

Abstract
Loss of the RNA-binding fragile X protein [fragile X mental retardation protein (FMRP)] results in a spectrum of cognitive deficits, the fragile X syndrome (FXS), while aging individuals with decreased protein levels present with a subset of these symptoms and tremor. The broad range of behavioral deficits likely reflects the ubiquitous distribution and multiple functions of the protein. FMRP loss is expected to affect multiple neuronal proteins and intracellular signaling pathways, whose identity and interactions are essential in understanding and ameliorating FXS symptoms. We used heterozygous mutants and targeted RNA interference-mediated abrogation in Drosophila to uncover molecular pathways affected by FMRP reduction. We present evidence that FMRP loss results in excess metabotropic glutamate receptor (mGluR) activity, attributable at least in part to elevation of the protein in affected neurons. Using high-resolution behavioral, genetic, and biochemical analyses, we present evidence that excess mGluR upon FMRP attenuation is linked to the cAMP decrement reported in patients and models, and underlies olfactory associative learning and memory deficits. Furthermore, our data indicate positive transcriptional regulation of the fly fmr1 gene by cAMP, via protein kinase A, likely through the transcription factor CREB. Because the human Fmr1 gene also contains CREB binding sites, the interaction of mGluR excess and cAMP signaling defects we present suggests novel combinatorial pharmaceutical approaches to symptom amelioration upon FMRP attenuation.
Introduction
Fragile X syndrome (FXS) patients present with developmental and behavioral deficits including mild to severe mental retardation, autism, anxiety, and aberrant attention, learning, and memory (Hagerman, 2002; Tsiouris and Brown, 2004; Jacquemont et al., 2007). FXS results from loss, or CGG triplet expansion-mediated silencing of the highly conserved FMR1 gene, encoding the RNA-binding protein fragile X mental retardation protein (FMRP) (O'Donnell and Warren, 2002; Bagni and Greenough, 2005). Alleles with expanded but unmethylated repeats, or expanded-methylated repeats but below the silencing threshold known as “premutations,” reduce but do not eliminate the protein (Pirozzi et al., 2011). Aging premutation carriers can develop a condition, fragile X-associated tremor/ataxia syndrome (FXTAS), which presents memory deficits along with emotional morbidity (Hagerman and Hagerman, 2004; Hunter et al., 2008a).
FMRP is ubiquitous in the CNS, where it associates with ∼4% of mammalian neuronal mRNAs (O'Donnell and Warren, 2002). It regulates RNA trafficking and metabolism and represses unregulated synaptic translation (Antar and Bassell, 2003; Ronesi and Huber, 2008). Hence, FMRP loss could result in elevation of proteins whose translation it negatively regulates. Loss of a ubiquitous translational regulator likely impacts multiple neuronal circuits, mediating deficits in distinct behavioral modules in patients and animal models (Mineur et al., 2002; Zarnescu et al., 2005; Mientjes et al., 2006; Gatto and Broadie, 2009), in agreement with the breadth of behavioral deficits. To understand these broad effects, it is critical to define the signaling systems altered upon FMRP attenuation and their impact.
There have been two main lines of thought on the major signaling mechanisms altered upon FMRP attenuation to account for FXS phenotypes. Bear et al. (2004) proposed that elevated signaling via the metabotropic glutamate receptor (mGluR), alters synaptic properties and dynamics in multiple neuronal circuits, and this underlies the FXS neurobehavioral deficits. Support for this hypothesis has emerged from various models (McBride et al., 2005; D'Hulst and Kooy, 2009; Gatto and Broadie, 2009; Choi et al., 2011), but little direct experimental evidence on the consequences of increased mGluR activity in different neuronal circuits is currently available. Conversely, altered cAMP metabolism noted in FXS patient-derived cultured cells and animal models has been proposed as a prime contributor to the phenotypes of the syndrome (Berry-Kravis and Huttenlocher, 1992; Kelley et al., 2008). This is supported by the importance of cAMP signaling for neuroplasticity, anxiety, and autism (Kelley et al., 2008). It has also been reported in animal FXS models (Kelley et al., 2007), but experimental tests of this hypothesis remain scant.
Drosophila contains a single Fmr1 ortholog (dfmr1), whose disruption precipitates deficits in neuronal development and function, reminiscent of those in patients (Zarnescu et al., 2005; Gatto and Broadie, 2009). We used the Drosophila model to investigate the relative contribution and possible functional relationships of the cAMP and mGluR signaling with regard to the reported learning and memory deficits (McBride et al., 2005; Bolduc et al., 2008). We provide evidence on a regulatory loop important for Drosophila olfactory learning and long-term memory involving cAMP and mGluR interactions and the effects of cAMP signaling on FMRP levels.
Materials and Methods
Drosophila culture and strains.
Drosophila were cultured at 25°C in 50–70% humidity in a 12 h light/dark (LD) cycle, as described previously (Acevedo et al., 2007). Mutant strains used in this study are described by Dockendorf and colleagues (Wan et al., 2000; Dockendorff et al., 2002). These flies and transgenic strains carrying the full genomic fragment (RES), the frameshift mutant (FS), and UAS-Fmr1 on the third chromosome were obtained from S. McBride and T. Jongens (University of Pennsylvania, Philadelphia, PA) (McBride et al., 2005). The UAS-dfmrRNAi (UAS-dfmr1-R) and the UAS-mGluRRNAi (UAS-mGluR-R) transgenic flies were obtained from Vienna Drosophila Resource Centre. The dnc1 and dncML mutants were obtained from the Bloomington Stock Center. The dc0B3 mutants were a kind gift from D. Kalderon (Columbia University, New York, NY) (Lane and Kalderon, 1993). The genetic background of all was normalized to that of the resident w1118 control flies by backcrossing for six generations.
Genetics.
UAS-dfmr1 and UAS-mGluR-R were recombined onto the third chromosome bearing the dfmr13 mutation in the same genetic background. For behavioral experiments, nonbalanced heterozygotes of a cross between the dfmr13/TM3Sb mutant males with w1118 females were selected for testing. To abrogate dFMRP levels, c772-Gal4, MB247-Gal4, NP1131-Gal4, c507-Gal4, Elav-Gal4;TubGal80ts, c772-Gal4;TubGal80ts virgin females were crossed en masse to UAS-dfmr1-R, UAS-mGluR-R/+; dfmr13/+, UAS-dfmr1/+; dfmr13/+ males. In each case, progeny of the Gal4 driver lines crossed with w1118 females were used as controls. Likewise, dc0B3/CyO males were crossed with w1118 to obtain dc0B3/+ used in all relevant experiments. All flies were reared at 18°C and resultant nonbalanced 4- to 6-d-old flies were used for testing. Flies were placed at 29°C for 48 h for maximal transgene induction and allowed 30 min recovery at 25°C before training. To generate dnc1/+; dfmr13/+ double heterozygotes, dnc1/Y males were mated to dfmr13/TM3Sb females.
Viability assays were conducted by placing 20 adult males of each genotype in food vials, changing the vials every 2 d and counting the remaining live flies. Each determination was performed in duplicate.
Behavioral analyses.
All experiments were performed in a balanced design, where all genotypes involved in an experiment were tested per day. The experimenter was blind to the genotype in experiments testing learning of mutant heterozygotes. Behavioral experiments were replicated at least once with flies from different crosses and a different time period (biological replicates). Olfactory learning and memory in the negatively reinforced paradigm coupling aversive odors as conditioned stimuli (CS+ and CS−) with the electric shock unconditioned stimulus (US) were performed essentially as described previously (Moressis et al., 2009). Hence, each shock stimulus was delivered within 5 s of continuous CS+ presentation, so six pairings were delivered within 30 s of odor presentation. The performance index (PI) was calculated as described previously (Pavlopoulos et al., 2008). Because the time between testing and the coupling of the CS+ with the US is 3 min, the initial tests represent 3 min memory, but will be referred to as learning.
Control olfactory and shock avoidance assays were performed as described previously (Pavlopoulos et al., 2008).
Spaced training for long-term memory (LTM) assessment was performed manually and used 12 US/CS pairings per round, and 5 or 10 such rounds of training with 15 min of rest between rounds (Pavlopoulos et al., 2008). After training, the flies were stored at 25°C in the dark for 3 h and then transferred in the dark to 18°C for the remaining period until testing as previously described (Pascual and Préat, 2001).
Data were analyzed parametrically with the JMP statistical package (SAS Institute) as described previously (Pavlopoulos et al., 2008; Moressis et al., 2009) and detailed in the text or figure legends.
Immunohistochemistry.
Frontal paraffin sections (5 μm) of heads were obtained and processed for immunohistochemistry or histology as described previously (Pavlopoulos et al., 2008). Sections from control and experimental heads were processed in parallel and photographed with same acquisition settings for each photograph. The sections were probed with monoclonal anti-dFMRP 5A11 (Developmental Studies Hybridoma Bank) and signal detected with diaminobenzidine.
Pharmaceutical administration.
2-Methyl-6-(phenylethynyl)pyridine (MPEP) was obtained from FRAXA. 4-(3-Cyclopentyloxy-4-methoxy-phenyl)pyrrolidin-2-one (Rolipram) was purchased from Sigma-Aldrich. The drugs were dissolved as per the manufacturer's instructions and used at the following concentrations: MPEP at 100 μm (McBride et al., 2005) and Rolipram at 200 μm (Hou et al., 2004; McBride et al., 2005). Solutions of these compounds were mixed with 1% sucrose/0.6% brewer's yeast in water. Approximately 50–60 flies were placed in plastic vials containing 500 μl of each solution on top of an agar matrix and allowed to feed ad libidum for 10 h for MPEP and 5 h for Rolipram before training. Vehicle treatment consisted of solvent added to 1% sucrose/0.6% brewer's yeast alone. Approximately 1 h before conditioning, flies were moved to plain food vials. After LTM training, flies were transferred in the dark in vials with drug for ∼22–23 h and were changed to plain food vials 1 h before testing.
Western blot analysis.
Monoclonal anti-dFMRP 5A11 was obtained from the Developmental Studies Hybridoma Bank. For Western blot analyses, two female fly heads were homogenized in standard Laemmli buffer and loaded on 10% acrylamide gels, transferred to PVDF membranes, probed with the anti-dFMRP antibody at 1:100 and anti-syntaxin (DSHB) at 1:2500, and detected with horseradish peroxidase-coupled secondary antibody at 1:4000.
For detection of DmGluRA in Western blot, 30 female heads were homogenized in 60 μl of RIPA buffer, which was then left on ice for 15 min to precipitate exoskeleton debris. The supernatant was collected and centrifuged for 3 min at 14,000 rpm. The supernatant was discarded, and the pellet was resuspended in 30 μl of Laemmli buffer, loaded on 8% acrylamide gels, transferred to PVDF membranes, probed with the anti-mGluR 7G11 (Panneels et al., 2003) at 1:10 and anti-tubulin at 1:1000, and detected with horseradish peroxidase-coupled secondary antibody at 1:30,000. Western Lightning Ultra (PerkinElmer) was used as chemiluminescence substrate.
Reverse transcription and real-time PCR.
Total RNA was isolated from Drosophila heads with Trizol (Invitrogen). The RNA samples were DNase treated and further purified with the RNeasy purification system (QIAGEN). RNA (5 μg) was used in reverse transcription reactions for 1 h at 37°C using SuperScript III reverse transcriptase (Invitrogen). Q-PCR was performed in diluted RT samples with the designated primer sets using the MiniOpticon System (Bio-Rad), with Platinum SYBR Green qPCR Supermix UDG (Invitrogen). Results presented are the average of three runs from four independent reverse transcription reactions. dfmr1 transcripts were amplified with the following primers: forward, 5′-CGCTACCACAACAATCGCC; reverse, 5′-AGCTGCCAGTGTTGTCACC.
Fluorescent cAMP assay.
The Bridge-It cAMP designer fluorescence assay system (Mediomics) was used to measure cAMP levels in all samples. Fifty heads from each genotype were homogenized in 100 μl of cold homogenization buffer (15 mm HEPES, pH 7.5, 10 mm KCl, 5 mm MgCl2, 0.1 mm EDTA, 0.1 mm EGTA, 5% sucrose, 1% protease inhibitor mixture). The suspension was allowed to settle on ice for 5 min to remove the chitin exoskeleton. Five milligrams of acid-rinsed charcoal was added to remove eye pigment, and the suspension was centrifuged four times at 500 × g for 2 min to remove the charcoal. The supernatant was centrifuged again at 15,000 × g for 15 min, and the pellet resuspended in 50 μl of Krebs'–Ringer's bicarbonate buffer (KRB) (1.19 m NaCl, 94 mm KCl, 50 mm CaCl2, 24 mm KH2PO4, 250 mm NaHCO3, 1 m d-glucose)–0.75 mm 3-isobutyl-1-methylxanthine (IBMX). Four micrograms of Drosophila head membranes were incubated in KRB–IBMX at 25°C for 20 min, and the suspension was centrifuged at 14,000 × g for 5 min. Supernatants were discarded and 90 μl of the cAMP designer assay solution was added to the membrane pellet, and cAMP levels were estimated per the manufacturer's specifications. Fluorescence intensity was evaluated with an Infinity 200 fluorescence plate reader (Tecan). Relative fluorescence change was measured from raw fluorescence values using the following formula, where RF is the relative fluorescence, F0 is the fluorescent intensity of the blank or buffer control, and F is the fluorescence of cAMP or sample: RF = (F0 − F)/F0. The RF values calculated this way are highly reproducible for the same cAMP concentrations and do not depend on the instrument used to read fluorescence. Using this modified method, we validated our results against those reported by Kelley et al. (2007) on the forskolin inducible cAMP fraction of heterozygotes and homozygotes. The forskolin inducible/basal ratios for the relevant genotypes were as follows: w1118, 1.818 ± 0.218; dfmr13/+, 2.602 ± 0.681; dfmr13/dfmr13, 0.968 ± 0.190.
Statistical analysis was conducted with the JMP statistical package (SAS Institute).
Results
dfmr1 mutant heterozygotes exhibit learning and memory deficits
Initially, we investigated whether dfmr1 mutant heterozygotes may present with learning and memory deficits because we aimed to establish a sensitive system amenable to their genetic or pharmacological modification. We hypothesized that this would be essential in revealing processes regulated by dFMRP and mechanisms regulating its levels, or function in the CNS. We have established sensitive methods of assessing Drosophila learning and memory in heterozygous mutants that do not exhibit phenotypes typically associated with complete loss of function (Moressis et al., 2009; Gouzi et al., 2011). This is particularly relevant, because homozygous dfmr1-null mutants are known to possess structurally aberrant mushroom bodies (MBs) (Michel et al., 2004; McBride et al., 2005). This developmental aberration may bias and interfere with interpretation of their behavioral deficits because these bilateral clusters of ∼2000–2200 neurons in each brain lobe are known to be essential for olfactory associative learning and memory in Drosophila and other insects (Heisenberg, 2003).
To establish sensitive conditions for genetic and pharmacological screens, we took advantage of the observation that homozygotes for the 7 kb intragenic deletion allele dfmr13, which removes the entire open reading frame (Dockendorff et al., 2002), maintained over a TM3Sb balancer exhibit increased lethality. This is apparent because a small number of viable mutant homozygotes are obtained, in contrast to the same mutation maintained over the TM6c balancer, which yields 40–45% of the expected number. In addition to highly penetrant MB defects, these homozygotes exhibited reduced adult longevity and severely suppressed locomotor activity, in contrast to dfmr13 heterozygotes, which presented increased locomotor activity (data not shown). Given these phenotypes, we wondered whether dfmr13/+ from TM3Sb-balanced parents might exhibit learning and memory deficits.
To determine whether learning and memory are compromised in such mutant heterozygotes, we trained them in the negatively reinforced olfactory learning paradigm (Tully and Quinn, 1985) with the previously established modifications (Pavlopoulos et al., 2008; Moressis et al., 2009). Clearly, dfmr13 heterozygotes exhibited a robust learning deficit regardless of whether were derived from TM3Sb or TM6c balanced parents (Fig. 1A). Ηeterozygotes for either balancer did not have adverse effects on associative learning, but nevertheless both balancers seem to enhance the performance deficit in trans with the dfmr13 deletion. Therefore, the balancers do not seem to have differential effects on learning as for lethality. However, in experiments assessing the levels of DmGluRA in mutant heterozygotes reported below, we uncovered that levels of the receptor were increased over those in w1118 2.899 ± 0.28 times in dfmr13/TM3Sb versus 1.691 ± 0.21 in dfmr13/TM6c. This significant difference may account for the enhanced lethality of homozygotes derived from TM3Sb-balanced parents, and it is not likely to involve the DmGluRA gene itself since it resides on the fourth chromosome.
Figure 1.

dfmr1 mutant heterozygotes are learning and memory deficient. Mean PIs and their SEMs (PI ± SEM) are shown for the behavioral experiments. A, The black bars represent the performance of the genetic control strain w1118, while other control genotypes are shown in the gray-shaded bars and the performance of mutant heterozygotes with the open bars. ANOVA indicated significant differences among the groups (p < 0.0001). We then used planned comparisons to uncover these differences. The performance of TM3 and TM6 heterozygotes was not significantly different from that of w1118 controls (p = 0.872 and p = 0.1852, respectively). In contrast, the performance of dfmr13/+ and dfmr13/TM3 were significantly different from that of +/TM3 (p < 0.0001). Similarly, the performance of dfmr13/+ and dfmr13/TM6 were significantly different from that of +/TM6 (p < 0.0001). Note that performances of dfmr13/+ animals were not different (p = 0.6648) whether their parents were TM6 or TM3-balanced. n ≥ 10. B, Controls are shown in black bars. Following initial ANOVA indicating significant differences (p < 0.0001), planned comparisons were used to compare performances of experimental groups (open bars) and relevant controls. Learning of dfmr13 mutant heterozygotes was significantly different (p < 0.0001) from that of w1118 controls, whereas that of dfmr13/+ animals carrying a transgenic genomic fragment bearing the dfmr1 gene was not (p = 0.109). In contrast, the performance of dfmr13/+ flies bearing a similar genomic transgene containing FS mutation remained significantly different (p < 0.0001) from controls (n ≥ 10 for all groups). C, The performance of dfmr13/+ immediately after training with five rounds of 12 CS/US pairings was not significantly different from that of controls (p = 0.784, Student's t; n ≥ 8). In contrast, 24 h memory (5× LTM) of the association was significantly impaired in the mutant heterozygotes compared with controls (p < 0.0001, Student's t; n ≥ 8). D, Twenty-four hour memory induced with 10 training rounds (10× LTM). The performance of dfmr13/+ animals remained significantly different from that of controls (p < 0.001, Student's t; n ≥ 7).
Importantly, these experiments uncovered a robust associative learning deficit for dfmr13 heterozygotes. To ascertain that the learning deficit was indeed a consequence of the dfmr1 mutation, we attempted transgenic reversal of the phenotype. In fact, the learning deficit in the heterozygotes was fully reversed by transgenes harboring the entire wild-type genomic region of the dfmr1 gene (RES), but not by an identical construct carrying a FS mutation in the coding region (McBride et al., 2005) (Fig. 1B).
Complete loss of dFMRP has been reported to result in LTM deficits in Drosophila (Bolduc et al., 2008; Banerjee et al., 2010). To determine whether dfmr13 heterozygotes are also LTM deficient, we subjected them to five cycles of spaced training as described previously (Pavlopoulos et al., 2008). Under these conditions, learning levels of mutant heterozygotes and controls were indistinguishable (Fig. 1C, Immediate), but nevertheless 24 h memory of the association was strongly impaired (Fig. 1C, LTM 5×). Because learning was normal after the spaced 12 pairing training, the memory deficit likely reflects an independent requirement for dFMRP in LTM. Interestingly, mutant heterozygotes were reported not to be LTM deficient after 10 cycle training (Bolduc et al., 2008). However, even after this extended training regime, the LTM of dfmr13/+ animals remained deficient compared with controls (Fig. 1D). Therefore, in our system, heterozygous animals exhibit memory deficits after 5 or 10 training rounds, probably a manifestation of the resolution and sensitivity of our methods (Pavlopoulos et al., 2008).
Mutant heterozygotes do not exhibit macroscopic MB deficits at the level of the transmission or confocal microscopy (data not shown). However, to determine whether the learning and memory deficits may be attributable to subtle developmental defects and as an independent method to abrogate dFMRP, we used RNA interference (RNAi), coupled with the temporal control afforded by the Gal80ts system (McGuire et al., 2003). Initially, dFMRP was attenuated specifically in the adult CNS to levels lower than the 50% reduction in mutant heterozygotes (Fig. 2A). Notably, adult-specific pan-neuronal dFMRP abrogation phenocopied the learning deficit of dfmr13 heterozygotes (Fig. 2B, white bar). Learning was normal if the transgene remained silent (18°C), while the induction conditions did not adversely affect learning in controls (Fig. 2B, black bar, 30°C). It must be noted that avoidance of the aversive CS odors and the footshock US was normal for all genotypes in this experiment and all genotypes tested below (Table 1). Therefore, the learning deficits of dfmr1-null heterozygotes are unlikely of developmental origin, and the protein appears to be required acutely for normal learning. In addition, given its apparent high efficiency (Fig. 2A), utilization of the RNAi-mediating transgene is equivalent or more severe than utilization of an independent mutant allele.
Figure 2.

dFMRP is required within the mushroom bodies for associative learning and memory. A, Western blot demonstrating the levels of dFMRP in adult head lysates of w1118 controls, dfmr13/+ heterozygotes, and animals expressing pan-neuronally the interfering UAS-dfmr1-R transgene. This represents one of four identical blots with similar results. dFMRP was revealed with the α-dfmrP 5A11 antibody, and Syntaxin (SYX) was used as loading control. B, Adult-specific pan-neuronal expression of UAS-dfmr1-R (30°C) yields significant (p < 0.0001, Dunnett's) learning deficits (open bars) compared with uninduced (18°C), transgene-alone and driver-alone (black bars) controls. n ≥ 8. All animals were heterozygous for the transgene Gal4 and Gal80ts. All animals were trained and tested at 25°C, regardless of the temperatures they were maintained before training to manipulate transgene expression as indicated. C, Spatially restricted abrogation of dFMRP. All animals were heterozygous for UAS-dfmr1-R and Gal4 drivers. (ANOVA, p < 0.0001). Attenuation specifically in α/β and γ MB lobes with c772 and MB247 (white bars) impaired learning (p < 0.0001 vs w1118/UAS-dfmr1-R controls; black bars) and phenocopies the dfmr13/+ mutant phenotype. In contrast, restricted dfmr1-R expression only within the γ lobes with NP1131, or to the ellipsoid body with c507 did not affect learning (gray bars; p = 0.023 and p = 0.367, respectively). n ≥ 10. D, A UAS-dfmr1 cDNA transgene recombined onto the chromosome harboring the dfmr13 mutation, fully rescues (darker gray bars) the dfmr13/+ learning deficit (white bars) when expressed in the MBs with c772 (p = 0.135 vs w1118; black bar) and MB247 (p = 0.667 vs w1118; black bar), but not when expressed γ lobes with NP1131, or to the ellipsoid body with c507 (lighter gray bars; p < 0.0001). All animals were heterozygous for the transgene and Gal4 drivers. n ≥ 12. E, Twenty-four hour memory induced with five rounds of spaced training was significantly impaired in dfmr13 heterozygotes (open bars) and animals with adult-specific, pan-neuronal, or MB-specific dfmr1-R expression (p < 0.0001 for all pairwise comparisons with their specific yoked controls; black bars). n ≥ 12. All animals were heterozygous for the transgene Gal4 and Gal80ts and were trained and tested at 25°C, regardless of the temperatures they were maintained before training to manipulate transgene expression as indicated. Error bars indicate SEM.
Table 1.
Task-relevant sensory behaviors
| Genotype | Benzaldehyde (1×) | Octanol (1×) | Electric shock (90 V) |
|---|---|---|---|
| w1118 | 83.39 ± 4.30 | 74.05 ± 2.73 | 94.17 ± 1.46 |
| dfmr13/+ | 94.09 ± 1.42 | 82.57 ± 4.09 | 89.95 ± 5.45 |
| dfmr13, RES/+ | 88.46 ± 2.78 | 80.28 ± 3.54 | 91.95 ± 2.05 |
| dfmr13, FS/+ | 90.82 ± 4.39 | 78.89 ± 5.18 | 94.11 ± 2.32 |
| dnc1/+ | 84.75 ± 1.60 | 89.78 ± 3.90 | 86.88 ± 7.16 |
| dnc1/+; dfmr13/+ | 83.46 ± 4.53 | 93.54 ± 1.77 | 89.77 ± 3.19 |
| dfmr13, mGluR-R/+ | 68.18 ± 2.19 | 80.71 ± 4.36 | 93.03 ± 1.50 |
| c772-Gal4/+; Gal80ts/dfmr13, mGluR-R | 69.41 ± 2.21 | 84.43 ± 1.46 | 89.16 ± 1.75 |
| elav-Gal4/+; Gal80ts/dfmr13, mGluR-R | 63.72 ± 1.89 | 89.40 ± 1.69 | 88.50 ± 3.05 |
| dfmr1-R/+ | 60.15 ± 1.73 | 88.24 ± 2.18 | 88.16 ± 2.63 |
| c772-Gal4/dfmr1-R +; Gal80ts/+ | 78.66 ± 4.26 | 83.49 ± 2.91 | 82.56 ± 1.65 |
| elav-Gal4/+; dfmr1-R/+; Gal80ts/+ | 73.88 ± 3.89 | 89.83 ± 1.70 | 88.12 ± 2.06 |
| dfmr1-R/+; MB247-Gal4/+ | 68.54 ± 3.84 | 89.42 ± 4.31 | 89.28 ± 2.73 |
| dfmr1-R/+; NP1131-Gal4/+ | 69.21 ± 2.78 | 79.14 ± 4.61 | 80.70 ± 1.76 |
| dfmr1-R/+; c507-Gal4/+ | 62.43 ± 4.09 | 82.30 ± 3.91 | 83.87 ± 4.26 |
| dfmr13, UAS-dfmr1/+ | 69.01 ± 4.61 | 88.91 ± 4.59 | 83.34 ± 3.21 |
Avoidance of benzaldehyde and octanol versus air and of the 90 V electric footshock. The performances of the experimental animals were not significantly lower than those of controls. The groupings indicate animals used for each comparison and their relevant controls. w1118 served as controls for experiments involving dnc1/+ as well.
We used the abrogating UAS-dfmr1-R transgene to determine the minimal neuronal subset requiring dFMRP for normal olfactory learning and targeted the MBs given their essential role in the process (Davis, 2005). The c772 and MB247 Gal4 drivers are expressed in 1800 and 1600 α/β and γ, respectively, of the1900 MB neurons per hemisphere marked with the Gal4 system (Aso et al., 2009). Abrogation of dFMRP with c772 and MB247 yielded significant learning deficits, phenocopying that of mutant heterozygotes (Fig. 2C). In contrast, UAS-dfmr1-R expression specifically within the γ neurons with NP1131 (Aso et al., 2009), or the ellipsoid body with c507 (Yang et al., 2000) did not affect learning. The performance of all driver heterozygotes was indistinguishable from that of transgene heterozygotes and w1118 (Table 2). Therefore, the learning deficits appear specific to the MBs and in particular the α/β lobes. In a complementary approach, driving a full-length cDNA transgene (UAS-dfmr1) within the MBs with c772 and MB247 in dfmr13/+ animals restored learning fully (Fig. 2D). In contrast, mutant heterozygotes expressing the transgene with NP1131 or c507 retained the learning deficit. Furthermore, adult-specific dFMRP abrogation either pan-neuronally or specifically within the α/β MB neurons resulted in defective LTM (Fig. 2E). Hence, dFMRP is required specifically within α/β neurons of the adult MBs for normal associative olfactory learning and memory.
Table 2.
Associative learning of driver heterozygotes and DmGluR RNAi-expressing flies
| Genotype | n | PI ± SEM |
|---|---|---|
| w1118 | 19 | 78.35 ± 1.84 |
| elav-Gal4; Gal80ts/+ | 18 | 79.30 ± 2.07 |
| c772-Gal4; Gal80ts/+ | 18 | 79.88 ± 1.70 |
| MB247-Gal4/+ | 13 | 83.07 ± 3.78 |
| c507-Gal4/+ | 18 | 79.96 ± 1.88 |
| NP1131-Gal4/+ | 12 | 80.48 ± 3.69 |
| elav-Gal4; Gal80ts/+ | 18 | 79.30 ± 2.07 |
| elav-Gal4; Gal80ts/UAS-mGluR-R | 11 | 70.39 ± 1.80 |
| c772-Gal4; Gal80ts/+ | 18 | 79.88 ± 1.70 |
| c772-Gal4; Gal80ts/UAS-mGluR-R | 12 | 71.67 ± 3.35 |
Associative learning performance (PI ± SEM) of the indicated control genotypes and flies with pan-neuronal and MB-specific abrogation of DmGluRA. None of the experimental genotypes performed significantly different than controls (w1118 in the top panel and the relevant driver heterozygotes in the bottom).
Learning aberrations in mutant heterozygotes are US/CS pairing specific
We performed a qualitative analysis of learning in the mutant heterozygotes aiming to determine whether the ability altogether or the rate of learning was deficient as described previously for other mutants (Moressis et al., 2009). Although conditioning with five rounds of 12 US/CS pairings resulted in normal performance (Fig. 1C), suggestive of a learning rate deficit in dfmr13/+, this intensive conditioning protocol yields “ceiling levels” of performance obscuring potential differences. Therefore, we systematically assessed learning after training with increasing number of US/CS pairings in mutants and animals with adult-specific dFMRP abrogation.
In agreement with previous reports (Moressis et al., 2009), the performance of control animals increased with the number of US/CS couplings to an apparent asymptote between 8 and 12 pairings. Surprisingly, however, the performance of dfmr13/+ was significantly higher than that of controls after 2 and 4 pairings, significantly lower after 6 pairings, and identical with controls after 12 pairings (Fig. 3A). This behavioral response profile was not specific to the mutant heterozygotes, because similar profiles were obtained with adult-specific pan-neuronal (Fig. 3B), or MB-limited (Fig. 3C) dFMRP reduction. It is notable that, in agreement with the results in Figure 1, the deficit in dfmr13/+ is maximal after 6 and eliminated with 12 US/CS pairings. This would be consistent with a deficit in the rate of learning as defined previously (Moressis et al., 2009). However, the elevated performance after two and four pairings does not allow us to make this conclusion. To our knowledge, this is the first time such an acquisition profile is reported and may reflect impairments in multiple associative and possibly nonassociative processes that potentially underlie the conditioned behavior measured in the assay. It is possible that the improved performance under limited pairings may reflect the reported increase of metabotropic glutamate receptor activity in the mutants (McBride et al., 2005; Bolduc et al., 2008; Gatto and Broadie, 2009). Interestingly, exaggerated responses during the initial sessions of a learning task have also been reported for Fmr1-null mice (Van Dam et al., 2000; Frankland et al., 2004; Moon et al., 2008) and may reflect similar phenomena. Alternatively, the enhanced performance under low pairings may be a consequence of the training procedure, possibly reflecting alterations in experience-dependent nonassociative responses (Acevedo et al., 2007), which may differentially alter the salience of the training or testing stimuli.
Figure 3.
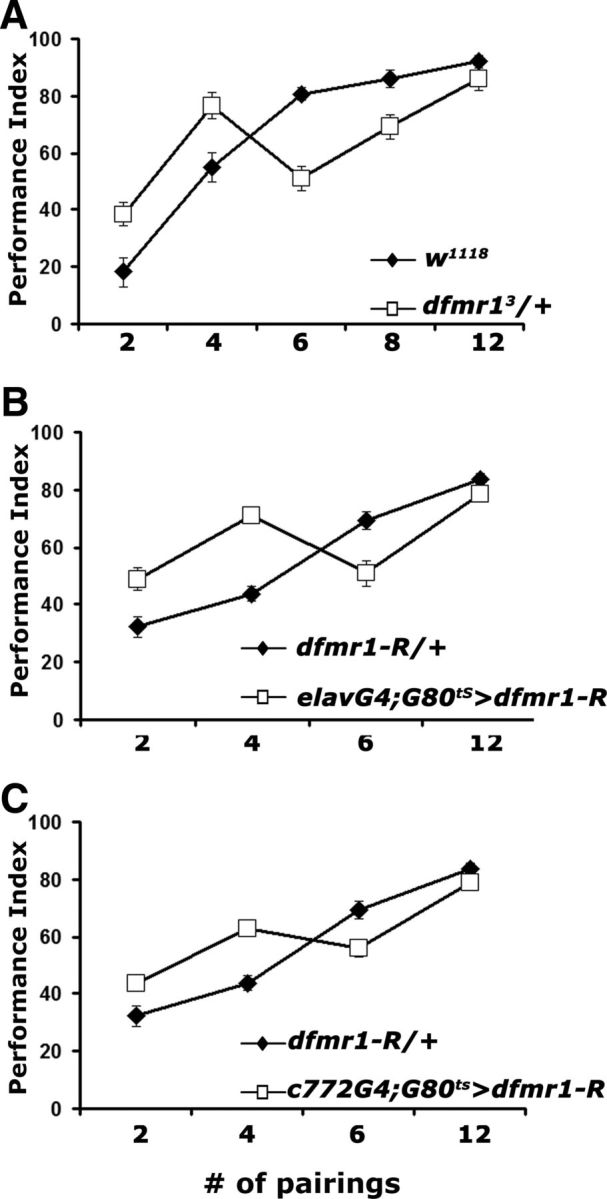
US/CS pairing specific learning deficits of dfmr1 mutant heterozygotes. Mean PIs and their SEMs (PI ± SEM) are shown for all experiments. Where appropriate, animals were heterozygous for all transgenes and Gal4 drivers. A, Performance immediately after training (learning) with the indicated number of pairings. n = 10 for all pairings. ANOVA indicated significant differences (p < 0.0001). Planned comparisons (contrast analysis) indicated that the performance of dfmr13/+ (open squares) was significantly different from that of w1118 (open squares) after conditioning with 2, 4, and 6 pairings, but not after 8 (p = 0.03) or 12 pairings (p = 0.25). B, Learning after training with the indicated number of pairings of animals with adult-specific pan-neuronal abrogation of dFMRP (open squares). Differences were indicated by ANOVA (p < 0.0001) and resolved as significant with contrast analysis after 2, 4, and 6 pairings (p < 0.0001), but not after 12 pairings (p = 0.12). n = 10 for all pairings. C, Adult-specific abrogation of dFMRP in α/β MB lobes with c772 (open squares) significantly impaired learning (p < 0.001) compared with transgene heterozygote controls (filled diamonds), except after 12 pairings (p = 0.01). n = 10 for all pairings.
To differentiate between these two possibilities, we aimed to block signaling via the metabotropic glutamate receptor (DmGluRA) (McBride et al., 2005) using the noncompetitive antagonist MPEP, previously shown to reverse the effects of dfmr1 loss (McBride et al., 2005). Feeding adult dfmr13/+ animals with 100 μm MPEP before conditioning eliminated their enhanced performance relative to controls after two and four pairings and fully restored the learning deficit after six US/CS (Fig. 4A). To further substantiate these results, we fed MPEP to animals with pan-neuronal adult-specific abrogation of dFMRP. Again, MPEP administration eliminated the enhanced performance under low pairings and restored normal learning after six US/CS (Fig. 4B). These data support the interpretation that the enhanced performance after conditioning with two and four pairings and the deficit after six pairing training are attributable to elevation of DmGluRA activity (McBride et al., 2005; Choi et al., 2010, 2011).
Figure 4.
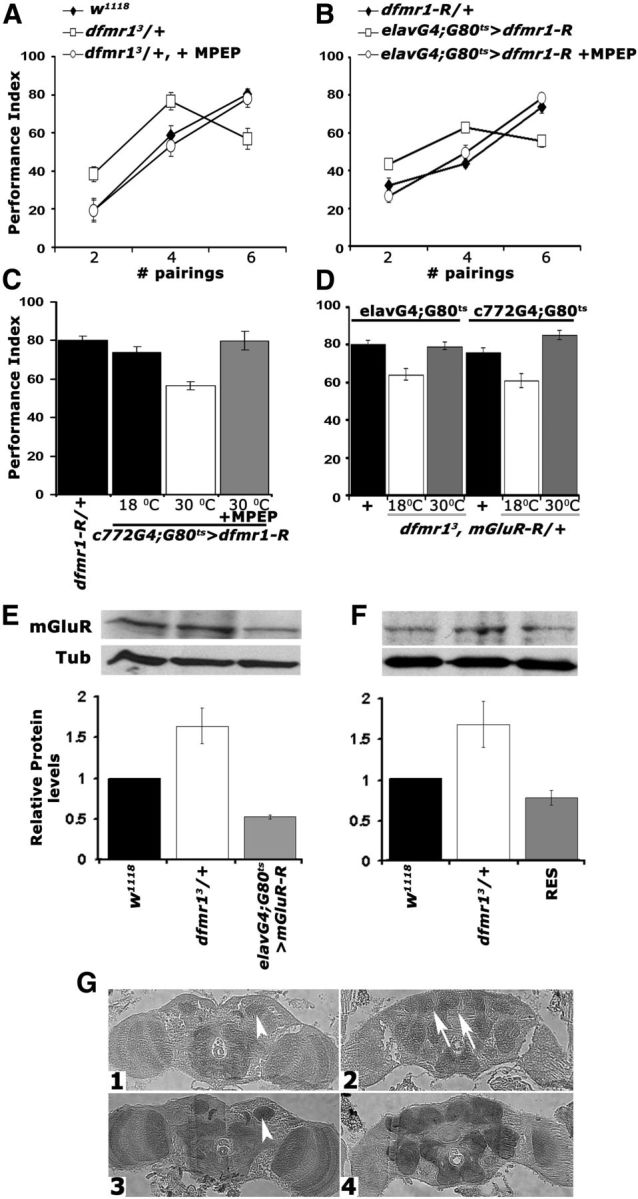
Genetic and pharmacological inhibition of DmGluRA restores the learning deficit of dfmr1 mutants. Mean PIs and their SEMs (PI ± SEM) are shown for all experiments. Where appropriate, animals were heterozygous for transgenes, Gal4 and Gal80ts. All animals were trained and tested at 25°C, regardless of the temperatures they were maintained before training to manipulate transgene expression as indicated. A, MPEP administration to dfmr13/+ reversed the aberrant learning of the heterozygotes (open squares) after two, four, and six pairings to control levels (filled diamonds). n ≥ 10. ANOVA indicated significant differences (p < 0.001) resolved by subsequent Dunnett's tests, which revealed that for two, four, and six pairings the performance of treated animals (open circles) was indistinguishable from controls (p = 0.55, p = 0.51, and p = 0.96, respectively), whereas that of untreated ones remained significantly different at all pairings (p < 0.0001). B, Pharmacological rescue (open circles) of learning in animals with pan-neuronal dfmr1 abrogation (open squares) to control levels (filled diamonds) of performance after training with two, four, and six pairings. The differences suggested by ANOVA (p < 0.001) were examined by Dunnett's tests and demonstrated that treated animals (open circles) and controls (filled diamonds) did not perform differently after two, four, and six pairings (p = 0.21, p = 0.57, and p = 0.23, respectively), whereas untreated animals performed significantly different from both regardless of pairings (p < 0.0001). C, Conditional abrogation (30°C) of dfmr1 in the MBs with c772 impairs learning (open bar) and can be rescued by MPEP (gray bar) to control levels (black bars). n = 10. The performance of animals carrying both transgenes but not expressing UAS-dfmr1-R (18°C) was used to compare that of experimental animals to (Dunnett's tests), after positive initial ANOVA (p < 0.0001). Transgene induction resulted in significant differences with control performance (p < 0.0001), but not after MPEP treatment (p = 0.54). D, Genetic rescue of the learning deficit in dfmr13/+ by pan-neuronal (left three bars) and MB limited conditional attenuation of DmGluRA. Heterozygotes for the Gal4 driver, Gal80ts with the w1118-derived chromosomes were used as controls (black bars) for Dunnett's tests in both cases. n ≥ 10. Maintaining the mGluR-R transgene inactive either pan-neuronally, or in the MBs of dfmr13 heterozygotes resulted in deficient six pairing learning (p < 0.0001). However, abrogation of DmGluRA pan-neuronally, or specifically in the MBs, resulted in performances similar to those of controls (p = 0.23 and p = 0.02, respectively). E, Semiquantitative Western blot analysis of mGluRA levels in adult head lysates from dfmr13/+ flies with pan-neuronal abrogation of DmGluRA and controls. A representative blot of four is shown above, and the quantification below includes all replicates. mGluRA levels relative to the Tubulin loading control were arbitrarily set to 1 for the w1118 controls, and the mean relative levels ± SEM in the experimental animals were estimated and plotted. Levels in the experimental animals were significantly different from those in controls (Dunnett's, p < 0.001). F, A representative semiquantitative Western blot of mGluRA levels in adult head lysates from w1118 controls, dfmr13/+, and their sibling dfmr13/+ heterozygotes carrying a single copy of the dfmr1 genomic transgene (RES). mGluRA levels relative to the Tubulin loading control were arbitrarily set to 1 for the w1118 controls and the mean relative levels ± SEM in the experimental animals were estimated and plotted. mGluRA levels in the experimental animals were not significantly different from those in controls, but were significantly different from those in dfmr13/+ (Dunnett's, p < 0.001). G, Five micrometer frontal paraffin sections from adult heads of w1118 (G1, G2) and dfmr13/+ (G3, G4) flies challenged with α-mGluRA. Sections are shown at the level of the MB calyces (G1, G3, arrowheads) in the posterior of the head and in the mid-anterior of the head (G2, G4). The arrows in G2 point to the MB axonal projections (lobes), which are not stained in agreement with prior reports (Devaud et al., 2008). DmGluRA elevation is apparent in dfmr13/+ brains, which were processed on the same histological slide as controls.
DmGluRA is present in the dendritic areas of adult MBs (calyces) (Devaud et al., 2008) (Fig. 4G1), suggesting potential involvement in signal integration within these neurons. To address this, dFMRP was attenuated specifically within adult MBs with the c772-Gal4 driver and significantly, the resultant learning deficit was readily reversed by feeding these animals with MPEP (Fig. 4C). It appears then that the orally delivered pharmaceutical must have reached the MBs and inhibited the receptor to fully rescue the six pairing learning deficit. This was confirmed by pan-neuronal, or MB-specific abrogation of DmGluRA in adult dfmr13 heterozygotes, which readily rescued their deficient learning upon six pairing conditioning (Fig. 4D). Interestingly, however, conditional DmGluRA attenuation pan-neuronally or specifically in adult MBs did not precipitate obvious learning deficits (Table 2), although a 50% reduction in protein levels was achieved (Fig. 4E). Collectively, these results demonstrate that upregulation of DmGluRA activity in the MBs of dfmr13/+ is responsible for their learning deficits.
Surprisingly, in the course of these experiments, a consistent and highly significant increase in DmGluRA levels was observed in dfmr13 heterozygotes over those in controls (Fig. 4E). Although consistent over different experiments, to ascertain that this result was not method specific, the DmGluRA distribution was examined histologically in controls and mutant heterozygotes. In sections processed, sectioned, and stained on the same slide, there was a clear increase in the levels of the receptor in the CNS of dfmr13/+ animals compared with controls (Fig. 4G), and this elevation included the calyces of the MBs (Fig. 4G3). Significantly, elevated DmGluRA in dfmr13/+ animals was restored to control levels upon expression of the genomic transgene carrying the dfmr1 gene (Fig. 4F). These observations are consistent with the MPEP-dependent reversal of the deficient learning in animals with dFMRP abrogated specifically in adult MBs (Fig. 4C) and rescued learning in dfmr13/+ upon attenuation of DmGluRA in their MBs (Fig. 4D). Furthermore, quantitative RT-PCR experiments did not reveal an increase in DmGluRA transcripts in these mutant heterozygotes (DmGluRA/Tubulin ratio set as 1 for w1118 vs 0.89 ± 0.07 for dfmr13/+). Taken together then, DmGluRA elevation in dfmr13/+ heterozygotes and upon dfmr1 RNAi induction indicates that, in Drosophila, the receptor is under negative translational regulation by dFMRP. It is likely then that the reported MPEP-ameliorated increase in DmGluRA activity in dfmr1 mutants (McBride et al., 2005; Choi et al., 2010) is a consequence of increased receptor levels. Given the lack of phenotype upon DmGluRA attenuation, these results demonstrate that upregulation of DmGluRA in the MBs of dfmr13/+ is likely responsible for their learning deficits and is consistent with the reversibility of the deficit upon abrogation of the receptor within these neurons (Fig. 4D).
cAMP elevation reverses the learning and memory deficits of dfmr1 heterozygotes
FMRP loss affects multiple signaling pathways and neuronal mechanisms (Bagni and Greenough, 2005; Gantois et al., 2006; Chang et al., 2008; Wang et al., 2008) and this may underlie at least some of the variable behavioral and cognitive symptoms in FXS patients (Tsiouris and Brown, 2004) and in models such as Drosophila. This along with the biphasic acquisition response of the mutants prompted us to perform a targeted pharmacological screen to determine whether defects in other molecular pathways in addition to the apparent DmGluRA upregulation may underlie these defects. The full results of this screen will be presented elsewhere. However, we describe herein a part of the screen focused on the cAMP pathway. Initially, this was based on reports that cultured cells from FXS patients and dfmr1 mutant Drosophila exhibited consistently low levels of the nucleotide (Kelley et al., 2007) and mostly older reports suggesting an association of cAMP and FMRP (Berry-Kravis and Huttenlocher, 1992; Berry-Kravis and Ciurlionis, 1998; McBride et al., 2005). Furthermore, many protein members of the cAMP cascade are expressed preferentially in the MBs and like dfmr1 exhibit learning and memory defects when mutated (Davis et al., 1995; Skoulakis and Grammenoudi, 2006).
To increase the levels of endogenous cAMP in dfmr13/+, we fed them for 5–6 h before conditioning the pyrrolidinone Rolipram, a potent PDE4 (phosphodiesterase-IV) inhibitor, known to be effective on its Drosophila ortholog Dnc (Davis et al., 1989; Henkel-Tigges and Davis, 1990; Hou et al., 2004). Interestingly, Rolipram completely reversed both the enhanced performance after two and four pairing training and the deficit after six pairings (Fig. 5A), much like that attained after DmGluRA inhibition with MPEP. Reversal of the six pairing learning deficit was independently confirmed by Rolipram treatment of animals harboring adult-specific pan-neuronal dFMRP abrogation (Fig. 5B). To ascertain the specificity of the effect, we used a complementary genetic approach using mutants in the cAMP-dependent phosphodiesterase-encoding gene dunce. The Dnc protein accumulates preferentially in the MBs and like dFMRP is required within these neurons for normal olfactory learning (Nighorn et al., 1991; Davis et al., 1995). Like dfmr13/+, dnc1 mutant heterozygotes exhibit a modest reduction in olfactory learning after six pairing training (Fig. 5C). However, to our surprise, learning in dnc1/+; dfmr13/+ flies was indistinguishable from that of controls (Fig. 5C), indicating that elevation of endogenous cAMP levels ameliorates the deficit of dfmr1 mutants. We confirmed this with a second allele dncML, which in trans-heterozygosity also rescued the deficient learning of dfmr13/+ animals (Fig. 5C). These results are congruent with the effect of Rolipram on animals with attenuated dFMRP (Fig. 5A,B), are consistent with previous reports of reduction in cAMP levels upon dFMRP loss (Kelley et al., 2007), and suggest that dFMRP and the cAMP pathway interact genetically.
Figure 5.
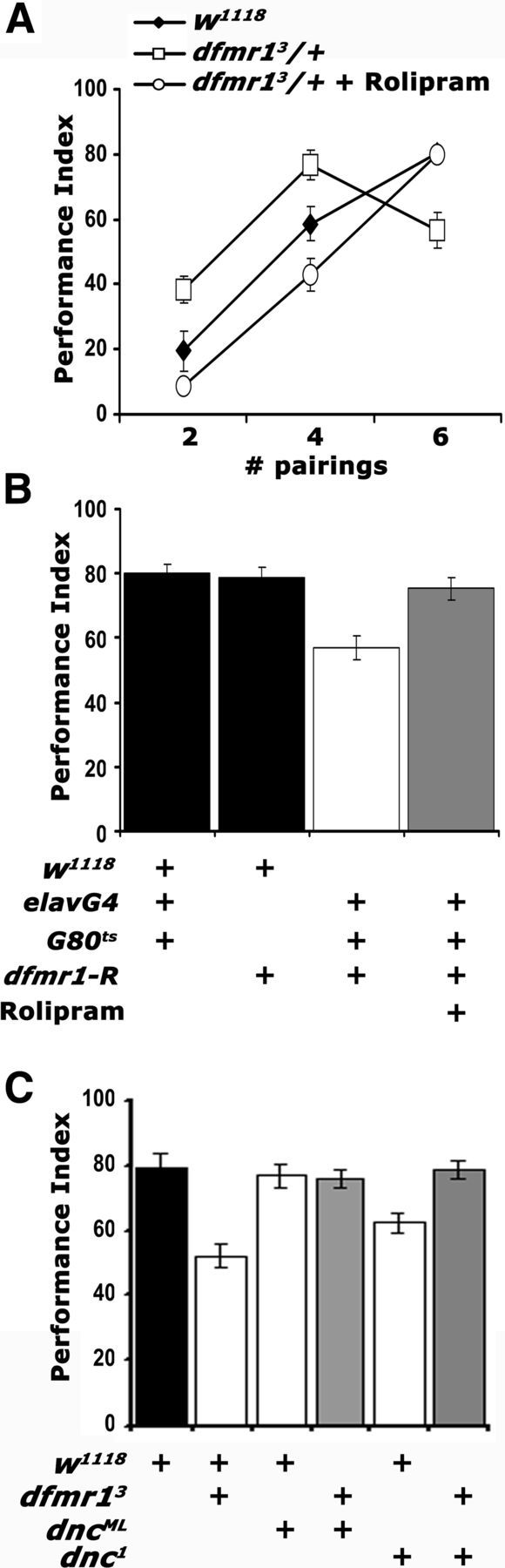
cAMP elevation in dfmr13/+ reverses their learning deficit. Mean PIs and their SEMs (PI ± SEM) are shown for all experiments. Where appropriate, animals were heterozygous for all transgenes, Gal4 drivers, and Gal80ts. A, Rolipram administration (open circles) resulted in performance indistinguishable from that of controls (filled diamonds) after two, four, and six pairing training (p = 0.16, p = 0.04, and p = 0.73, respectively). The performance of untreated animals (open squares) remained significantly different from both (p < 0.001). n ≥ 8. B, Rolipram administration (gray bar) to animals with pan-neuronally abrogated dfmr1 resulted in learning equivalent to that of control driver heterozygotes (p = 0.44) or UAS-dfmr1-R heterozygotes (p = 0.98). The performance of untreated animals (open bars) remained significantly different from all other groups (p < 0.0001). n ≥ 10. C, Genetic rescue of the dfmr13/+ learning deficit by reduction of functional DNC. The performances of dnc1/+; dfmr13/+ double heterozygotes and dncML/+; dfmr13/+ (gray bars) are not statistically distinguishable (p = 0.15 and p = 0.36, respectively) from that of w1118 controls (black bar). In contrast, the performances of dnc1 heterozygotes and dfmr13/+ (open bars) were significantly different from controls (p < 0.0001 for both). n ≥ 8.
Does this putative dFMRP/cAMP genetic interaction extend to LTM? Because complete rescue of the learning deficit was attained either by DmGluRA abrogation, or pharmacological and genetic elevation of cAMP, we tested whether both treatments also restore LTM in dfmr1 heterozygotes. As for learning, adult-specific DmGluRA attenuation in dfmr13/+ fully restored their LTM deficit (Fig. 6A, left). Furthermore, we assessed LTM in dnc1 heterozygotes alone and in combination with dfmr13/+. Interestingly, a LTM deficit for dnc1 heterozygotes was revealed, suggesting that 50% reduction in the phosphodiesterase does not support normal LTM. However, LTM was indistinguishable from that of controls in dnc1/+; dfmr13/+ animals (Fig. 6A, right). This striking result extends to LTM and at least partially confirms the proposed genetic interaction between cAMP and dFMRP.
Figure 6.
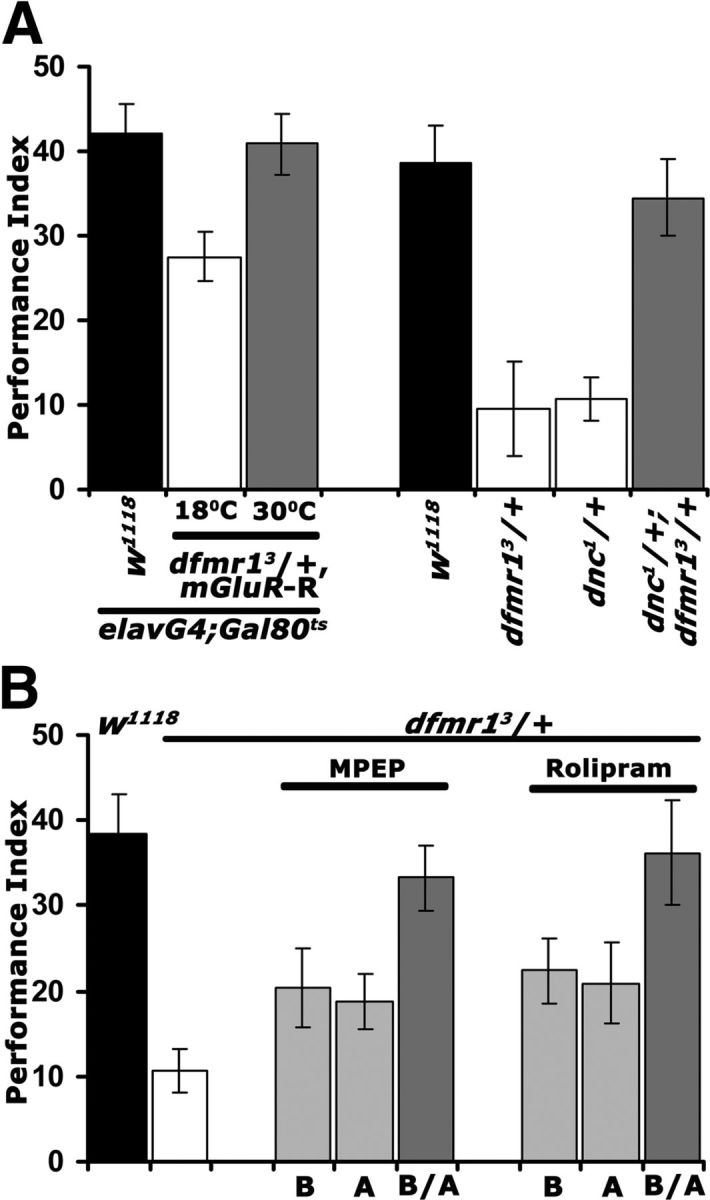
Rescue of the dfmr13/+ LTM deficit via DmGluRA inhibition and cAMP elevation. Mean PIs and their SEMs (PI ± SEM) are shown for all experiments. A, Conditional abrogation of DmGluRA reverses the LTM defect of dfmr13/+ animals (left side). Animals were heterozygous for all transgenes and Gal4 and Gal80ts as indicated. Repression of the abrogating transgene (18°C; open bar) revealed a significant (p < 0.0001) memory deficit compared with controls (black bar), whereas upon mGluR-R induction (30°C; gray bar) the performance was indistinguishable (p = 0.97) from that of controls. n ≥ 9. On the right side, the complementary genetic experiment confirms the conclusions. LTM of both dfmr13/+ and dnc1/+ single heterozygotes (open bars) were significantly different (p < 0.0001) than controls (black bar), whereas surprisingly that of dnc1/+; dfmr13/+ was not (p = 0.52). n ≥ 9. B, Similar temporal requirement for MPEP and Rolipram to rescue the LTM deficit of dfmr13/+. Pharmaceuticals were administered before (B), after training (A), or both prior and posttraining (B/A). Animals exposed to MPEP or Rolipram B/A (dark gray bars) did not perform different from controls (p = 0.78 and p = 0.99, respectively), whereas all other memory scores were significantly different (p < 0.0001) from those of controls.
To substantiate this interaction further, we attempted pharmacological rescue of the LTM deficit. We took advantage of the relatively short exposure to the pharmaceuticals required to rescue learning (Figs. 4A, 5A), to investigate whether DmGluRA inhibition and cAMP elevation have similar or different temporal requirements on LTM formation, consolidation, or expression/recall. Therefore, we treated dfmr13/+ animals with MPEP and Rolipram before (B) or after (A) conditioning and throughout the protocol (B/A). Both Rolipram and MPEP eliminated the LTM deficit if administered 6 h preconditioning and the following 24 h until testing. Feeding the drugs only before or specifically after conditioning and until testing, did not rescue the memory defect. Significantly then, pharmacological manipulation of DmGluRA activity, or cAMP elevation revert the deficient memory of dfmr13/+ animals with similar temporal requirements (Fig. 6A). Collectively, these results suggest that dFMRP reduction and the consequent DmGluRA elevation may be linked to the described (McBride et al., 2005; Kelley et al., 2007) reduction in cAMP levels. This may in fact be mediated by the DmGluRA receptor itself, which in Drosophila seems coupled to Gi/o signaling (Parmentier et al., 1996; McBride et al., 2005), but may also signal via the Gq system similar to its mammalian homolog (Gatto and Broadie, 2009). This interpretation suggests that the learning and memory deficits of dfmr1 mutants may, at least in part, be mediated by cAMP signaling perturbations. This seems particularly likely for LTM because of the importance of the cAMP-dependent transcription factor CREB in the process in both flies and vertebrates (Alberini, 2009; Benito and Barco, 2010).
Glutamate receptor inhibition restores cAMP and dFMRP levels
How does cAMP modulate the dFMRP-deficient behavioral phenotypes? Is it linked with DmGluRA inhibition? We addressed these questions to gain insights into the potential mechanisms underlying the cAMP/dFMRP genetic interactions. Initially, we monitored the levels of dFMRP in adult head lysates after pharmacological and genetic manipulations that modified the behavioral consequences of dfmr1 reduction. Semiquantitative Western blots verified the expected 50% dFMRP reduction in dfmr13/+. The relatively brief MPEP administration raised dFMRP to levels equivalent to that of controls, but surprisingly Rolipram yielded even higher protein amounts (Fig. 7A). A similar dFMRP increase to control levels was also observed upon DmGluRA abrogation in dfmr13/+ animals (Fig. 7B). We used conditions of adult-specific pan-neuronal expression of UAS-DmGluR-R instead of limiting it to the MBs because these neurons represent a small percentage of the CNS, hence do not afford the requisite resolution for this analysis. Also consistent with the effects of Rolipram, dFMRP levels were significantly higher in dnc1/+, while the levels of the protein in dnc1/+; dfmr13/+ animals were not significantly different from controls (Fig. 7C). Taken together, the pharmacological and genetic abrogation results are consistent with the notion that DmGluRA elevation (Fig. 4E,F) and the consequent increase in activity inhibits cAMP levels, potentially via the proposed Gi/o mechanism (McBride et al., 2005; Gatto and Broadie, 2009). Moreover, cAMP levels appear to modulate dFMRP in the adult fly CNS, consistent with their genetic interaction indicated by the behavioral data above. In fact, restoration of dFMRP levels by these pharmacological and genetic manipulations in dfmr13/+ animals explain the rescue of their learning and memory deficits (Figs. 5, 6).
Figure 7.

Genetic and pharmacological manipulation altering dFMRP levels also affect the levels cAMP. A–C, Representative blots probing dFMRP levels in the indicated genotypes (top) and quantitative representation of the means ± SEM of the results relative to Syntaxin (SYX) which served as loading control in three such independent blots per experimental set. The genotypes of each group are shown on the bottom. Relative dFMRP levels in control strains (black bars) were arbitrarily fixed as 1 and were used to compare the levels in the other genotypes (Dunnett's tests). The significant (p < 0.0001) 50% decrease of the protein in dfmr13/+ is evident (open bars). A, In contrast, MPEP and Rolipram administration restored dFMRP to control levels (p = 0.12 and p = 0.002, respectively). B, Similarly, DmGluRA abrogation resulted in significantly higher (p < 0.0001) dFMRP levels. Animals were heterozygous for all transgenes and Gal4 and Gal80ts as indicated. C, dFMRP levels are significantly elevated in dnc1/+ over that in controls (p < 0.0001), but the levels in dnc1/+; dfmr13/+ animals are not (p = 0.67). D–F, cAMP levels measured in head lysates of the indicated genotypes using the Bridge-IR Fluorescent assay as detailed in Materials and Methods. cAMP levels in controls were arbitrarily designated as 1, and all other measurements are presented relative to that. The means ± SEM of three independent measurements (each the mean of a duplicate) are shown. D, Following a significant result from ANOVA (p < 0.0001), Dunnett's tests showed that cAMP levels in dfmr13/+ are significantly different (p < 0.0001) than controls, but they are restored to normal levels upon MPEP treatment (p = 0.25). As expected, following Rolipram treatment cAMP levels are significantly higher (p < 0.0001) than in controls. E, Conditional abrogation of DmGluRA in dfmr13/+ restores cAMP to control levels (p = 0.45), whereas the nucleotide levels are significantly lower (p < 0.0001) than controls if the transgene remains uninduced. F, cAMP levels are significantly higher (p < 0.0001) than controls in dnc1 heterozygotes, but in dnc1/+; dfmr13/+ flies are not significantly different from in controls (p = 0.06).
To determine whether cAMP is in fact modulated by DmGluRA attenuation or pharmacological inhibition, we assessed the levels of the nucleotide in adult head lysates. In accord with previous reports (Kelley et al., 2007), total cAMP was reduced nearly 50% in dfmr13/+. As expected, administration of Rolipram increased cAMP levels in dfmr13/+ lysates even above those of controls, but this was also observed with MPEP administration, in agreement with the hypothesis (Fig. 7D). Similarly, adult-specific abrogation of DmGluRA in mutant heterozygotes (Fig. 7E), or heterozygosity for the dnc1 mutant allele (Fig. 7F), restored cAMP to control levels. These results strongly support the notion that it is the elevation of cAMP that reverses the behavioral deficits of dfmr1 reduction. Furthermore, they demonstrate that DmGluRA activation acts negatively on cAMP levels, providing in vivo evidence that the receptor indeed signals via a Gi/o mechanism in Drosophila. Therefore, upon dFMRP loss or functional reduction, DmGluRA elevation results in strongly depressed cAMP levels in neurons expressing the receptor and likely underlies the deficits in behavioral plasticity.
Transcriptional modulation of dfmr1 levels by cAMP
Elevation of dFMRP following pharmacological and genetic manipulations of cAMP levels could be the result of increased transcription, decreased protein turnover, increased protein or RNA stability, or a combination thereof. To differentiate between these possibilities, we initially quantified the level of dfmr1 transcripts in the heads of mutant heterozygotes under conditions that eliminate their behavioral deficits. As expected, there was a 50% reduction in dfmr1 mRNA in mutant heterozygotes, but its levels increased to match or even exceed those of controls after administration of either Rolipram or MPEP (Fig. 8A). This elevation must be acute given the relatively short time the animals were exposed to the pharmaceuticals. Similarly, acute pan-neuronal abrogation of DmGluRA in mutant heterozygotes resulted in significant elevation of transcripts from the wild-type dfmr1allele to match those of controls (Fig. 8B). This agrees with results from the larval neuromuscular junction (NMJ) of dfmr1-null mutants (Pan et al., 2008). Moreover, dnc1/+ harbored higher dfmr1 transcripts than controls and introducing this mutation into dfmr13/+ restored dfmr1 mRNA to control levels (Fig. 8C). In summary, these results indicate that dfmr1 transcription or RNA stability is responsive to cAMP levels, and this explains the observed dFMRP elevation under conditions that increase the nucleotide in the CNS. cAMP levels apparently are negatively modulated by DmGluRA, which in turn is subject to negative translational modulation by dFMRP. Therefore, our results uncovered a regulatory loop that appears responsive to dFMRP levels and is modulated by cAMP.
Figure 8.

Transcriptional regulation of dfmr1 by cAMP. The means ± SEM from three to four independent biological replicates of quantitative PCR probing the levels of dfmr1 mRNA relative to that in controls (black bars), in the indicated genotypes are shown. Following a significant ANOVA, all means were compared with controls, which were arbitrarily set to 1 (Dunnett's tests). A, MPEP and Rolipram administration restored the mRNA to levels equivalent to those in controls, or significantly higher (p = 0.002 and p < 0.0001, respectively). B, dfmr1 mRNA levels were also restored relative to controls (p = 0.43) upon adult-specific abrogation of DmGluRA. C, dfmr1 levels were significantly higher in dnc1/+ than controls (p < 0.0001), but not significantly different from controls in dnc1/+; dfmr13/+ flies (p = 0.06). D, dfmr1 mRNA levels in dc0B3/+ were significantly lower than in controls (p < 0.0001) and approximately the same as that in dfmr3/+. E, A representative Western blot demonstrating reduced levels of dFMRP in dc0B3/+ animals. dFMRP levels in this mutant do not respond to MPEP and Rolipram application. F, The mean levels ± SEM of dFMRP in dc0B3/+ and after pharmaceutical treatment from three independent experiments. Levels in untreated dc0B3/+ and after MPEP and Rolipram treatment were significantly different from those in controls (p < 0.0001). G, The genomic region upstream of the transcribed sequence of Drosophila dfmr1 is shown. Putative CREB binding sites are indicated by the bold underlined letters boxed in gray. The numbering of the sequence is per GenBank sequence 3R:5927142, 5935844.
Does cAMP modulate dfmr1 transcription or stability by activating the typical signaling cascade? To address this question, we assessed dfmr1 levels in heterozygotes for a null allele of the cAMP-activated protein kinase A catalytic subunit (dc0B3) (Lane and Kalderon, 1993). A 50% decrease in the catalytic subunit and presumably in cAMP signaling (Lane and Kalderon, 1993; Skoulakis et al., 1993) resulted in a similar decrease in dfmr1 mRNA as in dfmr13/+ (Fig. 8D). Furthermore, if the effects of MPEP and Rolipram were indeed upstream of PKA activity, then administration of the pharmaceuticals in dc0B3/+ would not rescue their low dfmr1 levels. As shown in the representative Western blot (Fig. 8E) and quantified in Figure 8F, neither MPEP nor Rolipram affected dfmr1 levels in dc0B3/+ mutants. These results suggest that cAMP modulation of dFMRP levels engages the typical signaling cascade, which likely leads to activation of the transcription factor CREB. In fact, the human Fmr1 gene has been reported to contain binding sites and to be regulated by CREB (Hwu et al., 1997; Smith et al., 2006). Interestingly, we also identified consensus CREB binding sites (Impey et al., 2004) in the putative promoter region of the dfmr1 gene (Fig. 8G), indicative of the plausibility of this model, which is currently under further investigation.
Discussion
Aberrant olfactory learning and memory upon dFMRP reduction
We have demonstrated robust learning and LTM deficits associated with 50% reduction in dFMRP and mapped the phenotype to adult MB α/β lobes with spatiotemporally controlled RNAi-mediated abrogation. The learning deficit of dfmr13-null allele heterozygotes was quantitatively similar (Fig. 1) to that of animals lacking significantly more of the protein due to RNAi-mediated dFMRP abrogation either pan-neuronally (Fig. 2B) or specifically in the MBs (Fig. 2C). This is surprising given the efficient attenuation of dFMRP with UAS-dfmr-R. Interestingly, a similar learning deficit was independently reported for null homozygotes as well (Bolduc et al., 2008).
The lack of enhanced learning and memory deficits upon further reduction than 50% in the heterozygotes indicates that the activity of the remaining dFMRP in these animals may be reduced to levels functionally approaching those in null homozygotes. This suggests that an essential posttranslational modification may be suppressed in the heterozygotes, effectively further reducing functional dFMRP. Ser499 phosphorylation by S6K1 is essential for the translational repressor function of vertebrate FMRP and phosphate removal or suppression inactivates it (Narayanan et al., 2008). Moreover, phosphorylation by casein kinase II at dFMRP Ser406 has been reported functionally important (Siomi et al., 2002). Hence, altered dFMRP phosphorylation in dfmr13/+ is a plausible explanation for the phenotypes in the heterozygotes, and this hypothesis is currently under investigation. Because near-complete abrogation of the protein does not completely eliminate olfactory learning (Fig. 2B,C), dFMRP does not appear to play an essential role in all molecular processes engaged within the MBs for associative olfactory learning.
Processes requiring normal dFMRP function appear to be involved in learning rate (Moressis et al., 2009), because dfmr13 heterozygotes and animals with attenuated protein in the MBs reach asymptotic performance levels like controls, but require additional US/CS pairings (Fig. 3A–C). The aberrantly elevated responses after training with two and four pairings may be akin to the heightened arousal reported to result in increased activity and exaggerated responses during the initial sessions of a learning task in mice lacking the Fmr1 gene (Moon et al., 2008). In addition, synaptic hyperexcitability, especially upon high-frequency stimulation, at the neuromuscular junction was reported for dfmr1-null larvae (Repicky and Broadie, 2009). These phenomena may be related to the elevated emotional reactivity and anxiety often associated with FXS patients (Hagerman and Sobesky, 1989; Hagerman, 2002). Excess DmGluRA activity appears to be involved in this exaggerated response, because it was eliminated after feeding mutant flies with MPEP (Fig. 4A,B) and, significantly, also with Rolipram (Fig. 5A). The known anxiolytic and antidepressant properties of Rolipram and other PDE inhibitors (Silvestre et al., 1999) support the interpretation that the aberrantly elevated responses after few US/CS pairings may reflect anxiety in flies as in mice. The mechanism of this performance enhancement is unclear at the moment but will be investigated in detail in the future.
DmGluRA and PDE inhibition rescue learning and memory by raising cAMP
Initially, we tested whether the mGluR elevation proposed to underlie many FXS behavioral deficits and validated in Drosophila with mutant homozygotes (McBride et al., 2005; Bolduc et al., 2008) was also applicable when dFMRP was reduced, but not eliminated. Our results indicate that, in mutant heterozygotes, it is not solely the activity of the mGluRA receptor that is increased but also the levels of the protein itself (Fig. 4E,F). Because we did not uncover evidence suggesting increased DmGluRA transcripts, this evidence suggests that it is translation of the receptor likely regulated by dFMRP. In the Drosophila larval NMJ, dFMRP has been reported to regulate the abundance of ionotropic glutamate receptor subclasses (Pan et al., 2008), and similarly it may regulate the levels of DmGluRA in the adult CNS. Moreover, a recent report indicated that several mGluRs are targets of FMRP-dependent translational regulation (Darnell et al., 2011). These results are consistent with the RNAi-mediated attenuation of the receptor, which reversed both learning (Fig. 4D) and memory (Fig. 6A) deficits in animals with abrogated dFMRP. Therefore, dFMRP function appears dosage sensitive since 50% reduction suffices to elevate DmGluRA.
DmGluRA is present in the MB dendrites (Devaud et al., 2008). Therefore, dFMRP abrogation in these neurons is expected to result in elevation of the receptor within them (Fig. 4G3) (data not shown). The fact that feeding MPEP reverses the learning deficits of animals with abrogated dFMRP specifically in the MBs strongly suggests that the pharmaceutical reaches these neurons and acts on the locally elevated DmGluRA.
Interestingly, inhibiting DmGluRA with MPEP or abrogating the receptor in the adult fly CNS rescued the low cAMP levels in dfmr13/+ (Fig. 7D,E). This indicates that cAMP levels are directly influenced by the level of DmGluRA. Furthermore, cAMP levels appear to lie downstream of the receptor, because elevation of the nucleotide by reducing the dosage of the PDE Dnc (Fig. 7F), or administration of Rolipram (Fig. 7D) resulted in complete reversal of the learning and LTM phenotypes of dfmr13/+ (Figs. 5, 6). Consistent with this observation, the mGluR antagonists LY341495 [2-[(1S,2S)-2-carboxycyclopropyl]-3-(9H-xanthen-9-yl)-d-alanine], MPPG [(RS)-α-methyl-4-phosphonophenylglycine], and MTPG [(RS)-α-methyl-4-tetrazolylphenylglycine], previously used to rescue the courtship learning defect of dfmr13 homozygotes (McBride et al., 2005), are known to also increase cAMP signaling in Drosophila (Kelley et al., 2008). Hence, the low cAMP levels reported for fly, mouse, and humans with compromised FMRP function (Kelley et al., 2007) are likely a consequence of enhanced levels of a Gi/o-linked glutamate receptor, which is thought to be the DmGluRA in Drosophila (McBride et al., 2005). These data strongly suggest this as the mechanism linking mGluR overactivity and/or levels and the proposed FMRP regulation of cAMP levels (Berry-Kravis and Huttenlocher, 1992; Berry-Kravis and Ciurlionis, 1998; Kelley et al., 2008). Consistently, use of group II mGluR antagonists rescued the LTD phenotype in a mouse FXS model (Choi et al., 2011). Therefore, at least in the Drosophila model and with respect to associative learning and memory, the mGluR (Bear et al., 2004) and cAMP (Berry-Kravis and Ciurlionis, 1998; Kelley et al., 2008) theories of accounting for the behavioral deficits of FXS seem to converge and describe different points of the same molecular interaction network.
The behavioral effects of reducing the dosage of Dnc by 50% are also noteworthy. First, dnc1 heterozygosity precipitates small but significant effects on learning (Fig. 5C), and much larger effects on LTM (Fig. 6A), suggesting exaggerated effects of elevated cAMP on consolidated memory. The significance of reestablishing cAMP balance within the MBs is likely reflected in the surprising complete rescue of the dfmr13/+ LTM defect by reducing the dosage of the PDE, thus elevating cAMP and dFMRP levels (Fig. 7C).
cAMP-driven regulation of dfmr1 transcription
Interestingly, pharmacological or genetic manipulations that raised cAMP levels in dfmr13/+ animals also resulted in increased levels of dFMRP (Fig. 7A–C). This response appears to be mediated entirely by a cAMP-dependent increase in dfmr1 transcription (Fig. 8A–C), is apparently transduced via PKA signaling (Fig. 8D–F), and likely engages the transcription factor CREB. A summary model of our observations are presented in Figure 9. A correlation between cAMP and FMRP levels had been noted previously (Berry-Kravis and Ciurlionis, 1998) with respect to regional variation in brain areas suggestive of interactions in a developmental context. In contrast, our results demonstrate an acute transcriptional response, as elevated cAMP and dfmr1 transcripts are apparent within a few hours of pharmaceutical administration (Figs. 7D, 8A). Therefore, dFMRP levels respond acutely to cAMP in the fly CNS and appear to reflect the abundance or activity of DmGluRA as a negative-feedback loop. In support of this notion, dFMRP levels were reduced in homozygous mutants for the Rut adenylyl cyclase (Bolduc et al., 2008), a situation where DmGluRA levels are presumed normal. Furthermore, given the function of dFMRP as a translational repressor, it is of interest to consider its elevation in dnc heterozygous and further increase in homozygous mutants (data not shown). Is then the etiology of the learning and memory deficits in dnc mutants cAMP elevation, or exaggerated translational repression within their MBs because of enhanced dFMRP levels therein?
Figure 9.

A schematic summary of the dFMRP interactions and regulation in Drosophila neurons described herein. The adenylyl cyclase (AC) at the cell membrane (top curved line) is hypothesized to be Rutabaga, based on the results of Bolduc et al. (2008). DmGluRA is shown to inhibit its activity, while itself is negatively regulated by dFMRP. Positive transcriptional regulation of dfmr1 by cAMP levels via protein kinase A is also indicated. The cell nucleus is indicated by the dotted line.
Collectively, our results suggest that pharmaceuticals that modulate cAMP signaling are promising routes to effectively ameliorate behavioral symptoms of “premutation” carriers (Hagerman and Hagerman, 2004; Jacquemont et al., 2007; Swanson and Orr, 2007). Amelioration due to transcriptional upregulation of FMRP in response to cAMP elevation is not possible in patients and models harboring deletions or functional silencing of the gene. However, point mutations that do not affect transcription of the gene, but rather particular functional domains were identified recently (Collins et al., 2010). Because these appear associated with some but not all behavioral deficits, they likely do not represent null but rather hypomorphic alleles. Perhaps Rolipram and MPEP-mediated increases in cAMP levels and FMRP transcription may be beneficial to such patients as well as those presenting FXTAS symptoms. Like in Drosophila, the human gene also appears to contain CREB sites in its putative promoter area, indicating that this approach is at least feasible.
Footnotes
This work was supported by the March of Dimes and FRAXA. We thank Sean M. J. McBride, Thomas C. Dockendorf, and Daniel Kalderon for fly stocks and reagents. We acknowledge the gift of MPEP from Dr. M. Tranfaglia and FRAXA, the Developmental Hybridoma Studies Bank for antibodies, the Bloomington Drosophila Stock Center for resources, Flybase for essential information, and Jean Y. Gouzi and Sean M. J. McBride for critical reading of this manuscript.
References
- Acevedo SF, Froudarakis EI, Tsiorva AA, Skoulakis EM. Distinct neuronal circuits mediate experience-dependent, non-associative osmotactic responses in Drosophila. Mol Cell Neurosci. 2007;34:378–389. doi: 10.1016/j.mcn.2006.11.011. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Alberini CM. Transcription factors in long-term memory and synaptic plasticity. Physiol Rev. 2009;89:121–145. doi: 10.1152/physrev.00017.2008. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antar LN, Bassell GJ. Sunrise at the synapse: the FMRP mRNP shaping the synaptic interface. Neuron. 2003;37:555–558. doi: 10.1016/s0896-6273(03)00090-4. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Aso Y, Grübel K, Busch S, Friedrich AB, Siwanowicz I, Tanimoto H. The mushroom body of adult Drosophila characterized by GAL4 drivers. J Neurogenet. 2009;23:156–172. doi: 10.1080/01677060802471718. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Bagni C, Greenough WT. From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat Rev Neurosci. 2005;6:376–387. doi: 10.1038/nrn1667. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Banerjee P, Schoenfeld BP, Bell AJ, Choi CH, Bradley MP, Hinchey P, Kollaros M, Park JH, McBride SM, Dockendorff TC. Short- and long-term memory are modulated by multiple isoforms of the fragile X mental retardation protein. J Neurosci. 2010;30:6782–6792. doi: 10.1523/JNEUROSCI.6369-09.2010. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Benito E, Barco A. CREB's control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci. 2010;33:230–240. doi: 10.1016/j.tins.2010.02.001. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Ciurlionis R. Overexpression of fragile X gene (FMR-1) transcripts increases cAMP production in neural cells. J Neurosci Res. 1998;51:41–48. doi: 10.1002/(SICI)1097-4547(19980101)51:1<41::AID-JNR4>3.0.CO;2-L. Medline. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Huttenlocher PR. Cyclic AMP metabolism in fragile X syndrome. Ann Neurol. 1992;31:22–26. doi: 10.1002/ana.410310105. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Bolduc FV, Bell K, Cox H, Broadie KS, Tully T. Excess protein synthesis in Drosophila fragile X mutants impairs long-term memory. Nat Neurosci. 2008;11:1143–1145. doi: 10.1038/nn.2175. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, Bray SM, Li Z, Zarnescu DC, He C, Jin P, Warren ST. Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat Chem Biol. 2008;4:256–263. doi: 10.1038/nchembio.78. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Choi CH, McBride SM, Schoenfeld BP, Liebelt DA, Ferreiro D, Ferrick NJ, Hinchey P, Kollaros M, Rudominer RL, Terlizzi AM, Koenigsberg E, Wang Y, Sumida A, Nguyen HT, Bell AJ, McDonald TV, Jongens TA. Age-dependent cognitive impairment in a Drosophila fragile X model and its pharmacological rescue. Biogerontology. 2010;11:347–362. doi: 10.1007/s10522-009-9259-6. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi CH, Schoenfeld BP, Bell AJ, Hinchey P, Kollaros M, Gertner MJ, Woo NH, Tranfaglia MR, Bear MF, Zukin RS, McDonald TV, Jongens TA, McBride SM. Pharmacological reversal of synaptic plasticity deficits in the mouse model of fragile X syndrome by group II mGluR antagonist or lithium treatment. Brain Res. 2011;1380:106–119. doi: 10.1016/j.brainres.2010.11.032. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SC, Bray SM, Suhl JA, Cutler DJ, Coffee B, Zwick ME, Warren ST. Identification of novel FMR1 variants by massively parallel sequencing in developmentally delayed males. Am J Med Genet A. 2010;152A:2512–2520. doi: 10.1002/ajmg.a.33626. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL. Olfactory memory formation in Drosophila: from molecular to systems neuroscience. Annu Rev Neurosci. 2005;28:275–302. doi: 10.1146/annurev.neuro.28.061604.135651. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Davis RL, Takayasu H, Eberwine M, Myres J. Cloning and characterization of mammalian homologs of the Drosophila dunce+ gene. Proc Natl Acad Sci U S A. 1989;86:3604–3608. doi: 10.1073/pnas.86.10.3604. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL, Cherry J, Dauwalder B, Han PL, Skoulakis E. The cyclic AMP system and Drosophila learning. Mol Cell Biochem. 1995;149–150:271–278. doi: 10.1007/978-1-4615-2015-3_31. [DOI] [PubMed] [Google Scholar]
- Devaud JM, Clouet-Redt C, Bockaert J, Grau Y, Parmentier ML. Widespread brain distribution of the Drosophila metabotropic glutamate receptor. Neuroreport. 2008;19:367–371. doi: 10.1097/WNR.0b013e3282f524c7. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- D'Hulst C, Kooy RF. Fragile X syndrome: from molecular genetics to therapy. J Med Genet. 2009;46:577–584. doi: 10.1136/jmg.2008.064667. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Dockendorff TC, Su HS, McBride SM, Yang Z, Choi CH, Siwicki KK, Sehgal A, Jongens TA. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron. 2002;34:973–984. doi: 10.1016/s0896-6273(02)00724-9. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Wang Y, Rosner B, Shimizu T, Balleine BW, Dykens EM, Ornitz EM, Silva AJ. Sensorimotor gating abnormalities in young males with fragile X syndrome and Fmr1-knockout mice. Mol Psychiatry. 2004;9:417–425. doi: 10.1038/sj.mp.4001432. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Gantois I, Vandesompele J, Speleman F, Reyniers E, D'Hooge R, Severijnen LA, Willemsen R, Tassone F, Kooy RF. Expression profiling suggests underexpression of the GABAA receptor subunit delta in the fragile X knockout mouse model. Neurobiol Dis. 2006;21:346–357. doi: 10.1016/j.nbd.2005.07.017. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Gatto CL, Broadie K. The fragile X mental retardation protein in circadian rhythmicity and memory consolidation. Mol Neurobiol. 2009;39:107–129. doi: 10.1007/s12035-009-8057-0. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouzi JY, Moressis A, Walker JA, Apostolopoulou AA, Palmer RH, Bernards A, Skoulakis EM. The receptor tyrosine kinase Alk controls neurofibromin functions in Drosophila growth and learning. PLoS Genet. 2011;7:e1002281. doi: 10.1371/journal.pgen.1002281. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman PJ, Hagerman RJ. The fragile-X premutation: a maturing perspective. Am J Hum Genet. 2004;74:805–816. doi: 10.1086/386296. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ. The physical and behavioral phenotype. In: Hagerman RJ, Hagerman PJ, editors. Fragile X syndrome: diagnosis, treatment and research. Baltimore: The Johns Hopkins UP; 2002. pp. 3–109. [Google Scholar]
- Hagerman RJ, Sobesky WE. Psychopathology in fragile X syndrome. Am J Orthopsychiatry. 1989;59:142–152. doi: 10.1111/j.1939-0025.1989.tb01641.x. Medline. [DOI] [PubMed] [Google Scholar]
- Heisenberg M. Mushroom body memoir: from maps to models. Nat Rev Neurosci. 2003;4:266–275. doi: 10.1038/nrn1074. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Henkel-Tigges J, Davis RL. Rat homologs of the Drosophila dunce gene code for cyclic AMP phosphodiesterases sensitive to rolipram and RO 20-1724. Mol Pharmacol. 1990;37:7–10. Medline. [PubMed] [Google Scholar]
- Hou J, Kuromi H, Fukasawa Y, Ueno K, Sakai T, Kidokoro Y. Repetitive exposures to nicotine induce a hyper-responsiveness via the cAMP/PKA/CREB signal pathway in Drosophila. J Neurobiol. 2004;60:249–261. doi: 10.1002/neu.20021. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Hunter JE, Allen EG, Abramowitz A, Rusin M, Leslie M, Novak G, Hamilton D, Shubeck L, Charen K, Sherman SL. Investigation of phenotypes associated with mood and anxiety among male and female fragile X premutation carriers. Behav Genet. 2008a;38:493–502. doi: 10.1007/s10519-008-9214-3. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwu WL, Wang TR, Lee YM. FMR1 enhancer is regulated by cAMP through a cAMP-responsive element. DNA Cell Biol. 1997;16:449–453. doi: 10.1089/dna.1997.16.449. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Impey S, McCorkle SR, Cha-Molstad H, Dwyer JM, Yochum GS, Boss JM, McWeeney S, Dunn JJ, Mandel G, Goodman RH. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell. 2004;119:1041–1054. doi: 10.1016/j.cell.2004.10.032. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Jacquemont S, Hagerman RJ, Hagerman PJ, Leehey MA. Fragile-X syndrome and fragile X-associated tremor/ataxia syndrome: two faces of FMR1. Lancet Neurol. 2007;6:45–55. doi: 10.1016/S1474-4422(06)70676-7. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Kelley DJ, Davidson RJ, Elliott JL, Lahvis GP, Yin JC, Bhattacharyya A. The cyclic AMP cascade is altered in the fragile X nervous system. PLoS One. 2007;2:e931. doi: 10.1371/journal.pone.0000931. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DJ, Bhattacharyya A, Lahvis GP, Yin JC, Malter J, Davidson RJ. The cyclic AMP phenotype of fragileX and autism. Neurosci Biobehav Rev. 2008;32:1533–1543. doi: 10.1016/j.neubiorev.2008.06.005. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane ME, Kalderon D. Genetic investigation of cAMP-dependent protein kinase function in Drosophila development. Genes Dev. 1993;7:1229–1243. doi: 10.1101/gad.7.7a.1229. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- McBride SM, Choi CH, Wang Y, Liebelt D, Braunstein E, Ferreiro D, Sehgal A, Siwicki KK, Dockendorff TC, Nguyen HT, McDonald TV, Jongens TA. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron. 2005;45:753–764. doi: 10.1016/j.neuron.2005.01.038. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- McGuire SE, Le PT, Osborn AJ, Matsumoto K, Davis RL. Spatiotemporal rescue of memory dysfunction in Drosophila. Science. 2003;302:1765–1768. doi: 10.1126/science.1089035. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Michel CI, Kraft R, Restifo LL. Defective neuronal development in the mushroom bodies of Drosophila fragile X mental retardation 1 mutants. J Neurosci. 2004;24:5798–5809. doi: 10.1523/JNEUROSCI.1102-04.2004. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mientjes EJ, Nieuwenhuizen I, Kirkpatrick L, Zu T, Hoogeveen-Westerveld M, Severijnen L, Rifé M, Willemsen R, Nelson DL, Oostra BA. The generation of a conditional Fmr1 knock out mouse model to study Fmrp function in vivo. Neurobiol Dis. 2006;21:549–555. doi: 10.1016/j.nbd.2005.08.019. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Mineur YS, Sluyter F, de Wit S, Oostra BA, Crusio WE. Behavioral and neuroanatomical characterization of the Fmr1 knockout mouse. Hippocampus. 2002;12:39–46. doi: 10.1002/hipo.10005. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Moon J, Ota KT, Driscoll LL, Levitsky DA, Strupp BJ. A mouse model of fragile X syndrome exhibits heightened arousal and/or emotion following errors or reversal of contigencies. Dev Psychobiol. 2008;50:473–485. doi: 10.1002/dev.20308. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Moressis A, Friedrich AR, Pavlopoulos E, Davis RL, Skoulakis EM. A dual role for the adaptor protein DRK in Drosophila olfactory learning and memory. J Neurosci. 2009;29:2611–2625. doi: 10.1523/JNEUROSCI.3670-08.2009. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan U, Nalavadi V, Nakamoto M, Thomas G, Ceman S, Bassell GJ, Warren ST. S6K1 phosphorylates and regulates fragile X mental retardation protein (FMRP) with the neuronal protein synthesis-dependent mammalian target of rapamycin (mTOR) signaling cascade. J Biol Chem. 2008;283:18478–18482. doi: 10.1074/jbc.C800055200. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nighorn A, Healy MJ, Davis RL. The cyclic AMP phosphodiesterase encoded by the Drosophila dunce gene is concentrated in the mushroom body neuropil. Neuron. 1991;6:455–467. doi: 10.1016/0896-6273(91)90253-v. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- O'Donnell WT, Warren ST. A decade of molecular studies of fragile X syndrome. Annu Rev Neurosci. 2002;25:315–338. doi: 10.1146/annurev.neuro.25.112701.142909. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Pan L, Woodruff E, 3rd, Liang P, Broadie K. Mechanistic relationships between Drosophila fragile X mental retardation protein and metabotropic glutamate receptor A signaling. Mol Cell Neurosci. 2008;37:747–760. doi: 10.1016/j.mcn.2008.01.003. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panneels V, Eroglu C, Cronet P, Sinning I. Pharmacological characterization and immunoaffinity purification of metabotropic glutamate receptor from Drososphila overexpressed in Sf9 cells. Protein Expr Purif. 2003;30:275–282. doi: 10.1016/s1046-5928(03)00100-1. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Parmentier ML, Pin JP, Bockaert J, Grau Y. Cloning and functional expression of a Drosophila metabotropic glutamate receptor expressed in the embryonic CNS. J Neurosci. 1996;16:6687–6694. doi: 10.1523/JNEUROSCI.16-21-06687.1996. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual A, Préat T. Localization of long-term memory within the Drosophila mushroom body. Science. 2001;294:1115–1117. doi: 10.1126/science.1064200. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Pavlopoulos E, Anezaki M, Skoulakis EM. Neuralized is expressed in the α/β lobes of adult Drosophila mushroom bodies and facilitates olfactory long-term memory formation. Proc Natl Acad Sci U S A. 2008;105:14674–14679. doi: 10.1073/pnas.0801605105. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirozzi F, Tabolacci E, Neri G. The FRAXopathies: definition, overview, and update. Am J Med Genet A. 2011;155A:1803–1816. doi: 10.1002/ajmg.a.34113. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Repicky S, Broadie K. Metabotropic glutamate receptor-mediated use-dependent down-regulation of synaptic excitability involves the fragile X mental retardation protein. J Neurophysiol. 2009;101:672–687. doi: 10.1152/jn.90953.2008. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi JA, Huber KM. Metabotropic glutamate receptors and fragile X mental retardation protein: partners in translational regulation at the synapse. Sci Signal. 2008;1:pe6. doi: 10.1126/stke.15pe6. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Silvestre JS, Fernández AG, Palacios JM. Effects of rolipram on the elevated plus-maze test in rats: a preliminary study. J Psychopharmacol. 1999;13:274–277. doi: 10.1177/026988119901300309. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Siomi MC, Higashijima K, Ishizuka A, Siomi H. Casein kinase II phosphorylates the fragile X mental retardation protein and modulates its biological properties. Mol Cell Biol. 2002;22:8438–8447. doi: 10.1128/MCB.22.24.8438-8447.2002. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoulakis EM, Grammenoudi S. Dunces and da Vincis: the genetics of learning and memory in Drosophila. Cell Mol Life Sci. 2006;63:975–988. doi: 10.1007/s00018-006-6023-9. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoulakis EM, Kalderon D, Davis RL. Preferential expression in mushroom bodies of the catalytic subunit of protein kinase A and its role in learning and memory. Neuron. 1993;11:197–208. doi: 10.1016/0896-6273(93)90178-t. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Smith KT, Nicholls RD, Reines D. The gene encoding the fragile X RNA-binding protein is controlled by nuclear respiratory factor 2 and the CREB family of transcription factors. Nucleic Acids Res. 2006;34:1205–1215. doi: 10.1093/nar/gkj521. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson MS, Orr HT. Fragile X tremor/ataxia syndrome: blame the messenger. Neuron. 2007;55:535–537. doi: 10.1016/j.neuron.2007.07.032. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Tsiouris JA, Brown WT. Neuropsychiatric symptoms of fragile X syndrome: pathophysiology and pharmacotherapy. CNS Drugs. 2004;18:687–703. doi: 10.2165/00023210-200418110-00001. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Tully T, Quinn WG. Classical conditioning and retention in normal and mutant Drosophila melanogaster. J Comp Physiol A. 1985;157:263–277. doi: 10.1007/BF01350033. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Van Dam D, D'Hooge R, Hauben E, Reyniers E, Gantois I, Bakker CE, Oostra BA, Kooy RF, De Deyn PP. Spatial learning, contextual fear conditioning and conditioned emotional response in Fmr1 knockout mice. Behav Brain Res. 2000;117:127–136. doi: 10.1016/s0166-4328(00)00296-5. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Wan L, Dockendorff TC, Jongens TA, Dreyfuss G. Characterization of dFMR1, a Drosophila melanogaster homolog of the fragile X mental retardation protein. Mol Cell Biol. 2000;20:8536–8547. doi: 10.1128/mcb.20.22.8536-8547.2000. CrossRef Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wu LJ, Kim SS, Lee FJ, Gong B, Toyoda H, Ren M, Shang YZ, Xu H, Liu F, Zhao MG, Zhuo M. FMRP acts as a key messenger for dopamine modulation in the forebrain. Neuron. 2008;59:634–647. doi: 10.1016/j.neuron.2008.06.027. CrossRef Medline. [DOI] [PubMed] [Google Scholar]
- Yang MY, Wang Z, MacPherson M, Dow JA, Kaiser K. A novel Drosophila alkaline phosphatase specific to the ellipsoid body of the adult brain and the lower Malpighian (renal) tubule. Genetics. 2000;154:285–297. doi: 10.1093/genetics/154.1.285. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarnescu DC, Shan G, Warren ST, Jin P. Come FLY with us: toward understanding fragile X syndrome. Genes Brain Behav. 2005;4:385–392. doi: 10.1111/j.1601-183X.2005.00136.x. CrossRef Medline. [DOI] [PubMed] [Google Scholar]