

Abstract
In addition to being a hallmark of neurodegenerative disease, axon degeneration is used during development of the nervous system to prune unwanted connections. In development, axon degeneration is tightly regulated both temporally and spatially. Here, we provide evidence that degeneration cues are transduced through various kinase pathways functioning in spatially distinct compartments to regulate axon degeneration. Intriguingly, glycogen synthase kinase-3 (GSK3) acts centrally, likely modulating gene expression in the cell body to regulate distally restricted axon degeneration. Through a combination of genetic and pharmacological manipulations, including the generation of an analog-sensitive kinase allele mutant mouse for GSK3β, we show that the β isoform of GSK3, not the α isoform, is essential for developmental axon pruning in vitro and in vivo. Additionally, we identify the dleu2/mir15a/16-1 cluster, previously characterized as a regulator of B-cell proliferation, and the transcription factor tbx6, as likely downstream effectors of GSK3β in axon degeneration.
Introduction
Neurons are distinctly polarized cells that extend neurites long distances to reach their targets. This unique spatial arrangement presents an intricate biological system in which survival or destruction of one cellular compartment, for example, axons, dendrites, or synapses, can be controlled independently from other compartments. Indeed, localized axon destruction is a common cellular mechanism for refinement of neuronal connections during development. A particularly robust example of spatial regulation of axon degeneration is the developmental pruning of mouse retinal ganglion cell (RGC) projections in the superior colliculus (SC). In the embryonic stage, RGC axons initially grow and extend beyond their targets. Shortly after birth, the axon portions that overshoot the termination zone are pruned through localized degeneration (McLaughlin et al., 2003). It is also apparent that many neurodegenerative diseases begin with the loss of synapses and/or axons before complete cellular death is observed (Raff et al., 2002). The spatial control of specific compartmental degeneration, however, remains poorly understood.
The molecular mechanisms underlying naturally occurring, developmental axon pruning are beginning to be unraveled. Genetic disruption of the ubiquitin proteasome system has been shown to block γ neuron pruning during Drosophila metamorphosis (Watts et al., 2003; Hoopfer et al., 2006). There is also evidence suggesting that developmental axon degeneration requires a transcriptional switch. For instance, the transcription factor Otx6 is required for pruning of cortical projections (Weimann et al., 1999; Zhang et al., 2002) and expression of the nuclear-localized ecdysone receptor is required for all known forms of Drosophila axon pruning (Lee et al., 2000). More recently, DLK (dual leucine zipper kinase) signaling via c-Jun N-terminal kinase (JNK) has also been implicated in axon degeneration as a naturally occurring mechanism during development (Ghosh et al., 2011).
Significant effort has been devoted to understanding axon degeneration by studying Wallerian degeneration, a process in which axons separated from the cell body rapidly bleb and fragment in a manner analogous to developmental axon degeneration. In the Wallerian degeneration slow (Wlds) mutant mouse, severed axons lacking trophic support from the cell body remain morphologically and physiologically intact up to 10-fold longer in mutants than in wild-type controls (Perry et al., 1990; Glass et al., 1993; Deckwerth and Johnson, 1994). Importantly, however, Wlds failed to block developmental axon degeneration, indicating that axon pruning and degeneration following injury do not proceed entirely through the same mechanisms (Hoopfer et al., 2006).
We used nerve growth factor (NGF) withdrawal in dorsal root ganglion (DRG) neurons as a model system for investigating the molecular pathways regulating developmental axon degeneration. Using this system, we provide evidence that glycogen synthase kinase-3 (GSK3) lies at the center of a concerted signaling program, functioning downstream of p38 mitogen-activated protein kinase (p38MAPK) and in parallel with JNK to fully execute axon degeneration. We also find that GSK3β acts largely in the cell body to regulate distal axon degeneration, likely via a transcriptional cascade that includes the dleu2/mir15a/16-1 cluster, and the transcription factor tbx6.
Materials and Methods
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde (PFA)/15% sucrose for 20–30 min and washed with PBS solution. Cells were then blocked and permeabilized in 5% bovine serum albumin (BSA)/0.3% Triton X-100 for 1 h at room temperature. Primary antibody was applied in block solution for 2 h at room temperature or overnight at 4°C. After multiple PBS washes, secondary antibody was applied in block solution for up to 1 h at room temperature. After additional PBS washes, no. 1.5 coverslips (VWR) were applied with Fluoromount G (Electron Microscopy Sciences) mounting medium.
Small-molecule screen
DRG explants were cultured from E13.5 CD-1 mouse (Charles River) embryos on poly-d-lysine (PDL)/laminin-coated glass chamber slides (BD Biosciences). Explants were cultured overnight in N3/F12 culture medium (Tessier-Lavigne et al., 1988) with 25 ng/ml 2.5S NGF (BD Biosciences). Before NGF deprivation, cells were pretreated with small-molecule inhibitors for 2 h at either 10 or 100 μm (Tocris), with a final concentration of 1% dimethyl sulfoxide (DMSO) (Sigma-Aldrich). NGF neutralizing antibody (Genentech) was then added directly to each condition for a final concentration of 25 μg/ml. Cells were deprived of NGF for 16–20 h before 4% PFA/15% sucrose fixation, followed by immunocytochemistry with phalloidin conjugated to Alexa Fluor 568 (1:40; Invitrogen), which labels actin, and anti-neuronal class III β-tubulin (Tuj1) (1:1000; Covance). Over 400 small molecules (Tocris) were manually evaluated on a scale from 1 (no protection) to 5 (complete protection) with raters blinded to the identity of the inhibitors.
Compartment-specific inhibition in Campenot chamber system
Campenot chambers were assembled essentially as described previously (Nikolaev et al., 2009). Vacuum grease (Dow Corning) was applied to Teflon dividers (Tyler Research) and mounted to PDL-precoated 35 mm dishes (BD Biosciences) with additional laminin coating (5 μg/ml; Invitrogen). Mouse CD-1 DRG neurons were dissociated after trypsin (Invitrogen) digestion and resuspended in Neurobasal/B27 (Invitrogen) with 50 ng/ml NGF and 3.5 g/L methylcellulose (Sigma-Aldrich). Approximately 200,000 cells were plated per chamber. After 3 d in culture, the outer medium was changed to medium containing 25 ng/ml NGF. After 5 d in culture, the medium in one axon compartment was replaced four times with NGF-free medium, with the final wash containing 50 μg/ml anti-NGF. The medium for the outer compartment was also replaced and the medium inside the slot, the location of the cell bodies, was replaced three times. For small-molecule application to the axon compartment, the inhibitor was included with the final wash. For cell body inhibition, the inhibitor was included in the outer compartment and the central slot washes. The final concentration of DMSO in both compartments was 0.5%. After 28 h of NGF deprivation, cells were fixed with PFA/sucrose and processed for immunocytochemistry as described above using the Tuj1 antibody. For Campenot chamber/ASKA inhibitor experiments, the culture medium was switched to methylcellulose-free medium beginning on day 3.
Quantification of percentage axon degeneration in Campenot chambers or explants
For explants from NGF withdrawal and axon lesion experiments, fluorescent images were taken in random locations, blind to treatment conditions and/or genotype. The images were cropped into 0.5-inch-wide slits in Adobe Photoshop to better distinguish individual axons. The total number of degenerated axons—defined as axons with fragmented, beaded, or rough surfaces—was determined for each image and divided by the total number of axons for the percentage degeneration. Axons from one to three images were pooled together for each condition. For genetic experiments, each n represents axons from a different animal.
For Campenot chambers, fluorescent images were taken from different “lanes” of the chamber. The percentage degeneration from six to eight images were pooled together for each chamber. Dividing the degenerated axons by the total axons gave the percentage degeneration. Each n represents a separate Campenot chamber.
Axon lesion of cultured DRG explants
E13.5 CD-1 mouse DRG explants were cultured in one-well plastic chamber slides (Nunc) for 3 d in N3/F12 supplemented with 25 ng/ml NGF, and 50 μm 5-fluorodeoxyuridine (FudR) (MP Biomedicals)/50 μm uridine (Sigma-Aldrich) to reduce contamination by non-neuronal cells. The culture medium was replaced 18 h before lesion, or immediately after lesion with medium containing small-molecule inhibitors or DMSO control. Explants (cell bodies) were cut free from the axons using a 15° ophthalmic scalpel (Feather Safety Razor). Twelve hours after axon lesion, axons were fixed and labeled for microtubules with the Tuj1 antibody.
Phosphorylation of GSK3α and GSK3β in the cell body compartment after local NGF deprivation
Dissociated DRG neurons were cultured in Campenot chambers as described above in the presence of 50 μm FudR/uridine. Both axon compartments were then deprived of NGF for varying periods of time. To isolate cell body protein, the Teflon divider was removed, and the cell bodies cut away using a scalpel. The isolated cell bodies were lysed and processed for Western blotting using a trichloroacetic acid (TCA) precipitation protocol, as previously described (Ghosh et al., 2011). Briefly, protein was precipitated with TCA in Tris–Triton X-100 lysis buffer (20 mm Tris, pH 7.5, 150 mm NaCl, 1% Triton with protease and phosphatase inhibitors; Roche). The pellet was washed in acetone and then suspended in 1× SDS NuPage loading buffer and reducing agent (Invitrogen). The primary antibodies targeting pGSK3α/β and total GSK3α/β (both Cell Signaling), and actin (BD Biosciences), were detected and quantified using infrared secondary antibodies and the LI-COR Odyssey Imaging system. The relative abundance of pGSK3α and pGSK3β were determined relative to total GSK3α and GSK3β levels, respectively.
Generation of GSK3β (V110I, L132A, F175L) knock-in mice
Targeting vectors
First targeting vector.
The V110I mutation was introduced into exon 3 of the first targeting vector. An additional silent mutation was inserted in exon 3 to generate a restriction site (BsaWI) for analytical purposes. The positive selection marker [neomycin resistance (NeoR)] was flanked by FRT sites and inserted into intron 3. An additional loxP site was inserted into the NeoR cassette for further characterization of clones with double targeting on the same chromosome. The distance between both targetings was ∼14 kb.
Second targeting vector.
The L132A and F175L mutations were introduced into exons 4 and 5 of the second targeting vector. In addition, exon 5 was flanked by loxP sites to generate a conditional knock-out (KO) allele. The positive selection marker [puromycin resistance (PuroR)] was flanked by F3 sites and was inserted into intron 4. Both targeting vectors were generated using BAC clones from the C57BL/6J RPCIB-731 BAC library.
Generation and verification of targeted C57BL/6 NTac ES cells
The C57BL/6NTac ES cell line was grown on a mitotically inactivated feeder layer comprised of mouse embryonic fibroblasts in DMEM high-glucose medium containing 20% FBS (PAN) and 1200 U/ml leukemia inhibitory factor (Millipore). The two targetings were performed with 107 cells and with 20 μg of linearized DNA vector (Bio-Rad Gene Pulser; 240 V and 500 μF) each. Selection was used according to the targeting strategy with 1 μg/ml puromycin and 200 μg/ml G418, respectively, starting on day 2 after electroporation. Counterselection with gancyclovir (2 μm) started on day 5 after electroporation. ES clones were isolated on day 8 after electroporation and analyzed by Southern blot and PCR: heterozygous and double heterozygous-targeted C57BL/6 NTac ES cell clones were verified by Southern blot analysis. Correct homologous recombination at the 5′ and 3′ sides and single integration were detected using external and internal probes, respectively. In addition, Southern blot analysis confirmed that both targetings occurred in cis- on the same chromosome. Insertion of all point mutations was verified by sequencing the PCR amplification products.
Generation of knock-in mice
GSK3β (V110I, L132A, F175L) mice were produced in the animal facility at TaconicArtemis, GmbH, in microisolator cages (Tecniplast Sealsave). Blastocysts were isolated from superovulated BALB/c Ola/Hsd mice. A flat tip, piezo-actuated microinjection pipette with an internal diameter of 12–15 μm was used to inject 10–15 targeted C57BL/6NTac ES cells into each blastocyst. After recovery, eight injected blastocysts were transferred to each uterine horn of 2.5 dpc pseudopregnant NMRI females. Chimerism was determined according to coat color. Highly chimeric mice were bred with mice carrying the Flpe recombinase transgene [C57BL/6-Tg(CAG-Flpe)2 Arte] to achieve germline transmission and to eliminate the selection marker. The resulting offspring, heterozygous for the analog-sensitive kinase allele (ASKA) (C57BL/6-GSK3βtm2138(V110I,L132A,F175L)Arte), was intercrossed to eliminate the Flpe recombinase transgene and to obtain homozygous animals.
Genotyping of mice by PCR
Genomic DNA was extracted from 1- to 2-mm-long tail tips using the NucleoSpin Tissue kit (Macherey-Nagel). Sample analysis was performed using a Caliper LabChip GX device. Insertion of all three point mutations was verified by sequencing of suitable PCR products.
Generation and in vitro characterization of GSK3β ASKA
Gatekeeper L132 of mouse GSK3β WT (NP_062801) was identified by sequence comparison. Because mutation of L132 to a smaller amino acid led to significant loss of activity, mutations at V110 and F175 were introduced to restore GSK3β L132A activity to ∼68% of WT. Mouse GSK3β WT, ASKA (V110I, L132A, F175L) and a kinase-deficient control (K85R) were immunoprecipitated from HEK cell lysate using a C-terminal V5-tag. A continuous fluorescent kinase assay (Omnia S/T peptide 13; Invitrogen) was used for determining activity and shown to be linear for time and kinase concentration. Inhibition by ASKA inhibitor 1-(tert-butyl)-3-(3-methylbenzyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine [3MB-PP1 (3MB)] was determined near Km ATP.
Genotyping of GSK3α and GSK3β knock-in animals for degeneration experiments
REDExtract-N-Amp Tissue PCR Kit (Sigma-Aldrich) was used for DNA preparation and PCR. Primers for GSK3β were as follows: 32638_43, 5′-TACTTACAGGCCAGATCTGTGG-3′, and 2638_44, 5′-GCTATGTACTCAGGTTCTAGACAGG-3′. Band sizes were 276 bp for wild type and 378 bp for knock-in. Primers for GSK3α were as follows: 23: 5′-CCATAGACCTGCGGCTTC-3′; 24: 5′-AAAGTACCGCAGCCTCACGATA-3′; 4: 5′-ACTTGGCTGAGCAGAGTAACAG-3′; and Neo3a: 5′-GCAGCGCATCGCCTTCTATC-3′. Band sizes were 270 bp for wild-type and 552 bp for knock-out. PCR conditions were as follows: 94°C, 4 min; 94°C, 1 min; 60°C, 30 s; 72°C, 1 min; steps 2–4, 30 cycles; 72°C, 10 min; 4°C, ∞.
shRNA knockdown of GSK3β in retinal ganglion cell populations by in utero electroporation
RGC populations were cotransfected with a CAGGs promoter-based expression plasmid encoding EGFP combined with either a mCherry-shLacZ or mCherry-shGSK3β (target, 5′-GTCAGTTACAGACACGAAA-3′) expression plasmid in utero via electroporation of RGC precursor cells as previously described (Garcia-Frigola et al., 2007). At E13.5, C56Bl7 time-pregnant females were anesthetized with 2% isoflurane and secured to a heating pad, and their uterine horns were exposed by incision through the abdominal dermis and body wall muscle. The right eye of individual embryos were injected through the uterine wall using a fine glass capillary needle (Humagen) connected to a Picospritzer pressure injector (Parker), calibrated to deliver ∼0.5 μl of 1 μg/μl plasmid DNA, then electroporated with a series of five square electrical pulses (36 V, 50 ms, 500 ms interval) passed through 3-mm-diameter paddle electrodes (Tweezertrode; BTX). After electroporation, the uterine horns were placed back into the abdominal cavity, and the incision was closed by suture. Dams were treated with an analgesic (0.1 mg/kg buprenorphine) before recovery from anesthesia. Approximately 6 d after surgery, electroporated pups were born expressing EGFP and mCherry in RGC populations located primarily in the central region of the retina, with coexpression efficiency >90% (D. Kallop and R. Weimer, unpublished observation).
At postnatal day 6, electroporated pups were killed, and their brains were fixed overnight in 4% PFA/PBS. After fixation, brains were immersed in PBS, stereotactically positioned under an objective lens (10×, NA 0.6 objective lens; Olympus) and a volume of ∼1 × 1 × 0.4 mm of the left superior colliculus was imaged en bloc by two-photon microscopy: 910 nm Mai Tai DeepSee pulsed laser (Spectra Physics), 510/60 nm bandpass filter (Chroma), Ultima IV two-photon laser-scanning microscope with resolution of 1.1 μm/pixel (Prairie Technologies). The extent of axon pruning was estimated by counting the number of green fluorescent protein (GFP)-expressing axons within 100 μm of the posterior edge of the imaged field of view, as previously described (Simon et al., submitted). Termination zone size per animal was estimated by calculating width at half-maximum corresponding to the distribution of EGFP-positive pixels along the anterior–posterior axis of the superior colliculus.
The specificity of shGSK3β knockdown was verified by qRT-PCR. E14 mouse hippocampal/cortical neurons were nucleofected with mCherry-shRNA expression constructs targeting LacZ or GSK3β (106 cells/transfection; 600 ng DNA/transfection; program CU-110) and grown for 2 d (see culture and transfection protocols below). Two million cells were plated per well in a six-well PDL/laminin plate (BD Biosciences). Worthington Papain Dissociation System was used to gently detach the cells, and mCherry-positive cells were sorted with a FACSAria system (BD Biosciences). RNA was extracted with the RNeasy Plus Mini kit (QIAGEN) following sorting, and relative expression of GSK3α and GSK3β were determined relative to GAPDH.
Automated quantification of axon degeneration
Ninety-six-well plates were imaged with the ImageXpress Micro system (Molecular Devices) with the 4× objective using laser-based and image-based focusing and 2× binning. A single well consisted of nine images that were stitched together in MetaXpress. Axon fragmentation was quantified using the MetaXpress built-in angiogenesis module, which detects filamentous structures with the minimum diameter set to 1 μm and maximum to 5 μm. Intensity above background was set to 15 gray levels. As axons degenerate and fragment, the average “tube length per set” approaches 0 μm because axons break apart into smaller pieces. The average tube length per set from all replicates of the degeneration condition (anti-NGF/siControl or GSK3βS9A/siControl) was divided by the mean tube length per set for each well to give a normalized relative degeneration value.
qRT-PCR by SYBR Green
Campenot chambers were prepared as described above. Axon compartments were deprived of NGF, and inhibitors [trans-4-[4-(4-fluorophenyl)-5-(2-methoxy-4-pyrimidinyl)-1H-imidazol-1-yl]cyclohexanol (SB239063) and N-[(4-methoxyphenyl)methyl]-N′-(5-nitro-2-thiazolyl)urea (AR-A014418)] were added to the axon compartment or cell body compartments. At 6 and 12 h after NGF withdrawal, the Teflon divider was removed and the dish was washed with PBS. RNA was extracted and purified by adding 750 μl of Trizol, followed by on-column purification using the RNeasy Micro Kit according to Appendix C in the manufacturer's handbook. Relative expression was determined using the Quantitect SYBR Green RT-PCR kit (QIAGEN) in a Stratagene MX3000P or an Applied Biosystems 7500 Real-Time PCR System. Relative expression was calculated from ΔΔCT, relative to GAPDH, as described in the QIAGEN user manual. Primer assays for bdnf, c-jun, GAPDH, both isoforms of GSK3, and tbx6, were purchased from QIAGEN. Dleu2 forward primer was 5′-CCAGTTCTGGAAAACAACCAA-3′, and the reverse primer was 5′-CCTCCCACTTTCGTTTGTGT-3′ (Klein et al., 2010).
siRNA target sequences and preparation
siRNA were all purchased from QIAGEN and resuspended in RNase-free water at a concentration of 300 ng/μl. siRNA target sequences are as follows: dleu2.1 (5′-ATGATAGGCGATTAAGGTTTA-3′); dleu2.2 (5′-AACGGGAATCAAACAAGTCTA-3′); dleu2.3 (5′-CAGAAACACGATACTTCTTGA-3′); tbx6.1 (5′-CAGAAGAAACTACAACATGTA-3′); tbx6.2 (5′-ACCCTGATTTGGATACTTCTA-3′); tbx6.3 (5′-CCCTAGGATCACACAGCTGAA-3′).
Gene expression profiling of axon degeneration
Campenot chambers were set as described in the text, with the addition of 50 μm FudR/uridine to reduce contamination by non-neuronal cells. On day 5, both axon compartments were washed three times with NGF-free medium and, on the fourth wash, replaced with medium containing NGF (control) or NGF antibodies. The cell body compartment was replaced with medium containing 30 μm GSK3 inhibitor AR-A014418 (Tocris) or 0.3% DMSO (Sigma-Aldrich). RNA was prepared with Trizol (Invitrogen) followed by the RNeasy Micro Kit processing with DNase I treatment (QIAGEN) as described in Appendix C of the RNeasy Micro Kit manual.
Agilent ratio data were collected as described. Expression ratios were were log2 transformed. Probes were only included in subsequent analysis if expression values were reported for at least four samples. Comparison of treatment groups was performed using the limma package (version 3.6.9) in Bioconductor (version 2.12.1). For each time point, two comparisons were made: control samples were compared with NGF-deprived samples, and NGF-deprived samples were compared with NGF-deprived/GSK3 inhibitor-treated samples. Counts of probes described in the text were generated using a cutoff of 0.5 logFC, where the logFC values were determined from the linear model fit in limma.
siRNA and NGF withdrawal axon degeneration assay
E13.5 DRGs were dissociated following trypsin digestion. siRNA was delivered to cells using the 96-well Amaxa Nucleofector System (Lonza). Approximately 200,000 cells were nucleofected with 600 ng of siRNA using the Mouse Basic Neuron kit according to the manufacturer's protocol (program DC-100). Cells were plated at a density of 25,000 cells per well in a 96-well PDL-precoated plate (BD Biosciences) coated with additional laminin (5 μg/ml; Invitrogen). Cells were grown overnight in N3/F12 with 25 ng/ml NGF before 20 h NGF deprivation by addition of NGF antibodies (25 μg/ml). Cells were fixed with PFA/sucrose and labeled for tubulin as described above.
siRNA and GSK3S9A hippocampal/cortical axon degeneration assay
Hippocampal/cortical tissue was removed from E19.5 Sprague Dawley rat embryos (Charles River) and dissociated after 0.1% trypsin digestion (30 min at 37°C). A total of 20,000 live cells in Nbactiv4 medium (BrainBits) was plated in each well of a 96-well PDL-precoated plate (BD Biosciences). After 5 d in culture, cells were transfected with vectors encoding a constitutively active version of GSK3βS9A or dsRed (Control), a GFP vector as a marker, and pooled siRNA. Cells were transfected with Neurofect (NF) (Genlantis) according to the manufacturer's protocol. Each well of a 96-well plate was transfected with 0.2 μl of NF, 160 ng of either Control or GSK3βS9A vector, 5 ng of GFP expression construct, and 80 ng of siRNA. After 1 and 3 d expression [6–8 d in vitro (DIV)], cells were fixed with 4% PFA/15% sucrose for 20 min, washed in PBS, and labeled with GFP primary antibody (1:500; Invitrogen) in 5% BSA/0.3% Triton X-100. Plates were then washed with PBS/0.1% Triton (PBST) and incubated with secondary antibody, Alexa Fluor 488 (1:800; Invitrogen). Cells were washed again in PBST, before switching to PBS and imaging.
Phosphorylation state of GSK3 and JNK after NGF withdrawal
Dissociated DRG cultures were prepared from E13.5 CD-1 mice as described above and plated at 105 cells/well in a 24-well plate. Neurons were cultured for 5 DIV in N3/F12 media containing 25 ng/ml NGF. Cells were then treated with fresh media containing 25 ng/ml NGF, 25 mg/ml anti-NGF, or 25 mg/ml anti-NGF in combination with different pharmacological inhibitors as indicated. After 6 h treatment, medium was removed and cells were lysed with Tris lysis buffer. Lysates (∼10 μg) were analyzed using the Phospho (Ser9)/Total GSK3β or Phospho (Thr183/Tyr185)/Total JNK Multiplex Assays (Meso Scale Diagnostics), and percentage phosphoprotein levels were determined as described in the manufacturer's protocol. Data were pooled from three biological samples from independent experiments, with duplicate measurements per sample.
Animal studies
Rodents and their embryos were handled in accordance with Genentech Institutional Animal Care and Use Committee guidelines. All animal procedures at TaconicArtemis, GmbH, were run according to German animal welfare laws.
Results
Identification of candidate pathways regulating NGF withdrawal-induced axon degeneration
Withdrawal of NGF from cultured DRG neurons resulted in stereotyped axon degeneration, characterized by growth cone collapse within 40 min, a lag period of 8–12 h, followed by widespread blebbing and fragmentation of axons before engulfment of axonal debris by non-neuronal cells (data not shown). Nearly four hundred small-molecule modulators of known pathways were individually applied to neurons before NGF withdrawal. Of the small molecules tested, 20 small molecules that are proposed to modulate nine unique pathways potently inhibited axon degeneration. Multiple inhibitors of the following kinase pathways were found to block axon degeneration: epidermal growth factor receptor/ErbB family (ErbB), p38MAPK (p38), and GSK3. Selected positive hits are shown in Figure 1 and summarized in Table 1.
Figure 1.
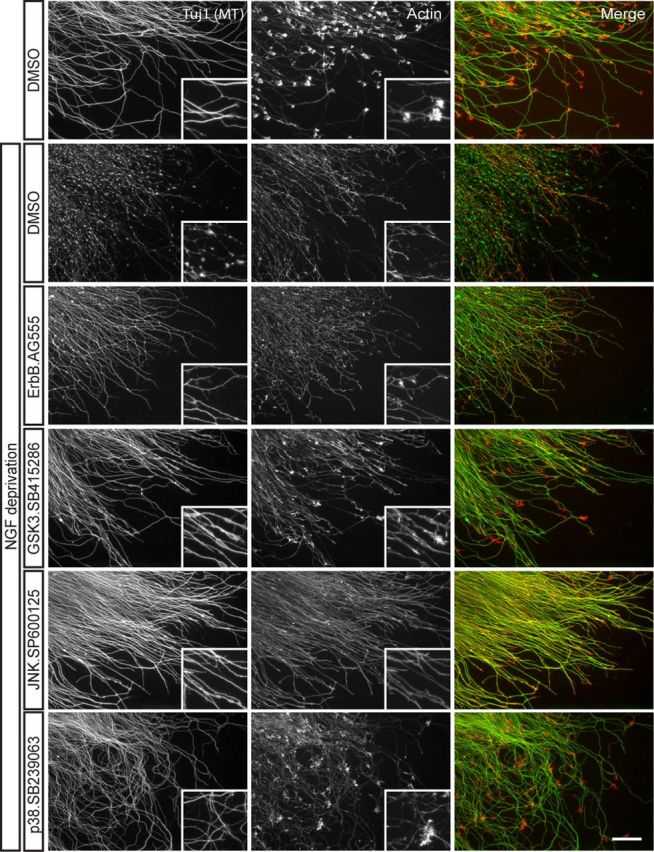
Identification of chemical compounds that inhibit axon degeneration. Small-molecule inhibitors were applied to cultured E13.5 mouse DRG explants for 2 h before adding NGF neutralizing antibodies. After 20 h of NGF deprivation, cells were fixed and labeled for microtubules (Tuj1; green), which are enriched in the axon shaft, and actin (red), a major component of growth cones and an indicator of NGF signaling. Selected positive hits are shown, including ErbB inhibitor AG555 (10 μm), GSK3 inhibitor SB415286 (30 μm), JNK inhibitor SP600125 (20 μm), and p38MAPK inhibitor SB239063 (30 μm). Images are representative of results obtained from at least three independent experiments. Scale bar, 50 μm. High-magnification insets are 123 μm squares.
Table 1.
Summary of chemical screen
| Proposed target | Compound |
|---|---|
| Adenylyl cyclase | Forskolin |
| NKH 477 | |
| p38MAPK | SB 202190 |
| SB 203580 | |
| SB 239063 | |
| GSK3β | AR-A014418 |
| SB 415286 | |
| TDZD-8 | |
| TWS119 | |
| α-4-Dibromoacetophenone | |
| JNK | SP 600125 |
| Ih cation channels | ZD 7288 |
| Transcription | Actinomycin D |
| EGFR/ErbB | AG 494 |
| AG 555 | |
| AG 556 | |
| PD168393 | |
| Tyrphostin B44 | |
| Bax, cytochrome c | Bax channel blocker |
| CaMKK | STO-609 acetate |
Shown is a summary of small molecules that inhibit axon degeneration following NGF withdrawal. A small-molecule library was tested in a growth factor withdrawal assay. Compounds that reduced axon degeneration are listed along with their proposed targets. All compounds shown were effective within the range of 1–100 μm.
Spatial differentiation of axon degeneration pathways into axon or cell body compartments
We were particularly interested in the mechanism of protection via inhibition of GSK3, a kinase often studied in the context of Alzheimer's disease and proposed to regulate microtubule stability through its interaction with microtubule-associated protein tau (Lovestone et al., 1996; Brion et al., 2001; Sang et al., 2001), and other pathways (Zhou et al., 2004; Zhou and Snider, 2005). Because disruption of the microtubule network is one of the earliest signs of axon degeneration in development and following injury (Watts et al., 2003; Zhai et al., 2003), we hypothesized that GSK3 drives degeneration by destabilizing the microtubule network directly at the site of degeneration.
To test our GSK3-microtubule destabilization hypothesis and begin to understand how other identified kinase pathways contribute to axon degeneration, we used the Campenot chamber system (Campenot, 1977) (Fig. 2A). The separation of the distal axon and cell body environments in the Campenot chamber permits selective manipulation of a single cellular compartment. Axons locally deprived of NGF in the Campenot chamber show signs of degeneration only at the site of NGF deprivation (Fig. 2B), as reported by others (Campenot, 1982; Nikolaev et al., 2009). Although axons extend continuously through a grease/Teflon divider, the barrier has been shown to restrict the activity of small molecules to a single compartment (Kuruvilla et al., 2000). By locally depriving axons of NGF, and then exposing either the axon or cell body compartment to inhibitors, we hoped to find where these kinases functioned in the process of axon degeneration (Fig. 2C). The concentration used was determined via dose titration until inhibition in either the axon or cell body compartment alone blocked axon degeneration.
Figure 2.
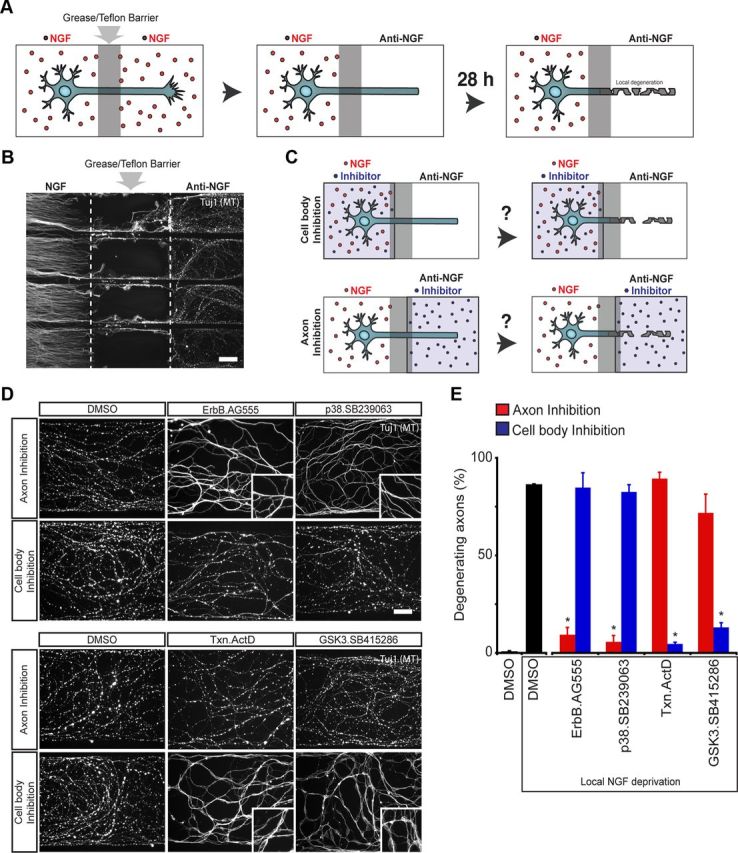
Compartment-specific roles for various pathways in axon degeneration. A, Diagram illustrating localized degeneration in the Campenot chamber. Dissociated DRGs neurons are plated in the “cell body” compartment, and axons extend under a “grease/Teflon” barrier into the “axon compartment.” Axons locally deprived of NGF degenerate over a period of 28 h. B, Low-magnification image showing degeneration of axons locally deprived of NGF. Proximal axons in the cell body compartment maintained in NGF and do not show signs of degeneration. C, Illustration of Campenot chamber assay used to determine functional localization of pathways. Concurrent with local NGF deprivation from axons, inhibitors were applied to either the axon or the cell body compartment. After 28 h, neurons were fixed and labeled for microtubules (Tuj1). D, Images of NGF-deprived axons with inhibitors applied to distal axons—at the site of degeneration—or in the cell body compartment. The p38MAPK and ErbB inhibitors are required at the site of degeneration, while GSK3 and transcription inhibitors are required at the cell body compartment to block axon degeneration. E, Quantification of axon degeneration in Campenot chambers (*p < 0.05, Student's t test; n = 3–7). Values represent mean ± SEM. Scale bars: B, 100 μm; D, 25 μm. High-magnification insets in D are 62 μm squares.
At high concentrations, some of these small molecules inhibited local axon degeneration when applied to either the axon or the cell body compartment. However, as the concentration was reduced, oftentimes these inhibitors showed efficacy only in a single compartment. We found the ErbB [10 μm (E)-2-cyano-3-(3,4-dihydroxyphenyl)-N-(3-phenylpropyl)-2-propenamide (AG555)] and p38MAPK (30 μm SB239063) inhibitors to be effective only when applied to the axon compartment and not the cell body compartment (Fig. 2D, top panel; E), suggesting these kinases function directly at the site of degeneration. JNK inhibitor [20 μm anthra[1–9-cd]pyrazol-6(2H)-one (SP600125)] is slightly more effective in the axon compartment, although the difference between axon inhibition and cell body inhibition is not as striking as with p38 and ErbB inhibitors (data not shown).
Unexpectedly, we found that the GSK3 inhibitor [30 μm 3-[(3-chloro-4-hydroxyphenyl)amino]-4-(2-nitrophenyl)-1H-pyrrole-2,5-dione (SB415286)] had to be applied to the cell body compartment to block distal axon degeneration, exactly the reverse of what we found with ErbB and p38 inhibitors, and in direct opposition to our original hypothesis. These data suggest that GSK3 may be regulating developmental axon degenerating at a distance, possibly through transcriptional regulation. We then tested a transcription inhibitor (15 μm ActD) and, consistent with this idea, found that it blocked distal axon degeneration only when applied to the cell body compartment (a reasonable observation considering transcription occurs in the cell body) (Fig. 2D, bottom panel; E). Although inhibition of either transcription or GSK3 effectively reduced degeneration and fragmentation of axons, the protected axons still formed large microtubule varicosities (Fig. 2D, bottom panel), a state rarely observed when degeneration was blocked with ErbB or p38MAPK inhibitors in the axon compartment (Fig. 2D, top panel). Evaluating the GSK3 inhibitor in the Campenot chamber system led us to consider whether GSK3 may be acting primarily through transcription, rather than directly on the axonal microtubule network to drive axon degeneration.
Preincubation of GSK3 inhibitor protects cut axons from degeneration
To determine whether the pathways we identified regulate both Wallerian and developmental degeneration, we added inhibitors immediately following axon transection. Lesioned axons degenerate over a period of ∼12 h, at which point axons show extensive blebbing and fragmentation (Fig. 3A). ErbB and p38MAPK inhibitors have no effect on Wallerian degeneration, whereas postlesion inhibition of GSK3 (30 μm SB415286) reduces Wallerian degeneration, as reported by others (Gerdts et al., 2011; Wakatsuki et al., 2011) (Fig. 3B). The level of protection was modest, however, as we detected only ∼20% less degeneration when GSK3 inhibitor was applied after axon lesion.
Figure 3.

Long-term GSK3 inhibition reduces Wallerian degeneration. A, Diagram illustrating lesion experiment. DRG explants were cultured for 3 d, lesioned, and fixed after 12 h of degeneration. Lesioned axons show extensive degeneration at the microtubule level. B, Either DMSO, EGFR/ErbB inhibitor (10 μm AG555), GSK3 inhibitor (30 μm SB415286), or p38MAPK inhibitor (30 μm SP239063) were applied immediately after axon lesion, and evaluated for microtubule degeneration 12 h later. Values represent mean ± SEM (*p < 0.05 compared with DMSO control, Student's t test; n = 3–4). Of the tested inhibitors, only GSK3 inhibitor SB415286 reduces Wallerian degeneration. C, GSK3 inhibitor (30 μm SB415286) or JNK inhibitor (20 μm SP600125) were individually applied after axon lesion and compared with 18 h of GSK3 inhibitor (30 μm SB415286) treatment before axon lesion. Long-term inhibition of GSK3 robustly inhibits axon degeneration following lesion. D, Quantification of percentage degenerating axons, normalized to DMSO controls. Values represent mean ± SEM (†p < 0.05 compared with GSK3 inhibition postlesion, Student's t test; n = 3). Scale bars: A, C, top panels, 400 μm; C, bottom panels, 200 μm.
Once separated, transcription occurring in the cell body can no longer regulate axon health or axonal sensitivity to degenerative cues. If GSK3 regulates axon degeneration through a transcriptional pathway, we reasoned that inhibitors would have to be added significantly before lesion to be maximally protective. In contrast to postlesion inhibition of GSK3, prelesion inhibition of GSK3 for 18 h potently inhibits Wallerian degeneration, to the same extent as applying JNK inhibitor immediately after axon lesion (Fig. 3C,D). Such a benefit after pretreatment is consistent with GSK3 regulating axon degeneration primarily through transcription. However, because the pathways regulating developmental axon degeneration and degeneration following injury do not fully overlap, we decided to focus our efforts on understanding the role of GSK3 in developmentally relevant axon degeneration.
GSK3β, not GSK3α, is the primary regulator of axon degeneration in vitro
GSK3 exists in two isoforms, α and β, which are highly homologous in the kinase domain (Woodgett, 1990). Despite their similarities, the biological functions of each GSK3 isoform are not fully redundant. For example, GSK3β knock-outs are embryonic lethal, showing extensive liver degeneration and hypersensitivity to TNF (tumor necrosis factor) toxicity (Hoeflich et al., 2000), while GSK3α knock-outs are fully viable and fertile (MacAulay et al., 2007). In Wnt/β-catenin signaling, however, GSK3α and GSK3β were shown to be redundant through gene deletion studies (Doble et al., 2007). We could not attribute the axonal protection observed via GSK3 inhibition to either GSK3α or GSK3β alone since most commercially available GSK3 inhibitors, including the ones used in this study, are not isoform specific (Meijer et al., 2004).
To address this, we first evaluated the localization and activation of both isoforms in DRG neurons. Confocal imaging revealed a high concentration of GSK3α/β protein in the cell body compared with the axon or growth cone in cultured DRG neurons (Fig. 4A). To demonstrate that GSK3 was activated in the cell body following NGF deprivation, we evaluated the phosphorylation of serine 21 on GSK3α and serine 9 on GSK3β. The PI3K (phosphatidylinositol 3-kinase)/PKB (protein kinase B) pathway is a key regulator of GSK3 phosphorylation and inactivation (Cross et al., 1995). We found that both isoforms were dephosphorylated/activated in cell bodies after local NGF withdrawal from the axon compartment (Fig. 4B).
Figure 4.
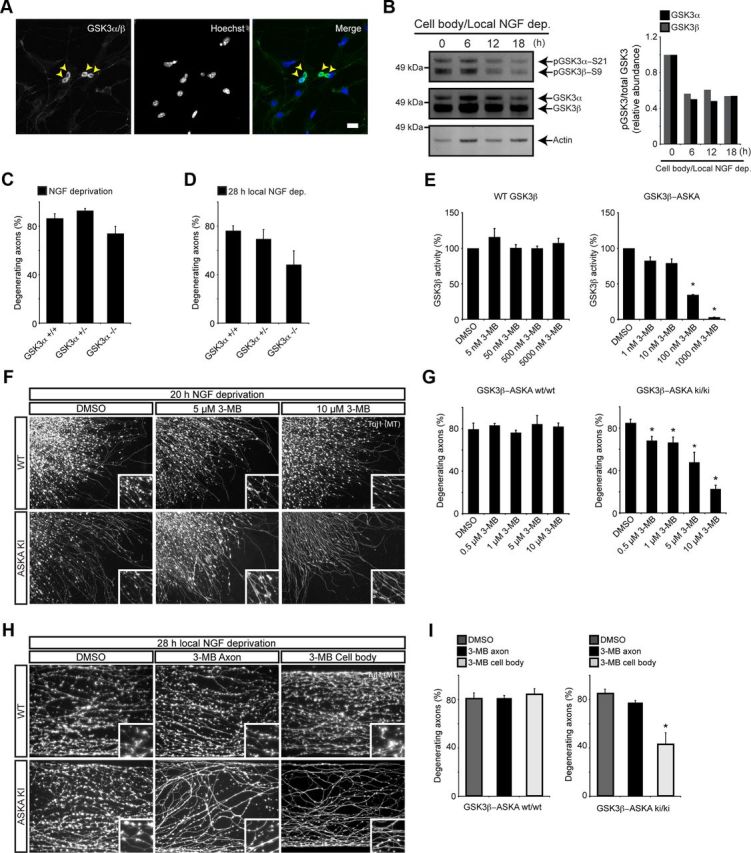
GSK3β, and not GSK3α, is a key regulator axon degeneration in vitro. A, Cultured DRG immunocytochemistry shows GSK3 enrichment in the soma. The yellow arrowheads point to neuronal cell bodies. B, Phosphorylation state of GSK3 isoforms in the cell body compartment following distal axon NGF deprivation. Over a period of 18 h, both isoforms of GSK3 are dephosphorylated, indicating elevated kinase activity. Representative blot from three independent experiments is shown. Relative abundance of pGSK3α/total GSK3α and pGSK3β/total GSK3β are shown in the adjacent graph. Note the difference in total protein loaded for each lane. C, E13.5 DRG explants from GSK3α+/+, GSK3α+/−, and GSK3α−/− embryos were cultured overnight, and then deprived of NGF for 20 h. Genetic deletion of GSK3α has no effect on axon degeneration following NGF deprivation. Values represent mean ± SEM (n ≥ 3). D, Dissociated DRG neurons from GSK3α+/+, GSK3α+/−, and GSK3α−/− were cultured for 5 d in Campenot chambers and then deprived of NGF for 28 h. Genetic deletion of GSK3α does not significantly reduce local axon degeneration. Values represent mean ± SEM (n ≥ 5). E, HEK293 cells were transfected with wild-type GSK3β or GSK3β V110I/L132A/F175L. Kinase activities were determined in the presence of varying concentrations of 3MB-PP1 using anti-V5 immunoprecipitates and a continuous fluorescent kinase assay. 3MB-PP1 potently inhibits GSK3β-ASKA activity but not wild-type activity. Values represent mean ± SEM (*p < 0.05, Wilcoxon's test, 25 μg of lysate, 20 μm ATP, n = 3). F, Images of DRG axons after 20 h NGF deprivation, cultured from ASKA mutants or wild-type animals. In the presence of ASKA inhibitor 3MB-PP1, ASKA mutants show significantly less degeneration. Wild-type explants degenerate with 3MB-PP1, indicating that GSK3β inhibition is specific. G, Quantification of reduced axon degeneration of GSK3β-ASKA knock-in DRGs in the presence of ASKA inhibitors. Values represent mean ± SEM (*p < 0.05, Student's t test; n = 9). H, Compartment-specific inhibition of GSK3β using chemical genetics. DRGs from GSK3β-ASKA knock-in embryos were cultured in Campenot chambers for 5 d. 3MB-PP1 (10 μm) was added to either the cell body or axon compartment immediately following local NGF deprivation. 3MB-PP1 potently inhibits axon degeneration when applied to the cell body compartment alone, but not the axon compartment alone. I, Quantification of reduced local axon degeneration, specifically in GSK3β-ASKA knock-in neurons exposed to 3MB-PP1 in the cell body compartment. Values represent mean ± SEM (*p < 0.05, Student's t test; n ≥ 6). Scale bar: A, 60 μm. The high-magnification insets in F and H are 62 μm squares.
To evaluate whether a single isoform could regulate axon degeneration, we tested the GSK3α knock-out described by MacAulay et al. (2007). Genetic deletion of GSK3α had no detectable effect on axon degeneration following global NGF deprivation in DRG explants (Fig. 4C). Because we had previously found the local NGF deprivation assay to be more sensitive for detecting small differences in axon degeneration, we also evaluated the GSK3α knock-out in the Campenot chamber system. Again, we were unable to find a significant difference in degeneration between wild type and GSK3α knock-outs following local NGF deprivation, although we did find a trend toward increased axonal protection in GSK3α knock-outs (p = 0.07; n = 5–7; Fig. 4D).
Because systemic deletion of GSK3β is early embryonic lethal, we sought out alternative methods for genetic analysis of GSK3β. Shokat and colleagues (Bishop et al., 2000) have recently elucidated powerful chemical–genetic tools for investigating kinase function, referred to as kinase switch or ASKAs. Only kinases harboring ASKA-specific mutations in their kinase domains—and not wild-type kinases—are sensitive to ASKA-specific inhibitors, allowing for highly selective inhibition. Based on the GSK3β gatekeeper residue, L132, two candidate ASKA mutations were generated: V110I/L132A and V110I/L132A/F175L. GSK3β V110I/L132A/F175L, from hereon referred to as GSK3β-ASKA, was used to generate knock-in mice because it showed acceptable baseline activity (68% that of wild-type GSK3β). In preliminary biochemical assays, GSK3β-ASKA was highly sensitive to the ASKA inhibitor 3MB-PP1, whereas wild-type GSK3β was unaffected (Fig. 4E).
We next investigated degeneration of DRG neurons harvested from homozygous GSK3β-ASKA knock-in embryos. Following NGF deprivation, application of 3MB-PP1 inhibited axon degeneration in knock-in DRGs but had no effect on degeneration of wild-type DRGs (Fig. 4F,G). These data show that, unlike GSK3α, GSK3β is essential for axon degeneration. Next, we wanted to verify that GSK3β activity is indeed required in the cell body for distal axon degeneration. Using the compartmentalized Campenot chamber system, we found 10 μm 3MB-PP1 inhibited distal axon degeneration when applied to the cell body compartment alone, but failed to block degeneration when applied to the axon compartment alone. Addition of 3MB-PP1 had no effect on degeneration of WT neurons in the compartmentalized culture system (Fig. 4H,I).
GSK3β, not GSK3α, is a key regulator of developmental axon pruning in vivo
Spatially regulated pruning of RGC axons in the superior colliculus occurs during postnatal days 1 and 6 in the mouse and represents an excellent model for testing spatially regulated axon degeneration during development (Fig. 5A). Pruning of RGC axons during development was tracked by expressing GFP in the retinas of E13.5 embryos via in utero electroporation (Fig. 5B,C). As expected from our in vitro data, genetic deletion of GSK3α had no effect on axon pruning (Fig. 5D,E).
Figure 5.

GSK3β, and not GSK3α, is a key regulator of naturally occurring developmental axon pruning in vivo. A, Diagram illustrating developmental axon pruning of RGC axons in the superior colliculus. At postnatal day 1 (P1), axons initially overshoot their target region and extend into the posterior superior colliculus (PSC). At P3, axons prune back through degeneration. By P6, axon terminals are confined to the termination zone (TZ), their final destination. B, Diagram illustrating in utero electroporation. Following DNA microinjection, DNA was electroporated by creating an electric field with paddle electrodes placed on either side of the embryo's head. C, Representative maximum intensity projection (MIP) images of GFP-positive axons within the SC at P1 and P6. D, Representative MIP images of GFP-positive axons within the SC at postnatal P6 from GSK3α+/+ and GSK3α−/− animals. The white dotted line outlines the region of the SC defined as the PSC, shown in higher magnification to the right. E, Quantification of axon pruning as measured by the number of GFP-positive axons in the PSC, size of the TZ, and the number of axons in the PSC normalized to the size of the TZ for GSK3α+/+ (n = 5) and GSK3α−/− (n = 6). Values represent mean ± SEM. No difference in RGC axon pruning was observed between GSK3α-null and wild-type littermate controls. F, GFP was coexpressed with either shLacZ or shGSK3β. Knockdown of GSK3β results in significantly more GFP-positive axons in the PSC compared with shLacZ control. G, Quantification of axon pruning as measured by the number of GFP-positive axons in the PSC, size of the TZ, and the number of axons in the PSC normalized to the size of the TZ for shLacZ (n = 15) and shGSK3β (n = 9) animals. Values represent mean ± SEM (*p < 0.05, Student's t test). Scale bar: B, C, 100 μm.
To circumvent toxicity associated with systemic inhibition of GSK3β during development, and to provide evidence of cell autonomy, we used an shRNA expression construct instead of the ASKA knock-in animals. To knockdown GSK3β and simultaneously label RGC projections, retinas of E13.5 embryos were coelectroporated with shRNA and GFP by in utero electroporation. Assessment of the selectivity of shRNA knockdown of GSK3β showed a >50% reduction in GSK3β mRNA as measured by RT-PCR (p < 0.05, Student's t test; n = 3), while a modest insignificant reduction in GSK3α expression was observed. Knockdown of GSK3β yielded robust RGC axon pruning deficits; compared with control shRNA, GSK3β knockdown produced approximately fivefold more axons persisting at the posterior edge of the superior colliculus (Fig. 5F,G).
Identification of GSK3-regulated pro-degeneration genes dleu2 and tbx6
As GSK3 regulates axon degeneration through activity in the cell body, likely via regulation of transcription, we set out to identify pro-degeneration genes that are regulated in a GSK3-dependent fashion. To accomplish this, we used an unbiased approach using microarrays to analyze RNA isolated from DRG neurons following NGF withdrawal in the presence or absence of GSK inhibitors. NGF deprivation was spatially limited to the axon using the Campenot chamber, thereby reducing or eliminating apoptosis signaling (Campenot, 1982; Mok et al., 2009) and highlighting genes most likely to function during axon degeneration. GSK3 inhibitor (30 μm AR-A014418) loses effectiveness between 6 and 12 h after local NGF deprivation (data not shown), suggesting that the gene changes required for axon degeneration downstream of GSK3 occur between 6 and 12 h after the loss of local NGF signaling in the axon. We therefore evaluated differential expression at these two time points. Finally, we evaluated gene changes following NGF withdrawal with and without GSK3 inhibitor, further reducing the candidate list to gene changes that are regulated by GSK3.
As expected, withdrawal of NGF from distal axons (mimicking loss of target-derived NGF) caused a decrease in the levels of brain-derived neurotrophic factor (bdnf) transcript (Deppmann et al., 2008) and an increase in jun transcript (Mok et al., 2009). We selected two genes identified in the microarray, transcription factor t-box 6 (tbx6) and deleted in lymphocytic leukemia 2 (dleu2), for further investigation. Probes for both of these genes revealed upregulated expression when comparing control to NGF-deprived neurons, and they also revealed downregulated expression when comparing NGF-deprived to NGF-deprived/GSK3-inhibited neurons (Fig. 6A). Dleu2 is a long noncoding RNA and host gene for two micro-RNAs, mir15a and mir16-1 (Liu et al., 1997; Migliazza et al., 2001), and tbx6 is involved in patterning and somite formation (Chapman and Papaioannou, 1998; Hadjantonakis et al., 2008). These results were verified by qRT-PCR, in which similar results were obtained except with bdnf, which showed similar trends but did not reach significance.
Figure 6.
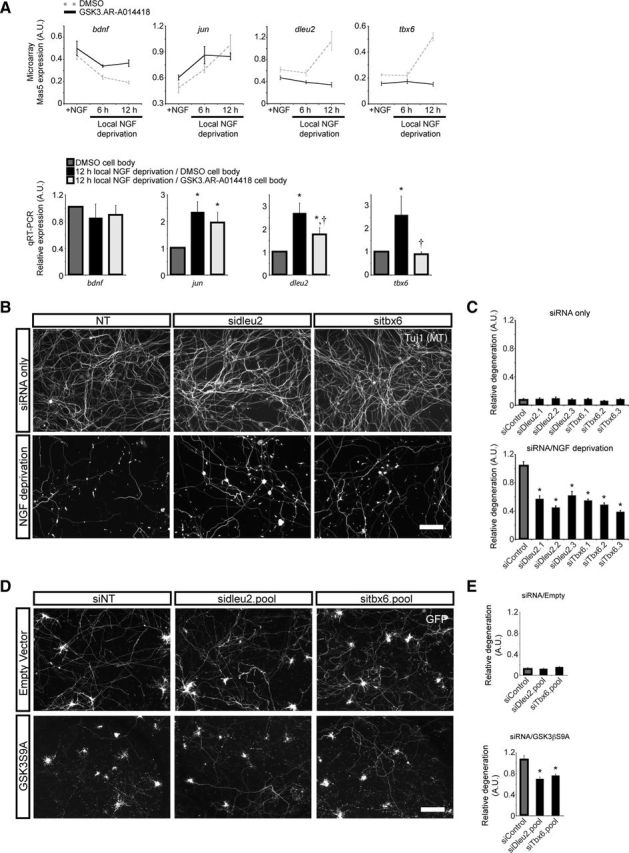
Time course microarray to identify candidate GSK3-driven axon degeneration genes. A, Expression profiling of genes following local NGF deprivation. RNA was isolated from neurons locally deprived of NGF for 6 and 12 h, with and without GSK3 inhibition (30 μm AR-A014418) and compared with neurons globally maintained in NGF. Values represent mean ± SEM (n = 5). bdnf mRNA decreases and jun mRNA increases after local NGF deprivation, as expected. Dleu2 and tbx6 meet the criteria for candidate axon degeneration genes. They are upregulated after 12 h of local NGF deprivation in a GSK3-dependent manner. Results from the microarray experiment were verified by qRT-PCR. Samples from NGF control were compared with 12 h NGF deprivation with and without GSK3 inhibition. Values represent mean ± SEM (*p < 0.05 compared with NGF control; †p < 0.05 compared with 12 h NGF deprivation, Wilcoxon's test; n ≥ 8). B, DIV2 siRNA-transfected DRG neurons maintained in NGF (siRNA only) or following 20 h NGF deprivation (NGF deprivation). siRNA targeting dleu2 and tbx6 do not affect baseline degeneration of cultured DRGs, but do reduce degeneration following NGF deprivation. C, Automated quantification of degeneration in NGF control and after 20 h NGF deprivation. Values are relative to the siControl/NGF deprivation condition and represent mean ± SEM (*p < 0.05 compared with siControl, Student's t test; n = 3 wells from 1 representative experiment). D, Cultured hippocampal/cortical neurons were transfected on DIV5 with siRNA and GFP, plus either Control vector or constitutively active GSK3 (GSK3βS9A or S9A). Neurons were fixed after 3 d of expression (8 DIV). The siRNA have no effect on baseline neurite degeneration when combined with Control vector. Both siDleu2 and siTbx6 reduce relative degeneration when cotransfected with constitutively active GSK3β. E, Automated quantification of degeneration after cotransfection with dleu2 or tbx6 siRNA and either Control vector or S9A (*p < 0.05, Student's t test; n ≥ 12 wells from a representative experiment). Values represent mean ± SEM. Scale bars: B, D, 100 μm.
To show that upregulation of dleu2 and tbx6 plays a functional role in axon degeneration, siRNA targeting dleu2 or tbx6 were nucleofected into dissociated E13.5 DRG neurons before plating. After 1 d in culture, neurons were either maintained in NGF for another 20 h, or deprived of NGF for 20 h with NGF neutralizing antibodies. Axon degeneration was then evaluated using an automated analysis system, which was designed to evaluate fragmentation of axons relative to the NGF deprivation/nontargeting siRNA condition. Smaller values indicate less fragmentation and/or more continuous axons. In control DRG neurons maintained in NGF, dleu2 and tbx6 siRNAs did not affect baseline axon degeneration. However, following NGF deprivation, siRNAs targeting either dleu2 or tbx6 significantly reduced axon degeneration (Fig. 6B,C). This effect was equivalent to ∼30% reduction in degenerating axons when determined manually (data not shown).
To more specifically evaluate the role of these genes in GSK3β-dependent degeneration, mixed hippocampal/cortical neurons were grown 5 d to allow neurite outgrowth, and then transfected with siRNA and a GSK3βS9A expression construct. GSK3βS9A is a mutant form of GSK3β that cannot be inactivated through phosphorylation at serine 9, thus representing a constitutively activated form of GSK3β. Cells were evaluated for neurite degeneration 3 d after transfection. GSK3βS9A overexpression resulted in a significant degeneration after 3 d, showing the GSK3 is not only necessary but also sufficient to drive axon degeneration. When combined with GSK3βS9A, both dleu2 and tbx6 siRNA pools significantly reduced neurite degeneration downstream of GSK3βS9A (Fig. 6D,E). As a control, we cotransfected siRNA with a control vector and found dleu2 and tbx6 siRNA pools to have no detectable effect on baseline neurite degeneration 3 d after transfection (8 DIV) (Fig. 6D,E). Degeneration was determined relative to GSK3βS9A/nontargeting siRNA condition. CNS neurons were chosen in place of DRG neurons due to limitations with our transfection reagents. Nevertheless, these results suggest dleu2 and tbx6 may function in a similar manner across a variety of neuronal subtypes.
Concerted activity of pathways functioning in spatially distinct compartments regulates axon degeneration
A number of pathways were identified in our small-molecule screen. To understand how these pathways interact, if at all, we asked whether inhibition of any of the pathways we identified could block GSK3 activation following NGF deprivation; such “initiator” pathways would lie upstream of a GSK3-mediated transcriptional axon degeneration program (Fig. 7A). NGF withdrawal drives GSK3β activation through dephosphorylation at serine 9 (Crowder and Freeman, 2000). We looked at various pathway inhibitors and found that the p38MAPK inhibitor (30 μm SB239063) and the CaMKK inhibitor [15 μm 7-oxo-7H-benzimidazo[2,1-a]benz[de]isoquinoline-3-carboxylic acid acetate (STO-609)] could prevent GSK3β dephosphorylation following NGF withdrawal (Fig. 7B).
Figure 7.

Concerted activity of kinases functioning in spatially distinct compartments is necessary for developmental degeneration. A, Pathways that are required for GSK3-dependent “priming” are described as “initiation” pathways. B, Dissociated DRG neurons were globally deprived of NGF for 6 h, with or without inhibitors of various pathways. Cell lysates were assayed for phospho-GSK3β (Ser9) and total GSK3β. Only CaMKK inhibitor (15 μm STO-609) and p38MAPK inhibitor (30 μm SB239063) reduce dephosphorylation of GSK3β following NGF deprivation. Values represent mean ± SEM (*,#p < 0.05 compared with NGF deprivation/DMSO control, Student's t test; n = 3). C, Local NGF deprivation/DMSO induces upregulation dleu2 and tbx6, and downregulation of bdnf. Applying the p38MAPK inhibitor (30 μm SB239063) in the axon compartment with local NGF deprivation prevents upregulation of dleu2 and tbx6 but does not reverse the downregulation of bdnf (*p < 0.05 compared with NGF control; †p < 0.05 compared with NGF deprivation/DMSO, Wilcoxon's test; n = 5). D, Does JNK act downstream of GSK3 or in parallel? E, Cell lysates from B were assayed for pJNK activity. GSK3 inhibitor (30 μm SB415286) does not prevent JNK phosphorylation after NGF withdrawal. Values represent mean ± SEM (*,#p < 0.05 compared with NGF deprivation/DMSO, Student's t test; n = 3).
As described above, the proposed p38MAPK inhibitor, SB239063, is only effective when applied directly to axons undergoing degeneration; we therefore reasoned that p38MAPK might be responsible for transducing the local trophic-deprivation signal to the cell body. To test this hypothesis, axons were locally deprived of NGF and either DMSO or SB239063 was applied to the axon compartment. Local NGF deprivation causes upregulation of dleu2 and tbx6, and a downregulation of bdnf, consistent with the microarray data. When local NGF deprivation is combined with local inhibition of p38MAPK with SB239063 in the axon compartment, the upregulation of dleu2 and tbx6 are potently inhibited (reversal of bdnf downregulation did not reach significance) (Fig. 7C).
JNK inhibitors block axon degeneration, yet the JNK inhibitor AS60125 failed to block GSK3β dephosphorylation after NGF withdrawal. This result might be explained by JNK acting downstream or in parallel with GSK3 to regulate axon degeneration (Fig. 7D). To test these two possibilities, we looked at JNK activation following 6 h NGF deprivation in the presence of small-molecule inhibitors. Phosphorylation at Thr183/Tyr185 served as a readout for JNK activation (Fig. 7E). As in the case of GSK3, we found evidence that p38MAPK is upstream of JNK, with SB239063 partially reversing JNK phosphorylation following NGF withdrawal. Interestingly, we found that GSK3 inhibitor SB415286 (30 μm) had no effect on JNK activation, suggesting JNK may act in parallel, as opposed to downstream of GSK3, to regulate axon degeneration.
Discussion
Axon degeneration requires signaling through various kinase pathways functioning in spatially distinct compartments. Working in combination with other kinases, GSK3 likely functions as a gatekeeper to a transcriptional axon degeneration program, driving gene changes such as upregulation of dleu2 and tbx6 to regulate distal axon degeneration.
Small-molecule screen identifies a number of pathways regulating axon degeneration
Many of the pathways identified in our screen have been previously investigated in the context of neurodegeneration, including GSK3 (Kaytor and Orr, 2002), p38MAPK (Munoz and Ammit, 2010), and JNK (Borsello and Forloni, 2007), while others such as CaMKK and EGFR/ErbB have limited prior evidence of playing a role in neurodegeneration.
It is difficult to validate these pathways through siRNA knockdown, as partial reduction in kinase levels may not be sufficient to block the amplifying effect of kinase activation. Thus, further genetic validation of the pathways we characterize in these studies, with the exception of GSK3 and JNK, would provide additional evidence that the small molecules being used are acting via the proposed pathways. However, genetic validation also has its difficulties as the majority of kinase pathways we describe have a number of isoforms or consist of multiple highly homologous genes. As an extreme example, validating a role for ErbB signaling in axon degeneration would require obtaining knock-outs for three of the four ErbB family members. Even when considering these important caveats, chemical inhibitors have served as unique entry points for understanding the molecular mechanisms of axon degeneration.
Localization of axon degeneration pathways to distinct cellular compartments
By investigating these pathways in the Campenot chamber system, we found that the ErbB and p38MAPK inhibitors were protective only at the site of degeneration. Unexpectedly, GSK3 inhibitors blocked distal axon degeneration only when applied to the cell body compartment, similar to a transcription inhibitor. In many instances in which axon degeneration was blocked with a GSK3 or transcription inhibitor, axons showed microtubule bulging/varicosities but were generally smooth contoured, in contrast to degenerating axons. This may result from blocking axon degeneration downstream of these local changes, leaving axons “frozen” in a degeneration-primed state. The overall spatial and morphological similarities observed between axons protected with transcription and GSK3 inhibition led us to consider whether GSK3 regulated axon degeneration acts via transcription.
Examples of GSK3-regulated transcription are abundant; for example, GSK3 has been shown to regulate a large number of transcription factors, including β-catenin, c-Jun, and CREB (cAMP response element-binding protein), to name a few (Hur and Zhou, 2010). Also, there are known processes in neurobiology in which events at a distal location require transcription for full execution. For instance, the immediate-early gene Arc/Arg3.1 is rapidly transcribed after long-term potentiation-inducing stimuli and then transported to the dendrite, where it is critical for the maintenance of synaptic potentiation (Bramham et al., 2008). Our data suggest GSK3 may regulate axon degeneration in similar manner, inducing transcriptional changes in the cell body to prime axons for degeneration.
Consistent with a model in which GSK3β activation drives the transcription of various genes to “prime” neurons for axon degeneration (Fig. 7), we showed that p38MAPK inhibitors added to the axon after local NGF deprivation blocked dleu2 and tbx6 transcriptional upregulation, suggesting that p38MAPK and possibly other pathways may function in the axon as an “initiators” upstream of GSK3. Nevertheless, it remains to be determined whether p38MAPK or other pathways are sufficient to drive axon degeneration, similar to GSK3. Although phosphorylation of GSK3β at Ser9 is only one means of regulating activity, our data nevertheless provide a tentative link between local degeneration signals acting in the axon and global transcriptional changes in the cell body that ultimately “execute” axon degeneration. In summary, the requirement of multiple stress pathways acting in distinct cellular compartments may provide spatial cues for restricting degeneration, as well as providing a safeguard against unwanted axon loss.
Although our data suggest GSK3 regulates developmental axon degeneration primarily through its activity in the cell body, it is important to note that this mechanism does not necessarily translate to Wallerian degeneration. GSK3 inhibition after axon lesion still reduces axon degeneration, indicating GSK3 can at least partially regulate degeneration directly at the site of degeneration in an injury paradigm. Additionally, our threshold for determining percentage degeneration was very low; any axons that showed rough edges at the microtubule level were deemed “degenerated.” It is possible that another measure axon degeneration, such as percentage axon fragmentation, would reveal an axon-specific role for GSK3 in developmental axon degeneration.
Evidence that dleu2 and tbx6 mediate axon degeneration downstream of GSK3
To provide additional evidence for this proposed transcriptional axon degeneration program, we set out to identify genes that may function downstream of GSK3 to regulate axon degeneration. In our microarray experiment, two GSK3-regulated axon degeneration genes were identified: dleu2 and tbx6. Functional analysis of these two genes provides an additional layer of evidence for our proposed GSK3-dependent axon degeneration program and identifies dleu2 and tbx6 as novel regulators of axon degeneration. Multiple siRNAs targeting either dleu2 or tbx6 modestly reduced axon degeneration, yielding ∼30% of the protection obtained with GSK3 small-molecule inhibitors in the growth factor withdrawal assay. These results were initially surprising as we suspected that individually modulating two genes from a genetic program, which may include the simultaneous upregulation and downregulation of a large number of genes, would not necessarily elicit any phenotype. Nevertheless, we found a reproducible protective effect with knockdown of either dleu2 or tbx6. Additionally, we used a constitutively active version of GSK3β (GSK3βS9A) to show that GSK3 signaling is sufficient to induce axon degeneration and to provide evidence that dleu2 and tbx6 lie downstream of GSK3-induced degeneration. Notably, we did not observe protection of axons from degeneration with either dleu2 or tbx6 knockdown after lesion-induced axon degeneration (data not shown). These data suggest that the ability of GSK to mediate axon degeneration in development and after injury may diverge. It also remains to be determined whether knocking down both genes, or other genes identified from our screen, will have an even more robust effect in modulating axon degeneration.
GSK3β, not GSK3α, is the primary regulator of developmental axon degeneration in vitro and in vivo
Despite using multiple, structurally unrelated inhibitors for each of our pathways, we could not rule out “off-target” effects (Bain et al., 2003). To address this issue, and also provide isoform-specific analysis of GSK3, we used gene deletion and chemical–genetics. Through these tools, we found that GSK3β is the primary regulator of developmental axon pruning, whereas GSK3α has little to no role in axon degeneration. By using GSK3β-ASKA knock-in neurons combined with ASKA-specific inhibitors, we were also able to verify that GSK3β acts primarily in the cell body to regulate axon degeneration following local trophic factor deprivation.
Pruning of RGC axons during development is a cell death-independent process that results in a substantial, spatially restricted loss of axonal material from P1 to P6. Consistent with our in vitro data, there was a pruning deficit with GSK3β knockdown but no detectable pruning phenotype with genetic deletion of GSK3α. It has previously been reported that inhibition of GSK3β can promote axon growth and branching (Zhou et al., 2004; Kim et al., 2006; Bilimoria et al., 2010). Based on our time-lapse experiments, there was negligible branching or axon growth following NGF deprivation/GSK3 inhibition (data not shown). We therefore speculate that the apparent RGC pruning deficit is indeed the result of blocking axon degeneration, rather than promoting axon branching/growth. Determining whether branching/growth contributes to the increase in axons at the posterior superior colliculus with GSK3β knockdown will require additional analysis of RGC pruning at more time points or at higher resolution.
It may seem inconsistent that GSK3β is the primary regulator of axon degeneration, even though both GSK3 isoforms are activated in the cell body to a similar extent following NGF deprivation, as determined by dephosphorylation at Ser21 or Ser9. However, regulation of GSK3 signaling is intricate. Formation of protein complexes, subcellular localization, and β isoform-specific inactivation by p38MAPK phosphorylation at Ser389 are examples of other factors that may regulate GSK3 isoform-specific activity (Hur and Zhou, 2010). Additionally, GSK3α is restricted from the nucleus by its N-terminal region, whereas GSK3β shuttles freely between the nucleus and cytoplasm (Azoulay-Alfaguter et al., 2011). Nuclear GSK3β is highly active, and its localization in the nucleus decreases following stimulation with growth factors and increases under apoptosis conditions (Bijur and Jope, 2001, 2003). These observations by others are consistent with a model in which GSK3β, not GSK3α, is competent to mediate a transcriptional cascade to regulate axon degeneration.
Footnotes
We thank Jeffrey Eastham Anderson, Meredith Sagolla, and Laszlo Kumovez for their assistance in microscopy. For helpful discussions and reagents, we thank Anatoly Nikolaev and Arundhati Senguptah-Ghosh. We also thank Zora Modrusan for her assistance with microarray processing, and Martin Garcia for breeding of the mouse lines used in this study.
K.B. and H.K. are employees of TaconicArtemis, GmbH. M.C., J.A.M., D.K., J.K.A., S.J.T., J.S.K., J.W.L., R.M.W., and R.J.W. are employees of Genentech, Inc.
References
- Azoulay-Alfaguter I, Yaffe Y, Licht-Murava A, Urbanska M, Jaworski J, Pietrokovski S, Hirschberg K, Eldar-Finkelman H. Distinct molecular regulation of glycogen synthase kinase-3alpha isozyme controlled by its N-terminal region: functional role in calcium/calpain signaling. J Biol Chem. 2011;286:13470–13480. doi: 10.1074/jbc.M110.127969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijur GN, Jope RS. Proapoptotic stimuli induce nuclear accumulation of glycogen synthase kinase-3 beta. J Biol Chem. 2001;276:37436–37442. doi: 10.1074/jbc.M105725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijur GN, Jope RS. Glycogen synthase kinase-3 beta is highly activated in nuclei and mitochondria. Neuroreport. 2003;14:2415–2419. doi: 10.1097/00001756-200312190-00025. [DOI] [PubMed] [Google Scholar]
- Bilimoria PM, de la Torre-Ubieta L, Ikeuchi Y, Becker EB, Reiner O, Bonni A. A JIP3-regulated GSK3beta/DCX signaling pathway restricts axon branching. J Neurosci. 2010;30:16766–16776. doi: 10.1523/JNEUROSCI.1362-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Borsello T, Forloni G. JNK signalling: a possible target to prevent neurodegeneration. Curr Pharm Des. 2007;13:1875–1886. doi: 10.2174/138161207780858384. [DOI] [PubMed] [Google Scholar]
- Bramham CR, Worley PF, Moore MJ, Guzowski JF. The immediate early gene arc/arg3.1: regulation, mechanisms, and function. J Neurosci. 2008;28:11760–11767. doi: 10.1523/JNEUROSCI.3864-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brion JP, Anderton BH, Authelet M, Dayanandan R, Leroy K, Lovestone S, Octave JN, Pradier L, Touchet N, Tremp G. Neurofibrillary tangles and tau phosphorylation. Biochem Soc Symp. 2001;2001:81–88. doi: 10.1042/bss0670081. [DOI] [PubMed] [Google Scholar]
- Campenot RB. Local control of neurite development by nerve growth factor. Proc Natl Acad Sci U S A. 1977;74:4516–4519. doi: 10.1073/pnas.74.10.4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campenot RB. Development of sympathetic neurons in compartmentalized cultures. II. Local control of neurite survival by nerve growth factor. Dev Biol. 1982;93:13–21. doi: 10.1016/0012-1606(82)90233-0. [DOI] [PubMed] [Google Scholar]
- Chapman DL, Papaioannou VE. Three neural tubes in mouse embryos with mutations in the T-box gene Tbx6. Nature. 1998;391:695–697. doi: 10.1038/35624. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Crowder RJ, Freeman RS. Glycogen synthase kinase-3 beta activity is critical for neuronal death caused by inhibiting phosphatidylinositol 3-kinase or Akt but not for death caused by nerve growth factor withdrawal. J Biol Chem. 2000;275:34266–34271. doi: 10.1074/jbc.M006160200. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Johnson EM., Jr Neurites can remain viable after destruction of the neuronal soma by programmed cell death (apoptosis) Dev Biol. 1994;165:63–72. doi: 10.1006/dbio.1994.1234. [DOI] [PubMed] [Google Scholar]
- Deppmann CD, Mihalas S, Sharma N, Lonze BE, Niebur E, Ginty DD. A model for neuronal competition during development. Science. 2008;320:369–373. doi: 10.1126/science.1152677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell. 2007;12:957–971. doi: 10.1016/j.devcel.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Frigola C, Carreres MI, Vegar C, Herrera E. Gene delivery into mouse retinal ganglion cells by in utero electroporation. BMC Dev Biol. 2007;7:103. doi: 10.1186/1471-213X-7-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Sasaki Y, Vohra B, Marasa J, Milbrandt J. Image-based screening identifies novel roles for IkappaB kinase and glycogen synthase kinase 3 in axonal degeneration. J Biol Chem. 2011;286:28011–28018. doi: 10.1074/jbc.M111.250472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AS, Wang B, Pozniak CD, Chen M, Watts RJ, Lewcock JW. DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J Cell Biol. 2011;194:751–764. doi: 10.1083/jcb.201103153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass JD, Brushart TM, George EB, Griffin JW. Prolonged survival of transected nerve fibres in C57BL/Ola mice is an intrinsic characteristic of the axon. J Neurocytol. 1993;22:311–321. doi: 10.1007/BF01195555. [DOI] [PubMed] [Google Scholar]
- Hadjantonakis AK, Pisano E, Papaioannou VE. Tbx6 regulates left/right patterning in mouse embryos through effects on nodal cilia and perinodal signaling. PLoS One. 2008;3:e2511. doi: 10.1371/journal.pone.0002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- Hoopfer ED, McLaughlin T, Watts RJ, Schuldiner O, O'Leary DD, Luo L. Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron. 2006;50:883–895. doi: 10.1016/j.neuron.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Hur EM, Zhou FQ. GSK3 signalling in neural development. Nat Rev Neurosci. 2010;11:539–551. doi: 10.1038/nrn2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaytor MD, Orr HT. The GSK3 beta signaling cascade and neurodegenerative disease. Curr Opin Neurobiol. 2002;12:275–278. doi: 10.1016/s0959-4388(02)00320-3. [DOI] [PubMed] [Google Scholar]
- Kim WY, Zhou FQ, Zhou J, Yokota Y, Wang YM, Yoshimura T, Kaibuchi K, Woodgett JR, Anton ES, Snider WD. Essential roles for GSK-3s and GSK-3-primed substrates in neurotrophin-induced and hippocampal axon growth. Neuron. 2006;52:981–996. doi: 10.1016/j.neuron.2006.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, Ambesi-Impiombato A, Califano A, Migliazza A, Bhagat G, Dalla-Favera R. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17:28–40. doi: 10.1016/j.ccr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- Kuruvilla R, Ye H, Ginty DD. Spatially and functionally distinct roles of the PI3-K effector pathway during NGF signaling in sympathetic neurons. Neuron. 2000;27:499–512. doi: 10.1016/s0896-6273(00)00061-1. [DOI] [PubMed] [Google Scholar]
- Lee T, Marticke S, Sung C, Robinow S, Luo L. Cell-autonomous requirement of the USP/EcR-B ecdysone receptor for mushroom body neuronal remodeling in Drosophila. Neuron. 2000;28:807–818. doi: 10.1016/s0896-6273(00)00155-0. [DOI] [PubMed] [Google Scholar]
- Liu Y, Corcoran M, Rasool O, Ivanova G, Ibbotson R, Grander D, Iyengar A, Baranova A, Kashuba V, Merup M, Wu X, Gardiner A, Mullenbach R, Poltaraus A, Hultstrom AL, Juliusson G, Chapman R, Tiller M, Cotter F, Gahrton G, et al. Cloning of two candidate tumor suppressor genes within a 10 kb region on chromosome 13q14, frequently deleted in chronic lymphocytic leukemia. Oncogene. 1997;15:2463–2473. doi: 10.1038/sj.onc.1201643. [DOI] [PubMed] [Google Scholar]
- Lovestone S, Hartley CL, Pearce J, Anderton BH. Phosphorylation of tau by glycogen synthase kinase-3 beta in intact mammalian cells: the effects on the organization and stability of microtubules. Neuroscience. 1996;73:1145–1157. doi: 10.1016/0306-4522(96)00126-1. [DOI] [PubMed] [Google Scholar]
- MacAulay K, Doble BW, Patel S, Hansotia T, Sinclair EM, Drucker DJ, Nagy A, Woodgett JR. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007;6:329–337. doi: 10.1016/j.cmet.2007.08.013. [DOI] [PubMed] [Google Scholar]
- McLaughlin T, Torborg CL, Feller MB, O'Leary DD. Retinotopic map refinement requires spontaneous retinal waves during a brief critical period of development. Neuron. 2003;40:1147–1160. doi: 10.1016/s0896-6273(03)00790-6. [DOI] [PubMed] [Google Scholar]
- Meijer L, Flajolet M, Greengard P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol Sci. 2004;25:471–480. doi: 10.1016/j.tips.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Migliazza A, Bosch F, Komatsu H, Cayanis E, Martinotti S, Toniato E, Guccione E, Qu X, Chien M, Murty VV, Gaidano G, Inghirami G, Zhang P, Fischer S, Kalachikov SM, Russo J, Edelman I, Efstratiadis A, Dalla-Favera R. Nucleotide sequence, transcription map, and mutation analysis of the 13q14 chromosomal region deleted in B-cell chronic lymphocytic leukemia. Blood. 2001;97:2098–2104. doi: 10.1182/blood.v97.7.2098. [DOI] [PubMed] [Google Scholar]
- Mok SA, Lund K, Campenot RB. A retrograde apoptotic signal originating in NGF-deprived distal axons of rat sympathetic neurons in compartmented cultures. Cell Res. 2009;19:546–560. doi: 10.1038/cr.2009.11. [DOI] [PubMed] [Google Scholar]
- Munoz L, Ammit AJ. Targeting p38 MAPK pathway for the treatment of Alzheimer's disease. Neuropharmacology. 2010;58:561–568. doi: 10.1016/j.neuropharm.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Perry VH, Brown MC, Lunn ER, Tree P, Gordon S. Evidence that very slow Wallerian degeneration in C57BL/Ola mice is an intrinsic property of the peripheral nerve. Eur J Neurosci. 1990;2:802–808. doi: 10.1111/j.1460-9568.1990.tb00472.x. [DOI] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Sang H, Lu Z, Li Y, Ru B, Wang W, Chen J. Phosphorylation of tau by glycogen synthase kinase 3beta in intact mammalian cells influences the stability of microtubules. Neurosci Lett. 2001;312:141–144. doi: 10.1016/s0304-3940(01)02206-6. [DOI] [PubMed] [Google Scholar]
- Tessier-Lavigne M, Placzek M, Lumsden AG, Dodd J, Jessell TM. Chemotropic guidance of developing axons in the mammalian central nervous system. Nature. 1988;336:775–778. doi: 10.1038/336775a0. [DOI] [PubMed] [Google Scholar]
- Wakatsuki S, Saitoh F, Araki T. ZNRF1 promotes Wallerian degeneration by degrading AKT to induce GSK3B-dependent CRMP2 phosphorylation. Nat Cell Biol. 2011;13:1415–1423. doi: 10.1038/ncb2373. [DOI] [PubMed] [Google Scholar]
- Watts RJ, Hoopfer ED, Luo L. Axon pruning during Drosophila metamorphosis: evidence for local degeneration and requirement of the ubiquitin-proteasome system. Neuron. 2003;38:871–885. doi: 10.1016/s0896-6273(03)00295-2. [DOI] [PubMed] [Google Scholar]
- Weimann JM, Zhang YA, Levin ME, Devine WP, Brûlet P, McConnell SK. Cortical neurons require Otx1 for the refinement of exuberant axonal projections to subcortical targets. Neuron. 1999;24:819–831. doi: 10.1016/s0896-6273(00)81030-2. [DOI] [PubMed] [Google Scholar]
- Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Q, Wang J, Kim A, Liu Q, Watts R, Hoopfer E, Mitchison T, Luo L, He Z. Involvement of the ubiquitin-proteasome system in the early stages of Wallerian degeneration. Neuron. 2003;39:217–225. doi: 10.1016/s0896-6273(03)00429-x. [DOI] [PubMed] [Google Scholar]
- Zhang YA, Okada A, Lew CH, McConnell SK. Regulated nuclear trafficking of the homeodomain protein otx1 in cortical neurons. Mol Cell Neurosci. 2002;19:430–446. doi: 10.1006/mcne.2001.1076. [DOI] [PubMed] [Google Scholar]
- Zhou FQ, Snider WD. Cell biology. GSK-3beta and microtubule assembly in axons. Science. 2005;308:211–214. doi: 10.1126/science.1110301. [DOI] [PubMed] [Google Scholar]
- Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD. NGF-induced axon growth is mediated by localized inactivation of GSK-3beta and functions of the microtubule plus end binding protein APC. Neuron. 2004;42:897–912. doi: 10.1016/j.neuron.2004.05.011. [DOI] [PubMed] [Google Scholar]