

Abstract
The number of functional transient receptor potential vanilloid 1 (TRPV1) channels at the surface, especially at the peripheral terminals of primary sensory neurons, regulates heat sensitivity, and increased surface localization of TRPV1s contributes to heat hyperalgesia. However, the mechanisms for regulating TRPV1 surface localization are essentially unknown. Here, we show that cyclin-dependent kinase 5 (Cdk5), a new player in thermal pain sensation, positively regulates TRPV1 surface localization. Active Cdk5 was found to promote TRPV1 anterograde transport in vivo, suggesting a regulatory role of Cdk5 in TRPV1 membrane trafficking. TRPV1-containing vesicles bind to the forkhead-associated (FHA) domain of the KIF13B (kinesin-3 family member 13B) and are thus delivered to the cell surface. Overexpression of Cdk5 or its activator p35 promoted and inhibition of Cdk5 activity prevented the KIF13B–TRPV1 association, indicating that Cdk5 promotes TRPV1 anterograde transport by mediating the motor–cargo association. Cdk5 phosphorylates KIF13B at Thr-506, a residue located in the FHA domain. T506A mutation reduced the motor–cargo interaction and the cell-permeable TAT–T506 peptide, targeting to the Thr-506, decreased TRPV1 surface localization, demonstrating the essential role of Thr-506 phosphorylation in TRPV1 transport. Moreover, complete Freund's adjuvant (CFA) injection-induced activation of Cdk5 increased the anterograde transport of TRPV1s, contributing to the development and possibly the maintenance of heat hyperalgesia, whereas intrathecal delivery of the TAT–T506 peptide alleviated CFA-induced heat hyperalgesia in rats. Thus, Cdk5 regulation of TRPV1 membrane trafficking is a fundamental mechanism controlling the heat sensitivity of nociceptors, and moderate inhibition of Thr-506 phosphorylation during inflammation might be helpful for the treatment of inflammatory thermal pain.
Introduction
Transient receptor potential vanilloid 1 (TRPV1), a calcium-permeable nonselective cation channel highly expressed in primary sensory neurons, is an important detector of noxious heat (>43°C) under normal conditions (Caterina et al., 1997, 2000) and is suggested as a central mediator of inflammatory heat hyperalgesia because multiple proinflammatory mediators, such as bradykinin, prostaglandin E2, NGF, and ATP, can potentiate its channel function (Szallasi et al., 2007).
As a transmembrane receptor, the number of functional TRPV1s at the plasma membrane must be a critical determinant of channel function, and potentiating channel function by increasing its levels at the cell surface is a potential mechanism underlying proinflammatory mediator-induced heat hyperalgesia (Ji et al., 2002; Van Buren et al., 2005; Zhang et al., 2005; Stein et al., 2006; Camprubí-Robles et al., 2009). In general, the number of receptors at the surface can be regulated by the membrane trafficking and insertion of receptors. To date, studies have shown that PKC activation-sensitized SNARE-dependent exocytosis and Src-dependent phosphorylation of TRPV1 are involved in inflammatory stimuli-induced rapid membrane insertion of TRPV1s from a ready-to-go vesicular pool (Morenilla-Palao et al., 2004; Zhang et al., 2005; Camprubí-Robles et al., 2009), but the mechanisms that regulate transport of TRPV1s from the Golgi apparatus to the plasma membrane of sensory neurons is essentially unclear. Kinesin superfamily motor proteins are responsible for the intracellular anterograde transport of most cargos, including transmembrane receptors (Hirokawa et al., 2009). However, the kinesin motor that anterogradely transports TRPV1-containing vesicles is also unclear.
Protein kinases play a pivotal role in modulating TRPV1 channel function (Huang et al., 2006). Recently, our group, along with Pareek's group, revealed that cyclin-dependent kinase 5 (Cdk5) in primary sensory neurons regulates heat sensitivity. Cdk5 activity is required for the basal response to noxious heat (Pareek et al., 2006, 2007), and activation of Cdk5 is involved in inflammatory heat hyperalgesia (Yang et al., 2007). In cultured DRG neurons, inhibition of Cdk5 results in a significant decrease in capsaicin-evoked calcium influx (Pareek et al., 2007), suggesting that Cdk5 might modulate TRPV1.
Cdk5 is involved in many cellular processes of the nervous system, including vesicle transport. Sustained Cdk5 activity is required for normal kinesin-driven motility in neurons (Ratner et al., 1998) and vesicle transport from the Golgi to neurites (Paglini et al., 2001). In the present study, we investigated whether and how Cdk5 regulates TRPV1 membrane trafficking. Through bioinformatics analysis, we find one potential phosphorylation site for Cdk5 at Thr-506 located in the forkhead-associated (FHA) domain of the kinesin-3 family motor protein GAKIN (guanylate kinase-associated kinesin)/KIF13B (kinesin-3 family member 13B). We demonstrate that KIF13B can indeed transport TRPV1s and Cdk5 promotes the trafficking process by mediating the motor–cargo association, which is at least partially dependent on Cdk5-mediated phosphorylation of KIF13B at Thr-506. We further investigated whether this regulatory mechanism contributes to inflammatory heat hyperalgesia. In vivo studies indicate that inflammation induces activation of Cdk5 which, in turn, increases TRPV1 trafficking to the periphery, whereas inhibition of Thr-506 phosphorylation during inflammation alleviates heat hyperalgesia.
Materials and Methods
Plasmids, antibodies, and chemicals.
The plasmids pcDNA3.1(+)–Cdk5, pcDNA3.1(+)–D144N–Cdk5, and pEGFP–C2–p35 were constructed as described previously (Wang et al., 2006; Yang et al., 2007). cDNA fragments encoding the corresponding domains of KIF13B were generated by PCR amplification from a rat brain cDNA pool and subcloned into pEGFP–C2 (Clontech) or pET–28a(+) (Novagen). Mutations (T506A, T506D) within the FHA domain were created with the QuickChange site-directed mutagenesis kit (Stratagene). All plasmids were verified by DNA sequencing.
Anti-KIF13B antiserum was raised by immunizing rabbits with a His6 fusion protein encoding amino acids 1451–1767 of rat KIF13B. The polyclonal anti-TRPV1 (P-19), anti-phospho-Thr (H-2), anti-GFP (FL), anti-Cdk5 (C-8), and anti-p35 (C-19) antibodies were purchased from Santa Cruz Biotechnology. The monoclonal anti-β-actin (TA-09), anti-N-cadherin (ZM-0094), and anti-His (TA-02) antibodies were purchased from Zhongshan Goldenbridge Biotechnology.
Roscovitine (R7772), complete Freund's adjuvant (CFA) (F5881), and incomplete Freund's adjuvant (IFA) (F5506) were from Sigma-Aldrich. The TAT fusion peptides were synthesized by GL Biochem.
Cell culture and transfection.
CHO cells stably expressing rat VR1 in a pTet off regulatory system (TRPV1–CHO cells) were cultured in Ham's F-12 medium containing 10% FBS, 0.25 mg/ml geneticin, and 1 μg/ml tetracycline as described previously (Szallasi et al., 1999; Wang et al., 2004). Expression of the rVR1 is induced after removal of tetracycline. F11 cells were routinely cultured in Ham's F-12 medium supplemented with 20% FBS and 1× HAT (sodium hypoxanthine, aminopterin, and thymidine; Sigma-Aldrich) as described previously (Francel et al., 1987a,b). Transfections of cells were performed with Lipofectamine 2000 (Invitrogen). For TRPV1–CHO cells, tetracycline was removed from the culture medium after transfection. Both cells were harvested 48 h after transfection.
DRG culture.
DRGs aseptically removed from neonatal rats (0–3 d) were digested with 0.25% trypsin (Sigma-Aldrich) for 35 min at 37°C, followed by trituration with a flame-polished Pasteur pipette. Dissociated cells were collected and resuspended in plating medium (DMEM containing 10% FBS), and 2 × 105 DRG neurons were plated onto a 35 mm dish coated with poly-d-lysine (Sigma-Aldrich). After 15 min, medium was replaced with Neurobasal medium supplemented with 2% B27, 2 mm GlutaMAX-I (Invitrogen), and 100 ng/ml mouse NGF (Promega). Five micromolar AraC (Sigma-Aldrich) was added to the culture 18–24 h after plating and maintained until the end of experiments. The cultures were infected with lentiviruses (LVs) on day 3 or treated with the TAT fusion peptides (10 μm) or roscovitine (30 μm) on day 7.
RNA interference.
RNA interference (RNAi) LVs coexpressing EGFP and shRNAs against rat KIF13B were purchased from Shanghai GeneChem. The target mRNA sequence was as follows: 5′-GGCCAUUGAAGUGUAUGGACAUAAA-3′ (Yoshimura et al., 2010). LVs expressing shRNAs targeting a nonspecific sequence were used as controls. To downregulate KIF13B expression in cultured DRG neurons, 1 × 107 lentiviral particles were added to one 35 mm dish (multiplicity of infection of ∼50), and the neurons were cultured for 4–5 d.
Western blot.
Tissues were homogenized in ice-cold lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1.5 mm MgCl2, 10% glycerol, 1% Triton X-100, 5 mm EGTA, 0.5 μg/ml leupeptin, 1 mm PMSF, 1 mm Na3VO4, 10 mm NaF, and proteinase inhibitor mixture) and rotated at 4°C for 1 h before the supernatant was extracted. LV-infected DRG neurons were lysed in lysis buffer, and the supernatant was extracted by centrifugation at 12,000 × g at 4°C for 5 min. Equal amounts of protein extracts were denatured and subjected to SDS-PAGE. After separation, proteins were transferred to nitrocellulose membranes (Bio-Rad). The membranes were blocked with 5% nonfat milk in TBST (25 mm Tris, pH 7.4, 137 mm NaCl, 2.7 mm KCl, and 0.05% Tween 20) for 1 h at room temperature and incubated with an anti-TRPV1 antibody (1:200 dilution) or anti-KIF13B antiserum (1:500 dilution) overnight at 4°C. After washing three times with TBST, the membranes were incubated with secondary antibody (1:2000 dilution) overnight at 4°C, then washed again, and finally developed with ECL solutions (Santa Cruz Biotechnology).
Immunoprecipitation.
For coimmunoprecipitation (Co-IP), protein extracts from transfected cells were prepared as described above. Extract containing 400–500 μg of protein was incubated at 4°C for 3 h with 2 μg of anti-TRPV1, anti-phospho-Thr, anti-Cdk5, or anti-p35 antibody before incubation with protein A-Sepharose CL-4B resin (GE Healthcare) overnight. Immunoprecipitates were washed five times with TBS containing 0.1% Triton X-100 and then immunoblotted with an anti-GFP antibody.
For immunoprecipitation, protein extracts from rat sciatic nerves and different parts of rat DRG were incubated with an anti-TRPV1 antibody, and the precipitated proteins were immunoblotted with an anti-TRPV1 antibody.
Pull-down assay.
His6 fusion proteins were expressed in Escherichia coli BL21 (DE3) cells and purified using nickel–nitrilotriacetic acid agarose beads (QIAGEN) according to the instructions of the manufacturer. For binding assays, eluted His6 fusion protein was added to the protein extract from TRPV1–CHO cells. The mixture was then incubated with an anti-TRPV1 antibody and protein A-Sepharose CL-4B resin overnight at 4°C. Precipitates were washed and immunoblotted with an anti-His antibody.
Cell surface biotinylation assay.
Cells were biotinylated with 500 μg/ml EZ-link sulfo-NHS-SS-biotin (Pierce) in PBS for 1 h at 4°C. After quenching with PBS containing 100 mm glycine, the cells were lysed in RIPA buffer (25 mm Tris, pH 7.4, 137 mm NaCl, 2.7 mm KCl, 1% Triton X-100, 0.1% SDS, and proteinase inhibitor mixture). Supernatants from cell lysates were incubated with Ultralink Plus immobilized streptavidin beads (Pierce) overnight at 4°C. After being washed with RIPA buffer five times, bound proteins were eluted by boiling for 5 min and analyzed by Western blot with an anti-TRPV1 antibody.
In vitro kinase assay.
The in vitro Cdk5 kinase assay was conducted as described previously (Yang et al., 2007). Briefly, rat DRG extracts were immunoprecipitated with an anti-Cdk5 antibody, and the immunoprecipitates were washed four times with TBS containing 0.1% Triton X-100 and twice with assay buffer (20 mm Tris, pH 7.5, 20 mm MgCl2, 1 mm EDTA, 1 mm EGTA, and 0.1 mm dithiothreitol) and resuspended to a final volume of 25 μl in assay buffer. To initiate the reaction, 10 μl of phosphorylation mix (assay buffer containing 5 μCi of [γ-32P]ATP and one of the following His6 fusion proteins: His6–motor FHA, His6–motor, His6–FHA, or His6–FHAT506A) was added. The mixture was incubated at 30°C for 30 min, and the reaction was terminated by the addition of SDS-PAGE sample buffer. After boiling for 5 min, the samples were subjected to SDS-PAGE. The gels were stained with Coomassie blue, dried, and exposed to x-ray film for autoradiography.
Animals.
Male Sprague Dawley rats (200–250 g) were used. All experiments were performed according to the guidelines of the Animal Care and Use Committee of Peking University. For the intrathecal delivery of drugs, a PE-10 polyethylene catheter was implanted into the intrathecal space reaching the lumbar enlargement of the spinal cord (Yang et al., 2007). Surgeries were performed under 10% chloral hydrate anesthesia (0.3 g/kg, i.p.), and rats were allowed 4–5 d for recovery. Five microliters of roscovitine (100 μg) were administered intrathecally 30 min before the injection of CFA (100 μl) into the plantar surface of the left hindpaw. DMSO (5 μl) was injected intrathecally as the vehicle control.
Behavioral assessment.
Heat sensitivity was assessed by measuring the paw-withdrawal latency in response to a radiant heat stimulus applied to the core of the plantar surface of the hindpaw (Hargreaves et al., 1988; Zhu et al., 2008). Mechanical sensitivity was tested by von Frey filaments (Stoelting). The paw-withdrawal threshold was calculated using the up–down method (Chaplan et al., 1994). The basal heat sensitivity and the basal mechanical sensitivity of rats that had recovered from catheter implantation surgery were tested before drug administration. Thirty microliters of TAT–T506 peptide (1 μg/μl) or TAT–T506A peptide (1 μg/μl) was intrathecally administered 30 min before the injection of 100 μl of 25% CFA (CFA was diluted with IFA) into the left hindpaw. The heat hyperalgesia and the mechanical allodynia of the rats were assessed 1 h, 2 h, 6 h, and 1 d after CFA injection. For the experiments assessing the effect of TAT–T506 peptide on the basal heat sensitivity, the same dose of TAT peptides were injected intrathecally, and the basal heat sensitivity was tested 1.5 h, 2.5 h, 6.5 h, and 1 d after intrathecal injection.
Isolation of DRG neurons and calcium imaging.
Isolation of DRG neurons and fura-2 AM-based Ca2+ imaging experiments were performed as described previously (Carlton et al., 2011). Rats were treated with intrathecal delivery of 30 μl of TAT–T506 peptide (1 μg/μl) or TAT–T506A peptide (1 μg/μl), followed by injection of 100 μl of 25% CFA into the left hindpaw. Two hours after CFA injection, the left L4 and L5 DRGs were rapidly picked out and treated with 1.5 mg/ml collagenase D (Sigma-Aldrich) for 45 min and 0.125% trypsin (Sigma-Aldrich) for 8 min at 37°C. Then the ganglia were washed three times with extracellular solution (ES) (Xu and Huang, 2002), followed by gentle trituration with a flame-polished Pasteur pipette. Dissociated cells suspended in 100 μl of ES were plated on a poly-d-lysine-coated confocal dish, and 2 ml of ES was added to each dish 15 min later. Neurons were maintained at room temperature for 1 h. Then, DRG neurons were loaded with 5 μm fura-2 AM (Biotium) in ice-cold ES for 40 min at room temperature. After loading, the neurons were washed with ES and left in ES at room temperature in the dark for 1 h. For calcium imaging, an inverted fluorescent microscope equipped with a 340 and 380 nm excitation filter changer (Olympus) and a computer with Metafluor software were used. Fluorescent images and the F340/F380 ratio were acquired every 5 s. TRPV1 activation was evoked by the addition of 1 μm capsaicin, and the cells were considered to be responsive if the ratio increased by 20% after capsaicin application. Non-responsive neurons that also failed to respond to 25 mm K+ were discarded from the analysis. A minimum of 50 cells derived from at least three separated isolations were analyzed.
Statistical analyses.
Data are expressed as mean ± SEM. Statistical analyses were performed using the Prism 4.0 software. Differences between groups were compared using either the Student's t test or one-way ANOVA, followed by Newman–Keuls post hoc tests or two-way repeated-measures ANOVA, followed by Bonferroni's post hoc tests. Statistical significance was set at p < 0.05. For Western blot, the immunoreactive bands were scanned and analyzed quantitatively by densitometry with the TotalLab software.
Results
Cdk5 activity regulates TRPV1 membrane localization
The function of TRPV1 is modulated by its gating properties and the total amount of receptor at the cell surface. It has been reported that Cdk5 can modulate capsaicin-induced calcium influx (Pareek et al., 2007). Here, we addressed whether Cdk5 can regulate the level of TRPV1 surface expression. Because Cdk5 displays constitutive kinase activity in the basal state, we first inhibited endogenous Cdk5 activity with roscovitine, a specific inhibitor of Cdk5, to investigate the issue. Cells of the following three types were treated with 30 μm roscovitine for 5 h: CHO cells stably expressing TRPV1 (TRPV1–CHO), TRPV1 transiently transfected DRG-derived F11 cells (mouse N18TG2 neuroblastoma × rat DRG, hybridoma), and primary cultured DRG neurons. Surface biotinylation assays revealed that treatment with roscovitine substantially decreased the surface TRPV1 levels in all three cell models (Fig. 1A–C). The ratio of surface to total TRPV1 was reduced to 58.7 ± 5.9% (p = 0.006, n = 4) of the control in roscovitine-treated TRPV1–CHO cells (Fig. 1A) and to 40.7 ± 1.9% (p = 0.001, n = 3) of the control in roscovitine-treated F11 cells (Fig. 1B) and to 51 ± 4.6% (p = 0.0087, n = 3) of the control in roscovitine-treated DRG neurons (Fig. 1C). In contrast, the number of surface NR1 subunits of the NMDA receptor in F11 cells was not reduced after the addition of roscovitine (Fig. 1D), indicating that roscovitine specifically reduces TRPV1 membrane localization. The total amount of TRPV1 was not significantly altered (Fig. 1A–C). These data show that the basal activity of Cdk5 is required for the surface localization of TRPV1s in basal conditions.
Figure 1.

Cdk5 plays a regulatory role in TRPV1 membrane localization. A–C, Roscovitine reduces the surface TRPV1 levels in TRPV1–CHO cells (A), F11 cells transfected with GFP–TRPV1 (B), and primary cultured DRG neurons (C). Cells were treated with 30 μm roscovitine for 5 h. The surface biotinylation assay was used to detect the amount of TRPV1 at the cell surface. The quantitative analyses show the ratio of surface to total TRPV1 normalized to the DMSO control and the total TRPV1 protein levels normalized to the DMSO control. Data were analyzed by two-tailed paired t test. **p < 0.01, ***p < 0.001 compared with DMSO control group, n = 3–4. Error bars indicate SEM. Ros, Roscovitine. D, The levels of surface NR1 subunits of the NMDA receptor in F11 cells are not reduced by roscovitine treatment (30 μm, 5 h). E, Overexpression of Cdk5 increases and overexpression of D144N–Cdk5 decreases the levels of surface TRPV1 in TRPV1–CHO cells. Cells were transfected with pcDNA3.1(+)–vector, pcDNA3.1(+)–Cdk5, or pcDNA3.1(+)–D144N-Cdk5. For the Cdk5+Ros group, cells expressing Cdk5 were treated with roscovitine (30 μm) for 5 h. Quantitative analysis show the ratio of surface to total TRPV1 normalized to the vector control (top) and the total TRPV1 protein levels normalized to the vector control (bottom). Data were analyzed by one-way ANOVA, followed by Newman–Keuls post hoc tests. **p < 0.01 compared with the vector group; ###p < 0.001 compared with the Cdk5 group, n = 5–6. Error bars indicate SEM. F, Overexpression of p35 increases the levels of surface TRPV1 in TRPV1–CHO cells. Cells were transfected with pEGFP–C2–p35 or pEGFP–C2–vector. For the GFP-p35+Ros group, cells expressing GFP–p35 were treated with 30 μm roscovitine for 5 h. Quantitative analysis of the ratio of surface to total TRPV1 normalized to the GFP control (top) and the total TRPV1 protein levels normalized to the GFP control (bottom) are shown. Data were analyzed by one-way ANOVA, followed by Newman–Keuls post hoc tests. **p < 0.01, ***p < 0.001 compared with the GFP group; ###p < 0.001 compared with the GFP–p35 group, n = 4–5. Error bars indicate SEM.
In an additional study, we transfected TRPV1–CHO cells with Cdk5, its activator p35, or a dominant-negative form of Cdk5 [D144N–Cdk5 (Nikolic et al., 1996)] and measured the surface TRPV1 levels. Overexpression of Cdk5 (Fig. 1E) or p35 (Fig. 1F), which elevates intracellular Cdk5 activity, significantly increased the ratio of surface to total TRPV1 by ∼35.9% (p < 0.01, vector vs Cdk5) or ∼50.4% (p < 0.001, GFP vs GFP–p35): this effect was not only blocked but reversed by exposure to roscovitine (30 μm, 5 h) (Fig. 1E; ∼28.2% reduction, vector vs Cdk5+Ros, p < 0.01; Fig. 1F; ∼34.4% reduction, GFP vs GFP-p35+Ros, p < 0.01). Furthermore, overexpression of D144N–Cdk5, which competitively inhibits endogenous Cdk5 activity, resulted in an effect similar to roscovitine treatment (Fig. 1E; the ratio of surface to total TRPV1 was reduced by ∼33.4% in cells expressing D144N–Cdk5, p < 0.01). TRPV1 total protein levels were comparable among the groups. Together, these results suggest that Cdk5 positively regulates membrane localization of TRPV1s, and the number of surface TRPV1s is directly proportional to Cdk5 activity.
Cdk5 activation increases TRPV1 anterograde transport after inflammation
Previous studies have shown that TRPV1s are normally transported from the DRG to peripheral terminals along the sciatic nerve (Guo et al., 1999), that TRPV1 production is increased after inflammation, and that inflammation-induced TRPV1s are also anterogradely transported to the periphery (Ji et al., 2002). Consistently, our data showed that the TRPV1 protein levels in the DRG somas, the peripheral processes, and the sciatic nerves are significantly increased 1 d after CFA-induced inflammation (Fig. 2A; 24.6% increase in the somas, p = 0.044; 53.9% increase in the peripheral processes, p = 0.0215; 40.5% increase in the sciatic nerves, p = 0.0481; n = 4), demonstrating that inflammation induces an increase in TRPV1 protein levels and TRPV1 anterograde transport. Together, these results indicate that TRPV1s are anterogradely transported from the cell body to the peripheral terminals both in the basal state and during inflammation. Because Cdk5 activity affects the levels of surface TRPV1 and its kinase activity is upregulated after inflammation (Yang et al., 2007) because of the increase in the p35 protein levels (Fig. 2A), we next investigated whether activated Cdk5 regulates TRPV1 anterograde transport after inflammation. Thirty minutes before an injection of CFA into a hindpaw, roscovitine (100 μg) was injected intrathecally to prevent the elevation of Cdk5 activity (Yang et al., 2007). Compared with the DMSO control, this treatment significantly decreased the amount of TRPV1 in the sciatic nerves (32.3% reduction, p = 0.0084, n = 7) 1 d after inflammation, the amount of TRPV1 in the peripheral processes of DRG is also reduced to some extent, although no significant difference is shown (12.2% reduction, p = 0.0869, n = 7) but a slight increase in TRPV1 protein levels in the cell body was concomitantly observed (∼8.7% increase, p = 0.464, n = 7) (Fig. 2B). These results suggest that Cdk5 regulates TRPV1 anterograde transport in vivo, and activation of Cdk5 increases TRPV1 peripheral transport after inflammation.
Figure 2.

Cdk5 activation induced by inflammation increases TRPV1 anterograde transport to the periphery. A, Western blot analysis showing alterations in the TRPV1 protein levels in the indicated tissues 1 d after CFA-induced inflammation. The quantitative analysis in the right shows the comparative levels of TRPV1 over the naive control after normalizing to β-actin. *p < 0.05, n = 4, two-tailed paired t test within each group. Error bars indicate SEM. Note that the p35 protein levels are primarily increased after inflammation, indicating the activation of Cdk5. B, Immunoprecipitation of TRPV1s from lysates of indicated tissues showing the effect of intrathecal delivery of roscovitine (Ros) on the amount of TRPV1 in these tissues. Either roscovitine (100 μg) or DMSO was delivered intrathecally to rats 30 min before the injection of CFA into a hindpaw. The quantitative analysis in the right shows the TRPV1 protein levels relative to the DMSO control. **p < 0.01, n = 7, two-tailed paired t test within each group. Error bars indicate SEM.
KIF13B mediates TRPV1 membrane trafficking
Cdk5 is a small proline-directed serine/threonine kinase and is implicated in the regulation of cellular processes by phosphorylating certain substrates (Dhavan and Tsai, 2001). In addition to an absolute requirement for proline in the +1 position, Cdk5 shows a marked preference for a basic residue in the +3 position and phosphorylates the consensus sequence (S/T)PX(K/H/R), where X is any amino acid (Beaudette et al., 1993; Songyang et al., 1996).
It is known that N-kinesins, which generally drive microtubule plus-end-directed cargo moving, are a critical determinant of anterograde transport toward the cell periphery or axon terminals in neurons (Hirokawa et al., 2009). Through sequence analysis, we found a “TPQK” sequence, which is consistent with the consensus phosphorylation sequence of Cdk5 (T/SPXK/H/R), in the FHA domain of KIF13B (Fig. 3A,B). This sequence is conserved across humans, rats, and mice (Fig. 3B), and the FHA domain of KIF13B has been reported to be a cargo binding domain (Horiguchi et al., 2006). Thus, we speculated that KIF13B might mediate the Cdk5-regulated anterograde transport of TRPV1s.
Figure 3.

KIF13B transports TRPV1 to the cell surface. A, Schematic diagram of the domain structure of KIF13B/GAKIN and the constructs used in the present study. B, Comparison of the rat, mouse, and human KIF13B sequences containing the putative phosphorylation site of Cdk5. Threonine-506 is conserved across the species shown here. * indicates identical residues. C, Co-IP of TRPV1 and GFP–motor FHA. TRPV1–CHO cells (left) were transfected with pEGFP–C2–motor FHA or pEGFP–C2–vector, and F11 cells (right) were cotransfected with GFP–TRPV1 and pEGFP–C2–motor FHA or pEGFP–C2–vector. TRPV1s were immunoprecipitated with an anti-TRPV1 antibody, and the immunoblots were probed with an anti-GFP antibody. Lysates of cells expressing GFP were used as controls. Inputs represent the direct immunoblotting of the lysates. D, In vitro pull-down assay. TRPV1s were immunoprecipitated from cell lysates containing His6–motor FHA, which was expressed in bacteria. The immunoblots were detected with an anti-His antibody. E, Western blot analysis demonstrating an efficient decrease in KIF13B protein levels in primary cultured DRG neurons after infection with LVs expressing KIF13B shRNAs. **p < 0.01, n = 3, two-tailed paired t test. Error bar indicates SEM. F, Effect of KIF13B knockdown on the surface targeting of TRPV1s in primary cultured DRG neurons. The amount of membrane localized TRPV1 was detected with a surface biotinylation assay. Detection of N-cadherin was used as a control for the equal loading of biotinylated protein. The quantitative analysis shows the ratio of surface to total TRPV1 normalized to the control. *p < 0.05, n = 3, two-tailed paired t test. Error bar indicates SEM. G, Effect of overexpression of GFP–motor FHA or GFP–FHA on the surface targeting of TRPV1s in TRPV1–CHO cells. The top shows the quantitative analysis of the ratio of surface to total TRPV1 normalized to the GFP control. The bottom shows the total TRPV1 protein levels normalized to the GFP control. **p < 0.01, ***p < 0.001 compared with the GFP control, n = 3, one-way ANOVA, followed by Newman–Keuls post hoc tests. Error bars indicate SEM. Experiments in C and D were repeated at least three times.
To determine whether KIF13B drives the anterograde transport of TRPV1s, we first tested whether KIF13B can associate with TRPV1. Co-IP studies using TRPV1–CHO cells expressing GFP–motor FHA and in F11 cells coexpressing GFP–motor FHA and GFP–TRPV1 revealed binding between TRPV1 and the KIF13B–motor FHA domain (Fig. 3C). TRPV1 immunoprecipitated from lysates of TRPV1–CHO cells also pulled down exogenously expressed His6–motor FHA (Fig. 3D). More significantly, the FHA domain seems to be the actual binding domain for TRPV1 (seen also Fig. 5D); this is also suggested indirectly by the lack of interaction between TRPV1 and the KIF13B–motor domain (Fig. 5E, left).
Figure 5.

The binding between TRPV1 and KIF13B is regulated by Cdk5 activity and requires Thr-506 phosphorylation. A, Roscovitine inhibits the Co-IP of TRPV1 and GFP–motor FHA. TRPV1–CHO cells (left) transfected with pEGFP–C2–motor FHA and F11 cells (right) cotransfected with GFP–TRPV1 and pEGFP–C2–motor FHA were treated with roscovitine (30 μm) or DMSO for 5 h. Cell lysates were immunoprecipitated with a TRPV1 antibody, and the immunoblots were detected with a GFP antibody. Inputs represent the direct immunoblotting of the lysates. B, Overexpression of Cdk5 promotes and overexpression of D144N–Cdk5 inhibits the Co-IP of TRPV1 and GFP–motor FHA. TRPV1–CHO cells were cotransfected with pEGFP–C2–motor FHA and pcDNA3.1(+)–vector, pcDNA3.1(+)–Cdk5, or pcDNA3.1(+)–D144N–Cdk5, and immunoprecipitation and immunoblot were performed as described in A. C, Overexpression of p35 promotes the Co-IP. TRPV1–CHO cells were cotransfected with pEGFP–C2–motor FHA and pEGFP–C2–p35 or pEGFP–C2–vector, and immunoprecipitation and immunoblot were performed as described in A. D, In vitro pull-down assay. TRPV1s were immunoprecipitated from cell lysates containing His6–FHA or His6–FHAT506A expressed in bacteria, and the immunoblots were detected with a His antibody. Input shows the equal addition of His6 fusion proteins. E, Co-IP assays showing that Thr-506 phosphorylation contributes to the association between TRPV1 and GFP–motor FHA. TRPV1–CHO cells were transfected with the indicated plasmids (left), and F11 cells were cotransfected with GFP–TRPV1 and the indicated plasmids (right). Anti-TRPV1 immunoprecipitates were analyzed by immunoblot with a GFP antibody; inputs represent the direct immunoblotting of the lysates. Note that TRPV1 binds to GFP–motor FHA but not GFP–motor. All above experiments were repeated at least three times.
Next, we used RNAi to knock down KIF13B expression and measured the effect on TRPV1 transport. KIF13B shRNAs efficiently knocked down endogenous KIF13B expression in cultured DRG neurons (Fig. 3E; 44.5 ± 4.8% of the control, p = 0.0074, n = 3). Biotinylation assays revealed that knockdown of KIF13B in DRG neurons resulted in a significant decrease in the number of surface TRPV1s (Fig. 3F; 74.4 ± 5.7% of the control, p = 0.0457, n = 3); this indicates that KIF13B is required for TRPV1 membrane trafficking. To further confirm that TRPV1 membrane trafficking is KIF13B dependent, we transfected GFP–motor FHA or GFP–FHA into TRPV1–CHO cells. Biotinylation assays showed that both treatments induced notable decreases in the content of surface TRPV1s compared with the transfection of GFP alone (Fig. 3G; the ratio of surface to total TRPV1 was reduced by ∼25.9% in cells expressing GFP–motor FHA, p < 0.01, and ∼38.1% in cells expressing GFP–FHA, p < 0.001, n = 3), indicating that both truncations disrupt the motor–cargo interaction. Together, the above results reveal that KIF13B mediates TRPV1 membrane trafficking, and the FHA domain of KIF13B functions as a TRPV1 binding domain.
Cdk5 binds to and phosphorylates KIF13B
As mentioned above, KIF13B possesses a conserved phosphorylation sequence for Cdk5 in the FHA domain (Fig. 3B); however, it remains to be clarified whether Cdk5 can phosphorylate KIF13B. To address this question, we first examined whether Cdk5 binds to KIF13B. GFP–motor FHA was coexpressed with Cdk5 or p35 in TRPV1–CHO cells, and Co-IP assays revealed that both Cdk5 and p35 were able to precipitate GFP–motor FHA (Fig. 4A). Then, an in vitro kinase assay was performed, and the results showed that Cdk5 phosphorylated the KIF13B–motor FHA and KIF13B–FHA domains in vitro but did not phosphorylate the KIF13B–motor domain (Fig. 4B, left). Based on the consensus phosphorylation sequence of Cdk5 (T/SPXK/H/R), we hypothesized that the Thr-506 residue located in the TPQK sequence is the phosphorylation site. To test this hypothesis, Thr-506 was mutated into the nonphosphorylatable alanine, and the mutant construct was subjected to a Cdk5 kinase assay. As expected, the mutation almost completely abolished Cdk5-mediated phosphorylation (Fig. 4B, right). These data indicate that Cdk5 and p35 interact with KIF13B, and Cdk5 directly phosphorylates KIF13B at Thr-506 in the FHA domain.
Figure 4.

Phosphorylation of KIF13B by Cdk5. A, Co-IP of GFP–motor FHA and Cdk5 (left) or GFP–p35 (right). Lysates of TRPV1–CHO cells coexpressing pEGFP–C2–motor FHA and pcDNA3.1(+)–Cdk5 (left) or pEGFP–C2–p35 (right) were immunoprecipitated with an anti-Cdk5 or an anti-p35 antibody, respectively. Lysates of cells coexpressing pEGFP–C2–vector and pcDNA3.1(+)–Cdk5 (left) or pEGFP–C2–p35 (right) were used as controls. Immunoblots were probed with an anti-GFP antibody. Inputs represent the direct immunoblotting of the lysates. B, In vitro kinase assay. Cdk5 was immunoprecipitated from rat DRG lysates and incubated with the indicated His6 fusion proteins in the presence of radioactive 32P. Left side of each panel, Autoradiogram of phosphorylated proteins (arrowheads indicate the nonspecific bands). Right side of each panel, The amount of recombinant His6 fusion protein (indicated by an asterisk) shown by Coomassie blue staining. C, Threonine phosphorylation of GFP–motor FHA in cells treated with roscovitine (30 μm) or DMSO for 5 h. Lysates of TRPV1–CHO cells expressing pEGFP–C2–motor FHA or F11 cells expressing pEGFP–C2–motor FHA were immunoprecipitated with a phosphothreonine-specific antibody, and the immunoblots were probed with a GFP antibody. Inputs represent the direct immunoblotting of the lysates. D, Threonine phosphorylation of GFP–motor FHA in cells expressing empty vector, Cdk5, or D144N–Cdk5. TRPV1–CHO cells were cotransfected with pEGFP–C2–motor FHA and pcDNA3.1(+)–vector, pcDNA3.1(+)–Cdk5, or pcDNA3.1(+)–D144N–Cdk5. Treatment of cells and detection of immunoblots were the same as in C. E, Threonine phosphorylation of GFP–motor FHA is reduced by T506A mutation. TRPV1–CHO cells and F11 cells were transfected with pEGFP–C2–motor FHA or pEGFP–C2–motor FHAT506A. Treatment of cells and detection of immunoblots were the same as in C. All above experiments were repeated at least three times.
To further investigate Cdk5-mediated Thr-506 phosphorylation in vivo, an anti-phospho-Thr antibody was used to immunoprecipitate proteins containing threonine phosphorylation from cells expressing GFP–motor FHA, and the immunoprecipitates were analyzed by immunoblot using an anti-GFP antibody. Phosphorylation at threonine residues in the KIF13B–motor FHA domain was decreased by roscovitine treatment (30 μm, 5 h) (Fig. 4C) or the overexpression of D144N–Cdk5 (Fig. 4D) and was increased by the overexpression of Cdk5 (Fig. 4D). These data indicate that Cdk5 is involved in threonine phosphorylation of the KIF13B–motor FHA domain. To test whether Thr-506 phosphorylation is a necessary component of threonine phosphorylation of the KIF13B–motor FHA, wild-type, and T506A mutant KIF13B–motor FHA domains were expressed in TRPV1–CHO and F11 cells. Threonine phosphorylation of the KIF13B–motor FHA in cells expressing the wild-type domain was much higher than that in cells expressing the T506A mutant (Fig. 4E). These in vivo results suggest that Cdk5-mediated Thr-506 phosphorylation is the main contributor to the threonine phosphorylation of the KIF13B–motor FHA domain.
Cdk5 mediates the association between TRPV1 and KIF13B, at least in part, by phosphorylating Thr-506
Because Cdk5 regulates TRPV1 anterograde transport (Figs. 1, 2) and phosphorylates KIF13B at Thr-506, which is located in the TRPV1 binding domain (Fig. 4), we hypothesized that Cdk5 may regulate the motor–cargo interaction. To test this hypothesis, TRPV1–CHO cells transfected with GFP–motor FHA and F11 cells cotransfected with GFP–TRPV1 and GFP–motor FHA were treated with roscovitine (30 μm, 5 h). Co-IP experiments revealed that roscovitine significantly decreased the number of TRPV1–KIF13B–motor FHA complexes (Fig. 5A). Conversely, overexpression of Cdk5 (Fig. 5B) or its activator p35 (Fig. 5C) in TRPV1–CHO cells remarkably increased the number of complexes. Overexpression of D144N–Cdk5, however, decreased the number of complexes (Fig. 5B). These results indicate that Cdk5 kinase activity is both necessary and sufficient for KIF13B–TRPV1 interaction and suggest that Cdk5 regulates TRPV1 membrane trafficking by mediating the KIF13B–TRPV1 association.
In the following studies, we examined the effect of Thr-506 phosphorylation of KIF13B on its association with TRPV1. TRPV1 immunoprecipitated from lysates of TRPV1–CHO cells pulled down exogenously expressed His6–FHA but not the T506A mutant (Fig. 5D). Co-IP studies in both TRPV1–CHO cells (Fig. 5E, left) and F11 cells (Fig. 5E, right) showed that binding between TRPV1 and the KIF13B–motor FHA was significantly inhibited by the T506A mutation. The phosphorylation-mimicking mutation T506D retained the ability to interact with TRPV1 but did not increase the interaction of KIF13B–motor FHA with TRPV1 over that of wild-type KIF13B–motor FHA (Fig. 5E, left). These data indicate that phosphorylation at Thr-506 is required for KIF13B–TRPV1 association. Thus, Cdk5 mediates KIF13B–TRPV1 association, at least partially, by phosphorylating Thr-506.
Phosphorylation at Thr-506 is necessary for TRPV1 membrane trafficking
To further investigate the role of Thr-506 phosphorylation in KIF13B-dependent TRPV1 membrane trafficking, we constructed a peptide with 15 aa of the KIF13B–FHA domain, which included the TPQK sequence, and the peptide was rendered cell permeable by fusion with the HIV TAT protein sequence (RKKRRQRRR) (termed TAT–T506). As a control peptide, Thr-506 was replaced with alanine (termed TAT–T506A) (Fig. 6A). The TAT–T506 peptide targeting Thr-506 of KIF13B was used as a dominant-negative approach to study the consequences of disrupting endogenous Thr-506 phosphorylation. Primary cultured DRG neurons were treated with 10 μm TAT fusion peptide for 3 h. Subsequently, biotinylation assays showed that the TAT–T506 peptide significantly decreased the amount of surface TRPV1 (Fig. 6B; 78.7 ± 3.4% of the control, p = 0.0081, n = 4). Thus, Thr-506 phosphorylation is indeed a critical determinant in KIF13B-mediated trafficking of TRPV1s to the cell surface.
Figure 6.

Construction of TAT–T506 fusion peptide and its negative effect on TRPV1 membrane trafficking. A, Schematic diagram of the TAT–T506 and control TAT–T506A peptides. Top, The letters on the dark background indicate the 15 aa sequence of the FHA domain of rat KIF13B, which contains Thr-506 (white letter). Nine amino acid residues (letters on the light background) of the HIV TAT protein were fused to this sequence. In the control peptide (bottom), Thr-506 was substituted with alanine. B, Effects of TAT peptides on the levels of surface TRPV1 in primary cultured DRG neurons. Neurons were treated with 10 μm TAT–T506 peptide or TAT–T506A peptide for 3 h and subjected to a surface biotinylation assay. Detection of N-cadherin was used as a control for the equal loading of biotinylated protein. The quantitative analysis in the right shows the comparative levels of surface TRPV1 over the TAT–T506A control after normalizing to surface N-cadherin levels; **p < 0.01, n = 4, two-tailed paired t test. Error bar indicates SEM.
Disruption of Thr-506 phosphorylation attenuates functional TRPV1 membrane targeting and the development of heat hyperalgesia after inflammation
Because Thr-506 phosphorylation is necessary for Cdk5-mediated regulation of TRPV1 (Figs. 5, 6), it might also be crucial for Cdk5-activation-induced modulation of TRPV1 channel function and heat hyperalgesia that develops after inflammation.
We first used calcium imaging to test the functional consequences of inhibiting Thr-506 phosphorylation after inflammation. The TAT–T506 peptide (30 μg) or the control peptide (30 μg) was injected intrathecally 30 min before the injection of CFA into a hindpaw. The uptake of TAT peptides into DRGs has been observed previously, and the peak concentration appears ∼3 h after delivery (Xie et al., 2009). Two hours after inflammation, ipsilateral L4 and L5 DRG neurons were acutely dissociated and subjected to fura-2 calcium imaging. Capsaicin (1 μm) evoked a rapid robust increase in intracellular Ca2+ concentration in neurons pretreated with the control peptide, whereas neurons pretreated with the TAT–T506 peptide showed delayed calcium signals and the intracellular Ca2+ concentration of these neurons increased apparently slower than that of the control neurons (Fig. 7A), although the maximum calcium signals are similar in the two groups. Measurements of the area under the curve within 2 min after the beginning of the stimulation revealed weaker capacities of the TAT–T506 peptide-pretreated neurons for the accumulation of intracellular Ca2+ (Fig. 7B; 43.8 ± 1.3 vs 52.3 ± 2.0 for TAT–T506, n = 104 vs TAT–T506A, n = 55, from three independent experiments, p = 0.0003), suggesting an obvious inhibition of TRPV1 channel function by the TAT–T506 peptide.
Figure 7.

Disruption of Thr-506 phosphorylation attenuates the function of TRPV1 and heat hyperalgesia induced by inflammation. A, B, Calcium imaging of acutely isolated DRG neurons undergoing channel activation by capsaicin (1 μm). Rats were pretreated with intrathecal delivery of 30 μg of TAT–T506 peptide or TAT–T506A peptide 30 min before the injection of CFA into a hindpaw. Neurons were isolated 2 h after CFA injection. A, Representative traces showing typical change patterns of intracellular Ca2+ in DRG neurons from each group. Application of capsaicin is shown with an arrow. B, Quantification of the area under the curve for 2 min after the beginning of capsaicin stimulation. For calculation, neurons (TAT–T506 peptide group, n = 104; TAT–T506A peptide group, n = 55) from three independent experiments were analyzed. The ratio of F340/F380 after stimulation was normalized to the baseline ratio. ***p < 0.001, two-tailed unpaired t test. Error bars indicate SEM. C, Response latencies for rats to a radiant heat stimulus applied to the hindpaw after intrathecal injection of 30 μg of TAT–T506 peptide or TAT–T506A peptide, n = 8–11. Error bars indicate SEM. D, Change in escape response latency to radiant heat applied to the hindpaw after interplanetary injection of CFA. Rats were pretreated with the TAT–T506 peptide (30 μg) or the TAT–T506A peptide (30 μg) 30 min before CFA injection. Data were analyzed by two-way repeated-measures ANOVA, followed by Bonferroni's post hoc tests. **p < 0.01, ***p < 0.001 compared with the corresponding time point of TAT–T506A peptide group. ###p < 0.001 indicates the extremely significant difference between two curves. n = 19–20. Error bars indicate SEM. E, Change in von Frey hair threshold after interplanetary CFA injection. Rats were pretreated with the TAT–T506 peptide (30 μg) or the TAT–T506A peptide (30 μg) 30 min before CFA injection. n = 10–13. Error bars indicate SEM.
We further determined the effects of disruption of Thr-506 phosphorylation on heat sensitivity after inflammation. We assessed heat-evoked behavioral responses after CFA injections preceded by intrathecal TAT–T506 peptide (30 μg) or TAT–T506A peptide (30 μg) delivery. Rats pretreated with the TAT–T506A peptide exhibited a significant decrease in radiant heat-evoked paw-withdrawal latency after inflammation, whereas inflammation-induced thermal hyperalgesia was relatively attenuated in rats pretreated with the TAT–T506 peptide at 1 and 2 h after the induction of inflammation (Fig. 7D; 7.6 ± 0.6 vs 4.9 ± 0.5 s for TAT–T506 vs TAT–T506A at 1 h, p < 0.01; 8.0 ± 0.7 vs 5.0 ± 0.4 s for TAT–T506 vs TAT–T506A at 2 h, p < 0.001; n = 19–20). The inhibition effect of the TAT–T506 peptide became weaker 6 h after inflammation and disappeared 1 d after inflammation (Fig. 7D). The basal heat-evoked paw-withdrawal latency of the rats treated with the TAT–T506 peptide is not significantly different from that of the control peptide-treated rats (Fig. 7C), and there is no significant difference in the withdrawal threshold to mechanical stimuli (von Frey hair) after inflammation between the two groups (Fig. 7E). These results suggest that disruption of Thr-506 phosphorylation with the TAT–T506 peptide during inflammation can alleviate heat hyperalgesia but not mechanical allodynia that develops after inflammation, and this treatment does not affect the basal heat sensitivity.
Discussion
We have investigated the regulatory mechanisms of fast anterograde transport of TRPV1s in DRG neurons. We demonstrate that Cdk5 positively regulates TRPV1 membrane trafficking by mediating KIF13B–TRPV1 association. Furthermore, we find that Cdk5-dependent phosphorylation of KIF13B at Thr-506 is necessary for Cdk5-mediated motor–cargo association and contributes to efficient TRPV1 transport (Fig. 8A).
Figure 8.
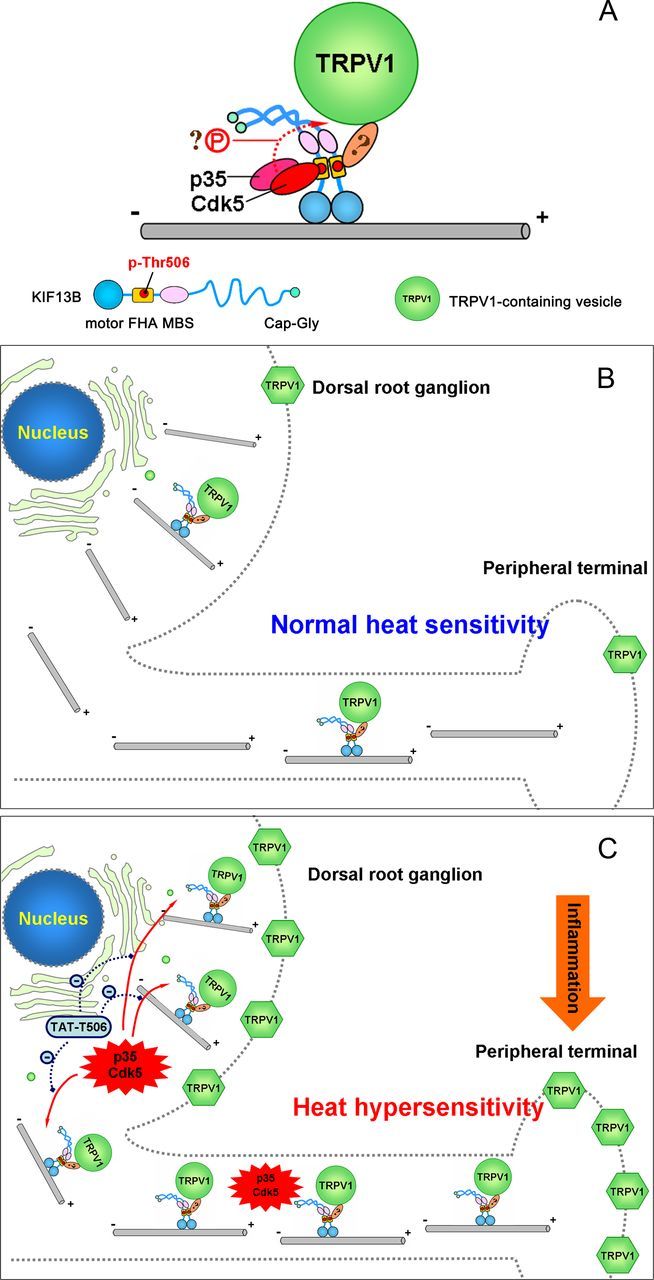
Schematic model representing Cdk5-mediated motor–cargo association (A) and Cdk5 activity-regulated TRPV1 membrane trafficking (B, C). A, Cdk5-mediated phosphorylation of KIF13B at Thr-506 is necessary for the KIF13B–TRPV1 association. Other Cdk5-dependent phospho-regulated mechanisms may also be required. Whether KIF13B binds to a TRPV1-carrying vesicle via adaptor proteins is unknown. B, Under normal conditions, basal Cdk5 activity in nociceptive neurons is low, and only a few TRPV1-carrying vesicles are loaded and transported by phosphorylated KIF13B from the Golgi apparatus to the neuronal surface, maintaining a low surface level for normal sensitivity to noxious heat. C, After peripheral inflammation, Cdk5 activity is greatly elevated, and more TRPV1-carrying vesicles are loaded onto the motor and delivered to the cell surface. As a result, the number of TRPV1s at the neuronal surface (the plasma membrane of the soma and the peripheral axon) increases, contributing to the development and possibly the maintenance of heat hyperalgesia. If, at this time, the activated Cdk5-induced phosphorylation of KIF13B is disrupted by TAT–T506 peptide, the increase in TRPV1 membrane trafficking is prevented and the heat hyperalgesia is relieved to some extent.
Previous studies showed that Cdk5 activity regulates thermal pain sensation (Pareek et al., 2006, 2007). Mice overexpressing p35, which exhibit elevated Cdk5 activity, show increased sensitivity to noxious heat (hyperalgesia). In contrast, p35 knock-out mice or nociceptor-specific Cdk5 conditional knock-out mice, which exhibit reduced Cdk5 activity, show impaired thermal nociception (hypoalgesia). Consistent with these results, we reveal that Cdk5 activity regulates the density of functional TRPV1s at the neuronal surface (Fig. 1), and this in turn affects the heat sensitivity of nociceptors. This finding provides insight into Cdk5 activity-dependent regulation of pain signaling and suggests that the basal activity of Cdk5 is important for the normal function of TRPV1.
In searching for the motor protein responsible for Cdk5-regulated TRPV1 membrane trafficking, we speculated that this motor should be modulated by Cdk5. Fortunately, we found a consensus phosphorylation sequence of Cdk5 in the FHA domain (a cargo binding domain) of KIF13B motor protein. The involvement of KIF13B in TRPV1 trafficking was confirmed by Co-IP with an anti-TRPV1 antibody, RNAi, and a dominant-negative approach in which the TRPV1 binding domain was overexpressed (Fig. 3C,F,G). However, we cannot rule out the possibility that other kinesins also contribute to Cdk5-regulated TRPV1 anterograde transport.
A feature that distinguishes kinesin-3 family from other kinesin families is that all members of this group contain an FHA domain. The FHA domain is present in a wide variety of proteins from both prokaryotes and eukaryotes and acts as a protein interaction domain (Hammet et al., 2003; Mahajan et al., 2008). Here, we find that the FHA domain of KIF13B serves as a binding domain of TRPV1-containing vesicles (Fig. 5D,E). We did not, however, investigate whether it is the only binding domain for TRPV1 or whether other domains, including the MAGUK binding stalk (MBS) domain, the C-terminal tail, and the stalk region between them, may also interact with TRPV1. The MBS domain is a binding domain for human Discs large (Dlg) tumor suppressor and might be involved in KIF13B/Dlg-mediated transport of some cargos (Yamada et al., 2007; Bolis et al., 2009). A recent study showed that multiple domains of KIF13B are involved in its association with CARD11, a critical scaffold protein in T-cell receptor signaling (Lamason et al., 2010). Thus, we cannot exclude the possibility that a TRPV1-containing vesicle may interact with other domains of KIF13B at the same time. Despite this, the interaction between TRPV1-containing vesicle and the FHA domain is definitely essential for TRPV1 transport because disruption of their interaction via the overexpression of exogenous KIF13B–motor FHA or KIF13B–FHA domain impaired TRPV1 membrane trafficking (Fig. 3G).
Kinesins usually bind to cargo vesicles via adaptor proteins or scaffold proteins that determine the specificity of cargos transported by each motor (Hirokawa et al., 2009). In another study, KIF13B was found to transport phosphatidylinositol-3,4,5-triphosphate (PIP3)-containing vesicles to neurite endings via the adaptor protein centaurin α1/PIP3 binding protein (Horiguchi et al., 2006; Tong et al., 2010), and the FHA domain is just the domain that directly interacts with centaurin α1, which, in turn, specifically binds to PIP3. In the case of TRPV1, the motor associates with the cargo in a phosphorylation-dependent manner; however, it is not known whether the FHA domain interacts with TRPV1 directly or indirectly because all binding assays were performed in the presence of other cellular proteins. Both patterns may be possible. TRPV1 has large N- and C-terminal cytosolic domains, both of which are outside of the vesicle and are used to interact with intracellular proteins. The N-terminal region contains six ankyrin repeats that often mediate protein–protein interactions (Lishko et al., 2007), whereas the C terminus can interact with scaffold protein AKAP79/150 (A-kinase anchoring protein 79/150) (Zhang et al., 2008). Therefore, additional studies are required to clarify the molecular details of the association between KIF13B and TRPV1-containing vesicles and how phosphorylation of KIF13B promotes the formation of the complex.
Kinesin phosphorylation appears to regulate the motor–cargo interaction. GSK3 (glycogen synthase kinase 3) phosphorylates kinesin light chains and leads to the removal of kinesin from vesicles (Morfini et al., 2002, 2004). CaMKII-dependent phosphorylation of KIF17 at Ser-1029 disrupts the KIF17–Mint1 association and results in the release of transported cargo from the phosphorylated molecular motor (Guillaud et al., 2008). JNK pathway kinase-mediated phosphorylation inhibits the interaction between kinesin light chains and the JIP1 (JNK interacting protein 1) adaptor (Horiuchi et al., 2007). Altogether, phosphorylation of kinesin acts as a molecular switch for motor–cargo dissociation. However, our results show that phosphorylation of KIF13B by Cdk5 contributes to motor–cargo association. This is the first demonstration of a positive role of kinesin phosphorylation in controlling motor–cargo interaction and provides insight into the dynamic control of intracellular transport in vivo. We find that phosphorylation at Thr-506 is necessary for KIF13B–TRPV1 association because mutation of Thr-506 to a nonphosphorylatable alanine specifically inhibited the interaction (Fig. 5D,E). However, and contrary to our expectations, mutation to aspartic acid, which mimics a phosphoamino acid, did not result in increased affinity to TRPV1 compared with wild-type KIF13B–motor FHA (Fig. 5E). One interpretation is that this mutation does not mimic phosphorylation, and perhaps a glutamic acid substitution would be better. Another interpretation is that Thr-506 phosphorylation is not sufficient for KIF13B–TRPV1 association, which suggests that other Cdk5-dependent regulatory mechanisms may coexist. Thus, we conclude that phosphorylation at Thr-506 is necessary, but not necessarily sufficient, for Cdk5-mediated KIF13B–TRPV1 association. More specifically, Cdk5 mediates the motor–cargo association, at least partially, by phosphorylating Thr-506.
Because TRPV1 plays a central role in mediating inflammatory heat hyperalgesia, a key issue to be addressed is whether proinflammatory mediators can sensitize TRPV1 by activating Cdk5-regulated TRPV1 peripheral transport. Our previous study demonstrated that peripheral inflammation (produced by CFA injection) induces the activation of Cdk5 in DRGs (Yang et al., 2007). Here, we find that preventing the elevation of Cdk5 activity by intrathecal delivery of roscovitine efficiently decreased TRPV1 anterograde transport (Fig. 2B). Thus, we conclude that Cdk5 activation can increase peripheral transport of TRPV1s. Consistent with this conclusion, Cdk5 activation contributes to inflammatory heat hyperalgesia (Yang et al., 2007). Moreover, inhibition of increased TRPV1 trafficking through preventing the Cdk5-dependent phosphorylation at Thr-506 alleviated inflammatory heat hyperalgesia (Fig. 7). Therefore, we propose a model in which Cdk5 activity regulates the magnitude of TRPV1 anterograde transport (Fig. 8B,C). When activated by peripheral inflammation, Cdk5 increases the number of KIF13B–TRPV1 complexes, in turn increasing the anterograde transport of TRPV1s, resulting in increased density of TRPV1s at the surface of nociceptive neurons and increased heat sensitivity (Fig. 8C). Although our study only demonstrates the role of activation of Cdk5-regulated TRPV1 membrane trafficking in the development phase of heat hyperalgesia (within 24 h after CFA injection) (Fig. 7D), activation of Cdk5-regulated TRPV1 trafficking may also contribute to the maintenance phase of heat hyperalgesia because activation of Cdk5 by CFA injection is sustained for at least 1 week (Yang et al., 2007).
Our results further suggest that Thr-506 of KIF13B might be a potential target for analgesic treatments (Fig. 8C). The TAT–T506 peptide, which could competitively disrupt the phosphorylation of endogenous KIF13B at Thr-506, had an analgesic effect on inflammatory pain and did not affect the basal heat sensitivity (Fig. 7C,D), indicating that the TAT–T506 peptide could be a promising approach for pain management.
In conclusion, this study establishes a Cdk5 activity-regulated TRPV1 membrane trafficking model in which peripheral inflammation-induced activation of Cdk5 and the subsequent upregulation of TRPV1 membrane trafficking play pivotal roles in the development and possibly in the maintenance of heat hyperalgesia. Moreover, phosphorylation of KIF13B at Thr-506 is quite important for Cdk5 regulation of TRPV1 membrane trafficking, and moderate inhibition of Thr-506 phosphorylation during inflammation will be a promising strategy for the treatment of inflammatory thermal pain.
Footnotes
This work was supported by National Natural Science Foundation of China Grants 30830044, 30925015, and 81161120497, Beijing Natural Science Foundation Grant 7092061, and Specialized Research Fund for Doctoral Programs of Higher Education Grants 200800011028 and 20060001121.
The authors declare no competing financial interests.
References
- Beaudette KN, Lew J, Wang JH. Substrate specificity characterization of a cdc2-like protein kinase purified from bovine brain. J Biol Chem. 1993;268:20825–20830. [PubMed] [Google Scholar]
- Bolis A, Coviello S, Visigalli I, Taveggia C, Bachi A, Chishti AH, Hanada T, Quattrini A, Previtali SC, Biffi A, Bolino A. Dlg1, Sec8, and Mtmr2 regulate membrane homeostasis in Schwann cell myelination. J Neurosci. 2009;29:8858–8870. doi: 10.1523/JNEUROSCI.1423-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camprubí-Robles M, Planells-Cases R, Ferrer-Montiel A. Differential contribution of SNARE-dependent exocytosis to inflammatory potentiation of TRPV1 in nociceptors. FASEB J. 2009;23:3722–3733. doi: 10.1096/fj.09-134346. [DOI] [PubMed] [Google Scholar]
- Carlton SM, Zhou S, Govea R, Du J. Group II/III metabotropic glutamate receptors exert endogenous activity-dependent modulation of TRPV1 receptors on peripheral nociceptors. J Neurosci. 2011;31:12727–12737. doi: 10.1523/JNEUROSCI.6558-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–759. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- Francel PC, Harris K, Smith M, Fishman MC, Dawson G, Miller RJ. Neurochemical characteristics of a novel dorsal root ganglion × neuroblastoma hybrid cell line, F-11. J Neurochem. 1987a;48:1624–1631. doi: 10.1111/j.1471-4159.1987.tb05711.x. [DOI] [PubMed] [Google Scholar]
- Francel PC, Miller RJ, Dawson G. Modulation of bradykinin-induced inositol trisphosphate release in a novel neuroblastoma × dorsal root ganglion sensory neuron cell line (F-11) J Neurochem. 1987b;48:1632–1639. doi: 10.1111/j.1471-4159.1987.tb05712.x. [DOI] [PubMed] [Google Scholar]
- Guillaud L, Wong R, Hirokawa N. Disruption of KIF17-Mint1 interaction by CaMKII-dependent phosphorylation: a molecular model of kinesin-cargo release. Nat Cell Biol. 2008;10:19–29. doi: 10.1038/ncb1665. [DOI] [PubMed] [Google Scholar]
- Guo A, Vulchanova L, Wang J, Li X, Elde R. Immunocytochemical localization of the vanilloid receptor 1 (VR1): relationship to neuropeptides, the P2X3 purinoceptor and IB4 binding sites. Eur J Neurosci. 1999;11:946–958. doi: 10.1046/j.1460-9568.1999.00503.x. [DOI] [PubMed] [Google Scholar]
- Hammet A, Pike BL, McNees CJ, Conlan LA, Tenis N, Heierhorst J. FHA domains as phospho-threonine binding modules in cell signaling. IUBMB Life. 2003;55:23–27. doi: 10.1080/1521654031000070636. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. 2009;10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- Horiguchi K, Hanada T, Fukui Y, Chishti AH. Transport of PIP3 by GAKIN, a kinesin-3 family protein, regulates neuronal cell polarity. J Cell Biol. 2006;174:425–436. doi: 10.1083/jcb.200604031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi D, Collins CA, Bhat P, Barkus RV, Diantonio A, Saxton WM. Control of a kinesin-cargo linkage mechanism by JNK pathway kinases. Curr Biol. 2007;17:1313–1317. doi: 10.1016/j.cub.2007.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Zhang X, McNaughton PA. Inflammatory pain: the cellular basis of heat hyperalgesia. Curr Neuropharmacol. 2006;4:197–206. doi: 10.2174/157015906778019554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- Lamason RL, Kupfer A, Pomerantz JL. The dynamic distribution of CARD11 at the immunological synapse is regulated by the inhibitory kinesin GAKIN. Mol Cell. 2010;40:798–809. doi: 10.1016/j.molcel.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lishko PV, Procko E, Jin X, Phelps CB, Gaudet R. The ankyrin repeats of TRPV1 bind multiple ligands and modulate channel sensitivity. Neuron. 2007;54:905–918. doi: 10.1016/j.neuron.2007.05.027. [DOI] [PubMed] [Google Scholar]
- Mahajan A, Yuan C, Lee H, Chen ES, Wu PY, Tsai MD. Structure and function of the phosphothreonine-specific FHA domain. Sci Signal. 2008;1:re12. doi: 10.1126/scisignal.151re12. [DOI] [PubMed] [Google Scholar]
- Morenilla-Palao C, Planells-Cases R, García-Sanz N, Ferrer-Montiel A. Regulated exocytosis contributes to protein kinase C potentiation of vanilloid receptor activity. J Biol Chem. 2004;279:25665–25672. doi: 10.1074/jbc.M311515200. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002;21:281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Brown H, Pant HC, Pigino G, DeBoer S, Beffert U, Brady ST. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J. 2004;23:2235–2245. doi: 10.1038/sj.emboj.7600237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolic M, Dudek H, Kwon YT, Ramos YF, Tsai LH. The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 1996;10:816–825. doi: 10.1101/gad.10.7.816. [DOI] [PubMed] [Google Scholar]
- Paglini G, Peris L, Diez-Guerra J, Quiroga S, Cáceres A. The Cdk5–p35 kinase associates with the Golgi apparatus and regulates membrane traffic. EMBO Rep. 2001;2:1139–1144. doi: 10.1093/embo-reports/kve250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareek TK, Keller J, Kesavapany S, Pant HC, Iadarola MJ, Brady RO, Kulkarni AB. Cyclin-dependent kinase 5 activity regulates pain signaling. Proc Natl Acad Sci U S A. 2006;103:791–796. doi: 10.1073/pnas.0510405103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareek TK, Keller J, Kesavapany S, Agarwal N, Kuner R, Pant HC, Iadarola MJ, Brady RO, Kulkarni AB. Cyclin-dependent kinase 5 modulates nociceptive signaling through direct phosphorylation of transient receptor potential vanilloid 1. Proc Natl Acad Sci U S A. 2007;104:660–665. doi: 10.1073/pnas.0609916104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner N, Bloom GS, Brady ST. A role for cyclin-dependent kinase(s) in the modulation of fast anterograde axonal transport: effects defined by olomoucine and the APC tumor suppressor protein. J Neurosci. 1998;18:7717–7726. doi: 10.1523/JNEUROSCI.18-19-07717.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songyang Z, Lu KP, Kwon YT, Tsai LH, Filhol O, Cochet C, Brickey DA, Soderling TR, Bartleson C, Graves DJ, DeMaggio AJ, Hoekstra MF, Blenis J, Hunter T, Cantley LC. A structural basis for substrate specificities of protein Ser/Thr kinases: primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin-dependent kinase II, CDK5, and Erk1. Mol Cell Biol. 1996;16:6486–6493. doi: 10.1128/mcb.16.11.6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein AT, Ufret-Vincenty CA, Hua L, Santana LF, Gordon SE. Phosphoinositide 3-kinase binds to TRPV1 and mediates NGF-stimulated TRPV1 trafficking to the plasma membrane. J Gen Physiol. 2006;128:509–522. doi: 10.1085/jgp.200609576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szallasi A, Blumberg PM, Annicelli LL, Krause JE, Cortright DN. The cloned rat vanilloid receptor VR1 mediates both R-type binding and C-type calcium response in dorsal root ganglion neurons. Mol Pharmacol. 1999;56:581–587. doi: 10.1124/mol.56.3.581. [DOI] [PubMed] [Google Scholar]
- Szallasi A, Cortright DN, Blum CA, Eid SR. The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-of-concept. Nat Rev Drug Discov. 2007;6:357–372. doi: 10.1038/nrd2280. [DOI] [PubMed] [Google Scholar]
- Tong Y, Tempel W, Wang H, Yamada K, Shen L, Senisterra GA, MacKenzie F, Chishti AH, Park HW. Phosphorylation-independent dual-site binding of the FHA domain of KIF13 mediates phosphoinositide transport via centaurin α1. Proc Natl Acad Sci U S A. 2010;107:20346–20351. doi: 10.1073/pnas.1009008107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Buren JJ, Bhat S, Rotello R, Pauza ME, Premkumar LS. Sensitization and translocation of TRPV1 by insulin and IGF-I. Mol Pain. 2005;1:17. doi: 10.1186/1744-8069-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kedei N, Wang M, Wang QJ, Huppler AR, Toth A, Tran R, Blumberg PM. Interaction between protein kinase Cmu and the vanilloid receptor type 1. J Biol Chem. 2004;279:53674–53682. doi: 10.1074/jbc.M410331200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Xie WY, He Y, Wang M, Yang YR, Zhang Y, Yin DM, Jordan-Sciutto KL, Han JS, Wang Y. Role of CDK5 in neuroprotection from serum deprivation by mu-opioid receptor agonist. Exp Neurol. 2006;202:313–323. doi: 10.1016/j.expneurol.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Xie WY, He Y, Yang YR, Li YF, Kang K, Xing BM, Wang Y. Disruption of Cdk5-associated phosphorylation of residue threonine-161 of the delta-opioid receptor: impaired receptor function and attenuated morphine antinociceptive tolerance. J Neurosci. 2009;29:3551–3564. doi: 10.1523/JNEUROSCI.0415-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu GY, Huang LY. Peripheral inflammation sensitizes P2X receptor-mediated responses in rat dorsal root ganglion neurons. J Neurosci. 2002;22:93–102. doi: 10.1523/JNEUROSCI.22-01-00093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KH, Hanada T, Chishti AH. The effector domain of human Dlg tumor suppressor acts as a switch that relieves autoinhibition of kinesin-3 motor GAKIN/KIF13B. Biochemistry. 2007;46:10039–10045. doi: 10.1021/bi701169w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YR, He Y, Zhang Y, Li Y, Li Y, Han Y, Zhu H, Wang Y. Activation of cyclin-dependent kinase 5 (Cdk5) in primary sensory and dorsal horn neurons by peripheral inflammation contributes to heat hyperalgesia. Pain. 2007;127:109–120. doi: 10.1016/j.pain.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Yoshimura Y, Terabayashi T, Miki H. Par1b/MARK2 phosphorylates kinesin-like motor protein GAKIN/KIF13B to regulate axon formation. Mol Cell Biol. 2010;30:2206–2219. doi: 10.1128/MCB.01181-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Huang J, McNaughton PA. NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J. 2005;24:4211–4223. doi: 10.1038/sj.emboj.7600893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Li L, McNaughton PA. Proinflammatory mediators modulate the heat-activated ion channel TRPV1 via the scaffolding protein AKAP79/150. Neuron. 2008;59:450–461. doi: 10.1016/j.neuron.2008.05.015. [DOI] [PubMed] [Google Scholar]
- Zhu H, Yang Y, Zhang H, Han Y, Li Y, Zhang Y, Yin D, He Q, Zhao Z, Blumberg PM, Han J, Wang Y. Interaction between protein kinase D1 and transient receptor potential V1 in primary sensory neurons is involved in heat hypersensitivity. Pain. 2008;137:574–588. doi: 10.1016/j.pain.2007.10.025. [DOI] [PubMed] [Google Scholar]