

Abstract
Huntington disease (HD) is an inherited progressive neurodegenerative disorder, characterized by motor, cognitive, and psychiatric deficits as well as neurodegeneration and brain atrophy beginning in the striatum and the cortex and extending to other subcortical brain regions. The genetic cause is an expansion of the CAG repeat stretch in the HTT gene encoding huntingtin protein (htt). Here, we generated an HD transgenic rat model using a human bacterial artificial chromosome (BAC), which contains the full-length HTT genomic sequence with 97 CAG/CAA repeats and all regulatory elements. BACHD transgenic rats display a robust, early onset and progressive HD-like phenotype including motor deficits and anxiety-related symptoms. In contrast to BAC and yeast artificial chromosome HD mouse models that express full-length mutant huntingtin, BACHD rats do not exhibit an increased body weight. Neuropathologically, the distribution of neuropil aggregates and nuclear accumulation of N-terminal mutant huntingtin in BACHD rats is similar to the observations in human HD brains. Aggregates occur more frequently in the cortex than in the striatum and neuropil aggregates appear earlier than mutant htt accumulation in the nucleus. Furthermore, we found an imbalance in the striatal striosome and matrix compartments in early stages of the disease. In addition, reduced dopamine receptor binding was detectable by in vivo imaging. Our data demonstrate that this transgenic BACHD rat line may be a valuable model for further understanding the disease mechanisms and for preclinical pharmacological studies.
Introduction
Huntington disease (HD) is an autosomal dominant, progressive neurodegenerative disorder commonly manifesting in adulthood (Harper, 1991). Clinical features include motor deficits, cognitive decline, and psychological disturbances (Vonsattel and DiFiglia, 1998). The hallmark of HD neuropathology is initial striatal atrophy, which expands at later stages to the cerebral cortex and other subcortical brain regions (Bruyn, 1979; Vonsattel et al., 1985). Interestingly, neuropil and nuclear huntingtin (htt) aggregates are abundant in the cortex, but are only sparsely observed in the striatum of HD-affected brains (Gutekunst et al., 1999; Kuemmerle et al., 1999).
Expansions of the CAG repeat (>38) in exon 1 of the gene encoding htt were first discovered in 1993 and represent to date the sole genetic cause for the disease (The Huntington's Disease Collaborative Research Group, 1993). A number of mouse models expressing full-length htt or truncated htt fragments were developed for therapeutic studies and to better understand the pathogenesis of disease (Menalled and Chesselet, 2002; Ehrnhoefer et al., 2009; Crook and Housman, 2011; Munoz-Sanjuan and Bates, 2011). However, mouse models are in general limited for studying certain functional and behavioral measurements (Tecott and Nestler, 2004; Rodriguiz and Wetsel, 2006; Herrmann et al., 2012). The limitations of mouse models as well as pharmacogenomic differences between the species (Toutain et al., 2010) necessitate either the laborious development of test designs appropriate for mouse models or the use of additional species to generate HD models in which these test designs are readily available.
Rat models have made substantial contributions to our understanding of biological function and behavior. Numerous rat disease models have successfully proven their utility for modeling the human condition (Hammer et al., 1990; von Hörsten et al., 2003; Yamada et al., 2004; Liu et al., 2008). Although learning and memory can be studied with some restrictions in mice, the current scientific knowledge concerning the complexity of learning and memory, as well as the multiplicity of brain systems supporting it, has come largely from behavioral research using rats (Report of the NIH Rat Model Priority Meeting, 1999, http://www.nhlbi.nih.gov/resources/docs/ratmtg.pdf). Compared to mice, rats show excellent learning abilities, a mandatory requirement for the identification of the subtle cognitive deficits that may be present in the early stages of HD. Another practical advantage of rats is their larger brain size, which facilitates direct invasive procedures. In addition, miniaturized physiological in vivo approaches, such as structural and functional imaging of small brain structures, are more difficult in mouse models due to size limitation.
We have previously generated an HD transgenic rat line (tgHD rats), which expresses a fragment of mutant htt (von Hörsten et al., 2003). This rat model demonstrated many aspects of HD, but lacking the full-length mutant htt (fl-mhtt) protein certain aspects of the human disease would be imperfectly replicated.
In this study, we generated and characterized a new bacterial artificial chromosome (BAC) HD transgenic rat model expressing fl-mhtt under the control of the human HTT promoter and all its regulatory elements to determine its similarity to the human condition across the molecular, behavioral, and anatomical domains.
Materials and Methods
Animals
Generation of transgenic rats.
BACs, containing human genomic DNA spanning the full-length HTT gene with 97 CAG/CAA repeats and including all regulatory elements (Gray et al., 2008), were microinjected into oocytes of Sprague Dawley rats. Genotyping and determination of BAC transgene integrity were performed via PCR analysis using genomic DNA extracted from ear biopsy tissue (High Pure PCR Template Preparation Kit; Roche). Two primer pairs were designed, one pair binding to exon 1 (FW: 5′-ATGGCGACCCTGGAAAAGC-3′; RV: 5′-AGGTCGGTGCAGAGGCTCCTCTG-3′) of both endogenous Htt and exogenous mutant HTT with different amplicon lengths, while the second primer pair binds specifically to the last exon of the HTT transgene (FW: 5′-TGTGATTAATTTGGTTGTCAAGTTTT-3′; RV: 5′-AGCTGGAAACATCACCTACATAGACT-3′).
To estimate relative transgene copy number of the BAC insertion and number of integration sites, probe-based real-time PCR (TaqMan PCR) was performed using genomic DNA. Briefly, primers and probe were designed to bind specifically to intron 29 of the HTT transgene (FW: 5′-ACCGACCTTCTGAAGCCTACTTCT-3′; RV: 5′-TTCTCCTCCAAAGGATCACAACTC-3′; probe: 5′FAM-CTAAGTGGCGCTGCGTAGTGCGAA-3′Bhq). The Ct values of all samples were normalized to β-actin serving as reference gene (FW: 5′-AGCCATGTACGTAGCCATCCA-3′; RV: 5′-TCTCCGGAGTCCATCACAATG-3′; probe: 5′CY-TGTCCCTGTATGCCTCTGGTCGTACCAC-3′Bhq). The relative copy number of all F1 rats was compared within and between each line.
RNA analysis.
Expression levels of mRNA from each line were analyzed via real-time PCR using QuantiTect SYBR Green PCR Kits (Qiagen). Total mRNA was extracted from whole brain using the RNeasy Lipid Tissue Midi Kit (Qiagen) and cDNA was synthesized using a QuantiTect Reverse Transcription Kit (Qiagen) following the manufacturer's instructions. The relative mRNA expression levels were compared with endogenous wild-type (WT) htt. The primers were designed for transgenic HTT (FW: 5′-GTGGAGGTTTGCTGAGCTG-3′; RV: 5′-GCAAAATTGCCAAAAGAAGC-3′) and rat Htt (FW: 5′-ATCTTGAGCCACAGCTCCAGCCA-3′; RV: 5′-TCTGAAAACGTCTGAGACTTCACCAGA-3′) spanning at least one exon–exon junction.
Measurement of the CAG-CAA repeat length in BACHD rats
To verify the conservation of the polyQ repeat length, we analyzed the PCR fragment length of DNA samples extracted from 100 peripheral tissues and different brain regions. DNA extractions were performed as described before and rat samples were collected considering different rat generations, gender, and ages of the rats. PCRs were performed using a Cy5labeled forward primer and a reverse primer amplifying a fragment of HTT exon 1 including the CAG-CAA repeats (forward primer: 5′-GAT GAA GGC CTT CGA GTC CCT CAA GTC CTT CT-3′, reverse primer: 5′-CGG CTG AGG CAG CAG CGG CTG T-3′). The fragment lengths of the amplicons were determined and analyzed using the fluorescence-based capillary electrophoresis Sequencer CEQ8800 and genetic analysis system software in a complete set from Beckman Coulter. CAG-CAA repeat numbers were calculated according to the individual fragment length and the human HTT sequence published on the NCBI database.
Identification of alternative HTT splicing variants in BACHD transgenic rats
Splicing variants may possess a considerable effect on protein function with two or more splicing products fulfilling distinct functions in the expressing cells. Therefore, BACHD transgenic rats carrying the genomic sequence with upstream and downstream flanking regions instead of cDNA sequence were generated. Two protein-encoding splicing variants of the human mRNA have been identified and listed in the Ensemble Genome Browser (http://www.ensembl.org/Homo_sapiens/Gene/Summary?g=ENSG00000197386;r=4:3076408-3245676) so far (HTT-001, transcript ID: ENST00000355072 and HTT-011, transcript ID: ENST00000509618). The longer one (HTT-001) includes all 67 exons encoding for the full-length huntingtin; the smaller one (HTT-011) consists only of 3 exons, the last 31 bases of exon 29, exon 30, and 31 as well as the 96 bases of the following intron 31–32. A forward primer was designed to bind to both transcripts in exon 31 of HTT-001 and reverse primers were chosen to bind specifically to each variant. For HTT-001, the reverse primer binds to exon 32 whereas for HTT-011, the primer was directed against a region within the last 96 nucleotides. Brain samples from the striatum, the cortex, and the cerebellum were taken and after purification of the mRNA subjected to real-time PCR using the QuantiTect SYBR Green PCR Kit (Qiagen).
Western blotting.
Expression levels of transgenic mhtt in different lines and different brain regions were quantified using Western blot and ImageJ (National Institutes of Health, NIH) analysis. One-month-old rat brains were homogenized with a homogenizer at a speed of 30,000 rpm in modified radioimmunoprecipitation assay buffer (Gray et al., 2008). The lysates were centrifuged at 4°C for 15 min at 16,200 × g, and the supernatant was removed and stored at −80°C for Western blot analysis. Western blot analysis was performed as described previously (Gray et al., 2008). The blots were probed with two monoclonal antibodies: MAB2166 (1:2000; Millipore) recognizing the N-terminal region of both human and rat htt, and 1C2 (1:2000; Millipore) binding to the expanded polyQ sequence of human htt but not to the nonexpanded polyQ sequence of the endogenous rat htt.
Behavioral assessment
Rats were group-housed with mixed genotypes in a constant temperature-humidity room (22 ± 1°C, 55 ± 10% relative humidity) with a 12 h light/dark cycle (lights on/off at 2:00 A.M./P.M.). Food and water were provided ad libitum. All behavioral tests were performed only with male rats during the dark phase, which is the physiological activity period of the rats. Controls were an equal mix of WT littermates from both lines and the experimenters were blind to the individual animals' genotype. All animal procedures were approved by the state government of Baden–Württemberg, Germany, and are in accordance with animal protection guidelines.
Rotarod test.
Rotarod experiments (Accelerated rotarod for rats 7750, Ugo Basile) were used to measure forelimb and hindlimb motor coordination. BACHD transgenic rats and WT littermates were trained on 3 consecutive days with four trials per day. Directly after training they were tested on 2 consecutive days with two trials per day and an interval of 1 h between individual trials. During the training period, the rats were placed on the rotating rod at a constant speed of 12 rpm for 2 min. Rats were returned to the rod after falling during the training period up to 10 falls per trial. Individual tests were assessed for a maximum of 5 min with accelerating speed from 4 to 40 rpm over a period of 4 min, and the latencies to fall were recorded. Rats from the same cohort (n = 12) were tested every month from 1 to 15 months of age. Three rats were excluded from the analysis because already at younger ages they fell off the rotarod immediately when placed on it, despite extensive training.
Footprint test.
We used a footprint test to analyze gait abnormalities in BACHD rats. BACHD transgenics and WT littermates were evaluated in this task at 14 months of age (n = 11). The front and hindpaws of the animals were painted with nontoxic paint of different colors. On the day before testing, the rats were trained in three sessions. The best performance out of three tests with each rat was selected for data analysis. The stride width of the hindpaw, the step length (left paw to left paw), and the overlap (distance between front and hindpaw) were measured in three consecutive steps, and the average was taken for further analysis.
Elevated plus maze.
An elevated plus maze was used to assess the anxiety of BACHD rats. To eliminate the possibility of habituation effects, different cohorts of rats were tested at 1 (n = 13 per genotype), 4 (n = 13 per genotype), and 12 (TG5:WT = 8:11) months of age as described previously (Nguyen et al., 2006). Rats were placed at the center of an elevated plus maze (with two open and two closed arms) facing an open arm and were monitored for 5 min. The time spent in the open arms was recorded as a percentage of the total time for analysis.
Locomotor activity and food intake.
Rats were monitored using the PhenoMaster system (TSE Systems), which represents a modular setup that screens rats in a home cage-like environment for their ambulatory activity and rearing as well as feeding and drinking behavior. The activity detection is achieved using infrared sensor pairs arranged in horizontal (x,y level for ambulatory activity) and vertical (z level for rearing) strips. Food and water consumption were recorded by two weighting sensors. The same cohort of animals (TG5:TG9:WT = 16:19:18) was individually screened for 22 h every 3 months until the age of 18 months. Data were automatically collected with 1 min intervals and analyzed either entirely or only for the dark (active) phase. Rats, which did not drink >3 ml of water within 24 h were excluded from the data analysis since this might have confounded feeding behavior and activity.
Light and electron immunohistochemistry
Rats were deeply anesthetized with ketamine/xylazine (100/10 mg/kg, i.p.) and transcardially perfused with 4% paraformaldehyde in 0.1 m sodium cacodylate buffer, pH 7.4, followed by postfixation of the brains in the same fixative overnight. For lightmicroscopical immunohistochemistry 16 rat brains were embedded in one gelatin block; 40 μm coronal sections were freeze-cut and collected into 24 series (NeuroScience Associates). Free-floating staining was performed as previously described (Osmand et al., 2006). Sections were incubated with the polyclonal S830 antibody (1:15,000; ∼10 ng/ml, kindly provided by G. Bates), EM48 (1:300, MAB5374; Millipore Bioscience Research Reagents) or the polyclonal anti-calbindin D-28K antibody (1:50,000; Swant Swiss antibodies) followed by the respective secondary antibodies: biotinylated rabbit anti-sheep IgG antibody (1:1000, BA-6000; Vector Laboratories), biotinylated goat anti-mouse IgG antibody (1:1000, BA-9200; Vector Laboratories), or biotinylated goat anti-rabbit IgG antibody (1:1000, BA-1000; Vector Laboratories). Then sections were treated with an avidin–biotin–peroxidase complex (Vector Laboratories), and exposed to nickel-DAB-H2O2 (0.6% nickel sulfate, 0.01% DAB, and 0.001% hydrogen peroxide) until a suitable staining intensity had developed. S830 staining was routinely amplified using a single round of biotinylated tyramine amplification before the final ABC step. Images were taken using an Axioplan 2 Microscope (Zeiss) with a digital camera (AxioCam MRm; Zeiss) and imaging acquisition software (AxioVision-6; Zeiss). Quantification was performed using ImageJ (NIH).
For electron microscopy BACHD and control rat brains (13 and 16.5 months of age) were adjusted in Plexiglas frames according to the coordinates of a rat brain atlas (Paxinos and Franklin, 2006), embedded in 2% agarose, and cut into 3 mm coronal brain blocks. Blocks were cut into series of 50 μm vibratome sections and immunostained with the monoclonal EM48 antibody (1:100, MAB5374; Millipore Bioscience Research Reagents) the specificity of which was controlled in WT rats. Immunostained sections were photodocumented for later detection of the reaction product and flat embedded in Araldite (Serva) as described previously (Petrasch-Parwez et al., 2007). Ultrathin sections (90 nm) were contrasted with 5% aqueous uranyl acetate and lead citrate (pH 12).
Quantitative assessment of morphological changes in the striosome compartment
Calbindin immunostaining of rats at 6 months of age was used (TG5:TG9:WT = 5:4:5) to perform a relative quantification of the striatal striosome compartment. Four striata of each rat were measured in coronal 40-μm-thick brain sections between bregma 1.44 and −0.24 mm (Paxinos and Franklin, 2006). The striatum was outlined medially adjacent to the lateral ventricle, dorsolaterally below the corpus callosum, and ventrally by a line through the ventral tip of the ventricle as the region of interest (ROI) using ImageJ (NIH). The striosomal area was determined within the ROI by outlining the faintly stained calbindin areas. The ratter was blind to the rat's genotype.
[11C]raclopride positron emission tomographic imaging
For longitudinal positron emission tomography (PET) experiments, transgenic BACHD and control rats were imaged at 6, 12, and 18 months of age (n = 6 of each genotype at each time point) using an Inveon dedicated small-animal PET scanner (Siemens Preclinical Solutions), yielding a spatial resolution of ∼1.3 mm in the reconstructed image. Conscious animals were lightly restrained and injected with 29.6 MBq [11C]raclopride via one of the lateral tail veins. A 60 min dynamic PET scan was obtained immediately after tracer injection followed by a 15 min attenuation correction. During imaging, the animals were anesthetized with a mixture of 1.5% isoflurane in 100% oxygen. The animals were centered in the field of view of the PET scanner. Anesthesia was monitored by measuring respiratory frequency, and the body temperature was kept at 37°C by a heating pad underneath the animal. PET data were acquired in list mode; graphed in time frames of 4 × 60 s, 3 × 120 s, 7 × 300 s and 2 × 450 s; and reconstructed using a filtered backprojection algorithm with a matrix size of 256 × 256 and a zoom factor of two. Image files were analyzed using PMOD and AsiPro software (Siemens Preclinical Solutions). The PMOD image fusion software allowed for linear transformation and rotation to overlay the PET and magnetic resonance (MR) template images. The fusioned PET/MR images were analyzed to calculate specific ROIs in different brain areas with reference to the stereotactic brain atlas of Paxinos and Franklin (2006). With the PMOD software we also analyzed the [11C]raclopride uptake in two brain areas including cerebellum and striatum. The cerebellum was chosen as a reference region, to correlate the unspecific uptake with the tracer uptake in the striatum.
Statistical analysis
Standard two-way ANOVA (data not matched) and repeated-measures two-way ANOVA (repeated or matched data) were conducted to assess the effects of genotype and age and genotype × age interaction. Bonferroni post hoc tests were conducted to compare individual genotype effects at individual ages. For the analysis of the rotarod test results, only data until 10 months were taken into account because most TG5 rats fell immediately from the rotarod during the final 5 months (from age 11 to 15 months) leading to a non-normal distribution of the data in the final 5 months. For data where only one time point was assessed (such as the footprint test and matrix/striosome analysis), one-way ANOVA was conducted to evaluate the effects of genotype, followed by Tukey's post hoc test for multiple comparisons. Data are presented as mean ± SEM. Differences were considered significant if p < 0.05.
Results
Generation and establishment of BACHD rats
BACHD rats expressing full-length mutant human htt, were generated with BACs containing human genomic DNA spanning the full-length HTT gene and the flanking genomic sequences of 20 kb upstream and 50 kb downstream, which included all regulatory elements (Yang et al., 1997; Kazantsev et al., 1999; Gray et al., 2008). WT HTT exon 1 was replaced by mutant HTT exon 1 containing 97 mixed CAA/CAG repeats flanked by two LoxP sites (Fig. 1A). Thereby, the BAC construct allows for a conditional and inducible elimination of the mutant HTT exon 1 by Cre recombinase activity. Following microinjection, 21 of 24 transgenic founder rats generated F1 progeny, which was used to evaluate the integrity of the transgene. Two different primer pairs were used in the PCR analysis elongating a fragment of the first and last exon of the HTT gene. Of 21 founder rats, 18 possessed the full-length HTT gene; three founders had to be withdrawn as they were lacking at least the last exon (data not shown). Genomic transgene copy number was analyzed using TaqMan real-time PCR (data not shown). Three lines had multiple insertion sites of the BAC constructs and were therefore excluded from further experimentation. mRNA expression levels were quantified with SYBR Green quantitative PCR, indicating that line LY.005 had the highest mRNA expression of mutant htt (data not shown). Htt protein levels of 1-month-old transgenic rats were quantified by Western blot analysis with the antibody MAB2166, which recognizes both human and rat htt. Highest protein expression was also found in line LY.005, consistent with the mRNA results (data not shown), whereas line LY.009 displayed a comparable level of mRNA and protein as the well characterized BACHD mice (Fig. 1B) (Gray et al., 2008). Consequently, these two lines, which contain a single insertion site and express intact full-length htt were selected for subsequent phenotypic characterization.
Figure 1.
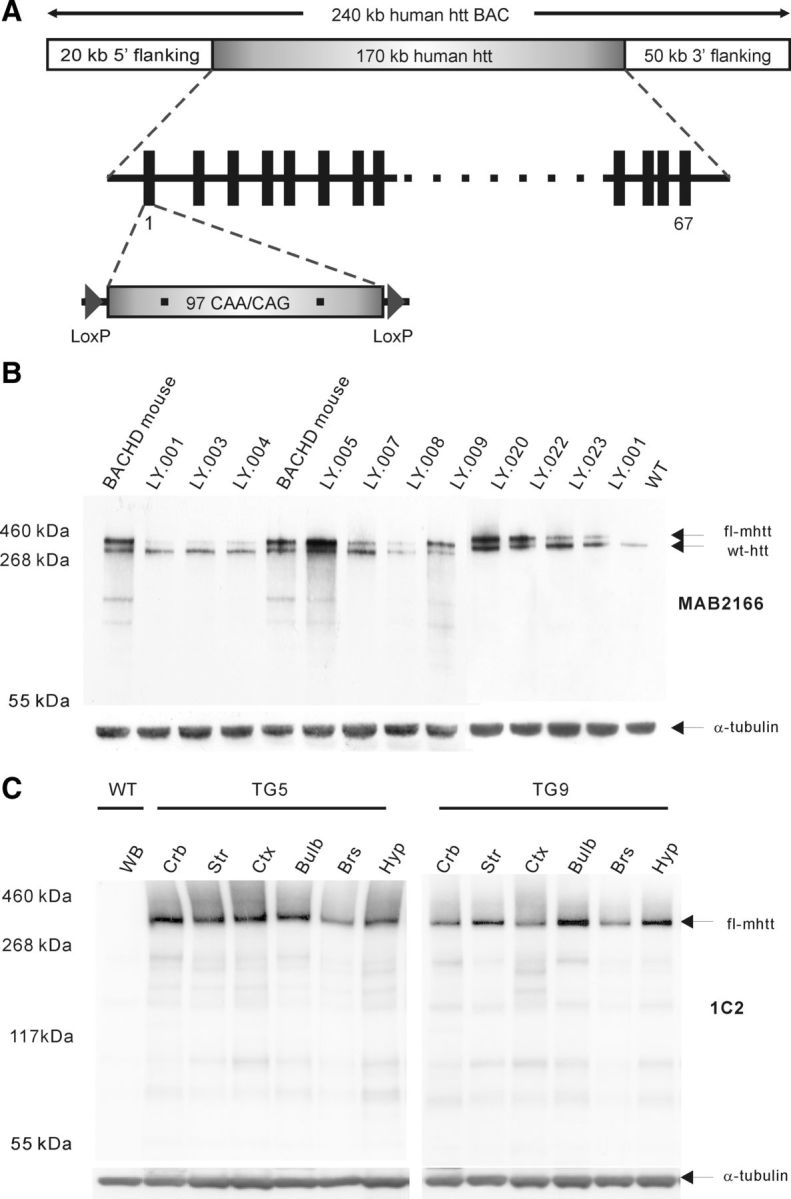
Generation of BACHD transgenic rats. A, BACHD construct was designed using a BAC containing the entire 170 kb of the HTT genomic locus with ∼20 kb upstream and 50 kb downstream flanking sequences. The mutant HTT exon 1 including 97 CAA-CAG trinucleotide repeats in place of endogenous HTT exon 1 is flanked by two loxP sites. B, Western blot analysis of mhtt expression in different transgenic rat lines and compared with BACHD mouse expression. Both mhtt and endogenous WT rat htt are recognized by MAB2166 showing bands of ∼360 and ∼330 kDa, respectively (arrows). Line 5 presents the highest mhtt expression and line 9 shows a comparable mhtt expression level to the BACHD mouse. C, Western blot analysis of mhtt expression in various brain regions in transgenic line 5 (TG5) and transgenic line 9 (TG9) using antibody 1C2. In TG5 rats mhtt is abundantly present in cerebellum (Crb), striatum (Str), and cortex (Ctx), whereas olfactory bulb (Bulb), brainstem (Brs), and hypothalamus (Hyp) show a relatively reduced expression of mhtt. In TG9 rats higher expression levels were found in striatum, olfactory bulb, and hypothalamus compared with cerebellum, cortex, and brainstem. WB, Whole brain.
The relative fold of transgene expression compared with endogenous rat htt was estimated by semiquantitative analysis of mhtt in each line with respect to the calibration line LY.001, which expressed the same level of mhtt mRNA as endogenous htt mRNA. The estimated expression level of mhtt is ∼4.5 times higher than the endogenous in the transgenic line LY.005 (TG5), and 2.5 times higher than endogenous htt in line LY.009 (TG9).
To assess the expression pattern of mhtt in different brain regions of the transgenic BACHD rats, protein extracts of 6 brain subregions (cerebellum, cortex, striatum, olfactory bulbs, brainstem, and hypothalamus) from 1-month-old TG5 and TG9 rats were analyzed using Western blot analysis (Fig. 1C). The blots were stained with antibody 1C2 to visualize the signal of mhtt. The results revealed that in TG5 rats mhtt is most expressed in the cerebellum, striatum, cortex, and olfactory bulb, whereas the hypothalamus and brainstem show a lower abundance (Fig. 1C). In TG9 rats higher expression levels were found in striatum, olfactory bulb, and hypothalamus compared with cerebellum, cortex, and brainstem.
Stable CAG repeat number and alternative splicing variants of mutant human huntingtin in BACHD rats
The stability of CAG repeat expansions is of critical importance for therapeutic studies, since the age of onset of HD inversely correlates with the number of CAG repeats (Duyao et al., 1993; Stine et al., 1993). In total, 100 samples from different brain regions or peripheral tissue were collected from transgenic rats with ages of up to 18 months of four generations and both genders to verify the stability of the polyglutamine stretch in our transgenic rat model. Our analysis revealed stability of the polyQ encoding sequence in both germlines and in different brain regions at different ages, gender, and rat generations as observed in BACHD mice (data not shown) (Gray et al., 2008).
Real-time PCR was performed to determine alternative splicing variants of the transgene mRNA. Samples of different brain regions of BACHD rats were analyzed for the presence of both mhtt protein-encoding variants, which have been previously identified (transcript HTT-001 and HTT-011 in Ensemble Genome Browser). Both the small (HTT-011) and the 67 exons spanning large transcript (HTT-001) were identified in all analyzed brain samples and the ratio of the transcript variants present in the different brain regions were similar (Fig. 2A,B).
Figure 2.

Alternative splicing variants in BACHD transgenic rats. Real-time PCR was performed using one common forward primer binding to both variants of HTT-001 and HTT-011; the reverse primers were chosen specifically to bind to each variant. A, Image of gel electrophoresis. The BACHD rats TG5 and TG9 exhibit both transcription variants HTT-001 and HTT-011 in cerebellum (Crb), cortex (Ctx), and striatum (Str). Whole-brain (WB) mRNA of a WT rat was taken as negative control. B, Quantification of the proportion of HTT-001/HTT-011 in different brain regions of both transgenic lines. The ratios of HTT-001 and HTT-011 vary between 2.49 and 4.40 in all brain regions investigated in both TG5 and TG9, except in the cortex of TG9 rats, where an equal expression of HTT-001 and HTT-011 is present.
BACHD rats do not exhibit an increased body weight compared with WT littermates
Since it has been shown that an increased expression of fl-mhtt in mice is associated with a dose-dependent increase in body weight (Van Raamsdonk et al., 2006), we have monitored body weight in both lines of BACHD rats and their WT littermates weekly (Fig. 3A). All rats gained body weight until 64 weeks of age with neither significant interaction between genotype and age, nor main effect of genotype (repeated-measures ANOVA, p > 0.05).
Figure 3.
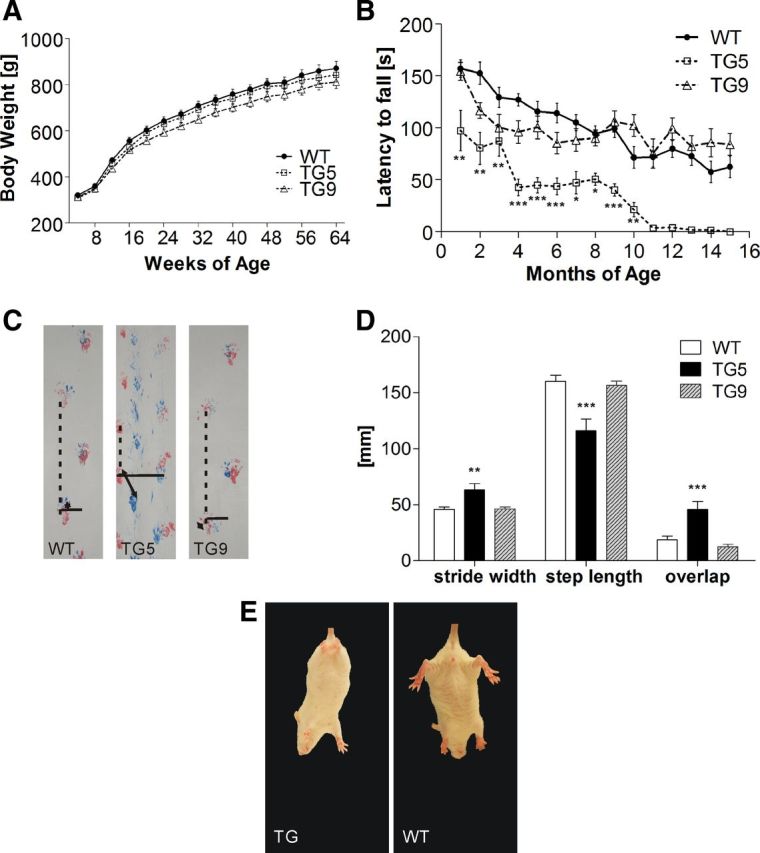
Motor function analysis of BACHD rat lines TG5 and TG9 compared with WT littermates. A, Comparison of body weights of BACHD rats and WT littermates (WT:TG5:TG9, N = 20:16:22). The body weight was measured once a week and was not significantly different between genotypes over an observation period of 64 weeks. B, Rats underwent four test sessions each month on an accelerating rod (4–40 rpm in 4 min). Mean latencies to fall (± SEM) were compared among different genotypes over 15 months (n = 12). TG5 rats displayed a progressive decrease in performance from 1 month of age, whereas TG9 showed an impaired performance at 3 and 4 months. Starting with the fifth test month, TG9 rats adapted an alternative strategy to remain on the rotating rod, which yielded comparable performances compared with WT rats. C, Footprints at 14 months of age. Stride width: continuous line; step length: dotted line; and overlap between front and hindlimbs: two directional arrows. D, Statistical analysis of footprints examined at 14 months of age. TG5 exhibited a significantly abnormal gait with increased stride width, decreased overlap as indicated by a greater distance between hind and front paw placement, and decreased step length. E, Hindlimb clasping in BACHD transgenic rats at 3 weeks of age. Data are expressed as means ± SEM, *p < 0.05; **p < 0.01; ***p < 0.0001.
Early onset and progressive motor deficits in BACHD rats
Motor deficits are an important clinical feature of HD patients including involuntary movements, motor dyscoordination, and gait disturbances. To assess motor dysfunction in our transgenic animals, we performed rotarod tests and footprint analyses and looked for clasping behavior.
Four trials on an accelerated rotarod were conducted with 1-month-old rats and repeated every month until the age of 15 months. The average latency until the animal fell off the rotating rod was measured and analyzed. Repeated-measures ANOVA revealed a highly significant main effect of genotype (F(2,270) = 22.86, p < 0.0001) and a statistically significant genotype × age interaction (F(18,270) = 1.67, p = 0.0448), reflecting a significant difference between the performance of BACHD rats and WT rats with increasing age. Subsequent post hoc analysis with Bonferroni tests demonstrated a progressive decline in rotarod performance in the TG5 group relative to both WT and TG9 animals (Fig. 3B). Significant differences were already evident at 1 month of age (p < 0.01). Here, TG5 rats exhibited significant difficulties in maintaining balance on the rod at higher rotation speeds and were unable to remain on the rod longer than 97.31 ± 19.45 s. In comparison, WT rats showed an average latency to fall of 156.88 ± 8.48 s. At 4 months of age, the performance of TG5 rats dropped drastically, but it was maintained over the next 4 months. A similar drastic reduction in performance was observed again between 9 and 11 months of age, when most of the TG5 rats were unable to walk on the rod even at the lowest velocity (4 rpm) (Fig. 3B).
Video recordings provided additional insight into the rats' performance on the rotarod. At young ages (<3 months), both transgenic and WT rats displayed similar walking behavior on the rotarod. Starting with 3 and 5 months of age, respectively, TG5 and TG9 transgenic rats adopted an abnormal walking strategy. After being placed on the rod, the transgenic animals turned 180° and instead of performing a coordinated walk, they started to jump backwards. The number of transgenic rats adapting this unusual strategy increased with age, which partly compensated for the decline in rotarod performance. None of the WT littermates displayed this abnormal movement strategy. Together, the results of the rotarod test demonstrated an early onset and progressive motor function deficit in BACHD transgenic rats of line TG5.
To investigate the gait of BACHD transgenic rats, footprints from all genotypes (n = 11 each) were analyzed at 14 months of age. The stride width of the hindlimbs as well as the step length and the overlap of hindlimbs and forelimbs of each individual rat were measured. One-way ANOVA revealed that 14-month-old transgenic TG5 rats made significantly shorter steps with forelimbs and hindlimbs (p < 0.0001) than TG9 and WT. Furthermore, TG5 rats showed an increased stride width (p < 0.01) and a reduced overlap between forelimb and hindlimb placement (p < 0.0001) (Fig. 3C,D) compared with the other groups, indicating that BACHD transgenic rats TG5 have gait abnormalities at older ages compared with WT rats.
Additionally, characteristic hindlimb clasping behavior was observed during tail suspension in both transgenic lines starting at 3 weeks of age (Fig. 3E).
Decreased anxiety, initial hypoactivity, and reduced food intake in BACHD rats
Additional clinical features of HD patients involve multiple psychiatric symptoms. To score the anxiety level of the BACHD transgenic rats, we used the elevated plus maze test. Independent cohorts of TG5 rats and WT littermates were used at 1, 4, and 12 months of age. One- and four-month- old BACHD TG5 rats spent significantly more time on the open arms compared with WT (10.30 and 9.55% at 1 and 4 months of age, respectively, p < 0.05 at each time point), (Fig. 4A). At 12 months of age, the difference between TG5 and WT rats further increased with a mean difference of 13.43% (p < 0.01). The significance of these observations was confirmed by two-way ANOVA, which indicated a highly significant main effect of genotype (F(1,65) = 25.56, p < 0.0001) as well as a main effect of age (F(2,65) = 37.7, p < 0.0001). However, there was no significant interaction between genotype and age (F(2,65) = 0.26, p = 0.7753) because the percentage of time spent on the open arms of the maze decreased in both genotypes with increasing age.
Figure 4.

Emotional changes, reduced food intake, and initial hypoactivity in BACHD rats. A, Independent cohorts of TG5 and WT control rats were tested in the elevated plus maze at 1, 4, and 12 months of age. Data are reported as mean ± SEM for the percentage of time spent in the open arms. TG5 rats exhibited significantly increased open-arm exploration compared with WT controls, which was evident at 4 months of age. The time spent in the open arms decreased in both genotypes throughout the study. Over an 18 month period with 3 month intervals, a cohort of TG5 (n = 16), TG9 (n = 19), and WT rats (n = 18) were measured for food intake (B), ambulatory activity (C), and rearing (D) during 12 h of a dark phase. We found significant decreased food consumption in both transgenic lines over a period of 18 months (asterisks), and a highly significant interaction effect between genotype and ambulatory activities. Both ambulatory activity and rearing decreased in all three genotypes over time, whereas significantly lower activity and rearing in both TG5 and TG9 compared with the WT rats were only observed up to 6 months of age. Data are expressed as means ± SEM, *p < 0.05; **p < 0.01; ***p < 0.0001.
Locomotor activity and food consumption of BACHD rats were registered in an automated, home cage-like environment (PhenoMaster; TSE Systems). Measurements were taken from one cohort of each rat line (TG5:TG9:WT = 16:19:18) every 3 months over a period of 15 months. At each time point, ambulatory activity over 22 h as well as rearing activity during the dark phase were analyzed. Both transgenic and WT rats showed a pronounced dark/light cycle in their behavioral activities with their main activity taking place during the dark phases. The activity pattern did not differ between different genotypes (data not shown).
Highly significant differences in ambulatory activity were found at early time points in both transgenic rat lines compared with WT (both TG5 and TG9 p < 0.001 at 3 and 6 months of age) (Fig. 4C,D). The reduction in rearing activity was only observed in TG5 compared with WT control rats at early time points (p < 0.05 at 3 months of age, p < 0.0001 at 6 months of age). The significance of these observations was confirmed by two-way ANOVA, which indicated a highly significant interaction effect (genotype × age: F(10,259) = 4.22, p < 0.0001) in ambulatory activity, a main effect of genotype in both ambulatory activity (F(2,259) = 22.32, p < 0.0001) and rearing activity (F(2,248) = 15.11, p < 0.0001) as well as a main effect of age (F(5,259) = 217.89, p < 0.0001 in ambulatory activity; F(5,248) = 23.23, p < 0.0001 in rearing activity).
The total food consumption during the dark phase was compared between the three genotypes over 15 months. Analysis with two-way ANOVA revealed a highly significant main effect of genotype (F(2,259) = 67.80, p < 0.0001). BACHD rats of both lines showed a reduced food intake throughout the study, which was due to reduction in food intake in both transgenic rat lines compared with WT littermates with a mean value of 8.79 g in TG5 and 4.90 g in TG9 (Fig. 4B) compared with 20.71 g in WT. However, there was no significant interaction between genotype and age (F(10,259) = 1.06, p = 0.3965).
Huntingtin aggregates increase over time and are widely distributed in BACHD rats
To investigate the regional distribution pattern of mhtt-positive aggregates in BACHD transgenic rats, we used the polyclonal sheep antibody S830 and the monoclonal mouse antibody EM48 in serial brain sections at 12 months of age (Fig. 5). Immunohistological staining with both antibodies demonstrated a similar distribution pattern in BACHD transgenic rats. However, mEM48 staining was weaker and may exhibit an unspecific immunoreactivity with blood vessels (Fig. 5h). For this reason, the results obtained with the S830 antibody are described in more detail in the following. In TG5 and TG9 brains, the aggregates were found to be widely distributed in all regions with prominent expression in the neocortex and in limbic areas including nucleus accumbens, hippocampus (specifically in the CA3 region), bed nucleus of the stria terminalis, and the amygdala (Fig. 5). A lower expression was observed in the hypothalamus and in most thalamic nuclei (data not shown). In comparison, very few aggregates were detected in the dorsolateral caudate–putamen, the lateral globus pallidus, and the substantia nigra (data not shown). The cerebellar cortex showed a few small aggregates throughout all three layers (data not shown). Both the size and number of aggregates increased with age, showing the largest and most abundant aggregates in amygdala, in the CA3 region of the hippocampus, and in the cerebral cortex, indicating that aggregate formation continued to progress in these regions (Fig. 6). Nuclear accumulation of N-terminal huntingtin was only observed in outer layers of cerebral cortex, in striatum, and in the granule cells of the dentate gyrus in older rats (after 9 months of age) and increased thereafter (Figs. 5, 6). Mhtt aggregates were more prominent in TG5 than in TG9 rats at all ages investigated. No immunoreactivity was observed in WT rats.
Figure 5.
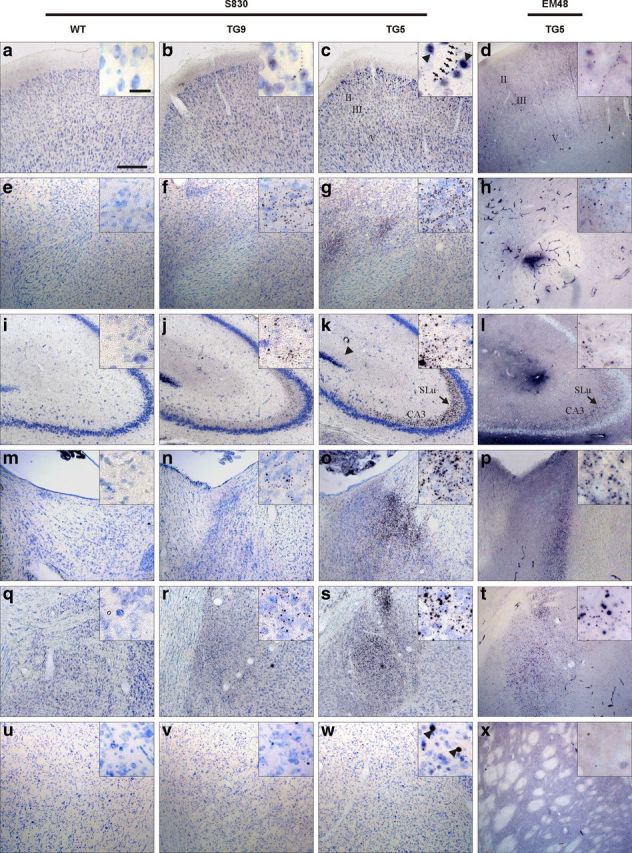
Htt aggregates were investigated using polyclonal sheep antibody S830 and monoclonal mouse antibody EM48 showing similar brain regional distribution pattern. Mutant htt immunoreactivity is widely distributed throughout the cerebrum of BACHD rats by 12 months of age. Nickel-DAB (black) visualizes immuno-activity of mhtt, and the counterstaining with thionin (blue) marks nuclei. In both TG5 and TG9 rats, aggregates varying in size and formation were abundant in the cortex (a–d), nucleus accumbens (e–h), and hippocampus (i–l) and strongly expressed in the stratum lucidum of CA3 area (arrow in k and l), bed nucleus of stria terminals (m–p), and amygdala (q–t). Few aggregates were found in caudate–putamen (u–x). Most aggregates were localized in the neuropil, some of which arranged in a linear array (arrows in the insert of c). Nuclear htt staining (arrowheads in the insert of c) was only detected in layer II/III of the cortex, a few in caudate–putamen, and in the granule cells of dentate gyrus at 12 months of age. Scale bars: inlay showing high-magnification images, 20 μm; in low-magnification images, 200 μm.
Figure 6.

Spatiotemporal accumulation of mhtt in amygdala (Amg) and cortex (Ctx) of TG5 rat brains. An increase in both number and size of neuropil aggregates (arrows) was observed in both regions during aging; nuclear accumulation of mhtt (arrowheads) appeared only in cortex at 9 months of age becoming more abundant at 15 months of age. Scale bar, 20 μm.
Subcellular localization of N-terminal htt aggregates and neurodegeneration
Using light microscopy, aggregates varying in size and form as well as chain-like structures were observed throughout the brain areas with abundant mhtt. These data indicate that mhtt aggregates are primarily located in the neuropil (Figs. 5, 6). Subcellular localization of N-terminal htt aggregates and neurodegeneration were further investigated ultrastructurally in TG5 rat brains at advanced ages by EM48 immunohistochemistry (Fig. 7) when aggregates were abundantly expressed. Electron microscopy confirmed that most aggregates were localized in the neuropil. Htt deposits were predominantly detected in axons (Fig. 7C,K) and synaptic terminals (Fig. 7D,G,L). Dark degenerating neurons and dark dendrites were also identified in these areas as documented in the nucleus accumbens (Fig. 7B), bed nucleus of the stria terminalis (Fig. 7F), and the cortex (Fig. 7J). Dark neurons with condensed nucleus and cytoplasm were often localized adjacent to normal light neurons (Fig. 7B,F). Dark degenerating terminals were seen in apposition to large pallidal dendrites (Fig. 7H). Some dark cortical neurons displayed a fine granular immunoreaction product (Fig. 7J) similar to that seen by light microscopy in the respective area (Fig. 6). The prominent S830 immunoreactivity in the stratum lucidum of the CA3 region of the hippocampus (Fig. 5) corresponded to immunopositive unmyelinated mossy fibers (Fig. 7K) and their terminals, some of which were engulfed by lamellated structures as a sign of degeneration (Fig. 7L).
Figure 7.

Neurodegeneration and subcellular localization of mhtt aggregates in TG5 brains (A, E, I). Vibratome sections (50 μm) show the areas investigated by EM48 immune-electron microscopy (B, F, J). Dark degenerated neurons (DN) and dendrites (B, arrowheads) were detected in the nucleus accumbens (Acb), bed nucleus of the stria terminalis (BNST), and cortex (Ctx), the latter with punctate nuclear htt reactivity as seen adjacent to the nucleolus (N) at higher enlargement (N, inlay in J). DNs were seen localized near normal neurons, MSN (B), or myelinated nerve fibers showing evidence of degeneration (arrow in J). Htt aggregates (*) were observed in axons (arrow in C) and synaptic terminals (T in D and G) often contacting dendrites (De). Dark terminals (DT in H) were seen in the globus pallidus (GP) apposing pallidal dendrites (PDe). In the hippocampal CA3 region of hippocampus mossy fiber bundles (MF) emerging from the dentate gyrus (DG) exhibited immunoreactive unmyelinated fibers (K, arrows); htt reactivity engulfed by lamellated structures was also observed in mossy fiber terminals (MFT in L), adjacent to neuronal dendrites (De). Caudate–putamen (CPu); anterior commissure (ac). Scale bars: (in A, E, I), 0.5 mm; (in B, F, J), 5 μm; (in C, H, K, L), 1 μm; (in D, G), 0.5 μm.
Reduced dopamine receptor binding potential in aged BACHD transgenic rats
A specific uptake of the D2-receptor antagonist [11C]raclopride was demonstrated in dynamic PET scans in the striatum of WT and BACHD TG5 rats, while there was no specific tracer uptake in the corresponding cerebella. Longitudinal measurements up to 18 months of age revealed a significant decrease in the striatal uptake of [11C]raclopride in BACHD rats (two-way ANOVA for the main effect of genotype, F(1,24) = 8.77, p < 0.01). This reduction of dopamine receptor binding potential was evident at 18 months of age (p < 0.01), as confirmed by Bonferroni post hoc multiple-comparison test (Fig. 8). However, there was no significant interaction between genotype and age (F(2,24) = 2.33, p = 0,1185).
Figure 8.
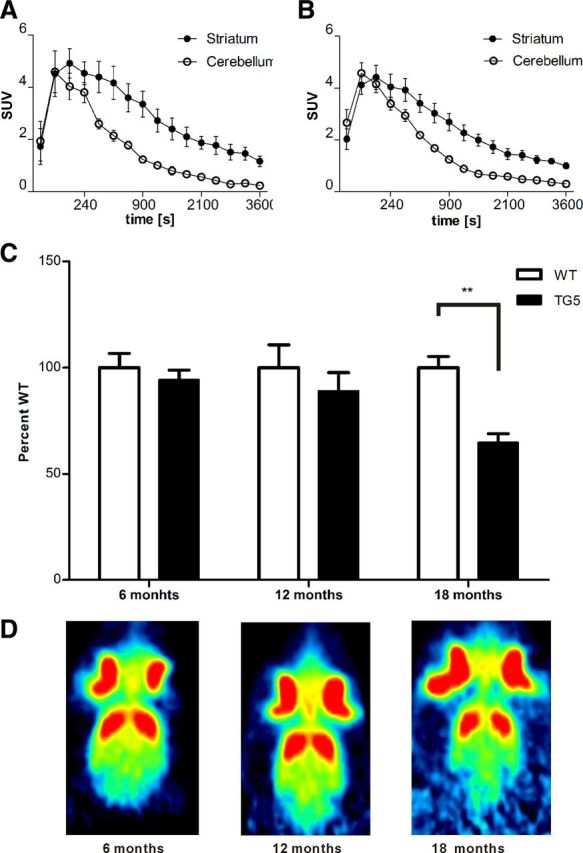
[11C]raclopride PET imaging reveals D2 receptor loss in striatum of BACHD rats TG5. [11C]raclopride uptake was measured as standard uptake volume (SUV) of different brain areas. PET time activity curves of target (striatum) and reference region (cerebellum) are plotted for the control (A) and BACHD rats (B). C, Corresponding binding potential (BPND) values are shown in the striatum. There was no significant difference in BPND between the first two time points in transgenic and control rats. After 18 months the BPND of BACHD was significantly lower than in control littermates. Representative images of [11C]raclopride uptake at each age is shown in D. Data are expressed as means ± SEM, **p < 0.01.
Imbalance of striosome and matrix compartments in early disease stages of BACHD rats
Calbindin immunostaining was performed to determine changes in the striosome and matrix compartments, as calbindin is strongly expressed in the matrix surrounding the Calbindin-poor striosomes (Fig. 9A). The intensity of the calbindin staining neither in the matrix nor in the irregular-shaped striosomes (Fig. 9A) was altered in BACHD rats compared with WT littermates. One-way ANOVA demonstrated that both total matrix area and number of the striosomes in BACHD transgenic animals did not differ from that of their WT littermates (Fig. 9B). However, the total striosome area (Fig. 9C) and the mean area of the striosomes (Fig. 9D) were significantly different among the three genotypes, which subsequent Tukey's multiple-comparison test determined was due to the reduction in total and mean striosomal area in TG5 transgenic rats compared with WT littermates (29% in total area, p = 0.0359; 34% in mean area, p = 0,0196). This imbalance in the striosome-matrix compartments in TG5 rats may affect the equilibrium of inhibitory and excitatory output from the striatum to downstream neurons.
Figure 9.

Striosome abnormalities in the striatum of BACHD rats. The ROI is outlined by a dotted black line. A, Calbindin immunostained sections of WT, TG5, and TG9 rats at 6 months of age show striosomes visible by faint calbindin staining (arrows and outlined by a continuous white line), while the intense staining visualizes the matrix surrounding the striosomes. Scale bar, 0.5 cm. B, The total matrix area and number of striosomes was similar in WT, TG5, and TG9 rats. A significant decrease in both total (C) and mean striosome areas (D) was only observed in TG5 rats compared with WT littermates. Data are expressed as means ± SEM, *p < 0.05.
Discussion
BACHD transgenic rats expressing fl-mhtt, exhibit several robust HD-like behavioral phenotypes as well as changes at the molecular and cellular levels (Fig. 10). Importantly, there was no significant difference in body weight between BACHD rats of both lines and WT littermates (Fig. 3A). This is in contrast to what has been reported in BACHD mice and YAC128 mice (Menalled et al., 2009), where overexpression of fl-mhtt is associated with a significantly increased body weight. It has been postulated that body weight is modulated by levels of fl-htt with increased levels of fl-wthtt or fl-mhtt leading to an increased body and organ weight in mice, while a decrease of fl-htt is associated with body weight loss (Van Raamsdonk et al., 2006). The increase of body weight in mice resulted from an increase of both total fat mass and fat-free mass and was associated with increased levels of plasma IGF-1 (Pouladi et al., 2010). Interestingly, we also observed an increase in body fat mass in BACHD rats. But in contrast to the published mouse data a decrease in organ weight in BACHD rats of line TG5 was found resulting in a comparable body weight between BACHD rats and WT rats (preliminary results, data not shown). This discrepancy is especially intriguing since the same construct was used to generate BACHD mice and rats. It might be attributed to species-specific differences and is less likely caused by integration site effects as we did not observe an increased body weight in any of the 18 full-length BACHD rat lines. Furthermore, there is a proportional correlation between the severity of the HD phenotype and the protein expression level in the two BACHD lines arguing against an integration site effect. As progressive rotarod deficits are readily apparent in BACHD rats of line TG5 these are very likely motor defects rather than a consequence of increased body weight. Also, we can exclude that differences in body weight confound other behavioral tests in BACHD rats rendering this model valuable for further phenotype studies.
Figure 10.

Progression of various phenotypes in BACHD TG5 rats. The findings of this study are summarized according to the earliest onset or appearance of each phenotype.
Another advantage of the BACHD rat is its strong phenotype. Compared with the already existing fragment tgHD rats BACHD rats show an earlier onset and a faster progression of motor deficits without the need to breed for homozygosity as heterozygous tgHD rats show only subtle deficits (Nguyen et al., 2006; Brooks et al., 2009). Recently, a milder phenotype in tgHD rats as originally described has been reported (Casteels et al., 2011; Antonsen et al., 2012; Blockx et al., 2012). Also, while striatal atrophy was found in some groups of old tgHD rats (von Hörsten et al., 2003; Kántor et al., 2006; Nguyen et al., 2006), other studies revealed little or no evidence for atrophy (Winkler et al., 2006; Bode et al., 2008; Blockx et al., 2011).
In addition, the expression of fl-mhtt compared with a fragment of mhtt may add important features. For example, transgenic HTT in BACHD rats undergoes natural splicing as confirmed by the presence of the two naturally occurring mRNA isoforms. Even though the small splicing variant does not contain a polyQ stretch, it may fulfill additional functions relevant for the disease process. Since both of these isoforms were found in humans (www.ensembl.org/Homo_sapiens/Gene/Summary?g=ENSG00000197386;r=4:3076408-3245676), the BACHD rat model closely resembles the HD patient condition in this respect.
In BACHD rats, we found a significantly decreased D2 receptor binding potential by [11C]raclopride PET, which is in accordance with findings in HD patients and has been proposed as a neuroimaging biomarker for HD (Pavese et al., 2003; van Oostrom et al., 2005). Due to the relatively large size of the rat brain, in vivo MRI and PET imaging are more practicable than in mice, thus providing an excellent tool to study disease progression. To our knowledge, this is the first study showing clear longitudinal changes by [11C]raclopride PET in an HD animal model, making the BACHD rats a valuable model for noninvasive neuroimaging studies with novel ligands to be subsequently translated into the clinics.
Remarkably, the BACHD rat model reflects many aspects of the aggregation pattern of mhtt found in HD patients. The aggregates occur more frequently in the cortex than in the striatum and neuropil aggregates appear earlier than mhtt accumulation in the nucleus (DiFiglia et al., 1997; Gutekunst et al., 1999). In contrast, R6/2 mice display mainly diffusible mhtt throughout the nucleus with intensively stained nuclear aggregates (Kosinski et al., 1999; Wang et al., 2008). In the fragment rat model for HD, cortical aggregates are far less expressed than in the limbic striatum (von Hörsten et al., 2003; Nguyen et al., 2006). BACHD mice display a similar mhtt aggregate distribution pattern as our BACHD rats with prevalence of neuropil aggregates and paucity of intranuclear inclusions, but both are less abundant than in BACHD rats and appear at a later time point; the diffuse nuclear mhtt accumulation was not detected until 18 months of age (Gray et al., 2008). In comparison, YAC128 mice display both neuropil aggregates and intranuclear inclusions, which were present at 15 months of age, particularly in ventral striatum, amygdala, and cortex (Bayram-Weston et al., 2012). Interestingly, neuropil aggregates are much more common in HD patients with an adult onset than in juvenile onset patients, and the number of neuropil aggregates correlates with the extent of disease. While intranuclear aggregates were not detected in the presymptomatic patient, neuropil aggregates were observed in the cortex of the same presymptomatic patient (DiFiglia et al., 1997; Gourfinkel-An et al., 1998; Gutekunst et al., 1999). This difference in the aggregation pattern might be explained by sequence differences in the human mutant HTT transgenes harbored by the BACHD models and the YAC128 mice, including single nucleotide polymorphisms as well as differences in the nature of CAA interruptions of the CAG tract (Pouladi et al., 2012).
However, whether the extent of aggregation or numbers of aggregates predict neuronal death remains controversial. The formation of neuropil aggregates can induce axonal degeneration (Li et al., 2001). Additionally, they can influence the mitochondrial transport along both axons and dendrites, thereby impairing energy supply within neuronal processes (Chang et al., 2006). Importantly, BACHD rats show mhtt deposits prominently expressed in axons and synaptic terminals (Fig. 7). In contrast, other studies have suggested that aggregation may exert beneficial effects by protecting against polyglutamine toxicity, since the aggregation of N-terminal htt reduces the amount of monomeric and oligomeric N-terminal htt, which possesses a higher toxicity (Arrasate et al., 2004; Lajoie and Snapp, 2010; Miller et al., 2010). In BACHD rats, numerous dark degenerating neurons and dendrites were found in brain regions, where prominent numbers of aggregates were present. Furthermore, dark neurons and dendrites containing aggregates were frequently observed together suggesting a connection between aggregate formation and neurodegeneration. However, further studies would be needed to investigate this association.
The striatum is composed of two interdependent compartments: the striosome (patch) and the matrix, which can be distinguished by the differential expression of neurotransmitter-related molecules (Goldman-Rakic, 1982; Graybiel et al., 1990; Holt et al., 1997). Discriminative input and output connections suggest that the striosome and matrix compartments participate in limbic-based and sensorimotor/associative forebrain circuits, respectively (Donoghue and Herkenham, 1986; Gerfen, 1992). In HD patients, an imbalanced loss of neurons in the striosome and matrix compartments has been described leading to a reduced total matrix area whereas the volume of the striosome is not affected (Ferrante et al., 1987; Seto-Ohshima et al., 1988; Hersch and Ferrante, 1997). Conversely, it has also been reported that this imbalance in neuron loss is initially found in the striosomes but affects both compartments equally in later stages of the disease (Reiner et al., 1988; Hedreen and Folstein, 1995; Lawhorn et al., 2008). Striosomes exchange information with the surrounding matrix, the integrated signal will be subsequently sent through D1 (excitatory) and D2 (inhibitory) medium spiny neurons (MSNs) via direct and indirect pathways to the thalamocortical circuits. However, the striosome and matrix compartments contain disproportionate amounts of D1 and D2 neurons and the imbalance of those two compartments influence the proportion of excitatory and inhibitory signaling in limbic-based and sensorimotor/associative forebrain circuits. Thus, a change in the established equilibrium, as is the case in HD, causes a disorder of the basal ganglia. In BACHD rats, we observed early changes in the pattern of striosome and matrix compartments, as well as a decrease in the total and mean striosome area. This could cause alterations in the basal ganglia and lead to neuronal dysfunction and clinical signs of HD. However, the exact mechanism behind this is unknown and how this is linked to the neuropil aggregates within the sensorimotor circuit and the limbic-based circuits still needs to be answered.
In summary, we have developed a novel transgenic rat model for Huntington disease, which expresses fl-mhtt with 97 polyQ repeats under the control of the human htt promoter and regulatory elements. These BACHD rats display a robust, early onset and progressive HD-like phenotype combined with characteristic neuropathological features of Huntington disease making them a valuable model for further understanding the disease mechanisms and for preclinical pharmacological studies.
Footnotes
This work was supported by the CHDI Foundation and this rat model is available to the HD community for HD research. We are grateful to Professor Gillian P. Bates for providing us the sheep polyclonal antibody S830. We thank Esteban Portal for the technical support in data analysis for the PhenoMaster experiments and Joachim Täger for the helpful discussions. The skillful technical assistance of Marlen Löbbecke-Schumacher and Hans-Werner Habbes is also gratefully acknowledged.
References
- Antonsen BT, Jiang Y, Veraart J, Qu H, Nguyen HP, Sijbers J, von Hörsten S, Johnson GA, Leergaard TB. Altered diffusion tensor imaging measurements in aged transgenic Huntington disease rats. Brain Struct Funct. 2012 doi: 10.1007/s00429-012-0427-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Bayram-Weston Z, Jones L, Dunnett SB, Brooks SP. Light and electron microscopic characterization of the evolution of cellular pathology in YAC128 Huntington's disease transgenic mice. Brain Res Bull. 2012;88:137–147. doi: 10.1016/j.brainresbull.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Blockx I, Van Camp N, Verhoye M, Boisgard R, Dubois A, Jego B, Jonckers E, Raber K, Siquier K, Kuhnast B, Dollé F, Nguyen HP, Von Hörsten S, Tavitian B, Van der Linden A. Genotype specific age related changes in a transgenic rat model of Huntington's disease. Neuroimage. 2011;58:1006–1016. doi: 10.1016/j.neuroimage.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Blockx I, De Groof G, Verhoye M, Van Audekerke J, Raber K, Poot D, Sijbers J, Osmand AP, Von Hörsten S, Van der Linden A. Microstructural changes observed with DKI in a transgenic Huntington rat model: evidence for abnormal neurodevelopment. Neuroimage. 2012;59:957–967. doi: 10.1016/j.neuroimage.2011.08.062. [DOI] [PubMed] [Google Scholar]
- Bode FJ, Stephan M, Suhling H, Pabst R, Straub RH, Raber KA, Bonin M, Nguyen HP, Riess O, Bauer A, Sjoberg C, Petersén A, von Hörsten S. Sex differences in a transgenic rat model of Huntington's disease: decreased 17beta-estradiol levels correlate with reduced numbers of DARPP32+ neurons in males. Hum Mol Genet. 2008;17:2595–2609. doi: 10.1093/hmg/ddn159. [DOI] [PubMed] [Google Scholar]
- Brooks S, Fielding S, Döbrössy M, von Hörsten S, Dunnett S. Subtle but progressive cognitive deficits in the female tgHD hemizygote rat as demonstrated by operant SILT performance. Brain Res Bull. 2009;79:310–315. doi: 10.1016/j.brainresbull.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Bruyn GW. Huntington's chorea. Tijdschr Ziekenverpl. 1979;32:101–105. [PubMed] [Google Scholar]
- Casteels C, Vandeputte C, Rangarajan JR, Dresselaers T, Riess O, Bormans G, Maes F, Himmelreich U, Nguyen H, Van Laere K. Metabolic and type 1 cannabinoid receptor imaging of a transgenic rat model in the early phase of Huntington disease. Exp Neurol. 2011;229:440–449. doi: 10.1016/j.expneurol.2011.03.014. [DOI] [PubMed] [Google Scholar]
- Chang DT, Rintoul GL, Pandipati S, Reynolds IJ. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol Dis. 2006;22:388–400. doi: 10.1016/j.nbd.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Crook ZR, Housman D. Huntington's disease: can mice lead the way to treatment? Neuron. 2011;69:423–435. doi: 10.1016/j.neuron.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Donoghue JP, Herkenham M. Neostriatal projections from individual cortical fields conform to histochemically distinct striatal compartments in the rat. Brain Res. 1986;365:397–403. doi: 10.1016/0006-8993(86)91658-6. [DOI] [PubMed] [Google Scholar]
- Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F, Frontali M, Folstein S, Ross C, Franz M, Abbott M. Trinucleotide repeat length instability and age of onset in Huntington's disease. Nat Genet. 1993;4:387–392. doi: 10.1038/ng0893-387. [DOI] [PubMed] [Google Scholar]
- Ehrnhoefer DE, Butland SL, Pouladi MA, Hayden MR. Mouse models of Huntington disease: variations on a theme. Dis Model Mech. 2009;2:123–129. doi: 10.1242/dmm.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Beal MF, Martin JB, Bird ED, Richardson EP., Jr Morphologic and histochemical characteristics of a spared subset of striatal neurons in Huntington's disease. J Neuropathol Exp Neurol. 1987;46:12–27. doi: 10.1097/00005072-198701000-00002. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 1992;15:133–139. doi: 10.1016/0166-2236(92)90355-c. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. Cytoarchitectonic heterogeneity of the primate neostriatum: subdivision into Island and Matrix cellular compartments. J Comp Neurol. 1982;205:398–413. doi: 10.1002/cne.902050408. [DOI] [PubMed] [Google Scholar]
- Gourfinkel-An I, Cancel G, Duyckaerts C, Faucheux B, Hauw JJ, Trottier Y, Brice A, Agid Y, Hirsch EC. Neuronal distribution of intranuclear inclusions in Huntington's disease with adult onset. Neuroreport. 1998;9:1823–1826. doi: 10.1097/00001756-199806010-00028. [DOI] [PubMed] [Google Scholar]
- Gray M, Shirasaki DI, Cepeda C, André VM, Wilburn B, Lu XH, Tao J, Yamazaki I, Li SH, Sun YE, Li XJ, Levine MS, Yang XW. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. 2008;28:6182–6195. doi: 10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, Ohta K, Roffler-Tarlov S. Patterns of cell and fiber vulnerability in the mesostriatal system of the mutant mouse weaver. I. Gradients and compartments. J Neurosci. 1990;10:720–733. doi: 10.1523/JNEUROSCI.10-03-00720.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer RE, Maika SD, Richardson JA, Tang JP, Taurog JD. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human beta 2m: an animal model of HLA-B27-associated human disorders. Cell. 1990;63:1099–1112. doi: 10.1016/0092-8674(90)90512-d. [DOI] [PubMed] [Google Scholar]
- Harper P. Huntington's disease. London: Saunders; 1991. [Google Scholar]
- Hedreen JC, Folstein SE. Early loss of neostriatal striosome neurons in Huntington's disease. J Neuropathol Exp Neurol. 1995;54:105–120. doi: 10.1097/00005072-199501000-00013. [DOI] [PubMed] [Google Scholar]
- Herrmann KH, Schmidt S, Kretz A, Haenold R, Krumbein I, Metzler M, Gaser C, Witte OW, Reichenbach JR. Possibilities and limitations for high resolution small animal MRI on a clinical whole-body 3T scanner. MAGMA. 2012;25:233–244. doi: 10.1007/s10334-011-0284-5. [DOI] [PubMed] [Google Scholar]
- Hersch S, Ferrante R. Neuropathology and pathophysiology of Huntington's disease. In: Watts RL, Koller WC, editors. New York: McGraw-Hill; 1997. [Google Scholar]
- Holt DJ, Graybiel AM, Saper CB. Neurochemical architecture of the human striatum. J Comp Neurol. 1997;384:1–25. doi: 10.1002/(sici)1096-9861(19970721)384:1<1::aid-cne1>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Kántor O, Temel Y, Holzmann C, Raber K, Nguyen HP, Cao C, Türkoglu HO, Rutten BP, Visser-Vandewalle V, Steinbusch HW, Blokland A, Korr H, Riess O, von Hörsten S, Schmitz C. Selective striatal neuron loss and alterations in behavior correlate with impaired striatal function in Huntington's disease transgenic rats. Neurobiol Dis. 2006;22:538–547. doi: 10.1016/j.nbd.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Kazantsev A, Preisinger E, Dranovsky A, Goldgaber D, Housman D. Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc Natl Acad Sci U S A. 1999;96:11404–11409. doi: 10.1073/pnas.96.20.11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosinski CM, Cha JH, Young AB, Mangiarini L, Bates G, Schiefer J, Schwarz M. Intranuclear inclusions in subtypes of striatal neurons in Huntington's disease transgenic mice. Neuroreport. 1999;10:3891–3896. doi: 10.1097/00001756-199912160-00031. [DOI] [PubMed] [Google Scholar]
- Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ. Huntington aggregates may not predict neuronal death in Huntington's disease. Ann Neurol. 1999;46:842–849. [PubMed] [Google Scholar]
- Lajoie P, Snapp EL. Formation and toxicity of soluble polyglutamine oligomers in living cells. PLoS One. 2010;5:e15245. doi: 10.1371/journal.pone.0015245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawhorn C, Smith DM, Brown LL. Striosome-matrix pathology and motor deficits in the YAC128 mouse model of Huntington's disease. Neurobiol Dis. 2008;32:471–478. doi: 10.1016/j.nbd.2008.08.006. [DOI] [PubMed] [Google Scholar]
- Li H, Li SH, Yu ZX, Shelbourne P, Li XJ. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington's disease mice. J Neurosci. 2001;21:8473–8481. doi: 10.1523/JNEUROSCI.21-21-08473.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Orozco IJ, Planel E, Wen Y, Bretteville A, Krishnamurthy P, Wang L, Herman M, Figueroa H, Yu WH, Arancio O, Duff K. A transgenic rat that develops Alzheimer's disease-like amyloid pathology, deficits in synaptic plasticity and cognitive impairment. Neurobiol Dis. 2008;31:46–57. doi: 10.1016/j.nbd.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled LB, Chesselet MF. Mouse models of Huntington's disease. Trends Pharmacol Sci. 2002;23:32–39. doi: 10.1016/s0165-6147(00)01884-8. [DOI] [PubMed] [Google Scholar]
- Menalled L, El-Khodor BF, Patry M, Suárez-Fariñas M, Orenstein SJ, Zahasky B, Leahy C, Wheeler V, Yang XW, MacDonald M, Morton AJ, Bates G, Leeds J, Park L, Howland D, Signer E, Tobin A, Brunner D. Systematic behavioral evaluation of Huntington's disease transgenic and knock-in mouse models. Neurobiol Dis. 2009;35:319–336. doi: 10.1016/j.nbd.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J, Arrasate M, Shaby BA, Mitra S, Masliah E, Finkbeiner S. Quantitative relationships between huntingtin levels, polyglutamine length, inclusion body formation, and neuronal death provide novel insight into huntington's disease molecular pathogenesis. J Neurosci. 2010;30:10541–10550. doi: 10.1523/JNEUROSCI.0146-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Sanjuan I, Bates GP. The importance of integrating basic and clinical research toward the development of new therapies for Huntington disease. J Clin Invest. 2011;121:476–483. doi: 10.1172/JCI45364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HP, Kobbe P, Rahne H, Wörpel T, Jäger B, Stephan M, Pabst R, Holzmann C, Riess O, Korr H, Kántor O, Petrasch-Parwez E, Wetzel R, Osmand A, von Hörsten S. Behavioral abnormalities precede neuropathological markers in rats transgenic for Huntington's disease. Hum Mol Genet. 2006;15:3177–3194. doi: 10.1093/hmg/ddl394. [DOI] [PubMed] [Google Scholar]
- Osmand AP, Berthelier V, Wetzel R. Imaging polyglutamine deposits in brain tissue. Methods Enzymol. 2006;412:106–122. doi: 10.1016/S0076-6879(06)12008-X. [DOI] [PubMed] [Google Scholar]
- Pavese N, Andrews TC, Brooks DJ, Ho AK, Rosser AE, Barker RA, Robbins TW, Sahakian BJ, Dunnett SB, Piccini P. Progressive striatal and cortical dopamine receptor dysfunction in Huntington's disease: a PET study. Brain. 2003;126:1127–1135. doi: 10.1093/brain/awg119. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The rat brain in stereotaxic coordinate. Ed 6. San Diego: Academic; 2006. [Google Scholar]
- Petrasch-Parwez E, Nguyen HP, Löbbecke-Schumacher M, Habbes HW, Wieczorek S, Riess O, Andres KH, Dermietzel R, Von Hörsten S. Cellular and subcellular localization of Huntingtin [corrected] aggregates in the brain of a rat transgenic for Huntington disease. J Comp Neurol. 2007;501:716–730. doi: 10.1002/cne.21272. [DOI] [PubMed] [Google Scholar]
- Pouladi MA, Xie Y, Skotte NH, Ehrnhoefer DE, Graham RK, Kim JE, Bissada N, Yang XW, Paganetti P, Friedlander RM, Leavitt BR, Hayden MR. Full-length huntingtin levels modulate body weight by influencing insulin-like growth factor 1 expression. Hum Mol Genet. 2010;19:1528–1538. doi: 10.1093/hmg/ddq026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouladi MA, Stanek LM, Xie Y, Franciosi S, Southwell AL, Deng Y, Butland S, Zhang W, Cheng SH, Shihabuddin LS, Hayden MR. Marked differences in neurochemistry and aggregates despite similar behavioural and neuropathological features of Huntington disease in the full-length BACHD and YAC128 mice. Hum Mol Genet. 2012;21:2219–2232. doi: 10.1093/hmg/dds037. [DOI] [PubMed] [Google Scholar]
- Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. 1988;85:5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguiz RM, Wetsel WC. In: Assessments of cognitive deficits in mutant mice, Chap 5. Levin ED, Buccafusco JJ, editors. Boca Raton, FL: CRC; 2006. [PubMed] [Google Scholar]
- Seto-Ohshima A, Emson PC, Lawson E, Mountjoy CQ, Carrasco LH. Loss of matrix calcium-binding protein-containing neurons in Huntington's disease. Lancet. 1988;1:1252–1255. doi: 10.1016/s0140-6736(88)92073-9. [DOI] [PubMed] [Google Scholar]
- Stine OC, Pleasant N, Franz ML, Abbott MH, Folstein SE, Ross CA. Correlation between the onset age of Huntington's disease and length of the trinucleotide repeat in IT-15. Hum Mol Genet. 1993;2:1547–1549. doi: 10.1093/hmg/2.10.1547. [DOI] [PubMed] [Google Scholar]
- Tecott LH, Nestler EJ. Neurobehavioral assessment in the information age. Nat Neurosci. 2004;7:462–466. doi: 10.1038/nn1225. [DOI] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Toutain PL, Ferran A, Bousquet-Melou A. Species differences in pharmacokinetics and pharmacodynamics. Handb Exp Pharmacol. 2010:19–48. doi: 10.1007/978-3-642-10324-7_2. [DOI] [PubMed] [Google Scholar]
- van Oostrom JC, Maguire RP, Verschuuren-Bemelmans CC, Veenma-van der Duin L, Pruim J, Roos RA, Leenders KL. Striatal dopamine D2 receptors, metabolism, and volume in preclinical Huntington disease. Neurology. 2005;65:941–943. doi: 10.1212/01.wnl.0000176071.08694.cc. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Gibson WT, Pearson J, Murphy Z, Lu G, Leavitt BR, Hayden MR. Body weight is modulated by levels of full-length huntingtin. Hum Mol Genet. 2006;15:1513–1523. doi: 10.1093/hmg/ddl072. [DOI] [PubMed] [Google Scholar]
- von Hörsten S, Schmitt I, Nguyen HP, Holzmann C, Schmidt T, Walther T, Bader M, Pabst R, Kobbe P, Krotova J, Stiller D, Kask A, Vaarmann A, Rothke-Hartlieb S, Schulz JB, Grasshoff U, Bauer I, Vieira-Saecker AM, Paul M, Jones L, et al. Transgenic rat model of Huntington's disease. Hum Mol Genet. 2003;12:617–624. doi: 10.1093/hmg/ddg075. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Wang CE, Tydlacka S, Orr AL, Yang SH, Graham RK, Hayden MR, Li S, Chan AW, Li XJ. Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington's disease. Hum Mol Genet. 2008;17:2738–2751. doi: 10.1093/hmg/ddn175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler C, Gil JM, Araújo IM, Riess O, Skripuletz T, von Hörsten S, Petersén A. Normal sensitivity to excitotoxicity in a transgenic Huntington's disease rat. Brain Res Bull. 2006;69:306–310. doi: 10.1016/j.brainresbull.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Yamada M, Iwatsubo T, Mizuno Y, Mochizuki H. Overexpression of alpha-synuclein in rat substantia nigra results in loss of dopaminergic neurons, phosphorylation of alpha-synuclein and activation of caspase-9: resemblance to pathogenetic changes in Parkinson's disease. J Neurochem. 2004;91:451–461. doi: 10.1111/j.1471-4159.2004.02728.x. [DOI] [PubMed] [Google Scholar]
- Yang XW, Model P, Heintz N. Homologous recombination based modification in Escherichia coli and germline transmission in transgenic mice of a bacterial artificial chromosome. Nat Biotechnol. 1997;15:859–865. doi: 10.1038/nbt0997-859. [DOI] [PubMed] [Google Scholar]