kit (Bio-Rad). Real-time PCR was performed on the Light Cycler 480 system (Roche) using the LightCycler 480 SYBR Green I Master (Roche). Expression levels were normalized to the expression of the two most stable housekeeping genes, which was determined for each organ using the geNorm Housekeeping Gene Selection Software (Vandesompele et al., 2002): 1) liver: ubiquitin (Ubc), hypoxanthine-guanine phosphoribosyltransferase (HPRT); 2) ileum: ribosomal protein (RPL), Ubc; 3) lung: GAPDH, HPRT; 4) kidney: GAPDH, RPL; 5) spleen: RPL, Ubc; 6) brain: GAPDH, RPL; and 7) CP: RPL, Ubc.
Bone marrow transplantation.
Mice were pretreated for 2 weeks with 0.2% neomycin sulfate in the drinking water and exposed to a radiation dose of 10 Gray. The next day, the mice were reconstituted by intravenous injection of 7 × 106 bone marrow cells obtained by flushing tibia and femur. After 6 more weeks of neomycin treatment, transplantation was evaluated by genotyping whole-blood DNA (Higuchi, 1989). The presence of donor blood cells in circulation in the transplanted mice was analyzed to determine the quality of the transplantations. Only mice without chimerism were included in the experiment.
Vascular permeability.
One hour before organ isolation, mice were injected intravenously with 75 mg/kg body weight of FITC-labeled dextran (4 kDa; Sigma). Mice were perfused with 0.9% saline to remove all labeled dextran in circulation. To extract the remaining FITC-labeled dextran from the tissues, formamide was added to the organs (8 ml of formamide to 1 g of tissue), which were then cut into little pieces. After overnight incubation at 37°C, the samples were centrifuged. Relative leakage into the organ tissue was determined by measurement of the fluorescence of the supernatant with λex/λem = 488/520 nm. Values were normalized to the lowest value per tissue.
Blood–CSF permeability.
One hour before CSF isolation, mice were injected intravenously with 75 mg/kg body weight of FITC-labeled dextran (4 kDa; Sigma). They were perfused with 0.9% saline to remove all labeled dextran in the circulation. A CSF sample of 2 μl, harvested from the fourth ventricle, was diluted 25-fold in sterile PBS, and leakage into this brain compartment was determined by measurement of fluorescence with λex/λem = 488/520 nm. To observe vascular leakage into the brain, lysine fixable TRITC–dextran (10 kDa; Sigma) was used. Brain tissue was isolated without perfusion, fixed in 4% paraformaldehyde (PFA) overnight, and embedded in 5% agarose (type VII low gelling temperature; Sigma) in PBS, pH 7.2. Sections of 80 μm were cut on a Leica VT1000S vibrating blade microtome, and leakage was visualized with a Leica TCS SP5 II confocal microscope.
Immunostainings.
Tissues were fixed with 4% PFA, embedded in paraffin, sectioned at 4 μm, dewaxed, and stained. For immunofluorescent labeling of MMP8 (ab78423; Abcam), collagen I (CL50151AP-1; Cedarlane) and collagen IV (ab6581; Abcam) sections were blocked for 1 h (10 mm Tris-HCl, pH 7.4, 0.1 m MgCl2, 0.5% Tween 20, 1% BSA, and 5% serum), followed by overnight incubation with primary antibody: MMP8 (1:250), collagen I (1:250), or biotinylated collagen IV (1:300). After rinsing with TBS/0.1% Tween 20, sections were incubated with fluorescently labeled secondary antibody: GAR-549 (1:300; Invitrogen), GAR-633 (1:300; Invitrogen), and Streptavidin-647 (1:500; Invitrogen), respectively. After another washing step, sections were counterstained with Hoechst, mounted, and visualized with a Leica TCS SP5 II confocal microscope. For immunoperoxidase labeling of glial fibrillary acidic protein (GFAP) (Z0334; Dako) and CD45 (550539; BD PharMingen), sections were incubated in H2O2/PBS for 10 min. After rinsing in PBS, sections were boiled in citrate buffer and again rinsed. Sections were then incubated with blocking buffer (PBS supplemented with 10% FCS and 1% Triton X-100) for 15 min, followed by overnight incubation with primary antibody: GFAP (1:200) or CD45 (1:100). After rinsing with PBS, sections were incubated with biotinylated secondary antibody (85-9043; Zymed) for 1 h at room temperature. Sections were again rinsed in PBS and incubated with streptavidin–HRP complex (85-9043; Zymed) for 45 min. After rinsing in PBS, sections were incubated in 3,30-diaminobenzidine (Merck) and H2O2/PBS for 6 min. Finally, sections were counterstained with hematoxylin, dehydrated, and mounted. “No primary antibody control” was taken along in all immunostainings.
Electron microscopy.
The excised CP was fixed in a solution of 0.3% glutaraldehyde and 2.5% formaldehyde dissolved in 0.1 m sodium cacodylate buffer containing 20 mg/100 ml CaCl2. Fixed specimens were dehydrated through a graded ethanol series and embedded in Spurr's resin. Ultrathin sections of a gold interference color were cut using an ultramicrotome (Leica EM UC6) and post-stained with uranyl acetate for 40 min and lead citrate for 7 min in a Leica EM AC20. Sections were collected on Formvar-coated copper slot grids and viewed with a transmission electron microscope (JEOL 1010).
Cytokine/chemokine measurements.
Quantification of cytokines and chemokines in serum and CSF was performed using the Bio-Plex cytokine assays (Bio-Rad) or the cytometric bead array (R & D Systems), according to the instructions of the manufacturer.
Corticosterone measurements.
Plasma corticosterone levels were determined using the Coat-A-Count rat corticosterone in vitro diagnostic test kit (Siemens Medical Solutions), according to the instructions of the manufacturer.
Histopathology lung, kidney, and small intestine.
Tissues were fixed with PFA, embedded in paraffin, sectioned at 4 μm, and stained with hematoxylin (Fluka) and eosin (Merck). The degree of damage was evaluated on entire organ sections by four neutral observers in a blinded manner. Lung damage is characterized by atelectasis, i.e., thickening of the alveolar wall and influx of inflammatory cells. Taking into account all histological features, a damage score ranging from 0 (normal) to 2 (abnormal) was given to each mouse. Kidney damage after renal I/R was assessed by the percentage of affected tubuli (characterized by tubule dilatation) and affected glomeruli (characterized by a widening of the urinary space of the Bowman's capsule and an increase in mesenchymal cells). Intestinal damage is characterized by decreased villus height, epithelial cell death at the villus top, and loss of mucus layer and goblet cells. Taking into account all histological features, a damage score ranging from 0 (normal) to 4 (abnormal) was given to each mouse.
Coagulation test (Lewis et al., 2001).
Prothrombin time was determined on citrated plasma of mice on a STA Compact coagulation analyzer (Diagnostica Stago) according to the instructions of the manufacturer. Data are expressed as time needed for coagulation.
Creatinine measurements.
Serum creatinine levels were determined by the compensated Jaffé method on a Cobas 6000-c501 analyzer (Roche).
Aspartate amino transferase measurements.
Serum aspartate amino transferase (AST) levels were determined using the Fluitest GPT ALAT test (Analyticon), according to the instructions of the manufacturer.
Statistics.
Data are presented as means ± SEM. Data were analyzed with an unpaired Mann–Whitney U test, unless mentioned differently. Survival curves were compared using a log-rank test, unless mentioned differently. Significances were calculated for differences from the corresponding 0 h time point and/or between MMP8+/+ and MMP8−/− mice, as indicated (*, 0.01 ≤ p < 0.05; **, 0.001 ≤ p < 0.01; ***, p < 0.001).
Results
MMP8 inhibition protects against systemic inflammation
To investigate the role of MMPs in systemic inflammation, we injected C57BL/6 mice with a lethal dose of LPS to induce SIRS, a TLR4-dependent model called endotoxemia. Several studies have confirmed the relevance of endotoxemia as a model for human sepsis (Cantaluppi et al., 2008; Cruz et al., 2008, 2009). Pretreatment of mice with the broad-spectrum MMP inhibitor BB-94 completely protected the mice against LPS-induced hypothermia and death (Fig. 1a,b). Because BB-94 inhibits most MMPs (Watson et al., 1995), we also investigated whether MMP8 mediates the lethal effects of LPS by testing the response of MMP8-deficient mice in the LPS model. MMP8−/− mice were significantly protected against hypothermia and death caused by LPS (Fig. 1c,d). Moreover, an MMP8-specific inhibitor (Biasone et al., 2007) completely prevented LPS-induced death in MMP8+/+ mice (Fig. 1e,f). Polymicrobial sepsis induced by CLP is the most frequently used model because it closely resembles the progression and characteristics of human sepsis (Dejager et al., 2011). Comparable with LPS challenge, significantly more MMP8−/− mice survived sepsis challenge compared with their MMP8+/+ counterparts, which also showed more severe hypothermia (Fig. 1g,h).
Figure 1.
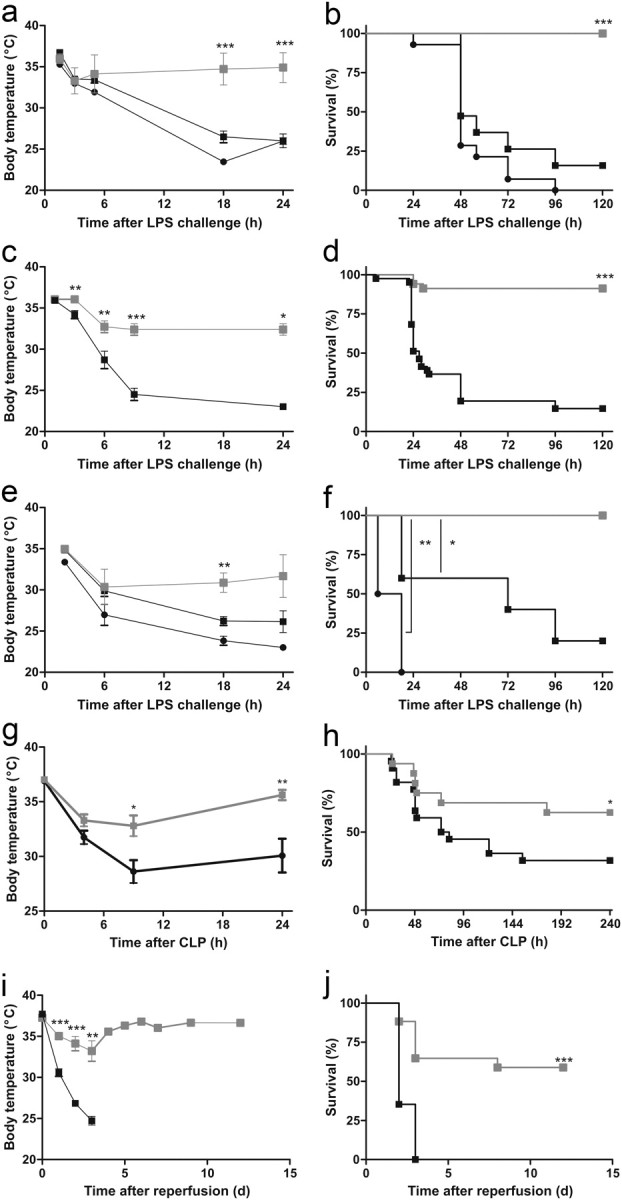
MMP8 inhibition protects against systemic inflammation induced by endotoxemia, renal I/R, and CLP. a, b, Hypothermia and survival in function of time after intraperitoneal injection of LPS (17 mg/kg; LD100) in mice pretreated with BB-94 ( , n = 16) or PBS/Tween 20 (■, n = 19) and in untreated mice (●, n = 14). Significance refers to the difference between each control group and the BB-94 group. c, d, Hypothermia and survival in function of time of MMP8−/− mice (, n = 34) and MMP8+/+ mice (■, n = 41) after intraperitoneal challenge with a lethal dose of LPS (17 mg/kg; LD100). e, f, Survival of MMP8+/+ mice pretreated with MMP8 inhibitor (, n = 5) or with vehicle (■, n = 4) after intraperitoneal LPS challenge (17 mg/kg; LD100), or left untreated (●, n = 5). Significance refers to the difference between each control group and the inhibitor-treated group. g, h, Hypothermia and survival of MMP8−/− mice (, n = 16) and MMP8+/+ mice (■, n = 22) in function of time after CLP. Data were analyzed with a χ2 test. i, j, Hypothermia and survival of male MMP8−/− mice (, n = 17) and MMP8+/+ mice (■, n = 17) after renal I/R.
, n = 16) or PBS/Tween 20 (■, n = 19) and in untreated mice (●, n = 14). Significance refers to the difference between each control group and the BB-94 group. c, d, Hypothermia and survival in function of time of MMP8−/− mice (, n = 34) and MMP8+/+ mice (■, n = 41) after intraperitoneal challenge with a lethal dose of LPS (17 mg/kg; LD100). e, f, Survival of MMP8+/+ mice pretreated with MMP8 inhibitor (, n = 5) or with vehicle (■, n = 4) after intraperitoneal LPS challenge (17 mg/kg; LD100), or left untreated (●, n = 5). Significance refers to the difference between each control group and the inhibitor-treated group. g, h, Hypothermia and survival of MMP8−/− mice (, n = 16) and MMP8+/+ mice (■, n = 22) in function of time after CLP. Data were analyzed with a χ2 test. i, j, Hypothermia and survival of male MMP8−/− mice (, n = 17) and MMP8+/+ mice (■, n = 17) after renal I/R.
LPS is known to activate the inflammatory cascade via the TLR4 receptor, which also recognizes several endogenous ligands (e.g., heparan sulfate, fibrinogen, and HMGB1) released during systemic inflammatory conditions, such as burns, trauma, and I/R. Hence, we tested the effect of MMP8 depletion in renal I/R, which is characterized by disruption of blood flow and diminished oxygen delivery and results in TLR4-dependent inflammation and organ damage (Schrier and Wang, 2004). Diminishing the activity of MMPs, especially MMP2 and MMP9, has been shown to improve the outcome of renal I/R injury in rats (Sutton et al., 2005). When male MMP8−/− and MMP8+/+ mice were subjected to standard renal I/R, the resulting hypothermia was less severe in the MMP8−/− mice (Fig. 1i). Moreover, all MMP8+/+ animals died within 3 days, whereas only a minority of the MMP8−/− animals died over 2 weeks (Fig. 1j).
MMP8 activity is detrimental to CNS barrier integrity
Shortly after LPS injection, de novo MMP8 expression was increased at least 50-fold in all organs tested (Fig. 2a–d). Because bone-marrow-derived cells, such as macrophages and neutrophils, are considered the most important LPS-responsive cells (Tobias et al., 1999) and MMP8 is constitutively present in azurophilic granules of neutrophils but can also be expressed by macrophages and non-myeloid cells (Van Lint and Libert, 2006; Dejonckheere et al., 2011a), we studied the importance of bone-marrow-derived cells in the resistance of MMP8−/− mice. Two independent bone marrow transplantation experiments yielded the same result (Fig. 2e): MMP8+/+ mice remained LPS sensitive after transplantation with MMP8−/− bone marrow. However, MMP8−/− mice partially lost their resistance after transplantation with MMP8+/+ bone marrow, but this occurred only at later time points. These data indicate that MMP8 produced by polymorphonuclear neutrophils or macrophages plays a role mainly in later stages of endotoxemia leading to death, whereas MMP8 from non-myeloid cells is important during the first hours of endotoxemia.
Figure 2.

a–d, RNA qPCR expression profile of MMP8 in liver (a), kidney (b), ileum (c), and lung (d) at different times after LPS challenge of MMP8+/+ mice (n = 5). e, Survival after LPS challenge of MMP8−/− and MMP8+/+ mice that underwent bone marrow transplantation. MMP8−/− mice reconstituted with MMP8−/− bone marrow ( , n = 7) or MMP8+/+ bone marrow (
, n = 7) or MMP8+/+ bone marrow ( , n = 6) versus control MMP8−/− mice (, n = 11). MMP8+/+ mice transplanted with MMP8−/− bone marrow (♦, n = 7) or MMP8+/+ bone marrow (●, n = 7) compared with control MMP8+/+ mice (■, n = 9). f, g, Permeability of the vascular endothelium of the kidney, lung, ileum, and liver in MMP8+/+ mice (■) and MMP8−/− mice () after LPS (f, n = 6) and renal I/R (g, n = 4). h, i, Permeability of the BCSFB determined by measuring leakage of fluorescently labeled dextran from the blood into the CSF during endotoxemia (h) and renal I/R (i) in MMP8−/− mice () and MMP8+/+ mice (■) (LPS, n = 9–15; I/R, n = 4; control and sham, n = 3–4) determined by measuring leakage of fluorescently labeled dextran from the blood into the CSF.
, n = 6) versus control MMP8−/− mice (, n = 11). MMP8+/+ mice transplanted with MMP8−/− bone marrow (♦, n = 7) or MMP8+/+ bone marrow (●, n = 7) compared with control MMP8+/+ mice (■, n = 9). f, g, Permeability of the vascular endothelium of the kidney, lung, ileum, and liver in MMP8+/+ mice (■) and MMP8−/− mice () after LPS (f, n = 6) and renal I/R (g, n = 4). h, i, Permeability of the BCSFB determined by measuring leakage of fluorescently labeled dextran from the blood into the CSF during endotoxemia (h) and renal I/R (i) in MMP8−/− mice () and MMP8+/+ mice (■) (LPS, n = 9–15; I/R, n = 4; control and sham, n = 3–4) determined by measuring leakage of fluorescently labeled dextran from the blood into the CSF.
Increased vascular permeability caused by endothelial cell injury and disruption of cellular junctions are essential steps in SIRS. Subsequent vascular leakage compromises tissue oxygenation and causes organ dysfunction. Indeed, preservation of vascular integrity was shown to be a potential therapeutic approach in sepsis (Groger et al., 2009; London et al., 2010). Evidence suggests that MMPs might play a critical role in endothelial function (Rodriguez et al., 2010). In our experiments, both MMP8+/+ and MMP8−/− mice displayed increased vascular permeability in kidney, lung, and ileum but not in liver during endotoxemia (Fig. 2f) and after I/R (Fig. 2g). This indicates that MMP8 is not the primary MMP responsible for the loss of vascular integrity in these organs during SIRS. However, barrier integrity in the CNS was severely affected. This was evidenced by leakage of fluorescently labeled dextran into the CSF as early as 4 h after LPS stimulation (Fig. 2h), as well as after kidney I/R (Fig. 2i). In contrast, MMP8−/− mice showed almost no sign of CNS barrier leakage after LPS or I/R for at least 8 h.
MMP8 depletion protects from BCSFB leakage after systemic inflammation
Three key interfaces form barriers between the blood and the CNS: the BBB, the BCSFB, and the arachnoid barrier (Abbott et al., 2010). MMP8 was recently implicated in disruption of the BBB by proteolytic cleavage of the brain endothelial tight junction (TJ) junction protein occludin (Schubert-Unkmeir et al., 2010). However, we did not observe fluorescence leakage from the vasculature into the brain tissue 8 h after challenge in MMP8+/+ mice (data not shown). Some speculation exists in the literature about the physiological relevance of MMPs in inflammatory breakdown of the BCSFB. It was shown recently that TNF-induced breakdown of the BCSFB in vitro can be partly blocked by the broad-spectrum MMP inhibitor GM-6001 (Zeni et al., 2007). This BCSFB consists of the CP, a richly vascularized invagination of specialized secretory epithelial cells in the lateral, third and fourth cerebral ventricles of the brain and is responsible for the secretion of CSF. To prevent paracellular transport of blood-borne substances into the ventricles, adjacent CP epithelial cells are connected by unique parallel TJs (Abbott et al., 2010). Early after LPS injection in MMP8+/+ mice, fluorescence leaked from the CP into the CSF and surrounding brain tissue (Fig. 3b). In contrast, in MMP8−/− mice (Fig. 3c) and in untreated mice (Fig. 3a), fluorescence was detected only in the highly fenestrated CP. Moreover, 4 h after LPS challenge, MMP8 was upregulated as both mRNA and protein specifically in the CP (Fig. 3c,d) but not in total brain (data not shown), which suggests that disruption of the BCSFB is MMP8 dependent.
Figure 3.

MMP8 depletion protects from BCSFB leakage after systemic inflammation. Visualization of BCSFB leakage (red) (a) before and after endotoxemia in MMP8+/+ mice (b) and MMP8−/− mice (c); the inset displays nuclear staining (blue) of the CP and the surrounding brain tissue by confocal microscopy. d, MMP8 mRNA expression in the CP before and 4 h after LPS challenge (n = 5–8). e, Confocal images of MMP8 (red) immunofluorescence after LPS challenge in the epithelial CP cells and ependymal cells.
Recent results from an in vitro model of bacterial meningitis showed that MMP8 proteolytically degrades the TJ protein occludin in brain endothelial cells. This led to speculation that this process might mediate the disruption of the BBB (Schubert-Unkmeir et al., 2010). Our in vivo data show that SIRS, in contrast to bacterial meningitis, is associated with rapid MMP8-dependent loss of the BCSFB, caused by leakage of the epithelial cell layer of the CP, whereas the BBB is unaffected. Based on the meningitis data (Schubert-Unkmeir et al., 2010), it is tempting to speculate that MMP8 might also induce disruption of the BCSFB by cleaving occludin. However, electron microscopic (EM) analysis of the CP did not reveal severe disruption of TJs (data not shown).
MMP8 induces morphological changes in the CP by cleavage of collagen I
The CP consists of a continuous monolayer of cuboidal to columnar epithelial cells attached to a core of connective tissue. On their basal side, CP epithelial cells form an extended basal labyrinth, i.e., intertwining of basolateral membranes (BMs) of adjacent cells, and make contact with the basal lamina [extracellular matrix (ECM)] delimiting the inner stroma of the highly vascularized connective tissue (Wolburg and Paulus, 2010). It has been shown that the composition of the ECM influences the behavior of CP epithelial cells, which indicates that the BM might promote organization of the CP epithelial cells into a functional epithelium and thus might maintain the integrity of the BCSFB (Stadler and Dziadek, 1996). Morphological EM analysis revealed that the basal labyrinth, which was intact in untreated mice (Fig. 4a–c), disappeared in MMP8+/+ mice 8 h after challenge with LPS (Fig. 4d–f) or by renal I/R (Fig. 4g–i). In contrast, the intertwining of BMs remained intact in MMP8−/− mice (Fig. 4j–o). Moreover, EM analysis also revealed that CP epithelial cells of challenged MMP8+/+ mice had lost their typical cuboidal morphology and had become round, resulting in a flattened epithelial cell layer (Fig. 4d,g,p,q) which was not observed in MMP8−/− mice (Fig. 4j,m,p,q).
Figure 4.
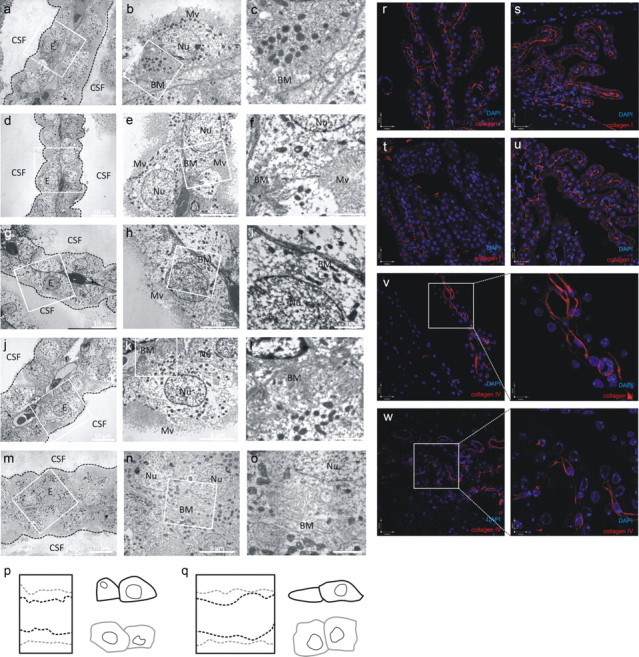
MMP8 induces morphological changes in the CP by cleavage of collagen I and not collagen IV. a–c, EM images of the CP of untreated mice. d–o, EM images of the CP of MMP8+/+ mice after LPS challenge (d–f) and ischemia reperfusion (g–i). EM images of the CP of MMP8−/− mice after LPS challenge (j–l) and ischemia reperfusion (m–o). E, Epithelial cell; Nu, nucleus; Mv, microvilli. p, q, Diagram overlay delineating the borders of the outer region of the CP and of two neighboring cells of MMP8+/+ (black) and MMP8−/− (gray) mice after LPS (p) and ischemia reperfusion (q). r–u, Collagen I staining (red) of the CP of unchallenged MMP8+/+ (r) and MMP8−/− (s) mice and MMP8+/+ (t) and MMP8−/− (u) mice 8 h after LPS stimulus. v, w, Representative confocal images of the CP of untreated (v) and LPS-stimulated (w) MMP8+/+ mice. Collagen IV is visualized in red and nuclei in blue.
Not much is known about the composition and function of the ECM deposited along the outer surface of the basal side of the CP epithelial cells. Based on in vitro and in vivo analyses, several matrix proteins, such as thrombospondin, fibronectin, laminin, and collagen types I, III, IV, and XVIII, are believed to be present (Gabrion et al., 1998; Wei et al., 2000; Haselbach et al., 2001; Utriainen et al., 2004; Thouvenot et al., 2006; Thanos et al., 2011). Because MMP8 has collagenolytic activity, we speculated that MMP8 modulates the CP epithelial BM by degradating collagen of the basal lamina. Indeed, collagen I was reduced in the basal lamina of MMP8+/+ mice after LPS challenge but was unaffected in MMP8−/− mice (Fig. 4r–u). In contrast, an MMP8-insensitive collagen, collagen IV, was unaffected by systemic inflammation (Fig. 4v,w).
MMP8 depletion reduces the effect of peripheral inflammation on the CNS
Peripheral inflammatory stimuli rapidly trigger the expression of different cytokines in the choroidal tissue. In contrast, the activation pattern in brain parenchyma is more restricted and delayed, suggesting that the CP mediates, via the BCSFB, interaction and/or signaling between the peripheral immune system and the brain (Quan et al., 1999; Konsman et al., 2002). MMPs might also promote inflammation by interfering with the ability of the CP to clear harmful compounds from the brain (Strazielle et al., 2003). In agreement with this and with the changes in basal labyrinth and morphology, CSF analysis revealed that the local inflammatory response in the brain of MMP8−/− mice was minor compared with MMP8+/+ mice, as reflected in the lower levels of several cytokines and chemokines (Fig. 5a). These cytokines can be produced either locally or by peripheral leukocytes entering the CNS through the disrupted BCSFB. Although we cannot fully exclude it, the absence of CD45 staining, in both MMP8+/+ and MMP8−/− mice, suggests that there is no infiltration of peripheral leukocytes into the brain tissue or CSF 8 h after challenge (Fig. 5b,c). However, in agreement with the literature, the increased cytokine levels in the CSF were correlated with an increase in cells positive for GFAP in the brain of MMP8+/+ mice after LPS challenge (Beurel and Jope, 2009) (Fig. 5d). GFAP is an intermediate filament protein and a marker of activated astrocytes. No increase in GFAP-positive cells was observed in MMP8−/− mice (Fig. 5e). The inducible isoform of nitric oxide synthase (iNOS) is a marker of activated microglia and astrocytes, known to be induced by LPS (Chang et al., 2008; Lu et al., 2010). iNOS is a well-established source of nitric oxide during inflammation of the CNS and was shown to play an important role in neurotoxicity after systemic LPS injection (Sierra et al., 2008). In agreement with this, iNOS expression in brain parenchyma showed a large increase after LPS challenge in MMP8+/+ mice but not in MMP8−/− mice (Fig. 5f). However, when the initial inflammatory activation of the CP itself was examined by determining iNOS and IL-6 levels 4 h after challenge, no difference between MMP8+/+ and MMP8−/− mice was observed (Fig. 5g,h).
Figure 5.
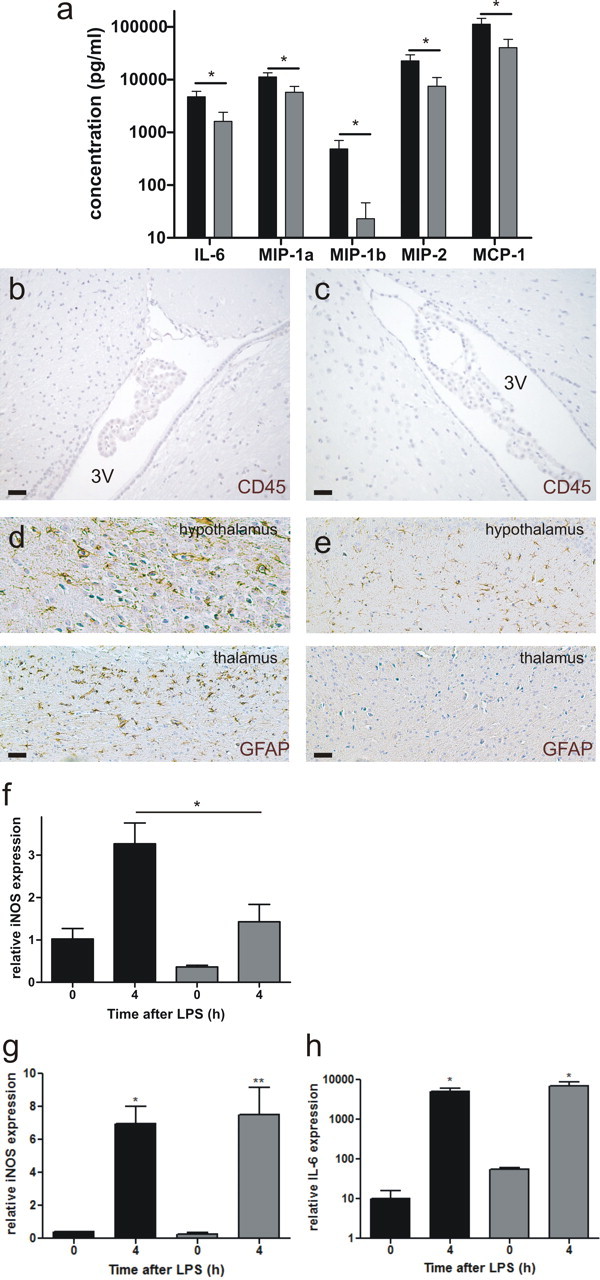
MMP8 depletion reduces the effect of peripheral inflammation on the CNS. a, Cytokine and chemokine profile in CSF isolated from MMP8+/+ mice (■, n = 8) and MMP8−/− mice (, n = 8) 8 h after LPS challenge. Data are expressed on a log10 scale. Cytokines and chemokines were determined by BioPlex assay. Levels in unstimulated mice were negligible. Absence of white blood cell influx in the brain tissue surrounding the third ventricle (3V) in MMP8+/+ (b) and MMP8−/− (c) mice 8 h after endotoxemia induction, determined by CD45 immunohistochemistry (brown). Scale bar, 20 μm. GFAP-positive cells (brown) in the hypothalamus (d) and thalamus (e) of LPS-stimulated mice, 8 h after stimulus. Scale bar, 20 μm. f, Relative iNOS expression in the brain, before and 4 h after LPS injection in MMP8+/+ mice (■, n = 5) and MMP8−/− mice (, n = 5). iNOS (g) and IL-6 (h) mRNA levels in the CP before and 4 h after LPS challenge in MMP8+/+ (■) and MMP8−/− () mice (0 h, n = 3; 4 h, n = 5).
Consequences of MMP8-dependent BCSFB disruption on peripheral toxicity via hypothalamic–pituitary–adrenal (HPA) axis disruption and consequent MOF
HPA axis activation is a consequence of systemic inflammation and is necessary for dampening of the inflammatory reaction of the body. To assess HPA axis activation in MMP8+/+ and MMP8−/− mice, we determined systemic corticosterone levels (Fig. 6a). Both groups of mice showed comparable increases in corticosterone early during endotoxemia, which rules out differences in initial HPA axis stimulation. However, the anti-inflammatory properties of corticosterone are closely dependent on glucocorticoid receptor (GR) levels. LPS challenge was shown to downregulate GR expression levels in microglia, which might be a prerequisite for suppression of the anti-inflammatory actions of endogenous steroid hormones on the immune system (Sierra et al., 2008). Indeed, we observed GR downregulation in the brain during endotoxemia in MMP8+/+ but not in MMP8−/− mice (Fig. 6b). These data indicate that the protection of the BCSFB in MMP8−/− mice limits local cytokine production. This ensures the maintenance of GR levels and proper anti-inflammatory responses, which subsequently should prevent a lethal systemic cytokine storm.
Figure 6.
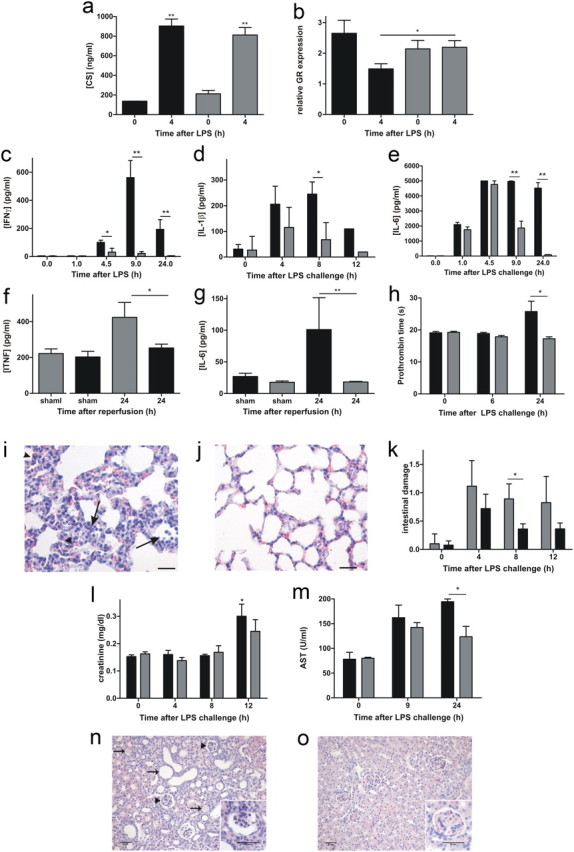
Consequences of MMP8-dependent BCSFB disruption on peripheral toxicity via HPA axis disruption and consequent MOF. a, Plasma corticosterone (CS) levels determined by RIA in MMP8−/− mice () and MMP8+/+ mice (■) (0 h, n = 2–3; 4 h, n = 4). Data were analyzed with an unpaired t test. b, GR levels determined by qPCR in brain lysate of MMP8+/+ (■, n = 4) and MMP8−/− (, n = 4) mice before and 4 h after LPS challenge. Data were analyzed with an unpaired t test. Serum levels of IFNγ (c, n = 7), IL-1β (d, n = 6) and IL-6 (e, n = 6) in MMP8−/− mice () and MMP8+/+ mice (■) in function of time after a lethal intraperitoneal LPS challenge. IFNγ was determined by Cytometrix bead assay and IL-1β by BioPlex assay. Serum TNF (f) and IL-6 (g) levels in male MMP8−/− mice (, n = 5) and MMP8+/+ mice (■, n = 5) 24 h after renal I/R. Cytokines were determined by BioPlex assay. h, Prothrombin time, measured 0, 6, and 24 h after intraperitoneal LPS challenge of MMP8+/+ mice (■, n = 6) and MMP8−/− mice (, n = 6). Data are expressed as percentage of normal based on a standard curve of normal pooled plasma. i, j, Histopathology (hematoxylin and eosin staining) of lung sections of MMP8+/+ (i) and MMP8−/− (j) mice, 12 h after LPS. LPS-induced lung injury was characterized by influx of inflammatory cells (arrows) and thickening of the alveolar wall and atelectasis (heads). k, Quantification of intestinal tissue damage in MMP8+/+ (■) and MMP8−/− () mice: four neutral observers evaluated hematoxylin and eosin stained sections (n = 3–5) in a blinded setup. LPS-induced intestinal damage is characterized by decreased villus height, disappearance of the mucus layer and Goblet cells along the villus, cell death at the villus top, and cell debris in the lumen. l, Creatinine accumulation in circulation of MMP8+/+ mice (■) and MMP8−/− mice () 0, 4, 8, and 12 h after LPS challenge (n = 4–5). m, AST levels in circulation of MMP8+/+ mice (■) and MMP8−/− mice () (n = 3–5). Data were analyzed with an unpaired t test. n, o, Histopathology (hematoxylin and eosin staining) of kidney sections of female MMP8+/+ mice (n) and MMP8−/− mice (o) 12 d after the induction of sublethal kidney I/R. Recovery from renal injury is shown by the presence of dilated tubuli (arrows), and hyperfiltration is shown by widening of the urinary space of Bowman's capsule and the increased number of mesenchymal cells in the glomeruli (heads and insert). Scale bar, 50 μm.
Hence, we studied cytokine levels in sera of challenged mice. Serum IFNγ, IL-1β, and IL-6 levels were significantly lower in MMP8−/− mice than in MMP8+/+ mice after LPS challenge (Fig. 6c–e), whereas the levels and kinetics of TNF were comparable (data not shown). After kidney I/R, the accumulation of TNF and IL-6 24 h after I/R was significantly lower in MMP8−/− than in MMP8+/+ mice (Fig. 6f,g). These results confirm the reduction of systemic inflammation in MMP8−/− mice.
Next, we analyzed the consequences of the increased systemic inflammation, such as coagulation and MOF. LPS-induced coagulation, which is believed to be a major cause of organ damage (Levi, 2007), was significantly less in MMP8−/− mice than in MMP8+/+ mice, as shown by the significantly shorter clotting times (Fig. 6h). MOF is most likely the direct cause of death in patients with SIRS. The organs that fail most frequently in sepsis patients are the lungs (Martin et al., 2003). Our histological analysis revealed that MMP8 deficiency protects against the progression of lung parenchymal damage during endotoxemia: 12 h after challenge, thickening of the alveolar wall, inflammatory cell influx, and atelectasis (collapsed alveoli) were more severe in MMP8+/+ mice (Fig. 6i) than in MMP8−/− mice (Fig. 6j). The “gut as motor of sepsis” hypothesis has been postulated based on the importance of the intestinal epithelium as a physical barrier between the intestinal lumen and the immune cells in the lamina propria (Carrico et al., 1986; Deitch and Berg, 1987). Indeed, in critically ill SIRS patients, intestinal permeability is correlated with bacterial translocation and subsequent MOF (Swank and Deitch, 1996; Faries et al., 1998). Morphological analysis of the small intestine showed some reduction in intestinal damage in MMP8−/− mice after LPS challenge (Fig. 6k). Mortality from sepsis is dramatically increased in the presence of acute kidney injury (Schrier and Wang, 2004). Histological analysis revealed no obvious kidney injury during the first 12 h of endotoxemia (data not shown), and creatinine levels increased only slightly in MMP8+/+ mice (Fig. 6l). Liver dysfunction is also common during sepsis. Our histological analysis showed no liver damage (data not shown), whereas AST levels increased in MMP8+/+ but not in MMP8−/− mice, especially 24 h after LPS challenge (Fig. 6m). Histopathology caused by sublethal I/R showed that MMP8−/− mice display significantly weaker signs of recovery from renal damage. In contrast to MMP8−/− mice, MMP8+/+ mice displayed glomerular hyperfiltration, as shown by widening of the urinary space of Bowman's capsule, increased number of mesenchymal cells in the glomeruli, and dilatation of tubuli, all of which are manifestations of previous tubular damage (Fig. 6n,o). These data show that, in endotoxemia and in renal I/R, systemic inflammation and consequent MOF is reduced in MMP8−/− mice relative to MMP8+/+ mice.
Discussion
Several studies indicate that neurological complications, such as SIRS-associated encephalopathy, frequently occur in SIRS patients and that the brain might be one of the first organs affected (Ebersoldt et al., 2007). This notion is supported by the association between brain dysfunction and higher mortality in SIRS patients: 49% mortality in septic patients with an acutely altered mental status and 26% mortality in those with normal status have been reported (Sprung et al., 1990; Ebersoldt et al., 2007). Hence, it is believed that brain dysfunction contributes to mortality in SIRS (Sprung et al., 1990).
The cellular and molecular mechanisms involved in SIRS-associated encephalopathy remain unknown (Flierl et al., 2010). Induction of a SIRS by LPS injection in mice and rats evokes microglia activation, cell death, metabolic changes, and loss of neurons in different regions of the brain (Semmler et al., 2005; Czapski et al., 2007; Qin et al., 2007; Semmler et al., 2008; Czapski et al., 2010). Similarly, kidney ischemia, another TLR4-dependent systemic disease resulting in SIRS, induces brain inflammation (Liu et al., 2008). Septic encephalopathy was shown to be associated with increased protein leakage from the circulation into the CSF (Young et al., 1992) as a result of breakdown of the CNS barriers, which leads to disruption of CNS homeostasis. Most research on CNS barriers, especially in light of systemic inflammation, focuses on the BBB, whereas disruption of the BCSFB is seldom examined. Nevertheless, the CP, the epithelial cell layer that prevents diffusion of blood-borne substances into the brain ventricles containing the CSF, is thought to act as a brain immune sensor involved in the communication between the peripheral immune system and the CNS (Marques et al., 2007). Our in vivo data show that the BCSFB is the first and most important barrier affected in systemic diseases such as SIRS.
Recent evidence suggests that MMP8 is involved in the pathogenesis of sepsis. Solan et al. (2012) reported that MMP8 inhibition can improve outcome in experimental sepsis. Moreover, serum MMP8 levels at admission to intensive care unit were high among sepsis patients with fatal outcome (Lauhio et al., 2011). In agreement with this, we show that genetic and pharmacologic inhibition of MMP8 improves survival in LPS-, CLP-, and kidney I/R-induced sepsis and SIRS. Moreover, we demonstrate that MMP8 is a critical component in the neurotoxic effects after systemic inflammation, and we show that the primary and detrimental role of MMP8 is manifested at the BCSFB. This is in agreement with a recent report showing that broad-spectrum MMP inhibition protects from TNF-dependent BCSFB leakage in vitro (Zeni et al., 2007). However, based on our bone marrow transplantation experiments, we do not exclude an MMP8-dependent effect in organs other than the brain. For example, MMP8−/− mice are reported to be protected against local LPS-induced inflammation in the skin, and this effect was attributed to MMP8 activation of LPS-induced CXC chemokine (Tester et al., 2007). However, in our systemic model, we found no evidence that neutrophil migration into tissues is disturbed in MMP8−/− mice (data not shown). MMPs can also have a direct effect on cytokine activity. MMP8 was shown to cleave pro-IL-1β into the mature form in vitro (Schonbeck et al., 1998), but we could not show this for bioactive MMP8 (data not shown). Moreover, the onset of brain dysfunction that we observed in MMP8+/+ mice, but not in MMP8−/− mice, precedes the systemic differences in cytokine levels and MOF between the two genotypes. This indicates that the observed brain dysfunction in SIRS is not a consequence of the different inflammation states of MMP8+/+ and MMP8−/− mice.
Recently, MMP8 was shown to proteolytically degrade the TJ protein occludin in an in vitro model of bacterial meningitis, which resulted in the disruption of the BBB (Schubert-Unkmeir et al., 2010). Consequently, we speculated that MMP8 might also induce disruption of the BCSFB by cleavage of occludin, but EM analysis of the CP did not reveal disruption of TJs (data not shown).
Peripheral inflammatory stimuli rapidly stimulate the expression of inflammatory mediators in the choroidal tissue (Strazielle and Ghersi-Egea, 2000). Indeed, we observed increased iNOS and IL-6 levels in the isolated CP early after challenge in both MMP8+/+ and MMP8−/− mice. However, we detected notable morphological changes in the CP after stimulation only in MMP8+/+ mice. The CP epithelial cells that form the BCSFB contain microvilli at the apical side and at the basal side an extended basal labyrinth that makes contact with the basal lamina. The tight attachment of cells to the underlying basement membrane is necessary for their functional differentiation (Merker, 1994). Moreover, proper function of the BBB has been shown to rely critically on sustained interaction with the adjacent ECM components (Engelhardt and Sorokin, 2009). Apparently, this also holds true for the BCSFB because we observed MMP8-dependent collagen I cleavage, which affects the composition of the basal lamina of the CP and thereby influences epithelial cell morphology and consequently also barrier integrity. We found LPS-induced upregulation of MMP8 expression on both mRNA and protein level specifically at the CP and the ependymal epithelial cells that line the CSF-filled ventricles in the brain. Collagen I, but not collagen IV, was clearly reduced in the CP basal lamina of LPS-treated MMP8+/+ mice; none of the collagens was affected in MMP8−/− mice. Detailed analysis of the CP epithelial cells by EM revealed that peripheral inflammation in the MMP8+/+ mice induced disappearance of the basal labyrinth, i.e., the intertwining of the BMs responsible for cell–matrix contact. Additionally, the epithelial cells lost their typical cuboidal morphology. In contrast, the CP of MMP8−/− mice was not affected by systemic inflammation.
Our data suggest that MMP8 aggravates, via BCSFB disruption, the response of the CNS to systemic inflammation. We further confirmed this hypothesis by analyzing CSF from MMP8+/+ and MMP8−/− mice after induction of systemic inflammation. These experiments demonstrated increased cytokine and chemokine levels in the CSF from LPS-injected MMP8+/+ mice, 8 h after stimulus. However, CD45 immunostaining revealed no apparent white blood cell influx at this time point. This was expected because there is no direct evidence for entry of immune cell into the CNS across the BCSFB (Engelhardt and Sorokin, 2009). However, based on GFAP (Beurel and Jope, 2009) and iNOS (Chang et al., 2008) measurements, we did observe an increase in activated astrocytes and microglia, respectively, in MMP8+/+ mice but not in MMP8−/− mice.
Activation of the HPA axis has a key role in the crosstalk between the immune and the neuroendocrine systems and is crucial in restoring physiological homeostasis after immune activation (Beishuizen and Thijs, 2003). Consequently, impairment of the anti-inflammatory activity of the HPA axis is a frequent complication of sepsis that contributes to morbidity and mortality (Sprung et al., 1990; Chrousos, 1995). Although corticosterone serum levels were comparable in MMP8+/+ and MMP8−/− mice, GR was downregulated only in the MMP8+/+ brain. GR downregulation might be a prerequisite for suppression of the anti-inflammatory actions of endogenous steroid hormones on the immune system (Sierra et al., 2008).
Based on their higher serum cytokine levels, MMP8+/+ mice display a reduced anti-inflammatory response, which results in more organ damage compared with MMP8−/− mice, especially at later time points. None of the detected differences in MOF correlate with the observed early effects of MMP8 deficiency in the brain. Hence, this observation confirms our hypothesis that the observed brain dysfunction in SIRS in MMP8+/+ mice leads, via perturbation of the HPA axis, to increased systemic inflammation with consequent organ failure and not the other way around.
Based on our findings, we hypothesize that, in systemic inflammation, MMP8 mediates disruption of the BCSFB via collagen I cleavage (Fig. 7). We believe that activation of MMP8 in the CP contributes to early onset of neuroinflammation before MOF in SIRS and sepsis. Subsequent brain inflammation impedes the regulation by the CNS of the inflammatory responses in the brain and periphery and thereby contributes to the exaggerated and lethal inflammatory response in endotoxemia and renal I/R.
Figure 7.

Effect of TLR4-mediated pathologies and MMP8 deficiency on BCSFB and lethality. 1, During a trigger, such as sepsis or kidney I/R, systemic cytokine levels are increased. 2, This increase results in MMP8 upregulation in the CP, a highly perfused organ in which a single epithelial cell layer separates the blood from the CSF. Importantly, the CP regulates homeostasis in the CNS but also participates in the interaction between the periphery and the brain. 3, The increase in MMP8 subsequently leads to collagen I cleavage, resulting in morphological changes of the choroidal epithelial cells: loss of cuboidal shape and loss of basal labyrinth. 4, Next, this injury of the CP causes impairment of the BCSFB and increases local cytokine and chemokine levels. 5, When these chemokines and cytokines leak into the surrounding brain tissue, this results in increased local inflammation and downregulation of the GR. Steps 3–5 are absent or reduced in the MMP8−/− mice. 6, Increased systemic cytokine levels also lead to activation of the HPA axis, which results in upregulation of corticosterone (CS) serum levels. 7, CS activates to the GR, which is an important feedback mechanism in inflammatory conditions, ensuring the appropriate dampening of the inflammatory response. 8, Downregulation of the GR in the brain after LPS challenge, detected in the MMP8+/+ but not in the MMP8−/− mice, reduces the anti-inflammatory effect of the corticosterone activated GR. 9, The resulting exaggerated immune response in the MMP8+/+ mice causes MOF and death, which is reduced in the MMP8−/− mice.
In conclusion, we identified MMP8 as a candidate new drug target in systemic inflammatory diseases, and we propose that the mechanism of action of MMP8 involves disruption of the integrity of the BCSFB.
Footnotes
This work was supported by the agency for the Innovation by Science and Technology in Flanders, the Research Council of Ghent University, the Research Foundation Flanders, and the Interuniversity Attraction Poles Program of the Belgian Science Policy (IAP-VI-18). Work at the laboratory of C.L.-O. was supported by grants from the Ministry of Education and Science of Spain, the M. Botín Foundation, the Lilly Foundation, and the European Union. Work at the laboratory of P.M. was supported by grants from the Dormeur Foundation. We thank Amin Bredan for editing the manuscript, British Biotech for providing BB-94, Dr. K. Devreese for coagulation tests, Dr. C. Guérin for suggestions concerning the confocal studies, Dr. W. Waelput for help with the histopathology, and Sophie Steeland for excellent technical assistance.
References
- Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- Balbín M, Fueyo A, Tester AM, Pendás AM, Pitiot AS, Astudillo A, Overall CM, Shapiro SD, López-Otín C. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat Genet. 2003;35:252–257. doi: 10.1038/ng1249. [DOI] [PubMed] [Google Scholar]
- Barie PS. The last Xigris® survivor. Surg Infect (Larchmt) 2011;12:423–425. doi: 10.1089/sur.2011.9909. [DOI] [PubMed] [Google Scholar]
- Beishuizen A, Thijs LG. Endotoxin and the hypothalamo-pituitary-adrenal (HPA) axis. J Endotoxin Res. 2003;9:3–24. doi: 10.1179/096805103125001298. [DOI] [PubMed] [Google Scholar]
- Beurel E, Jope RS. Lipopolysaccharide-induced interleukin-6 production is controlled by glycogen synthase kinase-3 and STAT3 in the brain. J Neuroinflammation. 2009;6:9. doi: 10.1186/1742-2094-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasone A, Tortorella P, Campestre C, Agamennone M, Preziuso S, Chiappini M, Nuti E, Carelli P, Rossello A, Mazza F, Gallina C. alpha-Biphenylsulfonylamino 2-methylpropyl phosphonates: enantioselective synthesis and selective inhibition of MMPs. Bioorg Med Chem. 2007;15:791–799. doi: 10.1016/j.bmc.2006.10.047. [DOI] [PubMed] [Google Scholar]
- Cantaluppi V, Assenzio B, Pasero D, Romanazzi GM, Pacitti A, Lanfranco G, Puntorieri V, Martin EL, Mascia L, Monti G, Casella G, Segoloni GP, Camussi G, Ranieri VM. Polymyxin-B hemoperfusion inactivates circulating proapoptotic factors. Intensive Care Med. 2008;34:1638–1645. doi: 10.1007/s00134-008-1124-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrico CJ, Meakins JL, Marshall JC, Fry D, Maier RV. Multiple-organ-failure syndrome. Arch Surg. 1986;121:196–208. doi: 10.1001/archsurg.1986.01400020082010. [DOI] [PubMed] [Google Scholar]
- Chang YC, Kim HW, Rapoport SI, Rao JS. Chronic NMDA administration increases neuroinflammatory markers in rat frontal cortex: cross-talk between excitotoxicity and neuroinflammation. Neurochem Res. 2008;33:2318–2323. doi: 10.1007/s11064-008-9731-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrousos GP. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med. 1995;332:1351–1362. doi: 10.1056/NEJM199505183322008. [DOI] [PubMed] [Google Scholar]
- Cruz DN, Perazella MA, Bellomo R, de Cal M, Polanco N, Corradi V, Lentini P, Nalesso F, Ueno T, Ranieri VM, Ronco C. Effectiveness of polymyxin B-immobilized fiber column in sepsis: a systematic review. Crit Care. 2007;11:R47. doi: 10.1186/cc5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz DN, Antonelli M, Fumagalli R, Foltran F, Brienza N, Donati A, Malcangi V, Petrini F, Volta G, Bobbio Pallavicini FM, Rottoli F, Giunta F, Ronco C. Early use of polymyxin B hemoperfusion in abdominal septic shock: the EUPHAS randomized controlled trial. JAMA. 2009;301:2445–2452. doi: 10.1001/jama.2009.856. [DOI] [PubMed] [Google Scholar]
- Czapski GA, Cakala M, Chalimoniuk M, Gajkowska B, Strosznajder JB. Role of nitric oxide in the brain during lipopolysaccharide-evoked systemic inflammation. J Neurosci Res. 2007;85:1694–1703. doi: 10.1002/jnr.21294. [DOI] [PubMed] [Google Scholar]
- Czapski GA, Gajkowska B, Strosznajder JB. Systemic administration of lipopolysaccharide induces molecular and morphological alterations in the hippocampus. Brain Res. 2010;1356:85–94. doi: 10.1016/j.brainres.2010.07.096. [DOI] [PubMed] [Google Scholar]
- Deitch EA, Berg R. Bacterial translocation from the gut: a mechanism of infection. J Burn Care Rehabil. 1987;8:475–482. [PubMed] [Google Scholar]
- Dejager L, Pinheiro I, Dejonckheere E, Libert C. Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol. 2011;19:198–208. doi: 10.1016/j.tim.2011.01.001. [DOI] [PubMed] [Google Scholar]
- Dejonckheere E, Vandenbroucke RE, Libert C. Matrix metalloproteinase8 has a central role in inflammatory disorders and cancer progression. Cytokine Growth Factor Rev. 2011a;22:73–81. doi: 10.1016/j.cytogfr.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Dejonckheere E, Vandenbroucke RE, Libert C. Matrix metalloproteinases as drug targets in ischaemia/reperfusion injury. Drug Discov Today. 2011b;16:762–778. doi: 10.1016/j.drudis.2011.06.009. [DOI] [PubMed] [Google Scholar]
- Ebersoldt M, Sharshar T, Annane D. Sepsis-associated delirium. Intensive Care Med. 2007;33:941–950. doi: 10.1007/s00134-007-0622-2. [DOI] [PubMed] [Google Scholar]
- Engelhardt B, Sorokin L. The blood-brain and the blood-cerebrospinal fluid barriers: function and dysfunction. Semin Immunopathol. 2009;31:497–511. doi: 10.1007/s00281-009-0177-0. [DOI] [PubMed] [Google Scholar]
- Faries PL, Simon RJ, Martella AT, Lee MJ, Machiedo GW. Intestinal permeability correlates with severity of injury in trauma patients. J Trauma. 1998;44:1031–1035. doi: 10.1097/00005373-199806000-00016. discussion 1035–1036. [DOI] [PubMed] [Google Scholar]
- Flierl MA, Rittirsch D, Huber-Lang MS, Stahel PF. Pathophysiology of septic encephalopathy—an unsolved puzzle. Crit Care. 2010;14:165. doi: 10.1186/cc9035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrion JB, Herbuté S, Bouillé C, Maurel D, Kuchler-Bopp S, Laabich A, Delaunoy JP. Ependymal and choroidal cells in culture: characterization and functional differentiation. Microsc Res Tech. 1998;41:124–157. doi: 10.1002/(SICI)1097-0029(19980415)41:2<124::AID-JEMT3>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Gröger M, Pasteiner W, Ignatyev G, Matt U, Knapp S, Atrasheuskaya A, Bukin E, Friedl P, Zinkl D, Hofer-Warbinek R, Zacharowski K, Petzelbauer P, Reingruber S. Peptide Bbeta(15–42) preserves endothelial barrier function in shock. PLoS One. 2009;4:e5391. doi: 10.1371/journal.pone.0005391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haselbach M, Wegener J, Decker S, Engelbertz C, Galla HJ. Porcine choroid plexus epithelial cells in culture: regulation of barrier properties and transport processes. Microsc Res Tech. 2001;52:137–152. doi: 10.1002/1097-0029(20010101)52:1<137::AID-JEMT15>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Higuchi R. Rapid, efficient DNA extraction for PCR from cells or blood. Amplifications. 1989;2:1–3. [Google Scholar]
- Hutchens MP, Dunlap J, Hurn PD, Jarnberg PO. Renal ischemia: does sex matter? Anesth Analg. 2008;107:239–249. doi: 10.1213/ane.0b013e318178ca42. [DOI] [PubMed] [Google Scholar]
- Konsman JP, Parnet P, Dantzer R. Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci. 2002;25:154–159. doi: 10.1016/s0166-2236(00)02088-9. [DOI] [PubMed] [Google Scholar]
- Lauhio A, Hästbacka J, Pettilä V, Tervahartiala T, Karlsson S, Varpula T, Varpula M, Ruokonen E, Sorsa T, Kolho E. Serum MMP-8, -9 and TIMP-1 in sepsis: high serum levels of MMP-8 and TIMP-1 are associated with fatal outcome in a multicentre, prospective cohort study. Hypothetical impact of tetracyclines. Pharmacol Res. 2011;64:590–594. doi: 10.1016/j.phrs.2011.06.019. [DOI] [PubMed] [Google Scholar]
- Levi M. Disseminated intravascular coagulation. Critical care medicine. 2007;35:2191–2195. doi: 10.1097/01.ccm.0000281468.94108.4b. [DOI] [PubMed] [Google Scholar]
- Lewis SM, Bain BJ, Bates I, editors. Dacie and Lewis practical haematology. London: Churchill Livingstone; 2001. [Google Scholar]
- Liu M, Liang Y, Chigurupati S, Lathia JD, Pletnikov M, Sun Z, Crow M, Ross CA, Mattson MP, Rabb H. Acute kidney injury leads to inflammation and functional changes in the brain. J Am Soc Nephrol. 2008;19:1360–1370. doi: 10.1681/ASN.2007080901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London NR, Zhu W, Bozza FA, Smith MC, Greif DM, Sorensen LK, Chen L, Kaminoh Y, Chan AC, Passi SF, Day CW, Barnard DL, Zimmerman GA, Krasnow MA, Li DY. Targeting Robo4-dependent Slit signaling to survive the cytokine storm in sepsis and influenza. Sci Transl Med. 2010;2:23ra19. doi: 10.1126/scitranslmed.3000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Ma L, Ruan L, Kong Y, Mou H, Zhang Z, Wang Z, Wang JM, Le Y. Resveratrol differentially modulates inflammatory responses of microglia and astrocytes. J Neuroinflammation. 2010;7:46. doi: 10.1186/1742-2094-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangoni NR, Melo GD, Moraes OC, Souza MS, Perri SH, Machado GF. Levels of matrix metalloproteinase-2 and metalloproteinase-9 in the cerebrospinal fluid of dogs with visceral leishmaniasis. Parasite Immunol. 2011;33:330–334. doi: 10.1111/j.1365-3024.2011.01285.x. [DOI] [PubMed] [Google Scholar]
- Marques F, Sousa JC, Correia-Neves M, Oliveira P, Sousa N, Palha JA. The choroid plexus response to peripheral inflammatory stimulus. Neuroscience. 2007;144:424–430. doi: 10.1016/j.neuroscience.2006.09.029. [DOI] [PubMed] [Google Scholar]
- Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Eng J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- Merker HJ. Morphology of the basement membrane. Microsc Res Tech. 1994;28:95–124. doi: 10.1002/jemt.1070280203. [DOI] [PubMed] [Google Scholar]
- Owens T, Bechmann I, Engelhardt B. Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol. 2008;67:1113–1121. doi: 10.1097/NEN.0b013e31818f9ca8. [DOI] [PubMed] [Google Scholar]
- Pagenstecher A, Stalder AK, Kincaid CL, Volk B, Campbell IL. Regulation of matrix metalloproteinases and their inhibitor genes in lipopolysaccharide-induced endotoxemia in mice. Am J Pathol. 2000;157:197–210. doi: 10.1016/S0002-9440(10)64531-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev. 2004;4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan N, Stern EL, Whiteside MB, Herkenham M. Induction of pro-inflammatory cytokine mRNAs in the brain after peripheral injection of subseptic doses of lipopolysaccharide in the rat. J Neuroimmunol. 1999;93:72–80. doi: 10.1016/s0165-5728(98)00193-3. [DOI] [PubMed] [Google Scholar]
- Rangel-Frausto MS, Pittet D, Costigan M, Hwang T, Davis CS, Wenzel RP. The natural history of the systemic inflammatory response syndrome (SIRS). A prospective study. JAMA. 1995;273:117–123. [PubMed] [Google Scholar]
- Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31–36. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta. 2010;1803:39–54. doi: 10.1016/j.bbamcr.2009.09.015. [DOI] [PubMed] [Google Scholar]
- Schönbeck U, Mach F, Libby P. Generation of biologically active IL-1 beta by matrix metalloproteinases: a novel caspase-1-independent pathway of IL-1 beta processing. J Immunol. 1998;161:3340–3346. [PubMed] [Google Scholar]
- Schrier RW, Wang W. Acute renal failure and sepsis. N Eng J Med. 2004;351:159–169. doi: 10.1056/NEJMra032401. [DOI] [PubMed] [Google Scholar]
- Schubert-Unkmeir A, Konrad C, Slanina H, Czapek F, Hebling S, Frosch M. Neisseria meningitidis induces brain microvascular endothelial cell detachment from the matrix and cleavage of occludin: a role for MMP-8. PLoS Pathog. 2010;6:e1000874. doi: 10.1371/journal.ppat.1000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semmler A, Okulla T, Sastre M, Dumitrescu-Ozimek L, Heneka MT. Systemic inflammation induces apoptosis with variable vulnerability of different brain regions. J Chem Neuroanat. 2005;30:144–157. doi: 10.1016/j.jchemneu.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Semmler A, Hermann S, Mormann F, Weberpals M, Paxian SA, Okulla T, Schäfers M, Kummer MP, Klockgether T, Heneka MT. Sepsis causes neuroinflammation and concomitant decrease of cerebral metabolism. J Neuroinflammation. 2008;5:38. doi: 10.1186/1742-2094-5-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Gottfried-Blackmore A, Milner TA, McEwen BS, Bulloch K. Steroid hormone receptor expression and function in microglia. Glia. 2008;56:659–674. doi: 10.1002/glia.20644. [DOI] [PubMed] [Google Scholar]
- Solan PD, Dunsmore KE, Denenberg AG, Odoms K, Zingarelli B, Wong HR. A novel role for matrix metalloproteinase-8 in sepsis*. Crit Care Med. 2012;40:379–387. doi: 10.1097/CCM.0b013e318232e404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprung CL, Peduzzi PN, Shatney CH, Schein RM, Wilson MF, Sheagren JN, Hinshaw LB. Impact of encephalopathy on mortality in the sepsis syndrome. The Veterans Administration Systemic Sepsis Cooperative Study Group. Crit Care Med. 1990;18:801–806. doi: 10.1097/00003246-199008000-00001. [DOI] [PubMed] [Google Scholar]
- Stadler E, Dziadek M. Extracellular matrix penetration by epithelial cells is influenced by quantitative changes in basement membrane components and growth factors. Exp Cell Res. 1996;229:360–369. doi: 10.1006/excr.1996.0381. [DOI] [PubMed] [Google Scholar]
- Stephenson JA, Gravante G, Butler NA, Sorge R, Sayers RD, Bown MJ. The Systemic Inflammatory Response Syndrome (SIRS)—number and type of positive criteria predict interventions and outcomes in acute surgical admissions. World J Surg. 2010;34:2757–2764. doi: 10.1007/s00268-010-0709-8. [DOI] [PubMed] [Google Scholar]
- Strazielle N, Ghersi-Egea JF. Choroid plexus in the central nervous system: biology and physiopathology. J Neuropathol Exp Neurol. 2000;59:561–574. doi: 10.1093/jnen/59.7.561. [DOI] [PubMed] [Google Scholar]
- Strazielle N, Khuth ST, Murat A, Chalon A, Giraudon P, Belin MF, Ghersi-Egea JF. Pro-inflammatory cytokines modulate matrix metalloproteinase secretion and organic anion transport at the blood-cerebrospinal fluid barrier. J Neuropathol Exp Neurol. 2003;62:1254–1264. doi: 10.1093/jnen/62.12.1254. [DOI] [PubMed] [Google Scholar]
- Sutton TA, Kelly KJ, Mang HE, Plotkin Z, Sandoval RM, Dagher PC. Minocycline reduces renal microvascular leakage in a rat model of ischemic renal injury. Am J Physiol Renal Physiol. 2005;288:F91–F97. doi: 10.1152/ajprenal.00051.2004. [DOI] [PubMed] [Google Scholar]
- Swank GM, Deitch EA. Role of the gut in multiple organ failure: bacterial translocation and permeability changes. World J Surg. 1996;20:411–417. doi: 10.1007/s002689900065. [DOI] [PubMed] [Google Scholar]
- Tester AM, Cox JH, Connor AR, Starr AE, Dean RA, Puente XS, López-Otín C, Overall CM. LPS responsiveness and neutrophil chemotaxis in vivo require PMN MMP-8 activity. PloS One. 2007;2:e312. doi: 10.1371/journal.pone.0000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanos C, Bintz B, Goddard M, Boekelheide K, Hall S, Emerich D. Functional modulation of choroid plexus epithelial clusters in vitro for tissue repair applications. Cell Transplant. 2011 doi: 10.3727/096368911X564985. [DOI] [PubMed] [Google Scholar]
- Thouvenot E, Lafon-Cazal M, Demettre E, Jouin P, Bockaert J, Marin P. The proteomic analysis of mouse choroid plexus secretome reveals a high protein secretion capacity of choroidal epithelial cells. Proteomics. 2006;6:5941–5952. doi: 10.1002/pmic.200600096. [DOI] [PubMed] [Google Scholar]
- Tobias PS, Tapping RI, Gegner JA. Endotoxin interactions with lipopolysaccharide-responsive cells. Clin Infect Dis. 1999;28:476–481. doi: 10.1086/515163. [DOI] [PubMed] [Google Scholar]
- Tsuge M, Yasui K, Ichiyawa T, Saito Y, Nagaoka Y, Yashiro M, Yamashita N, Morishima T. Increase of tumor necrosis factor-alpha in the blood induces early activation of matrix metalloproteinase-9 in the brain. Microbiol Immunol. 2010;54:417–424. doi: 10.1111/j.1348-0421.2010.00226.x. [DOI] [PubMed] [Google Scholar]
- Utriainen A, Sormunen R, Kettunen M, Carvalhaes LS, Sajanti E, Eklund L, Kauppinen R, Kitten GT, Pihlajaniemi T. Structurally altered basement membranes and hydrocephalus in a type XVIII collagen deficient mouse line. Hum Mol Genet. 2004;13:2089–2099. doi: 10.1093/hmg/ddh213. [DOI] [PubMed] [Google Scholar]
- Van Lint P, Libert C. Matrix metalloproteinase-8: cleavage can be decisive. Cytokine Growth Factor Rev. 2006;17:217–223. doi: 10.1016/j.cytogfr.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Vandenbroucke RE, Dejonckheere E, Libert C. A therapeutic role for MMP inhibitors in lung diseases? Eur Respir J. 2011;38:1200–1214. doi: 10.1183/09031936.00027411. [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlaere I, Libert C. Matrix metalloproteinases as drug targets in infections caused by gram-negative bacteria and in septic shock. Clin Microbiol Rev. 2009;22:224–239. doi: 10.1128/CMR.00047-08. Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson SA, Morris TM, Robinson G, Crimmin MJ, Brown PD, Hardcastle JD. Inhibition of organ invasion by the matrix metalloproteinase inhibitor batimastat (BB-94) in two human colon carcinoma metastasis models. Cancer Res. 1995;55:3629–3633. [PubMed] [Google Scholar]
- Wei W, Xin-Ya S, Cai-Dong L, Zhong-Han K, Chun-Peng C. Relationship between extracellular matrix both in choroid plexus and the wall of lateral ventricles and intraventricular hemorrhage in preterm neonates. Clin Anat. 2000;13:422–428. doi: 10.1002/1098-2353(2000)13:6<422::AID-CA5>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Wielockx B, Lannoy K, Shapiro SD, Itoh T, Itohara S, Vandekerckhove J, Libert C. Inhibition of matrix metalloproteinases blocks lethal hepatitis and apoptosis induced by tumor necrosis factor and allows safe antitumor therapy. Nat Med. 2001;7:1202–1208. doi: 10.1038/nm1101-1202. [DOI] [PubMed] [Google Scholar]
- Wolburg H, Paulus W. Choroid plexus: biology and pathology. Acta Neuropathol. 2010;119:75–88. doi: 10.1007/s00401-009-0627-8. [DOI] [PubMed] [Google Scholar]
- Young GB, Bolton CF, Austin TW, Archibald YM, Gonder J, Wells GA. The encephalopathy associated with septic illness. Clin Invest Med. 1990;13:297–304. [PubMed] [Google Scholar]
- Young GB, Bolton CF, Archibald YM, Austin TW, Wells GA. The electroencephalogram in sepsis-associated encephalopathy. J Clin Neurophysiol. 1992;9:145–152. doi: 10.1097/00004691-199201000-00016. [DOI] [PubMed] [Google Scholar]
- Zeni P, Doepker E, Schulze-Topphoff U, Huewel S, Tenenbaum T, Galla HJ. MMPs contribute to TNF-alpha-induced alteration of the blood-cerebrospinal fluid barrier in vitro. Am J Physiol Cell Physiol. 2007;293:C855–C864. doi: 10.1152/ajpcell.00470.2006. [DOI] [PubMed] [Google Scholar]