

Abstract
It is well known that NMDA receptors (NMDARs) can both induce neurotoxicity and promote neuronal survival under different circumstances. Recent studies show that such paradoxical responses are related to the receptor location: the former to the extrasynaptic and the latter to the synaptic. The phosphoinositide 3-kinase (PI3K)/Akt kinase cascade is a key pathway responsible for the synaptic NMDAR-dependent neuroprotection. However, it is still unknown how synaptic NMDARs are coupled with the PI3K/Akt pathway. Here, we explored the role of an adaptor protein—adaptor protein containing pH domain, PTB domain, and leucine zipper motif (APPL1)—in this signal coupling using rat cortical neurons. We found that APPL1 existed in postsynaptic densities and associated with the NMDAR complex through binding to PSD95 at its C-terminal PDZ-binding motif. NMDARs, APPL1, and the PI3K/Akt cascade formed a complex in rat cortical neurons. Synaptic NMDAR activity increased the association of this complex, induced activation of the PI3K/Akt pathway, and consequently protected neurons against starvation-induced apoptosis. Perturbing APPL1 interaction with PSD95 by a peptide comprising the APPL1 C-terminal PDZ-binding motif dissociated the PI3K/Akt pathway from NMDARs. Either the peptide or lentiviral knockdown of APPL1 blocked synaptic NMDAR-dependent recruitment and activation of PI3K/Akt pathway, and consequently blocked synaptic NMDAR-dependent neuroprotection. These results suggest that APPL1 contributes to connecting synaptic NMDARs with the intracellular PI3K/Akt cascade and the downstream prosurvival signaling pathway in rat cortical neurons.
Introduction
NMDA receptors (NMDARs) are cation channels gated by the neurotransmitter glutamate and are essential mediators in synaptic transmission and plasticity. NMDARs play important roles in both the physiology and pathophysiology of the CNS. Excessive NMDAR activation causes neuronal loss in acute trauma and certain neurodegenerative diseases. In contrast, physiological NMDAR activity promotes neuronal survival and is resistant to apoptotic or excitotoxic insults (Hardingham, 2009). In addition, accumulating evidence indicates that the location of NMDARs mainly influences whether they are coupled to prodeath or prosurvival signals (Hardingham and Bading, 2010). Synaptic NMDARs are neuroprotective, and their activity triggers different neuroprotective signaling pathways, such as activation of the phosphoinositide 3-kinase (PI3K)/Akt pathway, activation of nuclear CREB (cAMP response element-binding protein), and enhancement of the antioxidant defense system (Hardingham, 2009). Extrasynaptic NMDARs preferentially initiate lethal downstream reactions including mitochondrial dysfunction (Murphy and Fiskum, 1999; Wang et al., 1999), oxidative stress (Kinouchi et al., 1991), and nitrosative stress (Aarts et al., 2003; Uehara et al., 2006; Luo and Zhu, 2011). Despite the increase in our understanding of NMDAR functions and their downstream signaling pathways, the molecular mechanisms underlying the coupling of NMDARs with different downstream signaling pathways are still far from well understood.
The PI3K/Akt kinase cascade is a key pathway responsible for the prosurvival effects of synaptic NMDAR activity (Lafon-Cazal et al., 2002; Papadia et al., 2005). PI3K is strongly activated by NMDARs in many neuronal types (Perkinton et al., 2002; Sutton and Chandler, 2002) and recruits the kinase PDK1 (phosphoinositide-dependent protein kinase) (Alessi et al., 1997) and its substrate Akt to the membrane. Subsequently, Akt is phosphorylated and activated, which further phosphorylates and inhibits proapoptotic substrates, such as GSK3β (glycogen synthase kinase-3β) (Soriano et al., 2006), ASK1 (apoptosis signal-regulating kinase 1) (Kim et al., 2001), p53 (Yamaguchi et al., 2001), BAD (Bcl2-associated death promoter) (Downward, 1999), and FOXO (forkhead box O) (Soriano et al., 2006). It is still unclear how Akt is recruited to the cell membrane, and how the PI3K/AKT kinase cascade is coupled to NMDARs.
Adaptor protein containing pH domain, PTB domain, and leucine zipper motif (APPL) is a multifunctional endosomal adaptor protein with two isoforms, APPL1 and APPL2. APPL interacts with transmembrane receptors such as the EGF receptor (Miaczynska et al., 2004), adiponectin receptors (Mao et al., 2006), TrkA (Lin et al., 2006), the follicle-stimulating hormone receptor (Nechamen et al., 2004), and DCC (Liu et al., 2002). Furthermore, APPL interacts with, and regulates the activity of, Akt (Mitsuuchi et al., 1999; Schenck et al., 2008) and is involved in fundamental processes such as proliferation, survival, growth, and metabolism.
Here, we found that, in the CNS, a subfraction of APPL1 localized at the postsynaptic density (PSD) and associated with NMDAR complexes by interacting with PSD95, and therefore tethered PI3K and Akt to the NMDAR complex. Functionally, APPL1 is required for synaptic NMDAR-dependent Akt activation and synaptic NMDAR-dependent neuroprotection.
Materials and Methods
Antibodies.
The primary antibodies for Western blotting (WB) were APPL1 (Santa Cruz; sc-67402), PSD95 (Millipore; MAB1596), GluN2A (sc-1468), GluN2B (homemade mAb), GluN1 (BD Biosciences Pharmingen; 556308), Akt (9272), Rab5 (sc-46692), synaptophysin (Sigma-Aldrich; S5768), GluA2 (MAB397), clathrin (sc-12734), PI3K p110β (sc-602), phospho-Akt Ser473 (4060), Erk1/2 (4696), phospho-Erk1/2 (9101), and GFP (Abcam; ab1218). The secondary antibodies for WB were DyLight 680/800-conjugated IgG (Thermo Fisher Scientific). The antibodies for immunoprecipitation were APPL1 (as for WB), GluN1 and antigen peptide (sc-1467 and sc-1467p), PSD95 (ab18258), and GFP (as for WB). The primary antibodies for immunocytochemistry were APPL1 (Cell Signaling; 3858), and GluN1 and PSD95 (as for WB). The secondary antibodies for immunocytochemistry were Alexa Fluor 488 anti-mouse IgG and Alexa Fluor 546 anti-rabbit IgG (Invitrogen).
Peptides.
Tat-APPL1CT peptide (YGRKKRRQRRREEGKKRESEA) and Tat-sAPPL1CT peptide (YGRKKRRQRRREREKEESAKG) were synthesized by GL Biochem.
Tissue coimmunoprecipitation.
The protocol for tissue coimmunoprecipitation (co-IP) was as previously described (Luo et al., 1997) with some modifications. All animal experiments were performed in accordance with the ethical guidelines of the Zhejiang University Animal Experimentation Committee and were in complete compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Adult mouse cortex of either sex was homogenized with 10 vol of homogenization buffer (10 mm Tris-HCl, pH 7.4, 320 mm sucrose, 1 mm PMSF, 1 μg/ml aprotinin). The tissue homogenate was centrifuged at 700 × g for 10 min at 4°C. A 0.10 vol of 10% sodium deoxycholate in 500 mm Tris-HCl, pH 9.0, was added to the supernatant, and the preparation was incubated at 37°C for 20 min for membrane solubilization. A 0.10 vol of 1% Triton X-100, 50 mm Tris-HCl, pH 9.0, was then added, and the preparation was dialyzed against binding buffer (50 mm Tris-HCl, pH 7.4, 0.1% Triton) overnight at 4°C. After centrifugation at 37,000 × g at 4°C for 30 min, IP antibody was added to the supernatant and incubated overnight at 4°C. Protein A-Sepharose (GE Healthcare; for APPL1 and PSD95 rabbit polyclonal antibodies) or protein G-agarose (Thermo Fisher Scientific; for GluN1 goat polyclonal antibody) was then added. After incubation for 1 h at 4°C, the mixtures were washed four times with binding buffer, and the immunoprecipitates were eluted with 2× SDS-PAGE loading buffer by boiling at 100°C for 5 min.
Co-IP of cultured neurons.
Cultured cortical neurons [days in vitro (DIV) 12–14] were lysed by buffer containing 50 mm Tris-HCl, pH 7.4, 1% sodium deoxycholate, 0.1% Triton X-100, 1 mm PMSF, and 1 μg/ml aprotinin. The lysate was then dialyzed against binding buffer overnight at 4°C. All subsequent procedures were the same as the protocol for tissue co-IP.
Co-IP of transfected HEK cells.
After transfection with indicated plasmids for 24 h, HEK293 cells were lysed by buffer containing 20 mm Tris, pH 7.5, 150 mm NaCl, 1% Triton X-100, and protease inhibitor mixtures. Anti-APPL1 antibody or anti-GFP antibody was added to the lysate and incubated overnight at 4°C. All subsequent procedures were the same as for tissue co-IP.
Subcellular fractionation.
Subcellular fractionation was conducted on adult mouse cortex of either sex using an adapted protocol (Pacchioni et al., 2009).
Detection of fluorescence resonance energy transfer using three-cube fluorescence resonance energy transfer measurement.
The fluorescence imaging work station for fluorescence resonance energy transfer (FRET) and the FRET quantification method have been described previously (Qiu et al., 2005, 2009). Briefly, the fluorescence imaging workstation consisted of an inverted microscope (TE2000; Nikon), Dual-View (Optical Insights), and a SNAP-HQ-cooled CCD (Roper Scientific). MetaMorph, version 5.0, software was used for analysis of the cell image data. The FRET ratio (FR) was calculated according to the following equation:
 |
SCUBE(SPECIMEN) denotes an intensity measurement, where CUBE indicates the filter cube (CFP, YFP, or FRET) and SPECIMEN indicates whether the cell is expressing donor (D) (CFP), acceptor (A) (YFP), or both (DA). RD1 = SFRET(D)/SCFP(D), and RA1 = SFRET(A)/SYFP(A). FRET signals were acquired from intact HEK293 cells expressing the indicated fluorescent tag protein.
Cortical neuronal cultures.
Cortical tissue was harvested from embryonic day 17–19 rats of either sex, and then gently chopped and digested in 0.5% trypsin for 13 min at 37°C. Dissociated cells were plated at a density of 0.4 × 104/cm2 in a 35 mm dish with poly-l-lysine-coated coverslips in Neurobasal medium containing 1% horse serum, 0.5 mm glutamine, 1% antibiotic, and 2% B27, at 37°C under 5% CO2. After 3 d, the medium was replaced with Neurobasal medium containing 0.5 mm glutamine, 1% antibiotic, and 2% B27. Subsequently, the culture medium was replaced every 5 d. At 5 DIV, cytosine arabinofuranoside was added at a final concentration of 2.5 μm.
Immunocytochemistry.
For staining of endogenous APPL1, GluN1, and PSD95, cortical neurons (DIV 11–13) were fixed for 10 min with 4% paraformaldehyde. After fixation, neurons were washed three times with PBS and incubated with blocking buffer (0.4% Triton X-100, 2.5% BSA in PBS) for 30 min. Primary antibodies were then applied in blocking buffer for 1 h at room temperature. Neurons were washed three times with PBS. Secondary antibodies were applied in blocking buffer for 1 h at room temperature. After washing three times with PBS, neurons were mounted. The images were acquired with a confocal microscope (Fluoview FV1000; Olympus). Image-Pro Plus 6.0 software was used to analyze colocalization of clusters.
Lentivirus-mediated RNAi in cortical neurons.
pFUGW-APPL1 shRNA-GFP lentiviral vector targeted against rat APPL1 (5′-CTCACCTGACTTCGAAACT-3′) and nonspecific control shRNA lentiviral vector against the sequence 5′-TTCTCCGAACGTGTCACGT-3′ were from Shanghai GeneChem. Cultured cortical neurons at DIV 6 were infected by lentivirus. Five to 6 d after infection, neurons were used for synaptic NMDAR activation or the neuroprotective model.
Synaptic or extrasynaptic NMDAR activation.
For synaptic NMDAR activation, cortical neurons at DIV 12–14 were treated for 30 min with 50 μm bicuculline (Bic) and 250 μm 4-aminopyridine (4-AP). For peptide blocking, 10 μm Tat-APPL1CT or Tat-sAPPL1CT was applied to neurons and incubated for 1 h before Bic and 4-AP treatment. Extrasynaptic NMDAR activation was performed as previously established protocols (Hardingham et al., 2002). Briefly, synaptic NMDARs were irreversibly blocked by MK801 (50 μm), Bic (50 μm), and 4-AP (250 μm) for 5 min. After washout, extrasynaptic NMDARs were then selectively activated by NMDA (10 μm) for 1 h. Treated neurons were lysed by buffer containing 20 mm Tris, pH 7.5, 150 mm NaCl, 1% Triton X-100, and protease inhibitor mixtures. Protein concentrations were measured. Equivalent samples were loaded for SDS-PAGE and Western blot.
Neuroprotective model.
DIV 10 cortical neurons were subjected to trophic deprivation by transferring them from growth medium to a starvation medium containing 10% MEM and 90% salt–glucose–glycine medium (114 mm NaCl, 0.22% NaHCO3, 5.3 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 10 mm HEPES, 1 mm glycine, 30 mm glucose, 0.5 mm sodium pyruvate). For neuroprotection, 50 μm Bic and 250 μm 4-AP were added to the starvation medium. For peptide blocking, 10 μm Tat-APPL1CT or Tat-sAPPL1CT was added at the same time and readded every 24 h. After 3 d of trophic deprivation, neurons were either subjected to Hoechst staining or lysed for Western blotting to detect cleaved caspase-3.
Hoechst staining.
Hoechst staining was performed according to the instructions with the kit (Beyotime Biotech). Tau protein was immunostained to distinguish neurons from glia and help to determine the state of neurons.
Results
APPL1 exists in postsynaptic density and partially colocalizes with PSD95 and NMDA receptors
APPL1 protein is enriched in mouse brain and widely expressed in different brain regions (data not shown). We determined the synaptic location of APPL1 in cortex by fractionation of synaptosomes prepared as previously described (Pacchioni et al., 2009). Digestion of synaptosomes yielded a PSD-enriched and a non-PSD-enriched membrane fraction. As expected, Rab5 and synaptophysin were enriched in the non-PSD fraction, while PSD95, as well as GluN1, GluN2A, and GluN2B subunits, were enriched in the PSD fraction (Fig. 1A). APPL1 was distributed both in the non-PSD and PSD fractions (Fig. 1A); this extends our previous knowledge of APPL1 as a Rab5-interacting protein localized on endosomes, implicating function of APPL1 in synapses. In addition, the APPL1-binding protein Akt, a key survival signaling molecule, was also partly located in the PSD fraction (Fig. 1A). We further examined the expression pattern of endogenous APPL1 in cortical neurons at DIV 11–13 by double immunofluorescent staining. APPL1 exhibited puncta along the dendrites, either appearing to be associated with dendritic spines or located in the dendritic shafts. The punctate staining of APPL1 and PSD95 was partially colocalized (Fig. 1B), which is consistent with a previous report (Majumdar et al., 2011). APPL1 also partially colocalized with the GluN1 subunit of the NMDAR (Fig. 1C). Further quantification revealed that 32.3 ± 2.1% (mean ± SEM) of total APPL1 clusters colocalized with PSD95, versus 25.0 ± 0.9% of PSD95 clusters containing APPL1 (2000–3000 clusters of each protein from six separate experiments). In addition, 48.0 ± 2.8% of APPL1 colocalized with GluN1, while 33.7 ± 3.0% of GluN1 clusters contained APPL1 (2000–3000 clusters of each protein from five separate experiments). These data indicate that APPL1 plays a potential role in synaptic function.
Figure 1.

Subcellular localization of APPL1 protein in brain. A, APPL1 and Akt existed both in the non-PSD and PSD fractions. PSD95, GluN2A, GluN2B, and GluN1 are components of the PSD fraction. Rab5 is a component of the late endosome. Synaptophysin is a component of presynaptic structure. B, Cultured cortical neurons (DIV 11–13) coimmunostained for endogenous APPL1 and PSD95. APPL1 clusters partially colocalized with PSD95 clusters (arrows). Scale bar, 10 μm. C, Coimmunostaining of endogenous APPL1 and GluN1. APPL1 clusters partially colocalized with GluN1 clusters (arrows). Scale bar, 10 μm.
APPL1 is a binding partner within NMDA receptor multiprotein complexes
APPL was identified as a component of NMDAR multiprotein complexes by mass spectrometric analysis (Husi et al., 2000). Here, co-IPs were performed to assess the association of APPL1 and NMDARs in mouse cortical lysates. APPL1 antibody immunoprecipitated APPL1 protein efficiently and coprecipitated the scaffold protein of NMDARs, PSD-95 (Fig. 2A). In addition, a small but significant fraction of GluN1, GluN2A, and GluN2B coprecipitated with APPL1. GluA2, a subunit of the AMPA receptor, did not coprecipitate with APPL1 (Fig. 2A), which indicated that APPL1 specifically associated with the NMDAR but not the AMPA receptor. We did not detect an interaction of GIPC with APPL1 in mouse cortical lysates (Fig. 2A), although GIPC is reported to bind with APPL1 through PDZ interaction in PC12 cells and in sympathetic neurons (Lin et al., 2006). We also found that APPL1 antibody was unable to coprecipitate clathrin, which is in accord with previous reports showing a lack of colocalization of APPL1-positive structures with clathrin-coated vesicles (Miaczynska et al., 2004; Zoncu et al., 2009). Control nonimmune IgGs immunoprecipitated none of these proteins. In the opposite direction, GluN1 antibody coprecipitated GluN2B and faint but detectable amounts of APPL1 (Fig. 2B). In contrast, GluN1 antibody was unable to precipitate GluN1 or coprecipitate APPL1 or GluN2B when preblocked by excessive antigen peptide, which further indicated the specificity of APPL1 and GluN1 co-IP. Immunoprecipitation with PSD95 antibody also coprecipitated APPL1, GluN2A, and GluN2B, but did not coprecipitate GluA2 (Fig. 2C). Together, these results demonstrate that APPL1 is a binding partner of the NMDAR multiprotein complex in vivo.
Figure 2.

APPL1 associates with the NMDA receptor complex in brain. A, APPL1 coprecipitated PSD95, GluN1, GluN2A, and GluN2B, but did not coprecipitate GluA2, GIPC, and clathrin from lysate of mouse cortex. Immunoprecipitation with nonimmune rabbit IgG was included as a negative control. B, GluN1 coprecipitated APPL1 and GluN2B but not GluA2 from lysate of mouse cortex. Immunoprecipitation with GluN1 antibody preblocked by excess antigen peptide (antigen blocking) was included as a negative control. C, PSD95 coprecipitated APPL1, GluN2A, and GluN2B, but not GluA2 from lysate of mouse cortex.
APPL1 interacts with PSD95 through its C-terminal PDZ-binding motif
The results above indicate that APPL1 belongs to the NMDAR multiprotein complex. To further assess whether APPL1 interacts directly with the NMDAR, we transfected GFP-GluN1, GFP-GluN2A, or GFP-GluN2B into HEK293 cells, and assessed the interaction of endogenous APPL1 with the different transfected NMDAR subunits by co-IP. APPL1 antibody precipitated endogenous APPL1 efficiently in HEK293 cells but was unable to coprecipitate GFP-GluN1, GFP-GluN2A, or GFP-GluN2B (Fig. 3A). The GluN1, GluN2A, or GluN2B subunit, when expressed alone in heterogeneous cells, is retained in the ER, and unable to be expressed in the cell membrane. However, APPL1 is an endosomal adaptor protein and mainly involved in the endocytosis of receptors expressed in the cell membrane. In the following experiments, we cotransfected GluN1/GluN2A or GluN1/GluN2B into HEK293 cells and functional NMDARs were formed in the cell membrane. Under such conditions, APPL1 antibody was still unable to coprecipitate GluN1, GluN2A, or GluN2B (Fig. 3B). Together, these results indicated that APPL1 does not interact directly with NMDAR subtypes.
Figure 3.

APPL1 interacts with PSD95 but not with NMDAR subunits. A, Co-IP of transfected GFP-GluN1, GFP-GluN2A, or GFP-GluN2B with endogenous APPL1 in HEK293 cells. GFP-GluN1, GFP-GluN2A, or GFP-GluN2B did not coprecipitate with APPL1. B, GFP-GluN2A/GluN1 or GFP-GluN2B/GluN1 were cotransfected into HEK293 so that they could form functional NMDARs on the plasma membrane. APPL1 was still unable to coprecipitate GluN1, GluN2A, or GluN2B under such conditions. C, GFP-PSD95 was transfected into HEK293 cells. APPL1 coprecipitated GFP-PSD95. D, GFP-PSD95 or GFP was transfected into HEK293 cells. GFP-PSD95 but not GFP coprecipitate APPL1.
Our results showed that APPL1 had a relatively strong co-IP with the PDZ protein PSD95 in mouse cortex (Fig. 2A). In addition, APPL1 has a PDZ-binding motif (-ESEA) at its C terminus. Next, we assessed the interaction of transfected GFP-PSD95 with endogenous APPL1 in HEK293 cells by co-IP. We found that APPL1 coprecipitated GFP-PSD95 efficiently (Fig. 3C). In the reverse co-IP test, GFP-PSD95 coprecipitated APPL1 (Fig. 3D). To investigate whether the APPL1 C-terminal PDZ-binding motif determines the APPL1–PSD95 interaction, we cotransfected CFP-APPL1/PSD95 or CFP-APPL1Δ4 (the last 4 aa, the PDZ-binding domain of APPL1, were deleted)/PSD95 into HEK293 cells. Co-IP tests with anti-GFP antibody showed that CFP-APPL1 but not CFP-APPL1Δ4 interacted with PSD95 (Fig. 4A). FRET is another method to detect protein–protein interactions in vivo (Qiu et al., 2005, 2009). Here, we tagged APPL1 with CFP and PSD95 with YFP and examined the interaction of APPL1 with PSD95 using FRET (Fig. 4B). As the negative control, when CFP-APPL1 and YFP were cotransfected, no FRET signal was detected [FR = 1.01 ± 0.03 (mean ± SEM); n = 24]. Cotransfection of CFP-APPL1 and YFP-PSD95 yielded a positive FRET signal (FR = 1.16 ± 0.04; n = 24). However, CFP-APPL1Δ4 and YFP-PSD95 yielded a negative FRET signal (FR = 1.00 ± 0.04; n = 23) (Fig. 4B). This suggests that the PDZ-binding motif of APPL1 is necessary for APPL1–PSD95 interaction.
Figure 4.
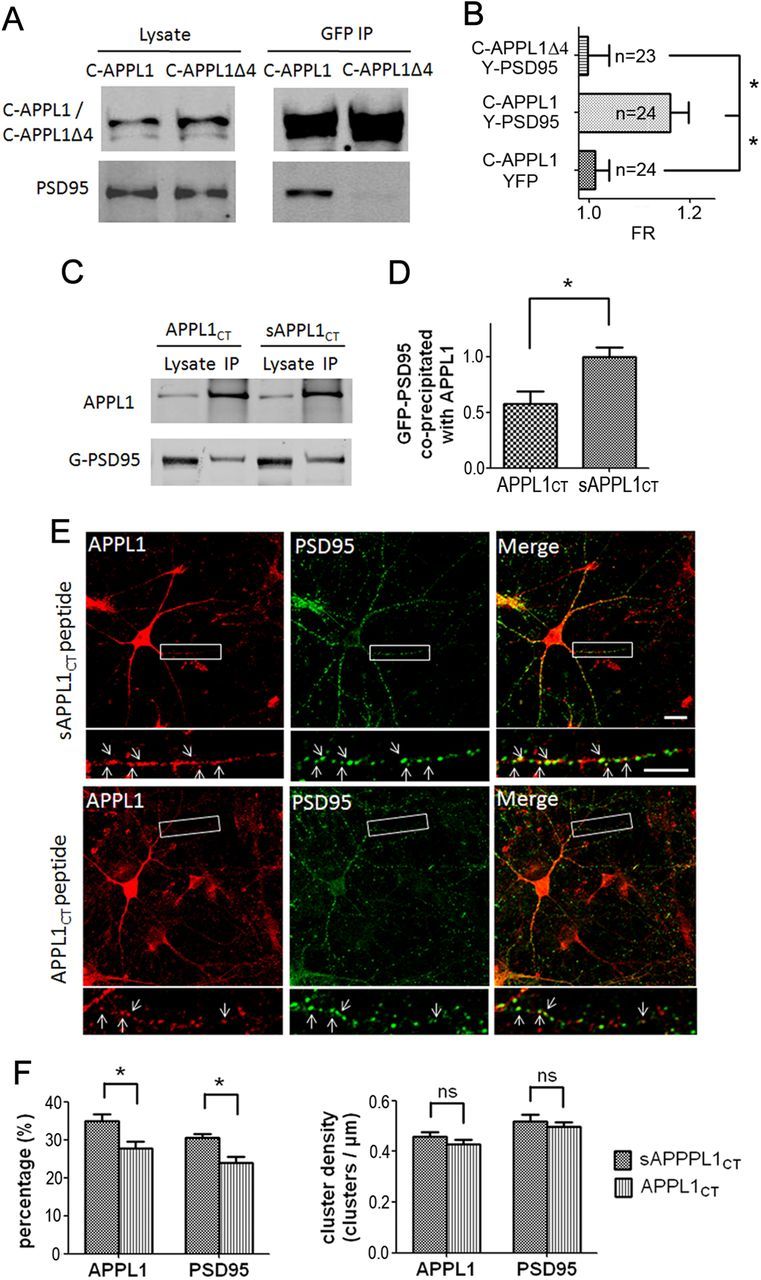
APPL1 interacts with PSD95 through its C-terminal PDZ-binding motif. A, CFP-APPL1/PSD95 or CFP-APPL1Δ4/PSD95 was cotransfected into HEK293. CFP-APPL1 but not CFP-APPL1Δ4 coprecipitated PSD95. B, Three-cube FRET measurements in HEK293 cells. Coexpression of CFP-APPL1 and YFP-PSD95 yielded positive FRET signals while CFP-APPL1 and YFP, CFP-APPL1Δ4, and YFP-PSD95 did not. The horizontal axis indicates FRET ratio (FR). *p < 0.01, two-tailed t test. C, Application of 100 μm Tat-APPL1CT peptide (APPL1CT) on GFP-PSD95 transfected HEK293 cells for 2 h before cell harvest perturbed the co-IP of GFP-PSD95 with APPL1, compared with that of Tat-sAPPL1CT peptide (sAPPL1CT). D, Statistical analysis of C. Western results of three independent co-IP tests were analyzed. The ratio of the band intensity of coprecipitated GFP-PSD95 to precipitated APPL1 was compared between the APPL1CT group and the sAPPL1CT group. All ratios were normalized (divided by the average ratio of sAPPL1CT group) before t test (*p < 0.05, two-tailed t test; n = 3). E, Application of 10 μm Tat-APPL1CT peptide for 1 h on cultured cortical neurons perturbed the colocalizaion of endogenous APPL1 and PSD95, compared with that of Tat-sAPPL1CT peptide. Scale bar, 10 μm. F, Statistical analysis of E. Left, Tat-APPL1CT significantly decreased the percentage of APPL1 clusters that colocalized with PSD95 clusters and the percentage of PSD95 that colocalized with APPL1 (∼1000 clusters of each protein from three separate experiments; *p < 0.05, two-tailed t test). Right, Density of APPL1 or PSD95 clusters (clusters per micrometer of dendrite branch) was not affected by Tat-APPL1CT peptide (ns, no significant difference; two-tailed t test; n = 3). Error bars indicate SEM.
We further generated the cell membrane-permeable peptide by fusion of the 10 C-terminal residues of APPL1 (EEGKKRESEA) to the cell membrane transduction domain of the HIV-1 Tat protein (YGRKKRRQRRR) (Tat-APPL1CT). The peptide of the scrambled APPL1 C-terminal sequence fused to Tat protein (Tat-sAPPL1CT) was also constructed as the negative control. Co-IP showed that incubation of transfected HEK293 cells with Tat-APPL1CT peptide for 2 h before cell harvest perturbed the interaction between APPL1 and GFP-PSD95, while Tat-sAPPL1CT peptide did not (Fig. 4C). The ratio of coprecipitated GFP-PSD95 to precipitated APPL1 in the APPL1CT group was ∼40% lower than that in the sAPPL1CT group (Fig. 4D). We further detected the effect of Tat-APPL1CT peptide treatment on the colocalization of endogenous APPL1 with PSD95 in cultured cortical neurons by immunostaining. The results showed that application of Tat-APPL1CT peptide slightly but significantly decreased the colocalization of APPL1 cluster with PSD95 cluster, without affecting the density of either APPL1 or PSD95 clusters (Fig. 4E,F). Together, these results suggest that APPL1 interacts with PSD95 through its C-terminal PDZ-binding motif.
APPL1 couples the PI3K/Akt pathway with NMDA receptor complexes
APPL1 was first identified as an Akt-interacting protein (Mitsuuchi et al., 1999). It directly binds Akt through its PTB domain and regulates the activity of Akt (Schenck et al., 2008; Cleasby et al., 2011; Majumdar et al., 2011). APPL1 also directly binds to the PI3K p110 subunit (Mitsuuchi et al., 1999; Tan et al., 2010a). This indicates that APPL1 acts as an adaptor protein coupling PI3K with Akt. In cultured cortical neurons, we found that GluN1 antibody coprecipitated PSD95 and APPL1, together with Akt and the PI3K p110β subunit (Fig. 5A). To assess the role of APPL1 in this NMDAR/PSD95/APPL1/PI3K/Akt protein complex, we used the Tat peptide to perturb the interaction of APPL1 with PSD95. Cultured cortical neurons were treated with Tat-APPL1CT or Tat-sAPPL1CT peptide for 2 h, and the protein interactions with the NMDAR complex were tested by co-IP (Fig. 5B,C). Western blots of the precipitates revealed that application of Tat-APPL1CT efficiently uncoupled APPL1 from the NMDAR complex (∼40% decrease), while the Tat-sAPPLCT had no effect. Coprecipitated PSD95 showed no changes in the Tat-APPL1CT group, suggesting that the interaction of NMDAR and PSD95 was not disturbed by Tat-APPL1CT. Importantly, Akt and p110β were also partially uncoupled from NMDARs when the cortical neurons were treated with Tat-APPL1CT (Fig. 5B,C), suggesting that APPL1 is involved in coupling Akt and PI3K with NMDAR complexes. To test the specificity of Tat-APPL1CT, we also assessed its effects on the PDZ interaction between the GluN2B subunit and PSD95, since the PDZ-binding motifs of GluN2B (-ESDV) and APPL1 (-ESEA) both belong to class I PDZ-binding motifs (Sheng and Sala, 2001). The results showed that Tat-APPL1CT did not affect the interaction between GluN2B and PSD95 (Fig. 5D,E), suggesting that Tat-APPL1CT specifically disrupts the PDZ interaction between APPL1 and PSD95. The above results indicate that APPL1 acts as an adaptor to form a signaling platform that couples the PI3K/AKT signaling pathway to NMDARs.
Figure 5.

Disruption of APPL1 and NMDAR complex interaction dissociates PI3K and Akt from NMDARs. A, GluN1 coprecipitated PSD95, APPL1, Akt, and p110β but not GluA2 from lysate of cultured cortical neurons. Immunoprecipitation with GluN1 antibody preblocked by excess antigen peptide (antigen blocking) was included as the negative control. B, Incubation of cultured cortical neurons with Tat-APPL1CT peptide for 2 h partially blocked the co-IP of APPL1, Akt, and p110β with GluN1, but not the co-IP of PSD95 with GluN1. Treatment with Tat-sAPPL1CT peptide did not have the blocking effect. C, Statistical analysis of three independent co-IP tests. The ratio of the band intensity of coprecipitated PSD95, APPL1, Akt, or p110β to precipitated GluN1 was compared. All ratios were normalized (divided by the average ratio of control group) before t test (*p < 0.05; ns, no significant difference; two-tailed t test; n = 3). D, Co-IP of GluN2B with PSD95 was not perturbed by Tat-APPL1CT peptide. E, Statistical analysis of D (ns, no significant difference; two-tailed t test; n = 3). Error bars indicate SEM.
APPL1 mediates synaptic NMDA receptor-induced recruitment of Akt and PI3K
Next, we assessed whether the association of this receptor signaling complex is regulated by synaptic NMDAR activity. We induced synaptic NMDAR activation by treatment with the GABAA receptor antagonist Bic (50 μm) and the K+ channel blocker 4-AP (250 μm) (Papadia et al., 2005). After treatment for 30 min, neurons were lysed and processed for the co-IP test. Synaptic NMDAR activation did not affect GluN1–GluN2B or GluN1–PSD95 interaction, but significantly upregulated the interaction between NMDAR and APPL1/Akt/p110β (Fig. 6A) (also see Fig. 6B,C). The upregulation was blocked by the NMDAR antagonist MK801 (Fig. 6A). These results suggest that synaptic NMDAR activity recruits more APPL1 along with PI3K/Akt to the NMDAR complex.
Figure 6.

Synaptic NMDAR activity increases the association of APPL1/PI3K/Akt to the NMDAR complex. A, Treatment of cultured cortical neurons with 50 μm Bic and 250 μm 4-AP for 30 min increased the co-IP of APPL1, Akt, and p110β with GluN1, but not the co-IP of GluN2B or PSD95 with GluN1. MK801 (50 μm) blocked the Bic- and 4-AP-induced increase of interaction. B, Preincubation of cortical neurons with Tat-APPL1CT peptide for 1 h blocked the Bic- and 4-AP-induced increase of interaction. Tat-sAPPL1 CT peptide did not have the blocking effect. C, Statistical analysis of three independent co-IP tests. ns, No significant difference by one-way ANOVA (GluN2B, F(3,8) = 0.04, n = 3, p > 0.05; PSD95, F(3,8) = 0.15, n = 3, p > 0.05). *p < 0.05 by Bonferroni's multiple-comparisons test after significant one-way ANOVA (APPL1, F(3,8) = 31.55, n = 3, p < 0.05; Akt, F(3,8) = 7.62, n = 3, p < 0.05; p110β, F(3,8) = 8.30, n = 3, p < 0.05). Error bars indicate SEM.
Preincubation with Tat-APPL1CT peptide for 1 h before the application of Bic and 4-AP blocked the recruitment of APPL1/Akt/p110β to NMDARs, while Tat-sAPPL1CT did not have a blocking effect (Fig. 6B,C). To verify the results of Tat-APPL1CT peptide, we generated APPL1 RNAi lentivirus. Cultured cortical neurons were infected by APPL1 RNAi or control RNAi virus at DIV 6. Six days after infection, APPL1 was efficiently knocked down (Fig. 7A). The expression level of APPL1 after RNAi underwent a 60–70% decrease (Fig. 7B). Immunofluorescent staining of infected neurons by APPL1 antibody indicated that APPL1 RNAi virus-infected (GFP-positive) neurons showed less APPL1 expression, compared with noninfected (GFP-negative) neurons or control virus-infected neurons (Fig. 7C). Knockdown of APPL1 did not affect the expression of Akt or p110β, but blocked Bic- and 4-AP-induced recruitment of Akt and p110β to NMDARs (Fig. 7D,E). These results indicate that APPL1 mediates synaptic NMDAR activity-dependent recruitment of Akt/PI3K pathway to NMDAR signaling complex.
Figure 7.
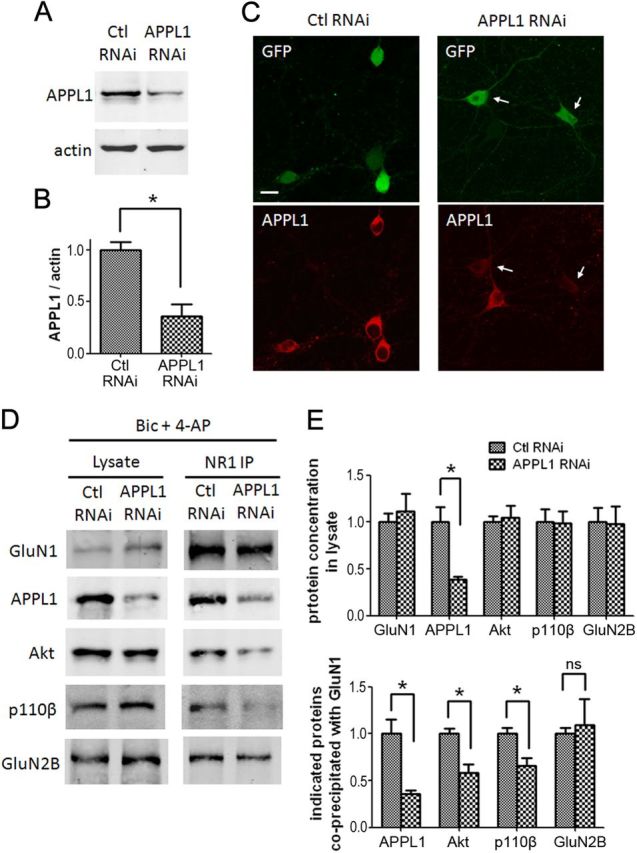
Lentiviral RNAi of APPL1 blocks synaptic NMDAR-induced recruitment of Akt and PI3K to NMDARs. A, Cultured cortical neurons (DIV 6) were infected by APPL1 RNAi or control RNAi lentivirus. Six days after infection, neurons were harvested for Western blot. B, Quantification of blots from three independent experiments (*p < 0.05; two-tailed t test; n = 3). C, Immunofluorescent staining of infected neurons by APPL1 antibody. Neurons expressing GFP fluorescent indicate virus expression. The arrows indicate APPL1 knockdown neurons. Scale bar, 10 μm. D, Six days after lentiviral infection, neurons were treated for 30 min with Bic and 4-AP and processed for co-IP. E, Statistical analysis of three independent co-IP. Top, APPL1 knocking down did not affect the concentration of GluN1, Akt, p110β, and GluN2B in lysate. Bottom, APPL1 knocking down blocked the co-IP of APPL1, Akt, and p110β with GluN1, but did not affect the co-IP of GluN2B with GluN1 (*p < 0.05; ns, no significant difference; two-tailed t test; n = 3). Error bars indicate SEM.
APPL1 mediates synaptic NMDA receptor-induced Akt activation
Next, we assessed whether APPL1 is involved in NMDAR-dependent Akt activation. Previous studies reported that synaptic NMDAR activation increases the phosphorylation of Akt in cultured hippocampal neurons (Papadia et al., 2005). We showed that, when cultured cortical neurons were treated with Bic and 4-AP for 30 min, phosphorylation of Akt was significantly increased, and this effect was blocked by the NMDAR antagonist MK801 (Fig. 8A,B) and the PI3K inhibitor 2-morpholin-4-yl-8-phenylchromen-4-one (LY294002) (Fig. 8D,E). This suggests that the stimulation of Akt is dependent on NMDAR activity and PI3K. Bic and 4-AP treatment also induced MAP kinase/extracellular signal-regulated kinase (ERK1/2) activation (Fig. 8A,C), which was blocked by MK801, suggesting that synaptic NMDAR activity also induced ERK1/2 activation. To further test whether Akt pathway and ERK1/2 pathway is specifically coupled to synaptic NMDARs but not extrasynaptic NMDARs, we used an established method to selectively activate extrasynaptic NMDARs (see Materials and Methods). Extrasynaptic stimulation did not induce Akt or ERK1/2 activation (Fig. 8F--H), consistent with the previous report (Ivanov et al., 2006; Papadia et al., 2008).
Figure 8.

Synaptic NMDARs but not extrasynaptic NMDARs induce Akt and ERK activation. A, Cultured cortical neurons (DIV 12–14) were treated for 30 min with 50 μm Bic and 250 μm 4-AP. MK801 (50 μm) was used to test whether the activation was NMDAR dependent. The levels of phospho-Akt Ser473 (pAkt), Akt, phospho-ERK1/2 (pErk), and ERK1/2 (Erk) were detected by Western blot. B, The ratio of phospho-Akt to Akt was compared between three groups. All ratios were normalized (divided by the average ratio of control group) before one-way ANOVA. *p < 0.05 by Bonferroni's multiple-comparisons test after significant one-way ANOVA (F(2,11) = 6.40; n = 5; p < 0.05). C, The ratio of phospho-ERK1/2 to ERK1/2 was compared between three groups. *p < 0.05 by Bonferroni's test after significant one-way ANOVA (F(2,6) = 11.50; n = 3; p < 0.05). D, Pretreatment with the PI3K inhibitor LY294002 (50 μm) for 5 min blocked the increase of phospho-Akt by Bic plus 4-AP treatment. E, Statistical analysis of D (*p < 0.05; two-tailed t test; n = 3). F, Extrasynaptic NMDARs were selectively activated by NMDA (10 μm), following irreversible blockade of synaptic NMDARs. G, H, Statistical analysis of F. Extrasynaptic stimulation did not increase pAkt/Akt or pERK/ERK (ns, no significant difference; two-tailed t test; n = 3). Error bars indicate SEM.
Next, we treated the cultured cortical neurons with 10 μm Tat-APPL1CT or Tat-sAPPL1CT for 1 h before application of Bic and 4-AP. Pretreatment with Tat-APPL1CT, but not Tat-sAPPL1CT, blocked the Bic- and 4-AP-stimulated Akt activation (Fig. 9A,B). Tat-APPL1CT did not affect ERK1/2 activation (Fig. 9A,C). We then use APPL1 RNAi lentivirus to verify the involvement of APPLI in NMDAR–Akt pathway. Knockdown of APPL1 in cortical neurons did not affect the basal level of phospho-Akt (Fig. 9D,E), which is consistent with the finding from APPL1 KO mice that APPL1 is dispensable for the basal level of Akt activity in thymic T-cells (Tan et al., 2010b) and in murine embryonic fibroblasts (Tan et al., 2010a). However, knockdown of APPL1 effectively blocked the Akt activation induced by synaptic NMDAR activity. Control virus did not block the NMDAR-induced Akt activation (Fig. 9D,E). Knockdown of APPL1 did not block the NMDAR-dependent ERK1/2 activation (Fig. 9D,F). These results suggest that either disrupting APPL1–NMDAR association or knocking down APPL1 uncouples NMDA receptors from the downstream PI3K/AKT signaling pathway, but not from the ERK pathway.
Figure 9.

APPL1 is necessary for synaptic NMDAR-mediated AKT activation. A–C, Tat-APPL1CT peptide specifically blocked synaptic NMDAR-mediated AKT activation but not ERK1/2 activation. A, Cultured cortical neurons were preincubated with 10 μm Tat-APPL1CT or Tat-sAPPL1CT peptide for 1 h before treatment with Bic and 4-AP. The levels of pAkt, Akt, pErk, and Erk were detected by Western blot. B, Statistical analysis of A. Tat-APPL1CT, but not Tat-sAPPL1CT blocked the increase of phospho-Akt by Bic and 4-AP treatment. *p < 0.05 by Bonferroni's test after significant one-way ANOVA (F(3,8) = 11.68; n = 3; p < 0.05). C, Tat-APPL1CT or Tat-sAPPL1CT did not affect the increase of phospho-ERK1/2 by Bic and 4-AP treatment. ns, p > 0.05 by Bonferroni's test after significant one-way ANOVA (F(3,8) = 8.38; n = 3; p < 0.05). D–F, Lentiviral RNAi of APPL1 specifically blocked synaptic NMDAR-mediated AKT activation but not ERK1/2 activation. D, Lentivirus-infected neurons were treated for 30 min with Bic and 4-AP and processed for Western blot. E, Statistical analysis of D. APPL1 RNAi blocked Akt activation induced by Bic and 4-AP, while control RNAi did not (*p < 0.05; ns, no significant difference; two-tailed t test; n = 3). F, Neither APPL1 RNAi nor control RNAi affected ERK1/2 activation induced by Bic and 4-AP (*p < 0.05; two-tailed t test; n = 3). Error bars indicate SEM.
APPL1 is required for NMDA receptor-dependent neuroprotection
Our results above indicated that APPL1 is involved in PI3K/Akt activation by NMDARs. The PI3K/Akt kinase cascade is an important signaling pathway that contributes to the prosurvival effects of NMDAR activity. Previous studies reported that synaptic NMDAR activation activates Akt and protects neurons from starvation-induced apoptosis (Papadia et al., 2005). Here, we further assessed whether APPL1 is involved in NMDAR-dependent neuroprotective function using a similar model. DIV 10 cortical neurons switched from growth medium to serum-free medium underwent apoptosis after 4 d, as indicated by enhanced DNA fragmentation. The condensation and/or fragmentation of nuclei as assessed by Hoechst 33258 staining indicated an apoptotic process (Fig. 10A). Application of Bic and 4-AP to neurons subjected to trophic deprivation improved cell survival and reduced the proportion of Hoechst-positive neurons from 50.9 ± 2.2% (mean ± SEM) (Fig. 10A,B, starvation group) to 26.7 ± 4.0% (starvation plus Bic plus 4-AP group). However, application of 10 μm Tat-APPL1CT along with Bic and 4-AP blocked their neuroprotective effect, with the proportion of apoptotic neurons being 46.4 ± 4.0% (starvation plus Bic plus 4-AP plus APPL1CT group). In contrast, Tat-sAPPL1CT did not affect the neuroprotective effect of Bic and 4-AP, the proportion being 27.4 ± 4.2% (starvation plus Bic plus 4-AP plus sAPPL1CT group).
Figure 10.

APPL1 is required for synaptic NMDA receptor-dependent neuroprotection. A, Cultured cortical neurons (DIV 10) were subjected to trophic deprivation (starvation) for 3 d. Incubation of starved neurons with 50 μm Bic and 250 μm 4-AP protected them from apoptosis. Tat-APPL1CT or Tat-sAPPL1CT peptide was applied at 10 μm along with Bic and 4-AP to test whether it blocked the neuroprotective effect of Bic and 4-AP. Hoechst staining was then used to quantify the death rate of neurons. Hoechst brightly stains the condensed or fragmented nuclei of apoptotic neurons and more dimly stains the normal nuclei of healthy neurons. Tau protein was simultaneously immunostained to reveal the morphology of neurons. Note that the Tau staining of Hoechst-positive neurons was unclear, suggesting the unhealthy state of these neurons. Scale bar, 10 μm. B, Quantification data of A. Tat-APPL1CT but not Tat-sAPPL1CT increased the proportion of apoptotic neurons, suggesting that Tat-APPL1CT blocked the neuroprotective effect of Bic and 4-AP. *p < 0.05 by Bonferroni's test after significant one-way ANOVA (F(4,10) = 28.78; n = 3; p < 0.05); 300–500 cells for each group in one independent experiment. C, Neurons at DIV 6 were infected by APPL1 RNAi or control RNAi lentivirus. Five days after infection, neurons were subjected to trophic deprivation for another 3 d, with or without the addition of Bic and 4-AP. The proportion of apoptotic neurons was analyzed by Hoechst staining. Neurons expressing GFP fluorescence indicate virus expression. D, Quantification data of C. APPL1 RNAi blocked the neuroprotective effect of Bic and 4-AP, while control RNAi did not (*p < 0.05; ns, no significant difference; two-tailed t test; n = 3; 300–500 cells for each group in one independent experiment). Error bars indicate SEM.
Finally, lentiviral RNAi was used to verify the involvement of APPL1 in NMDA receptor-dependent neuroprotection. Knockdown of APPL1 did not affect the neuronal survival under basal conditions (6.7 ± 1.6% under basal condition of APPL1 RNAi and 7.0 ± 2.5% under basal condition of control RNAi; Fig. 10C,D). For the neuroprotection test, infected neurons were subjected to trophic deprivation for 3 d with or without the addition of Bic and 4-AP. APPL1 RNAi blocked the neuroprotective effect of Bic and 4-AP. The apoptosis rate in the starvation plus Bic plus 4-AP group was not significantly decreased (40.8 ± 2.6%), compared with that in the starvation group (47.7 ± 2.9%) (Fig. 10C,D). In contrast, control RNAi did not affect the neuroprotective effect. The apoptosis rate in the starvation plus Bic plus 4-AP group was decreased to 22.3 ± 2.0%, compared with that in the starvation group (45.2 ± 1.2%) (Fig. 10C,D). The above results suggest that either uncoupling the connection between APPL1 and NMDARs or knockdown of APPL1 blocks the synaptic NMDAR-mediated neuroprotective effect.
Discussion
This study aimed at determining the potential involvement of APPL1 in NMDAR-dependent neuroprotection. We found that APPL1 associated with NMDARs through binding to PSD95 at its C-terminal PDZ-binding motif. Furthermore, APPL1 specifically coupled NMDARs to the downstream PI3K/AKT signaling pathway and mediated the synaptic NMDAR-dependent neuroprotective function.
APPL1 associates with NMDA receptor complexes through binding to PSD95
APPL1, a multifunctional adaptor protein, binds to various transmembrane receptors in various cell types and is involved in their intracellular signal transduction. Here, we found that APPL1 is a component of the NMDAR complex, but it does not directly interact with NMDAR subunits. Further analysis using co-IP and FRET indicated that APPL1 interacts with PSD95. Consistently, such interaction between APPL1 and PSD95 was interrupted either by deletion of the APPL1 C-terminal PDZ-binding motif or by treatment with the peptide against this domain. These results strongly suggested that APPL1 associates with NMDARs by binding directly with PSD95 at its C-terminal PDZ-binding motif. We noted that another PDZ domain-containing protein GIPC has been reported to interact with APPL1 (Lin et al., 2006) and to interact with NMDA receptors (Yi et al., 2007). These results imply that GIPC may be another potential mediator between APPL1 and NMDARs. However, we did not find an interaction between APPL1 and GIPC by co-IP from mouse cortex (Fig. 2A). This difference may come from the different experimental system. Moreover, we did not find an interaction of APPL1 with SAP102, another PDZ domain-containing scaffold protein of NMDARs, in transfected HEK293 cells (data not shown). This indicates a selective binding of APPL1 among PDZ-containing proteins.
APPL1 couples NMDA receptors to the PI3K/AKT kinase cascade
Previous studies showed that APPL1 interacts with Akt and regulates Akt activity. Here, we found that both APPL1 and Akt are partially located in the postsynaptic density and are components of NMDAR complexes. Synaptic NMDAR activity increases the association of APPL1/PI3K/Akt to NMDARs, the mechanism of which is unknown. Disturbance of the interaction between APPL1 and PSD95 by Tat-APPL1CT peptide either uncoupled NMDARs from PI3K and Akt under basal conditions, or blocked the synaptic NMDAR activity-induced recruitment and activation of PI3K/Akt kinase cascade. Consistent with this, APPL1 knockdown blocked the synaptic NMDAR-dependent recruitment and activation of PI3K/Akt. This indicates that APPL1, acting as an adaptor protein, mediates the activity-regulated coupling of NMDARs with the downstream PI3K/Akt kinase cascade. This organization may facilitate the signal transduction initiated by local Ca2+ influx via NMDARs (Fig. 11). APPL1 has been reported to connect some receptors with different signaling pathways, such as the FSH (follicle-stimulating hormone) receptors with Akt2, adiponectin receptors with MAPK/AMPK, and TrkA with its downstream signaling pathway. This kind of protein complex organization of receptors, adaptor proteins, and signaling molecules likely facilitates the signal transduction induced by receptors.
Figure 11.

Schematic illustration of the involvement of APPL1 in synaptic NMDA receptor-dependent neuroprotection. Left, Synaptic NMDAR activity induces the recruitment of APPL1/PI3K/Akt to the NMDAR complex. APPL1 binds to the NMDAR complex through the PDZ interaction with PSD95. The formation of this complex facilitates the signaling from NMDARs to the Akt pathway. Local Ca2+ influx via NMDARs activates calmodulin. Calmodulin directly binds to and activates PI3K (Joyal et al., 1997) or indirectly activates PI3K through the Ras pathway (Sutton and Chandler, 2002). PI3K then phosphorylates and activates Akt, and phospho-Akt phosphorylates and regulates many intracellular substrates implicated in neuroprotection. Right, Treatment with Tat-APPL1CT peptide that blocks the APPL1–PSD95 interaction (1) or knockdown of APPL1 (2) dissociates PI3K and Akt from the NMDAR complex. Under this circumstance, synaptic NMDAR activity does not activate the PI3K/Akt cascade or the downstream prosurvival pathway.
Previous studies reported that APPL1 binds to Rab5 and forms a endosomal signaling platform to “channel” endocytic receptors to downstream signaling pathways (Miaczynska et al., 2004; Varsano et al., 2006; Zoncu et al., 2009). However, our results from synaptosomal fractionation showed that APPL1, but not Rab5, was located in the PSD-enriched fraction. This indicates that APPL1 in the PSD may function other than a key component of signaling endosomes, but directly relay signals from membrane receptors to downstream signaling molecules that can be recruited to the PSD by APPL1. However, our results also showed both APPL1 and Rab5 are expressed at high levels in the non-PSD fraction of mouse cortex, which suggests that APPL1 may also play an important role in the formation of signaling endosomes and mediating endocytic receptor signaling in the CNS.
APPL1 is involved in synaptic NMDA receptor-dependent neuroprotective effect
Evidence indicates that NMDAR location influences whether the receptor is coupled to prodeath or prosurvival signals (Hardingham and Bading, 2010). Synaptic NMDAR activity promotes neuronal survival through different signaling pathways under different circumstances. Among these, the PI3K/Akt kinase cascade is a key pathway responsible for the neuroprotective effects induced by synaptic NMDAR activity. Here, we found that APPL1 is involved in the synaptic NMDAR-dependent neuroprotective effect. Tat-APPL1CT peptide or APPL1 knockdown blocked the synaptic NMDAR-dependent activation of Akt and, in turn, blocked the synaptic NMDAR-dependent neuroprotective effect (Fig. 11). As mentioned previously, synaptic and extrasynaptic NMDARs have dichotomous roles in prosurvival and prodeath signaling. As for the Akt pathway, our result and other report (Papadia et al., 2008) indicated that synaptic NMDAR activity promotes sustained activation of Akt, leading to the phosphorylation and inhibition of the proapoptotic activity of FOXO, while activation of extrasynaptic NMDARs did not sustainably activate Akt, which, in turn, did not inhibit FOXO. However, the mechanism underlying this dichotomous signaling remains unclear. Our data indicate that APPL1 associates with NMDARs through PSD95, a scaffold protein highly accumulating in postsynaptic but not in extrasynaptic sites. So it is possible that APPL1 preferentially tethers the PI3K/Akt cascade to the NMDA receptor in synaptic but not in extrasynaptic sites, so that only the synaptic NMDARs can efficiently and sustainably activate the Akt pathway.
In summary, we reveal a novel function of adaptor protein APPL1 in mediating a neuroprotective effect through coupling synaptic NMDARs and the PI3K/Akt kinase cascade. Investigating the regulation of this pathway under physiological or pathophysiological conditions should be informative and may lead to potential intervention strategies for the treatment of acute trauma and neurodegenerative diseases.
Footnotes
This work was supported by National Basic Research Program of China Grant 2010CB912002, National Natural Science Foundation of China Grants 30730038 and 30800308, the Fundamental Research Funds for the Central Universities of China, and Natural Science Foundation of Zhejiang Province, China, Grant Y2100425. We thank Dr. James A. Dias for providing the human full-length APPL1 plasmid.
References
- Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. doi: 10.1016/s0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- Cleasby ME, Lau Q, Polkinghorne E, Patel SA, Leslie SJ, Turner N, Cooney GJ, Xu A, Kraegen EW. The adaptor protein APPL1 increases glycogen accumulation in rat skeletal muscle through activation of the PI3-kinase signalling pathway. J Endocrinol. 2011;210:81–92. doi: 10.1530/JOE-11-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J. How BAD phosphorylation is good for survival. Nat Cell Biol. 1999;1:E33–E35. doi: 10.1038/10026. [DOI] [PubMed] [Google Scholar]
- Hardingham GE. Coupling of the NMDA receptor to neuroprotective and neurodestructive events. Biochem Soc Trans. 2009;37:1147–1160. doi: 10.1042/BST0371147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci. 2000;3:661–669. doi: 10.1038/76615. [DOI] [PubMed] [Google Scholar]
- Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben-Ari Y, Medina I. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J Physiol. 2006;572:789–798. doi: 10.1113/jphysiol.2006.105510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyal JL, Burks DJ, Pons S, Matter WF, Vlahos CJ, White MF, Sacks DB. Calmodulin activates phosphatidylinositol 3-kinase. J Biol Chem. 1997;272:28183–28186. doi: 10.1074/jbc.272.45.28183. [DOI] [PubMed] [Google Scholar]
- Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol. 2001;21:893–901. doi: 10.1128/MCB.21.3.893-901.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinouchi H, Epstein CJ, Mizui T, Carlson E, Chen SF, Chan PH. Attenuation of focal cerebral ischemic injury in transgenic mice overexpressing CuZn superoxide dismutase. Proc Natl Acad Sci U S A. 1991;88:11158–11162. doi: 10.1073/pnas.88.24.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon-Cazal M, Perez V, Bockaert J, Marin P. Akt mediates the anti-apoptotic effect of NMDA but not that induced by potassium depolarization in cultured cerebellar granule cells. Eur J Neurosci. 2002;16:575–583. doi: 10.1046/j.1460-9568.2002.02124.x. [DOI] [PubMed] [Google Scholar]
- Lin DC, Quevedo C, Brewer NE, Bell A, Testa JR, Grimes ML, Miller FD, Kaplan DR. APPL1 associates with TrkA and GIPC1 and is required for nerve growth factor-mediated signal transduction. Mol Cell Biol. 2006;26:8928–8941. doi: 10.1128/MCB.00228-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yao F, Wu R, Morgan M, Thorburn A, Finley RL, Jr, Chen YQ. Mediation of the DCC apoptotic signal by DIP13 alpha. J Biol Chem. 2002;277:26281–26285. doi: 10.1074/jbc.M204679200. [DOI] [PubMed] [Google Scholar]
- Luo CX, Zhu DY. Research progress on neurobiology of neuronal nitric oxide synthase. Neurosci Bull. 2011;27:23–35. doi: 10.1007/s12264-011-1038-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Wang Y, Yasuda RP, Dunah AW, Wolfe BB. The majority of N-methyl-d-aspartate receptor complexes in adult rat cerebral cortex contain at least three different subunits (NR1/NR2A/NR2B) Mol Pharmacol. 1997;51:79–86. doi: 10.1124/mol.51.1.79. [DOI] [PubMed] [Google Scholar]
- Majumdar D, Nebhan CA, Hu L, Anderson B, Webb DJ. An APPL1/Akt signaling complex regulates dendritic spine and synapse formation in hippocampal neurons. Mol Cell Neurosci. 2011;46:633–644. doi: 10.1016/j.mcn.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Kikani CK, Riojas RA, Langlais P, Wang L, Ramos FJ, Fang Q, Christ-Roberts CY, Hong JY, Kim RY, Liu F, Dong LQ. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol. 2006;8:516–523. doi: 10.1038/ncb1404. [DOI] [PubMed] [Google Scholar]
- Miaczynska M, Christoforidis S, Giner A, Shevchenko A, Uttenweiler-Joseph S, Habermann B, Wilm M, Parton RG, Zerial M. APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment. Cell. 2004;116:445–456. doi: 10.1016/s0092-8674(04)00117-5. [DOI] [PubMed] [Google Scholar]
- Mitsuuchi Y, Johnson SW, Sonoda G, Tanno S, Golemis EA, Testa JR. Identification of a chromosome 3p14.3-21.1 gene, APPL, encoding an adaptor molecule that interacts with the oncoprotein-serine/threonine kinase AKT2. Oncogene. 1999;18:4891–4898. doi: 10.1038/sj.onc.1203080. [DOI] [PubMed] [Google Scholar]
- Murphy AN, Fiskum G. Bcl-2 and Ca2+-mediated mitochondrial dysfunction in neural cell death. Biochem Soc Symp. 1999;66:33–41. doi: 10.1042/bss0660033. [DOI] [PubMed] [Google Scholar]
- Nechamen CA, Thomas RM, Cohen BD, Acevedo G, Poulikakos PI, Testa JR, Dias JA. Human follicle-stimulating hormone (FSH) receptor interacts with the adaptor protein APPL1 in HEK 293 cells: potential involvement of the PI3K pathway in FSH signaling. Biol Reprod. 2004;71:629–636. doi: 10.1095/biolreprod.103.025833. [DOI] [PubMed] [Google Scholar]
- Pacchioni AM, Vallone J, Worley PF, Kalivas PW. Neuronal pentraxins modulate cocaine-induced neuroadaptations. J Pharmacol Exp Ther. 2009;328:183–192. doi: 10.1124/jpet.108.143115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Stevenson P, Hardingham NR, Bading H, Hardingham GE. Nuclear Ca2+ and the cAMP response element-binding protein family mediate a late phase of activity-dependent neuroprotection. J Neurosci. 2005;25:4279–4287. doi: 10.1523/JNEUROSCI.5019-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Soriano FX, Léveillé F, Martel MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, McKenzie G, Craigon M, Corriveau R, Ghazal P, Horsburgh K, Yankner BA, Wyllie DJ, Ikonomidou C, Hardingham GE. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci. 2008;11:476–487. doi: 10.1038/nn2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkinton MS, Ip JK, Wood GL, Crossthwaite AJ, Williams RJ. Phosphatidylinositol 3-kinase is a central mediator of NMDA receptor signalling to MAP kinase (Erk1/2), Akt/PKB and CREB in striatal neurones. J Neurochem. 2002;80:239–254. doi: 10.1046/j.0022-3042.2001.00699.x. [DOI] [PubMed] [Google Scholar]
- Qiu S, Hua YL, Yang F, Chen YZ, Luo JH. Subunit assembly of N-methyl-d-aspartate receptors analyzed by fluorescence resonance energy transfer. J Biol Chem. 2005;280:24923–24930. doi: 10.1074/jbc.M413915200. [DOI] [PubMed] [Google Scholar]
- Qiu S, Zhang XM, Cao JY, Yang W, Yan YG, Shan L, Zheng J, Luo JH. An endoplasmic reticulum retention signal located in the extracellular amino-terminal domain of the NR2A subunit of N-methyl-d-aspartate receptors. J Biol Chem. 2009;284:20285–20298. doi: 10.1074/jbc.M109.004960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenck A, Goto-Silva L, Collinet C, Rhinn M, Giner A, Habermann B, Brand M, Zerial M. The endosomal protein Appl1 mediates Akt substrate specificity and cell survival in vertebrate development. Cell. 2008;133:486–497. doi: 10.1016/j.cell.2008.02.044. [DOI] [PubMed] [Google Scholar]
- Sheng M, Sala C. PDZ domains and the organization of supramolecular complexes. Annu Rev Neurosci. 2001;24:1–29. doi: 10.1146/annurev.neuro.24.1.1. [DOI] [PubMed] [Google Scholar]
- Soriano FX, Papadia S, Hofmann F, Hardingham NR, Bading H, Hardingham GE. Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J Neurosci. 2006;26:4509–4518. doi: 10.1523/JNEUROSCI.0455-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton G, Chandler LJ. Activity-dependent NMDA receptor-mediated activation of protein kinase B/Akt in cortical neuronal cultures. J Neurochem. 2002;82:1097–1105. doi: 10.1046/j.1471-4159.2002.01031.x. [DOI] [PubMed] [Google Scholar]
- Tan Y, You H, Wu C, Altomare DA, Testa JR. Appl1 is dispensable for mouse development, and loss of Appl1 has growth factor-selective effects on Akt signaling in murine embryonic fibroblasts. J Biol Chem. 2010a;285:6377–6389. doi: 10.1074/jbc.M109.068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, You H, Coffey FJ, Wiest DL, Testa JR. Appl1 is dispensable for Akt signaling in vivo and mouse T-cell development. Genesis. 2010b;48:531–539. doi: 10.1002/dvg.20657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- Varsano T, Dong MQ, Niesman I, Gacula H, Lou X, Ma T, Testa JR, Yates JR, 3rd, Farquhar MG. GIPC is recruited by APPL to peripheral TrkA endosomes and regulates TrkA trafficking and signaling. Mol Cell Biol. 2006;26:8942–8952. doi: 10.1128/MCB.00305-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- Yamaguchi A, Tamatani M, Matsuzaki H, Namikawa K, Kiyama H, Vitek MP, Mitsuda N, Tohyama M. Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J Biol Chem. 2001;276:5256–5264. doi: 10.1074/jbc.M008552200. [DOI] [PubMed] [Google Scholar]
- Yi Z, Petralia RS, Fu Z, Swanwick CC, Wang YX, Prybylowski K, Sans N, Vicini S, Wenthold RJ. The role of the PDZ protein GIPC in regulating NMDA receptor trafficking. J Neurosci. 2007;27:11663–11675. doi: 10.1523/JNEUROSCI.3252-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Perera RM, Balkin DM, Pirruccello M, Toomre D, De Camilli P. A phosphoinositide switch controls the maturation and signaling properties of APPL endosomes. Cell. 2009;136:1110–1121. doi: 10.1016/j.cell.2009.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]