

Abstract
Remodeling of dendritic spines through regulation of actin dynamics is a key event in activity-dependent structural plasticity. However, the molecular mechanism underlying this process is poorly understood. Here, we show that activity-dependent modulation of Abl interactor 1–Ca2+/calmodulin-dependent kinase IIα (Abi1–CaMKIIα) interaction, and thereby their activity, is important for regulation of spine morphology in cultured rat hippocampal neurons. Abi1 interacts with CaMKIIα at resting conditions through Abi1's tSNARE (target membrane-associated SNARE), which harbors striking homology with CaMKIIα regulatory domain. The interaction of the two proteins, Abi1 and CaMKIIα, results in their simultaneous inhibition, inhibition of CaMKIIα activity, and also inhibition of Abi1-dependent Rac activation. Their functional impediment is released when they dissociate from each other by calmodulin binding through glutamate receptor activation. Before dissociation, Abi1 is phosphorylated by CaMKIIα at serine 88, which may involve in regulation of Rac activation and spine maturation. Our results suggest that modulation of the interaction between Abi1 and CaMKIIα, through the glutamate receptor pathway, may be a molecular mechanism underlying activity-regulated structural plasticity in rat hippocamapal neurons.
Introduction
Dendritic spines, which are small, highly specialized, and dynamic structures in neurons, receive and relay synaptic information in response to synaptic activity by undergoing rapid morphological alterations (Nimchinsky et al., 2002). Reorganization of the actin cytoskeleton, a major cytoskeleton in the synapse, is a key event in modulation of the structural modifications of excitatory synapses (Dillon and Goda, 2005; Cingolani and Goda, 2008).
Ca2+/calmodulin-dependent kinase II (CaMKII) is a predominant serine/threonine kinase in the postsynaptic density of excitatory synapses and has been suggested to be a key molecule in regulating functional and structural synaptic plasticity. CaMKII activation is necessary and sufficient for induction of activity-dependent spine plasticity (Maletic-Savatic et al., 1999; Toni et al., 1999; Matsuzaki et al., 2004). CaMKII also has been shown to control activity-dependent filopodial growth and branching, as well as spine formation (Wu and Cline, 1998; Jourdain et al., 2003; Andersen et al., 2005).
Because of its switch-like properties and abundance in the brain, CaMKII has come to be considered a crucial enzyme in the brain and CaMKII's regulatory mechanisms have been intensively studied. NMDA subunit (NR2B) and Drosophila Eag potassium channel have been found to recruit CaMKII to synapses and prolong CaMKII activity after Ca2+/CaM release from the enzyme (Bayer et al., 2001; Wang et al., 2002; Sun et al., 2004). Thus far, many groups have focused on the mechanisms for retention of CaMKIIα activation. However, the intermolecular mechanisms of regulating basal CaMKIIα activity have not been reported yet.
Abl interactor 1 (Abi1) acts as a cofactor essential for specifying Sos1 guanine nucleotide exchange factor activity for Rac GTPase when in complex with Eps8 and PI3K (Scita et al., 1999; Innocenti et al., 2003). Also, Abi1 was recently reported to directly bind to Rac (Dubielecka et al.). In addition, the stability of WAVE1 and WAVE2 complexes which are important in regulation of Arp2/3 complexes, have also been reported to be mediated by Abi1 (Innocenti et al., 2003; Leng et al., 2005). Abi family proteins have also been implicated in neuronal morphogenesis. Overexpression and knockdown of Abi1 in hippocampal neurons at 3 and 10 DIV demonstrate an essential function of Abi1 in dendritic morphology (Proepper et al., 2007).
Here, we examined the roles of Abi1 and CaMKIIα in structural plasticity and the molecular mechanisms underlying their regulation. We found that binding of Abi1 to CaMKIIα inhibits its Thr 286 autophosphorylation, which is crucial for kinase activation. Abi1 inhibits CaMKIIα by mimicking its autoinhibitory domain through sequence homology. This interaction is regulated in a glutamate-dependent manner in which Ca2+-loaded CaM disrupts Abi1–CaMKIIα binding. In addition, Abi1 is phosphorylated by CaMKIIα and this phosphorylation is essential for maturation of dendritic spines through Rac activation. These data not only provide new insights into the molecular mechanisms underlying glutamate-dependent structural plasticity in dendritic spines, but also indicate novel functions of tSNARE (target membrane-associated SNARE) domain in neurons.
Materials and Methods
Antibodies and reagents.
Antibodies purchased were anti-CaMKIIα (clone CBα-2, Zymed Laboratories), anti-Abi1 pAb (clone T-15, Santa Cruz Biotechnology), anti-Abi1 mAb (Medical & Biological Laboratories), anti-GFP (Santa Cruz Biotechnology), anti-Flag (Sigma-Aldrich), anti-phospho-Thr 286 CaMKIIα (Santa Cruz Biotechnology), anti-actin (Sigma-Aldrich), anti-Myc (Santa Cruz Biotechnology), anti-calmodulin (Millipore Bioscience Research Reagents), anti-HA (Sigma-Aldrich), anti-pSer (clone PSR45, Sigma-Aldrich), anti-Rac (BD Biosciences), anti-vGlut1 (Synaptic Systems), anti-NMDAR (Millipore Bioscience Research Reagents), and anti-PSD95 (Affinity BioReagents). Reagents purchased were KN92, KN93, and ionomycin (Calbiochem).
DNA constructs.
The murine Abi1 cDNA used in this study was cloned from the Matchmaker Mus musculus brain cDNA Library (Clontech Laboratories). It is identical to Mus musculus Abi1, transcript variant 1 (GenBank Accession No. BC004657). The coding region of murine Abi1 was subcloned into pcDNA3.1/MycHis vector (Invitrogen), pEGFP-C1 (Clontech Laboratories), and pcDNA-HA vectors. Glutathione S-transferase (GST)-R84E, GST-S88A, GST-T96A, GST-TT113,114VA, GFP-Abi1S88A, and GFP-Abi1S88D mutants were cloned using Stratagene Mutagenesis kit. GST-83-114 construct was used as the template for GST-R83E, GST-S88A, GST-T96A, and GST-TT113,114VA. GFP-CaMKIIα and GFP-CaMKIIα-containing T286D mutation constructs were kind gifts from T. Meyer (Stanford University). GFP-tagged CaMKII-deletion mutants were generated by using GFP-CaMKIIα as the template and by inserting stop codons at the ends of each domain (stop codons at His 273 for GFP-Ca and Gly 315 for GFPCaRe) using a Stratagene Mutagenesis kit. GFP-CaMKIIαK42E, T286A mutant was generated using the GFP-CaMKIIαT286A construct as the template. Knockdown construct pSIREN-Abi1shRNA was constructed using primers targeting nucleotides 135–141. The shRNA-resistant GFP-Abi1 (GFP-Abi1shRNAresis) construct was generated using a Stratagene Mutagenesis kit. Five consecutive amino acids targeted by Abi1 shRNA were mutated to a different codon translating the same amino acid. Mutagenesis of all constructs was confirmed by DNA sequencing. SEP-NR2B was provided by Robert Malinow (Addgene plasmid 23998).
Cell culture, transfection, and immunoblotting.
HeLa cells were maintained in DMEM (Invitrogen) supplemented with 10% certified fetal bovine serum (c-FBS, Invitrogen) and penicillin-streptomycin (Invitrogen) in 5% CO2 at 37°C. For Abi1-knockdown stable cell lines, HeLa cells were transfected with pSIREN-DsRed as control or pSIREN-DsRed-Abi1shRNA using LipofectAMINEPLUS (Invitrogen). Cells stably expressing the vectors were selected by addition of 400 μg/ml of G418 (Sigma-Aldrich) in growth media. DsRed-expressing cells were collected using FACS. The cells were then cultured into single cell colonies, each giving rise to a stable cell line. Transfection of cells with mammalian expression vectors were performed with LipofectAMINEPLUS (Invitrogen) according to the manufacturer's instructions. Cells were washed twice with PBS and lysed with SDS-lysis buffer (100 mm Tris, pH 6.8, 2% SDS, and 10% glycerol). Protein concentration was determined with BCA reagent (Pierce). Equal amounts of protein were resolved by SDS-PAGE and transferred to a PVDF membrane. Blots were blocked with 5% skim milk in PBS-T (0.1% Triton X-100 in PBS) for 30 min. The blots were incubated with primary antibodies for 1 h and washed with PBS-T. Then the blots were incubated with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) and analyzed by enhanced chemiluminenesence.
GST pull-down assays and immunoprecipitation.
GST pull-down assays were performed with GST fusion proteins expressed in E. coli (BL21) cells and purified by glutathione Sepharose beads (GE Healthcare Pharamia Biotech). The bound fusion proteins were then eluted by glutathione elution buffer (20 mm glutathione, 100 mm Tris-HCl, pH 8.0, 120 mm NaCl). Purified proteins were quantified by Bradford quantification method (Bio-Rad). For isolation of GST-tSNARE-binding proteins, four brains of 4-week-old male mice (strain C57BL/6) were homogenized in 8 ml of homogenization buffer (50 mm Tris-HCl, pH 8.5, 1 mm EDTA, 1 mm EGTA, 150 mm NaCl, 2 mm Na3VO4, 15 mm NaF, 1 μl/ml leupeptin, and 1 μl/ml pepstatin) with a Polytron homogenizer and incubated at 4°C for 1 h after addition of 1% Triton X-100. The homogenates were centrifuged at 15,000 rpm for 30 min at 4°C. The supernatant was used as brain lysate for GST pull-down assays. One milligram of brain lysate was incubated with 4 μg of GST or GST-tSNARE for 2 h at 4°C in immunoprecipitation buffer (50 mm Tris-HCl, pH 7.4, 1% Triton X-100, 150 mm NaCl, 15 mm NaF, 1 μl/ml leupeptin, 1 μl/ml pepstatin, and 1 μl/ml aprotinin). Glutathione Sepharose beads were added to the samples for 1 h at 4°C and washed with immunoprecipitation buffer three times to remove unbound proteins. Proteins bound to beads were denatured in Laemelli sample buffer and subjected to SDS-PAGE and Western blotting. Fourteen days in vitro rat cortical neurons were incubated in Tyrode solution (119 mm NaCl, 2.5 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 25 mm HEPES, pH 7.4, and 30 mm glucose) for 30 min before treatment with 50 μm glutamate and 5 μm glycine for 5 min.
For in vitro binding assays, 30 ng of purified rat brain CaMKII (Calbiochem) was incubated for 10 min at 30°C with or without 0.3 mm Ca2+, 2 μm CaM, and 1 μm ATP in kinase buffer (50 mm HEPES, pH 7.4, and 5 mm MgCl2). Then CaMKII, loaded with Ca2+/CaM and/or ATP, was mixed with 2 μg of GST-47-114 and incubated for 1 h at 4°C in pull-down buffer (50 mm HEPES, pH 7.4, 5 mm MgCl2, 1% Triton X-100, 150 mm NaCl, 15 mm NaF, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 1 μg/ml pepstatin). Glutathione beads were then added and incubated for 1 h at 4°C. The beads were then washed and the bead-bound proteins were separated by SDS-PAGE and immunoblotted for CaMKIIα and phospho-Thr 286-CaMKIIα. The fusion proteins were visualized by Coomassie Blue staining.
Neuron culture and transfection.
Rat hippocampal and cortical neuron cultures were prepared from embryonic day 18 Sprague Dawley rat embryos of either sex as previously described (Chang and De Camilli, 2001). Dissociated hippocampi and cortex tissues were treated with papain (20 μg/ml) and DNase (10 U/μl) for 40 min at 37°C. The tissues were then mechanically dissociated by titration with a glass Pasteur pipette. Hippocampal neurons (2 × 105 cells/60 mm dish) and cortical neurons (2 × 105 cells/ml) were plated in MEM (JBI) supplemented with 0.6% glucose, 1 mm sodium pyruvate, 1% penicillin-streptomycin (Invitrogen), 2 mm l-glutamine, and 10% certified FBS (Invitrogen) for 4 h before exchange with Neurobasal medium (Invitrogen) containing 0.5 mm l-glutamine and B27 supplement (Invitrogen). The cells were maintained in a 5% CO2 incubator at 37°C for up to 21 d. Every 4–7 d, half of the original media was discarded and replaced with fresh Neurobasal media supplemented with 0.5 mm glutamine and B27 supplement. Hippocampal neurons were transiently transfected at 6–8 DIV with a modified calcium phosphate method using CalPhos Transfection Kit (Calbiochem) (Ryan et al., 2005). Original media was exchanged with fresh Neurobasal media containing 25 mm HEPES, pH 7.35; 148 mm CaCl2 was mixed with 10 μg of DNA; and distilled water was added dropwise to the mixture with vortexing, making a final volume of 100 μl. This mixture was then added dropwise to 100 μl of 2× HEPES-buffered solution while vortexing. The solution was left at room temperature for 2 min and then added dropwise to each 60 mm dish. Precipitate formed within 15 min. After formation of precipitate, the cells were washed twice with fresh Neurobasal media and then incubated for 5 min at 37°C. Following two more subsequent washings, the cells were returned to their original media.
Immunocytochemistry.
Rat hippocampal neurons at 14 DIV seeded on 18 mm coverslips were fixed for 15 min in fixative (4% paraformaldehyde and 4% sucrose in PBS). The neurons were permeabilized with 0.5% Triton X-100 in PBS for 10 min and then incubated in blocking solution [10% c-FBS (Invitrogen), 0.5% gelatin, 0.1% Triton X-100 in PBS] for 30 min. The coverslips were then incubated with primary antibodies diluted in blocking solution for 1 h at room temperature. After washing with PBS-T, coverslips were stained with fluorescein isothiocyanate-conjugated or tetramethyl rhodamine isothiocyanate-conjugated anti-mouse or anti-rabbit IgG (Jackson ImmunoResearch Laboratories) for 1 h. Following incubation, coverslips were washed with PBS-T, then mounted and observed with a Zeiss Axiovert fluorescence microscope equipped with a 100×, 1.4 NA plan-apochromat objective lens (see Figs. 4C, 7C) or with a Zeiss LSM710 confocal microscope equipped with 40×, 1.20 NA C-Apochromat objective (see Fig. 1E). Where indicated, cells were treated with 10 μm KN93 in Tyrode solution for 30 min before fixation.
Figure 4.

Knockdown of Abi1 in HeLa cells and rat hippocampal neurons elicit increased CaMKIIα autophosphorylation. A, HeLa cells stably expressing pSIREN-DsRed (P1) and pSIREN-DsRed Abi1shRNA (S1) were transfected with GFP-Abi1 or shRNA-resistant GFP-Abi1 (GFP-Abi1shRNAresis) and immunoblotted (IB) with indicated antibodies. Tubulin was used as the loading control. B, HeLa cells stably expressing pSIREN-Dsred (P1) and pSIREN-DsRed Abi1shRNA (S1, S2, and S3) were transfected with GFP-CaMKIIα. CaMKIIα autophosphorylation was examined by immunoblotting. Changes in P-CaMKIIα were quantified and expressed in arbitrary units as changes in P-CaMKIIα/GFP-CaMKIIα compared with control cells. C, 14 DIV neurons transfected with pSIREN-DsRed or p-SIREN-DsRed-Abi1shRNA (Abi1shRNA) were treated with or without 10 μm KN93 or 10 μm KN92. Representative neurites for each condition are shown. Scale bars, 5 μm. D, E, Results of subjecting 7310 dendritic protrusions from 120 neurons to morphometric analysis. Protrusion tip/shaft fluorescence intensity ratio of P-CaMKIIα in neurons was quantified for each condition (D). Compared with pSIREN-DsRed-transfected neurons, ratio of P-CaMKIIα increased in control and KN92-treated pSIREN-DsRed-Abi1shRNA neurons (mean ± SEM, *p = 0.001, **p < 0.00001; ns, not significant; Student's t test). Protrusion lengths were measured for each condition (E).
Figure 7.
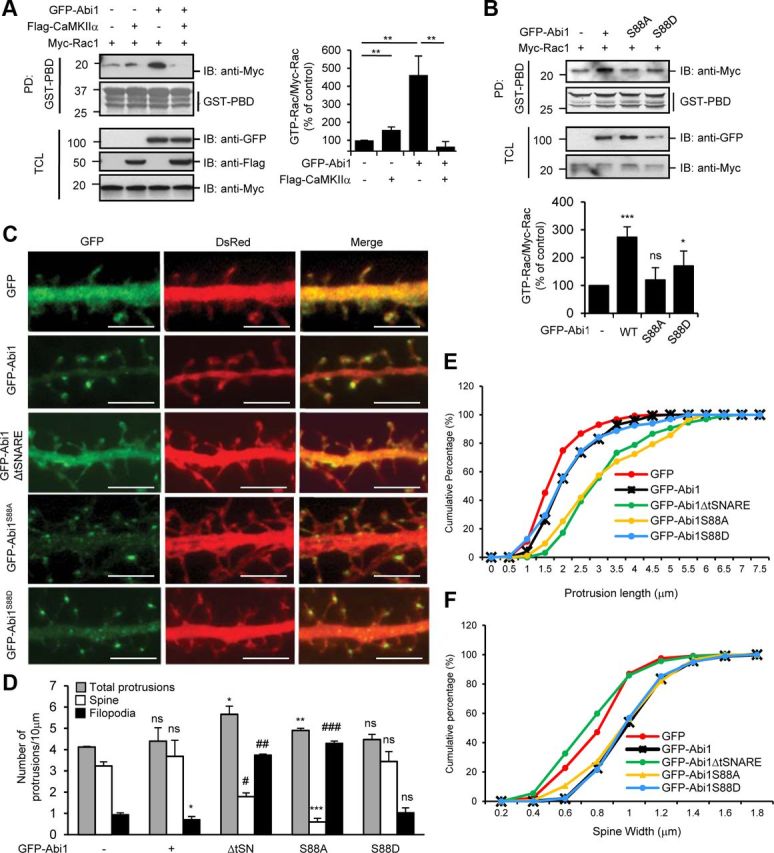
Phosphorylation of Abi1 at the serine 88 is critical for Rac activation and spine maturation. A, HeLa cells were cotransfected with Flag-CaMKIIα, Myc-Rac1, or GFP-Abi1 as indicated. B, HeLa cells were cotransfected with Myc-Rac1 with either GFP-Abi1, GFP-Abi1S88A, or GFP-Abi1S88D. In A and B, cell lysates were subjected to GST-PBD (p21-binding domain) pull-down (PD) and immunoblotted (IB) with anti-Myc antibodies. The ratio of GTP-Rac (bound Rac to GST-PBD)/Myc-Rac was quantified and expressed as the percentage of the amount in control cells from three independent experiments in both A and B (mean ± SD, *p = 0.04, **p = 0.02, ***p = 0.007; ns, not significant; Student's t test). C, Rat hippocampal neurons were transfected with GFP, GFP-Abi1, GFP-Abi1ΔtSNARE, GFP-Abi1S88A, and GFP-Abi1S88D at 7 DIV and fixed at 14 DIV. DsRed was also coexpressed to show overall morphology of the neurons. Representative neurites for each condition are shown. Scale bars, 5 μm. D–F, Results after 1500 10 μm dendritic segments from 100 neurons were subjected to morphometric analysis. D, Number of total protrusions, spines, and filopodia per 10 μm dendritic segments in hippocampal neurons transfected as indicated in C were quantified and compared with control cells using Student's t test (mean ± SEM, *p = 0.04, **p = 0.02, ***p = 0.009, #p = 0.005, ##p = 0.004, ###p = 0.001; ns, not significant; Student's t test). E, Protrusion length was measured for neurons transfected as indicated. F, Spine widths were measured for neurons transfected as indicated. TCL, Total cell lysate.
Figure 1.

Abi1 tSNARE domain interacts with CaMKIIα catalytic domain. A, Mouse brain lysates were incubated with purified GST or GST-tSNARE protein, and the bound proteins to GST fusion proteins were pulled-down (PD) by glutathione Sepharose beads and immunoblotted (IB) with anti-CaMKIIα antibodies. GST fusion proteins were visualized by Coomassie Blue staining. B, Lysates from 14 DIV rat cortical neurons were immunoprecipitated (IP) with control polyclonal IgGs or anti-Abi1 antibodies (anti-Abi1) and immunoblotted with anti-CaMKIIα antibodies and anti-Abi1 antibodies. C, Lysates from HeLa cells expressing Flag-CaMKIIα and either GFP-Abi1 or GFP-Abi1ΔtSNARE were immunoprecipitated with anti-Abi1 antibodies and immunoblotted with anti-GFP antibodies and anti-Flag antibodies. D, Lysates from HeLa cells expressing GFP-CaMKIIα (wild-type), GFP-CaRe (catalytic domain and regulatory domain), or GFP-Ca (catalytic domain) were immunoprecipitated with anti-Abi1 antibodies and immunoblotted with anti-GFP antibodies and anti-Abi1 antibodies. Arrowheads (▶) indicate coimmunoprecipitated CaMKIIα proteins with Abi1. Asterisks in C and D indicate heavy chains of IgGs in the immunoprecipitates. E, Immunocytochemistry of 14 DIV rat hippocampal neurons in the top panels shows partial colocalization of the endogenous Abi1 (red) and CaMKIIα (blue) at synapses visualized with anti-vGlut1 antibodies (green). Bottom panels show P-CaMKIIα (blue) localization with Abi1 (red) and vGlut1 (green). White arrowheads indicate regions of vGlut1 staining in each panel. Scale bar, 10 μm. TCL, Total cell lysate.
Analysis of neuronal morphology and imaging.
After image acquisition, images were cropped and processed using Adobe Photoshop. Image processing for colocalization assay was performed using colocalization plugin in ImageJ (NIH) software. For morphometric analysis of neurons, protrusion length and spine width were measured using Axiovision (Zeiss) software. All morphometric experiments were repeated in at least three independent experiments with an n > 7 per condition in each experiment. Protrusion lengths were defined as the length from the base of the neck to the farthest end of the spine head. Protrusions were classified into spines and filopodia as previously described (Zhang and Macara, 2006). Filopodia were defined as thin protrusions without a distinguishable head, and spines were defined as protrusions with a distinguishable head. Spine heads were defined as those with head/neck ratio of >1.2. Spine head widths were measured as the spine diameter perpendicular to the spine neck. Protrusion tip/shaft fluorescence intensity ratio of Thr 286 autophosphorylated CaMKIIα (P-CaMKIIα) was analyzed by line-scan analyses using ImageJ software. A one-pixel wide line was drawn across the protrusion into the dendritic shaft and the fluorescence intensity profile along the line was measured and profiles were transferred to Microsoft Excel. To quantify protrusion tip/shaft fluorescence intensity ratio, the mean fluorescence intensities of protrusion tips and shafts were measured.
In vitro kinase assay.
For phosphorylation assays, 1 ng of purified rat brain CaMKII (Calbiochem) was activated in buffer containing 50 mm HEPES, pH 7.4, 5 mm MgCl2, 2 μm CaM, 0.3 mm CaCl2, and 0.5 mm [γ-32P]ATP (10 Ci/mmol) for 10 min at 30°C. As the control, the same buffer, but without 2 μm CaM and 0.3 mm CaCl2, was used. The reactions were stopped on ice and then incubated with 4 μg of GST fusion proteins for 30 min at 30°C. The reactions were then stopped with 5× Laemelli sample buffer and subjected to SDS-PAGE. The acrylamide gels were stained with Coomassie Blue staining solution to observe the total amount of GST fusion proteins and then dried on 3M Whatman paper. Exposure of the gel to film showed phosphorylation of the GST fusion proteins.
Statistics.
All analyses were completed from a minimum of three independent experiments. Statistical significances for measurements were calculated using Student's t tests compared with control cells unless indicated. The actual p values are indicated in the figure legends. All histograms representing Western blots are represented as mean ± SD and all histograms analyzing morphogenic changes in neurons are represented as mean ± SEM.
Results
Abi1 tSNARE binds to CaMKIIα catalytic domain
Abi1 contains a tSNARE domain (Fig. 2A) generally known to be involved in membrane fusion (Echarri et al., 2004). This domain has been reported to bind to Syntaxin-1 (Echarri et al., 2004) and to be involved in regulating nuclear shuttling of Abi1 (Proepper et al., 2007). Its function, however, remains elusive. To investigate novel pathways involving the tSNARE domain of Abi1, we searched for binding partners to this domain in brain lysates of 4-week-old mice (strain C57BL/6) using a GST fusion protein of the Abi1 tSNARE domain (GST-tSNARE) as bait. Protein bands specific for GST–tSNARE binding were excised and subjected to matrix-assisted laser desorption ionization time-of-flight analysis (data not shown). One of the bands identified was CaMKIIα. We confirmed this interaction by GST pull-down assay (Fig. 1A). This binding was also corroborated in 14 DIV rat cortical neurons by coimmunoprecipitation with anti-Abi1 antibody (Fig. 1B). Transient overexpression of wild-type GFP-Abi1 and tSNARE deletion mutant of Abi1 (GFP-Abi1ΔtSNARE) in HeLa cells shows binding of CaMKIIα to only GFP-Abi1, verifying that their interaction is through the tSNARE (Fig. 1C). Abi1 binds to CaMKIIα through its catalytic domain (Fig. 1D). Although wild-type CaMKIIα was more efficiently coimmunoprecipitated with Abi1 than the deletion mutants, coimmunoprecipitation still took place, an indication that the presence of CaMKIIα's catalytic domain represents the minimum requirement for CaMKIIα and Abi1 to bind CaMKIIα is known to oligomerize into a dodecamer through its association domain (Rosenberg et al., 2005). Due to this oligomerization, wild-type CaMKIIα would be more efficiently sedimented with Abil in comparison to mutants lacking the association domain. Comparison of the other two deletion mutants shows that Abi1 seems to bind with higher affinity to the deletion mutant comprised of only its catalytic domain (GFP-Ca). Colocalization of endogenous Abi1 and CaMKIIα was also observed in 14 DIV rat hippocampal neurons (Fig. 1E). Colocalization was observed in not all synapses but in a subset of synapses. Arrowheads indicate excitatory synapses identified by anti-vGlut1 immunostaining. The top panels in Figure 1E show CaMKIIα and Abi1 localization while the bottom panels show localization of Abi1 with P-CaMKIIα. Strong correlation of Abi1 and CaMKIIα localization is seen in synapses. However, P-CaMKIIα shows weak correlation with Abi1 localization. Notably, colocalization of CaMKIIα and Abi1 is not seen in the dendritic shafts.
Figure 2.

Abi1 tSNARE has high homology to CaMKIIα regulatory domain and mediates Abi1 binding to CaMKIIα. A, Schematic representation of Abi1 and sequence alignment of Abi1, NR2B, and Eag channel to CaMKIIα. P-CaMKIIα sites are indicated with black dot. Amino acids conserved between CaMKIIα and other proteins are highlighted in black, and conservative substitutions are in gray. B, Top, Schematic diagram of GST-tSNARE deletion mutants. Middle, In vitro GST pull-down (PD) assay. Purified rat brain CaMKII were incubated with indicated GST fusion proteins and analyzed for binding to CaMKII by immunoblotting (IB). GST fusion proteins were visualized by Coomassie Blue staining. Bottom, Results from three independent experiments were quantified, normalized to amount of GST fusion proteins, and expressed in arbitrary units (a. u.). Pro, Proline-rich domain.
Abi1 tSNARE domain has homology with CaMKIIα regulatory domain
In studying the interaction of the two proteins, Abi1 and CaMKIIα, sequence analyses showed a striking similarity between the Abi1 tSNARE domain and the CaMKIIα regulatory domain. In a region of the Abi1 tSNARE domain (amino acids 83–114), 44.1% of the amino acids are identical to those in the regulatory domain (amino acids 273–306) of CaMKIIα (Fig. 2A). Homology of this nature has also been reported in NR2B, an NMDA receptor subunit (8.8% identity) (Bayer et al., 2001), and in the Drosophila Eag potassium channel (11.8% identity) (Wang et al., 2002; Sun et al., 2004). Binding of CaMKIIα to this region in NR2B (amino acids 1290–1315) is implemented as a mechanism in which NR2B association retains CaMKIIα activity even after dissociation of Ca2+/CaM (Bayer et al., 2001). This mechanism has also been reported for Drosophila Eag potassium channel (Wang et al., 2002; Sun et al., 2004). One notable difference between Abi1 and these molecules is that Abi1 has high homology in the CaM-binding region of CaMKIIα in addition to the autoinhibitory region. Pull-down assays with GST-tSNARE deletions including and excluding the region of homology show that this region (amino acids 83–114) is essential for interaction with CaMKIIα (Fig. 2B). Quantification of CaMKIIα binding to GST deletion mutants shows similar binding affinity of mutants containing the region of homology.
Abi1 negatively regulates CaMKIIα Thr 286 autophosphorylation in a Ca2+-dependent manner
Because NR2B and Eag channel affect CaMKIIα activation, we investigated whether Abi1 also affects CaMKIIα activation by examining the level of Thr 286 autophosphorylation, a hallmark of CaMKIIα activation (Fig. 3A). When GFP-Abi1 was overexpressed with Flag-CaMKIIα, basal level Thr 286 P-CaMKIIα was attenuated by ∼60%. However, GFP-Abi1ΔtSNARE, which does not bind to CaMKIIα, increased basal level of Thr 286 autophosphorylation by 70%. Increasing levels of GFP-Abi1 expression with Flag-CaMKIIα resulted in a gradual decrease in Thr 286 autophosphorylation (Fig. 3B). Thus, Abi1 and NR2B/Eag channel elicit an opposite effect on CaMKIIα activation. To observe the effect of calcium on Abi1 inhibition of P-CaMKIIα, 30 μm ionomycin, a calcium ionophore to induce Ca2+ influx into cells, was treated for 15 min (Fig. 3C). The inhibitory effects of Abi1 on CaMKIIα at basal states were relieved upon ionomycin treatment. To further confirm that binding of CaMKIIα to Abi1 has a negative effect on P-CaMKIIα, we compared the effects of Abi1 and NR2B on P-CaMKIIα. NR2B has been reported to sustain CaMKIIα activity through its region of homology (Bayer et al., 2001). As expected, expression with Abi1 decreased P-CaMKIIα compared with control cells and expression with SEP-NR2B increased P-CaMKIIα (Fig. 3D). In ionomycin-treated cells, cotransfection with SEP-NR2B slightly increased P-CaMKIIα compared with control cells and GFP-Abi1-expressing cells. Although both proteins, Abi1 and NR2B, have homology to CaMKIIα, they regulate CaMKIIα in opposite ways. These results suggest that Abi1 may be a negative regulator of CaMKIIα during basal states before an increase in intracellular calcium.
Figure 3.

Binding of Abi1 inhibits P-CaMKIIα. A, Levels of CaMKIIα Thr 286 autophosphorylation were examined by immunoblotting (IB) with anti-phospho-Thr 286 CaMKIIα antibodies (P-CaMKIIα) in HeLa cells transfected with indicated constructs. Results from three independent experiments were quantified, normalized to GFP-CaMKIIα, and expressed as percentage of the amount in control cells (mean ± SD, *p = 0.003, **p = 0.00003, Student's t test). B, HeLa cells were transfected with the increasing levels of GFP-Abi1. Anti-Flag blot was used as the loading control. Results from three independent experiments were quantified, normalized to Flag-CaMKIIα, and expressed as the percentage of the amount in control cells (mean ± SD; ns, not significant; *p = 0.05, **p = 0.004, Student's t test). C, HeLa cells transfected with GFP-CaMKIIα and GFP-Abi1 were treated with or without 30 μm ionomycin and 2 mm CaCl2 for 10 min and examined by immunoblotting with indicated antibodies. Changes in P-CaMKIIα were quantified and expressed in arbitrary units as changes in P-CaMKIIα/GFP-CaMKIIα compared with control cells. D, HeLa cells were transfected with GFP-CaMKIIα with or without GFP-Abi1 or SEP-NR2B. Cells were treated with or without 30 μm ionomycin for 10 min and immunoblotted with indicated antibodies.
Next we used Abi1 shRNA to reduce Abi1 expression in HeLa cells and hippocampal neurons and to investigate the effects of Abi1 knockdown on P-CaMKIIα. First, the specificity of the Abi1 shRNA was tested in HeLa cells stably expressing scrambled shRNA (P1) or Abi1 shRNA (S3) by transfecting wild-type (GFP-Abi1) or shRNA-resistant Abi1 (GFP-Abi1shRNAresis) (Fig. 4A). P1 cells show no change in expression of both GFP constructs, whereas S3 cells exhibit decreased expression of wild-type GFP-Abi1 but not GFP-Abi1shRNAresis. Anti-Abi1 immunoblot shows that endogenous Abi1 is decreased in S3 cells in both cases. Using these cells, we demonstrate that knockdown of Abi1 in HeLa cells stably expressing pSIREN-DsRed-Abi1 shRNA increased Thr 286 autophosphorylation in CaMKIIα compared with control cells (Fig. 4B). To observe these effects in neurons, 14 DIV rat hippocampal neurons transfected with pSIREN-DsRed and pSIREN-DsRed-Abi1 shRNA were treated with or without 10 μm KN93, a CaMKII-specific inhibitor, or 10 mm KN92, an inactive compound of KN93 (Fig. 4C). Consistent with previously reported data (Proepper et al., 2007), Abi1 knockdown in 14 DIV hippocampal neurons showed immature dendritic spines and consist mostly of long filopodia. In control neurons, Thr 286 autophosphorylation in CaMKIIα was present at low levels and dispersed throughout dendrites, but in Abi1-knockdown neurons, the ratio of P-CaMKIIα fluorescence intensity in protrusion tip to shaft was ∼60% increased, depicting a marked induction of P-CaMKIIα in dendritic spines (Fig. 4D). The increase of P-CaMKIIα was reversed when treated with KN93 and unaffected by KN92 treatment. The localization of phospho-Thr 286 CaMKIIα at the tips of the filopodial extensions is consistent with previous reports showing translocation of active CaMKIIα to the synapse (Shen and Meyer, 1999; Bayer et al., 2001). In addition, length of protrusions did not change significantly after KN93 or KN92 treatment (Fig. 4E). Together, Abi1 negatively regulates CaMKIIα activity in dendritic spines in a Ca2+-dependent manner by binding to its catalytic domain.
Abi1–CaMKIIα interaction is modulated in a glutamate-dependent manner via CaM
Because intracellular calcium modulates CaMKIIα activity, and because increased intracellular calcium inhibits Thr 286 autophosphorylation by Abi1 (Fig. 3C), we investigated the effect of excitatory stimuli on CaMKIIα and Abi1 interaction. Given that CaMKIIα is known to be activated through glutamate receptor stimulation, the interaction of Abi1 and CaMKIIα was examined in 21 DIV cortical neurons with glutamate and glycine, NMDA, and AMPA treatments. KCl and bicuculline were also used to induce depolarization of the neurons. Abi1 showed reduced binding affinity to CaMKIIα after all the above stimulations (Fig. 5A).
Figure 5.

Interaction between Abi1 and CaMKIIα is disrupted by excitatory stimulation through Ca2+/CaM binding to Abi1. A, 14 DIV rat cortical neurons were treated with 50 μm glutamate and 5 μm glycine for 5 min, 30 μm NMDA for 5 min, 50 μm AMPA for 5 min, 90 mm KCl for 3 min, or 20 μm bicuculline for 30 min. Lysates were immunoprecipitated (IP) with anti-Abi1 antibodies, subjected to SDS-PAGE, and then immunoblotted (IB) with anti-CaMKIIα antibodies. B, Purified CaMKII was incubated with ATP and/or 0.3 mm Ca2+/ 2 mm CaM at 30°C for 10 min, to promote activation of CaMKII, and then the purified GST or GST-Abi1 tSNARE (GST-47–114) protein was added to the mixture and GST pull-down (PD) assays were performed. The interaction of Abi1 tSNARE with CaMKII was analyzed by immunoblotting with anti-CaMKIIα antibodies. C, HeLa cells were cotransfected with HA-Abi1 and GFP-CaMKIIα constructs (wild-type, autophosphorylation mimetic GFP-CaMKIIαT286D, or autophosphorylation mimetic kinase-dead GFP-CaMKIIαK42E,T286D), and the lysates were immunoprecipitated with anti-HA antibodies and immunoblotted with anti-GFP antibodies. Ratio of bound GFP-CaMKIIα mutants to HA-Abi1 were quantified and expressed as arbitrary units compared with wild type. D, CaM Sepharose beads were used for pull-down of GST-83-114 in the presence of 1 mm EGTA or 2 mm CaCl2. Pull-down assays were analyzed by Western blot as indicated. E, 1 or 10 mm CaM, and 0.3 mm CaCl2 were incubated with purified CaMKII and GST-47-114. Binding of CaMKII and CaM to GST-47-114 was observed by immunoblotting with indicated antibodies. TCL, Total cell lysate.
To determine whether binding of Abi1 to CaMKIIα is inhibited by CaM-dependent conformational change or by change in kinase activity, purified GST-Abi1 (amino acids 47–114) was incubated with purified rat CaMKII preincubated with or without Ca2+/CaM, and/or ATP (Fig. 5B). In the presence of Ca2+/CaM, interaction between CaMKII and GST-Abi1 (amino acids 47–114) was significantly attenuated (Fig. 5B, lane 4) and completely abolished when CaMKII is autophosphorylated by the addition of ATP (Fig. 5B, lane 5). The binding of Ca2+/CaM to CaMKIIα elicits three events: (1) a change in conformation where the catalytic domain is released from the inhibitory domain, (2) activation of the catalytic domain, and (3) last Thr 286 autophosphorylation (Lisman et al., 2002). So to confirm that Abi1–CaMKIIα interaction is disrupted by CaM and not by the downstream effects of CaM binding to CaMKIIα, binding of Abi1 to GFP-CaMKIIαT286D (a mutant that mimics autophosphorylation and therefore has open conformation) and GFP-CaMKIIαK42E, T286D (a mutant that has open conformation but lacking kinase activity) were examined (Fig. 5C). Compared with wild-type CaMKIIα, the mutants did not have significant differences in their interaction with Abi1. These results indicate that CaM binding, and not alterations in conformation or kinase activity, prohibit Abi1 binding to CaMKIIα. Given that Abi1 has homology in the CaM-binding domain of CaMKIIα (Fig. 2A) and that the presence of Ca2+/CaM disrupts Abi1–CaMKIIα interaction, we tested whether Abi1 can bind directly to CaM. Direct interaction of Abi1 (GST-83-114) to Ca2+/CaM was observed in vitro (Fig. 5D) in the presence of calcium. In addition we confirmed the interaction with an in vitro competition assay where increasing amounts of Ca2+/CaM were added to GST-47-114 and CaMKII. Binding of CaMKII and CaM to GST-47-114 were observed (Fig. 5E). Increasing amounts of Ca2+/CaM showed subsequent decrease in CaMKII binding to GST-47-114. Therefore, Abi1–CaMKIIα interaction is regulated in a Ca2+/CaM-dependent manner in which Ca2+/CaM binds to Abi1 and initiates dissociation of Abi1 from CaMKIIα.
Phosphorylation of Abi1 tSNARE at Serine 88 by CaMKIIα increases its affinity for Ca2+/CaM
Sequence analysis of Abi1 and CaMKIIα demonstrates that Abi1 has a high degree of homology with the CaMKIIα inhibitory domain, including sites of autophosphorylation (Fig. 2A). So we tested whether Abi1 is phosphorylated by CaMKIIα. Immunoprecipitation of phosphoserine proteins in 14 DIV cortical neuron lysates showed serine phosphorylation of Abi1 after treatment with 50 μm glutamate and 5 μm glycine and this phosphorylation was not detected when 10 μm KN93 was pretreated (Fig. 6A). There was no change in threonine phosphorylation of Abi1 under the same conditions (data not shown). These results suggest that activation of CaMKIIα through the glutamate receptor may induce serine phosphorylation of Abi1. Several mutants were generated using the GST-83-114 (amino acids 83–114 of Abi1) construct as the template and kinase assays were performed (Fig. 6B). Of the mutants, GST-R84E and GST-S88A showed ablation of phosphorylation, whereas GST-T96A and GST-TT113,114VA showed a slight decrease in phosphorylation. Since the binding of GST-R84E and GST-S88A to CaMKIIα was not impaired (Fig. 6C), lack of phosphorylation does not seem due to inability of these mutants to interact with CaMKIIα. Lack of phosphorylation in GST-R84E suggests that this site may be needed as a part of CaMKIIα phosphorylation consensus sequence [RXX(S/T)]. Although Abi1 has homology with CaMKIIα in three major autophosphorylation sites (Fig. 2A), Abi1 does not mimic phosphorylation in these amino acids, but instead Ser 88 of Abi1 seems to be a major phosphorylation site in the tSNARE domain by CaMKIIα. NR2B and Eag channel are also phosphorylated in the regions with homology with CaMKIIα but, because they are phosphorylated in the serine that aligns with the Thr 286 autophosphorylation site, they differ from Abi1 phosphorylation (Bayer et al., 2001; Sun et al., 2004). To determine whether Ser 88 is the major site of Abi1 phosphorylation by CaMKIIα, we transfected HeLa cells with constitutively active GFP-CaMKIIαT286D and GFP-Abi1 or GFP-Abi1S88A (Fig. 6D). GFP-Abi1S88A mutant shows a marked decrease in serine phosphorylation compared with GFP-Abi1, indicating that Ser 88 is a major site of phosphorylation by CaMKIIα.
Figure 6.

Abi1 is phosphorylated at serine 88 by CaMKIIα in vitro and in vivo and this phosphorylation enhances Abi1 binding to CaM. A, We treated 14 DIV cortical neurons with 50 μm glutamate and 5 μm glycine for 5 min with or without the pretreatment of 10 μm KN93 for 30 min. The lysates were immunoprecipitated (IP) with antiphosphoserine antibodies (anti-P-Ser) and immunoblotted (IB) with anti-Abi1 antibodies. Immunoprecipitated Abi1 is indicated with an arrowhead. B, In vitro, serine 88 in Abi1 tSNARE domain is a major phosphorylation site by CaMKII. Before pull-down (PD) assays, CaMKII was incubated with γ-P32-ATP and/or Ca+2/CaM at 30°C for 10 min to initiate CaMKII activation, and then aliquoted to samples containing wild-type (GST-83–114) or mutant GST-tSNARE proteins (GST-R84E, GST-S88A, and GST-TT113,114VA) and incubated at 30°C for 30 min. Phosphorylation of the GST fusion proteins and CaMKII was observed by autoradiography. C, Purified CaMKII was incubated with the wild-type and the mutant GST-tSNARE proteins and GST pull-down precipitates were analyzed by Western blot. D, Lysates of HeLa cells cotransfected with constitutively active GFP-CaMKIIαT286D and wild-type GFP-Abi1 or GFP-Abi1S88A were immunoprecipitated with anti-P-Ser antibodies and immunoblotted with anti-GFP antibodies. E, HeLa cells transfected with GFP-Abi1, GFP-Abi1S88A, or GFP-Abi1S88D were incubated with CaM Sepharose beads in the presence of 2 mm EGTA or 0.2 mm CaCl2. Pull-down precipitates were subjected to SDS-PAGE and immunoblot with indicated antibodies. TCL, Total cell lysate.
Since Abi1 was found to bind to Ca2+/CaM, we investigated whether Abi1 phosphorylation changes its affinity for Ca2+/CaM (Fig. 6E). Through pull-down assays with CaM Sepharose beads, we found that GFP-Abi1S88D mutant binds with higher affinity to Ca2+/CaM than wild-type and GFP-Abi1S88A mutant. This result suggests that Abi1 binds to Ca2+/CaM and that Ser 88 phosphorylation may regulate its binding affinity to Ca2+/CaM.
Abi1 serine 88 phosphorylation is essential for spine maturation via Rac pathway
Abi1 is known to be essential for Rac-dependent regulation of actin dynamics through receptor tyrosine kinase pathways (Scita et al., 1999; Innocenti et al., 2004). Activated Rac GTPase in neurons, besides fostering the development and maintenance of dendritic spines through regulation of actin cytoskeleton (Tashiro and Yuste, 2004), increases dendritic branch stability, according to reports (Li et al., 2002). Hence, we investigated the effects of CaMKIIα binding on Abi1-dependent activation of Rac through pull-down assays with GST-PBD (p21-binding domain) (Fig. 7A). Rac activation by GFP-Abi1 overexpression was suppressed by cotransfection with Flag-CaMKIIα. These results suggest that the binding of Abi1 to CaMKIIα sequesters Abi1 function to activate Rac. Next, we examined the effects of Abi1 Ser 88 phosphorylation on Abi1-dependent Rac activation (Fig. 7B). Interestingly, Abi1-dependent Rac activation was not observed in phosphorylation-ablated GFP-Abi1S88A. In contrast, phosphorylation-mimicking GFP-Abi1S88D somewhat retained Rac-activating ability although with lower efficiency than wild-type Abi1. To observe the effects of Abi1 phosphorylation in formation of dendritic spines, primary rat hippocampal neurons were transfected with the indicated constructs as shown in Figure 7C. GFP-Abi1 is localized specifically to spine heads as shown by the punctate staining, whereas GFP-Abi1ΔtSNARE is dispersed throughout neurite shafts and protrusions, indicating an essential role of the tSNARE domain for specific localization of Abi1 to spine heads. Previously, it was reported that Shank family proteins are crucial for trafficking of Abi1 to dendritic spines by interaction with Abi1 SH3 domain (Proepper et al., 2007). They showed that deletion of the Abi1 SH3 domain loses its postsynaptic localization and the mutant Abi1 is found only in the neurite shafts. Our study shows that the tSNARE domain of Abi1 is also important for its localization to synapses. The tSNARE domain may be important for retention of Abi1 in synapses because GFP-Abi1ΔtSNARE shows localization in both neurite shafts and spines. GFP-Abi1S88A shows punctate staining not only in dendritic spines but also in dendritic shafts. GFP-Abi1S88D has more pronounced punctate staining in dendritic spines, but also can be seen in the shafts as well. Morphometric analysis of neurons transfected as indicated in Figure 7C shows changes in the number of protrusions, spines, and filopodia (Fig. 7D). Overexpression of GFP-Abi1ΔtSNARE and GFP-Abi1S88A in hippocampal neurons results in an increase in protrusions and also a change in number of spines and filopodia compared with GFP-transfected control neurons. In control neurons, the majority of protrusions were spines, but in GFP-Abi1ΔtSNARE-transfected and GFP-Abi1S88A-transfected neurons, the majority of protrusions were filopodia. In addition, the protrusions in these neurons were longer (Fig. 7E). In contrast, overexpression of phosphorylation-mimicking GFP-Abi1S88D in neurons shows phenotype similar to overexpression of wild-type GFP-Abi1 (Fig. 7D,E). The increase in spine width as seen in GFP-Abi1-overexpressed neurons was not affected by overexpression of phosphomutants GFP-Abi1S88A and GFP-Abi1S88D but was affected only by GFP-Abi1ΔtSNARE (Fig. 7F). These results suggest that the tSNARE is essential for Abi1's function to modulate spine morphology and that Abi1 phosphorylation at Ser 88 by CaMKIIα plays a central role in this regulation.
Discussion
Regulation of CaMKIIα activity is of substantial interest because of its implications in various areas such as LTP, LTD, experience-based learning, memory, and structural plasticity (Silva et al., 1992; Maletic-Savatic et al., 1999; Toni et al., 1999; Lisman et al., 2002; Matsuzaki et al., 2004). In addition, model mice of neurological diseases were reported to have elevated levels of CaMKII activity or autophosphorylation. Ube3a maternal-deficient mice, a mouse model strain for Angelman syndrome, a disorder characterized by mental retardation and seizures, show increased levels of P-CaMKIIα in the hippocampus (Weeber et al., 2003). Increased P-CaMKII and activity was also observed in a mouse model for α-thalassemia X-linked mental retardation (ATX-R) (Shioda et al., 2011). Our understanding of these neurological diseases, each of which are associated with abnormal dendritic spine formation, may benefit from these studies showing the importance of CaMKIIα activity in spine regulation. However, the underlying molecular mechanisms are largely unknown. Abi1–CaMKIIα interaction provides a molecular mechanism in which CaMKIIα activity may be regulated at basal states. Our results show that knockdown of Abi1 in hippocampal neurons results in an increase in CaMKIIα Thr 286 autophosphorylation and that this upregulation coincides with the increase of P-CaMKII to the tips of dendritic protrusions (Fig. 4C). Dendritic protrusions in Abi1-knockdown neurons are characterized as mostly long protrusions lacking stable mushroom-shaped spines (Proepper et al., 2007). Injection of active CaMKIIα in hippocampal neurons also results in the formation of filopodia (Jourdain et al., 2003). The morphological changes seen in Abi1-knockdown neurons are similar to the phenotypes of neurons with upregulated CaMKIIα activity. It is possible that the changes in dendritic spine morphology related to CaMKIIα activity may be due to disruption of its regulation by Abi1.
Not only does Abi1 knockdown and GFP-Abi1ΔtSNARE overexpression phenotype resemble activated CaMKIIα in neurons, but also Rac activation. When the interaction between Abi1 and CaMKIIα is blocked, Rac may become activated in an irregular manner, resulting in an increase of filopodial growth rather than spine enlargement. Compared with spine-specific localization of wild-type GFP-Abi1, GFP-Abi1ΔtSNARE is dispersed throughout the dendrite (Fig. 7C). Therefore, GFP-Abi1ΔtSNARE may activate Rac throughout the length of the dendrite, leading to increased protrusion density rather than spine enlargement. Abi1–CaMKIIα interaction supports previous studies that demonstrate the importance of the proteins containing homology regions with CaMKIIα regulatory domain in the regulation of CaMKIIα activity. Previous studies have reported channels, such as the Eag sodium channel and the NR2B, to have homology to CaMKIIα regulatory domain (Bayer et al., 2001; Wang et al., 2002; Sun et al., 2004). The interaction of CaMKIIα with these homology regions has been found to have a role in sustaining CaMKIIα activity after glutamate stimulation. Because both the Eag sodium channel and NR2B are membrane proteins and abundant in synaptic membranes, they serve as anchors to sustain CaMKIIα activation at the membranes after excitation. Similar to Eag sodium channel and NR2B, Abi1 binds to CaMKIIα through a region with homology to the regulatory domain. On the other hand, as a soluble protein, Abi1 binds to CaMKIIα and inhibits its kinase activity in the postsynaptic density before glutamate stimulation, which may serve as a mechanism ensuring stimulus-dependent spatiotemporal regulation of kinase activation. In addition, the three proteins—Eag sodium channel, NR2B, and Abi1—exhibit different modes of regulation by glutamate signaling. NR2B and Eag potassium channel bind to CaMKIIα after glutamate signaling and enhance CaMKIIα activity, while Abi1 binds to CaMKIIα before glutamate signaling and inhibits CaMKIIα activity. Last, the region of homology in Abi1 extends to the CaM-binding domain in CaMKIIα. On the contrary, NR2B and Eag channel exhibit little similarity to the CaM-binding domain (Fig. 2A). We demonstrate that through this region in Abi1, Abi1 binds to CaM and this binding regulates Abi1–CaMKIIα interaction. Not only does CaM bind to CaMKIIα to initiate its kinase activity, but CaM also binds to Abi1 and disrupts its binding to CaMKIIα. Although CaMKIIα homology regions in various proteins may have differences in regulating CaMKIIα activity, having homology to the regulatory domain may serve as a guide for finding other endogenous regulators of CaMKIIα activity.
Numerous studies show that CaMKIIα is crucial for glutamate-dependent structural plasticity (Yamasaki et al., 2008; Yamagata et al., 2009). In CaMKIIα kinase-dead knock-in mice, tetanic stimulation failed to sustain spine enlargement, which is characteristic of tetanus-induced LTP (Yamagata et al., 2009). In addition, mice heterozygous for null mutation of CaMKIIα display phenotypes similar to schizophrenia and other neurological diseases, such as bipolar disease (Yamasaki et al., 2008; Matsuo et al., 2009). These mice display immature dentate gyrus and severe deficits in working memory. However, specific mechanisms explaining the role of CaMKIIα in modulating morphological changes in dendritic spines have not been defined. In addition, upstream signaling activating Rac in a glutamate-dependent manner has been poorly understood (Li et al., 2002; Tashiro and Yuste, 2004). Abi1–CaMKIIα interaction provides a molecular link in which glutamate stimulation regulates both Rac-dependent actin dynamics and also CaMKIIα signaling. The interaction between the two proteins results in the dual inhibition of each other's function and this impediment is resolved after glutamate treatment, resulting in formation of Ca+2/CaM that binds to both Abi1 and CaMKIIα and in activation of Rac and CaMKII pathways.
We identified a phosphorylation site in Abi1 by CaMKIIα that is crucial for Abi1's function in dendritic spine regulation. Transfection of the phosphorylation-ablated Abi1 shows dendritic morphology similar to Abi1 knockdown neurons and neurons transfected with a tSNARE deletion mutant of Abi1, which implies the significance of this phosphorylation in modulating spine morphology. Our results suggest that Abi1 phosphorylation by CaMKIIα is critical for Rac activation, which in turn regulates actin dynamics in the spine. A previous report shows that Abi1 translocates to the nucleus within 1 h of 30 μm NMDA treatment (Proepper et al., 2007). It would be interesting to see whether Abi1 Ser 88 phosphorylation has a role in nuclear translocation.
In this study, we discuss the role of Abi1 and CaMKIIα in dendritic spines. However, Abi1 has also been reported to be targeted to growth cones and early synaptic contacts during neuronal development (Courtney et al., 2000; Proepper et al., 2007; Liebau et al., 2011). Since both structures also undergo Ca2+-dependent actin remodeling, the molecular mechanism we described pertaining to dendritic spines may also be applied to regulation of growth cones and early synaptic contacts. Although we observed a later stage of development, it would be interesting to see whether Abi1–CaMKIIα interaction could modulate actin dynamics at various stages of neuronal development.
In summary, Abi1 during basal conditions inhibits CaMKIIα by binding to the kinase through its tSNARE domain, and this CaMKIIα binding sequesters Abi1-dependent Rac activation. Glutamate receptor-mediated calcium influx induces Ca2+/CaM-dependent activation of CaMKIIα, allowing Abi1 phosphorylation at Ser 88. Binding of Ca2+/CaM to Abi1 induces Abi1 dissociation from CaMKIIα, and phosphorylated Abi1 is able to activate the Rac pathway, initiating structural remodeling of dendritic spines. CaMKIIα liberated from inhibition by Abi1 is activated and phosphorylates its numerous substrates in the spine (Fig. 8). Thus, Abi1 and CaMKIIα may act synergistically to promote dynamic changes in dendritic spine morphology. We postulate that in dendritic spines the two proteins inhibit each other's function until excitatory signaling, such as activation of glutamate receptors, is initiated. Interaction of CaMKIIα with Abi1 demonstrates a novel mechanism in which CaMKIIα is inhibited by an intermolecular interaction through regions of homology, rather than an intramolecular mechanism through inhibitory domains of the holoenzyme. This interaction also makes it possible for Abi1 and CaMKIIα, two important regulators of synaptic plasticity, to keep each other in check until neuronal activation occurs. Furthermore, the interaction between these two proteins through the tSNARE domain of Abi1 implies a novel role of tSNARE domains in not only membrane fusion but also in signaling pathways. The findings in this study suggest that the activity-regulated interaction between Abi1 and CaMKIIα may be a molecular mechanism underlying activity-dependent structural plasticity in dendritic spines.
Figure 8.

Proposed model for the roles of activity-dependent interaction between Abi1 and CaMKIIα in spine regulation. At basal levels, Abi1 is bound to CaMKII and this interaction results in the mutual inhibition of their activity. Once glutamate receptors are activated, there is an increase in intracellular calcium, which induces Ca2+/CaM to trigger CaMKII activity. Active CaMKII phosphorylates Abi1 and binding of Ca2+/CaM to Abi1 elicits dissociation of Abi1 from CaMKII. Phosphorylated Abi1 then binds to Rac complex and activates Rac.
Footnotes
This work was supported by Grant M103KV010013-04K2201-01310 from the Brain Research Center, funded by the Korean Government, to D.P.
The authors declare no competing financial interests.
References
- Andersen R, Li Y, Resseguie M, Brenman JE. Calcium/calmodulin-dependent protein kinase II alters structural plasticity and cytoskeletal dynamics in Drosophila. J Neurosci. 2005;25:8878–8888. doi: 10.1523/JNEUROSCI.2005-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- Chang S, De Camilli P. Glutamate regulates actin-based motility in axonal filopodia. Nat Neurosci. 2001;4:787–793. doi: 10.1038/90489. [DOI] [PubMed] [Google Scholar]
- Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci. 2008;9:344–356. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- Courtney KD, Grove M, Vandongen H, Vandongen A, LaMantia AS, Pendergast AM. Localization and phosphorylation of Abl-interactor proteins, Abi-1 and Abi-2, in the developing nervous system. Mol Cell Neurosci. 2000;16:244–257. doi: 10.1006/mcne.2000.0865. [DOI] [PubMed] [Google Scholar]
- Dillon C, Goda Y. The actin cytoskeleton: integrating form and function at the synapse. Annu Rev Neurosci. 2005;28:25–55. doi: 10.1146/annurev.neuro.28.061604.135757. [DOI] [PubMed] [Google Scholar]
- Dubielecka PM, Cui P, Xiong X, Hossain S, Heck S, Angelov L, Kotula L. Differential regulation of macropinocytosis by Abi1/Hssh3bp1 isoforms. PloS One. 2010;5:e10430. doi: 10.1371/journal.pone.0010430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echarri A, Lai MJ, Robinson MR, Pendergast AM. Abl interactor 1 (Abi-1) wave-binding and SNARE domains regulate its nucleocytoplasmic shuttling, lamellipodium localization, and wave-1 levels. Mol Cell Biol. 2004;24:4979–4993. doi: 10.1128/MCB.24.11.4979-4993.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocenti M, Frittoli E, Ponzanelli I, Falck JR, Brachmann SM, Di Fiore PP, Scita G. Phosphoinositide 3-kinase activates Rac by entering in a complex with Eps8, Abi1, and Sos-1. J Cell Biol. 2003;160:17–23. doi: 10.1083/jcb.200206079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocenti M, Zucconi A, Disanza A, Frittoli E, Areces LB, Steffen A, Stradal TE, Di Fiore PP, Carlier MF, Scita G. Abi1 is essential for the formation and activation of a WAVE2 signalling complex. Nat Cell Biol. 2004;6:319–327. doi: 10.1038/ncb1105. [DOI] [PubMed] [Google Scholar]
- Jourdain P, Fukunaga K, Muller D. Calcium/calmodulin-dependent protein kinase II contributes to activity-dependent filopodia growth and spine formation. J Neurosci. 2003;23:10645–10649. doi: 10.1523/JNEUROSCI.23-33-10645.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng Y, Zhang J, Badour K, Arpaia E, Freeman S, Cheung P, Siu M, Siminovitch K. Abelson-interactor-1 promotes WAVE2 membrane translocation and Abelson-mediated tyrosine phosphorylation required for WAVE2 activation. Proc Natl Acad Sci U S A. 2005;102:1098–1103. doi: 10.1073/pnas.0409120102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Aizenman CD, Cline HT. Regulation of rho GTPases by crosstalk and neuronal activity in vivo. Neuron. 2002;33:741–750. doi: 10.1016/s0896-6273(02)00621-9. [DOI] [PubMed] [Google Scholar]
- Liebau S, Steinestel J, Linta L, Kleger A, Storch A, Schoen M, Steinestel K, Proepper C, Bockmann J, Schmeisser MJ, Boeckers TM. An SK3 channel/nWASP/Abi-1 complex is involved in early neurogenesis. PloS One. 2011;6:e18148. doi: 10.1371/journal.pone.0018148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- Matsuo N, Yamasaki N, Ohira K, Takao K, Toyama K, Eguchi M, Yamaguchi S, Miyakawa T. Neural activity changes underlying the working memory deficit in alpha-CaMKII heterozygous knockout mice. Front Behav Neurosci. 2009;3:20. doi: 10.3389/neuro.08.020.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky EA, Sabatini BL, Svoboda K. Structure and function of dendritic spines. Annu Rev Physiol. 2002;64:313–353. doi: 10.1146/annurev.physiol.64.081501.160008. [DOI] [PubMed] [Google Scholar]
- Proepper C, Johannsen S, Liebau S, Dahl J, Vaida B, Bockmann J, Kreutz MR, Gundelfinger ED, Boeckers TM. Abelson interacting protein 1 (Abi-1) is essential for dendrite morphogenesis and synapse formation. EMBO J. 2007;26:1397–1409. doi: 10.1038/sj.emboj.7601569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg OS, Deindl S, Sung RJ, Nairn AC, Kuriyan J. Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell. 2005;123:849–860. doi: 10.1016/j.cell.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Ryan XP, Alldritt J, Svenningsson P, Allen PB, Wu GY, Nairn AC, Greengard P. The Rho-specific GEF Lfc interacts with neurabin and spinophilin to regulate dendritic spine morphology. Neuron. 2005;47:85–100. doi: 10.1016/j.neuron.2005.05.013. [DOI] [PubMed] [Google Scholar]
- Scita G, Nordstrom J, Carbone R, Tenca P, Giardina G, Gutkind S, Bjarnegård M, Betsholtz C, Di Fiore PP. EPS8 and E3B1 transduce signals from Ras to Rac. Nature. 1999;401:290–293. doi: 10.1038/45822. [DOI] [PubMed] [Google Scholar]
- Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- Shioda N, Beppu H, Fukuda T, Li E, Kitajima I, Fukunaga K. Aberrant calcium/calmodulin-dependent protein kinase II (CaMKII) activity is associated with abnormal dendritic spine morphology in the ATRX mutant mouse brain. J Neurosci. 2011;31:346–358. doi: 10.1523/JNEUROSCI.4816-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- Sun XX, Hodge JJ, Zhou Y, Nguyen M, Griffith LC. The eag potassium channel binds and locally activates calcium/calmodulin-dependent protein kinase II. J Biol Chem. 2004;279:10206–10214. doi: 10.1074/jbc.M310728200. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Yuste R. Regulation of dendritic spine motility and stability by Rac1 and Rho kinase: evidence for two forms of spine motility. Mol Cell Neurosci. 2004;26:429–440. doi: 10.1016/j.mcn.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Toni N, Buchs PA, Nikonenko I, Bron CR, Muller D. LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature. 1999;402:421–425. doi: 10.1038/46574. [DOI] [PubMed] [Google Scholar]
- Wang Z, Wilson GF, Griffith LC. Calcium/calmodulin-dependent protein kinase II phosphorylates and regulates the Drosophila eag potassium channel. J Biol Chem. 2002;277:24022–24029. doi: 10.1074/jbc.M201949200. [DOI] [PubMed] [Google Scholar]
- Weeber EJ, Jiang YH, Elgersma Y, Varga AW, Carrasquillo Y, Brown SE, Christian JM, Mirnikjoo B, Silva A, Beaudet AL, Sweatt JD. Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J Neurosci. 2003;23:2634–2644. doi: 10.1523/JNEUROSCI.23-07-02634.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GY, Cline HT. Stabilization of dendritic arbor structure in vivo by CaMKII. Science. 1998;279:222–226. doi: 10.1126/science.279.5348.222. [DOI] [PubMed] [Google Scholar]
- Yamagata Y, Kobayashi S, Umeda T, Inoue A, Sakagami H, Fukaya M, Watanabe M, Hatanaka N, Totsuka M, Yagi T, Obata K, Imoto K, Yanagawa Y, Manabe T, Okabe S. Kinase-dead knock-in mouse reveals an essential role of kinase activity of Ca2+/calmodulin-dependent protein kinase IIα in dendritic spine enlargement, long-term potentiation, and learning. J Neurosci. 2009;29:7607–7618. doi: 10.1523/JNEUROSCI.0707-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki N, Maekawa M, Kobayashi K, Kajii Y, Maeda J, Soma M, Takao K, Tanda K, Ohira K, Toyama K, Kanzaki K, Fukunaga K, Sudo Y, Ichinose H, Ikeda M, Iwata N, Ozaki N, Suzuki H, Higuchi M, Suhara T, et al. Alpha-CaMKII deficiency causes immature dentate gyrus, a novel candidate endophenotype of psychiatric disorders. Mol Brain. 2008;1:6. doi: 10.1186/1756-6606-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Macara IG. The polarity protein PAR-3 and TIAM1 cooperate in dendritic spine morphogenesis. Nat Cell Biol. 2006;8:227–237. doi: 10.1038/ncb1368. [DOI] [PubMed] [Google Scholar]