

Abstract
The integration of neurons within the developing cerebral cortex is a prolonged process dependent on a combination of molecular and physiological cues. To examine the latter we used laser scanning photostimulation (LSPS) of caged glutamate in conjunction with whole-cell patch-clamp electrophysiology to probe the integration of pyramidal cells in the sensorimotor regions of the mouse neocortex. In the days immediately after postnatal day 5 (P5) the origin of the LSPS-evoked AMPA receptor (AMPAR)-mediated synaptic inputs were diffuse and poorly defined with considerable variability between cells. Over the subsequent week this coalesced and shifted, primarily influenced by an increased contribution from layers 2/3 cells, which became a prominent motif of the afferent input onto layer 5 pyramidal cells regardless of cortical region. To further investigate this particular emergent translaminar connection, we alternated our mapping protocol between two holding potentials (−70 and +40 mV) allowing us to detect exclusively NMDA receptor (NMDAR)-mediated inputs. This revealed distal MK-801-sensitive synaptic inputs that predict the formation of the mature, canonical layer 2/3 to 5 pathway. However, these were a transient feature and had been almost entirely converted to AMPAR synapses at a later age (P16). To examine the role of activity in the recruitment of early NMDAR synapses, we evoked brief periods (20 min) of rhythmic bursting. Short intense periods of activity could cause a prolonged augmentation of the total input onto pyramidal cells up until P12; a time point when the canonical circuit has been instated and synaptic integration shifts to a more consolidatory phase.
Introduction
Our current view of the mechanisms that underlie the integration and consolidation of neural networks is that promiscuous connections are initially supported by spontaneous intrinsic activity and subsequently pruned by emergent sensory input (Katz and Shatz, 1996). Spontaneous activity is evident in many developing neuronal circuits (Blankenship and Feller, 2010) including the neocortex (Garaschuk et al., 2000; Khazipov and Luhmann, 2006; Allène et al., 2008; Golshani et al., 2009). However, there is remarkably little understanding of the development of pathways intrinsic to the neocortex that convey such early activity, with previous studies focusing primarily on subplate neurons (Zhao et al., 2009), the period of sensory awakening in the ferret visual cortex (Dalva and Katz, 1994) or the feedforward pathways in the barrel field of the mouse somatosensory cortex (Bureau et al., 2004; Ashby and Isaac, 2011).
Neocortical pyramidal cells are generated in the embryonic telencephalon over a period of several days (E11.5–E15.5) through a process controlled via a complex genetic cascade (Molyneaux et al., 2007). They undergo radial migration to form, in an inside-out pattern, the layers of the emergent cerebral cortex (Angevine and Sidman, 1961; Rakic, 1974). In the immediate days following layer formation, signaling is mediated primarily through gap junction coupling between pyramidal cells clustered in local domains (Yuste et al., 1992; Montoro and Yuste, 2004; Dupont et al., 2006; Yu et al., 2012). From postnatal day 4 (P4)–P5 onward, there is an emergence of long-range synchrony (Golshani et al., 2009), paralleled by increased synaptic activity. However, the time course and mechanisms that drive the network transition from local domains toward the mature circuit have not been described.
Here we employ laser scanning photostimulation (LSPS) in acute in vitro mouse neocortical slices to assess gross excitatory connectivity throughout the P5–P21 postnatal time window. We find that one of the key features of this early network are long-range NMDA receptor (NMDAR)-only-mediated inputs (Isaac et al., 1995; Liao et al., 1995) that appear to predict the subsequent development of the canonical cortical circuit (Gilbert and Wiesel, 1983; Douglas and Martin, 2004). Evoking brief periods of spike activity could reshape synaptic input onto a proportion of pyramidal cells; however, this property was only apparent during a finite temporal window (≤P12). This provides support for the notion that early pyramidal cells constantly sample the milieu of activity causing them to oscillate between quiescent and integrated synaptic states (Hanse et al., 2009; Kwon and Sabatini, 2011). P12 represents a critical juncture in the development of the neocortex. Beyond this point activity can no longer facilitate the gross physiological integration of cells and there is a concomitant collapse in the number of silent, NMDAR-dependent synaptic input (Rumpel et al., 2004). As such, this would appear to represent the time point when the relay of sensory input via the supragranular layers to the output layers of the canonical cortical circuit is firmly established (Golshani et al., 2009; Ashby and Isaac, 2011; Rochefort et al., 2011).
Materials and Methods
Slice preparation.
All animal experiments were approved by the local ethical review committee and conducted in accordance with Home Office personal and project (70/6767) licenses under the UK Animals (Scientific Procedures) 1986 Act. CD1 mice of either sex, bred in the local animal facility, were deeply anesthetized with 4% isoflurane (in 100% O2) before decapitation and dissection of the brain in ice-cold, normal artificial CSF [ACSF; composition (in mm): 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, 20 glucose; pH equilibrated with 95%O2/5% CO2; all chemicals were sourced from Sigma unless otherwise specified]. Coronal slices (375–400 μm) containing the somatosensory regions of the neocortex were cut in ice-cold ACSF using a vibratome (Vibratome 3000 Plus; The Vibratome Company) before being individually transferred to an incubation chamber containing normal ACSF maintained at room temperature, where they were stored for a minimum of 1 h before recording.
Whole-cell patch-clamp electrophysiology.
Electrophysiological analysis was performed on mice (P5–P21) similar to the method previously described (Butt et al., 2005). Whole-cell tight-seal patch-clamp recordings were made from randomly selected pyramidal cells located in layers 2/3 and 5 of the motor (M1, M2; Paxinos et al., 2007) and adjacent primary somatosensory cortices (S1HL, S1Tr, S1BF). Cells (>50 μm below the surface) were selected in areas that showed good preservation of the columnar structure; assessed by the presence of apical dendrites of layer 5 pyramidal cells extending up to layer 2. Patch electrodes were made from borosilicate glass (5–8 MΩ; Harvard Apparatus) and filled with a solution containing the following (in mm): 128 K-gluconate, 4 NaCl, 0.3 Li-GTP, 5 Mg-ATP, 0.0001 CaCl2 (it should be noted that the Ca2+ concentration can be higher due to contamination with Ca2+ ions present in the other salts), 10 HEPES, and 1 glucose. To facilitate immunohistochemistry and morphological reconstruction, 0.2% Lucifer yellow was also included. For silent synapse input maps the K+ in the electrode solution was replaced with Cs+ (Rumpel et al., 2004) and the ECl adjusted to ∼0 mV.
The intrinsic maturity of the recorded cells was ascertained in normal ACSF using current-clamp configuration (MultiClamp 700B; Molecular Devices) and analyzed off-line (Clampfit v10.1). Standard electrophysiological protocols were followed throughout. Access resistance and passive membrane properties were ascertained shortly after rupturing the patch and periodically during the course of the experiments to ensure that there was no significant deterioration in the health of the cell. Cells that showed significant rundown were discarded. Intrinsic membrane properties were ascertained using short-duration (500 ms) depolarizing and hyperpolarizing current steps (0.1–0.2 Hz). All parameters were measured on at least 3 occasions for each cell.
Laser scanning photostimulation.
LSPS was performed using a ultraviolet (UV)-laser (DPSL-355/30) and accompanying UGA-40 targeting module (Rapp OptoElectronic GmbH), focused through a Zeiss Axioskop FS2-plus microscope and 10× UPLFLN objective (Olympus). Slices were preincubated for a minimum of 6 min before UV photostimulation with high divalent cation (HDC) ACSF, which was identical in composition to normal ACSF with the exception that it contained raised levels (4 mm) of MgCl2 and CaCl2, and in addition, 100 μm methyl 1-[5-(4-amino-4-carboxybutanoyl)]-7-nitroindoline-5-acetate hydrate-caged glutamate (Sigma). The direct response of the recorded cell to glutamate uncaging was assessed under current-clamp configuration. Maximum power obtained at the specimen plane was ∼2 mW. UV laser power was calibrated to evoke ≥2 action potentials when the laser was fired directly at the cell soma, but minimize suprathreshold responses elicited at distal sites. On a few occasions (n = 5) laser power was calibrated in cell attached mode before whole-cell patch-clamp configuration and only a slight difference observed compared with whole-cell recordings in the same cells. Neurons recorded in cell attached mode were marginally more excitable with 1–2 additional action potentials elicited in response to LSPS target to the cell soma. However, the ability to evoke action potentials were still restricted to the immediate layer. The direct, somatic suprathreshold glutamate response dynamics were determined for each cell and used to calculate the putative monosynaptic response interval for the subsequent mapping experiments performed under voltage clamp. The EPSC response interval was defined as running from the average onset of the first elicited action potential to 100 ms after the last; the exception being those mapped using the Cs+-based intracellular electrode solution when the laser power and response interval for these cells was based on the average for cells previously mapped at the same developmental stage using standard intracellular solution. For all the glutamatergic synaptic input mapping experiments the uncaging grid was organized with 50 μm between laser target points, which were triggered in a pseudorandom pattern to prevent sequential stimulation of adjacent sites (Shepherd et al., 2003). In addition the laser was fired at 1 Hz, with no stimulation at adjacent sites within 4 s. The width of the grid was kept constant at 450 μm, while the length (650–850 μm) varied according to developmental age. The first run through of the targeting protocol was conducted in current clamp to confirm the direct response profile of the cell and the absence of distal suprathreshold responses. Cells were then mapped a further 5–8 times under voltage clamp (holding potential: −60 mV). Current traces were analyzed offline using Minianalysis 6.0 (Synaptosoft Inc.) and all EPSCs logged. In instances where multiple, summating EPSCs were observed within the event detection window (see Fig. 3B, trace 3), the minianalysis multipeak extrapolation function was used to ensure an accurate representation of total input. The number and amplitude of EPSCs that occurred within the response interval were subsequently extracted using a customized Matlab R2011 script (The MathWorks). To generate the synaptic input map the sum amplitude of the time-locked EPSCs was calculated for each target spot per run, and then averaged across all sweeps. A photomicrograph was taken of the targeting grid relative to the acute in vitro slice using the Axiovision v4.6 module of the UGA-40 to allow direct correlation of the laser target points and the layer boundaries.
Figure 3.
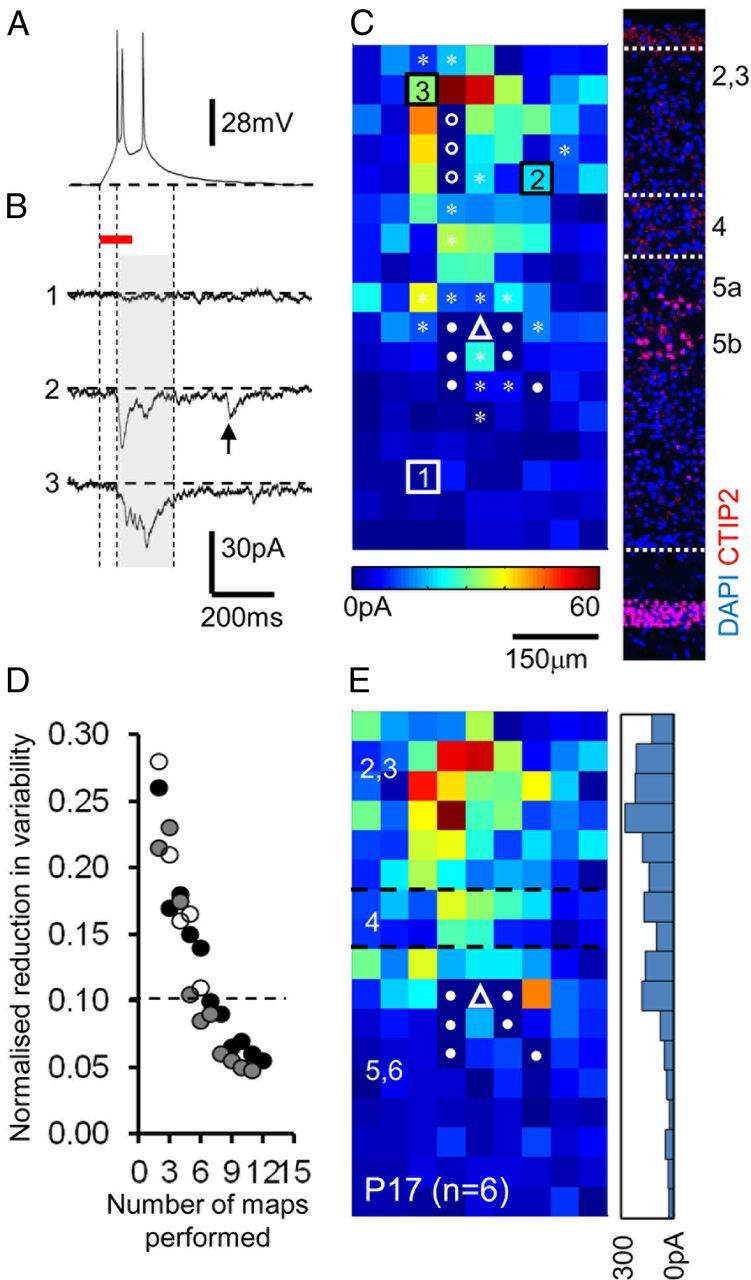
Application of LSPS to map emergent connectivity of neocortical pyramidal cells across layers 2/3 and 5. A, A tuned (only evoking 2–3 action potentials), direct current-clamp response to LSPS evoked by targeting the UV laser at the soma of the recorded pyramidal cell. B, Voltage-clamp responses observed after the laser was fired at spots distal to the recorded pyramidal cell. The onset of the laser pulse is indicated by the left vertical dashed line, the duration (100 ms) is indicated by the horizontal red line. The onset of the analysis window (shaded gray) is marked with the middle vertical dashed line and the offset—which occurs 100 ms after the last action potential from the direct response profile (shown in A)—is marked by the right vertical dashed line. The arrow, trace 2, indicates an EPSC that would be excluded from the data analysis. C, A single-sweep, raw EPSC input map for a P17 layer pyramidal cell (the location of the soma denoted by the white triangle). Numbered squares refer to the traces shown in B; squares with circles indicate target sites with large-amplitude, direct glutamate-uncaging responses; open circles indicate points at which EPSCs were observed after repeat runs of the same map; sites with low-amplitude direct responses in which EPSCs were included are indicated by the white asterisks. Inset, Corresponding DAPI (4′,6′-diamidino-2-phenylindole dihydrochloride) stain (blue) of the somatosensory cortex shown to scale with immunohistochemistry for CTIP2 (red) to delineate approximate layer boundaries. D, The reduction in variability (error) associated with repeated mapping for 3 cells mapped at P11–P12. E, Average map (n = 6 sweeps) for the cell shown in A–C; approximate (to the nearest 50 μm on the vertical axis) layer boundaries delineated by white dashed lines. The location of the recorded cell is shown by the white triangle; squares with filled circles indicate target sites with large-amplitude, direct glutamate-uncaging responses that precluded the analysis of EPSCs at this target spot. Inset, Histogram showing the distribution of synaptic input (summed for each 50 μm grid line) across the vertical orientation.
Previous reports have demonstrated differences in the source of synaptic input onto the two major physiological subtypes of layer 5 pyramidal cells (Agmon and Connors, 1989; Schubert et al., 2001, 2006). Given the late emergence of the intrinsic bursting properties of these cells (Flint et al., 1997) data from layer 5 pyramidal cells was pooled regardless of putative subtype. Recovered morphologies suggest that the vast majority were of the simple dendritic arbor, regular spiking subtype (Schubert et al., 2001)
Technical consideration for LSPS mapping of putative NMDA receptor-mediated afferent input.
LSPS mapping of NMDA receptor (NMDAR)-mediated afferent input presented a number of technical challenges. (1) The increased sensitivity of the postsynaptic cell to glutamate via NMDARs meant that we encountered additional direct (short latency, long duration) responses at +40 mV (see Fig. 8D), where previously we had observed none at −70 mV. However, these were often of sufficiently low amplitude that they did not obscure EPSCs elicited as a result of glutamatergic stimulation of adjacent pyramidal cells. (2) We had to be able to account for the contribution of GABAergic input to the PSCs observed at +40 mV. We used a modified cesium chloride electrode solution that gave an approximate reversal potential for chloride (ECl) of 0 mV, meaning that any GABAA receptor-mediated conductances would contribute at both holding potentials; using standard intracellular electrode solution IGABA was barely detectable at a holding potential of −70 mV. The success of our strategy was confirmed by both mapping at a holding potential of 0 mV, which revealed no detectable conductances across the extent of the uncaging grid (data not shown) and also UV laser uncaging of 100 μm α-carboxy-2-nitrobenzyl ester-caged GABA (Life Technologies) in proximity of the recorded cell. The latter caused a picrotoxin-sensitive conductance at −70 and +40 mV but no response at 0 mV (data not shown). This allowed us to attribute sites where PSCs were observed solely at +40 mV as those possessing inputs carried by putative NMDARs. (3) To confirm these novel inputs were synaptic in origin, and caused by NMDAR-dependent currents, slices were incubated in a 5 μm concentration of the open channel NMDAR blocker MK-801(5H-dibenzo[a,d]cyclohepten-5,10-imine; dizocilpine maleate) and 4 μm bicuculline methiodide for ∼45 min. This approach has been used previously to dissociate the role of synaptic and extrasynaptic NMDARs (Hardingham et al., 2002). Slices were then maintained in normal ACSF+MK-801 briefly before recording and mapping in HDC ACSF plus caged glutamate.
Figure 8.
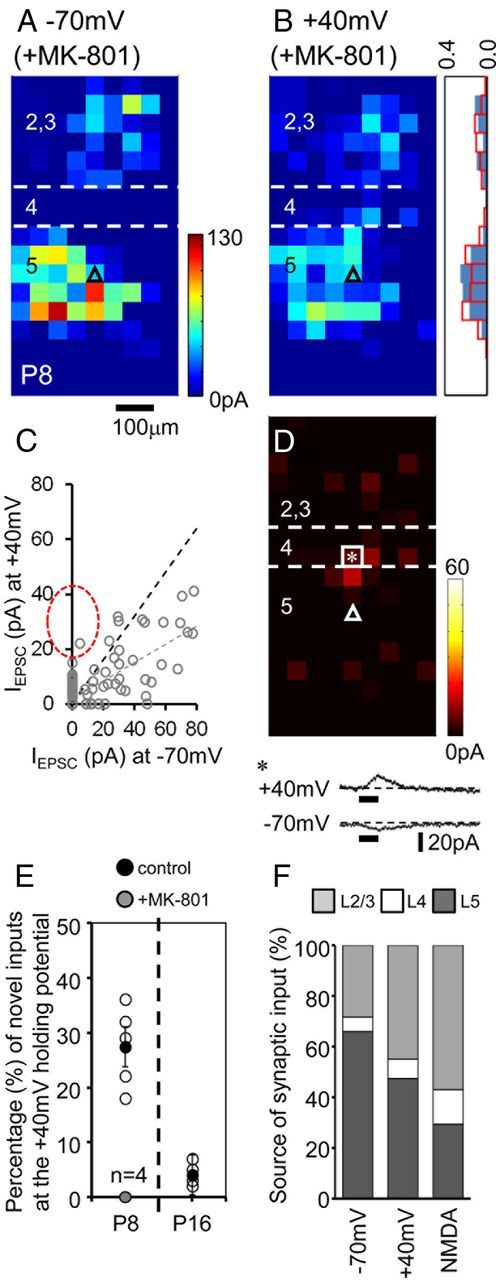
Synaptic nature and source of the NMDA receptor-mediated input in the early (P8) neocortex. A, B, LSPS maps recorded from a layer 5 somatosensory pyramidal cell at holding potentials of −70 mV (A) and −40 mV (B) which had previously been incubated for 45 min in 4 μm bicuculline and 5 μm MK-801. Inset, Histogram showing the normalized input across the vertical axis. C, Plot comparing the amplitude of the evoked EPSCs for each laser target spot. The dashed black line indicates the ratio expected from the observed somatic direct glutamate; the gray dashed line, linear regression for all the observed synaptic responses; the red dashed oval, expected distribution for exclusively NMDAR responses observed under control conditions. D, Plot of laser target points with evoked synaptic responses biased toward that observed at the +40 mV holding potential; a direct glutamate response that is more prominent at +40 mV is shown below and was identified from the laser target point highlighted by the white square and asterisk. E, Plot of the percentage of novel synaptic inputs observed at a holding potential of +40 mV in control cells (white circles for individual cells; average indicated by the black circle ±SEM) mapped at either P8 (n = 5) or P16 (n = 4); light gray data points indicate the equivalent data obtained from P8 cells preincubated in MK-801 (n = 4). F, Histogram showing the average percentage afferent input from each layer impacting on P8 layer 5 pyramidal cells recorded at the different holding potentials (n = 5); NMDA, the layer source of the novel input observed at +40 mV.
Pharmacological stimulation of network activity.
Immediately after LSPS cells were returned to normal ACSF and the viability of the cell assessed. To globally stimulate activity in the acute in vitro slices we incubated them in either elevated K+ (4 mm) ACSF to elicit cGDPs (Allène et al., 2008) or nominally zero Mg2+ concentration (0-Mg2+) ACSF (Silva et al., 1991). After a period of 20 min the cells were returned temporarily to normal ACSF until residual activity had diminished to baseline levels before being remapped in HDC ACSF as described above.
Morphological reconstruction of cells.
After completion of the electrophysiology, slices containing Lucifer yellow-filled neurons were transferred to 4% paraformaldehyde and fixed for a maximum of 3 h. at 4°C under weighted fine gauze, to prevent distortion of the tissue. The layer 5 boundary was defined at various developmental ages using a rat anti-Ctip2 antibody (1:400; Abcam plc) incubated overnight at 4°C and then visualized using Goat anti-Rat Cy3- or Cy5-conjugated secondary antibodies (both 1:200; Millipore). After fluorescent image capture, the Lucifer yellow was converted to a dense, dark-colored diaminobenzidine content to facilitate detailed histological reconstruction as described previously (Butt et al., 2005).
Statistical analysis.
All statistical analysis was performed using Matlab R2011. The normality of the data was tested using a combination of Lilliefors test (lillietest) and normplot function. Differences in population data conforming to a normal distribution were compared using a Student's t test. For individual cells one-way ANOVA (anova1 function, Matlab R2011) was used to look for systematic differences in the level of synaptic input across the depth of the cortical column; the null hypothesis being that the input was evenly distributed across this axis. To compare afferent input maps for the same cell recorded under two conditions (e.g., synaptic input at a holding potential of −70 mV or +40 mV) we performed two-tailed Wilcoxon signed-rank test. For all statistical tests the difference was deemed significant if p < 0.05. All data are reported as mean ± SEM; percentage data was arcsine transformed before calculation of the SEM and reported as +SEM%, −SEM% where a difference in the SEM percentage value ≥1% was calculated.
Results
Photostimulation uncaging of glutamate in the developing neocortex
One of the challenges presented when using photostimulation technologies to probe connectivity over a prolonged early developmental time window is the shift in the intrinsic membrane properties and excitability of the target cells (Kasper et al., 1994; Luhmann et al., 2000; Maravall et al., 2004; Oswald and Reyes, 2008). To control for this, our approach was to calibrate the intensity of the UV LSPS according to the direct glutamate response of the recorded pyramidal cell recorded in the mouse neocortex acute in vitro slice. Our objective was to consistently evoke two or more action potentials in the recorded cell only when the laser was fired directly at the soma. In an initial series of experiments (n = 6 cells) we targeted the immediate proximity of the recorded cell soma with a 25 μm spaced pseudorandom grid, and over a number of repeats (n ≥ 3 per laser setting) varied the laser intensity (Fig. 1A) until our criterion of ≥2 action potentials at the fewest spots was fulfilled (25% laser setting in the example shown in Fig. 1A,B). For all six cells we could not resolve to less than two adjacent target spots suggesting that the effective resolution of our LSPS protocol was on the order of 50 μm diameter.
Figure 1.
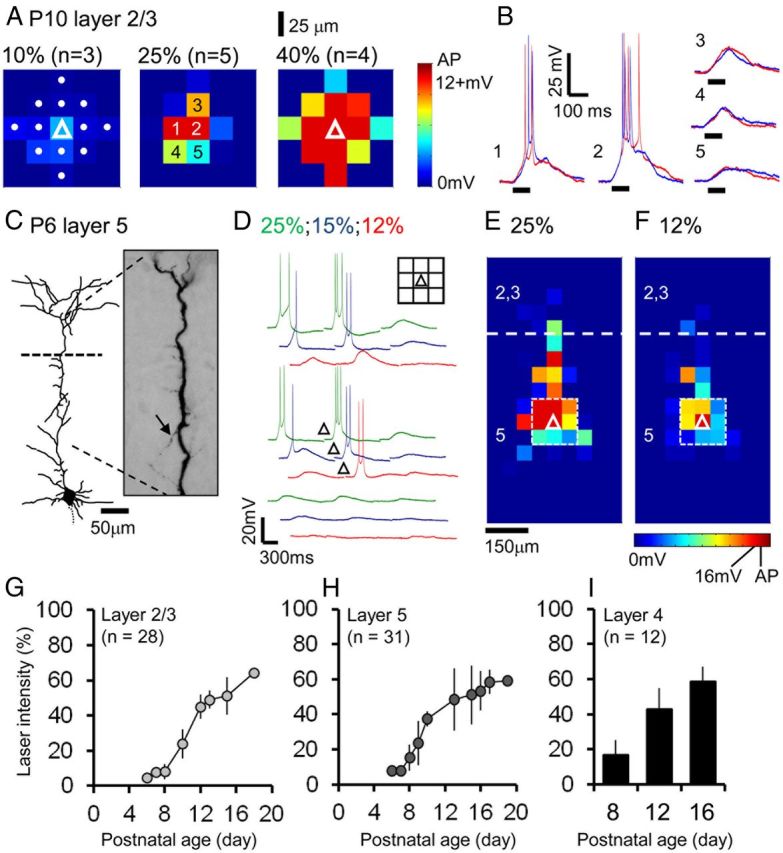
Adjustment and calibration of laser intensity according to postnatal developmental time point. A, To establish the approximate area excited by LSPS-evoked glutamate uncaging, the UV laser (355 nm; 100 ms duration) was repeatedly fired at a range of intensities (from left to right: 10%, 25%, 40% of maximal laser power at the slice interface) in the immediate vicinity of the recorded P10 pyramidal cell (white triangle, left panel) at target points spaced at a distance of 25 μm (white dots, left panel). Action potentials (red squares) were consistently evoked at two or more spots at higher intensities. Raw current-clamp traces from the numbered laser target points at 25% laser power (middle panel) are shown in B. B, Superimposed traces from two repeats (red and blue traces) at 25% laser power. Direct responses matched the duration (100 ms) of the laser pulse (bold black line under the traces). C, Reconstructed morphology of a P6, layer 5 pyramidal cell. At early ages the relatively simple dendritic arbors with numerous small neurites (arrow) required careful adjustment of the laser intensity. D, Direct stimulation profiles of the cell (C) at a range of laser powers; only spots in the immediate vicinity of the recorded cell are shown for clarity; full averaged response maps are shown in E and F. Inset, Diagram of the 150 × 150 μm grid with the location of the cell soma target spot indicated by the triangle. E, F, Complete direct stimulation maps of the cell shown in C and D at 25 and 12% laser power, respectively. Calibration of the laser intensity ensured that action potentials (red squares) were only elicited when the laser was fired directly at the cell soma (indicated by the white triangles) during the columnar LSPS mapping (50 μm spaced grid). The laser spots enclosed by the white dashed box correspond to those points shown in D; the layer 2/3 to 5 boundary (white dashed line) is approximated to the nearest 50 μm. Calibration of the laser intensity required to evoke action potentials at the cell soma revealed increased power for layer 2/3 (G), layer 5 (H), and a smaller sample of layer 4 (I) pyramidal cells over the developmental period studied; all values ±SEM.
To map the translaminar synaptic input onto the developing pyramidal cells we used an extended 50 μm spaced pseudorandom uncaging grid programmed using the laser targeting UGA-40 system. Great care was taken at the start of all subsequent recordings to calibrate the power of the UV laser using a similar approach to that mentioned above, but now controlled for the entire extent of the dendritic arbor. This was particularly problematic at early ages when the relatively simple dendritic arbor of the pyramidal cells was dominated by a broad apical dendrite (Fig. 1C) and numerous thin neurites. The most effective strategy—similar to that reported for 2-photon glutamate uncaging (Ashby and Isaac, 2011), was to use low intensity (<2 MW in the plane of the slice), long-duration (100 ms) pulses that reduced distal LSPS-evoked spikes (Fig. 1D–F), without compromising the ability to reliably generate action potentials at the cell soma (Fig. 1D, red traces, F). A plot of the calibrated laser power over development revealed a pronounced shift around P9–P12 in the excitability of the pyramidal cells in both layer 2/3 and 5 (Fig. 1G,H). A difference in the excitability of layer 4 neurons to short-duration, high-power LSPS at early postnatal ages has previously been reported (Bureau et al., 2004). Mindful that such a difference could cause distortions in our excitatory input maps, we recorded from layer 4 pyramidal cells at three time points around P12 (Fig. 1I). These data suggest that the response of pyramidal cells to our LSPS protocol is fairly uniform across all the layers of the cortex, not long after the inside-out development of the neocortex has ceased (Rakic, 1974; Miller, 1988).
Our approach accounted for variation in the sensitivity of pyramidal cells to LSPS-evoked glutamate uncaging over development, which in turn reduced action potential generation in response to distal LSPS, thus maintaining the spatial resolution of the maps. However, the requirement for long-duration (100 ms) laser pulses, necessary to achieve the latter, resulted in increased temporal variability in the onset and time course of action potential generation (Fig. 1B). To establish the putative monosynaptic event window parameters for the calibrated laser pulse, we repeatedly stimulated (0.05 Hz) action potentials in a number of cells recorded in HDC ACSF either at the beginning (P5–P8, n = 46 evoked bursts of action potentials from 4 cells) or end of our developmental time frame (P17–P21, n = 59 from 4 cells). Recorded cells had similar spike distributions at both developmental time points (Fig. 2A,B), with a high probability of the first spike occurring toward the end of the 100 ms laser pulse, while the last spike was triggered over a broader time frame culminating ≤200 ms after termination of LSPS. Afferent input onto the cells were then mapped using our standard LSPS protocol (in HDC ACSF), and all the detected EPSCs pooled for the 4 cells recorded at each age group. The temporal distribution of the EPSCs at early (Fig. 2C) and late (Fig. 2D) ages revealed an increase in the number of EPSCs toward the end of the 100 ms laser pulse, around the time of the average first spike. This peaked shortly thereafter and then tailed off to background levels. Based on our analysis of the spike and EPSC distributions we decided to use the average onset of the first spike and an arbitrary end point of 100 ms after the average last spike to identify and collect putative monosynaptic EPSCs for the maps (gray histogram bars, Fig. 2C,D). Regardless of the developmental age this time window captured on the order of 65–70% of the recorded EPSCs (Fig. 2E,F).
Figure 2.

Capture of putative monosynaptic EPSCs elicited in response to reliable, long-duration photostimulation. A, B, Frequency histograms of the first (black) and last (gray) action potentials recorded in current-clamp mode in response to firing the calibrated laser pulses (100 ms; bold black line above the graphs) directly at the cell soma for both early (A) and late (B) time points (n = 46 and 59 photostimulation events, respectively). C, D, The number of EPSCs per 10 ms bin recorded under voltage-clamp conditions (in HDC ACSF) in response to firing the laser across the whole extent of the mapping grid. The arbitrary window used to pull out putative monosynaptic EPSCs (see text for full details) is highlighted by the gray histogram bars. E, F, A similar proportion of the recorded EPSCs was captured at both ages using our arbitrary criteria as revealed by the cumulative frequency plots.
LSPS mapping of glutamatergic synaptic input
Our analysis of glutamatergic synaptic input onto pyramidal cells commenced at P5, a time point that coincides with the emergence of synaptically driven oscillations in the neocortex (Dupont et al., 2006; Allène et al., 2008), and also when we could first observe consistent EPSCs in response to LSPS. We divided the time frame of our investigation into four arbitrary time windows: P5–P8, P9–P12, P13–P16, and P17–P21. To reduce background synaptic activity and attenuate LSPS-evoked polysynaptic events we perfused the slices with HDC ACSF before and during photostimulation (Shepherd et al., 2003; Jin et al., 2006). For all cells the individual cell-specific putative monosynaptic window was established by first calibrating the laser to fire two or more (typically 2–4) action potentials when targeted at the cells soma recorded in current-clamp mode (Fig. 3A). Synaptic input was subsequently mapped with the pyramidal cells voltage-clamped at −60 mV (Fig. 3B,C). Firing of the calibrated UV-laser at a give spot in the tissue (and resultant localized uncaging of glutamate) evoked one of three responses in the recorded cell: (1) no response at all (Fig. 3B, trace 1), indicative that that particular target point did not contain any pyramidal cells forming afferent connections onto the recorded cell; (2) a large inward current that commenced from the moment the UV-laser fired and gradually increased in amplitude over the duration of photostimulation which corresponded to a direct glutamate response (data not shown; see, however, Fig. 1B); (3) time-locked EPSCs that occurred at a delay from laser onset (Fig. 3B, traces 2, 3) indicative that presynaptic cells were present at that target point. In contrast to direct responses, EPSCs had rapid rise times and were of short duration. EPSCs that occurred within the defined monosynaptic event interval (Fig. 3B) were extracted and the amplitude measured for each pixel across the entire grid. The sum amplitude of the EPSCs was then calculated and plotted to generate a single, raw input map (Fig. 3C). EPSCs coincident with, but not obscured by, low amplitude direct responses were included in the analysis (Fig. 3C, asterisks). To be consistent in our allocation of layers over the broad developmental time window examined, we related the position of the recorded, Lucifer yellow-filled neuron to the deep layer (predominant layer 5) marker CTIP2 (Fig. 3C, inset) in addition to the photomicrograph and anatomical boundaries in the recovered morphology (data not shown). Layers were approximated to the nearest pixel. We examined the variability between individual map by repeatedly recording (≥5 min between each mapping epoch) from the same cell for as long as the whole-cell patch clamp was stable (n = 3 cells; P11 and P12). We then used an ad hoc criterion of the difference between the running average map and the latest epoch, divided by the variances of those maps, to study how similar a newly recorded map was compared with the current estimate. Repeated mapping (Fig. 3D) greatly reduced the potential for erroneous inputs. We settled on a ∼10% error in the final synaptic input map, for which we needed to collect and average at least 5 individual maps (Fig. 3E). This represented a fair trade off between maintaining stable whole-cell patch clamp and the reliability of our reported data.
Development of excitatory synaptic input in motor and adjacent somatosensory cortices
Previous studies on the development of excitatory synaptic input have focused on the subplate (Zhao et al., 2009; Viswanathan et al., 2012), as well as local and feedforward pathways in the highly specialized whisker barrel cortex (Bureau et al., 2004; Ashby and Isaac, 2011) and layer 2/3 of the visual cortex (Dalva and Katz, 1994). We were intrigued by the possibility that there might be early, stereotypical connections conserved across cortical regions independent of thalamic input; connections that provide the conduit for the spontaneous, intrinsically generated, activity observed in early cortical circuits (Garaschuk et al., 2000; Allène et al., 2008; Golshani et al., 2009). All the experiments were performed on coronal slices, severing any subcortical input. We mapped the excitatory synaptic inputs onto a total of 70 pyramidal cells, 36 of which were located in motor regions (Fig. 4) of the neocortex and 34 in adjacent somatosensory areas (Fig. 5). At early ages the observed LSPS-evoked EPSCs were invariably of small amplitude (<10 pA) and could be triggered in a relatively diffuse manner across the extent of the LSPS grid (Figs. 4A,E, 5A,E), with considerable variability between cells in terms of the layer providing the predominant input (Figs. 4D,H, 5D,H). From the end of the first postnatal week onward, more prominent sources of excitatory input became apparent in both of the cortical regions examined (Figs. 4B, 5B,F). These continued to develop (Fig. 4F) so that by the later time points (Figs. 4C,G, 5C,G) we could observe afferent input consistent with the canonical circuit that has previously been described for the mammalian neocortex (Gilbert and Wiesel, 1983; Thomson and Bannister, 2003; Douglas and Martin, 2004).
Figure 4.
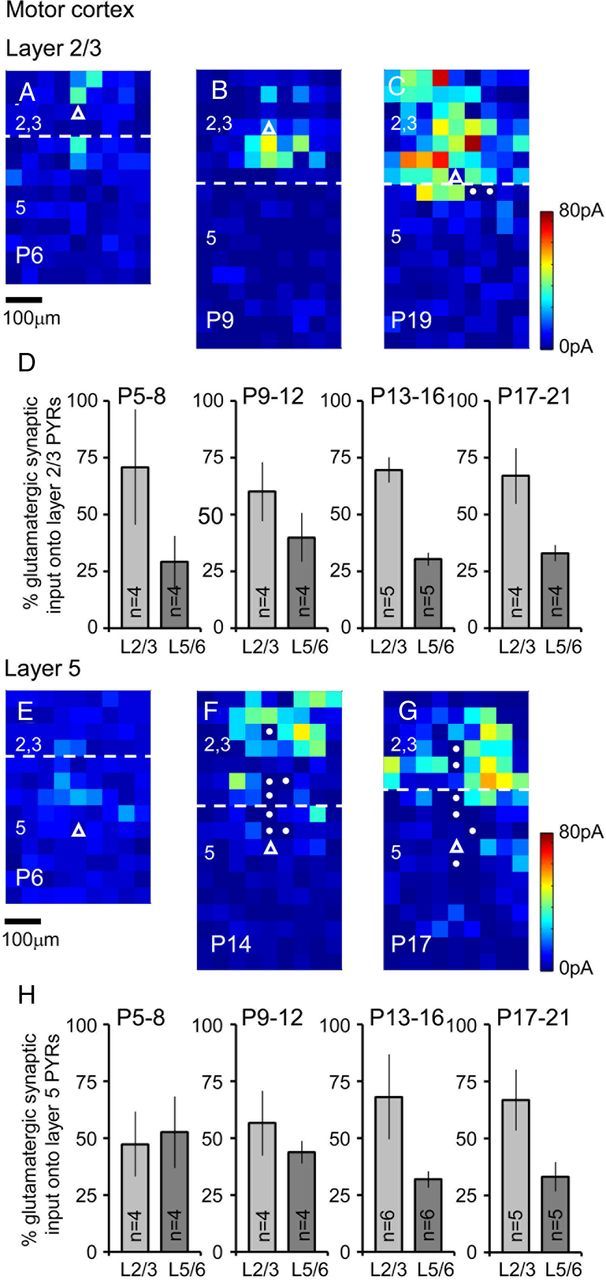
LSPS-derived synaptic input maps for motor cortex pyramidal cells throughout early postnatal development. A–C, Synaptic input maps (n ≥ 5 sweeps per cell) for pyramidal cells located in layer 2/3 of the mouse motor cortex at P6 (A), P9 (B), and P19 (C). Layer boundaries are marked with dashed white lines and the location of the recorded cells highlighted by the white open triangles; target spots with filled circles indicate those where the size of the direct glutamate-uncaging responses precluded analysis of the synaptic input. D, Percentage input (±SEM) onto layer 2/3 motor cortex pyramidal cells; light gray histogram bars, input from layer 2/3; dark gray, input from layer 5/6. E–G, Example synaptic input maps for layer 5 motor cortex pyramidal cells recorded at P6 (E), P14 (F), and P17 (G). H, Percentage input similar to that shown in D but for layer 5 pyramidal cells. Amplitude (pA) calibrations for A–C and E–G are shown adjacent to C and G, respectively. All maps are shown to the same micrometer scale (indicated under A and E).
Figure 5.
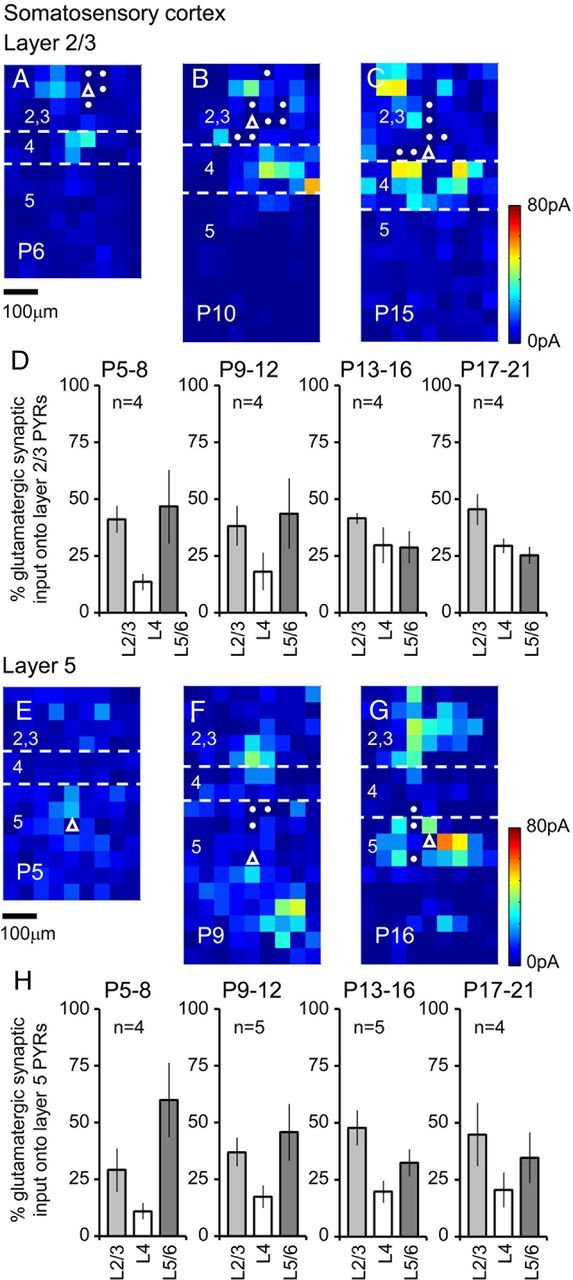
LSPS-derived synaptic input maps for somatosensory cortex pyramidal cells throughout early postnatal development. A–C, Synaptic input maps (n ≥ 5 sweeps per cell) for pyramidal cells located in layer 2/3 of the mouse somatosensory cortex at P6 (A), P10 (B), and P15 (C). Layer boundaries are marked with dashed white lines and the location of the recorded cells highlighted by the white open triangles; target spots with filled circles indicate those where the size of the direct glutamate-uncaging responses precluded analysis of the synaptic input. D, Percentage input (±SEM) onto layer 2/3 somatosensory cortex pyramidal cells; light gray histogram bars, input from layer 2/3; white bars, input from layer 4; dark gray, input from layer 5/6. Example synaptic input maps for layer 5 somatosensory cortex pyramidal cells recorded at P5 (E), P9 (F), and P16 (G). H, The percentage input onto the recorded cells is shown, similar to D but for afferent input onto layer 5 pyramidal cells. Amplitude (pA) calibrations for A–C and E–G are shown adjacent to C and G, respectively. All maps are shown to the same micrometer scale (indicated under A and E).
The overall development of afferent connectivity was similar across the two cortical regions examined (Fig. 6). We pooled the data from the somatosensory cortices as there was no significant difference in total synaptic input between age-matched S1BF and non-S1BF regions (data not shown). There was a more gradual increment in total input on pyramidal cells in somatosensory areas (Fig. 6A), with gross connectivity showing a significant increase around the moment when the circuit first experiences concerted feedforward sensory input (Bureau et al., 2004). In contrast, pyramidal cells of the motor cortex showed a significant increase in glutamatergic input later in our developmental time frame (Fig. 6B; for representative example, see Fig. 4C). We also examined the degree to which afferent input was focused across the layers of the cortex. One-way-ANOVA was performed to test the null hypothesis that the source of synaptic input was evenly distributed across the vertical/ layer axis (Fig. 6C,D). This revealed that toward the end of our period of analysis (P17–P22) all somatosensory pyramidal cells received focused input (p < 0.05; ANOVA), whereas not all layer 5 motor pyramidal cells did (Fig. 6D). These differences aside, it was evident that regardless of the layer or region, pyramidal cells were increasing dominated by layer 2/3 afferent input over time (Hooks et al., 2011). This was particularly noticeable in layer 5a/upper 5b (marked with Ctip2) pyramidal cells (see also Anderson et al., 2010) when compared side-by-side across development (Fig. 6E,F). In the days immediately after P12 there was a significant increase in the total amount of layer 2/3 input onto layer 5 somatosensory pyramidal cells (Fig. 6G) with a similar pattern observed in the motor cortex (Fig. 6H). An almost identical timeline was observed in layer 2/3, somatosensory cells showed a significant increase in local afferent input from layer 2/3 after P12 (p = 0.02; P9–P12, average of 601 ± 91 pA; P13–P16, 986 ± 133 pA) whereas the increase was delayed in motor cortex until P17–P21 (p < 0.01; P13–P16, average of 826 ± 63 pA; P17–P21, 1661 ± 154 pA).
Figure 6.
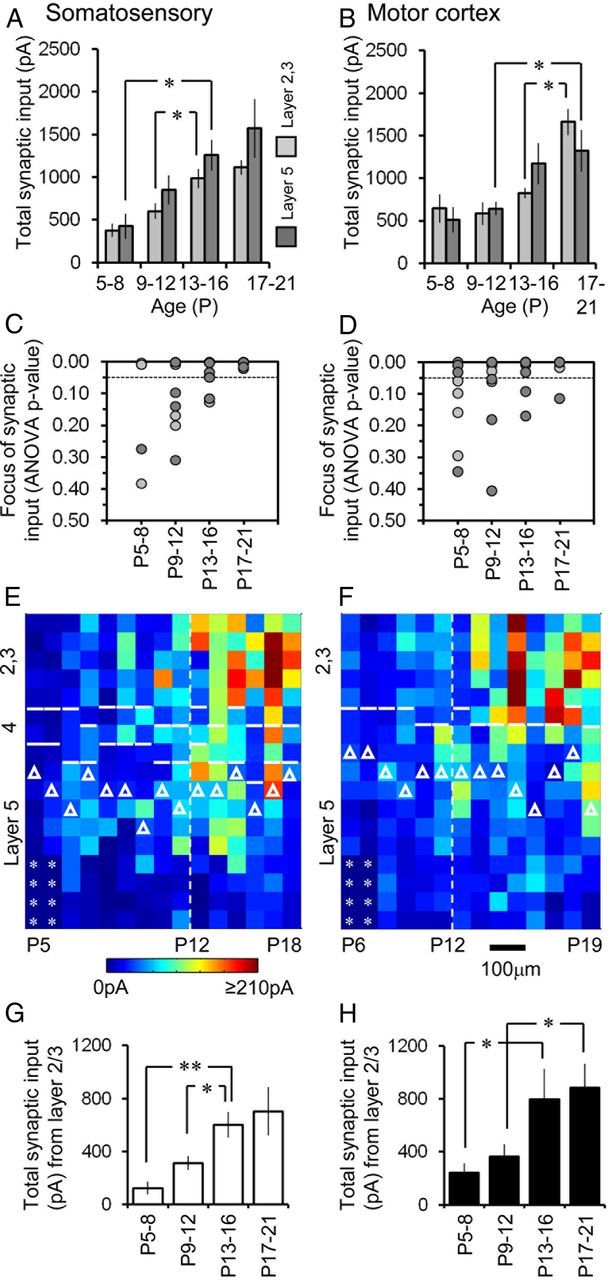
Development of synaptic input onto somatosensory and motor cortex pyramidal cells. A, Histogram showing the average (±SEM) total synaptic input onto layer 2/3 (light gray) and 5 (dark gray) somatosensory pyramidal cells over development (*p < 0.05, significant difference in the population data; Student's t test). B, Corresponding data for motor cortex. C, The degree to which synaptic input was distributed across the vertical orientation of map was determined using a one-way ANOVA with a null hypothesis that the input onto the recorded somatosensory cell was evenly distributed across this axis. Each data point represents a single cell located in either layer 2/3 (light gray) or 5 (dark gray); the null hypothesis was rejected when p < 0.05; p = 0.05 is indicated by the dashed line. D, Corresponding data for motor cortex. E, Side view of a group of input maps for somatosensory cortex layer 5 (predominantly layer 5a) pyramidal cells over development, ordered according to increasing age from left to right (the age of each cell has not been annotated for clarity). Cell body location indicated by the white triangles; layer boundaries indicated by the white lines; asterisks indicate points not targeted during LSPS in shortened P5–P6 postnatal maps. F, Side view of motor cortex layer 5 pyramidal cell input maps similar to that shown for somatosensory cortex in E; amplitude (pA) calibration for E and F shown under E; micrometer scale shown under F. G, H, Development of synaptic input on somatosensory (white histogram bars) and motor (black histogram bars) cortex layer 5 pyramidal cells, respectively. All values reported ±SEM; indicated by asterisk, *p < 0.05, **p < 0.01, significant difference (Student's t test) in the populations.
NMDAR-mediated contribution to early synaptic input
The shift in inputs onto layer 5 pyramidal cells independent of cortical region occurs at a time that is consistent with the progressive maturation of the canonical cortical circuit (Bureau et al., 2004; Chittajallu and Isaac, 2010; Ashby and Isaac, 2011) and provides an intriguing insight into the dynamics of this emergent multilayered network. It is unclear what the substrate for this process is. However, the fairly rapid emergence of the circuitry post-P12 suggests that some form of template must already be in place. Previous experiments detailing the development of glutamatergic input have focused primarily on that mediated by AMPA receptors (AMPARs) (Bureau et al., 2004). However, it is widely accepted that NMDAR-mediated synaptic transmission is important at early developmental time points and required for cortical early network oscillations (cENOs; Allène and Cossart, 2010).
To gain an understanding of the spatial distribution of NMDAR-mediated inputs onto pyramidal cells over development, we mapped layer 5 pyramidal cells located in the primary somatosensory cortex at P8 (n = 5) and a later age (P16; n = 4). We alternated between two holding potentials (−70 mV, Fig. 7A,D,H; and +40 mV, Fig. 7B,E,I), using a Cs+ replacement intracellular electrode solution (Rumpel et al., 2004). It was apparent from the raw traces that while at most target points PSCs observed at −70 mV were replicated at +40 mV (Fig. 7A,B, top two traces), there were on occasion novel PSC responses at the depolarized holding potential (Fig. 7A,B bottom two traces). Analysis of the 10–90% rise time of the mapped PSCs at both holding potentials (Fig. 7C) revealed the presence of a second PSC population with slower dynamics at the depolarized potential. LSPS mapping of afferent input onto P8 cells at −70 mV (Fig. 7D) revealed the pattern of synaptic input similar to that we had previously observed, namely that at early ages a fair proportion of the input came from the immediate vicinity of the cell. This distribution was altered at +40 mV input (Fig. 7E) with an increase in input from layer 2/3 (histogram, Fig. 7E). To gain a better understanding of the extent of the NMDA contribution we compared the average PSC amplitude observed at a holding potential of +40 mV with that detected at −70 mV across all the uncaging sites (Fig. 7F). This revealed a cluster of novel responses observed at +40 mV. We performed a Wilcoxon signed-rank test to test the null hypothesis that there was no difference in the distribution of the synaptic input measured at the two holding potentials. The null hypothesis was rejected for all five P8 cells. However, it was possible that this was influenced by the rectification in synaptic EPSC amplitude measured at +40 mV (Kumar et al., 2002). For comparison we also determined the corresponding ratio for the somatic glutamate response under control conditions [Fig. 7F, solid black line, inset trace (i)] and in the presence of a 20 μm concentration of the NMDAR antagonist AP-5 [Fig. 7F, dashed black line, inset trace (ii)]. The somatic response did not exhibit such pronounced rectification. We used the AMPA-ratio derived from the somatic glutamate response to calculate the expected AMPA-only EPSC amplitude at +40 mV for each laser point (data not shown). Across all P8 cells test the distribution of the predicted and actual, observed +40 mV response amplitude was significantly different in favor of the latter (two-tailed Wilcoxon signed-rank test; p < 0.01 for the example shown, Fig. 7F). A plot of the points with an increased amplitude or entirely novel response at the holding potential of +40 mV (Fig. 7G, asterisk) revealed a prominent skew toward layer 2/3 target points that was consistently observed across all the P8 cells recorded (n = 5). In contrast, mapping P16 inputs in this manner (Fig. 7H,I) revealed similar afferent input maps at both holding potentials, with no difference (Wilcoxon signed rank test; p = 0.549 for the example shown) in the distribution of the inputs between the two holding potential (Fig. 7J,K).
Figure 7.
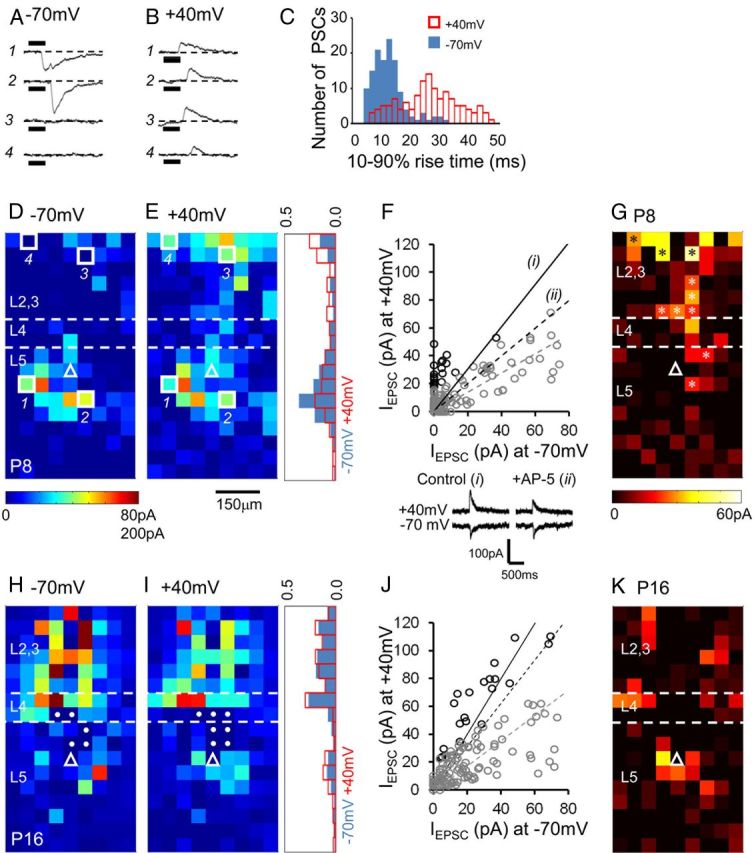
The presence of distal, putative NMDAR-mediated afferent inputs mapped in the early postnatal cortex. A, B, Voltage-clamp recordings of synaptic input elicited in response to LSPS at four separate laser target points in a P8, layer 5 pyramidal cell recording (numbers correspond to the sites indicated in D, E). C, Distribution of EPSC 10–90% rise times obtained from −70 mV and +40 mV holding potentials for the same cell. D, E, Complete synaptic input map of the P8, layer 5 pyramidal cell recorded at a holding potential of −70 mV (D; calibration maximum 80 pA) and +40 mV (E). The histogram adjacent to E indicates the normalized input measured across the vertical axis for the two holding potentials (blue bars, −70 mV; red open bars, +40 mV). F, Plot comparing the amplitude of the evoked EPSCs for each laser target spot. Black circles, The top ranked (Wilcoxon signed rank test) observations with a bias toward the response observed at +40 mV. The black line (i) corresponds to the expected glutamate response ratio calculated from the recorded somatic direct glutamate response (see inset below the graph). Dashed, black line (ii), The AMPA glutamate response ratio measured in the presence of 20 μm AP-5; gray dashed line, linear regression for all the observed synaptic responses. G, Difference in the observed response between −70 mV and +40 mV holding potentials; asterisks highlight novel +40 mV responses. H, Synaptic inputs mapped onto a P16 layer 5 pyramidal cell at a holding potential of −70 mV; pixels with a filled circle indicate target sites with large-amplitude, direct glutamate-uncaging responses that precluded the analysis of EPSCs at this target spot. I, Corresponding inputs onto the same cell as in G but at +40 mV. Inset, Histogram showing the normalized input across the vertical axis for both holding potentials. J, Plot comparing the amplitude of the evoked EPSCs for each laser target spot as shown in F but for a P16 pyramidal cell. K, Pixel map showing the location of the target points with a pronounced +40 mV (NMDAR) contribution identified in the P16 pyramidal cell mapped in H and I.
The delayed onset and time course of the LSPS-evoked slower PSCs elicited at +40 mV suggest that they were synaptic in origin; however, an alternative possibility is that the responses observed were due to activation of extrasynaptic receptors which form a sizeable proportion of the NMDAR pool throughout early postnatal ages (Harris and Pettit, 2007). To test the latter we used an established protocol (Hardingham et al., 2002) to block synaptic NMDARs using the anticonvulsant NMDAR antagonist MK-801 (Halliwell et al., 1989). P8 acute in vitro slices were preincubated in bicuculline and MK-801, before mapping afferent input onto layer 5 pyramidal cells (n = 4) as before, at both −70 mV and +40 mV (Fig. 8A,B, respectively). In contrast to our previous control observations at P8 (Fig. 7B), analysis of the MK-801-incubated afferent input maps revealed similar distribution at both holding potentials (histogram, Fig. 8B). Again, we calculated the somatic AMPA ratio and tested whether or not there was a significant difference in the predicted (for AMPA alone) versus observed +40 mV response (Fig. 8C). Our analysis revealed that in two cases there was a significant difference (both p < 0.05; two-tailed Wilcoxon signed-rank test); however, it was a decrease in the observed response, likely attributed to the lack of rectification in the recorded somatic AMPAR response. Furthermore there were no pronounced novel sources of input at +40 mV, with only a few target spots dispersed across the mapping grid having a more pronounced input onto the recorded cell when recorded at the +40 mV holding potential (Fig. 8D). Also evident, but excluded from our PSC analysis, were direct, low amplitude glutamate responses (Fig. 8D; laser target spot marked with asterisk) observed when the cells were held at +40 mV in the presence of MK-801 (Fig. 8D, inset). The absence of novel EPSCs at +40 mV in MK-801-incubated slices suggests that the distal sources of input ordinarily observed onto P8 pyramidal cells (Fig. 7E,G) are synaptic in nature and caused by NMDAR activation. These accounted for a good proportion (27 ± 4%; n = 5) of the laser target points that provided synaptic input at P8, but were almost entirely absent at 6 (4 + 2%, −1%, Fig. 8E). Identification of target points that provided exclusively NMDAR-mediated input enabled us to calculate the relative amplitude distribution across cortical layers at this early time point (Fig. 8F). This revealed that the predominant layer 5 input observed in P8 pyramidal cells voltage-clamped at −70 mV shifted to a more even distribution when held at +40 mV, due to a pronounced layer 2/3 bias in the source of novel NMDAR-mediated PSC inputs (termed NMDA, Fig. 8F). Together these data suggest that early pyramidal cells do receive a considerable amount of input from beyond the immediate layers, in line with the eventual emergence of the canonical network. However, it is initially mediated by NMDARs imparting a requirement for coincident activation of layer 2/3 and 5 networks for signaling to occur in postsynaptic layer 5 pyramidal cells.
The impact of burst activity on the integration states of neonatal pyramidal cells
Low-frequency, synchronous activity are common to developing neural networks in the mammalian cerebral cortex (Leinekugel et al., 2002; Golshani et al., 2009) and could facilitate the physiological integration of neurons in the neocortex via the awakening of silent synapses (Kasyanov et al., 2004; Sivakumaran et al., 2009). Given the considerable amount of NMDA-dependent glutamatergic input evident at early ages we examined whether short-term exposure to synchronous activity could influence integration as assessed by LSPS. Regardless of ages our recorded neurons were largely devoid of spontaneous suprathreshold activity so our initial strategy was to evoke cortical giant depolarizing potentials (cGDPs) in young (<P9) acute in vitro cortical slices by elevating the extracellular K+ concentration. However, we were unable to evoke consistent suprathreshold spike activity in coronal slices (n = 7) as previously reported (Allène et al., 2008). Therefore we decided to pursue a more robust approach and perfused the acute in vitro slices with nominally zero magnesium (0-Mg2+) ACSF (Fig. 9A; Silva et al., 1991; Flint et al., 1997). Slices were perfused with 0-Mg2+ ACSF following completion of the initial control mapping (Fig. 9B,G) and once the HDC ACSF had been washed off (minimum of 6 min). This manipulation resulted in an increase in baseline activity regardless of the age of the slice (Fig. 9A). To avoid excessive stimulation we only exposed the slices to 20 min of 0-Mg2+ ACSF so as to limited our chances of eliciting any long-duration, ‘seizure-like state’ events (Kawaguchi, 2001); a single cell that exhibited such a response during the 20 min period was discarded from our subsequent analysis. The level of activity elicited (for which measures included average and maximum instantaneous spike frequency, number of action potentials, number of bursts and sustained (>200 ms) subthreshold depolarizations) was remarkably similar across development, the only significant difference being the proportion of action potentials associated with burst (defined as ≥2 action potentials at a frequency of ≥5 Hz; inset, Fig. 9A) which dropped over development (data not shown).
Figure 9.
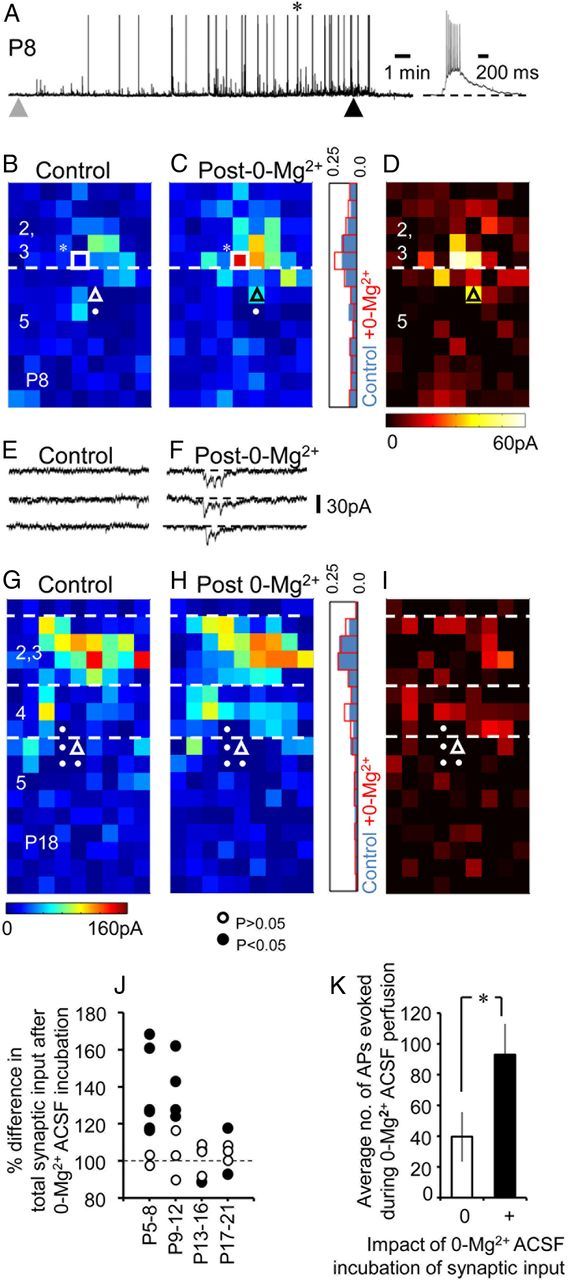
Activity-dependent enhancement of synaptic input onto early (≤P12) postnatal pyramidal cells. A, Impact of 20 min 0-Mg2+ ACSF incubation on sub- and suprathreshold activity levels in a P8 pyramidal cell; gray and black arrowheads, onset and end of 0-Mg2+ ACSF perfusion, respectively. Inset, A burst of action potentials marked with an asterisk in the main trace shown on an expanded time scale. B, C, Average synaptic input maps onto a P8, layer 5 motor cortex pyramidal cell under baseline conditions (Control; B) and after recovery from 20 min incubation in 0-Mg2+ ACSF (Post-0-Mg2+; C). The histogram adjacent to C indicates the normalized sum of inputs across the vertical axis for the control (blue) and post-activity (red line) maps. The white square and asterisk indicates the laser target spot from which 3 repeat voltage-clamp traces are shown below the maps for each condition (E, F). D, Plot showing laser target points that showed an increase in synaptic input following 0-Mg2+ ACSF perfusion. G–I, Data presented in a manner similar to the cell shown in B–D but for a P17, layer 5 somatosensory pyramidal cell. J, A plot showing the impact of 20 min incubation with 0-Mg2+ ACSF on the total synaptic input onto pyramidal cells plotted over the development time window examined. Black circles indicate a significant (two-tailed Wilcoxon signed-rank test; p < 0.05) difference in total synaptic input between control and post-0-Mg2+ maps. K, Histogram showing the average number of action potentials (APs) recorded during the 20 min 0-Mg2+ ACSF perfusion for all the P5–P12 pyramidal cells that showed no (white bar) or alternatively a significant change (p < 0.05) in total synaptic input.
One advantage of using 0-Mg2+ ACSF perfusion was that burst activity could be rapidly terminated on switching back to normal ACSF (Fig. 9A), enabling us to remap the afferent input in a timely manner once a stable baseline had been achieved. Exposure to activity did not alter the LSPS-evoked direct glutamate response of the cell recorded in current clamp (n = 26) across all the ages tested, be that either the number of spikes elicited by firing the laser directly at the cell soma or the amplitude of distal subthreshold direct glutamate responses (data not shown). In early preparations (before P12; n = 17) LSPS performed after the cessation of activity revealed an increase in the EPSCs elicited (Fig. 9C) with some previously quiescent target points now revealing robust, active afferent synaptic connections (Fig. 9D–F). In the cases where we could track the prolonged impact of 0-Mg2+ ACSF perfusion this elevated synaptic input remained stable even after further washout in normal ACSF (up to 30 min, n = 4). At later ages (>P12, n = 10; Figs. 9G–I) exposure to 0-Mg2+ ACSF-evoked activity caused no significant shift in the overall distribution or amplitude of the majority (7 of 10) of the input maps (Fig. 9G,H). In these cells examination of the individual laser target points (Fig. 9I) revealed that there were only small fluctuations in the sum of EPSC amplitudes per pixel that were invariably in equilibrium within the layers examined (histogram, Fig. 9H). Of the three cells that did show a significant difference, two showed reduced synaptic input (Fig. 9J).
Analysis revealed that in the majority of cells recorded before P12 (11 of 17; Fig. 9J) there was a significant change in afferent input following incubation with 0-Mg2+ ACSF (Wilcoxon signed-rank test, p < 0.05, black circles), and that in all cases this represented an enhancement of total synaptic input. Assessment of the control and post-0-Mg2+ EPSC properties revealed similar distribution in terms of rise time and amplitude; the only noticeable difference being an increase in the numbers of events detected. Six early postnatal cells (<P12) did not exhibit a significant shift in input map in response to our manipulation. These cells had similar (p > 0.05) baseline levels of input (746 ± 88 pA; range 441–991 pA) to cells that did show a response (628 ± 127 pA; range 297–1275 pA) and were indistinguishable in all their passive and active attributes, with the exception of the number of action potential elicited during 0-Mg2+ ACSF perfusion (Fig. 9K; p = 0.04). Control, early (P8–P10) pyramidal cells incubated in normal ACSF for the 20 min (n = 5) showed spike activity and no significant change in total synaptic input (Wilcoxon signed-rank test, p > 0.1 for all 5 cells). Thus it would appear that active participation during even relatively brief periods of synchronized burst activity have the potential to alter the level of glutamatergic input onto pyramidal cells up until midway through the second postnatal week.
Discussion
We have used laser scanning photostimulation of caged glutamate to define the time course for the gross physiological integration of pyramidal projection neurons in the motor and adjacent somatosensory regions of the mouse neocortex. Our evidence points to a coordinated development of the glutamatergic system during the first three postnatal weeks that undergoes a shift around postnatal day (P)12 in somatosensory cortex and a few days later in adjacent motor cortex. At this critical juncture, a switch is seen in the profile of the synaptic input onto pyramidal cells from a labile integrative phase to that of a more consolidatory period. Post-P12 the physiological architecture of the glutamatergic system largely conforms to that previously described for the canonical cortical circuit. We suggest that the prolonged nature of this integrative phase is a requirement for the successful progressive recruitment of cortical circuitry by emergent sensory input.
LSPS as a tool to dissect emergent connectivity in the cerebral cortex
LSPS has been used previously to characterize mature excitatory inputs onto neocortical pyramidal cells and local GABAergic interneurons located in layers 2/3 (Yoshimura and Callaway, 2005; Barbour and Callaway;, 2008) and 5 of the neocortex (Schubert et al., 2001, 2006; Anderson et al., 2010; Apicella et al., 2012). However, only a few studies have used LSPS to study the development (Dalva and Katz, 1994; Bureau et al., 2004; Ashby and Isaac, 2011; Viswanathan et al., 2012) and plasticity (Jin et al., 2006, 2011; Bureau et al., 2008; Rosselet et al., 2011) of neocortical synaptic inputs. The majority of experiments have used LSPS on neurons held at near resting membrane potential to identify predominantly AMPAR-mediated afferent input. However, it has also been used on a number of occasions to look at the balance of excitatory and inhibitory input onto pyramidal cells (Schubert et al., 2001) and GABAergic interneurons (Xu and Callaway, 2009). Given the importance of NMDAR-dependent neurotransmission in the developing cortex, and the increased prevalence of silent NMDAR-mediated synapses at early ages, we decided to extend the approach used previously to map inputs onto the recorded, postsynaptic pyramidal cell held at +40 mV (see also Ashby and Isaac, 2011). Glutamatergic synapses containing only postsynaptic NMDARs have been termed silent synapses and are thought to be critical to synaptic plasticity in immature neuronal circuits. Previous studies have shown that the number of silent synapses drops throughout development, paralleled by the increase presence of active AMPAR synapses after the end of the first postnatal week (Isaac et al., 1997; Rumpel et al., 2004). By using LSPS at holding potential of −70 mV and +40 mV we could effectively map NMDA-mediated input across the breadth of the cortical column thus gain a more in-depth profile of the number and the laminar source of these synapses. Our data suggest a role for exclusively NMDAR-mediated synapses in the development of the layer 2/3 to 5 pathway.
Activity-dependent integration of neurons
Activity is known to be critical for the maturation (Voigt et al., 2005; Wang and Kriegstein, 2008), maintenance (Turrigiano et al., 1998), and modification (Markram et al., 1997) of neuronal networks. In vivo assessment has predominantly focused on changes in connectivity resulting from emergent sensory input, notably in the visual cortex (Katz and Shatz, 1996; Rochefort et al., 2009). Whereas the majority of in vitro experiments have taken advantage of the ability to control presynaptic and postsynaptic neurons, to dissect the impact of tightly controlled pairing paradigms on the strength of connectivity between cells (Sjöström et al., 2001). In reality the immature neurons of the neocortex encounter a variety of oscillatory activities present within the developing cortex (Golshani et al., 2009) as well as from subcortical structures (Yang et al., 2009; Colonnese and Khazipov, 2010; Minlebaev et al., 2011). A variety of methods for stimulating burst activity in acute in vitro slices of the cortex have been identified, and this activity has been shown to bear hallmarks of the activity observed in vivo (Allène et al., 2008; Hanganu et al., 2009). However, these protocols require the use of a more intact preparation that would rule out effective mapping of inputs across the cortical layers or, alternatively, prolonged incubation of the slices beyond the time frame that one could reasonably extract meaningful data. As such we decided to adopt a robust approach and use nominally 0-Mg2+ concentration ACSF to facilitate burst activity by removal of the voltage dependency of NMDAR activation (Silva et al., 1991); a method that has proven effective in eliciting synchronized activity in neonatal slices (Flint et al., 1997).
Pyramidal cells of the neonatal brain encounter an ebb-and-flow of activity in vivo (Golshani et al., 2009; Yang et al., 2009). One can envisage that waves of synchronized activity may act in an almost constant manner to maintain cells in a more integrated state (Hanse et al., 2009). By slicing the brain we effectively remove this global, rhythmic influence and as a consequence, start a process by which pyramidal cells revert to a pseudo-dormant state (Xiao et al., 2004). Not until we elicit activity through 0-Mg2+ ACSF, or as in the case of other studies through manipulation of extracellular K+ concentrations (Bonifazi et al., 2009), is it possible to awaken, and thereby assess, the full extent of connectivity (Abrahamsson et al., 2008). The ability to evoke gross physiological integration was only apparent up until P12, which suggests that this is a property of emergent networks (Burrone et al., 2002).
A time line for early pyramidal cell synaptic integration
Our experimental conditions were initially set to identify predominantly AMPAR-mediated EPSCs. Under these conditions we could not reliably detect LSPS-evoked postsynaptic currents in layer 2/3 and 5 pyramidal cells before P5. This time point coincides with a switch in the activity of the network from the cENOs, which broadly recruit neurons through volume transmission, to more focused synaptically driven activity (Allène and Cossart, 2010 and references therein). The mechanism underlying this switch has not been fully elucidated but a decrease in the influence of the subplate and a recruitment of NMDAR signaling are likely factors (Dupont et al., 2006).
During the next few days, up until P12, there is an acceleration of intrinsic electrophysiological properties (including input resistance (Rin), membrane time constant (τ), spike half-width, and maximum firing frequency), paralleled by a phase during which AMPAR-containing synapses become more prevalent. The period from P5–P8 is dictated by a number of pronounced physiological events such as peak spindle burst activity in vivo (Yang et al., 2009) and reliable production of cGDPs in vitro (Allène et al., 2008). These cGDPs represent synchronized activity among local neuronal clusters, which in line with their hippocampal correlates, could act to stabilize synaptic interactions (Mohajerani and Cherubini, 2006) by facilitating the recruitment of AMPARs to previously weak or silent synapses, through activation of NMDARs.
Our data show a sizeable contribution of layer 5 afferent input onto layer 5 pyramidal cells at early time points when compared with later ages. Although these inputs are relatively weak when compared with that observed in the more mature brain, the reciprocal nature of the connections would assist the synchronization of deep layer pyramidal cells. Perfusion with 0-Mg2+ ACSF evokes burst activity that originates in deep layers and then spreads to more superficial cortical layers (Silva et al., 1991). The burst nature of this activity would mimic high-frequency stimulation that has been shown to trigger transient potentiation of synaptic inputs in immature neurons (Groc et al., 2006). Thus one can envisage why evoking activity using this method could provide the ideal climate for integration. Why this manipulation fails to evoke the same effect after P12 is not entirely clear. It might be advantageous at early ages to instate transient integrated states to facilitate initial binding of sensory input to the rapidly developing cortex (Minlebaev et al., 2011). There is evidence to suggest that once cells are established within a network, homeostatic mechanisms develop to regulate synaptic input (Burrone et al., 2002), which might explain why overt integration is dampened beyond P12. A lack of putative silent synapses at P16 likely reflects that the cells are by that time point established in the network. It also supports the idea put forward by a number of other studies (Rumpel et al., 2004; Ashby and Isaac, 2011) that these NMDAR-mediated synapses act as a substrate for synaptic integration. Thus the transition around P12 is likely dependent on two conditions: first, the conversion of labile synapses to permanent ones through prolonged exposure to rhythmic activity. Second, that the early, simple burst activity of cGDPs could be disrupted by the gradual integration of sensory input leading to a collapse of broad, synchronized activity (Golshani et al., 2009; Rochefort et al., 2009). The emergence of this more sensory-orientated network (Bureau et al., 2004; Ashby and Isaac, 2011) would likely alter the kinetics of the system from slow, burst-dependent integration (Butts et al., 2007) to a more precise temporal encoding environment (Doischer et al., 2008; Larsen et al., 2010).
Footnotes
Research in the laboratory is funded by a Human Frontiers Science Program Organization career development grant awarded to S.J.B.B. (CDA0023/2008) and a grant from the Oxford University Press John Fell Fund. P.G.A. is the recipient of an Imperial College London-funded PhD studentship. We thank Drs. Kenneth Harris, Tom Mrsic-Flogel, and members of the laboratory for their comments on a previous version of the manuscript, and Drs. Chris Holmes and Pierre Yger for statistical advice and data analysis, respectively. We also acknowledge the support and mentoring provided by Prof. Richard Reynolds to P.G.A. over the last few years.
References
- Abrahamsson T, Gustafsson B, Hanse E. AMPA silencing is a prerequisite for developmental long-term potentiation in the hippocampal CA1 region. J Neurophysiol. 2008;100:2605–2614. doi: 10.1152/jn.90476.2008. [DOI] [PubMed] [Google Scholar]
- Agmon A, Connors BW. Repetitive burst-firing neurons in the deep layers of mouse somatosensory cortex. Neurosci Lett. 1989;99:137–141. doi: 10.1016/0304-3940(89)90278-4. [DOI] [PubMed] [Google Scholar]
- Allène C, Cossart R. Early NMDA receptor-driven waves of activity in the developing neocortex: physiological or pathological network oscillations? J Physiol. 2010;588:83–91. doi: 10.1113/jphysiol.2009.178798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allène C, Cattani A, Ackman JB, Bonifazi P, Aniksztejn L, Ben-Ari Y, Cossart R. Sequential generation of two distinct synapse-driven network patterns in developing neocortex. J Neurosci. 2008;28:12851–12863. doi: 10.1523/JNEUROSCI.3733-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CT, Sheets PL, Kiritani T, Shepherd GM. Sublayer-specific microcircuits of corticospinal and corticostriatal neurons in motor cortex. Nat Neurosci. 2010;13:739–744. doi: 10.1038/nn.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angevine JB, Jr, Sidman RL. Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature. 1961;192:766–768. doi: 10.1038/192766b0. [DOI] [PubMed] [Google Scholar]
- Apicella AJ, Wickersham IR, Seung HS, Shepherd GM. Laminarly orthogonal excitation of fast-spiking and low-threshold-spiking interneurons in mouse motor cortex. J Neurosci. 2012;32:7021–7033. doi: 10.1523/JNEUROSCI.0011-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashby MC, Isaac JT. Maturation of a recurrent excitatory neocortical circuit by experience-dependent unsilencing of newly formed dendritic spines. Neuron. 2011;70:510–521. doi: 10.1016/j.neuron.2011.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour DL, Callaway EM. Excitatory local connections of superficial neurons in rat auditory cortex. J Neurosci. 2008;28:11174–11185. doi: 10.1523/JNEUROSCI.2093-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship AG, Feller MB. Mechanisms underlying spontaneous patterned activity in developing neural circuits. Nat Rev Neurosci. 2010;11:18–29. doi: 10.1038/nrn2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifazi P, Goldin M, Picardo MA, Jorquera I, Cattani A, Bianconi G, Represa A, Ben-Ari Y, Cossart R. GABAergic hub neurons orchestrate synchrony in developing hippocampal networks. Science. 2009;326:1419–1424. doi: 10.1126/science.1175509. [DOI] [PubMed] [Google Scholar]
- Bureau I, Shepherd GM, Svoboda K. Precise development of functional and anatomical columns in the neocortex. Neuron. 2004;42:789–801. doi: 10.1016/j.neuron.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Bureau I, Shepherd GM, Svoboda K. Circuit and plasticity defects in the developing somatosensory cortex of FMR1 knock-out mice. J Neurosci. 2008;28:5178–5188. doi: 10.1523/JNEUROSCI.1076-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone J, O'Byrne M, Murthy VN. Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature. 2002;420:414–418. doi: 10.1038/nature01242. [DOI] [PubMed] [Google Scholar]
- Butt SJ, Fuccillo M, Nery S, Noctor S, Kriegstein A, Corbin JG, Fishell G. The temporal and spatial origins of cortical interneurons predict their physiological subtype. Neuron. 2005;48:591–604. doi: 10.1016/j.neuron.2005.09.034. [DOI] [PubMed] [Google Scholar]
- Butts DA, Kanold PO, Shatz CJ. A burst-based “Hebbian” learning rule at retinogeniculate synapses links retinal waves to activity-dependent refinement. PLoS Biol. 2007;5:e61. doi: 10.1371/journal.pbio.0050061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittajallu R, Isaac JT. Emergence of cortical inhibition by coordinated sensory-driven plasticity at distinct synaptic loci. Nat Neurosci. 2010;13:1240–1248. doi: 10.1038/nn.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonnese MT, Khazipov R. “Slow activity transients” in infant rat visual cortex: a spreading synchronous oscillation patterned by retinal waves. J Neurosci. 2010;30:4325–4337. doi: 10.1523/JNEUROSCI.4995-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalva MB, Katz LC. Rearrangements of synaptic connections in visual cortex revealed by laser photostimulation. Science. 1994;265:255–258. doi: 10.1126/science.7912852. [DOI] [PubMed] [Google Scholar]
- Doischer D, Hosp JA, Yanagawa Y, Obata K, Jonas P, Vida I, Bartos M. Postnatal differentiation of basket cells from slow to fast signaling. J Neurosci. 2008;28:12956–12968. doi: 10.1523/JNEUROSCI.2890-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas RJ, Martin KA. Neuronal circuits of the neocortex. Annu Rev Neurosci. 2004;27:419–451. doi: 10.1146/annurev.neuro.27.070203.144152. [DOI] [PubMed] [Google Scholar]
- Dupont E, Hanganu IL, Kilb W, Hirsch S, Luhmann HJ. Rapid developmental switch in the mechanisms driving early cortical columnar networks. Nature. 2006;439:79–83. doi: 10.1038/nature04264. [DOI] [PubMed] [Google Scholar]
- Flint AC, Maisch US, Kriegstein AR. Postnatal development of low [Mg2+] oscillations in neocortex. J Neurophysiol. 1997;78:1990–1996. doi: 10.1152/jn.1997.78.4.1990. [DOI] [PubMed] [Google Scholar]
- Garaschuk O, Linn J, Eilers J, Konnerth A. Large-scale oscillatory calcium waves in the immature cortex. Nat Neurosci. 2000;3:452–459. doi: 10.1038/74823. [DOI] [PubMed] [Google Scholar]
- Gilbert CD, Wiesel TN. Functional organization of the visual cortex. Prog Brain Res. 1983;58:209–218. doi: 10.1016/S0079-6123(08)60022-9. [DOI] [PubMed] [Google Scholar]
- Golshani P, Gonçalves JT, Khoshkhoo S, Mostany R, Smirnakis S, Portera-Cailliau C. Internally mediated developmental desynchronization of neocortical network activity. J Neurosci. 2009;29:10890–10899. doi: 10.1523/JNEUROSCI.2012-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groc L, Gustafsson B, Hanse E. AMPA signalling in nascent glutamatergic synapses: there and not there! Trends Neurosci. 2006;29:132–139. doi: 10.1016/j.tins.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Halliwell RF, Peters JA, Lambert JJ. The mechanism of action and pharmacological specificity of the anticonvulsant NMDA antagonist MK-801: a voltage clamp study on neuronal cells in culture. Br J Pharmacol. 1989;96:480–494. doi: 10.1111/j.1476-5381.1989.tb11841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanganu IL, Okabe A, Lessmann V, Luhmann HJ. Cellular mechanisms of subplate-driven and cholinergic input-dependent network activity in the neonatal rat somatosensory cortex. Cereb Cortex. 2009;19:89–105. doi: 10.1093/cercor/bhn061. [DOI] [PubMed] [Google Scholar]
- Hanse E, Taira T, Lauri S, Groc L. Glutamate synapse in developing brain: an integrative perspective beyond the silent state. Trends Neurosci. 2009;32:532–537. doi: 10.1016/j.tins.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Harris AZ, Pettit DL. Extrasynaptic and synaptic NMDA receptors form stable and uniform pools in rat hippocampal slices. J Physiol. 2007;584:509–519. doi: 10.1113/jphysiol.2007.137679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooks BM, Hires SA, Zhang YX, Huber D, Petreanu L, Svoboda K, Shepherd GM. Laminar analysis of excitatory local circuits in vibrissal motor and sensory cortical areas. PLoS Biol. 2011;9:e1000572. doi: 10.1371/journal.pbio.1000572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Crair MC, Nicoll RA, Malenka RC. Silent synapses during development of thalamocortical inputs. Neuron. 1997;18:269–280. doi: 10.1016/s0896-6273(00)80267-6. [DOI] [PubMed] [Google Scholar]
- Jin X, Prince DA, Huguenard JR. Enhanced excitatory synaptic connectivity in layer v pyramidal neurons of chronically injured epileptogenic neocortex in rats. J Neurosci. 2006;26:4891–4900. doi: 10.1523/JNEUROSCI.4361-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Huguenard JR, Prince DA. Reorganization of inhibitory synaptic circuits in rodent chronically injured epileptogenic neocortex. Cereb Cortex. 2011;21:1094–1104. doi: 10.1093/cercor/bhq181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper EM, Larkman AU, Lübke J, Blakemore C. Pyramidal neurons in layer 5 of the rat visual cortex. II. Development of electrophysiological properties. J Comp Neurol. 1994;339:475–494. doi: 10.1002/cne.903390403. [DOI] [PubMed] [Google Scholar]
- Kasyanov AM, Safiulina VF, Voronin LL, Cherubini E. GABA-mediated giant depolarizing potentials as coincidence detectors for enhancing synaptic efficacy in the developing hippocampus. Proc Natl Acad Sci U S A. 2004;101:3967–3972. doi: 10.1073/pnas.0305974101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y. Distinct firing patterns of neuronal subtypes in cortical synchronized activities. J Neurosci. 2001;21:7261–7272. doi: 10.1523/JNEUROSCI.21-18-07261.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khazipov R, Luhmann HJ. Early patterns of electrical activity in the developing cerebral cortex of humans and rodents. Trends Neurosci. 2006;29:414–418. doi: 10.1016/j.tins.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Kumar SS, Bacci A, Kharazia V, Huguenard JR. A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J Neurosci. 2002;22:3005–3015. doi: 10.1523/JNEUROSCI.22-08-03005.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HB, Sabatini BL. Glutamate induces de novo growth of functional spines in developing cortex. Nature. 2011;474:100–104. doi: 10.1038/nature09986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen RS, Rao D, Manis PB, Philpot BD. STDP in the developing sensory neocortex. Front Synaptic Neurosci. 2010;2:9. doi: 10.3389/fnsyn.2010.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinekugel X, Khazipov R, Cannon R, Hirase H, Ben-Ari Y, Buzsáki G. Correlated bursts of activity in the neonatal hippocampus in vivo. Science. 2002;296:2049–2052. doi: 10.1126/science.1071111. [DOI] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Luhmann HJ, Reiprich RA, Hanganu I, Kilb W. Cellular physiology of the neonatal rat cerebral cortex: intrinsic membrane properties, sodium and calcium currents. J Neurosci Res. 2000;62:574–584. doi: 10.1002/1097-4547(20001115)62:4<574::AID-JNR12>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Maravall M, Stern EA, Svoboda K. Development of intrinsic properties and excitability of layer 2/3 pyramidal neurons during a critical period for sensory maps in rat barrel cortex. J Neurophysiol. 2004;92:144–156. doi: 10.1152/jn.00598.2003. [DOI] [PubMed] [Google Scholar]
- Markram H, Lübke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- Miller MW. Development of projection and local circuit neurons in neocortex. In: Peters A, Jones EG, editors. Cerebral cortex, Vol 7: Development and maturation of cerebral cortex. Ed 2. New York: Plenum; 1988. pp. 133–175. [Google Scholar]
- Minlebaev M, Colonnese M, Tsintsadze T, Sirota A, Khazipov R. Early γ oscillations synchronize developing thalamus and cortex. Science. 2011;334:226–229. doi: 10.1126/science.1210574. [DOI] [PubMed] [Google Scholar]
- Mohajerani MH, Cherubini E. Role of giant depolarizing potentials in shaping synaptic currents in the developing hippocampus. Crit Rev Neurobiol. 2006;18:13–23. doi: 10.1615/critrevneurobiol.v18.i1-2.30. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8:427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- Montoro RJ, Yuste R. Gap junctions in developing neocortex: a review. Brain Res Rev. 2004;47:216–226. doi: 10.1016/j.brainresrev.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Oswald AM, Reyes AD. Maturation of intrinsic and synaptic properties of layer 2/3 pyramidal neurons in mouse auditory cortex. J Neurophysiol. 2008;99:2998–3008. doi: 10.1152/jn.01160.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Halliday G, Koutcherov Y, Wang H, Watson C. Atlas of the developing mouse brain at E17.5, P0, and P6. San Diego: Academic; 2007. [Google Scholar]
- Rakic P. Neurons in the Rhesus monkey visual cortex: systematic relationship between time of origin and eventual disposition. Science. 1974;183:425–427. doi: 10.1126/science.183.4123.425. [DOI] [PubMed] [Google Scholar]
- Rochefort NL, Garaschuk O, Milos RI, Narushima M, Marandi N, Pichler B, Kovalchuk Y, Konnerth A. Sparsification of neuronal activity in the visual cortex at eye-opening. Proc Natl Acad Sci U S A. 2009;106:15049–15054. doi: 10.1073/pnas.0907660106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochefort NL, Narushima M, Grienberger C, Marandi N, Hill DN, Konnerth A. Development of direction selectivity in mouse cortical neurons. Neuron. 2011;71:425–432. doi: 10.1016/j.neuron.2011.06.013. [DOI] [PubMed] [Google Scholar]
- Rosselet C, Fieschi M, Hugues S, Bureau I. Associative learning changes the organization of functional excitatory circuits targeting the supragranular layers of mouse barrel cortex. Front Neural Circuits. 2011;4:126. doi: 10.3389/fncir.2010.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpel S, Kattenstroth G, Gottmann K. Silent synapses in the immature visual cortex: layer-specific developmental regulation. J Neurophysiol. 2004;91:1097–1101. doi: 10.1152/jn.00443.2003. [DOI] [PubMed] [Google Scholar]
- Schubert D, Staiger JF, Cho N, Kötter R, Zilles K, Luhmann HJ. Layer-specific intracolumnar and transcolumnar functional connectivity of layer V pyramidal cells in rat barrel cortex. J Neurosci. 2001;21:3580–3592. doi: 10.1523/JNEUROSCI.21-10-03580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D, Kötter R, Luhmann HJ, Staiger JF. Morphology, electrophysiology and functional input connectivity of pyramidal neurons characterizes a genuine layer Va in the primary somatosensory cortex. Cereb Cortex. 2006;16:223–236. doi: 10.1093/cercor/bhi100. [DOI] [PubMed] [Google Scholar]
- Shepherd GMG, Pologruto TA, Svoboda K. Circuit analysis of experience-dependent plasticity in the developing rat barrel cortex. Neuron. 2003;38:277–289. doi: 10.1016/s0896-6273(03)00152-1. [DOI] [PubMed] [Google Scholar]
- Silva LR, Amitai Y, Connors BW. Intrinsic oscillations of neocortex generated by layer 5 pyramidal neurons. Science. 1991;251:432–435. doi: 10.1126/science.1824881. [DOI] [PubMed] [Google Scholar]
- Sivakumaran S, Mohajerani MH, Cherubini E. At immature mossy-fiber-CA3 synapses, correlated presynaptic and postsynaptic activity persistently enhances GABA release and network excitability via BDNF and cAMP-dependent PKA. J Neurosci. 2009;29:2637–2647. doi: 10.1523/JNEUROSCI.5019-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB. Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron. 2001;32:1149–1164. doi: 10.1016/s0896-6273(01)00542-6. [DOI] [PubMed] [Google Scholar]
- Thomson AM, Bannister AP. Interlaminar connections in the neocortex. Cereb Cortex. 2003;13:5–14. doi: 10.1093/cercor/13.1.5. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Viswanathan S, Bandyopadhyay S, Kao JP, Kanold PO. Changing microcircuits in the subplate of the developing cortex. J Neurosci. 2012;32:1589–1601. doi: 10.1523/JNEUROSCI.4748-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt T, Opitz T, de Lima AD. Activation of early silent synapses by spontaneous synchronous network activity limits the range of neocortical connections. J Neurosci. 2005;25:4605–4615. doi: 10.1523/JNEUROSCI.3803-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DD, Kriegstein AR. GABA regulates excitatory synapse formation in the neocortex via NMDA receptor activation. J Neurosci. 2008;28:5547–5558. doi: 10.1523/JNEUROSCI.5599-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao MY, Wasling P, Hanse E, Gustafsson B. Creation of AMPA-silent synapses in the neonatal hippocampus. Nat Neurosci. 2004;7:236–243. doi: 10.1038/nn1196. [DOI] [PubMed] [Google Scholar]
- Xu X, Callaway EM. Laminar specificity of functional input to distinct types of inhibitory cortical neurons. J Neurosci. 2009;29:70–85. doi: 10.1523/JNEUROSCI.4104-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JW, Hanganu-Opatz IL, Sun JJ, Luhmann HJ. Three patterns of oscillatory activity differentially synchronize developing neocortical networks in vivo. J Neurosci. 2009;29:9011–9025. doi: 10.1523/JNEUROSCI.5646-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura Y, Callaway EM. Fine-scale specificity of cortical networks depends on inhibitory cell type and connectivity. Nat Neurosci. 2005;8:1552–1559. doi: 10.1038/nn1565. [DOI] [PubMed] [Google Scholar]
- Yu YC, He S, Chen S, Fu Y, Brown KN, Yao XH, Ma J, Gao KP, Sosinsky GE, Huang K, Shi SH. Preferential electrical coupling regulates neocortical lineage-dependent microcircuit assembly. Nature. 2012;486:113–117. doi: 10.1038/nature10958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Peinado A, Katz LC. Neuronal domains in developing neocortex. Science. 1992;257:665–669. doi: 10.1126/science.1496379. [DOI] [PubMed] [Google Scholar]
- Zhao C, Kao JP, Kanold PO. Functional excitatory microcircuits in neonatal cortex connect thalamus and layer 4. J Neurosci. 2009;29:15479–15488. doi: 10.1523/JNEUROSCI.4471-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]