

Abstract
αB-crystallin is a member of the heat shock protein family that exerts cell protection under several stress-related conditions. Recent studies have revealed that αB-crystallin plays a beneficial role in a mouse model of multiple sclerosis, brain ischemia, and Alexander disease. Whether αB-crystallin plays a role in modulating the secondary damage after CNS trauma is not known. We report here that αB-crystallin mediates protective effects after spinal cord injury. The levels of αB-crystallin are reduced in spinal cord tissue following contusion lesion. In addition, administration of recombinant human αB-crystallin for the first week after contusion injury leads to sustained improvement in locomotor skills and amelioration of secondary tissue damage. We also provide evidence that recombinant human αB-crystallin modulates the inflammatory response in the injured spinal cord, leading to increased infiltration of granulocytes and reduced recruitment of inflammatory macrophages. Furthermore, the delivery of recombinant human αB-crystallin promotes greater locomotor recovery even when the treatment is initiated 6 h after spinal cord injury. Our findings suggest that administration of recombinant human αB-crystallin may be a good therapeutic approach for treating acute spinal cord injury, for which there is currently no effective treatment.
Introduction
Injury to the spinal cord results in immediate damage caused directly by the trauma, followed by a secondary phase of tissue degeneration that occurs over a period of several weeks, which contributes significantly to functional impairment (Schwab and Bartholdi, 1996; Oyadomari et al., 2002). Multiple mechanisms contribute to secondary damage, not all of which are fully defined (Kwon et al., 2004; Popovich and Longbrake, 2008). In addition to mechanisms that actively induce secondary damage, lack of effective protective responses that block secondary damage or mediate repair can also contribute to tissue damage and functional loss (Kwon et al., 2004; Popovich and Longbrake, 2008). The final outcome after injury will therefore depend on the balance between the harmful and protective responses. Activating endogenous protective mechanisms in spinal cord injury (SCI) can therefore be expected to reduce secondary damage and minimize functional deficits.
Heat Shock Proteins (Hsp) are among the most highly expressed cellular proteins across all species (Lindquist and Craig, 1988). Hsp act as molecular chaperones and protect cells by preventing protein misfolding and aggregation (Morange, 2005). In the CNS, some members of the Hsp family are upregulated and exert protection (Muchowski and Wacker, 2005). The beneficial functions of Hsp in the CNS include prevention of protein aggregation, refolding of partially denatured proteins, inhibiting cell death pathways, and modulating the inflammatory response (Muchowski and Wacker, 2005; Brown, 2007).
αB-crystallin (CRYAB) is a small Hsp family member closely related to Hsp27 (Ingolia and Craig, 1982). CRYAB is constitutively expressed in many tissues and is especially abundant in eye lens, heart, and muscles (Arrigo et al., 2007). Several studies have demonstrated that CRYAB mediates cell protection under various stress conditions (Arrigo et al., 2007). Moreover, human mutations in the CRYAB gene have been associated with cataracts (Safieh et al., 2009; Chen et al., 2010) and several myopathies (Goldfarb and Dalakas, 2009; Del Bigio et al., 2011). In the CNS, CRYAB is constitutively expressed in oligodendrocytes; nevertheless, its expression is found in other glial cells in multiple sclerosis and brain ischemia (Piao et al., 2005; Ousman et al., 2007). It is also found in inclusion bodies in many protein conformation disorders such as Alexander disease, Alzheimer disease, and Parkinson disease (Muchowski and Wacker, 2005; Arrigo et al., 2007). Little is known about the role of CRYAB in the CNS. Recent studies have revealed a protective role for CRYAB in a mouse model of multiple sclerosis [experimental autoimmune encephalomyelitis (EAE)], stroke (Arac et al., 2011), and Alexander disease (Ousman et al., 2007; Hagemann et al., 2009). In EAE, CRYAB acts as a negative modulator of inflammation and prevents demyelination and disease symptoms (Ousman et al., 2007). In Alexander disease, however, beneficial functions of CRYAB are mediated by preventing protein misfolding and aggregation (Hagemann et al., 2009). At present, the expression and role of CRYAB after CNS trauma has not been reported. Here, we report for the first time whether CRYAB confers protection in SCI in mice.
Materials and Methods
Spinal cord contusion injury.
All surgical procedures were approved by the Universitat Autònoma de Barcelona Animal Care Committee and the McGill University Animal Care Committee, and followed the guidelines of the European Commission on Animal Care and the Canadian Council on Animal Care. Adult (8–10 weeks old) female C57BL/6 mice (Charles River) were anesthetized with ketamine:xylazine:acepromazine (100:10:3 mg/kg). After performing a laminectomy at the 11th thoracic vertebrae, the exposed spinal cord was contused using the Infinite Horizon Impactor device (Precision Scientific Instrumentation). Injuries were made using a force of 60 kdynes and tissue displacement ranging between 500 and 700 μm.
Recombinant human αB-crystallin (rhCRYAB; US Biologicals) was diluted in sterile saline and mice injected intravenously with 10 μg of rhCRYAB in 100 μl of total solution. This dose was previously shown to be effective in EAE and stroke (Ousman et al., 2007; Arac et al., 2011). The total amount of endotoxins administrated intravenously per mouse was 0.5 EU/kg body weight/d, lower than the maximum endotoxin levels set by the FDA for injectable drugs (5 EU/kg body weight/h). Control mice were injected with an equal volume of sterile saline. Mice were first injected 1, 6, or 12 h after SCI and injections were repeated daily thereafter until day 7.
To trace which cells uptake CRYAB in SCI, another group of mice received 10 μg of recombinant human GST-tagged CRYAB in 100 μl of saline.
Functional assessment.
Locomotor recovery was evaluated in an open-field test using the nine-point Basso Mouse Scale (BMS) (Basso et al., 2006), which was specifically developed for locomotor testing after contusion injuries in mice. The BMS analysis of hindlimb movements and coordination was performed by two independent assessors and the consensus score taken. The final score is presented as mean ± SEM.
Western blotting.
A 5 mm length of spinal cord tissue containing the lesion site was collected from uninjured mice and at 1, 3, 7, 14, 21, and 28 d postinjury (dpi) (n = 3 per time point). To reduce variability in protein solubilization, samples were homogenized simultaneously in 10% NP-40, 1% deoxycholate, 0.1% SDS in PBS containing Complete Protease and Phosphatase inhibitor Cocktail (Roche Applied Science). After centrifugation at 15,000 g for 20 min at 4°C, supernatants were isolated and protein concentrations determined using the Bio-Rad Protein Assay. Protein samples (30 μg) were separated by electrophoresis on a 10–15% polyacrylamide gel and transferred onto PVDF membranes (Millipore). Each polyacrylamide gel was loaded with individual protein samples from each of the different time points.
In addition, injured spinal cords were obtained at 12, 24, and 72 h postinjury from mice treated with rhCRYAB or saline (n = 4 per group and time point) and were homogenized using the same protocol.
The membranes were incubated with rabbit antibodies against CRYAB (1:250; Assay Designs), NF-kB p50/105 (1:200; Cell Signaling Technology), Iκ-Bα (1:200; Cell Signaling Technology), and C/EBP homologous protein (CHOP; 1:100; Santa Cruz Biotechnology). Bands were detected using Chemiluminescence (Immobilon Western Chemiluminescence HRP reagent; Millipore) and the data quantified by densitometry using Gene Snap and Gene Tools software in a Gene Genome apparatus (Syngene). β-actin (1:10,000; Sigma Aldrich) was used to ensure equal loading of samples.
Histology.
Naive and injured mice were perfused with 4% paraformaldehyde in 0.1 m-phosphate buffer (PB) at 12 h and 3 and 28 dpi. A 5 mm length of spinal cord containing the lesion site was removed, cryoprotected with 30% sucrose in 0.1 m PB at 4°C, and six series of 15-μm-thick section were picked up on glass slides. Adjacent sections on the same slide were therefore 90 μm apart. For immunofluorescence, tissue sections were rehydrated in PBS and blocked in 5% serum in TBS with 0.5% Triton X-100 for 1 h. The sections were then incubated overnight at 4°C with rabbit anti-CRYAB (1:500; Assay Designs) or rabbit anti-GST (1:250; Abcam) combined with antibodies against GFAP (for astrocytes; 1:500; Zymed), NeuN (for neurons; 1:200; Millipore), CC1 (for oligodendrocytes; 1:50; Calbiochem), Iba1 (for macrophage/microglia; 1:250; Abcam), Gr1 (for granulocytes; 1:100; Invitrogen), and CD31 (for endothelial cells; 1:100; Acris Antibodies). After several washes in PBS, sections were incubated for 1 h at room temperature with the appropriate Alexa 594- or Alexa 488-conjugated secondary antibodies (1:500; Invitrogen) and then coverslipped in Mowiol containing DAPI to label nuclei.
CRYAB+ oligodendrocytes were manually quantified in tissue cross-sections in a 7.5 × 104 μm2 area in the lateral and dorsal columns from five tissue sections of uninjured and contused spinal cord at day 7 postinjury and the percentage of CC1+ oligodendrocytes expressing CRYAB were averaged from these sections (n = 3 per time point). Quantification of CC1+ and CRYAB+ cells in the contused spinal cord was done at the epicenter and in cord sections located at 270 and 570 μm rostral and caudal, to the injury epicenter. Quantification of cells that have taken up rhCRYAB was performed from saggital spinal cord tissue sections harvested at 12 h postinjury from mice injected with GST-tagged rhCRYAB. Cells double-stained with GST+ cells and GFAP, NeuN, or CC1 were manually counted in an area of 7.5 × 104 μm2 at both lateral and rostrocaudal edges of the lesion. Three spinal cord tissue sections were analyzed per mouse (n = 3). Histopathological analyses of the injured spinal cord were performed from tissue samples harvested at day 28. Images were acquired with the aid of a digital camera attached to the microscope (Olympus) and then analyzed using NIH ImageJ software (NIH, Bethesda, MD). Tissue sparing was calculated by delineating the GFAP-stained areas (GFAP-positive tissue), and neuronal survival assessed by manually counting the NeuN-positive cells in the ventral horn. Double immunostaining against NF (1:1000; Millipore) and MBP (1:100; Abcam) was performed to assess axonal and myelin sparing. Assessment of axonal sparing was performed by counting the axons in the dorsal columns at the injury epicenter. Myelin sparing or demyelination in the dorsal column was assessed by counting the fibers double-stained for NF and MBP at the lesion epicenter. Assessment of astrogliosis was done by calculating the area of GFAP immunoreactivity in high-magnification images (×40) taken from lateral white matter at the injury epicenter and rostral and caudal regions, for a total length of 1800 μm. Two images from the lateral white were taken from each spinal cord section immunostained for GFAP, and the area of GFAP immunoreactivity was averaged from both images. Eight mice per group were used in all the histopathological analyses.
Cytokine protein expression.
A 5 mm length of spinal cord centered on the lesion was collected at 12 h after surgery from mice treated with rhCRYAB or saline (n = 4 each group) and snap-frozen. Spinal cords were homogenized in Tissue Extraction Reagent I (Invitrogen), and protein concentration was determined using the DC Protein Assay (Bio-Rad). Samples were concentrated using MicroCon centrifugation filters (Millipore) and the protein concentration redetermined. All samples were diluted to 3.7 μg/μl to ensure equal amounts of protein. The protein levels of 20 cytokines and chemokines (FGF, GM-CSF, IFN-γ, IL-1α, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12p40/p70, IL-13, IL-17, IP-10, CXCL1/KC, CCL2/MCP-1, MIG, CCL3/MIP-1α, TNF-α, and VEGF) were then analyzed using the BioSource Mouse Cytokine 20-PlexMultiplex Bead Immunoassay (Invitrogen) on a Luminex-100LS system as per the manufacturers' protocol. Results were analyzed using Beadview multiplex data analysis software (UpState Biotechnology).
Flow cytometry.
Immune cells from the injured spinal cord were analyzed by flow cytometry at 1, 3, and 7 dpi as described previously (Stirling and Yong, 2008). Briefly, spinal cords were cut in little pieces and passed through a cell strainer of 40 μm (BD Falcon) and the cell suspension was centrifuged twice at 300 g for 10 min at 4°C. After cell counts, samples were incubated with anti-CD16/CD32 (eBioscience) for 15 min at room temperature to block the nonspecific binding of immunoglobulins to the Fc receptors. Samples were divided, and cells alone and isotype-matched control samples were generated to control for nonspecific binding of antibodies and for autofluorescence. Isotype control antibodies were purchased from eBioscience and included phycoerythrin (PE)-labeled rat IgG2b, APC-labeled rat IgG2b, and peridinin chlorophyll-a protein (PerCP)-conjugated rat IgG2b, fluorescein isothiocyanate-conjugated (FITC) rat IgG2b, and IgG2M. The following antibodies were also purchased from eBioscience: CD45-PerCP, GR1-FITC, CD11b-PE, Ly-6C-FITC, and F4/80-APC. After 30 min of incubation with combinations of antibodies at 4°C, the samples were washed and fixed in 1% paraformaldehyde. Granulocytes were identified as CD45high, CD11b+, and Gr-1high (Stirling and Yong, 2008). Macrophage/microglia subsets (CD45+, CD11b+, F4/80+) were further differentiated based on Ly6C expression (Arnold et al., 2007; Nahrendorf et al., 2007). At least 3000 CD45+ cells were analyzed per spinal cord sample using FlowJo software on a FACSCanto flow cytometer (BD Biosciences).
Statistical analyses.
Data are shown as mean ± SEM. Changes in the expression of CRYAB in SCI assessed by Western blot were analyzed by using one-way ANOVA with post hoc Dunnett's test. BMS and histological data were performed by using two-way repeated-measures ANOVA with post hoc Tukey's test for multiple comparisons. Student's t test was used for the Western blot and FACS analyses comparing control and experimental groups. Differences were considered significant at p < 0.05.
Results
Expression of CRYAB in the injured spinal cord
We first assessed the protein levels of CRYAB in the uninjured spinal cord and at 1, 3, 7, 14, 21, and 28 d after contusion injury. Western blot analyses revealed that CRYAB protein levels were reduced ∼40% at day 1 after SCI, and remained lower than normal levels for up to day 28 postinjury (Fig. 1A,B). We next assessed cellular localization of CRYAB in naive and injured spinal cord tissue sections. Double immunofluorescence labeling revealed that CRYAB is expressed in 53.7 ± 2.3% of CC1-positive oligodendrocytes in the uninjured spinal cord (Fig. 1C–E). At day 7 following contusion, when levels for CRYAB peaked after injury, spinal cord tissue sections from 540 μm rostral to 540 μm caudal to the injury epicenter revealed that CRYAB is expressed in 44.5 ± 3.8% of CC1+ cells. We did not observed constitutive expression of CRYAB in astrocytes (Fig. 1F–H). Following lesion, however, CRYAB was detected in GFAP-positive astrocytes forming the glial scar (Fig. 1I–K). The death of oligodendrocytes that occurs after SCI (Crowe et al., 1997) and probably the failure of the remaining oligodendrocytes to upregulate CRYAB after injury might be responsible for the reduction of CRYAB levels in injured spinal cord seen by Western blotting. CRYAB was not observed in macrophages, granulocytes, neurons, or endothelial cells (data not shown).
Figure 1.
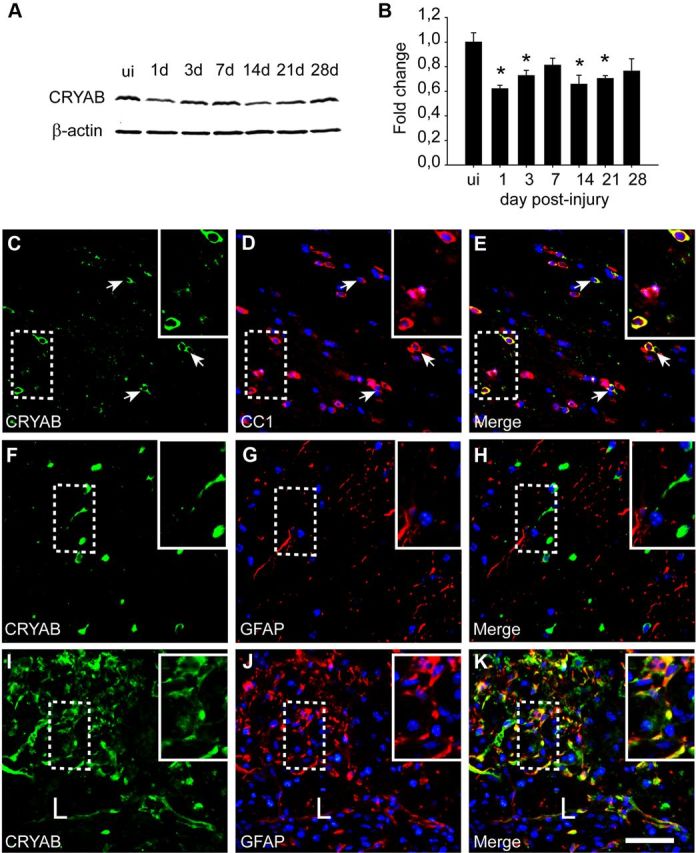
Expression of CRYAB in spinal cord injury. A, B, Western blot analysis shows reduced CRYAB protein levels after spinal cord injury starting at day 1. β-actin is used as a loading control (n = 3 mice per time point). ui, Uninjured. C–E, Double immunofluorescence labeling of a cross-section of the uninjured spinal cord for CC1 and CRYAB. Note the colocalization of CRYAB (green) in oligodendrocytes (red). F–K, Double immunofluorescence labeling for GFAP and CRYAB of a cross-section of uninjured (F–H) and injured spinal cord (I–K) harvested at day 7 postinjury. Note that astrocytes do not express CRYAB in the uninjured cord. However, there is strong CRYAB immunoreactivity (green) in reactive astrocytes (red) at the border of the lesion (L). The areas outlined in the boxes are shown in higher magnification in the insets. Scale bar, 50 μm. *p < 0.05. Error bars indicate SEM.
Administration of recombinant human CRYAB enters into the injured spinal cord
We hypothesized that the loss of CRYAB in the spinal cord tissue after injury contributes to secondary damage after SCI, and that exogenous administration of CRYAB may lead to protective effects. Since injury to the spinal cord leads to blood vessel disruption and blood–brain barrier breakdown, we assessed whether intravenous injection of rhCRYAB 1 h postinjury could enter the injured spinal cord. Western blot analysis of injured spinal cords revealed that 11 h postinjection (12 h postinjury), protein levels for CRYAB were approximately twofold greater in mice injected with rhCRYAB compared with those receiving saline (p < 0.05; Fig. 2A,B). However, CRYAB protein levels in mice treated with rhCRYAB were still reduced ∼20% compared with physiological levels in normal uninjured spinal cord (p < 0.05; Fig. 2A,B). To further confirm that the exogenously administered rhCRYAB entered the injured spinal cord and assess which cell types had taken it up, we injected GST-tagged rhCRYAB. The GST tag allowed us to distinguish endogenous CRYAB from the rhCRYAB administered. GST staining was observed in tissue sections from injured spinal cords but not in those harvested from uninjured animals, suggesting that rhCRYAB is only able to reach the injured CNS (Fig. 2C,D). Histological sections from spinal cord tissue harvested at 11 h postinjection (12 h postinjury) immunostained against GST revealed that rhCRYAB was located in the white matter at the injury epicenter, and in white and gray matter in adjacent areas, for a total rostrocaudal length of ∼3 mm. In these areas, double immunofluorescence labeling for GST and cell type-specific antibodies revealed that rhCRYAB was taken up by 30.7 ± 4.7% of oligodendrocytes, 38.8 ± 5.2% of astrocytes, and 45.1 ± 3.1% of neurons (Fig. 2E–G). rhCRYAB was not found in microglial cells, granulocytes, or endothelial cells (data not shown).
Figure 2.

Recombinant human CRYAB reaches the injured spinal cord when given intravenously. A, B, Western blot from spinal cords harvested at 12 h following lesion shows increased CRYAB protein levels in mice injected with recombinant human CRYAB. β-actin is used as a loading control (n = 4 per group). C–E, Double immunofluorescence for GST and GFAP (C), NeuN (D), and CC1 (E) at 12 h after injury. The areas outlined in the boxes are shown in higher magnification in the insets. Note the presence of the recombinant human CRYAB in astrocytes, neurons, and oligodendrocytes. GFAP and CC1 images were taken from the ventral white matter, while NeuN staining was taken from the ventral horn of spinal cord tissue sections taken at 270 μm rostral in relation to injury epicenter. Scale bar, 50 μm. *p < 0.05. Error bars indicate SEM.
Treatment with rhCRYAB promotes recovery after spinal cord injury
We next examined whether administration of rhCRYAB enhances locomotor recovery after SCI. Compared with the saline-treated mice, those treated with rhCRYAB showed significant improvement in locomotor recovery [p < 0.05; two-way repeated-measure (RM)-ANOVA with post hoc Tukey's test; n = 8 for both groups; Fig. 3]. Post hoc analysis revealed significant differences in the BMS score starting at day 5 after injury and remaining significantly enhanced for the 4 weeks of the follow up. At 28 d postinjury, vehicle-treated mice showed no plantar stepping (score, 2.8) but showed extensive ankle movements or plantar placement of the paw with or without weight support. In contrast, all the mice treated with rhCRYAB displayed occasional (∼40%) or frequent plantar stepping (∼60%) (score, 4.6) but lacked coordination.
Figure 3.

Time course of locomotor recovery in mice treated with recombinant human CRYAB starting 1 h after SCI. Locomotor recovery was assessed after spinal cord contusion injury in mice treated with saline or recombinant human CRYAB (n = 8 per group) using the BMS. Administration of recombinant human CRYAB show significant improvement in locomotor performance compared with wild-type mice as early as 5 d after injury. *p < 0.05. Error bars indicate SEM.
We then assessed whether the locomotor improvement observed after rhCRYAB administration was associated with amelioration of secondary tissue damage. Histological sections of the spinal cord labeled with anti-GFAP antibodies revealed that rhCRYAB treatment reduced tissue loss at the epicenter of the injury and in adjacent regions (p < 0.05; two-way RM-ANOVA with post hoc Tukey's test; Fig. 4A–C). We also assessed axonal loss and demyelination at the dorsal column of the spinal cord at the epicenter of the injury (Fig. 4D–H). Spinal cord sections immunolabeled with anti-NF showed that rhCRYAB treatment resulted in an approximately twofold greater number of spared axons in the dorsal columns at the epicenter of the lesion (p = 0.02; t test; Fig. 4D,E,G). Furthermore, double immunostaining against NF and MBP revealed that rhCRYAB administration led to approximately fivefold greater number of myelinated axons (p = 0.007; t test; Fig. 4D,F,H). We also evaluated whether rhCRYAB treatment protected against neuronal loss. Quantification of NeuN-positive cells in the ventral horn of the spinal cord indicated that mice treated with rhCRYAB showed improved neuronal survival in regions rostral to the injury epicenter for a total length of 1000 μm rostral to the epicenter (p < 0.05; two-way RM-ANOVA with post hoc Tukey's test; Fig. 4I–M). Overall, these data demonstrate that treatment with rhCRYAB has multiple protective effects in spinal cord injury.
Figure 4.
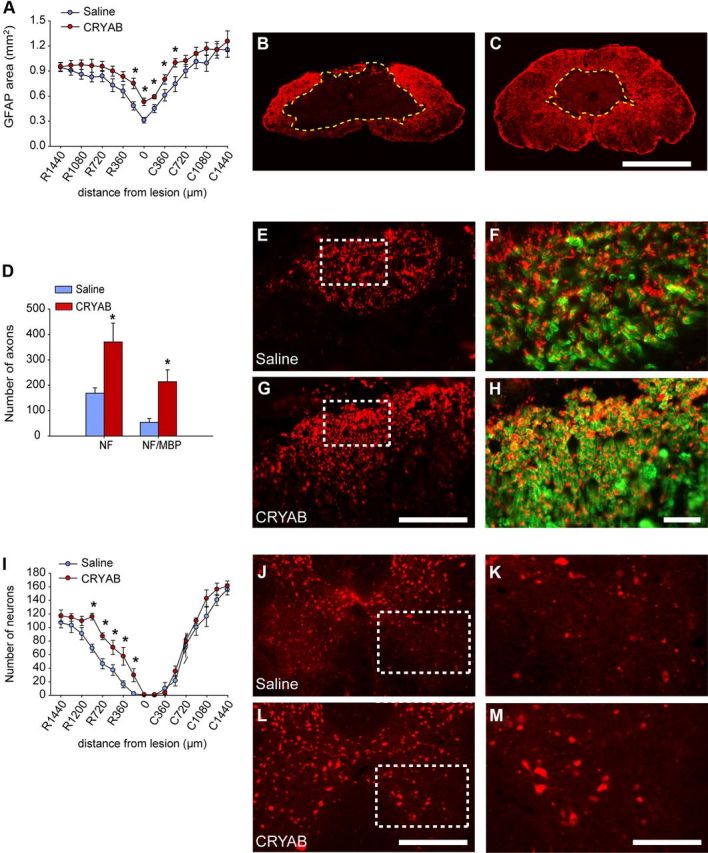
Recombinant human CRYAB treatment ameliorates tissue damage. A, Quantification of tissue sparing at various distances rostral (R) and caudal (C) to the injury epicenter revels significant reduction in tissue loss in mice treated with recombinant human CRYAB at the epicenter of the injury and adjacent sections. B, C, Representative micrographs showing tissue sparing at the injury epicenter in mice treated with saline (B) or recombinant human CRYAB (C). D, Quantification of axon and myelin sparing in the dorsal column at the epicenter of the injury. Treatment with recombinant human CRYAB resulted in greater axon and myelin preservation. E–H, Representative micrographs of the dorsal columns stained against NF (E, G) and MBP (F, H) from mice treated with saline (E, F) and recombinant human CRYAB (G, H). Note the greater preservation of axons and myelinated fibers in mice administered with recombinant human CRYAB. I, Quantification of ventral horn neuron survival at various distances rostral and caudal to the injury epicenter reveals significantly greater neuronal survival in mice treated with recombinant human CRYAB. J–L, Representative micrographs showing sparing of ventral horn neurons in mice administered with saline (J) and recombinant human CRYAB (L) in sections stained against NeuN at 600 μm rostral to the injury epicenter. J, L, The areas outlined in the boxes are shown in higher magnification in K and E, respectively. Scale bars: B, C (in C), 500 μm; E, G (in G), 125 μm; F, H (in H), 25 μm; J, L (in L), 250 μm; K, M (in M), 75 μm. *p < 0.05. Error bars indicate SEM.
Treatment with rhCRYAB modulates inflammatory responses in spinal cord injury
Previous studies have shown that CRYAB mediates protection against protein misfolding and aggregation, which is a pathological hallmark of many neuropathological disorders (Arrigo et al., 2007; Hagemann et al., 2009; Bousette et al., 2010). Misfolded and aggregated proteins induce cell death, in part, by triggering the accumulation of the transcription factor CHOP, also known as GADD153, an endoplasmic reticulum stress inducer of apoptosis (Zinszner et al., 1998; Oyadomari et al., 2002). We therefore assessed whether rhCRYAB reduced CHOP levels in SCI at 3 d following injection, when CHOP levels peak after SCI (Penas et al., 2007; Ohri et al., 2011). Our results revealed that rhCRYAB treatment failed to reduce CHOP levels in SCI (data not shown). These data suggest that the protective mechanisms of rhCRYAB in SCI are unlikely to be mediated by reduction of stress response triggered by protein misfolding and aggregation.
Recent studies suggest that CRYAB modulates inflammatory response in multiple sclerosis and its mouse model (EAE) and after brain stroke (Ousman et al., 2007; van Noort et al., 2010; Arac et al., 2011). We therefore assessed whether rhCRYAB administration was able to modulate inflammation in SCI. We first examined activation of NF-κB in SCI, a key nuclear factor in triggering the activation of the neuroinflammatory response. We found that at 12 h after SCI, mice treated with rhCRYAB displayed greater levels of the activated NF-κB p50 subunit compared with saline-treated mice (p < 0,05; t test), while the level of its negative regulator, Iκ-Bα, remained unchanged (Fig. 5A,B). We then studied whether the increased NF-κB levels observed after rhCRYAB treatment altered the expression of inflammatory cytokines and chemokines in the spinal cord at 12 h following contusion (Fig. 5C). We found that injured spinal cords from mice treated with rhCRYAB had a marked increase in expression of four chemokines (CCL1/KC, CCL2/MCP-1, CCL3/MIP-1α, CXCL10/IP-10) and two cytokines (IL-5, IL-6), when compared with saline-treated mice (n = 4 for each group; Student's t test; Fig. 5C). Since cytokines and chemokines promote the infiltration and activation of immune cells, we used FACS analyses to study whether treatment with rhCRYAB modulated the infiltration of myeloid cells in SCI. CD45 was used as a marker for peripheral immune cells and microglia to sort cells obtained from injured spinal cords from rhCRYAB and saline-treated mice. The combination of antibodies against CD45, CD11b, Gr-1, F4/80, and Ly6C allowed us to discriminate among granulocytes (CD45+, CD11bhigh, Gr1high), inflammatory macrophages (CD45+, CD11b+, F4/80+, Ly6C+), and resident macrophages (CD45+, CD11b+, F4/80+, Ly6C−) from blood (Arnold et al., 2007; Nahrendorf et al., 2007). Our data show a significant increase of granulocytes in the spinal cord parenchyma of mice injected with rhCRYAB at day 1 following contusion compared with those mice treated with saline (n = 4; p = 0.004, Student's t test; Fig. 6A). No differences were found with regards to the two types of monocyte populations at this time point (Fig. 6B). Analyses performed at day 3 after injury revealed that there was no difference in the proportion of granulocytes and resident macrophages in the spinal cord between both experimental groups (Fig. 6). However, the proportion of inflammatory macrophages was significantly reduced in mice treated with rhCRYAB (n = 4; p = 0.03, t test; Fig. 6B). We did not observe any differences regarding these three immune cell populations at day 7 postinjury (data not shown).
Figure 5.

Treatment with recombinant human CRYAB modulates inflammatory response. A, Western blots showing NF-kB p50 and IkB-α expression at 12 h following injury in mice treated with saline of recombinant human CRYAB. β-actin is used as a loading control (n = 4 per group). B, Densitometric values of the Western blot reveal increased expression of NF-kB p50 after treatment with recombinant human CRYAB. C, Changes in chemokine and cytokine expression in the spinal cord at 12 h following contusion injury in mice administered with saline or recombinant human CRYAB. Note that treatment with recombinant human CRYAB resulted in greater expression of IL5, IL6, KC, IP10, MCP1, and MIG. *p < 0.05. Error bars indicate SEM.
Figure 6.

Altered recruitment of immune cells in the injured spinal cord after treatment with recombinant human CRYAB. A, B, Representative density plots of FACS analysis showing the infiltration of granulocytes (CD45 high, CD11b+, Gr1 high; A) and type I inflammatory macrophages (CD45+, CD11b high, F4/80+, Ly6C+) and type II anti-inflammatory macrophages (CD45+, CD11b high, F4/80+, Ly6C−) (B) at days 1 and 3 following contusion injury. Note that treatment with recombinant human CRYAB results in increased infiltration of granulocytes at day 1 but not at day 3 following injury compared with mice treated with saline. Administration of recombinant human CRYAB also leads to significant reduction of inflammatory macrophages at day 3 after SCI but does not altered the anti-inflammatory macrophage population (n = 4 per group and time point). *p < 0.05. Error bars indicate SEM.
We also assessed whether rhCRYAB had any effect on astrogliosis. Tissue sections from spinal cord taken at day 28 postinjury revealed that treatment with rhCRYAB led to significant reduction in astroglia reactivity at the injury epicenter and in adjacent sections (p < 0.05; two-way RM-ANOVA with post hoc Tukey's test; n = 8 for both groups; Fig. 7). Therefore, administration of rhCRYAB in SCI modulates immune cell infiltration, promoting greater entry of granulocytes at day 1 following lesion and reducing the recruitment of inflammatory macrophages at day 3, and reduces astrogliosis.
Figure 7.

Effects of recombinant human CRYAB treatment on astrogliosis. A, Graph showing quantitative analysis of GFAP immunoreactivity area at the lateral white matter of the spinal cord at day 28 postinjury. B, C, Representative micrographs of the lateral funiculus stained for GFAP from mice treated with saline (B) or CRYAB (C) taken at 180 μm rostral to injury epicenter.
Effects of delayed treatment with rhCRYAB in SCI
We assessed the effectiveness of delaying the onset of rhCRYAB treatment beyond 1 h after SCI. Two delay treatments were tested, either starting at 6 or 12 h postinjury. We observed significant enhancement in locomotor recovery when rhCRYAB treatment was initiated 6 h following contusion. Significant differences were seen starting at day 7 following injury and remained enhanced until day 28 (n = 7 for each group; p < 0.05; two-way RM-ANOVA with post hoc Tukey's test; Fig. 8A). However, this enhancement in locomotor performance when rhCRYAB treatment was delayed for 6 h was less than that seen when treatment was started 1 h postlesion (Fig. 3). The beneficial effects of rhCRYAB in SCI were further reduced when treatment was delayed for 12 after SCI (Fig. 8B). Mice receiving rhCRYAB 12 h postinjury show significant improvement in locomotor control from days 5 to 14 but not at later time points (n = 9 for each group; p < 0.05; two-way RM-ANOVA with post hoc Tukey's test; Fig. 8B). Histological analysis of tissue sections taken at day 28 postinjury revealed that rhCRYAB promotes tissue sparing when the treatment is delayed for 6 but not 12 h postinjury (Fig. 8C,D).
Figure 8.

Locomotor recovery and tissue sparing in mice treated with recombinant human CRYAB starting 6 and 12 h after SCI. A, BMS scores of locomotor skills show that recombinant human CRYAB treatment started after a delay of 6 h following SCI also promoted significant improvement in locomotor recovery. B, After a 12 h delay in the start of treatment, there was a significant improvement in locomotor score from day 5 to14, but this effect was not sustained at later time points. C, D, Quantification of tissue sparing in mice treated with rhCRYAB at 6 h (C) or 12 h (D) postinjury. *p < 0.05. Error bars indicate SEM.
Discussion
Stress conditions triggered by injury or disease lead to increase protein misfolding and aggregation (Morange, 2005). This is of importance since the accumulation of misfolded and aggregated proteins is a pathological hallmark of several neurodegenerative diseases. Protein misfolding also occurs in SCI, which triggers the accumulation of CHOP in neurons and oligodendrocytes, and subsequently their death (Penas et al., 2007; Ohri et al., 2011). Previous studies have highlighted the protective role for CRYAB in some CNS conditions where protein misfolding occurs, such as in mouse models of Alexander disease, multiple sclerosis, brain ischemia, and Alzheimer′s disease (Ousman et al., 2007; Hagemann et al., 2009; Bousette et al., 2010; Arac et al., 2011; Ojha et al., 2011). Unexpectedly, we observed that protein levels of CRYAB are reduced following SCI. This loss of CRYAB may be due to oligodendrocyte cell death that occurs following lesion (Crowe et al., 1997), as this is the only cell type that expresses CRYAB in the uninjured spinal cord. However, the absence of CRYAB in neurons, and probably the failure of oligodendrocytes to increase CRYAB levels following SCI, may make these cells more vulnerable to death. The loss of CRYAB in the CNS has been also showed after brain ischemia (Piao et al., 2005), and in theTg2576 mouse (Ojha et al., 2011), a mouse model of Alzheimer disease. Interestingly, CRYAB-null Tg2576 mice showed greater exacerbation of protein aggregation and locomotion impairment compared with Tg2576, suggesting that the loss of CRYAB may contribute to the progression of Alzheimer disease (Ojha et al., 2011). Similarly, CRYAB−/− mice underwent greater tissue damage after stroke (Arac et al., 2011). Here we show that administration of rhCRYAB, which is taken up in the spinal cord parenchyma by oligodendrocytes, neurons, and astrocytes, reduces demyelination and neuronal cell death following SCI. rhCRYAB treatment, however, did not reduce CHOP levels after SCI, suggesting that the beneficial effects of rhCRYAB in SCI may be independent of protein misfolding and aggregation. Apart from its chaperone function, some Hsps modulate inflammation (Quintana and Cohen, 2005), which is one the major contributors to tissue damage after SCI (Popovich and Longbrake, 2008; Kwon et al., 2011). Recent studies revealed that CRYAB might be a negative regulator of inflammation, since mice lacking CRYAB showed exacerbated inflammatory response and functional deficits in EAE (Ousman et al., 2007) and after stroke (Arac et al., 2011). Administration of rhCRYAB, however, failed to reduce adaptive immune response after brain ischemia, whereas the effects on innate immune response were not assessed (Arac et al., 2011). Although it is unknown how the absence of CRYAB may exacerbate inflammation, it could be due, in part, to the greater susceptibility of cells to stress due to the lack of this chaperone, since CRYAB−/− astrocytes have augmented cell death, ERK, and NF-κB signaling upon TNF-α stimulation, but also under resting conditions (Ousman et al., 2007). Recent observations suggest that CRYAB may activate the innate immune response, since microglia become activated upon CRYAB stimulation in vitro (van Noort et al., 2010). These apparently diverging effects of CRYAB on inflammation may be due to the ability of this Hsp to act as an extracellular signal, as shown in other Hsp, such as HSP27 and HSP70 (Brown, 2007). Indeed, several studies demonstrate that α-crystallin family members are agonist of several receptors involved in the initiation of inflammation, such as protease-activated receptors (PARs) (Li et al., 2009; Li and Reiser, 2011) and toll-like receptors (TLRs) (Ohashi et al., 2000; Asea et al., 2002). Although the role of PARs in SCI is unknown, PAR-1 and PAR-2 exert protection in brain ischemia (Jin et al., 2005; Thiyagarajan et al., 2008). Similarly, TLR2 and TLR4-null mice showed greater functional deficits and myelin loss after SCI (Kigerl et al., 2007) and impaired axon regeneration after sciatic nerve injury (Boivin et al., 2007).
Astrocytes play an important role in initiating the inflammatory response following SCI (Pineau et al., 2010), as they are one of the major contributors to cytokines and chemokine synthesis (Pineau and Lacroix, 2007; Pineau et al., 2010). Activation of the immune response by astrocytes appears to be mediated, in part, by NF-κB signaling since inactivation of astroglial NF-κB in SCI results in reduced cytokine expression and inflammation (Brambilla et al., 2005). Here we show that astrocytes take up rhCRYAB when administered intravenously after SCI. Interestingly, astrocytes express PARs and TLRs (Kigerl et al., 2009; Li et al., 2009). Spinal cords harvested at 12 h postinjury revealed that rhCRYAB treatment led to greater levels in the expression of the active subunit NF-kB p50, and certain cytokines and chemokines (IL-5, IL-6, KC/CXCL1, MIG/CXCL9, IP-10/CXCL10, and MCP-1/CCL2), which are mainly synthesized by astrocytes in SCI (Pineau and Lacroix, 2007; Pineau et al., 2010). Our data suggest that exogenous rhCRYAB may stimulate astrocytes to increase the expression of cytokines and chemokines in SCI, as previously demonstrated in microglia (van Noort et al., 2010). Although we did not observe rhCRYAB in microglial cells, we cannot exclude the possibility that microglia may uptake low amounts of rhCRYAB after SCI and contribute to the increased levels of cytokines and chemokines observed after rhCRYAB administration. We also observed that mice treated with rhCRYAB had less astrogliosis at day 28 postinjury. Whether this effect is mediated by the direct action of CRYAB on astrocytes is not known; however, transgenic expression of CRYAB in astrocytes is reported to reduce astrocyte hypertrophy in Alexander's disease (Hagemann et al., 2009). Thus, our data suggest that administration of rhCRYAB enhances early activation of astrocytes following SCI, but reduces astrogliosis at late stages. Similar effects have been observed in after SCI in TLR4-null mice (Kigerl et al., 2007). Although the lack of TLR4 led to a marked reduction in the expression of some inflammatory triggers such as IL-1β early after SCI, it also resulted in greater astrogliosis (Kigerl et al., 2007).
Cytokines and chemokines trigger the recruitment and activation of immune cells. Our results show rhCRYAB treatment resulted in an increase of granulocytes at day 1 postlesion, but not at later time points. The contribution of granulocytes in SCI is a subject of debate. Several studies have reported that granulocytes play a detrimental role after SCI because blocking their recruitment or activity following lesion leads to reduced tissue damage (Taoka et al., 1997; Díaz-Ruiz et al., 2002; Naruo et al., 2003). However, since these treatments do not exclusively reduce granulocytes, but also monocytes and microglia, it is difficult to attribute these beneficial effects entirely to granulocytes. Recently, depletion of granulocytes with anti-Ly6G/Gr-1 antibody was reported to impair wound healing and lead to greater tissue damage and functional deficits after SCI, suggesting a protective role for neutrophils in SCI (Stirling et al., 2009). Granulocytes and reactive astrocytes are the main cell sources of secretory leukocyte protease inhibitor at early stages after SCI, an enzyme that promotes tissue healing and dampens inflammation (Taggart et al., 2005), and that mediates protection in SCI (Ghasemlou et al., 2010), which may explain, in part, the beneficial effects of granulocytes. However, a recent work shows that depletion of granulocytes before injury by Ly6G/Gr-1 antibody administration had no effect on functional and histological outcomes (Lee et al., 2011). Interestingly, the same study revealed that depletion of neutrophils before SCI promoted neurological recovery only if monocytes were also depleted from circulation (Lee et al., 2011). These studies therefore suggest that neutrophils may exert either helpful, harmful, or no effects in SCI depending on the presence or their interactions with other immune cells such as macrophages. However, since the Ly6G/Gr-1 antibody also recognizes Ly6C found on inflammatory but not in resident monocyte subsets (David and Kroner, 2011), it is possible that the divergent effects of the Ly6G/Gr-1 antibody may be due to a difference in the efficacy of the treatment to deplete the Ly6C+ inflammatory macrophages when given before and after SCI.
Studies performed in muscle injury have shown that Ly6C+ macrophages exhibit phagocytic, proteolytic, and inflammatory functions, whereas Ly6C− macrophages release anti-inflammatory cytokines and promote wound healing and repair (Arnold et al., 2007; Nahrendorf et al., 2007). Interestingly, prevention of the accumulation of Ly6C+ inflammatory macrophages in sites of inflammation reduces infarct size after coronary artery occlusion, prolongs normoglycemia in diabetic mice after pancreatic islet transplantation, and results in reduced tumor volumes (Leuschner et al., 2011), suggesting a crucial role for inflammatory macrophages in the course of many pathological conditions. These two populations of macrophages have been also described in SCI (Pineau et al., 2010). We observed that rhCRYAB treatment reduced the recruitment of Ly6C+ inflammatory macrophages into the injured spinal cord at 3 d following lesion, when this population peaked, but did not alter the accumulation of resident macrophage subset. Recent studies also revealed that a subset of macrophages, which express LyC6 but share some features of the resident macrophages, such as the expression of CX3CR1, have anti-inflammatory and neuroprotective properties (Shechter et al., 2009; London et al., 2011). Herein we observe an increase in granulocytes and a reduction of Ly6C+ inflammatory macrophages after rhCRYAB treatment. Therefore, the beneficial effects of CRYAB could be due to the effects on one or both of these Gr1+ cell populations.
Although further studies are needed to determine the optimal dose of CRYAB, our data provides evidence that administration of rhCRYAB after SCI modulates the inflammatory response and results in reduced secondary damage and functional impairment. In contrast to current anti-inflammatory therapies such as methylprednisolone or minocycline, among others, which broadly block the inflammatory response (Kwon et al., 2011), the administration of rhCRYAB seems to increase some early aspects of inflammation, such as cytokine/chemokine production and granulocytes recruitment, while reducing some delayed aspects, such as the infiltration of inflammatory macrophages. Since rhCRYAB treatment improved functional recovery when given within 1–6 h after SCI, but not when delayed longer, early changes in cytokine and chemokine expression observed upon rhCRYAB treatment may be crucial to conferring protection.
Overall, this study shows for the first time that CRYAB plays a beneficial role in SCI. Since rhCRYAB is effective even when treatment is delayed for 6 h after contusion injury, it could be a good therapeutic candidate for acute SCI in human, for which there is currently no effective treatment.
Footnotes
This work was supported by grants from Fundación Mapfre, Wings for Life Foundation, Marie-Curie Initial Training Network (AXREGEN, FP7-MC-214003-2), and by funds from the Fondo de Investigación Sanitaria of Spain (TERCEL and CIBERNED) to R.L.-V. and X.N.; and from the Canadian Institutes of Health Research to S.D. A.R. and E.S.-N. were funded by a grant from the Wings for Life Foundation (to S.D.). A.K. is a recipient of an AXREGEN fellowship.
References
- Arac A, Brownell SE, Rothbard JB, Chen C, Ko RM, Pereira MP, Albers GW, Steinman L, Steinberg GK. Systemic augmentation of alphaB-crystallin provides therapeutic benefit twelve hours post-stroke onset via immune modulation. Proc Natl Acad Sci U S A. 2011;108:13287–13292. doi: 10.1073/pnas.1107368108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, Chazaud B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med. 2007;204:1057–1069. doi: 10.1084/jem.20070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigo AP, Simon S, Gibert B, Kretz-Remy C, Nivon M, Czekalla A, Guillet D, Moulin M, Diaz-Latoud C, Vicart P. Hsp27 (HspB1) and alphaB-crystallin (HspB5) as therapeutic targets. FEBS Lett. 2007;581:3665–3674. doi: 10.1016/j.febslet.2007.04.033. [DOI] [PubMed] [Google Scholar]
- Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma. 2006;23:635–659. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- Boivin A, Pineau I, Barrette B, Filali M, Vallières N, Rivest S, Lacroix S. Toll-like receptor signaling is critical for Wallerian degeneration and functional recovery after peripheral nerve injury. J Neurosci. 2007;27:12565–12576. doi: 10.1523/JNEUROSCI.3027-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousette N, Chugh S, Fong V, Isserlin R, Kim KH, Volchuk A, Backx PH, Liu P, Kislinger T, MacLennan DH, Emili A, Gramolini AO. Constitutively active calcineurin induces cardiac endoplasmic reticulum stress and protects against apoptosis that is mediated by alpha-crystallin-B. Proc Natl Acad Sci U S A. 2010;107:18481–18486. doi: 10.1073/pnas.1013555107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med. 2005;202:145–156. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown IR. Heat shock proteins and protection of the nervous system. Ann N Y Acad Sci. 2007;1113:147–158. doi: 10.1196/annals.1391.032. [DOI] [PubMed] [Google Scholar]
- Chen Q, Yan M, Xiang F, Zhou X, Liu Y, Zheng F. Characterization of a mutant R11H alphaB-crystallin associated with human inherited cataract. Biol Chem. 2010;391:1391–1400. doi: 10.1515/BC.2010.143. [DOI] [PubMed] [Google Scholar]
- Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med. 1997;3:73–76. doi: 10.1038/nm0197-73. [DOI] [PubMed] [Google Scholar]
- David S, Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci. 2011;12:388–399. doi: 10.1038/nrn3053. [DOI] [PubMed] [Google Scholar]
- Del Bigio MR, Chudley AE, Sarnat HB, Campbell C, Goobie S, Chodirker BN, Selcen D. Infantile muscular dystrophy in Canadian aboriginals is an alphaB-crystallinopathy. Ann Neurol. 2011;69:866–871. doi: 10.1002/ana.22331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Ruiz A, Ibarra A, Pérez-Severiano F, Guizar-Sahagún G, Grijalva I, Ríos C. Constitutive and inducible nitric oxide synthase activities after spinal cord contusion in rats. Neurosci Lett. 2002;319:129–132. doi: 10.1016/s0304-3940(01)02540-x. [DOI] [PubMed] [Google Scholar]
- Ghasemlou N, Bouhy D, Yang J, López-Vales R, Haber M, Thuraisingam T, He G, Radzioch D, Ding A, David S. Beneficial effects of secretory leukocyte protease inhibitor after spinal cord injury. Brain. 2010;133:126–138. doi: 10.1093/brain/awp304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb LG, Dalakas MC. Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest. 2009;119:1806–1813. doi: 10.1172/JCI38027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann TL, Boelens WC, Wawrousek EF, Messing A. Suppression of GFAP toxicity by alphaB-crystallin in mouse models of Alexander disease. Hum Mol Genet. 2009;18:1190–1199. doi: 10.1093/hmg/ddp013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia TD, Craig EA. Four small Drosophila heat shock proteins are related to each other and to mammalian alpha-crystallin. Proc Natl Acad Sci U S A. 1982;79:2360–2364. doi: 10.1073/pnas.79.7.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin G, Hayashi T, Kawagoe J, Takizawa T, Nagata T, Nagano I, Syoji M, Abe K. Deficiency of PAR-2 gene increases acute focal ischemic brain injury. J Cereb Blood Flow Metab. 2005;25:302–313. doi: 10.1038/sj.jcbfm.9600021. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, Lai W, Rivest S, Hart RP, Satoskar AR, Popovich PG. Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. J Neurochem. 2007;102:37–50. doi: 10.1111/j.1471-4159.2007.04524.x. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon BK, Tetzlaff W, Grauer JN, Beiner J, Vaccaro AR. Pathophysiology and pharmacologic treatment of acute spinal cord injury. Spine J. 2004;4:451–464. doi: 10.1016/j.spinee.2003.07.007. [DOI] [PubMed] [Google Scholar]
- Kwon BK, Okon E, Hillyer J, Mann C, Baptiste D, Weaver LC, Fehlings MG, Tetzlaff W. A systematic review of non-invasive pharmacologic neuroprotective treatments for acute spinal cord injury. J Neurotrauma. 2011;28:1545–1588. doi: 10.1089/neu.2009.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SM, Rosen S, Weinstein P, van Rooijen N, Noble-Haeusslein LJ. Prevention of both neutrophil and monocyte recruitment promotes recovery after spinal cord injury. J Neurotrauma. 2011;28:1893–1907. doi: 10.1089/neu.2011.1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G, Lee KM, Kim JI, Markmann JF, Marinelli B, Panizzi P, Lee WW, Iwamoto Y, Milstein S, Epstein-Barash H, Cantley W, Wong J, Cortez-Retamozo V, Newton A, Love K, et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29:1005–1010. doi: 10.1038/nbt.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Reiser G. Phosphorylation of Ser45 and Ser59 of alphaB-crystallin and p38/extracellular regulated kinase activity determine alphaB-crystallin-mediated protection of rat brain astrocytes from C2-ceramide- and staurosporine-induced cell death. J Neurochem. 2011;118:354–364. doi: 10.1111/j.1471-4159.2011.07317.x. [DOI] [PubMed] [Google Scholar]
- Li R, Rohatgi T, Hanck T, Reiser G. Alpha A-crystallin and alpha B-crystallin, newly identified interaction proteins of protease-activated receptor-2, rescue astrocytes from C2-ceramide- and staurosporine-induced cell death. J Neurochem. 2009;110:1433–1444. doi: 10.1111/j.1471-4159.2009.06226.x. [DOI] [PubMed] [Google Scholar]
- Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- London A, Itskovich E, Benhar I, Kalchenko V, Mack M, Jung S, Schwartz M. Neuroprotection and progenitor cell renewal in the injured adult murine retina requires healing monocyte-derived macrophages. J Exp Med. 2011;208:23–39. doi: 10.1084/jem.20101202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morange M. What history tells us. II. The discovery of chaperone function. J Biosci. 2005;30:461–464. doi: 10.1007/BF02703718. [DOI] [PubMed] [Google Scholar]
- Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naruo S, Okajima K, Taoka Y, Uchiba M, Nakamura T, Okabe H, Takagi K. Prostaglandin E1 reduces compression trauma-induced spinal cord injury in rats mainly by inhibiting neutrophil activation. J Neurotrauma. 2003;20:221–228. doi: 10.1089/08977150360547125. [DOI] [PubMed] [Google Scholar]
- Ohashi K, Burkart V, Flohé S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. 2000;164:558–561. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- Ohri SS, Maddie MA, Zhao Y, Qiu MS, Hetman M, Whittemore SR. Attenuating the endoplasmic reticulum stress response improves functional recovery after spinal cord injury. Glia. 2011;59:1489–1502. doi: 10.1002/glia.21191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousman SS, Tomooka BH, van Noort JM, Wawrousek EF, O'Connor KC, Hafler DA, Sobel RA, Robinson WH, Steinman L. Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature. 2007;448:474–479. doi: 10.1038/nature05935. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002;109:525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penas C, Guzmán MS, Verdú E, Forés J, Navarro X, Casas C. Spinal cord injury induces endoplasmic reticulum stress with different cell-type dependent response. J Neurochem. 2007;102:1242–1255. doi: 10.1111/j.1471-4159.2007.04671.x. [DOI] [PubMed] [Google Scholar]
- Piao CS, Kim SW, Kim JB, Lee JK. Co-induction of alphaB-crystallin and MAPKAPK-2 in astrocytes in the penumbra after transient focal cerebral ischemia. Exp Brain Res. 2005;163:421–429. doi: 10.1007/s00221-004-2197-2. [DOI] [PubMed] [Google Scholar]
- Pineau I, Lacroix S. Proinflammatory cytokine synthesis in the injured mouse spinal cord: multiphasic expression pattern and identification of the cell types involved. J Comp Neurol. 2007;500:267–285. doi: 10.1002/cne.21149. [DOI] [PubMed] [Google Scholar]
- Pineau I, Sun L, Bastien D, Lacroix S. Astrocytes initiate inflammation in the injured mouse spinal cord by promoting the entry of neutrophils and inflammatory monocytes in an IL-1 receptor/MyD88-dependent fashion. Brain Behav Immun. 2010;24:540–553. doi: 10.1016/j.bbi.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Longbrake EE. Can the immune system be harnessed to repair the CNS? Nat Rev Neurosci. 2008;9:481–493. doi: 10.1038/nrn2398. [DOI] [PubMed] [Google Scholar]
- Quintana FJ, Cohen IR. Heat shock proteins as endogenous adjuvants in sterile and septic inflammation. J Immunol. 2005;175:2777–2782. doi: 10.4049/jimmunol.175.5.2777. [DOI] [PubMed] [Google Scholar]
- Safieh LA, Khan AO, Alkuraya FS. Identification of a novel CRYAB mutation associated with autosomal recessive juvenile cataract in a Saudi family. Mol Vis. 2009;15:980–984. [PMC free article] [PubMed] [Google Scholar]
- Schwab ME, Bartholdi D. Degeneration and regeneration of axons in the lesioned spinal cord. Physiol Rev. 1996;76:319–370. doi: 10.1152/physrev.1996.76.2.319. [DOI] [PubMed] [Google Scholar]
- Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, Rolls A, Mack M, Pluchino S, Martino G, Jung S, Schwartz M. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 2009;6:e1000113. doi: 10.1371/journal.pmed.1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling DP, Yong VW. Dynamics of the inflammatory response after murine spinal cord injury revealed by flow cytometry. J Neurosci Res. 2008;86:1944–1958. doi: 10.1002/jnr.21659. [DOI] [PubMed] [Google Scholar]
- Stirling DP, Liu S, Kubes P, Yong VW. Depletion of Ly6G/Gr-1 leukocytes after spinal cord injury in mice alters wound healing and worsens neurological outcome. J Neurosci. 2009;29:753–764. doi: 10.1523/JNEUROSCI.4918-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taggart CC, Cryan SA, Weldon S, Gibbons A, Greene CM, Kelly E, Low TB, O'Neill SJ, McElvaney NG. Secretory leucoprotease inhibitor binds to NF-kappaB binding sites in monocytes and inhibits p65 binding. J Exp Med. 2005;202:1659–1668. doi: 10.1084/jem.20050768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taoka Y, Okajima K, Uchiba M, Murakami K, Kushimoto S, Johno M, Naruo M, Okabe H, Takatsuki K. Role of neutrophils in spinal cord injury in the rat. Neuroscience. 1997;79:1177–1182. doi: 10.1016/s0306-4522(97)00011-0. [DOI] [PubMed] [Google Scholar]
- Thiyagarajan M, Fernández JA, Lane SM, Griffin JH, Zlokovic BV. Activated protein C promotes neovascularization and neurogenesis in postischemic brain via protease-activated receptor 1. J Neurosci. 2008;28:12788–12797. doi: 10.1523/JNEUROSCI.3485-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Noort JM, Bsibsi M, Gerritsen WH, van der Valk P, Bajramovic JJ, Steinman L, Amor S. Alphab-crystallin is a target for adaptive immune responses and a trigger of innate responses in preactive multiple sclerosis lesions. J Neuropathol Exp Neurol. 2010;69:694–703. doi: 10.1097/NEN.0b013e3181e4939c. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]