

Abstract
The ability of neurons to modulate synaptic strength underpins synaptic plasticity, learning and memory, and adaptation to sensory experience. Despite the importance of synaptic adaptation in directing, reinforcing, and revising the behavioral response to environmental influences, the cellular and molecular mechanisms underlying synaptic adaptation are far from clear. Brain-derived neurotrophic factor (BDNF) is a prime initiator of structural and functional synaptic adaptation. However, the signaling cascade activated by BDNF to initiate these adaptive changes has not been elucidated. We have previously shown that BDNF activates mitogen- and stress-activated kinase 1 (MSK1), which regulates gene transcription via the phosphorylation of both CREB and histone H3. Using mice with a kinase-dead knock-in mutation of MSK1, we now show that MSK1 is necessary for the upregulation of synaptic strength in response to environmental enrichment in vivo. Furthermore, neurons from MSK1 kinase-dead mice failed to show scaling of synaptic transmission in response to activity deprivation in vitro, a deficit that could be rescued by reintroduction of wild-type MSK1. We also show that MSK1 forms part of a BDNF- and MAPK-dependent signaling cascade required for homeostatic synaptic scaling, which likely resides in the ability of MSK1 to regulate cell surface GluA1 expression via the induction of Arc/Arg3.1. These results demonstrate that MSK1 is an integral part of a signaling pathway that underlies the adaptive response to synaptic and environmental experience. MSK1 may thus act as a key homeostat in the activity- and experience-dependent regulation of synaptic strength.
Introduction
A prominent theme in neurobiology is the dramatic extent to which synapses are modified by experience (Rosenzweig and Bennett, 1996; Markham and Greenough, 2004). At the cellular level, these modifications underpin synaptic plasticity (Citri and Malenka, 2008; Ho et al., 2011), and in the whole animal support learning and memory (Morris, 2006; Neves et al., 2008) and its response to the environment (Nithianantharajah and Hannan, 2006; Baroncelli et al., 2010). These changes involve both functional and structural adaptations in terms of intrinsic excitability (Davis, 2006), postsynaptic glutamate receptor complement (Kerchner and Nicoll, 2008; Kessels and Malinow, 2009), neurotransmitter release (Citri and Malenka, 2008), and in the shape and density of dendritic spines, axonal arbors, and synaptic boutons (Holtmaat and Svoboda, 2009; Hübener and Bonhoeffer, 2010).
Brain-derived neurotrophic factor (BDNF) has emerged as a prime mediator of synaptic adaptation and plasticity. Increased BDNF mRNA and protein have been observed in rodents in response to environmental enrichment (Nithianantharajah and Hannan, 2006; Baroncelli et al., 2010), which, via the BDNF TrkB receptor, promotes dendritic growth and increased spine density in an ERK1/2-dependent manner (Cowansage et al., 2010). BDNF is also implicated in a form of synaptic adaptation known as homeostatic synaptic scaling in which suppression of neuronal firing in vitro by tetrodotoxin (TTX) (Turrigiano et al., 1998) causes a reduction in BDNF levels and a compensatory increase in synaptic strength, an observation that can be mimicked by BDNF TrkB receptor antagonists (Rutherford et al., 1998). This form of non-Hebbian plasticity may underlie the ability of neuronal networks to “self-tune” and maintain stable outputs in the face of LTP and LTD-driven Hebbian synaptic changes, while still allowing relative differences in synaptic strength to persist and continue to encode information (Turrigiano, 2008; Pozo and Goda, 2010).
In neurons, BDNF stimulates the ERK1/2 MAP kinase (MAPK) cascade, resulting in the activation of mitogen- and stress-activated protein kinase 1 (MSK1) (Arthur, 2008; Frenguelli and Corrêa, 2012). We have previously shown that MSK1 is required for MAPK-induced CREB and histone H3 phosphorylation, and the induction of Cre-dependent immediate-early genes (Soloaga et al., 2003; Arthur et al., 2004). Subsequently, MSK1 has been implicated in circadian clock gene expression (Butcher et al., 2005), visual cortical plasticity (Putignano et al., 2007), drug addiction (Brami-Cherrier et al., 2005), neurodegeneration (Roze et al., 2008), and learning and memory (Chwang et al., 2007; Chandramohan et al., 2008).
In the present study, we demonstrate that mice carrying an inactivating kinase-dead (KD) knock-in point mutation of the MSK1 N-terminal kinase domain fail to show synaptic adaptation in response to either environmental enrichment in vivo or chronic deprivation of synaptic activity in vitro. Furthermore, MSK1 KD neurons fail to downregulate activity-regulated cytoskeleton-associated protein/activity-regulated gene 3.1 (Arc/Arg3.1), a protein implicated in the trafficking of AMPA receptors in homeostatic synaptic plasticity (Chowdhury et al., 2006; Shepherd et al., 2006). Our data implicate MSK1 as key regulator of both synaptic and experience-dependent synaptic adaptation, actions that likely revolve around the ability of MSK1 to directly influence transcription via its downstream targets CREB and histone H3.
Materials and Methods
Materials.
The following antibodies (source; catalog number) were used in this study: Arc/Arg3.1 (Synaptic Systems; 156 003), GluA2 N-terminal (Santa Cruz; sc-7611), GAPDH (Abcam; AB8245), total ERK1/2 (Cell Signaling Technology; 9102), phospho-ERK1/2 (Cell Signaling Technology; 9101), total MSK1 [in-house (Wiggin et al., 2002)], phospho-Thr581 MSK1 (Cell Signaling Technology; 9595), and total RSK2 (Cell Signaling Technology; 9340). The GluA1 N-terminal (NT) antibody was a kind gift from Prof. Elek Molnár (University of Bristol, Bristol, UK) (Molnár et al., 1993; Richmond et al., 1996). The GFP-MSK1 plasmid has been described previously (Soloaga et al., 2003).
Animals.
All animal experiments were conducted in accordance with the United Kingdom Animals (Scientific Procedures) Act 1986, and with local ethical review procedures. A kinase-dead MSK1 mouse was generated by mutating Asp194 to Ala in the endogenous MSK1 gene. The knock-in was produced by TaconicArtemis using standard targeting methods in C57BL/6 ES cells using the strategy summarized in Figure 1A. Routine genotyping was performed by PCR using the primers 5′-CGGCCATGTGGTGCTGACAGC-3′ and 5′-GGGTCAGAGGCCTGCACTAGG-3′, which gives 378 and 529 bp products for wild-type and targeted alleles, respectively. MSK1 knock-out (KO) mice have been described previously (Wiggin et al., 2002). Gross morphology of MSK1 KD and wild-type mouse brains was assessed with propidium iodide staining of permeabilized 40 μm brain sections.
Figure 1.

Characterization of MSK1 kinase dead mice. A, Schematic of the kinase-dead (KD) knock-in strategy. MSK1 KD mice were generated by mutating Asp194 in the MSK1 gene to Ala. C57BL/6 ES cells were modified using the targeting vector, and homologous recombination confirmed by Southern blotting. Targeted ES cells were injected into blastocysts to generate chimeric mice. Those chimeras that demonstrated germline transmission were crossed to Flp recombinase transgenic mice to excise the neo cassette. MSK1 KD mice were subsequently crossed away from the Flp transgene. B, Gross brain morphology as assessed using propidium iodide staining of parasagittal brain sections was similar in wild-type (top panels) and MSK1 KD (bottom panels) mice. An enlargement of the hippocampus is shown in the right panels. C, Protein was extracted from the hippocampus or cerebellum from wild-type, MSK1 KD, or MSK1 KO mice and immunoblotted for total MSK1 or ERK1/2. D, Embryonic fibroblasts were prepared from wild-type, MSK1 KD, or MSK1 KO mice. Following serum starvation for 16 h, cells were stimulated with 400 ng/ml PMA for 30 min as indicated. Levels of MSK1, p-T581 MSK1, p-ERK1/2, or total ERK1/2 were determined by immunoblotting. In C and D, the correct MSK1 band was confirmed with tissue from MSK1 KO mice. E, MSK1 activity was determined by immunoprecipitation kinase assays. The white bars represent unstimulated embryonic fibroblast lysates and gray bars PMA-stimulated cells. Error bars represent the SEM of four independent stimulations.
All the mice used in this study were on a C57BL/6 genetic background.
Cell culture.
Hippocampal cultures were prepared from P0–P1 mouse pups of either sex as described previously (Corrêa et al., 2009). The cells were plated onto 22 mm glass coverslips coated with poly-l-lysine and used after 15–18 d in vitro (DIV). Transfection of hippocampal cultures with wild-type GFP-MSK1 cDNA (Soloaga et al., 2003) for rescue experiments was performed using Lipofectamine 2000 (Invitrogen). Neurons were used 15–18 h after transfection. The hippocampal cultures were also used for immunocytochemistry, immunoblotting, and mEPSC recordings. Embryonic fibroblasts were isolated from day 13.5 embryos and used for kinase assays (Wiggin et al., 2002).
Immunocytochemistry, microscopy, and data analysis.
Surface expression of endogenous GluA1 and GluA2 subunits was measured in hippocampal neurons grown in vitro for 15–18 d and treated with either vehicle (dH2O) or 1 μm TTX (Ascent Scientific) for 24 h in their conditioned medium. After treatment, cells were washed three times with warm HEPES-buffered saline (HBS) of the following composition (in mm): 119 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 25 HEPES, 30 glucose at pH 7.2, fixed in warm 4% paraformaldehyde (PFA) in 0.1 m PBS, pH 7.4, for 5 min at room temperature and incubated with either rabbit anti-GluA1 N-terminal or mouse anti-GluA2 N-terminal antibodies (diluted in 10% BSA) for 80 min at room temperature. Coverslips were then washed with HBS and incubated with Alexa 568-labeled secondary antibodies (1: 100) and mounted on slides using Mowiol.
Immunofluorescent staining was observed using a 63× oil-immersion lens on an inverted Leica SP5 confocal microscope. Fluorophores were excited at 561 nm and emission from a single confocal plane was detected through a 560 nm long-pass filter. All cells were imaged as Z stacks using the same acquisition parameters. Images comprising a Z stack were exported using Leica Microsystem LAS AF6000 software as a TIFF file and processed using Adobe Photoshop 8.0. For each experimental condition, at least three cells were imaged and analyzed using hippocampal cultures from three different preparations. The quantification of the intensity of fluorescent puncta was performed with ImageJ software (http://rsb.info.nih.gov/ij/index.html). The images displaying the endogenous surface GluA1 and GluA2 protein were first calibrated according to the magnification at which they were acquired. A region of diffuse fluorescence near the cell body was selected as background and the threshold was set at twice this background level. Once the threshold was set, a dendrite arising from the cell body was selected using the brush tool and all the puncta marked using the default settings. The selection of individual puncta was then made by hand comparing the grayscale and color images. After correcting the background in the grayscale images, the pixel intensity of the selected puncta was calculated.
Dendritic spine analysis in hippocampal culture.
Cultured hippocampal neurons grown for 21 DIV from wild-type and MSK1 KD mice were transfected with an eGFP construct using Lipofectamine 2000 (Invitrogen) as described previously (Corrêa et al., 2009). After 15–18 h transfection, the cells were fixed using 4% PFA and mounted on coverslips using Mowiol. The cells were then imaged on an inverted SP5 Leica confocal microscope using a 100× oil-immersion lens. Spine morphology analysis was performed in Neuronstudio (Wearne et al., 2005; Rodriguez et al., 2008). The confocal images were exported as compressed Z stacks, opened in Neuronstudio and calibrated according to the magnification parameters for spine and dendrite measurements. The spines were identified manually while the length of the dendrites were traced automatically. The spine volume (in cubic micrometers), spine head width (in micrometers), and spine length (in micrometers) were measured, as well as the spine density per 10 μm of dendrite (n = 20–25 neurons from 3–4 wild-type and MSK1 KD primary neuronal preparations). Data were analyzed using one-way ANOVA.
Western blotting.
Dissociated hippocampal neurons from both wild-type and MSK1 KD mice were cultured for 15 DIV and treated with either vehicle (dH2O) or 1 μm TTX (Ascent Scientific) for 24 h in their conditioned medium. After incubation, cells were lysed using lysis buffer (which contained the following: 1 mm EDTA, 1 m Tris-HCl, pH 7.5, 1% Triton X-100, 1 mm Na3VO4, 50 mm NaF, 5 mm Na4P2O7, 0.27 m sucrose, 20% NaN3, 0.1% β-mercaptoethanol, and protease inhibitor mixture tablets), centrifuged, and protein separated using SDS-PAGE electrophoresis system. Proteins were then transferred onto Hybond-ECL membrane (GE Healthcare), and blots were incubated overnight at 4°C with primary antibodies including rabbit anti-Arc/Arg3.1 (1:1000) and mouse anti-GAPDH (1:5000). Blots were then incubated with appropriate secondary antibodies (affinity-purified HRP- and AP-conjugated anti-IgG; 1:20,000; Jackson ImmunoResearch) for 1 h at room temperature and developed using ECL reagents. The bands were analyzed using Fiji software (http://fiji.sc/wiki/index.php/Fiji). GAPDH was used as a loading control (n = 3 for both wild-type and MSK1 KD). Data were analyzed using one-way ANOVA. Significance was taken at p < 0.05.
Kinase assays.
Kinase assays in embryonic fibroblasts were performed as described previously (Wiggin et al., 2002). One unit of activity was that amount of enzyme that incorporates 1 nmol of phosphate into the peptide substrate in 1 min.
Whole-cell mEPSC recordings.
Whole-cell patch-clamp recordings were made from primary hippocampal cultures (15–18 DIV) and from CA1 neurons in 350 μm hippocampal slices [prepared from male mice as described previously for neocortical slices (Pankratov et al., 2007)] in the presence of 1 μm TTX and 100 μm picrotoxin and with patch pipettes (4 MΩ) filled with the following intracellular solution (in mm): 50 KCl, 55 K-gluconate, 10 NaCl, 10 HEPES, 2 MgATP, 10 EGTA, pH 7.35, at 29–31°C. The membrane potential was set at −80 mV. The series and the input resistances were 4–8 and 500–1300 MΩ, respectively, and varied by <20% in the cells accepted for analysis. Currents were monitored using an AxoPatch200B patch-clamp amplifier (Molecular Devices) filtered at 2 kHz and digitized at 4 kHz. Experiments were controlled by PCI-6229 data acquisition board (National Instruments) and WinWCP software (Strathclyde Electrophysiology Software).
Spontaneous mEPSCs recorded in hippocampal neurons were analyzed off-line using methods described previously (Pankratov and Krishtal, 2003; Pankratov et al., 2007). Briefly, inward transmembrane currents of amplitude >2 SD of baseline noise were selected for the initial detection of spontaneous events. Every single spontaneous event thereafter was analyzed within a 140 ms time window, and its amplitude and kinetics were determined by fitting a model curve with single exponential rise and decay phases. As a rule, mean square error of fit amounted to 5–20% of peak amplitude depending on the background noise. Whenever the error of fit exceeded 25%, spontaneous currents were discarded from further analysis. The amplitude distributions of spontaneous and evoked currents were analyzed with the aid of probability density functions and likelihood maximization techniques, as described previously (Pankratov and Krishtal, 2003; Pankratov et al., 2007). All amplitude distributions shown were calculated as probability density functions.
Environmental enrichment.
Environmental enrichment was provided via the rearing of wild-type and MSK1 KD mice in large cages (Ferplast Mini Duna Hamster Cage: 39 cm width × 55 cm length × 27 cm height) containing bedding and toys arranged on two levels. To provide novelty, toys were moved around and new toys introduced every few days. Two to three pregnant dams were placed in these cages to provide additional mothering (D'Amato et al., 2011) and larger groups for social interactions. At weaning (3 weeks), females were removed and the males remained in the enriched environment for a further 3–4 weeks until brain slices were prepared for electrophysiological and morphological analysis. Age-matched male mice maintained in standard housing [one dam (until 3 weeks), bedding, a cardboard tube, two to three mice] served as controls for environmental enrichment.
Analysis of dendritic spines after exposure to standard and enriched housing.
A flame-polished patch pipette coated with ∼0.5 mg of dextran-conjugated Alexa 488 (Invitrogen) was inserted into the CA1 pyramidal layer of a 350 μm hippocampal slice (prepared as per the electrophysiological recordings described above), which was then returned to an incubation chamber for 3 h to allow uptake and transport of the dye (Delaney, 2010). Analysis of Z stacks of fluorescent images of CA1 stratum radiatum from 20 μm sections yielded the number of dendritic spines per unit length of dendrite. Data were obtained from 55 dendrites and 2151 dendritic spines under enriched conditions (two MSK1 KD and three wild-type mice), and 35 dendrites and 1536 dendritic spines under standard housing conditions (three MSK1 KD and three wild-type mice).
Statistical analysis.
Data were analyzed using unpaired or paired t tests and ANOVA as appropriate. Significance was achieved at p < 0.05. Unless stated otherwise, data are presented as mean ± SEM.
Results
Generation and characterization of MSK1 kinase-dead mice
To examine the potential role of MSK1 in synaptic function, we generated an MSK1 KD knock-in mouse by mutating Asp194 to alanine in the N-terminal kinase domain DFG motif (Fig. 1A). These mice were viable and had no apparent developmental problems or obvious differences in brain structure (Fig. 1B). Despite the presence of MSK1 protein (Fig. 1C), and its ability to be phosphorylated by MAPK at the key threonine 581 residue following stimulation of mouse embryonic fibroblasts with the phorbol ester phorbol 12-myristate 13-acetate (PMA) (Fig. 1D), MSK1 KD mice lacked detectable MSK1 activity (Fig. 1E). Western blots of the related RSK2 kinase showed no compensatory increases between wild-type and MSK1 KD mice in either neonatal (P0; n = 5–6) or adult (∼3 months of age; n = 6 of each genotype) hippocampal tissue when expressed as a ratio of total ERK1/2 (data not shown).
MSK1 is required for synaptic adaptation to environmental enrichment
Given the role of MSK1 in transducing neurotrophin receptor activation into CREB phosphorylation (Arthur et al., 2004), its role in mediating the phosphorylation of histone H3 in peripheral tissue and neurons (Arthur, 2008; Frenguelli and Corrêa, 2012), and the importance of both neurotrophins and transcriptional regulation in developmental and experience-dependent adaptation (Cowansage et al., 2010), we hypothesized that MSK1 may be important in regulating adaptive, or homeostatic responses to sensory and synaptic stimulation.
Accordingly, we exposed wild-type and MSK1 KD mice to environmental enrichment from birth to 6–8 weeks, at which point we studied synaptic transmission and dendritic spine density in hippocampal CA1 neurons from mice housed under enriched conditions compared with mice reared in standard housing.
In keeping with previous observations of the effects of environmental enrichment on synaptic transmission (Nithianantharajah and Hannan, 2006), we observed an increase in the amplitude of miniature EPSCs (mEPSCs) in wild-type mice exposed to environmental enrichment. This was evidenced by a clear rightward shift in both the amplitude distribution of individual mEPSCs (Fig. 2A, left panel) and the cumulative distribution of mEPSC amplitudes (Fig. 2B, left panel), resulting in an increase in the mean amplitude of mEPSCs (standard housing: 12.2 ± 1.3 pA, n = 10; enriched environment: 18.3 ± 2.1 pA, n = 16; unpaired t test, p < 0.05; Fig. 2C). In contrast, mEPSCs from MSK1 KD mice exposed to environmental enrichment were no different in their amplitude distribution (Fig. 2A,B, right panels) or mean amplitude to those recorded from MSK1 KD mice raised in standard housing (standard housing: 10.7 ± 1.4 pA, n = 10; enriched environment: 10.9 ± 1.2 pA, n = 15; Fig. 2C).
Figure 2.

MSK1 regulates environmental enrichment-induced synaptic upscaling in vivo. Electrophysiological analysis of mEPSCs revealed an enriched environment-induced enhancement of hippocampal synaptic transmission in wild-type, but not MSK1 KD mice. A, Top traces, Examples of mEPSCs from wild-type (left) and MSK1 KD neurons (right) in standard (left traces) and enriched environments (right traces) and, bottom graphs, corresponding amplitude distributions recorded in the same neurons. Note that the unitary quantal size, indicated by the position of the main peak in the amplitude distribution, undergoes a considerable increase, suggesting a postsynaptic effect of environmental enrichment in wild-type, but not MSK1 KD mice. Calibration: 20 ms, 5 pA. B, Cumulative distribution of mEPSC amplitude in standard housing (solid line ± SD in gray) and enriched environment (broken line ± SD in gray) in wild-type (left) and MSK1 KD neurons (right). C, Mean mEPSC amplitude across the two genotypes and housing conditions showing an increase (*p < 0.05) in wild-type, but not MSK1 KD mice. Data reflect measurements from between 10 and 16 neurons from four to six mice. Error bars indicate SEM.
To determine whether environmental enrichment was associated with structural as well as functional neuronal changes, we labeled CA1 neurons in hippocampal slices with a fluorescent dextran (Fig. 3A). Confocal imaging of dendritic spines in stratum radiatum revealed that environmental enrichment significantly increased spine density in wild-type neurons from 0.97 ± 0.08 to 1.64 ± 0.08 spines/μm (n = 3 mice for each condition; p < 0.001; Fig. 3B). In contrast, the increase in spine density on MSK1 KD neurons was blunted, increasing from 1.21 ± 0.08 spines/μm under standard housing to 1.50 ± 0.09 spines/μm following environmental enrichment (n = 2–3 MSK1 KD mice for each condition; p < 0.05; Fig. 3B), a value that was not different from the spine density in enriched wild-type mice.
Figure 3.

MSK1 regulates environmental enrichment-induced spine growth. A, Fluorescent dextran labeling of dendrites and spines in stratum radiatum of area CA1 in standard housing (top images) and enriched environment (bottom images) in wild-type (left) and MSK1 KD neurons (right). B, Spine density was significantly increased in wild-type mice (***p < 0.001) following environmental enrichment, but to a smaller extent in MSK1 KD (*p < 0.05), which showed increased basal spine density (+p = 0.044). Data reflect measurements on 12–27 dendrites from two to three mice for each condition. Scale bar, 10 μm. Error bars indicate SEM.
Interestingly, we observed a small, but significant increase in basal spine density in the MSK1 KD neurons (1.21 ± 0.08 spines/μm) compared with wild-type neurons (0.97 ± 0.08 spines/μm; p = 0.044). This potentially reflects an increase in spine size in the MSK1 KD mutant neurons allowing greater access of the high-molecular-weight 10 kDa fluorescently labeled dextran. To examine this possibility in more detail we prepared primary hippocampal neuronal cultures from neonatal (P0–P1) wild-type and MSK1 KD mice and transfected them with eGFP after 21 DIV (Fig. 4). Although these studies revealed that there were no obvious differences in spine head width, spine length, or spine density (Fig. 4A–C, respectively), there was an increase in the overall spine volume in neurons prepared from MSK1 KD mice (Fig. 4D), potentially explaining the increased spine density in fluorescent dextran-treated CA1 neurons (Fig. 3).
Figure 4.

MSK1 regulates spine volume. Top panels, Representative regions of eGFP-transfected dendrites in hippocampal neurons cultured for 21 DIV from wild-type (WT) (left) and MSK1 KD (KD) (right) mice. Scale bar, 10 μm. No significant differences were observed in spine head width (A), in spine length (B), or in the density of spines (C). However, an increase in spine volume (D) was observed between wild-type (black line) and MSK1 KD (gray line) neurons (p < 0.001). Data from 20–25 neurons from three to four wild-type and MSK1 KD primary neuronal preparations are shown. Error bars indicate SEM.
MSK1 is required for homeostatic synaptic plasticity
Given our observation of the importance of MSK1 in experience-dependent synaptic plasticity (Figs. 2, 3), and previous reports of the role of BDNF in the induction of homeostatic synaptic scaling in vitro (Turrigiano, 2008; Pozo and Goda, 2010), we hypothesized that MSK1 might regulate the adaptive synaptic response to activity deprivation.
Accordingly, we exposed primary hippocampal neurons from wild-type and MSK1 KD mice to 1 μm TTX for 24 h, a manipulation that is known to inhibit BDNF release and results in a homeostatic upregulation of excitatory synaptic transmission (Rutherford et al., 1998). Wild-type neurons showed a robust rightward shift in the individual (Fig. 5A, left panel) and cumulative (Fig. 5B, left panel) amplitude distributions of mEPSCs and an increase in the mean amplitude of mEPSCs following application of TTX (mean amplitude from 13.9 ± 0.8 to 29.0 ± 2.8 pA; n = 21 and 16 cells, respectively; p < 0.005; Fig. 5C). In stark contrast, hippocampal neurons from MSK1 KD mice, while demonstrating larger basal mEPSC amplitude, showed no change in the distribution of mEPSC amplitudes (Fig. 5A,B, right panels), or in the mean amplitude of mEPSCs in response to TTX (17.8 ± 1.1 to 17.3 ± 1.6 pA; n = 14 and 10 cells, respectively; Fig. 5C).
Figure 5.
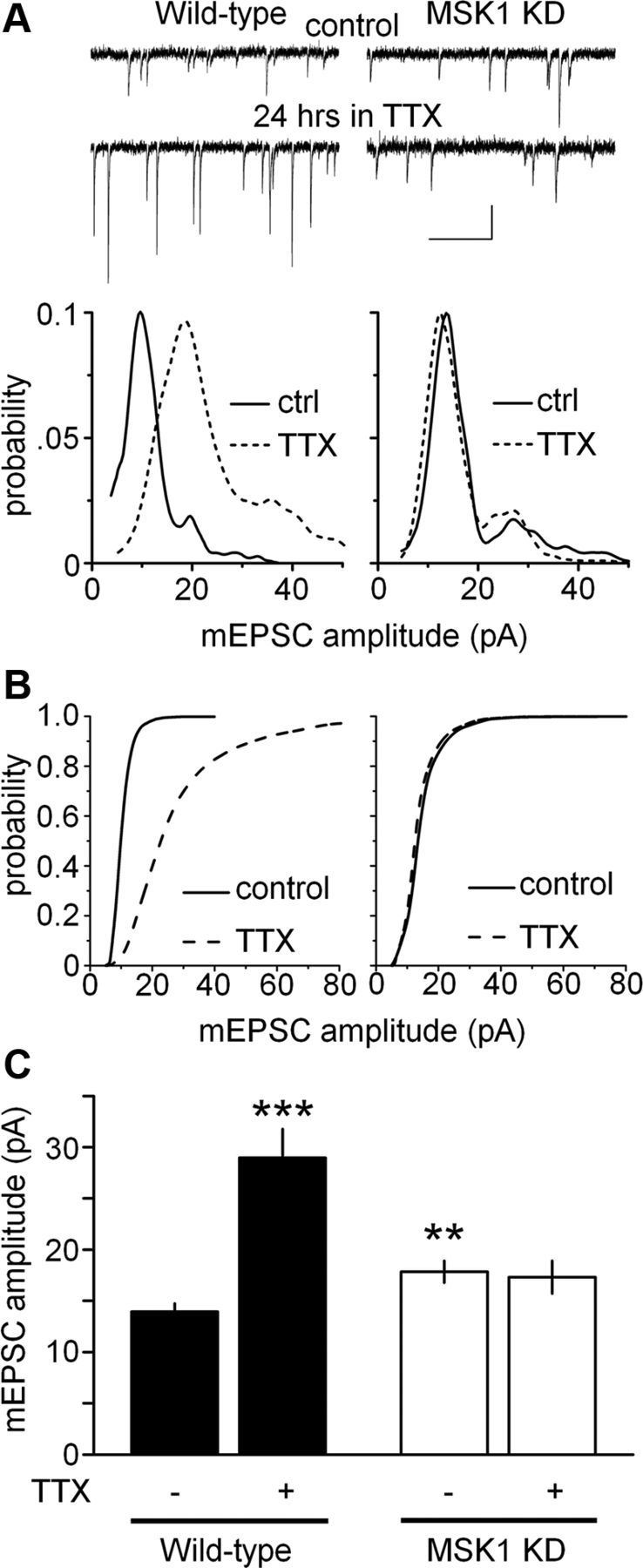
MSK1 is required for homeostatic synaptic scaling of mEPSCs in hippocampal neurons. A, Top traces, Representative mEPSCs from wild-type (left) and MSK1 KD (right) neurons in control (top traces) and after 24 h in 1 μm TTX (bottom traces). Calibration: 20 pA, 500 ms. Bottom graphs, The amplitude distributions of individual mEPSCs recorded from the neurons presented in the top panels showing an increase in quantal amplitude in wild-type, but not MSK1 KD neurons. B, Mean cumulative distributions of mEPSC amplitude for wild-type (left) and MSK1 KD neurons (right) in control (solid line) and TTX-treated conditions (broken line). C, Bar graph of mean mEPSC amplitude across the two genotypes (black, wild type; white, MSK1 KD) and in control (−) and TTX-treated (+) primary hippocampal cultures. Data are presented as mean ± SEM and are taken from between 7 and 21 cells from three to five independent preparations. **p < 0.01 and ***p < 0.005, when compared with wild type in the absence of TTX.
To confirm the dependence of these changes on MSK1, we transfected GFP-tagged wild-type MSK1 cDNA into dissociated wild-type neurons or neurons from either MSK1 KD or KO mice (Fig. 6). Transfection of GFP alone had no effect on mEPSC amplitude compared with wild-type control neurons [13.3 ± 0.8 pA (n = 6) vs 13.7 ± 1.3 pA (n = 6), respectively] (data not shown). Overexpression of wild-type MSK1 in wild-type neurons caused a significant decrease in the amplitude of mEPSCs (10.4 ± 0.7 pA; n = 8; p < 0.02 when compared with untransfected and GFP-transfected neurons) (data not shown). This is consistent with the loss of MSK1 kinase activity in the MSK1 KD mutants causing an increase in mEPSC amplitude (Fig. 5) and furthermore implicates MSK1 as an important regulator of basal synaptic strength.
Figure 6.
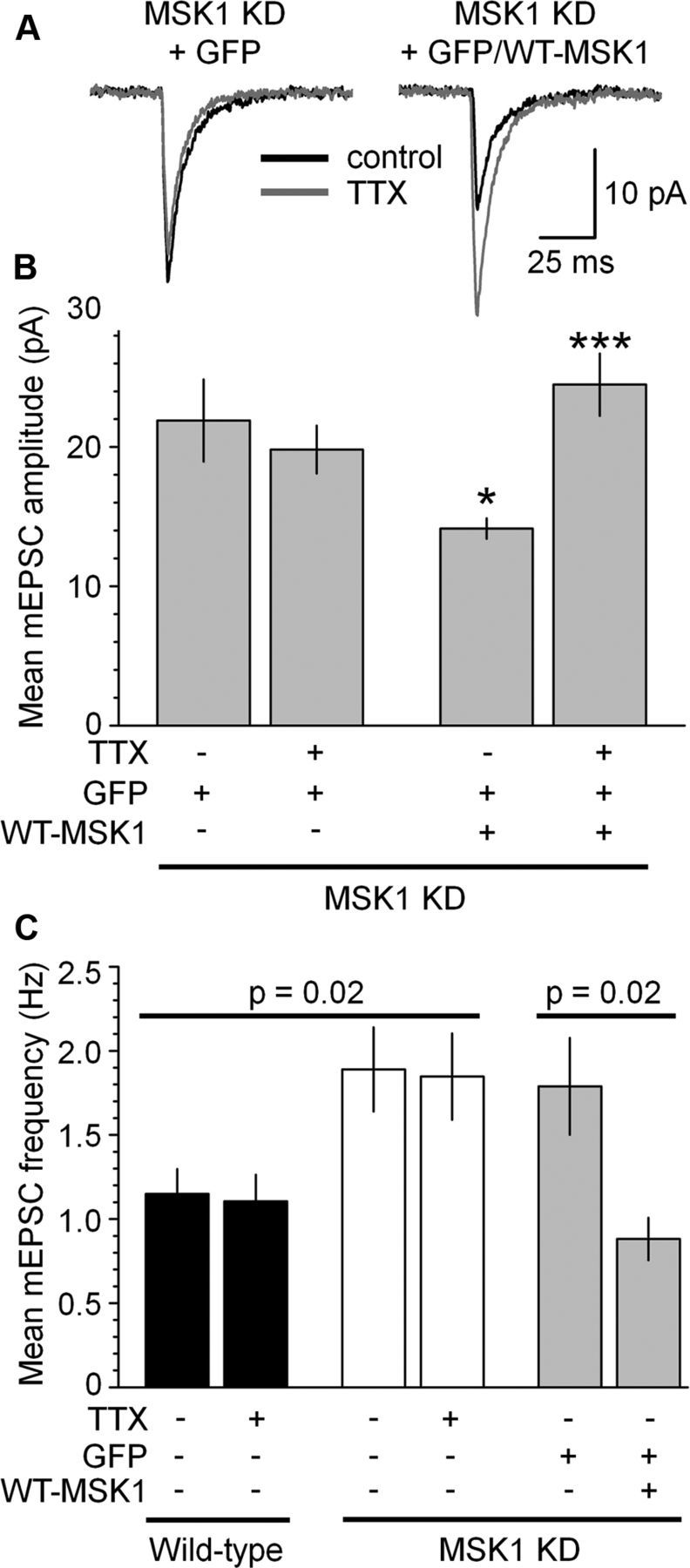
Rescue of mEPSC amplitude, frequency, and synaptic scaling by wild-type MSK1 in MSK1 mutant neurons. A, mEPSCs recorded in the absence (black traces) and after exposure to TTX (gray traces) from GFP-transfected MSK1 KD neurons (left) and MSK1 KD neurons transfected with GFP-tagged wild-type MSK1 (right). B, Pooled data of mean mEPSC amplitude from MSK1 mutant neurons in control conditions (GFP-transfected) and after the introduction of GFP-tagged wild-type MSK1, in the absence or presence of TTX. C, Pooled data of mEPSC frequency in wild-type (black bars), MSK1 KD (white bars), and MSK1 KD neurons transfected with GFP or GFP-tagged wild-type MSK1 (gray bars). mEPSC frequency is not affected by synaptic scaling induced by TTX in wild-type neurons (despite the increase in mEPSC amplitude; Fig. 5) but is increased in basal conditions in MSK1 KD neurons (white bars) or GFP-transfected MSK1 KD neurons (gray bars). Reintroduction of wild-type MSK1 into MSK1 KD neurons (gray bars; + WT-MSK1) reverses the increase in basal mEPSC frequency. Since there was no difference between MSK1 KD and KO neurons, data from the two genotypes were pooled in B and C. Shown are data from 6–21 neurons from three to five preparations. *p < 0.05 compared to GFP-transfected MSK1 KD neurons; ***p < 0.005 compared to GFP/WT-MSK1-transfected MSK1 KD neurons in the absence of TTX. Error bars represent SEM.
Reintroduction of wild-type MSK1 into dissociated MSK1 mutant neurons rescued the MSK1 KD phenotype (Fig. 6A) and resulted in a return of basal mEPSC amplitude to that of wild-type neurons (from 21.9 ± 3.0 to 14.1 ± 0.7 pA; n = 8 and 7 cells, respectively; p < 0.05; Fig. 6B) and furthermore permitted TTX-induced homeostatic synaptic upscaling (from 14.1 ± 0.7 to 24.5 ± 2.2 pA; data from 7 cells; p < 0.005; Fig. 6B).
In addition to the effects on the amplitude of mEPSCs, we observed an increase in basal mEPSC frequency in neurons from MSK1 KD mice compared with wild-type (1.89 ± 0.25 and 1.15 ± 0.15 Hz, respectively; Fig. 6C), which was not influenced by TTX treatment in either MSK1 KD or wild-type neurons (1.85 ± 0.26 and 1.10 ± 0.16 Hz, respectively; Fig. 6C). Transfection of MSK1 into wild-type neurons had no significant effect on mEPSC frequency [from 1.18 ± 0.12 Hz (12 GFP-transfected and untransfected cells) to 0.92 ± 0.11 Hz (8 wild-type MSK1-transfected cells)] (data not shown), potentially due to a floor effect on mEPSC frequency. In contrast, reintroduction of wild-type MSK1 into MSK1 KD neurons reversed the enhancement of basal mEPSC frequency observed in MSK1 mutant neurons (from 1.79 ± 0.29 to 0.88 ± 0.13 Hz; Fig. 6C).
These findings demonstrate that MSK1 is a key regulator of basal quantal amplitude and homeostatic scaling at individual synapses, but in addition has the capacity to regulate the number of active synapses.
Changes in GluA1 cell surface expression underlie MSK1-dependent homeostatic synaptic plasticity
Electrophysiological observations in cultured MSK1 KD neurons revealed an increase in both the amplitude (Fig. 5) and frequency (Fig. 6C) of basal mEPSCs. Given that homeostatic synaptic plasticity involves changes in the cell surface expression of glutamate AMPA receptor (AMPAR) subunits (O'Brien et al., 1998; Turrigiano, 2008; Pozo and Goda, 2010), we investigated the cell surface expression of two AMPAR subunits in wild-type and MSK1 KD neurons under basal conditions and following exposure to TTX.
Consistent with the increase in the amplitude and frequency of basal mEPSCs in MSK1 KD neurons, we observed an increase in the cell surface expression of the AMPAR GluA1 subunit in MSK1 KD neurons under control conditions (Fig. 7A), which was reflected in a rightward shift in the pixel intensity distribution (Fig. 7B), and in terms of the mean pixel intensity (Fig. 7C) when compared with wild-type neurons from parallel cultures.
Figure 7.
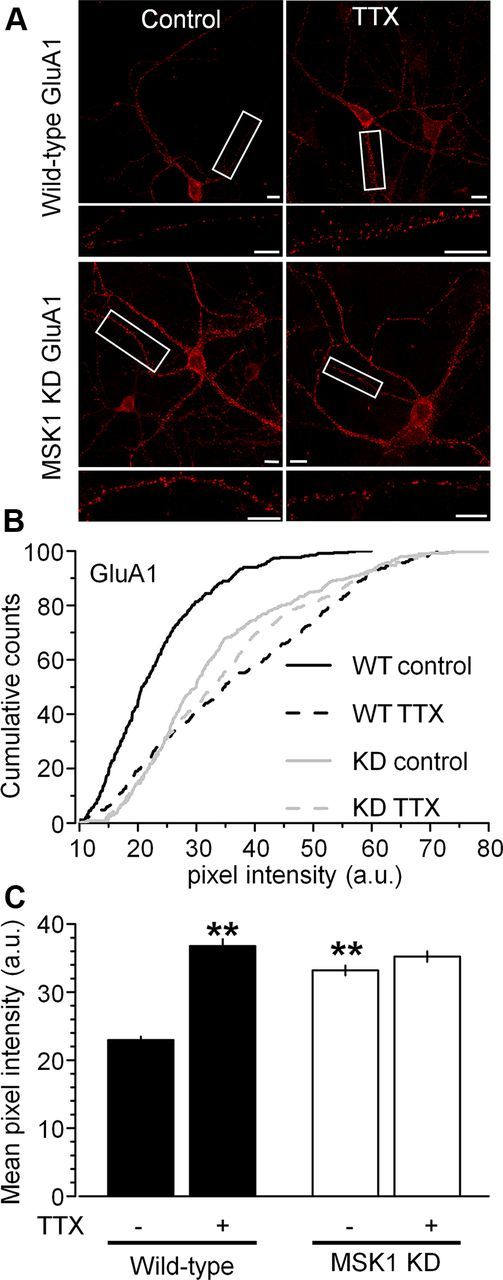
MSK1 is necessary for cell surface GluA1 expression during homeostatic synaptic upscaling in hippocampal neurons. A, Immunostaining of nonpermeabilized hippocampal neurons (15–18 DIV) with an antibody against the N-terminal of the GluA1 subunit showed increased cell surface expression of GluA1 subunits in wild-type neurons (top images) treated with TTX (1 μm) for 24 h (right panel) when compared with vehicle-treated control cells (left panel). However, no changes in GluA1 cell surface expression were observed between control and TTX-treated MSK1 KD neurons (bottom images). Note increased GluA1 staining in MSK1 KD neurons compared with wild-type neurons under control conditions. The boxed dendritic region is enlarged in the inset below the image of the neuron. Scale bars, 10 μm. B, Cumulative distributions of pixel intensity (in arbitrary units) showing rightward shift in TTX in wild-type, but not MSK1 KD neurons. Pixel intensity distribution is shifted to the right in MSK1 KD neurons under control conditions. C, Mean pixel intensity across the two genotypes (black, wild-type; white, MSK1 KD) and two experimental conditions (control and TTX). Data are presented as mean ± SEM and are taken from three cells from three independent preparations. **p < 0.01, when compared with wild-type neurons in the absence of TTX.
In keeping with the TTX-induced increase in mEPSC amplitude in wild-type neurons, TTX caused an upregulation of GluA1 cell surface expression (Fig. 7A), a rightward shift in pixel intensity (Fig. 7B), and an increase in the mean pixel intensity in wild-type neurons (from 23.0 ± 0.5 to 36.8 ± 1.0 a.u.; p < 0.01; Fig. 7C). In contrast, and consistent with the electrophysiological observations, TTX-treated MSK1 KD neurons showed no increase in GluA1 cell surface expression (Fig. 7A), pixel intensity distribution (Fig. 7B), or mean pixel intensity (from 33.2 ± 0.7 to 35.2 ± 0.8 a.u.; Fig. 7C).
To determine whether changes in cell surface AMPAR subunit expression were specific to GluA1 subunits or also involved GluA2 subunits, we performed similar immunocytochemical analysis of GluA2 cell surface expression. In contrast to our observations with the GluA1 subunit, there was no appreciable difference in basal expression of the GluA2 subunit between wild-type and MSK1 KD neurons, and no influence of TTX in either genotype (Fig. 8), suggesting that the effects of TTX and MSK1 are specific to the GluA1 subunit.
Figure 8.
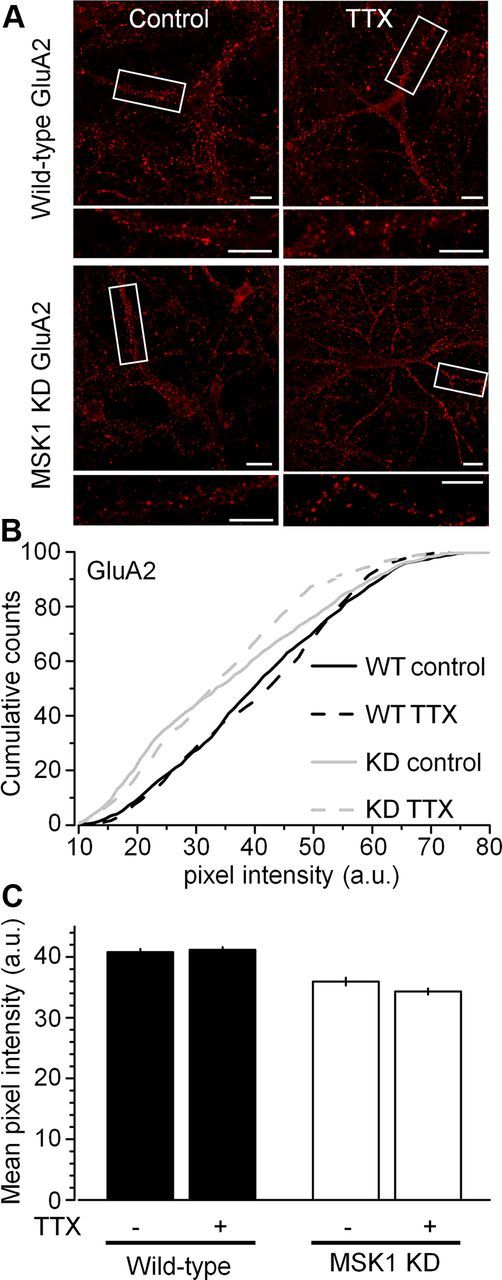
No influence of TTX or MSK1 on GluA2 surface expression. Immunostaining of nonpermeabilized hippocampal dissociated neurons (15–18 DIV) from wild-type and MSK1 KD mice with an antibody against the N-terminal of the GluA2 subunit showed no changes in GluA2 cell surface expression (A), no changes in pixel intensity distribution (B), and no change in mean pixel intensity (C) across genotype (black, wild type; white, MSK1 KD) or across TTX treatment (control and TTX). Scale bars, 10 μm. Error bars indicate SEM.
The BDNF/MAPK pathway regulates homeostatic synaptic plasticity via MSK1
Although several signaling molecules have been identified as mediating synaptic scaling (Turrigiano, 2008; Pozo and Goda, 2010), the MAPK cascade has yet to be examined. Since the BDNF-dependent activation of MSK1 requires ERK1/2 (Arthur et al., 2004), the synaptic scaling deficit in the MSK1 KD mice predicts a role for ERK1/2 in the homeostatic regulation of synaptic strength.
Accordingly, to confirm that the homeostatic synaptic scaling we observed in response to TTX was dependent on decreased BDNF levels and reduced activity of TrkB and ERK1/2, we incubated wild-type cultured neurons in BDNF (50 ng/ml), BMS-SH722 (SH722; 500 nm), a Trk inhibitor that has been shown to block BDNF-induced ERK1/2 activation (Martin et al., 2011), or the MEK1/2 inhibitor 2-[(2-chloro-4-iodophenyl)amino]-N-cyclopropylmethoxy)-3,4-difluorobenzamide (PD 184352; 2 μm), which prevents ERK1/2 activation (Fig. 9A). As predicted (Rutherford et al., 1998) BDNF reduced mEPSC amplitude in wild-type neurons (to 10.0 ± 1.0 pA; p < 0.05). In contrast, the TrkB and MEK1/2 inhibitors enhanced mEPSCs (23.5 ± 2.8 pA; p < 0.01 and 37.3 ± 6.0 pA; p < 0.005, respectively), identifying TrkB-mediated ERK1/2 activation as an initiator of homeostatic synaptic scaling.
Figure 9.
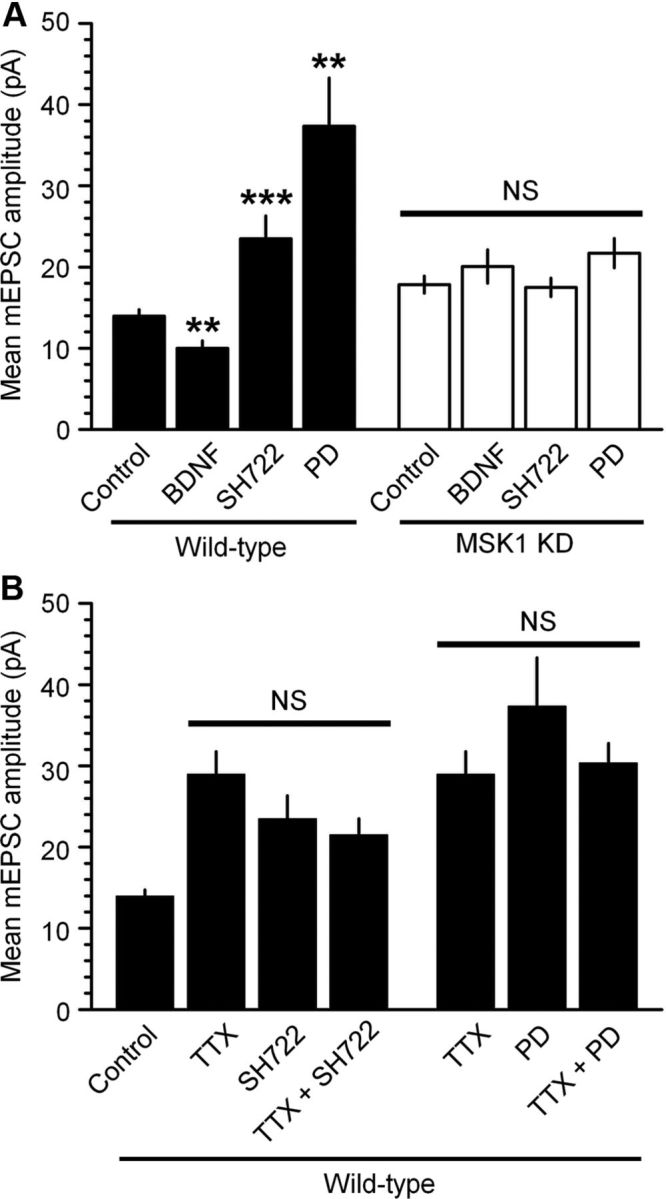
The BDNF/TrkB/MAPK pathway bidirectionally regulates homeostatic synaptic scaling via MSK1. A, Cultured hippocampal neurons from wild-type (black bars) or MSK1 KD (white bars) mice were incubated with BDNF (50 ng/ml), the TrkB inhibitor SH722 (500 nm), or the MEK1/2 inhibitor PD 184352 (PD; 2 μm), which blocks ERK1/2 activation, for 24 h, and mEPSCs were measured thereafter. Similar observations were made with a second, structurally unrelated TrkB inhibitor, BMS-SHN753 (Martin et al., 2011) (500 nm) (data not shown). Data are presented as mean ± SEM and are taken from between 6 and 21 cells from two to five independent preparations. NS, p > 0.05; **p < 0.01; ***p < 0.005. B, TTX- and BDNF-dependent synaptic scaling share a common intracellular signaling cascade. Dissociated hippocampal neurons from wild-type mice were exposed to TTX (1 μm) in the presence or absence of the TrkB inhibitor SH722 (500 nm) or the MEK1/2 inhibitor PD 184352 (PD; 2 μm) for 24 h and mEPSCs were measured using whole-cell patch clamping. While each manipulation resulted in a significant increase in mean mEPSC amplitude (p < 0.05), no additional synaptic scaling was observed when TTX was combined with either the BDNF TrkB receptor antagonist SH722 or the MEK inhibitor PD 184352.
To test whether these observations of the influence of the BDNF/TrkB/MAPK cascade in homeostatic synaptic involved MSK1, we repeated these experiments in neurons prepared from MSK1 KD mice. Neither application of BDNF, inhibition of TrkB receptors, nor inhibition of ERK1/2 activation affected mEPSC amplitude in MSK1 KD neurons (Fig. 9A). These data suggest the convergence of the BDNF/TrkB/MAPK signaling pathway upon MSK1, which ultimately influences synaptic strength.
To confirm that TTX- and MAPK-dependent synaptic scaling recruited a common pathway, we performed occlusion experiments in wild-type neurons, the rationale being that should two pathways cause synaptic scaling by independent means their effects will be additive, while if they recruit a common mechanism, their effects will not be greater than each individually. Accordingly, coapplication of TTX and the TrkB inhibitor SH722 caused synaptic scaling no different to that evoked by each individually (Fig. 9B). Similarly, coapplication of TTX and the MEK1/2 inhibitor PD 184352 had no additional effect on synaptic scaling (Fig. 9B). These results are consistent with the hypothesis that TTX induces synaptic upscaling via the inhibition of BDNF release, with the concomitant reduction in activation of both TrkB receptors and the MAPK cascade, and that this can be recapitulated by inhibition of TrkB receptors (SH722) and MEK1/2 (PD 184352).
Arc/Arg3.1 as the molecular effector of MSK1-dependent homeostatic synaptic plasticity
Arc/Arg3.1 has risen to prominence as an important regulator of homeostatic synaptic scaling by virtue of its ability to influence cell surface AMPAR expression via interaction with the endocytotic proteins dynamin and endophilin (Chowdhury et al., 2006; Rial Verde et al., 2006; Peebles et al., 2010). Moreover, Arc/Arg3.1-dependent endocytosis of AMPAR is regulated by synaptic activity: suppression of synaptic activity with TTX causes a downregulation of Arc/Arg3.1, a reduction in AMPAR endocytosis, and results in an increase in synaptic strength (Shepherd et al., 2006). In addition, several reports have described the BDNF-dependent induction of Arc/Arg3.1 (Yin et al., 2002) and that this involves TrkB-mediated activation of the MAPK cascade (Ying et al., 2002; Alder et al., 2003; Rao et al., 2006).
To test whether Arc/Arg3.1 expression during synaptic scaling was regulated by MSK1, we exposed hippocampal cultures from wild-type and MSK1 KD neurons to TTX for 1 and 24 h and measured Arc/Arg3.1 protein expression using Western blotting (Fig. 10). TTX resulted in a time-dependent decrease in Arc/Arg3.1 protein expression in wild-type neurons (Fig. 10A), which reached statistical significance at the 24 h time point (Fig. 10B). In contrast, no such pattern was observed in neurons prepared from MSK1 KD mice (Fig. 10A,B), suggesting that Arc/Arg3.1 expression changes during homeostatic synaptic scaling require MSK1.
Figure 10.

TTX-induced changes in Arc/Arg3.1 expression require MSK1. A, Western blots showing Arc/Arg3.1 protein expression in control (C) and after 1 or 24 h TTX (1 μm) exposure in wild-type or MSK1 KD dissociated hippocampal neurons (14–18 DIV). B, Quantification of Western blots showing (*p < 0.05) time-dependent reduction in Arc/Arg3.1 expression in wild-type neurons (black bars), with no significant effect (NS) in the MSK1 KD neurons (white bars). n = 3 for each condition. Error bars represent SEM.
Discussion
Our observations implicate MSK1 as a key regulator of both activity- and experience-dependent synaptic adaptation. As such, MSK1 serves as a molecular homeostat regulating synaptic strength in the face of prevailing synaptic and sensory activity. MSK1, positioned between TrkB receptors and transcriptional regulation mediated by CREB and histone H3 phosphorylation, defines a MAPK-dependent signaling cascade capable of matching BDNF TrkB receptor activation with the appropriate induction of key plasticity-related proteins, such as Arc/Arg3.1 (Fig. 11).
Figure 11.
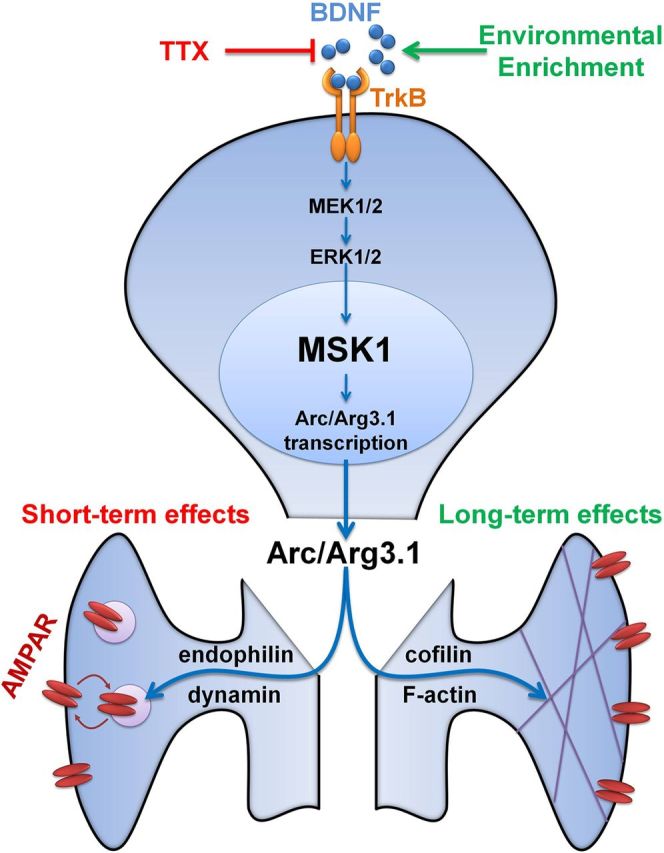
Working model for the role of MSK1 in homeostatic and experience-dependent synaptic scaling. BDNF stimulates the TrkB receptor that activates the ERK1/2 pathway, resulting in the activation of MSK1. MSK1 induces Arc/Arg3.1 transcription, potentially via the phosphorylation of one or both of its downstream targets, CREB and histone H3. Arc/Arg3.1, together with endophilin and dynamin, acutely regulates cell surface expression of AMPA GluA1 subunits, while interactions with cofilin and F-actin may result in longer term functional and morphological effects. TTX treatment, which blocks activity-dependent BDNF release, causes reduced MSK1 activation and a decrease in Arc/Arg3.1 induction. This results in reduced AMPAR endocytosis and therefore increased synaptic AMPAR levels and hence synaptic upscaling of mEPSC amplitudes. In the longer term, environmental enrichment, which increases levels of BDNF, may stimulate MSK1 and hence regulate Arc/Arg3.1 levels to influence the structure of dendritic spines and the complement of synaptic AMPA receptors to induce sensory experience-dependent morphological and functional plasticity.
Role of MSK1 in neuronal function
Following observations that MSK1 represents the predominant neurotrophin-activated CREB kinase in the mammalian brain (Arthur et al., 2004), and regulates histone H3 phosphorylation and chromatin remodeling during learning and memory (Chwang et al., 2007), MSK1 has been implicated in a range of neuronal processes ranging from circadian clock gene expression to neurodegeneration (Arthur, 2008; Frenguelli and Corrêa, 2012). This information has been largely gleaned from studies on MSK1 knock-outs, which have shown that MSK1 KOs have no discernible basal behavioral, sensory, or morphological abnormalities that might explain these observations (Frenguelli and Corrêa, 2012), nor do they have compensatory changes in related kinases such as RSK2 (Arthur et al., 2004).
However, the assumption in MSK1 knock-out studies is that phenotypes attributed to MSK1 reflect the loss of kinase activity rather than some structural aspect of MSK1 required for signaling. Given the recent observation that MSK1 may form a physical signaling complex with ERK1/2 and glucocorticoid receptor (Gutièrrez-Mecinas et al., 2011), this assumption may not hold in all cases. Accordingly, to selectively eliminate the kinase activity of MSK1, we created a knock-in KD point mutation of MSK1 by mutating Asp194 to Ala in the endogenous MSK1 gene.
MSK1 KD mice were fertile, viable, and demonstrated no obvious behavioral or morphological brain abnormalities. While there were no changes in protein levels of the related RSK2, levels of MSK1 protein were somewhat reduced in hippocampus and cerebellum, which may reflect some instability in the mutated protein. Indeed, we have previously observed reduced expression of MSK1 protein in mutants affecting MSK1 kinase activity (McCoy et al., 2005, 2007). Accordingly, at this stage, we cannot attribute the synaptic and experience-dependent phenotype of the MSK1 KD mutant exclusively to a loss of kinase activity. However, the efficacy of the kinase-dead mutation was confirmed by the lack of MSK1 kinase activity in tissue from MSK1 KD mice, a finding consistent with that in tissue obtained from MSK1 KOs, and despite the fact that both upstream ERK1/2 signaling and the phosphorylation of MSK1 at the key T581 residue were intact in the kinase-dead mutant. Thus, despite the caveat of somewhat lower MSK1 expression, the MSK1 KD mutant represents an advance in the tools available to study the kinase role of MSK1 in the mammalian CNS.
MSK1 is required for synaptic enhancement in response to environmental enrichment
A considerable literature exists demonstrating the beneficial effects of enriched or complex environments on neuronal structure and function (Rosenzweig and Bennett, 1996; Nithianantharajah and Hannan, 2006), and a common theme is the importance of BDNF in these processes (Cowansage et al., 2010). In keeping with this body of evidence, we observed an increase in excitatory synaptic transmission and spine density in wild-type mice raised from birth in an enriched environment. In contrast, MSK1 KD mice showed no such increase in the amplitude of mEPSCs, while the increase in spine density was blunted. Of note, however, is the observation that there was a small increase in basal spine density in the MSK1 KD, which may have been due to preferential access of the fluorescent dextran to larger spines in the MSK1 mutant. Analysis of eGFP-labeled spines in wild-type and MSK1 KD cultured hippocampal neurons confirmed an increase in spine volume in the mutants, suggesting that MSK1 may regulate the size of dendritic spines, and that loss of MSK1 kinase activity favors larger spines.
Interestingly, an increase in the number of larger, mushroom spines was observed in the Arc/Arg3.1 KO (Peebles et al., 2010), while the visual cortex of Arc/Arg3.1 KO mice was unresponsive to sensory experience or deprivation (McCurry et al., 2010). Given the Arc/Arg3.1 deficit observed in MSK1 KD neurons in response to TTX treatment (Fig. 10), MSK1 may regulate spine morphology and the neuronal response to sensory experience via regulation of Arc/Arg3.1 expression. Arc/Arg3.1 has been suggested to influence spine morphology and plasticity via the cytoskeletal proteins cofilin and F-actin (Messaoudi et al., 2007; Bramham et al., 2010; Shepherd and Bear, 2011). It is therefore possible that reduced Arc/Arg3.1 expression and cofilin/F-actin interaction in the MSK1 KD mice may underlie the deficits in neuronal structure and function observed after environmental enrichment (Fig. 11).
MSK1 regulates homeostatic synaptic plasticity in vitro
Neurons inversely modulate synaptic strength in the face of prevailing synaptic activity. This phenomenon of homeostatic synaptic plasticity is regarded as a key compensatory mechanism to avoid runaway synaptic excitation or depression occasioned by activity-dependent Hebbian plasticity induced by LTP and LTD, and a number of candidate molecules have been implicated (Turrigiano, 2008; Pozo and Goda, 2010). We found that while wild-type neurons exhibited the expected enhanced synaptic response to activity deprivation (TTX), neurons from MSK1 KD mice did not, indicating that MSK1 kinase activity is necessary to translate synaptic activity into adaptive or homeostatic responses.
MSK1 KD neurons did, however, show an increase in basal synaptic transmission and in the frequency of mEPSCs. This likely reflects the selective increase in cell surface GluA1 staining observed in MSK1 KD neurons compared with wild-type neurons. These observations are also reminiscent of the findings in the Arc/Arg3.1 KO in which cultured hippocampal neurons showed greater GluA1 expression, larger basal mEPSCs, but no modulation of synaptic strength by TTX (Chowdhury et al., 2006; Shepherd et al., 2006). Subsequent studies have confirmed the specificity of the interaction between Arc/Arg3.1 and GluA1 (Peebles et al., 2010) and are consistent with the findings reported here that GluA1, but not GluA2, expression was influenced by TTX and MSK1.
To confirm that the electrophysiological phenotype of the MSK1 KD neurons was due to a loss of MSK1 kinase activity, we performed rescue experiments in which wild-type MSK1-GFP was transfected into MSK1 mutant neurons. This manipulation had three effects: it returned basal mEPSC amplitude to that of wild-type neurons, it returned mEPSC frequency to that of wild-type neurons, and third, it also restored the ability of neurons from MSK1 mutant mice to demonstrate homeostatic synaptic scaling in response to activity deprivation. Thus, MSK1 represents a key regulator of quantal amplitude, basal synaptic transmission, synaptic adaptation, and potentially the number of active synapses.
MSK1 as the apex of the BDNF/MAPK signaling cascade regulating synaptic homeostasis
BDNF has been shown to activate MSK1 via stimulation of TrkB receptors and activation of ERK1/2 (Arthur et al., 2004). To establish the involvement of both this pathway and MSK1 in synaptic scaling, we performed two sets of experiments: stimulation or inhibition of the BDNF/MAPK pathway in wild-type and MSK1 KD neurons, and occlusion experiments in wild-type neurons. In wild-type neurons, application of BDNF induced synaptic downscaling, while inhibition of either TrkB receptors or of the upstream ERK1/2 kinase, MEK1/2, caused synaptic upscaling. In contrast, these manipulations were without effect in MSK1 KD neurons, suggesting that MSK1 is an integral feature of the BDNF/MAPK pathway in regulating synaptic adaptation. To confirm that TTX-induced synaptic downscaling invoked a similar BDNF/MAPK-dependent pathway, we coapplied TTX with either the TrkB inhibitor SH722 or the MEK1/2 inhibitor (PD 184352). This did not result in additional synaptic scaling, suggesting the recruitment of a common pathway involving TrkB receptors and MAPK.
Arc/Arg3.1 as a mediator of MSK1-dependent homeostatic synaptic scaling
Arc/Arg3.1 has risen to prominence as a regulator of cell surface AMPA receptor expression via its ability to interact with the endocytotic proteins dynamin and endophilin (Chowdhury et al., 2006; Rial Verde et al., 2006; Peebles et al., 2010), as a mediator of homeostatic synaptic plasticity (Shepherd et al., 2006; Béïque et al., 2011), in shaping spine morphology (Peebles et al., 2010) and in LTP and learning and memory (Ying et al., 2002; Plath et al., 2006; Shepherd and Bear, 2011). BDNF has been reported to induce Arc/Arg3.1 expression via activation of TrkB and MAPK (Ying et al., 2002; Alder et al., 2003), and both BDNF and Arc/Arg3.1 have independently been implicated in the induction of homeostatic synaptic plasticity (Turrigiano, 2008; Pozo and Goda, 2010). Our observations suggest that MSK1 may serve as a mechanistic link between the activation of BDNF TrkB receptors and the induction of Arc/Arg3.1 expression, at least under conditions of activity deprivation, resulting in the appropriate compensatory changes in cell surface GluA1 expression.
In conclusion, we have shown that MSK1 occupies a prime position in the CNS in translating both synaptic activity and sensory experience into enduring forms of functional and structural plasticity. MSK1 may thus represent a signaling focus for neurotrophin-directed neuronal development and adaptation, and potentially a therapeutic target for the promotion of neuronal and synaptic growth and recovery.
Footnotes
This work was supported by the Medical Research Council (MRC) (R.G.M.M., B.G.F., J.S.C.A.), FP6 (Sirocco, J.S.C.A.), and Biotechnology and Biological Sciences Research Council (BBSRC) (Y.P., S.A.L.C.). S.A.L.C. is a Warwick University Research Fellow and is in receipt of Research Development Funding from the University of Warwick. S.C.W. was supported by a GlaxoSmithKline/BBSRC CASE PhD Studentship. C.J.H. was supported by a Wellcome Trust PhD Studentship. K.M. was supported by an Engineering and Physical Sciences Research Council/Molecular Organisation and Assembly in Cells PhD Studentship. We thank Prof. Nick Dale, Prof. Graham Collingridge, and Dr. Roberto Malinow for input on this manuscript, Prof. Elek Molnár (MRC Centre for Synaptic Plasticity, University of Bristol, Bristol, UK) for the N-terminal GluA1 antibody, Natalia Shpiro for the synthesis of the MEK1/2 and Trk inhibitors, Julia Carr for genotyping, and the staff of the Biomedical Services Units of Dundee and Warwick Universities for their excellent animal husbandry and colony management.
The authors declare no competing financial interests.
References
- Alder J, Thakker-Varia S, Bangasser DA, Kuroiwa M, Plummer MR, Shors TJ, Black IB. Brain-derived neurotrophic factor-induced gene expression reveals novel actions of VGF in hippocampal synaptic plasticity. J Neurosci. 2003;23:10800–10808. doi: 10.1523/JNEUROSCI.23-34-10800.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur JS. MSK activation and physiological roles. Front Biosci. 2008;13:5866–5879. doi: 10.2741/3122. [DOI] [PubMed] [Google Scholar]
- Arthur JS, Fong AL, Dwyer JM, Davare M, Reese E, Obrietan K, Impey S. Mitogen- and stress-activated protein kinase 1 mediates cAMP response element-binding protein phosphorylation and activation by neurotrophins. J Neurosci. 2004;24:4324–4332. doi: 10.1523/JNEUROSCI.5227-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroncelli L, Braschi C, Spolidoro M, Begenisic T, Sale A, Maffei L. Nurturing brain plasticity: impact of environmental enrichment. Cell Death Differ. 2010;17:1092–1103. doi: 10.1038/cdd.2009.193. [DOI] [PubMed] [Google Scholar]
- Béïque JC, Na Y, Kuhl D, Worley PF, Huganir RL. Arc-dependent synapse-specific homeostatic plasticity. Proc Natl Acad Sci U S A. 2011;108:816–821. doi: 10.1073/pnas.1017914108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR, Alme MN, Bittins M, Kuipers SD, Nair RR, Pai B, Panja D, Schubert M, Soule J, Tiron A, Wibrand K. The Arc of synaptic memory. Exp Brain Res. 2010;200:125–140. doi: 10.1007/s00221-009-1959-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brami-Cherrier K, Valjent E, Hervé D, Darragh J, Corvol JC, Pages C, Arthur SJ, Simon AJ, Girault JA, Caboche J. Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. J Neurosci. 2005;25:11444–11454. doi: 10.1523/JNEUROSCI.1711-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher GQ, Lee B, Cheng HY, Obrietan K. Light stimulates MSK1 activation in the suprachiasmatic nucleus via a PACAP-ERK/MAP kinase-dependent mechanism. J Neurosci. 2005;25:5305–5313. doi: 10.1523/JNEUROSCI.4361-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramohan Y, Droste SK, Arthur JS, Reul JM. The forced swimming-induced behavioural immobility response involves histone H3 phospho-acetylation and c-Fos induction in dentate gyrus granule neurons via activation of the N-methyl-d-aspartate/extracellular signal-regulated kinase/mitogen- and stress-activated kinase signalling pathway. Eur J Neurosci. 2008;27:2701–2713. doi: 10.1111/j.1460-9568.2008.06230.x. [DOI] [PubMed] [Google Scholar]
- Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron. 2006;52:445–459. doi: 10.1016/j.neuron.2006.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chwang WB, Arthur JS, Schumacher A, Sweatt JD. The nuclear kinase mitogen- and stress-activated protein kinase 1 regulates hippocampal chromatin remodeling in memory formation. J Neurosci. 2007;27:12732–12742. doi: 10.1523/JNEUROSCI.2522-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Malenka RC. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. 2008;33:18–41. doi: 10.1038/sj.npp.1301559. [DOI] [PubMed] [Google Scholar]
- Corrêa SA, Müller J, Collingridge GL, Marrion NV. Rapid endocytosis provides restricted somatic expression of a K+ channel in central neurons. J Cell Sci. 2009;122:4186–4194. doi: 10.1242/jcs.058420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowansage KK, LeDoux JE, Monfils MH. Brain-derived neurotrophic factor: a dynamic gatekeeper of neural plasticity. Curr Mol Pharmacol. 2010;3:12–29. doi: 10.2174/1874467211003010012. [DOI] [PubMed] [Google Scholar]
- D'Amato FR, Zanettini C, Sgobio C, Sarli C, Carone V, Moles A, Ammassari-Teule M. Intensification of maternal care by double-mothering boosts cognitive function and hippocampal morphology in the adult offspring. Hippocampus. 2011;21:298–308. doi: 10.1002/hipo.20750. [DOI] [PubMed] [Google Scholar]
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annu Rev Neurosci. 2006;29:307–323. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- Delaney KR. Loading neurons with dextran-conjugated calcium indicators in intact nervous tissue. Curr Protoc Neurosci. 2010;50:2.5.1–2.5.11. doi: 10.1002/0471142301.ns0205s20. [DOI] [PubMed] [Google Scholar]
- Frenguelli BG, Corrêa SA. Regulation and role of MSK in the mammalian brain. In: Vermeulen L, Arthur JS, editors. MSKs. Austin, TX: Landes Bioscience; 2012. in press. [Google Scholar]
- Gutièrrez-Mecinas M, Trollope AF, Collins A, Morfett H, Hesketh SA, Kersanté F, Reul JM. Long-lasting behavioral responses to stress involve a direct interaction of glucocorticoid receptors with ERK1/2-MSK1-Elk-1 signaling. Proc Natl Acad Sci U S A. 2011;108:13806–13811. doi: 10.1073/pnas.1104383108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho VM, Lee JA, Martin KC. The cell biology of synaptic plasticity. Science. 2011;334:623–628. doi: 10.1126/science.1209236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Hübener M, Bonhoeffer T. Searching for engrams. Neuron. 2010;67:363–371. doi: 10.1016/j.neuron.2010.06.033. [DOI] [PubMed] [Google Scholar]
- Kerchner GA, Nicoll RA. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat Rev Neurosci. 2008;9:813–825. doi: 10.1038/nrn2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham JA, Greenough WT. Experience-driven brain plasticity: beyond the synapse. Neuron Glia Biol. 2004;1:351–363. doi: 10.1017/s1740925x05000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KJ, Shpiro N, Traynor R, Elliott M, Arthur JS. Comparison of the specificity of Trk inhibitors in recombinant and neuronal assays. Neuropharmacology. 2011;61:148–155. doi: 10.1016/j.neuropharm.2011.03.021. [DOI] [PubMed] [Google Scholar]
- McCoy CE, Campbell DG, Deak M, Bloomberg GB, Arthur JS. MSK1 activity is controlled by multiple phosphorylation sites. Biochem J. 2005;387:507–517. doi: 10.1042/BJ20041501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy CE, Macdonald A, Morrice NA, Campbell DG, Deak M, Toth R, McIlrath J, Arthur JS. Identification of novel phosphorylation sites in MSK1 by precursor ion scanning MS. Biochem J. 2007;402:491–501. doi: 10.1042/BJ20061183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCurry CL, Shepherd JD, Tropea D, Wang KH, Bear MF, Sur M. Loss of Arc renders the visual cortex impervious to the effects of sensory experience or deprivation. Nat Neurosci. 2010;13:450–457. doi: 10.1038/nn.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messaoudi E, Kanhema T, Soulé J, Tiron A, Dagyte G, da Silva B, Bramham CR. Sustained Arc/Arg3.1 synthesis controls long-term potentiation consolidation through regulation of local actin polymerization in the dentate gyrus in vivo. J Neurosci. 2007;27:10445–10455. doi: 10.1523/JNEUROSCI.2883-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnár E, Baude A, Richmond SA, Patel PB, Somogyi P, McIlhinney RA. Biochemical and immunocytochemical characterization of antipeptide antibodies to a cloned GluR1 glutamate receptor subunit: cellular and subcellular distribution in the rat forebrain. Neuroscience. 1993;53:307–326. doi: 10.1016/0306-4522(93)90198-o. [DOI] [PubMed] [Google Scholar]
- Morris RG. Elements of a neurobiological theory of hippocampal function: the role of synaptic plasticity, synaptic tagging and schemas. Eur J Neurosci. 2006;23:2829–2846. doi: 10.1111/j.1460-9568.2006.04888.x. [DOI] [PubMed] [Google Scholar]
- Neves G, Cooke SF, Bliss TV. Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat Rev Neurosci. 2008;9:65–75. doi: 10.1038/nrn2303. [DOI] [PubMed] [Google Scholar]
- Nithianantharajah J, Hannan AJ. Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat Rev Neurosci. 2006;7:697–709. doi: 10.1038/nrn1970. [DOI] [PubMed] [Google Scholar]
- O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21:1067–1078. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- Pankratov YV, Krishtal OA. Distinct quantal features of AMPA and NMDA synaptic currents in hippocampal neurons: implication of glutamate spillover and receptor saturation. Biophys J. 2003;85:3375–3387. doi: 10.1016/S0006-3495(03)74757-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankratov Y, Lalo U, Verkhratsky A, North RA. Quantal release of ATP in mouse cortex. J Gen Physiol. 2007;129:257–265. doi: 10.1085/jgp.200609693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peebles CL, Yoo J, Thwin MT, Palop JJ, Noebels JL, Finkbeiner S. Arc regulates spine morphology and maintains network stability in vivo. Proc Natl Acad Sci U S A. 2010;107:18173–18178. doi: 10.1073/pnas.1006546107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plath N, Ohana O, Dammermann B, Errington ML, Schmitz D, Gross C, Mao X, Engelsberg A, Mahlke C, Welzl H, Kobalz U, Stawrakakis A, Fernandez E, Waltereit R, Bick-Sander A, Therstappen E, Cooke SF, Blanquet V, Wurst W, Salmen B, et al. Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron. 2006;52:437–444. doi: 10.1016/j.neuron.2006.08.024. [DOI] [PubMed] [Google Scholar]
- Pozo K, Goda Y. Unraveling mechanisms of homeostatic synaptic plasticity. Neuron. 2010;66:337–351. doi: 10.1016/j.neuron.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putignano E, Lonetti G, Cancedda L, Ratto G, Costa M, Maffei L, Pizzorusso T. Developmental downregulation of histone posttranslational modifications regulates visual cortical plasticity. Neuron. 2007;53:747–759. doi: 10.1016/j.neuron.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Rao VR, Pintchovski SA, Chin J, Peebles CL, Mitra S, Finkbeiner S. AMPA receptors regulate transcription of the plasticity-related immediate-early gene Arc. Nat Neurosci. 2006;9:887–895. doi: 10.1038/nn1708. [DOI] [PubMed] [Google Scholar]
- Rial Verde EM, Lee-Osbourne J, Worley PF, Malinow R, Cline HT. Increased expression of the immediate-early gene arc/arg3.1 reduces AMPA receptor-mediated synaptic transmission. Neuron. 2006;52:461–474. doi: 10.1016/j.neuron.2006.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond SA, Irving AJ, Molnár E, McIlhinney RA, Michelangeli F, Henley JM, Collingridge GL. Localisation of the glutamate receptor subunit GluR1 on the surface of living and within cultured hippocampal neurones. Neuroscience. 1996;75:69–82. doi: 10.1016/0306-4522(96)00217-5. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger DB, Dickstein DL, Hof PR, Wearne SL. Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One. 2008;3:e1997. doi: 10.1371/journal.pone.0001997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig MR, Bennett EL. Psychobiology of plasticity: effects of training and experience on brain and behavior. Behav Brain Res. 1996;78:57–65. doi: 10.1016/0166-4328(95)00216-2. [DOI] [PubMed] [Google Scholar]
- Roze E, Betuing S, Deyts C, Marcon E, Brami-Cherrier K, Pagès C, Humbert S, Mérienne K, Caboche J. Mitogen- and stress-activated protein kinase-1 deficiency is involved in expanded-huntingtin-induced transcriptional dysregulation and striatal death. FASEB J. 2008;22:1083–1093. doi: 10.1096/fj.07-9814. [DOI] [PubMed] [Google Scholar]
- Rutherford LC, Nelson SB, Turrigiano GG. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron. 1998;21:521–530. doi: 10.1016/s0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- Shepherd JD, Bear MF. New views of Arc, a master regulator of synaptic plasticity. Nat Neurosci. 2011;14:279–284. doi: 10.1038/nn.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52:475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soloaga A, Thomson S, Wiggin GR, Rampersaud N, Dyson MH, Hazzalin CA, Mahadevan LC, Arthur JS. MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J. 2003;22:2788–2797. doi: 10.1093/emboj/cdg273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Wearne SL, Rodriguez A, Ehlenberger DB, Rocher AB, Henderson SC, Hof PR. New techniques for imaging, digitization and analysis of three-dimensional neural morphology on multiple scales. Neuroscience. 2005;136:661–680. doi: 10.1016/j.neuroscience.2005.05.053. [DOI] [PubMed] [Google Scholar]
- Wiggin GR, Soloaga A, Foster JM, Murray-Tait V, Cohen P, Arthur JS. MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol Cell Biol. 2002;22:2871–2881. doi: 10.1128/MCB.22.8.2871-2881.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Edelman GM, Vanderklish PW. The brain-derived neurotrophic factor enhances synthesis of Arc in synaptoneurosomes. Proc Natl Acad Sci U S A. 2002;99:2368–2373. doi: 10.1073/pnas.042693699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TV, Bramham CR. Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci. 2002;22:1532–1540. doi: 10.1523/JNEUROSCI.22-05-01532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]