

Abstract
Ketamine, an NMDA receptor antagonist, has been shown to induce behavioral abnormalities in humans that mimic the positive, negative, and most importantly cognitive deficits observed in schizophrenia. Similar cognitive deficits have been observed in nonhuman primates after a subanesthetic dose of ketamine, including an impairment in their ability to perform the antisaccade task, which requires the suppression of a prosaccade toward a flashed stimulus and the generation of a saccade in the opposite direction. The neural basis underlying these cognitive impairments remains unknown. Here, we recorded single-neuron activity in the lateral prefrontal cortex of macaque monkeys before and after the administration of subanesthetic doses of ketamine during the performance of randomly interleaved prosaccade and antisaccade trials. Ketamine impeded the monkeys' ability to maintain and apply the correct task rule and increased reaction times of prosaccades and antisaccades. These behavioral changes were associated with an overall increase in activity of PFC neurons and a reduction in their task selectivity. Our results suggest that the mechanism underlying ketamine-induced cognitive abnormalities may be the nonspecific increase in PFC activity and the associated reduction of task selectivity.
Introduction
The use of NMDA receptor antagonists to induce a preclinical animal model of schizophrenia has been gaining traction over recent years. Acute doses of ketamine, a noncompetitive NMDA antagonist, have been shown to induce short-lived behavioral profiles that include the positive, negative, and cognitive symptoms of schizophrenia in humans (Krystal et al., 1994; Lahti et al., 1995; Adler et al., 1999; Newcomer et al., 1999; Taffe et al., 2002). Further, a subanesthetic dose of ketamine can often trigger a psychotic episode in patients already suffering from the disease (Malhotra et al., 1997; Lahti et al., 2001).
The ketamine-induced preclinical model of schizophrenia generates robust cognitive impairments as demonstrated by tasks probing working memory and the suppression of prepotent responses to stimuli (Tsai et al., 1995; Olney et al., 1999; Javitt, 2009). Reduced cognitive function is considered to be the most debilitating aspect of schizophrenia as the severity of these symptoms directly relates to the patient's quality of life and current pharmaceutical interventions provide minimal improvement (Elvevåg and Goldberg, 2000; Goldman-Rakic et al., 2004; Goeree et al., 2005; van Os and Kapur, 2009). Nonhuman primates also show similar cognitive deficits following systemic subanesthetic doses of ketamine as patients with schizophrenia in a number of behavioral tasks (Condy et al., 2005; Stoet and Snyder, 2006). An example is the antisaccade paradigm, which requires the inhibition of a prepotent prosaccade toward a flashed peripheral stimulus in favor of the generation of a saccade away from the stimulus toward the opposite direction (Everling and Fischer, 1998; Munoz and Everling, 2004). Following ketamine injections, nonhuman primates exhibit increased reaction times and error rates on antisaccade trials (Condy et al., 2005). These impairments seem to mimic the deficits observed in patients with schizophrenia (Fukushima et al., 1988; McDowell et al., 2002) and patients with prefrontal cortex (PFC) lesions (Guitton et al., 1985; Pierrot-Deseilligny et al., 1991).
The behavioral profile of cognitive deficits following subanesthetic doses of ketamine has been well documented; however, the neural mechanisms in the primate PFC responsible for these changes are still unknown. Although rodent studies have reported an increase in frontal cortex neural activity following acute ketamine administration (Jackson et al., 2004; Homayoun and Moghaddam, 2007), rodents lack a granular PFC, which is characteristic for lateral, ventral, medial, and frontopolar prefrontal areas in primates (Povinelli and Preuss, 1995; Preuss, 2000; Wise, 2008). To directly investigate the effects of ketamine on task-selective neural activity in the primate lateral PFC, we recorded single-neuron activity in macaque monkeys before and after the administration of subanesthetic doses of ketamine during the performance of randomly interleaved prosaccade and antisaccade trials.
Materials and Methods
All experiments were performed in accordance with the Canadian Council of Animal Care policy on the use of laboratory animals and all procedures were approved by the Animal Use Subcommittee of the University of Western Ontario Council on Animal Care. Two male rhesus monkeys (Macaca mulatta) weighing 7 kg (monkey O) and 10 kg (monkey W) participated in the study. Both animals had a plastic head restraint and plastic recording chambers implanted above their lateral PFC as previously described by our laboratory (Johnston and Everling, 2006b). Animals received analgesics and antibiotics postoperatively and were closely monitored by a university veterinarian. Following implantation surgery, we obtained anatomical MR images of both monkeys using an actively shielded 7 tesla 68 cm horizontal bore scanner (Varian) and a Siemens AC84 gradient subsystem. Plastic grids filled with betadine were inserted into the recording chambers during the scan, which allowed for precise electrode targeting and subsequent reconstruction of recording locations.
Behavioral task.
Both animals were trained on rule-visible and rule-memorized variants of the prosaccade/antisaccade task (Fig. 1). The rule for the current trial was presented following an initial 100 ms fixation period by changing the central white fixation spot to either green or red, with each color indicating whether the animal had to perform a prosaccade (green fixation cue) or antisaccade (red fixation cue) in response to the ensuing peripheral stimulus. On rule-visible trials, a peripheral stimulus appeared 8° to either the left or right following a 1000–1200 ms fixation period on the rule cue. To challenge the animal's working memory, the rule-memorized trials presented the rule cue for only 200 ms, after which the color of the cue returned to white for 800–1000 ms. Following this delay period, the peripheral stimulus was presented in a manner identical with the rule-visible trials. To obtain a juice reward, the animal was required to look to the correct target location within a 8° circular window. On prosaccade trials, this required a saccade toward the peripheral stimulus, whereas on antisaccade trials, the correct target location was diametrically opposite of the peripheral stimulus. Every block performed consisted of a pseudorandomized ordering of the eight trial variants: prosaccade left and right; antisaccade left and right; with each of these four conditions appearing as either rule-visible or rule-memorized. Error trials were not repeated. The animals' eye positions were recorded and digitized at 120 Hz using an ISCAN infrared pupillary tracking system (ISCAN).
Figure 1.

Experimental paradigm. A, Rule-visible task. Each trial began with a fixation point (FP) signaling, by its color, a prosaccade or antisaccade trial. A stimulus then appeared either 8° to either the left or right. B, Rule-memorized task. Same as A, but the color of the FP changed to white 800–1000 ms before stimulus presentation. This required the monkey to memorize the task rule.
Multielectrode recording.
Each behavioral testing session was accompanied by electrophysiological recordings from the left or right lateral PFC in monkey O and the left lateral PFC in monkey W. Initial recordings were performed with 20 screw mini-microdrives (Miller Laboratory, Massachusetts Institute of Technology, Boston, MA), each equipped with two tungsten electrodes (FHC). Neural activity was combined with performance data and eye position data into a single recording file using a multiacquisition processor system (Plexon). Recorded neurons were sorted off-line using 2-D and 3-D principal component analysis. In later recording sessions, we used a custom-designed semichronic screw microdrive recording system that contained 32 recording electrodes and 2 reference electrodes (Neuronitek). Initial insertion of the semichronic drive consisted of lowering individual electrodes through both a thin silicone membrane and the animal's dura until stable units were isolated on a maximal number of channels. Each subsequent recording day only required reconnecting the 32 channel headstage (Plexon) and minimal depth adjustments to isolate new units to record for that day's experimental session. The microdrive system remained implanted for 2 weeks before it was removed for cleaning and maintenance.
Drug administration.
Each experimental session began with a 15 min period of baseline activity during which the animal performed ∼200–250 trials in a pseudorandomized order. Both animals were trained extensively at the task and performed at a <20% error rate during these preinjection periods. After 15 min of trials, the experiment was briefly paused and the animal received a single intramuscular injection of ketamine (0.4 mg/kg, monkey O; 0.8 mg/kg, monkey W) diluted in sterile saline to 0.4 ml into their right triceps brachii muscle. The injection process only interrupted the animal's experimental session for <15 s after which the monkeys would continue the behavioral task. We previously performed titration experiments with each monkey to determine animal-specific concentrations of ketamine (between 0.2 and 1.0 mg/kg) for the experiment, as these values have previously been found to elicit cognitive deficits with minimal anesthetic effects (Condy et al., 2005). Monkey W had more exposure to ketamine before this study for surgeries and routine veterinary procedures and developed a greater tolerance to the drug (Pouget et al., 2010). The behavioral effects of ketamine became apparent after ∼5 min (50–75 trials). Control experiments involving the intramuscular injection of saline only were also performed. No changes in behavior (error rates and reaction times) or single-neuron activity between the preinjection and postinjection periods were observed (data not shown here). Experimental sessions were separated by at least 4 d to ensure the ketamine had a sufficient washout period and thus avoid cumulative dosing effects.
Data analysis.
Behavioral data were calculated with the preinjection periods defined as 15 min before injection and the postinjection period defined as 5 min after injection until 25 min after injection, as this was the time period that displayed the strongest behavioral effects of the drug. Directional errors were trials in which the animal made a saccade in the opposite direction of a correct response (antisaccade on a prosaccade trial and vice versa). No response trials were those in which the animal maintained fixation during the response period, while fixation errors consisted of trials in which the animal did not maintain fixation until the stimulus onset. Significance was calculated between preinjection and postinjection values for each category using paired t tests. Later, time course behavioral data were calculated by combining all experimental sessions for both monkeys and sorting the data into 5 min bins for error rates and saccadic reaction times. A one-way ANOVA followed by a post hoc Dunnett's test was used to probe each trial type for significant differences between the animal's preinjection performance and their performance at each binned time point. For the analysis of neural data, we included correct and error trials. The calculation of indexed values for the change in the discharge frequency of each neuron included neural activity from the entire trial period defined as 1000 ms preceding stimulus presentation until 500 ms after stimulus presentation.
Neurons were subjected to further analysis to highlight changes in task selectivity following ketamine administration. Task selectivity was calculated as follows: (x̄preferred − x̄nonpreferred)/(x̄preferred + x̄nonpreferred), using the mean discharge rate from each neuron during a prestimulus epoch and a stimulus/response epoch. Preference was defined as either task preference (prosaccade vs antisaccade) or direction preference (contralateral stimulus vs ipsilateral stimulus). Whichever comparison exhibited a greater selectivity was used for the preinjection versus postinjection scatter plot defined by epoch and rule-visible or rule-memorized conditions. Statistical significance for each neuron was tested using a Mann–Whitney U test for changes in task selectivity caused by the ketamine administration.
To define cells as either narrow-spiking (putative interneurons) or broad-spiking neurons (putative pyramidal neurons), we calculated mean trough-to-peak times for the extracellular waveform of each neuron and constructed a histogram of the resulting values as previously described (Johnston et al., 2009). In accordance with this previous study, which used the same recording system and the same type of microelectrodes, we defined neurons with waveform durations of <270 μs as narrow-spiking neurons and any neurons with waveform durations of >270 μs as broad-spiking neurons.
Finally, a sliding receiver operating characteristic (ROC) curve was computed using the neurons that were shown to exhibit selectivity in either of the two epochs so that any differences in the significance of task selectivity across the entire trial period both preinjection and postinjection would be apparent. The ROC value was calculated using a sliding 100 ms window beginning 1000 ms before peripheral stimulus onset, advancing in 1 ms increments, and extending 500 ms beyond stimulus presentation. Significance cutoff values for the 2.5th and 97.5th percentile were calculated using a bootstrap method (Koval et al., 2011).
Results
Behavioral deficits
Both animals displayed acute increases in response errors, fixation errors, and no response trials when attempting to perform both prosaccade and antisaccade trials following ketamine administration (Fig. 2A). The effect lasted ∼20 min in monkey O and 35 min in monkey W, with performance returning to preinjection error rates afterward in monkey O. Error rates decreased in monkey W over time after the initial ketamine effect, but never returned to preinjection levels in the time span of each experimental session. The increased error rate on prosaccade trials for both rule-visible and rule-memorized conditions reached a maximum of 10% in monkey O and 30% in monkey W. Ketamine caused a much stronger effect on the antisaccade task in monkey O, with response errors increasing threefold to 30% in the rule-visible task and to almost 60% in the rule-memorized task.
Figure 2.

Behavioral effects of ketamine administration. A, Effect of ketamine on the directional error rates (blue bars), response rates (green bars), and fixation error rates (red bars). The darker shaded foreground bars represent preinjection values, and the lighter background bars are postinjection values. Data for monkey O were averaged from 8 experiment sessions; data for monkey W were averaged from 10 experiment sessions. B, Effect of ketamine on saccade reaction times. The format is the same as in A, with blue bars representing saccade reaction times on correct trials and red bars representing saccade reaction times on (directional) error trials. Error bars indicate SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Error rates on the antisaccade task highlighted the additional challenge that the rule-memorized paradigm introduced. Both monkeys made more errors in the rule-memorized condition than in the rule-visible condition. A similar separation between rule-visible and rule-memorized tasks was not observed on the prosaccade task for either monkey. These changes were accompanied by a weak postsaccade nystagmus, similar to those reported in previous studies involving ketamine and saccadic eye movements (Radant et al., 1998; Condy et al., 2005; Shen et al., 2010).
Saccade reaction times (SRTs) were also increased across all conditions in both animals (Fig. 2B). When analyzing the SRT for error trials only, we found that these were also increased and that the errors being made were not express saccades (Fischer et al., 1984). Ketamine had a similar lengthening effect on SRTs of both correct and error responses.
Ketamine increases activity but decreases task selectivity of PFC neurons
We recorded the activity of 115 neurons in the lateral PFC from two monkeys (97 from O; 18 from W) in 18 recording sessions in which we injected subanesthetic doses of ketamine (Fig. 3). Figure 4 illustrates the effect of ketamine on a single PFC neuron. Preinjection (left panel), the neuron exhibited higher levels of activity before peripheral stimulus onset on prosaccade trials than on antisaccade trials. These rule-related differences were present at the time of stimulus presentation on both rule-visible (Fig. 4A, left panel) and rule-memorized trials (Fig. 4B, left panel). In the postinjection period, the neuron displayed an overall increase in activity and a decrease in rule selectivity at the time of stimulus presentation, which was especially evident on rule-memorized trials (Fig. 4B, right panel).
Figure 3.

MRI reconstruction of recording locations. L, Left hemisphere; R, right hemisphere; m, medial; a, anterior; p, posterior; l, lateral.
Figure 4.
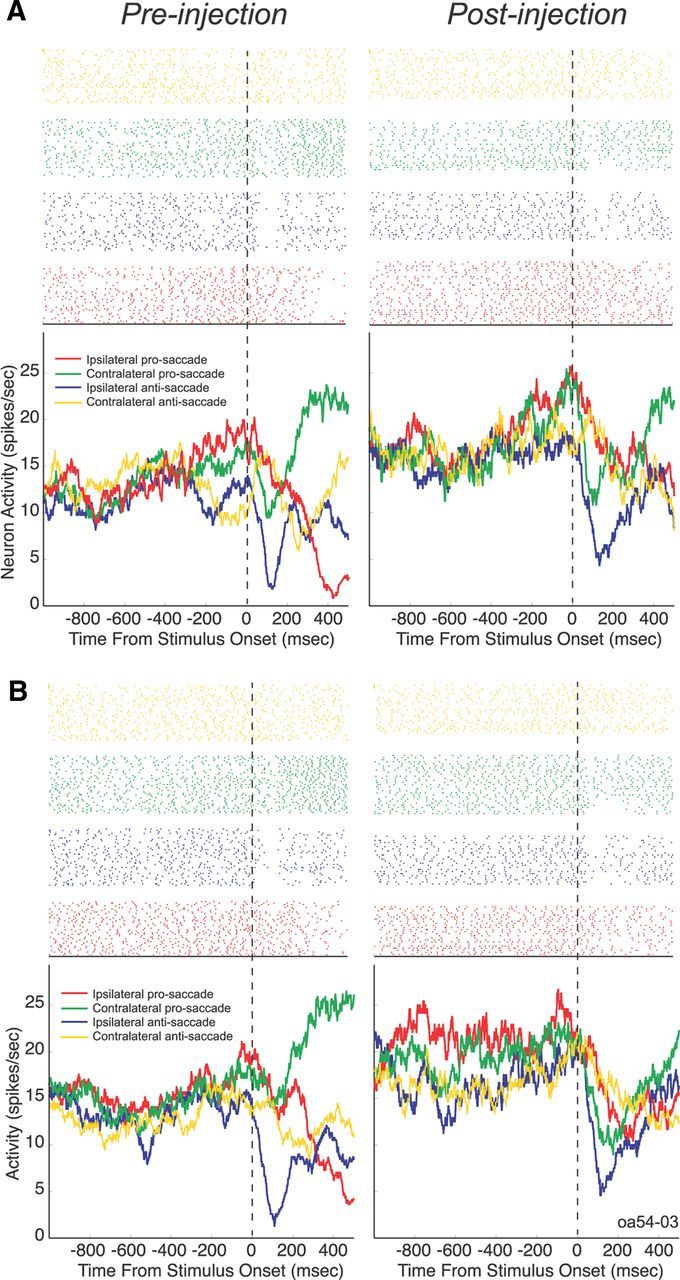
Effect of ketamine on a single PFC neuron. The neuron was recorded from monkey O in the dorsal bank of the principal sulcus in the right hemisphere. A, Activity on rule-visible trials. B, Activity on rule-memorized trials. The top panel shows a raster plot with each dot indicating the time of an action potential relative to stimulus presentation, and each row represents one trial.
Figure 5 shows a scatter plot comparing indexed values of the change in the overall neural activity with indexed values of the change in the task selectivity of each cell. A significant majority of neurons (n = 75; 65%) were found to be located in the bottom right quadrant, indicating an increase in neural activity but a decrease in task selectivity (χ2 test, p < 0.001). Calculations were also performed to define each neuron as either narrow-spiking (filled symbols; n = 20) or broad-spiking (hollow symbols; n = 95) neural activity, but there appeared to be no difference between the two groups in their distribution across the scatter plot.
Figure 5.

Effect of ketamine on change in discharge rate and change in task selectivity. Scatter plot depicts the change in discharge rates of neurons (interval from 1000 ms before to 500 ms after stimulus onset) after ketamine injection on the x-axis and the change in task selectivity (same analysis interval as x-axis) on the y-axis. The circles indicate neurons recorded from monkey O, the squares indicate neurons recorded from monkey W, filled represent narrow-spiking neurons, and hollow points represent broad-spiking neurons.
To further examine the effects of ketamine on neural activity, we analyzed mean discharge rates during the prestimulus period (500 ms before to stimulus onset) with a three-way ANOVA with the factors RUL (prosaccade or antisaccade), MEM (rule-visible or rule-memorized), and DRUG (preinjection and postinjection). This analysis showed that the factor DRUG had the strongest effect on prestimulus activity, with ∼60% (n = 69) of neurons showing significant differences in activity between preinjection and postinjection periods (Fig. 6A). Furthermore, 35 neurons (33 from monkey O; 2 from monkey W) displayed interaction effects of DRUG with MEM and/or RUL. Next, we examined the effects of ketamine on neural activity during the stimulus/response period (defined as the period 100–400 ms following stimulus onset) with a three-way ANOVA with the factors DIR (contralateral or ipsilateral stimulus location), RUL (prosaccade or antisaccade), and DRUG (preinjection and postinjection). The results are shown in Figure 6B. Overall, 40% (n = 46) of PFC neurons showed a main effect of DRUG. In addition, 49 neurons (43 from monkey O; 6 from monkey W) showed significant interactions of DRUG with DIR and/or RUL. For the remainder of the analysis, we focus on those PFC neurons that exhibited interaction effects of DRUG (pre- vs post-ketamine injection) during the prestimulus or stimulus/response period.
Figure 6.

Results of three-way ANOVA on differences in neural activity. A, Results during prestimulus epoch. B, Results during stimulus/response epoch. RUL, Rule (prosaccade vs antisaccade); MEM, memory condition (rule-visible vs rule-memorized condition); DIR, direction (ipsilateral vs contralateral stimulus location); DRUG, ketamine status (preinjection vs postinjection).
Ketamine-induced decrease in neural task selectivity is synchronous with behavioral changes
Figure 7 shows the time course of the behavioral changes as well as the task selectivity of these PFC neurons during the recording sessions. Shortly after ketamine administration, error rates (Fig. 7A) and SRTs (Fig. 7B) significantly increase from preinjection levels. Following the same time course, task selectivity (Fig. 7C) decreased during the prestimulus period (dashed line) and the stimulus/response period (solid line). For each of the neurons selected from the ANOVAs, we calculated a preinjection and postinjection selectivity index for the prestimulus (Fig. 8A) and stimulus/response period (Fig. 8B). The population of neurons exhibited a significant decrease in memory selectivity (rule-visible vs rule-memorized) (p < 0.001, Wilcoxon's signed rank test) and rule selectivity (prosaccade vs antisaccade) (p < 0.01, Wilcoxon's signed rank test) during the prestimulus period and in rule (p < 0.001, Wilcoxon's signed rank test) and direction selectivity (contralateral vs ipsilateral) (p < 0.001, Wilcoxon's signed rank test) during the stimulus/response period. Neurons were again delineated as either narrow-spiking neurons (filled symbols) or broad-spiking neurons (hollow symbols). Both groups of neurons showed a reduction in task selectivity following ketamine injection.
Figure 7.
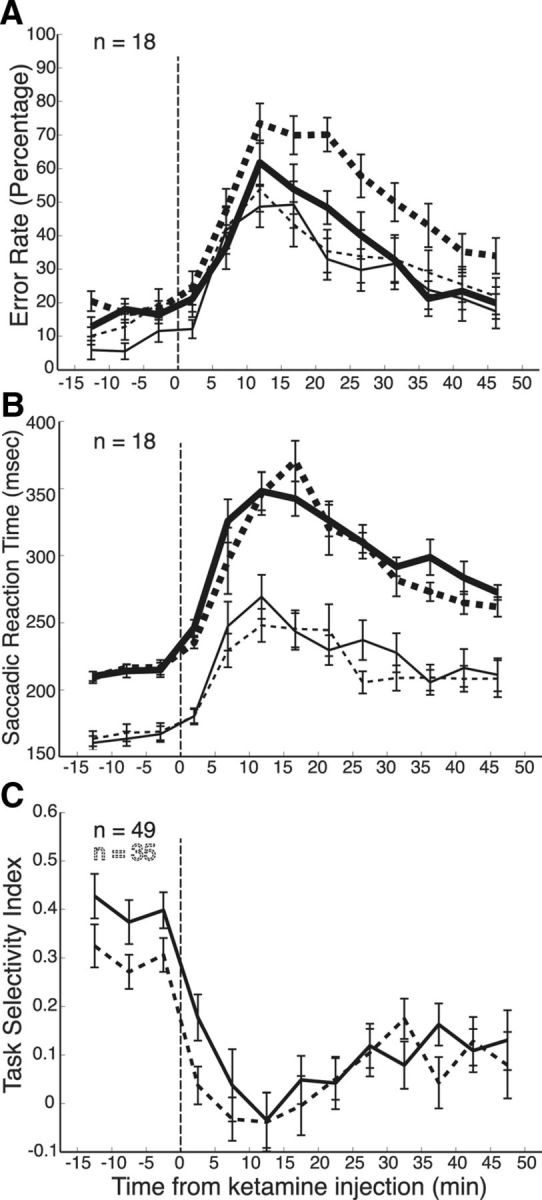
Time course of ketamine administration. A, Transient increase of error rates in prosaccade (thin lines) and antisaccade trials (thick lines) in rule-visible (solid lines) and rule-memorized (dashed lines) conditions. B, Transient increase in saccade reaction times, same format as A. C, Reduction of task selectivity following ketamine administration. Task selectivity was calculated for significant neurons for the prestimulus (dashed line) and stimulus/response (solid line) epochs. The time of ketamine injection is indicated by a vertical dashed line.
Figure 8.

Effects of ketamine on task selectivity. A, Task selectivity for the individual 35 neurons is plotted during the preinjection period against task selectivity during the postinjection period. Rule indicates neurons (squares) that showed maximal task selectivity between prosaccades and antisaccades. Memory indicates neurons (circles) that showed maximal task selectivity between the rule-visible and rule-memorized conditions. Dashed line, Unity line (slope = 1). B, Same as A, but for the 49 neurons in the stimulus/response epoch. Rule indicates neurons (squares) that showed maximal task selectivity between prosaccades and antisaccades. Direction indicates neurons (circles) that showed maximal task selectivity between ipsilateral and contralateral stimulus presentations. The solid points in both plots represent narrow-spiking neurons, and the hollow points represent broad-spiking neurons.
If the decrease in performance is related to the decrease in task selectivity of prefrontal neurons following ketamine administration, one would expect that task selectivity would be higher on correct trials than on error trials during this postinjection period. We tested this prediction by first identifying the preferred and nonpreferred condition for each neuron (highest and lowest discharge rate, respectively) during the preinjection period and then comparing task selectivity following ketamine injection between correct trials and error trials. Neurons that were recorded from sessions in which the animal completed fewer than five of any task type correctly were excluded from this analysis. In both the prestimulus period (Fig. 9A) and the stimulus/response period (Fig. 9B), neurons showed a significantly lower task selectivity during erroneous trials than correct trials (p < 0.01, Wilcoxon's signed rank test). This analysis demonstrates that prefrontal task selectivity during the prestimulus and stimulus/response period is correlated with the animals' task performance.
Figure 9.

Changes in task selectivity for error trials and correct trials. Change in task selectivity of neurons during trials completed correctly after ketamine administration is plotted on the x-axis, while change in task selectivity of neurons during error trials is plotted on the y-axis. A, Differences of correct trials versus error trials during the prestimulus epoch. Data points below the unity line (dashed line) indicate a greater loss in task selectivity during error trials. B, Differences of correct trials versus error trials during the stimulus/response epoch (same format as A). The solid points in both plots represent narrow-spiking neurons; the hollow points represent broad-spiking neurons.
To illustrate the changes in task selectivity following ketamine injection, we determined for each neuron that displayed interaction effects of DRUG during the prestimulus or stimulus/response period the preferred task (highest discharge rate) and nonpreferred task (lowest discharge rate) of the neuron during the preinjection period and constructed mean population activity plots across the trial both before (blue lines) and after ketamine administration (red lines) (Fig. 10). Although the mean discharge rate increased across the entire trial, the differences between the preferred (solid lines) and nonpreferred condition (dashed lines) were reduced considerably following ketamine injection.
Figure 10.
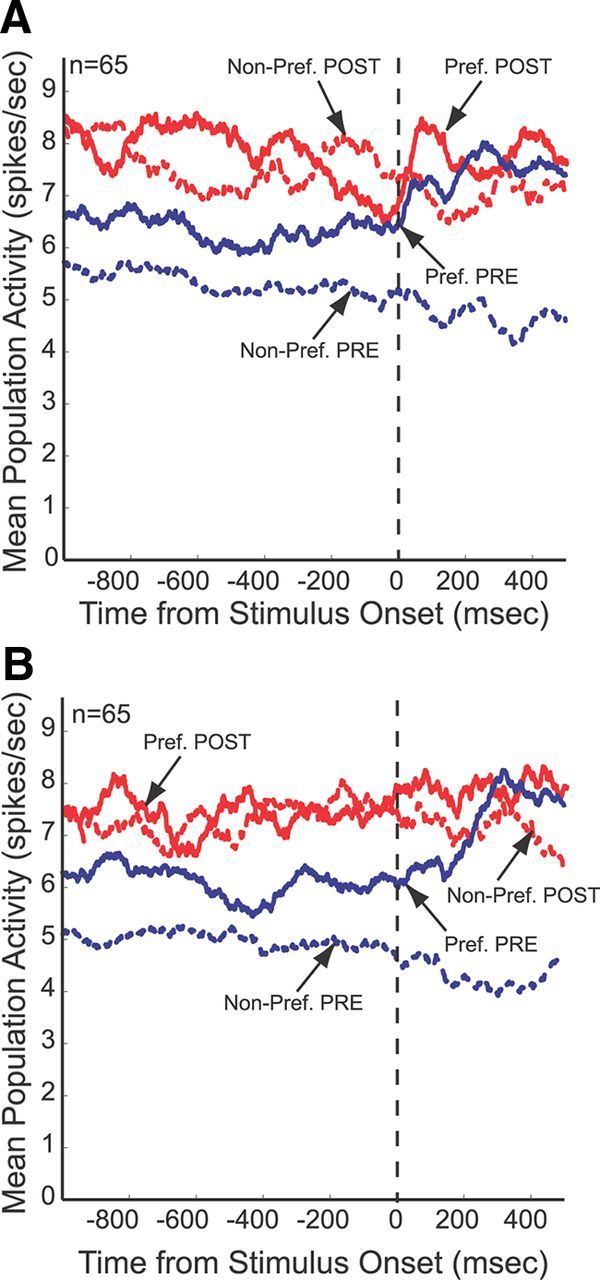
Effects of ketamine on population activity of task-selective neurons. In both rule-visible (A) and rule-memorized (B) conditions, the population activity shows a large separation between the preferred and nonpreferred condition before ketamine administration. This selectivity is reduced after ketamine.
Last, to highlight the importance of both epochs to our study, we constructed ROC curves testing task selectivity significance levels for rule-visible (Fig. 11A) and rule-memorized (Fig. 11B) tasks. The prestimulus period displayed multiple time points during which the selectivity achieved significance; however, the strength of task selectivity in the stimulus/response period was much more prominent. The ROC values became nonsignificant following ketamine administration during both epochs.
Figure 11.

ROC curve of task selectivity before and after ketamine administration. A, ROC values for rule-visible trials. Preinjection (blue solid line) and postinjection (red solid line) task selectivity ROC values with accompanying 2.5 and 97.5% (dashed lines) significance cutoff values as calculated by bootstrap analysis. B, ROC values for rule-memorized trials (same format as A).
Discussion
The ability of ketamine to induce a schizophrenia-like endophenotype was initially attributed to the antagonism of NMDA receptors in the PFC and an assumed downregulation of glutamatergic activity in this area (Flohr, 1992; Krystal et al., 1994; Malhotra et al., 1996). Electrophysiological studies in rodents, however, have shown that acute, subanesthetic doses of ketamine induce hyperactivity in the frontal cortex while still producing the cognitive deficits required for a model of this disease (Jackson et al., 2004; Homayoun and Moghaddam, 2007). Our results confirm this finding in the lateral PFC of nonhuman primates and demonstrate that subanesthetic doses of ketamine reduce task selectivity of PFC neurons. Our findings suggest that an increase in activity may be impairing task-selective outputs from the PFC by decreasing the task selectivity of neurons and thus reducing the ability of the PFC to exert cognitive control.
Ketamine impairs prosaccades and antisaccades
A previous study that examined the behavioral consequences of acute ketamine administration in green monkeys found a dose-dependent increase in reaction times for both prosaccades and antisaccades (Condy et al., 2005). Condy et al. (2005) tested prosaccade and antisaccade trials in separate blocks and reported that ketamine increased errors by up to 60–70% on antisaccade trials, whereas it did not impair the performance of prosaccade blocks. Our data confirm the increases in SRTs and increased errors on antisaccade trials following subanesthetic doses of ketamine but also show that ketamine impairs the performance on prosaccade trials when these are randomly interleaved with antisaccade trials. Similar increases in SRT and error rates have been reported in a cued task switch paradigm following ketamine administration (Stoet and Snyder, 2006).
The increased error rates by ketamine on antisaccade trials have been interpreted as a loss of behavioral inhibitory control over the prepotent stimulus-triggered saccade, potentially by interrupting dorsolateral PFC function (Condy et al., 2005). Our data do not support this hypothesis. While ketamine did increase the error rate on antisaccade trials, it also led to a greater number of errors on prosaccade trials (i.e., the monkeys generated antisaccades on prosaccade trials). The effects were prolonged on antisaccade trials that contained a working memory component, and following ketamine administration both monkeys frequently discontinued fixation or did not respond. Furthermore, the errors on antisaccade trials had reaction times well above the range of automatic express saccades (80–125 ms), which are directly triggered by the incoming visual stimulus in the superior colliculus (Dorris et al., 1997; Everling et al., 1999). Interestingly, although often described as automatic or prepotent responses, errors of schizophrenic patients in the antisaccade task are typically not in the express saccade range (R. Lencer, personal communication). We propose that ketamine increased errors on prosaccade and antisaccade trials because it impaired the animal's ability to selectively maintain, or apply the two task sets. This type of deficit is common in patients with frontal lobe damage and has been termed goal neglect (Duncan et al., 1996). Duncan et al. (2008) have suggested that frontal and possibly parietal brain areas are involved in organizing relevant facts, rules, and requirements into a “task model.” This idea is supported by many experiments on single neurons in the primate lateral PFC in behaving monkey. These studies have found that a large proportion of PFC neurons code various aspects of whatever task a monkey has been trained to perform (stimuli, responses, rules, rewards) and that the coding changes when the animal must perform a different task (Hoshi et al., 1998; Rainer et al., 1998; White and Wise, 1999; Asaad et al., 2000; Everling and DeSouza, 2005; Everling et al., 2006; Johnston and Everling, 2006a).
Ketamine increases activity of PFC neurons
To directly test whether ketamine interfered with the coding of task-relevant information in the PFC, we recorded the activity of single neurons in the lateral PFC before and after the administration of ketamine. Rodent studies showed that subanesthetic, systemic doses of ketamine are capable of increasing frontal cortex metabolic (Duncan et al., 1999; Dawson et al., 2011) and neural discharge activity (Jackson et al., 2004; Homayoun and Moghaddam, 2007). When administered to human subjects, acute doses of ketamine can also increase neural activity as measured by fMRI in working memory tasks (Honey et al., 2004) and regional cerebral blood flow (Holcomb et al., 2005). Here, we found that subanesthetic, systemic administration of ketamine in the behaving primate also acutely increases the activity of neurons in the lateral PFC. While the behavioral findings alone provide support for a ketamine-centric preclinical model of schizophrenia, there may be paradoxical reversal when the model is compared with the disease at the neurophysiological level.
The glutamate model of schizophrenia posits that many of the symptoms of the disease (including the cognitive deficits) arise from a hypofrontality and hypoglutamatergic state in the PFC (Olney et al., 1999). While chronic administrations of NMDA antagonists can induce a similar hypofrontality (Morris et al., 2005; Mouri et al., 2007), the present study in primates and previous studies in rodents (Jackson et al., 2004; Homayoun and Moghaddam, 2007) found that acute treatments of NMDA antagonists increase frontal activity. It should be noted here that the hypofrontality in the PFC of schizophrenic patients is inferred from EEG, PET, and fMRI studies but that it is unknown whether the activity of single PFC neurons is actually reduced in schizophrenic patients (Ragland et al., 2007; Marek et al., 2010).
It has been suggested that ketamine acts preferentially on PFC interneurons and therefore produces a localized disinhibition of pyramidal neurons (Homayoun and Moghaddam, 2007). This specificity for GABAergic interneurons parallels postmortem studies in humans that found patients with schizophrenia often exhibited reduced levels of interneuron markers (Benes et al., 1991; Lewis et al., 1999). However, a more recent study has found that this selectivity may not be this straightforward, as NMDA antagonists have been shown to exhibit a lowered affinity for fast-spiking interneurons (Rotaru et al., 2011). These newer data support the hypothesis that the mechanism behind the increased activity in the PFC may be an effect of NMDA receptor blockade in regions of the brain that have inhibitory projections to the PFC (Kiss et al., 2011) since localized injections of NMDA antagonists into the PFC were unable to increase PFC glutamatergic activity in rodents (Suzuki et al., 2002; Lorrain et al., 2003). To test whether ketamine had different effects on interneurons and pyramidal cells, we separated the recorded neurons based on their spike widths (Mitchell et al., 2007; Johnston et al., 2009). The results suggest that systemic ketamine administration had similar effects on the two groups; however, these results should be interpreted with caution because of the low number of putative interneurons.
One might have expected that an increase in activity would increase task selectivity of PFC neurons, but our data clearly show that the increase in activity was accompanied by a decrease in selectivity for the prosaccade/antisaccade task and response direction. Moreover, we could show that task selectivity was lower on error trials than correct trials following ketamine administration. This finding demonstrates that task selectivity of PFC neurons correlates with the animal's performance. This nondiscriminatory increase in neural activity that decreases task selectivity, would decrease the task-selective signals that the PFC sends to other areas like the superior colliculus (Johnston and Everling, 2006b, 2009). Without these task-selective signals, the flow of task-related activity in the saccade generation network may be impaired. Interestingly, an increased prefrontal activation has recently been shown using fMRI in the dorsolateral PFC of schizophrenic patients during the performance of prosaccades and antisaccades (Fukumoto-Motoshita et al., 2009).
It should be noted that the behavioral deficits in task performance are much stronger in this study that induced PFC hyperactivity than they were in a previous study, in which we induced acute PFC hypoactivity via surgically implanted cryoloops in the principal sulcus (Koval et al., 2011). Animals performing the antisaccade task exhibited only mild impairments in the rule-visible task when the principal sulcus was cryogenically deactivated; however, performance in the rule-memorized task was impaired to a much greater degree. The strong impairments on the rule-visible task following ketamine injections seen here may have been caused by a disruption of task selectivity in the ventral PFC, which is critical for cued stimulus–response associations (Bussey et al., 2001, 2002; Buckley et al., 2009).
Due to the systemic administration method, we cannot rule out that the behavioral effects by ketamine on prosaccades and antisaccades are mediated by areas outside of the lateral PFC, as task selectivity for prosaccades and antisaccades has also been found in the globus pallidus, thalamus, lateral intraparietal area, frontal eye fields, and supplementary eye fields (Schlag-Rey et al., 1997; Gottlieb and Goldberg, 1999; Everling and Munoz, 2000; Zhang and Barash, 2000; Ford and Everling, 2009; Watanabe and Munoz, 2009; Yoshida and Tanaka, 2009; Kunimatsu and Tanaka, 2010). In fact, it has previously been demonstrated that ketamine can have a significant effect on superior colliculus neurons (Populin, 2005); however, that study used doses of ketamine well within the anesthetic range.
In summary, our data show that ketamine increases the activity of PFC neurons in primates, while it reduces at the same time task selectivity in these neurons. The nondiscriminate increase in PFC activity may mask the efferent task-related signal required by downstream cortical and subcortical regions for correct task performance, effectively exporting irrelevant noise.
Footnotes
This work was supported by grants from the Ontario Mental Health Foundation and the Canadian Institutes of Health Research. We thank S. Hughes and B. Soper for excellent surgical and technical assistance. We are also grateful to J. Gati for help in MR imaging.
References
- Adler CM, Malhotra AK, Elman I, Goldberg T, Egan M, Pickar D, Breier A. Comparison of ketamine-induced thought disorder in healthy volunteers and thought disorder in schizophrenia. Am J Psychiatry. 1999;156:1646–1649. doi: 10.1176/ajp.156.10.1646. [DOI] [PubMed] [Google Scholar]
- Asaad WF, Rainer G, Miller EK. Task-specific neural activity in the primate prefrontal cortex. J Neurophysiol. 2000;84:451–459. doi: 10.1152/jn.2000.84.1.451. [DOI] [PubMed] [Google Scholar]
- Benes FM, McSparren J, Bird ED, SanGiovanni JP, Vincent SL. Deficits in small interneurons in prefrontal and cingulate cortices of schizophrenic and schizoaffective patients. Arch Gen Psychiatry. 1991;48:996–1001. doi: 10.1001/archpsyc.1991.01810350036005. [DOI] [PubMed] [Google Scholar]
- Buckley MJ, Mansouri FA, Hoda H, Mahboubi M, Browning PG, Kwok SC, Phillips A, Tanaka K. Dissociable components of rule-guided behavior depend on distinct medial and prefrontal regions. Science. 2009;325:52–58. doi: 10.1126/science.1172377. [DOI] [PubMed] [Google Scholar]
- Bussey TJ, Wise SP, Murray EA. The role of ventral and orbital prefrontal cortex in conditional visuomotor learning and strategy use in rhesus monkeys (Macaca mulatta) Behav Neurosci. 2001;115:971–982. doi: 10.1037//0735-7044.115.5.971. [DOI] [PubMed] [Google Scholar]
- Bussey TJ, Wise SP, Murray EA. Interaction of ventral and orbital prefrontal cortex with inferotemporal cortex in conditional visuomotor learning. Behav Neurosci. 2002;116:703–715. [PubMed] [Google Scholar]
- Condy C, Wattiez N, Rivaud-Péchoux S, Gaymard B. Ketamine-induced distractibility: an oculomotor study in monkeys. Biol Psychiatry. 2005;57:366–372. doi: 10.1016/j.biopsych.2004.10.036. [DOI] [PubMed] [Google Scholar]
- Dawson N, Morris BJ, Pratt JA. Subanaesthetic ketamine treatment alters prefrontal cortex connectivity with thalamus and ascending subcortical systems. Schizophr Bull. 2011 doi: 10.1093/schbul/sbr144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorris MC, Paré M, Munoz DP. Neuronal activity in monkey superior colliculus related to the initiation of saccadic eye movements. J Neurosci. 1997;17:8566–8579. doi: 10.1523/JNEUROSCI.17-21-08566.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan GE, Miyamoto S, Leipzig JN, Lieberman JA. Comparison of brain metabolic activity patterns induced by ketamine, MK-801 and amphetamine in rats: support for NMDA receptor involvement in responses to subanesthetic dose of ketamine. Brain Res. 1999;843:171–183. doi: 10.1016/s0006-8993(99)01776-x. [DOI] [PubMed] [Google Scholar]
- Duncan J, Emslie H, Williams P, Johnson R, Freer C. Intelligence and the frontal lobe: the organization of goal-directed behavior. Cogn Psychol. 1996;30:257–303. doi: 10.1006/cogp.1996.0008. [DOI] [PubMed] [Google Scholar]
- Duncan J, Parr A, Woolgar A, Thompson R, Bright P, Cox S, Bishop S, Nimmo-Smith I. Goal neglect and Spearman's g: competing parts of a complex task. J Exp Psychol Gen. 2008;137:131–148. doi: 10.1037/0096-3445.137.1.131. [DOI] [PubMed] [Google Scholar]
- Elvevåg B, Goldberg TE. Cognitive impairment in schizophrenia is the core of the disorder. Crit Rev Neurobiol. 2000;14:1–21. [PubMed] [Google Scholar]
- Everling S, DeSouza JF. Rule-dependent activity for prosaccades and antisaccades in the primate prefrontal cortex. J Cogn Neurosci. 2005;17:1483–1496. doi: 10.1162/0898929054985455. [DOI] [PubMed] [Google Scholar]
- Everling S, Fischer B. The antisaccade: a review of basic research and clinical studies. Neuropsychologia. 1998;36:885–899. doi: 10.1016/s0028-3932(98)00020-7. [DOI] [PubMed] [Google Scholar]
- Everling S, Munoz DP. Neuronal correlates for preparatory set associated with pro-saccades and anti-saccades in the primate frontal eye field. J Neurosci. 2000;20:387–400. doi: 10.1523/JNEUROSCI.20-01-00387.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everling S, Dorris MC, Klein RM, Munoz DP. Role of primate superior colliculus in preparation and execution of anti-saccades and pro-saccades. J Neurosci. 1999;19:2740–2754. doi: 10.1523/JNEUROSCI.19-07-02740.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everling S, Tinsley CJ, Gaffan D, Duncan J. Selective representation of task-relevant objects and locations in the monkey prefrontal cortex. Eur J Neurosci. 2006;23:2197–2214. doi: 10.1111/j.1460-9568.2006.04736.x. [DOI] [PubMed] [Google Scholar]
- Fischer B, Boch R, Ramsperger E. Express-saccades of the monkey: effect of daily training on probability of occurrence and reaction time. Exp Brain Res. 1984;55:232–242. doi: 10.1007/BF00237274. [DOI] [PubMed] [Google Scholar]
- Flohr H. Qualia and brain processes. In: Beckermann A, Flohr H, Kim J, editors. Emergence or reduction? Essays on the prospects of nonreductive phyiscalism. Berlin: De Gruyter; 1992. pp. 220–238. [Google Scholar]
- Ford KA, Everling S. Neural activity in primate caudate nucleus associated with pro- and antisaccades. J Neurophysiol. 2009;102:2334–2341. doi: 10.1152/jn.00125.2009. [DOI] [PubMed] [Google Scholar]
- Fukumoto-Motoshita M, Matsuura M, Ohkubo T, Ohkubo H, Kanaka N, Matsushima E, Taira M, Kojima T, Matsuda T. Hyperfrontality in patients with schizophrenia during saccade and antisaccade tasks: a study with fMRI. Psychiatry Clin Neurosci. 2009;63:209–217. doi: 10.1111/j.1440-1819.2009.01941.x. [DOI] [PubMed] [Google Scholar]
- Fukushima J, Fukushima K, Chiba T, Tanaka S, Yamashita I, Kato M. Disturbances of voluntary control of saccadic eye movements in schizophrenic patients. Biol Psychiatry. 1988;23:670–677. doi: 10.1016/0006-3223(88)90050-9. [DOI] [PubMed] [Google Scholar]
- Goeree R, Farahati F, Burke N, Blackhouse G, O'Reilly D, Pyne J, Tarride JE. The economic burden of schizophrenia in Canada in 2004. Curr Med Res Opin. 2005;21:2017–2028. doi: 10.1185/030079905X75087. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, Castner SA, Svensson TH, Siever LJ, Williams GV. Targeting the dopamine D1 receptor in schizophrenia: insights for cognitive dysfunction. Psychopharmacology (Berl) 2004;174:3–16. doi: 10.1007/s00213-004-1793-y. [DOI] [PubMed] [Google Scholar]
- Gottlieb J, Goldberg ME. Activity of neurons in the lateral intraparietal area of the monkey during an antisaccade task. Nat Neurosci. 1999;2:906–912. doi: 10.1038/13209. [DOI] [PubMed] [Google Scholar]
- Guitton D, Buchtel HA, Douglas RM. Frontal lobe lesions in man cause difficulties in suppressing reflexive glances and in generating goal-directed saccades. Exp Brain Res. 1985;58:455–472. doi: 10.1007/BF00235863. [DOI] [PubMed] [Google Scholar]
- Holcomb HH, Lahti AC, Medoff DR, Cullen T, Tamminga CA. Effects of noncompetitive NMDA receptor blockade on anterior cingulate cerebral blood flow in volunteers with schizophrenia. Neuropsychopharmacology. 2005;30:2275–2282. doi: 10.1038/sj.npp.1300824. [DOI] [PubMed] [Google Scholar]
- Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honey RA, Honey GD, O'Loughlin C, Sharar SR, Kumaran D, Bullmore ET, Menon DK, Donovan T, Lupson VC, Bisbrown-Chippendale R, Fletcher PC. Acute ketamine administration alters the brain responses to executive demands in a verbal working memory task: an FMRI study. Neuropsychopharmacology. 2004;29:1203–1214. doi: 10.1038/sj.npp.1300438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi E, Shima K, Tanji J. Task-dependent selectivity of movement-related neuronal activity in the primate prefrontal cortex. J Neurophysiol. 1998;80:3392–3397. doi: 10.1152/jn.1998.80.6.3392. [DOI] [PubMed] [Google Scholar]
- Jackson ME, Homayoun H, Moghaddam B. NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc Natl Acad Sci U S A. 2004;101:8467–8472. doi: 10.1073/pnas.0308455101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC. When doors of perception close: bottom-up models of disrupted cognition in schizophrenia. Annu Rev Clin Psychol. 2009;5:249–275. doi: 10.1146/annurev.clinpsy.032408.153502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston K, Everling S. Neural activity in monkey prefrontal cortex is modulated by task context and behavioral instruction during delayed-match-to-sample and conditional prosaccade-antisaccade tasks. J Cogn Neurosci. 2006a;18:749–765. doi: 10.1162/jocn.2006.18.5.749. [DOI] [PubMed] [Google Scholar]
- Johnston K, Everling S. Monkey dorsolateral prefrontal cortex sends task-selective signals directly to the superior colliculus. J Neurosci. 2006b;26:12471–12478. doi: 10.1523/JNEUROSCI.4101-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston K, Everling S. Task-relevant output signals are sent from monkey dorsolateral prefrontal cortex to the superior colliculus during a visuospatial working memory task. J Cogn Neurosci. 2009;21:1023–1038. doi: 10.1162/jocn.2009.21067. [DOI] [PubMed] [Google Scholar]
- Johnston K, DeSouza JF, Everling S. Monkey prefrontal cortical pyramidal and putative interneurons exhibit differential patterns of activity between prosaccade and antisaccade tasks. J Neurosci. 2009;29:5516–5524. doi: 10.1523/JNEUROSCI.5953-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss T, Hoffmann WE, Scott L, Kawabe TT, Milici AJ, Nilsen EA, Hajós M. Role of thalamic projection in NMDA receptor-induced disruption of cortical slow oscillation and short-term plasticity. Front Psychiatry. 2011;2:14. doi: 10.3389/fpsyt.2011.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koval MJ, Lomber SG, Everling S. Prefrontal cortex deactivation in macaques alters activity in the superior colliculus and impairs voluntary control of saccades. J Neurosci. 2011;31:8659–8668. doi: 10.1523/JNEUROSCI.1258-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr, Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- Kunimatsu J, Tanaka M. Roles of the primate motor thalamus in the generation of antisaccades. J Neurosci. 2010;30:5108–5117. doi: 10.1523/JNEUROSCI.0406-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahti AC, Koffel B, LaPorte D, Tamminga CA. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 1995;13:9–19. doi: 10.1016/0893-133X(94)00131-I. [DOI] [PubMed] [Google Scholar]
- Lahti AC, Weiler MA, Tamara Michaelidis BA, Parwani A, Tamminga CA. Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology. 2001;25:455–467. doi: 10.1016/S0893-133X(01)00243-3. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Pierri JN, Volk DW, Melchitzky DS, Woo TU. Altered GABA neurotransmission and prefrontal cortical dysfunction in schizophrenia. Biol Psychiatry. 1999;46:616–626. doi: 10.1016/s0006-3223(99)00061-x. [DOI] [PubMed] [Google Scholar]
- Lorrain DS, Baccei CS, Bristow LJ, Anderson JJ, Varney MA. Effects of ketamine and N-methyl-d-aspartate on glutamate and dopamine release in the rat prefrontal cortex: modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience. 2003;117:697–706. doi: 10.1016/s0306-4522(02)00652-8. [DOI] [PubMed] [Google Scholar]
- Malhotra AK, Pinals DA, Weingartner H, Sirocco K, Missar CD, Pickar D, Breier A. NMDA receptor function and human cognition: the effects of ketamine in healthy volunteers. Neuropsychopharmacology. 1996;14:301–307. doi: 10.1016/0893-133X(95)00137-3. [DOI] [PubMed] [Google Scholar]
- Malhotra AK, Pinals DA, Adler CM, Elman I, Clifton A, Pickar D, Breier A. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology. 1997;17:141–150. doi: 10.1016/S0893-133X(97)00036-5. [DOI] [PubMed] [Google Scholar]
- Marek GJ, Behl B, Bespalov AY, Gross G, Lee Y, Schoemaker H. Glutamatergic (N-methyl-d-aspartate receptor) hypofrontality in schizophrenia: too little juice or a miswired brain? Mol Pharmacol. 2010;77:317–326. doi: 10.1124/mol.109.059865. [DOI] [PubMed] [Google Scholar]
- McDowell JE, Brown GG, Paulus M, Martinez A, Stewart SE, Dubowitz DJ, Braff DL. Neural correlates of refixation saccades and antisaccades in normal and schizophrenia subjects. Biol Psychiatry. 2002;51:216–223. doi: 10.1016/s0006-3223(01)01204-5. [DOI] [PubMed] [Google Scholar]
- Mitchell JF, Sundberg KA, Reynolds JH. Differential attention-dependent response modulation across cell classes in macaque visual area V4. Neuron. 2007;55:131–141. doi: 10.1016/j.neuron.2007.06.018. [DOI] [PubMed] [Google Scholar]
- Morris BJ, Cochran SM, Pratt JA. PCP: from pharmacology to modelling schizophrenia. Curr Opin Pharmacol. 2005;5:101–106. doi: 10.1016/j.coph.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Mouri A, Noda Y, Enomoto T, Nabeshima T. Phencyclidine animal models of schizophrenia: approaches from abnormality of glutamatergic neurotransmission and neurodevelopment. Neurochem Int. 2007;51:173–184. doi: 10.1016/j.neuint.2007.06.019. [DOI] [PubMed] [Google Scholar]
- Munoz DP, Everling S. Look away: the anti-saccade task and the voluntary control of eye movement. Nat Rev Neurosci. 2004;5:218–228. doi: 10.1038/nrn1345. [DOI] [PubMed] [Google Scholar]
- Newcomer JW, Farber NB, Jevtovic-Todorovic V, Selke G, Melson AK, Hershey T, Craft S, Olney JW. Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology. 1999;20:106–118. doi: 10.1016/S0893-133X(98)00067-0. [DOI] [PubMed] [Google Scholar]
- Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. 1999;33:523–533. doi: 10.1016/s0022-3956(99)00029-1. [DOI] [PubMed] [Google Scholar]
- Pierrot-Deseilligny C, Rivaud S, Gaymard B, Agid Y. Cortical control of reflexive visually-guided saccades. Brain. 1991;114:1473–1485. doi: 10.1093/brain/114.3.1473. [DOI] [PubMed] [Google Scholar]
- Populin LC. Anesthetics change the excitation/inhibition balance that governs sensory processing in the cat superior colliculus. J Neurosci. 2005;25:5903–5914. doi: 10.1523/JNEUROSCI.1147-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouget P, Wattiez N, Rivaud-Pechoux S, Gaymard B. Rapid development of tolerance to sub-anaesthetic dose of ketamine: an oculomotor study in macaque monkeys. Psychopharmacology (Berl) 2010;209:313–318. doi: 10.1007/s00213-010-1797-8. [DOI] [PubMed] [Google Scholar]
- Povinelli DJ, Preuss TM. Theory of mind: evolutionary history of a cognitive specialization. Trends Neurosci. 1995;18:418–424. doi: 10.1016/0166-2236(95)93939-u. [DOI] [PubMed] [Google Scholar]
- Preuss TM. Taking the measure of diversity: comparative alternatives to the model-animal paradigm in cortical neuroscience. Brain Behav Evol. 2000;55:287–299. doi: 10.1159/000006664. [DOI] [PubMed] [Google Scholar]
- Radant AD, Bowdle A, Cowley DS, Kharasch ED, Roy-Byrne PP. Does ketamine-mediated N-methyl-D-aspartate receptor antagonism cause schizophrenia-like oculomotor abnormalities? Neuropsychopharmacology. 1998;19:434–444. doi: 10.1016/S0893-133X(98)00030-X. [DOI] [PubMed] [Google Scholar]
- Ragland JD, Yoon J, Minzenberg MJ, Carter CS. Neuroimaging of cognitive disability in schizophrenia: search for a pathophysiological mechanism. Int Rev Psychiatry. 2007;19:417–427. doi: 10.1080/09540260701486365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainer G, Asaad WF, Miller EK. Memory fields of neurons in the primate prefrontal cortex. Proc Natl Acad Sci U S A. 1998;95:15008–15013. doi: 10.1073/pnas.95.25.15008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotaru DC, Yoshino H, Lewis DA, Ermentrout GB, Gonzalez-Burgos G. Glutamate receptor subtypes mediating synaptic activation of prefrontal cortex neurons: relevance for schizophrenia. J Neurosci. 2011;31:142–156. doi: 10.1523/JNEUROSCI.1970-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlag-Rey M, Amador N, Sanchez H, Schlag J. Antisaccade performance predicted by neuronal activity in the supplementary eye field. Nature. 1997;390:398–401. doi: 10.1038/37114. [DOI] [PubMed] [Google Scholar]
- Shen K, Kalwarowsky S, Clarence W, Brunamonti E, Pare M. Beneficial effects of the NMDA antagonist ketamine on decision processes in visual search. J Neurosci. 2010;30:9947–9953. doi: 10.1523/JNEUROSCI.6317-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoet G, Snyder LH. Effects of the NMDA antagonist ketamine on task-switching performance: evidence for specific impairments of executive control. Neuropsychopharmacology. 2006;31:1675–1681. doi: 10.1038/sj.npp.1300930. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Jodo E, Takeuchi S, Niwa S, Kayama Y. Acute administration of phencyclidine induces tonic activation of medial prefrontal cortex neurons in freely moving rats. Neuroscience. 2002;114:769–779. doi: 10.1016/s0306-4522(02)00298-1. [DOI] [PubMed] [Google Scholar]
- Taffe MA, Davis SA, Gutierrez T, Gold LH. Ketamine impairs multiple cognitive domains in rhesus monkeys. Drug Alcohol Depend. 2002;68:175–187. doi: 10.1016/s0376-8716(02)00194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai G, Passani LA, Slusher BS, Carter R, Baer L, Kleinman JE, Coyle JT. Abnormal excitatory neurotransmitter metabolism in schizophrenic brains. Arch Gen Psychiatry. 1995;52:829–836. doi: 10.1001/archpsyc.1995.03950220039008. [DOI] [PubMed] [Google Scholar]
- van Os J, Kapur S. Schizophrenia. Lancet. 2009;374:635–645. doi: 10.1016/S0140-6736(09)60995-8. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Munoz DP. Neural correlates of conflict resolution between automatic and volitional actions by basal ganglia. Eur J Neurosci. 2009;30:2165–2176. doi: 10.1111/j.1460-9568.2009.06998.x. [DOI] [PubMed] [Google Scholar]
- White IM, Wise SP. Rule-dependent neuronal activity in the prefrontal cortex. Exp Brain Res. 1999;126:315–335. doi: 10.1007/s002210050740. [DOI] [PubMed] [Google Scholar]
- Wise SP. Forward frontal fields: phylogeny and fundamental function. Trends Neurosci. 2008;31:599–608. doi: 10.1016/j.tins.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida A, Tanaka M. Enhanced modulation of neuronal activity during antisaccades in the primate globus pallidus. Cereb Cortex. 2009;19:206–217. doi: 10.1093/cercor/bhn069. [DOI] [PubMed] [Google Scholar]
- Zhang M, Barash S. Neuronal switching of sensorimotor transformations for antisaccades. Nature. 2000;408:971–975. doi: 10.1038/35050097. [DOI] [PubMed] [Google Scholar]