

Abstract
In addition to triggering the birthing process and milk release, the hypothalamic neuropeptide oxytocin (OXT) plays an important role in the regulation of complex social cognition and behavior. Previous work has shown that OXT can regulate hippocampal synaptic plasticity and improve hippocampus-dependent cognitive functions in the female mice, but the underlying mechanisms remain largely unclear. Here, we demonstrate that OXT promotes the maintenance of long-term potentiation (LTP) induced by one train of tetanic stimulation (TS) in the CA1 region of hippocampal slices from both nulliparous female and male rats through a previously unknown mechanism involving OXT receptor (OXTR)-dependent and epidermal growth factor receptor (EGFR)-mediated local translation of an atypical protein kinase C isoform, protein kinase Mζ (PKMζ), in dendrites. Using pharmacological and biochemical approaches, we show that both the conventional OXTR-associated signaling pathway (Gq/11-coupled phospholipase C) and the transactivated EGFR downstream signaling pathways (phosphatidylinositol 3 kinase and extracellular signal-regulated kinase 1/2) are involved in the regulation of OXT. In addition, OXT stimulates local dendritic PKMζ mRNA translation via activation of a mammalian target of rapamycin-regulated mechanism. Furthermore, blockade of OXTR results in a modest decrease in the ability to maintain late-phase LTP induced by three trains of TS. These results reveal a novel OXTR-to-EGFR communication to regulate the new synthesis of PKMζ, which functions to promote the maintenance of LTP at hippocampal CA1 synapses.
Introduction
The neuropeptide oxytocin (OXT) is primarily synthesized by magnocellular neurons in the paraventricular and supraoptic nuclei of the hypothalamus (Gainer and Wray, 1992; Lee et al., 2009). In addition to neurosecretory projections to the posterior pituitary, OXT also travels along the axonal projections from parvocellular neurons of the hypothalamus to discrete brain regions, including the hippocampus, amygdala, striatum, suprachiasmatic nucleus, bed nucleus of stria terminalis (BNST), and brainstem, where it acts as a neuromodulator or neurotransmitter and thereby influences neurotransmission in these regions (Buijs and Swaab, 1979; Sofroniew, 1983; Young and Gainer, 2003; Meyer-Lindenberg et al., 2011). Since the first discovery that central OXT can induce maternal behavior in virgin rats (Pedersen and Prange, 1979), growing evidence has revealed a significant contribution of brain OXT in the regulation of various social behaviors, including social recognition, aggression, and affiliation, as well as nonsocial behaviors, such as learning and memory, anxiety, depression, and stress (Neumann, 2008; Neumann and Landgraf, 2008; Lee et al., 2009; Meyer-Lindenberg et al., 2011). Furthermore, the biological actions of OXT are primarily mediated by the activation of OXT receptors (OXTR), which belong to the class I family of G-protein-coupled receptors (GPCR) and are coupled to phospholipase C (PLC) through Gαq/11 (Gimpl and Fahrenholz, 2001). OXTR is widely expressed throughout the brain and is especially prominent in the hippocampus, amygdala, olfactory bulb, BNST, nucleus accumbens, and ventromedial hypothalamic nucleus (Insel et al., 1991; Veinante and Freund-Mercier, 1997).
The hippocampus is one of the most susceptible brain areas to the effects of OXT. Bilateral injections of OXT in the dorsal hippocampus have been shown to significantly attenuate the neuroendocrine and behavioral responses to stress (Cohen et al., 2010). More interestingly, prior evidence has indicated that OXT can enhance long-term potentiation (LTP) at Schaffer collateral–CA1 synapses and improve long-lasting spatial memory function during motherhood (Tomizawa et al., 2003); however, the molecular details of how it does so remain unclear. Moreover, a recent study has revealed that OXT could also act as a neurotrophic agent to stimulate cell proliferation and adult neurogenesis in the hippocampus even under conditions of stress or elevated glucocorticoids (Leuner et al., 2012).
The autonomously active protein kinase C isoform, protein kinase Mζ (PKMζ), has been found to be both necessary and sufficient for the maintenance of LTP and memory storage in the hippocampus (Sacktor et al., 1993; Ling et al., 2002; Pastalkova et al., 2006; Serrano et al., 2008; Hardt et al., 2010). Therefore, PKMζ could potentially mediate the enhancement effect of OXT on LTP. In this study, we provide novel evidence that OXT promotes the maintenance of LTP in hippocampal CA1 region via a PKMζ-dependent mechanism. De novo PKMζ protein synthesis at the synapses is required for OXT-induced enhancement of LTP. We further demonstrate that the OXT-regulated PKMζ protein production relies on a PLC-dependent transactivation of epidermal growth factor receptor (EGFR), which in turn activates its downstream phosphatidylinositol 3 kinase (PI3K) and extracellular signal-regulated kinase 1/2 (ERK1/2) signaling, thereby stimulating dendritic translation of PKMζ.
Materials and Methods
Hippocampal slice preparations and electrophysiology.
All experimental procedures were conducted in adherence to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 80–23, revised 1996) and were approved by the Institutional Animal Care and Use Committee of National Cheng Kung University. Hippocampal slices were prepared from 28- to 35-d-old young female or male Sprague Dawley rats as described previously (Lin et al., 2006). In brief, rats were killed by decapitation under deep isoflurane anesthesia, and hippocampal slices (400 μm) were prepared using Leica VT1200S vibrating blade microtome. The slices were placed in a storage chamber of artificial CSF (aCSF) oxygenated with 95% O2/5% CO2 and kept at room temperature (23−25°C) for at least 1 h before recording. The composition of the aCSF solution was as follows (in mm): 117 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, 1.2 NaH2PO4, and 11 glucose at pH 7.3–7.4.
For recording, one slice was transferred to a submerged recording chamber continually perfused with oxygenated aCSF at a flow rate of 2–3 ml/min at 32.0 ± 0.5°C. The extracellular field potential recordings were made using an Axoclamp-2B amplifier (Molecular Devices). Microelectrodes were pulled from microfiber 1.0 mm capillary tubing on a Brown-Flaming electrode puller (Sutter Instruments). The responses were low-pass-filtered at 2 kHz, digitally sampled at 10 kHz, and analyzed using pCLAMP software (Version 8.0; Molecular Devices). Postsynaptic responses were evoked in CA1 stratum radiatum by stimulation of Schaffer collateral/commissural afferents at 0.033 Hz with a bipolar tungsten stimulating electrode. The stimulation strength was set to elicit response for which the amplitude was 30–40% of the maximum spike-free response. Field EPSPs (fEPSPs) were recorded with a glass pipette filled with 1 m NaCl (2–3 mΩ resistance) and the fEPSP slope was measured from ∼20–70% of the rising phase using a least-squares regression. LTP was induced by tetanic stimulation (TS), at the test pulse intensity, consisting of either one 1 s train of TS at 100 Hz or three 1 s trains of TS at 100 Hz separated by an intertrain interval of 5 min. The magnitudes of LTP were averaged from the responses recorded during the last 10 min of the recording and normalized to 10 min of baseline before LTP induction. In some experiments, LTP was induced by one train of TS in the presence of GABAA receptor antagonist picrotoxin (PTX, 100 μm). To reduce epileptiform activity during blockade of GABAergic inhibition, a surgical cut was made at the border between the CA1 and CA3 areas.
Preparation of synaptoneurosomes.
Synaptoneurosomes were prepared by using a procedure adapted from the studies by Huttner et al. (1983) and Rao and Steward (1991). Briefly, hippocampal CA1 region was minced in ice-cold homogenization buffer (0.32 m sucrose, 0.5 mm EGTA, and 10 mm Tris; pH 7.4 with KOH) and homogenized using a glass tissue grinder with a Teflon pestle. The homogenate was centrifuged at 1000 × g for 10 min to remove nuclei and cell debris, and the supernatant was transferred to a new tube and centrifuged at 10,000 × g for 30 min to generate a crude synaptoneurosomal fraction. The crude synaptoneurosomal fraction was washed once in HBS buffer [in mm: 10 HEPES-KOH, 142 NaCl, 2.4 KCl, 1 MgCl2, 5 glucose, 0.1 EGTA, and 0.3 phenylmethylsulfonyl fluoride (PMSF); pH 7.5] and centrifuged once more at 13,000 × g for 15 min. It was then lysed in 10 volume of ice-cold H2O containing 0.3 mm PMSF for 30 min, buffered with 1 m HEPES-KOH, pH 7.4, and centrifuged at 25,000 × g for 30 min to generate the synaptoneurosomal fraction. The synaptoneurosomal pellet was gently resuspended in the same buffer at a protein concentration of 2 mg/ml. All procedures were performed at 4°C.
Establishment and treatment of hippocampal neuronal cultures.
Primary neuronal cultures were prepared from the hippocampi of embryonic day 18 Sprague Dawley rats as described previously (Balkowiec and Katz, 2002; Huang et al., 2010). Briefly, embryos were removed and their hippocampi were dissected out in ice-cold HBSS (Life Technologies) and freed from meninges. Tissues were enzymatically digested with 0.25% trypsin (Sigma-Aldrich) for 15 min at 37°C. Cells were disaggregated by trituration and plated on poly-l-lysine-coated Petri dishes in Neurobasal-A medium containing B27 serum-free supplement (Invitrogen), 0.5 mm l-glutamine, and antibiotics (50 U/ml penicillin and 50 μg/ml streptomycin). Cultures were incubated at 37°C, under 5% CO2/95% air and 90% relative humidity. Half of the growth medium was replaced every 3 d. Very few glial cells were observed in these cultures (Huang et al., 2010).
Hippocampal neurons were maintained for 18–19 d in vitro (DIV) before treatment with OXT. Cultured hippocampal neurons were starved from B27 for 6 h before performing any treatment. Either 1-[4-[(1-acetyl-4-piperidinyl)oxy]-2-methoxybenzoyl]-4-(2-oxo-2H-3,1-benzoxazin-1(4H)-yl)piperidine (L-371257), N-(3-chlorophenyl)-6,7-dimethoxy-4-quinazolinanine hydrochloride (AG1478), 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002), 1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene (U0126), or rapamycin was applied 30 min before the addition of OXT (1 μm) for 30 min.
Western blotting.
For each experimental group, homogenates from at least two slices were pooled. The microdissected CA1 subregions were homogenized in ice-cold Tris-HCl buffer solution (TBS; pH 7.4) containing a mixture of protein phosphatase and proteinase inhibitors (50 mm Tris-HCl, 100 mm NaCl, 15 mm sodium pyrophosphate, 50 mm sodium fluoride, 1 mm sodium orthovanadate, 5 mm EGTA, 5 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 1 μm microcystin-LR, 1 μm okadaic acid, 0.5% Triton X-100, 2 mm benzamidine, 60 μg/ml aprotinin, and 60 μg/ml leupeptin) to avoid dephosphorylation and degradation of proteins, and ground with a pellet pestle (Kontes Glassware). Samples were sonicated and spun down at 15,000 × g at 4°C for 10 min. The supernatant was then assayed for total protein concentration using Bio-Rad Bradford Protein Assay Kit. Each sample from tissue homogenate or synaptoneurosome was separated using 8% or 10% SDS-PAGE gel. Following the transfer on nitrocellulose or polyvinylidene fluoride membranes, blots were blocked in buffer solution containing 5% milk and 0.1% Tween-20 in PBS (in mm: 124 NaCl, 4 KCl, 10 Na2HPO4 and 10 KH2PO4; pH 7.2) for 1 h and then blotted for 2 h at room temperature with the antibodies that recognize PKMζ (1:1000; Santa Cruz Biotechnology), pEGFR (Tyr1068; 1:1000; Cell Signaling Technology), pAkt (Ser473; 1:1000; Cell Signaling Technology), pAkt (Thr308; 1:1000; Cell Signaling Technology), pERK1/2 (Thr202/Tyr204; 1:1000; Cell Signaling Technology), or β-actin (1:2000, Sigma-Aldrich). It was then probed with HRP-conjugated secondary antibody for 1 h and developed using the enhanced chemiluminescence immunoblotting detection system (GE Healthcare), according to manufacturer's instructions. The immunoblots using phosphorylation site-specific antibodies were subsequently stripped and reprobed with the following antibodies: anti-Akt antibody (1:1000) or anti-ERK1/2 antibody (1:1000) that were purchased from Cell Signaling Technology, and anti-EGFR antibody (1:1000) that was obtained from Cell Signaling Technology. Immunoblots were analyzed by densitometry using Bio-profil BioLight PC software (Vulber Lourmat). Only film exposures that were not saturated were used for quantification analysis. The expression level of PKMζ was evaluated relative to that for β-actin. Background correction values were subtracted from each lane to minimize the variability across membranes.
Drugs.
All drugs were applied by manually switching the superfusate. Drugs were diluted from stock solutions just before application. L-371257, 1-[6-[[(17β)-3-methoxyestra-1, 3, 5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2, 5-dione (U73122), 1-[6-[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-2,5-pyrrolidinedione (U73343), cycloheximide, anisomycin, actinomycin-D, AG1478, α-cyano-(4-hydroxy)dihydrocinnamonitrile (AG43), LY294002, U0126, (9S,10R,12R)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid methyl ester (K252a), rapamycin, ascomycin, and N-[(2R)-2-(hydroxamidocarbonylmethyl)-4-methylpentanoyl]-l-tryptophan methylamide (GM6001) were dissolved in dimethylsulfoxide (DMSO) stock solutions and then diluted to their final concentration in aCSF just before application. The final concentration of DMSO in the perfusion medium did not exceed 0.1%, which alone had no effect on basal synaptic transmission (Huang and Hsu, 2006). OXT, Myr-Ser-Ile-Tyr-Arg-Arg-Gly-Ala-Arg-Arg-Trp-Arg-Lys-Leu (ZIP), and Myr-Arg-Leu-Tyr-Arg-Lys-Ile-Trp-Arg-Ser-Ala-Gly-Arg (scrambled ZIP; Scr-ZIP) were dissolved in distilled water. The respective drug concentrations were selected on the basis of previously published and our pilot studies (Tomizawa et al., 2003; Chiu et al., 2005; Serrano et al., 2005; Huang and Hsu, 2006). U73343, ascomycin, K252a, PTX, and heparin were purchased from Sigma-Aldrich, OXT, L-371257, U73122, ZIP, Scr-ZIP, AG1478, rapamycin, cycloheximide, anisomycin, actinomycin D, U0126, and LY294002 were obtained from Tocris Cookson and AG43 and GM6001 were purchased from Calbiochem. The functional anti-EGF antibody (catalog #MAB3214) was purchased from R&D Systems.
Statistical analysis.
The data for each experiment were normalized relative to baseline, and are presented as mean ± SEM. Number of animals used is indicated by n. For LTP experiments, statistical analysis was performed using the non-parametric Mann–Whitney U test. The significance of the difference between the groups was calculated by one-way ANOVA with Bonferroni's post hoc analyses or Student's t test where appropriate. Probability values (P) of <0.05 were considered to represent significant differences.
Results
OXT enhances the maintenance of LTP in the CA1 region of the hippocampus
We first confirmed the ability of OXT to promote LTP at Schaffer-collateral CA1 synapses in hippocampal slices from nulliparous female rats (Tomizawa et al., 2003). LTP was induced by one 1 s train of TS at 100 Hz, a protocol that normally produced only an early, short-term phase of LTP. In dorsal hippocampal slices without OXT application, one train of TS produced only a short-term synaptic potentiation that was stable for ∼1 h (50–60 min after TS: 124.4 ± 4.3% of baseline, n = 12; p < 0.05; Mann–Whitney U test), but gradually declined to baseline by 2 h (110–120 min after TS: 106.7 ± 3.9% of baseline, n = 12; p > 0.05; Mann–Whitney U test). When OXT (1 μm) was applied concomitantly with TS, the persistence of LTP was significantly enhanced (110–120 min after TS: 122.8 ± 3.5% of baseline, n = 16; p < 0.05; Mann–Whitney U test; Fig. 1A,D). Moreover, a similar result was also observed in slices prepared from the ventral portion of the hippocampus. In the presence of OXT, one train of TS induced a late-phase LTP, whereas in the absence of OXT, synaptic potentiation was returned to baseline by 2 h (110–120 min after TS: control, 114.3 ± 3.3% of baseline, n = 8; OXT, 135.6 ± 4.5% of baseline, n = 6; p < 0.05; unpaired Student's t test; Fig. 1B,D). To test for the presence of sexually dimorphic effects of OXT on LTP, we compared the magnitude of LTP in dorsal hippocampal slices from male rats in the presence or absence of OXT. We found that, similar to those observed in slices from female rats, OXT significantly enhanced the maintenance of LTP in slices from male rats (110–120 min after TS: control, 101.3 ± 3.2% of baseline, n = 4; OXT, 121.4 ± 5.3% of baseline, n = 5; p < 0.05; unpaired Student's t test; Fig. 1C,D). There was no significant difference in the effect of OXT on LTP between male and female rats (p > 0.05; unpaired Student's t test; Fig. 1D). Furthermore, application of OXT alone (1 μm) for 30 min had no significant effect on basal synaptic transmission in slices from female and male rats (data not shown). These results support the notion that OXT can enhance the maintenance of LTP in hippocampal CA1 region of both sexes.
Figure 1.

The effect of OXT on the induction of LTP in the CA1 region of rat hippocampal slices. A, Summary of experiments showing pairing one train of 1 s TS at 100 Hz with OXT (1 μm) application induced LTP of fEPSPs in slices from female rat dorsal hippocampus. Pairing TS with vehicle induced only a transient increase in synaptic strength. B, Summary of experiments showing the effect of OXT treatment on the induction of LTP by one train of TS in slices from female rat ventral hippocampus. C, Summary of experiments showing the effect of OXT treatment on the induction of LTP by one train of TS in slices from male rat dorsal hippocampus. D, Summary histogram depicting the effect of oxytocin on the induction of LTP by one train of TS in slices from female or male hippocampus. The magnitudes of LTP were measured 110–120 min after TS. E, Summary of experiments showing the effect of OXT treatment on the induction of LTP by one train of TS in the presence of PTX (100 μm) in slices from female rat dorsal hippocampus. Representative traces of fEPSPs were taken at the time indicated by number. Dash lines show level of baseline. The total number of animals examined is indicated by n in parenthesis. Data are represented as mean ± SEM. *p < 0.05 compared with vehicle-treated group.
OXTR agonists have been shown to enhance inhibitory synaptic transmission in hippocampal CA1 pyramidal neurons by activating interneurons in stratum pyramidale (Zaninetti and Raggenbass, 2000). Maternal OXT has also been reported to trigger a transient excitatory-to-inhibitory switch of GABAergic signaling in the fetal brain during delivery (Tyzio et al., 2006). Given that GABA-mediated inhibition is known to be an important regulatory factor during LTP induction in hippocampal CA1 region (Wigström and Gustafsson, 1983), one possibility is that OXT promotes LTP by altering the strength of GABAergic inhibition to CA1 pyramidal neurons. To test this possibility, we examined the effect of OXT on LTP in the presence of GABAA receptor antagonist PTX (100 μm). We found that the enhancement of LTP by OXT was not significantly affected by PTX coapplication (110–120 min after TS: control, 103.2 ± 5.7% of baseline, n = 5; OXT, 126.4 ± 5.4% of baseline, n = 5; p < 0.05; unpaired Student's t test; Fig. 1E). These results suggest that the observed OXT-induced enhancement of hippocampal CA1 LTP is not caused by changes in GABAA receptor function.
The role of OXTR-mediated PLC activation on OXT-induced enhancement of LTP
The action of OXT is thought to be primarily mediated by its binding to OXTR, which activates the predominant downstream effector PLC mainly through the Gq/11 (Gimpl and Fahrenholz, 2001). We therefore explored the role of OXTR-mediated PLC activation in OXT-induced enhancement of LTP. To this end, a selective OXTR antagonist L-371257 and a broad spectrum PLC blocker U73122 were used. As shown in Figure 2, A and C, L-371257 dose-dependently prevented OXT-induced enhancement of LTP [110–120 min after TS: vehicle + OXT, 124.5 ± 5.3% of baseline, n = 8; L-371257 (1 μm) + OXT, 113.6 ± 4.5% of baseline, n = 4; L-371257 (3 μm) + OXT, 101.6 ± 6.7% of baseline, n = 8; p < 0.05; unpaired Student's t test]. In addition, pretreatment of the hippocampal slices with U73122 (10 μm), but not its inactive structural analog U73343 (10 μm), completely blocked OXT-induced enhancement of LTP (110–120 min after TS: U73122, 104.5 ± 3.4% of baseline, n = 5; U73343, 128.5 ± 4.3% of baseline, n = 5; Fig. 2B,C). These results suggest that the OXTR-mediated PLC activation is required for OXT-induced enhancement of LTP.
Figure 2.
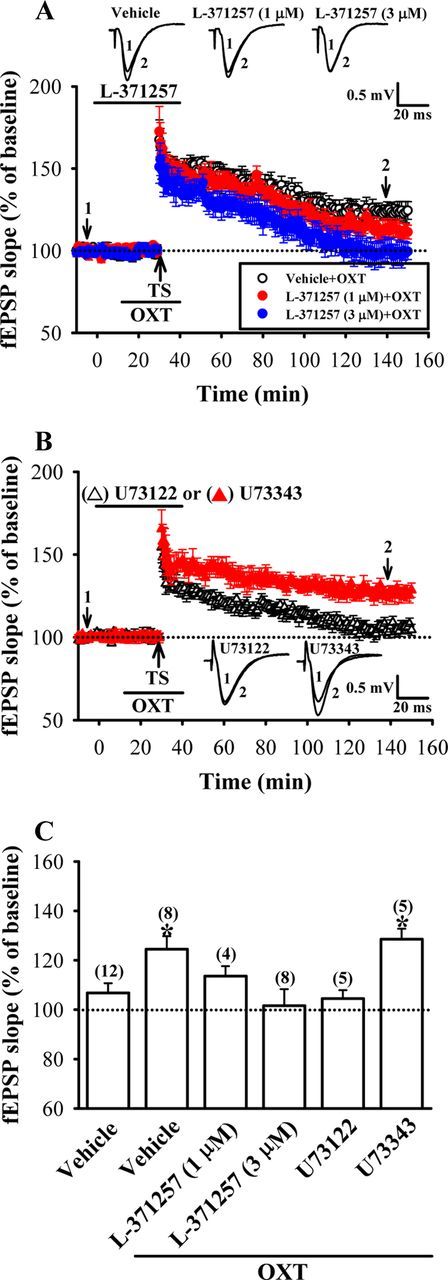
OXTR-coupled PLC signaling pathway mediates the enhancement of LTP by OXT. A, Summary of experiments showing pretreatment of hippocampal slices with OXTR antagonist L-371257 (1 or 3 μm) dose-dependently blocked OXT (1 μm)-induced enhancement of LTP. B, Summary of experiments showing the enhancement of OXT on LTP was blocked by pretreatment of the hippocampal slices with the PLC inhibitor U73122 (10 μm) but not by its inactive analog U73343 (10 μm). C, Summary histogram depicting the effect of L-371257, U73122, and U73343 on OXT-induced enhancement of LTP in slices from female dorsal hippocampus. The magnitudes of LTP were measured 110–120 min after TS. Representative traces of fEPSPs were taken at the time indicated by number. Dash lines show level of baseline. The total number of animals examined is indicated by n in parenthesis. Data are represented as mean ± SEM. *p < 0.05 compared with vehicle-treated group.
New protein synthesis is required for the enhancement of LTP by OXT
At hippocampal Schaffer collateral-CA1 synapses, the transition from early- to late-phase LTP requires gene expression and de novo protein synthesis (Stanton and Sarvey, 1984; Deadwyler et al., 1987; Nguyen et al., 1994). To investigate whether the enhancement of LTP by OXT depends upon protein and RNA synthesis, we tested the effects of two structurally different translational inhibitors, cycloheximide and anisomycin, and transcriptional inhibitor, actinomycin D, on OXT-induced enhancement of LTP. As shown in Figure 3, A and D, pretreatment of hippocampal slices with either cycloheximide (50 μm) or anisomycin (20 μm), concentrations that have been shown to be effective in inhibiting >80% of protein synthesis (Frey et al., 1988), completely blocked OXT-induced enhancement of LTP (110–120 min after TS: cycloheximide, 108.9 ± 5.3% of baseline, n = 8; anisomycin, 103.5 ± 7.1% of baseline, n = 5). In contrast to the translational inhibitors, actinomycin D (25 μm), a concentration that has been shown to block transcription by >70% in hippocampal slices (Nguyen et al., 1994), pretreatment had no effect on the enhancement of LTP by OXT (122.6 ± 6.6% of baseline, n = 8; p > 0.05; unpaired Student's t test; Fig. 3B,D). In addition, none of the cycloheximide, anisomycin, or actinomycin D treatment alone had an effect per se on the maintenance of synaptic transmission following one 1 s train of TS at 100 Hz (data not shown). The synaptic potentiation returned to baseline within 2 h after TS. These results suggest that OXT-induced enhancement of LTP is dependent on protein synthesis, but not gene transcription.
Figure 3.

LTP induced by pairing one train of TS with OXT application requires de novo protein synthesis. A, Summary of experiments showing pretreatment of hippocampal slices with protein synthesis inhibitors, cycloheximide (Chx, 50 μm) or anisomycin (Ani, 20 μm) blocked OXT (1 μm)-induced enhancement of LTP. B, Summary of experiments showing the effect of transcription inhibitor actinomycin D (Act-D, 25 μm) on OXT-induced enhancement of LTP in slices from female dorsal hippocampus. C, Summary of experiments showing the enhancement of OXT on LTP was blocked by pretreatment of the hippocampal slices with rapamycin (Rap, 200 nm) but not by its inactive analog ascomycin (Asc, 200 nm). D, Summary histogram depicting the effect of cycloheximide, anisomycin, actinomycin D, rapamycin, and ascomycin on OXT-induced enhancement of LTP in slices from female dorsal hippocampus. The magnitudes of LTP were measured 110–120 min after TS. Representative traces of fEPSPs were taken at the time indicated by number. Dash lines show level of baseline. The total number of animals examined is indicated by n in parenthesis. Data are represented as mean ± SEM. *p < 0.05 compared with vehicle-treated group.
The protein kinase mammalian target of rapamycin (mTOR) regulates multiple components of the translational machinery in mammalian cells (Brown and Schreiber, 1996), and mTOR activity has been shown to be required for the induction of late-phase LTP in the hippocampus (Tang et al., 2002; Tsokas et al., 2005; Stoica et al., 2011). We therefore investigated whether the enhancement of LTP by OXT may occur through activation of mTOR-mediated regulation of protein synthesis, so the mTOR inhibitor rapamycin was used. As expected, pretreatment of the hippocampal slices with rapamycin (200 nm), a concentration which has previously been shown to effectively block the expression of late-phase LTP and BDNF-induced synaptic potentiation in hippocampal CA1 region (Tang et al., 2002), but not its inactive analog ascomycin (200 nm), completely blocked OXT-induced enhancement of LTP (110–120 min after TS: rapamycin, 104.5 ± 5.3% of baseline, n = 6; ascomycin, 130.7 ± 7.2% of baseline, n = 5; Fig. 3C,D). Neither rapamycin nor ascomycin treatment alone affected the effect of TS on synaptic transmission (data not shown). Thus, mTOR-mediated regulation of protein synthesis is required for the enhancement of LTP by OXT.
Local dendritic PKMζ synthesis is crucial for OXT-induced enhancement of LTP
What kind of protein biosynthesis could be responsible for mediating the enhancement of LTP by OXT? Because translation of the dendritically localized PKMζ mRNA is importantly involved in the maintenance of late-phase LTP (Muslimov et al., 2004; Kelly et al., 2007), we therefore analyzed the contribution of PKMζ in OXT-induced enhancement of LTP by using a selective, cell-permeable PKMζ inhibitor ZIP (Laudanna et al., 1998; Ling et al., 2002). We found that pretreatment of the hippocampal slices with ZIP at 2 μm, a concentration sufficient to inhibit PKMζ-mediated synaptic potentiation (Serrano et al., 2005), but not its inactive scrambled peptide (Scr-ZIP, 2 μm), completely blocked OXT-induced enhancement of LTP (110–120 min after TS: ZIP, 104.2 ± 5.5% of baseline, n = 5; Scr-ZIP, 128.6 ± 6.2% of baseline, n = 5; Fig. 4A). Neither ZIP nor Scr-ZIP treatment alone affected the effect of TS on synaptic transmission (data not shown). If PKMζ is required for the maintenance of OXT-induced LTP, the established LTP should be reversed when ZIP is applied during its maintenance phase. Consistent with this idea, when ZIP (2 μm) was applied 80 min after conjoint OXT and one train of TS, synaptic potentiation returned toward baseline levels (110–120 min after TS: 104.6 ± 4.7% of baseline, n = 5; Fig. 4B). Thus, the persistent activity of PKMζ maintains OXT-induced enhancement of LTP.
Figure 4.
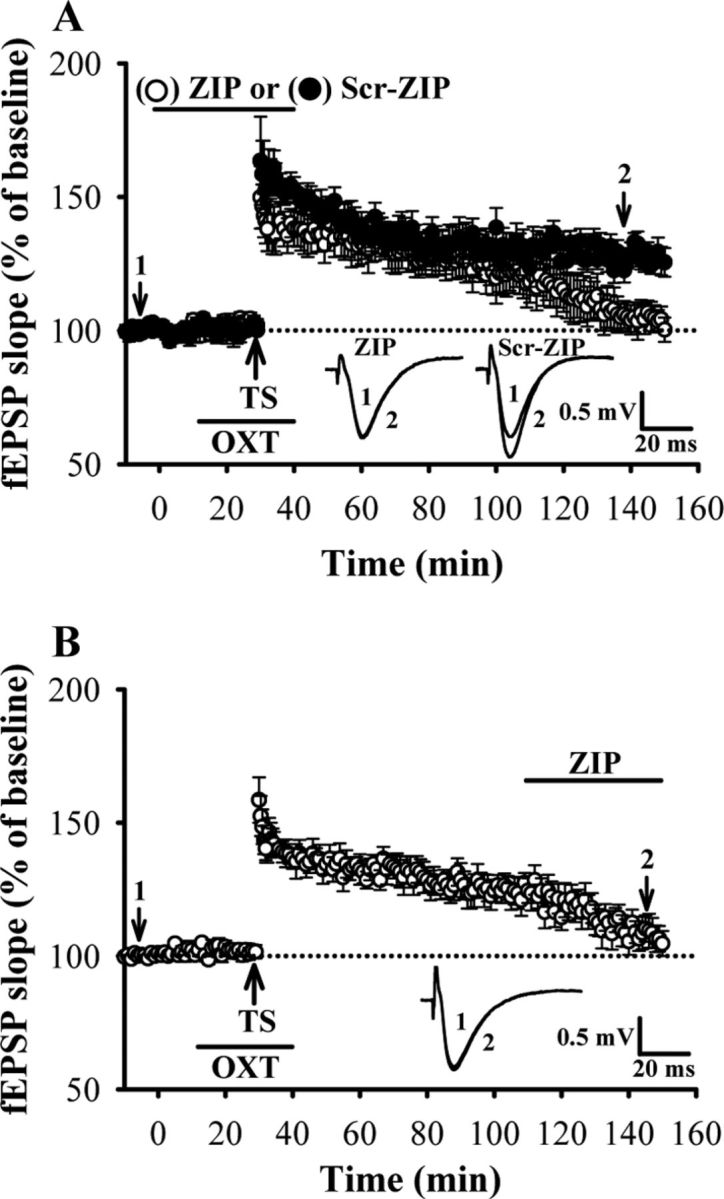
PKMζ is critical for OXT-induced enhancement of LTP. A, Summary of experiments showing the enhancement of OXT on LTP was blocked by pretreatment of the hippocampal slices with the PKMξ inhibitor ZIP (2 μm) but not its inactive analog scrambled ZIP (Scr-ZIP, 2 μm). B, Summary of experiments showing ZIP (2 μm) reversed synaptic potentiation when applied 80 min after conjoint OXT and one train of TS in slices from female dorsal hippocampus. Representative traces of fEPSPs were taken at the time indicated by number. Dash lines show level of baseline. Data are represented as mean ± SEM.
To further confirm the role of PKMζ in the enhancement of LTP by OXT, in another set of experiments, we examined the effect of OXT on PKMζ expression using Western blot analysis. Incubation of hippocampal slices with OXT (1 μm) for 10–60 min resulted in a time-dependent increase in levels of PKMζ protein in the whole tissue lysates of hippocampal CA1 region (Fig. 5A). Because application of OXT (1 μm) for 30 min consistently increased PKMζ expression, we chose this treatment protocol for all subsequent experiments designed to identify the mechanisms underlying this event. As shown in Figure 5B, application of one train of TS in the presence of OXT (1 μm) significantly increased PKMζ levels. A significant increase in PKMζ levels was also seen in slices stimulated with KCl (50 mm) for 3 min or three trains of TS (Fig. 5B). In addition, OXT-induced increases in PKMζ expression were completely blocked by cycloheximide (50 μm) and anisomycin (20 μm), respectively, but not by actinomycin D (25 μm) (Fig. 5C), thus confirming that OXT stimulates PKMζ expression through a translation-dependent mechanism. None of the actinomycin D, cycloheximide, or anisomycin treatments alone had an effect on basal levels of PKMζ (Fig. 5C).
Figure 5.

OXT stimulates PKMξ synthesis in hippocampal CA1 region. A, Representative immunoblot and summary histogram showing OXT (1 μm) time-dependently induces increases in PKMξ expression in the whole tissue lysates of hippocampal CA1 region. B, Representative immunoblot and summary histogram showing the effect of vehicle, OXT (1 μm), one train of TS, KCl (50 mm), and three trains of TS treatment on the expression of PKMξ in the whole tissue lysates of hippocampal CA1 region. C, Representative immunoblot and summary histogram showing OXT (1 μm)-induced increases in PKMξ expression were blocked by pretreatment with cycloheximide (Chx, 50 μm) or anisomycin (Ani, 20 μm) but not by actinomycin D (Act-D, 25 μm). D, Representative immunoblot and summary histogram showing pretreatment of the synaptoneurosomes with rapamycin (Rap, 200 nm) but not ascomycin (Asc, 200 nm) blocked OXT (1 μm)-induced increases in PKMξ synthesis. The total number of experiments is indicated by n in parenthesis. Data are represented as mean ± SEM. *p < 0.05 compared with vehicle-treated group.
Although there is evidence that PKMζ mRNA can be translated in hippocampal CA1 pyramidal cell dendrites (Muslimov et al., 2004; Kelly et al., 2007), the major site of protein synthesis is the cell body. To specifically explore the presence and the possible synthesis of PKMζ protein from the synaptic fractions, we prepared synaptoneurosome, a biochemical preparation that enriches for synaptic fractions (Hollingsworth et al., 1985), from microdissected CA1 subfield samples. We first validated whether PKMζ mRNA located to the synaptic compartment could be under the same translational control as we detected in intact slices. As shown in Figure 5D, an increase in PKMζ protein expression was observed after OXT (1 μm) treatment for 30 min. Pretreatment of the synaptoneurosomes with rapamycin (200 nm) completely blocked this event, whereas ascomycin had no effect on OXT-induced increases in PKMζ protein expression. Neither rapamycin nor ascomycin treatments alone had an effect on basal PKMζ protein levels (Fig. 5D). These results suggest that OXT-induced increases in PKMζ expression occurs, at least in part, in dendrites.
Transactivation of EGFR is required for OXT-induced enhancement of LTP and PKMζ protein synthesis
A series of studies were performed to identify the cellular mechanisms underlying the OXT-induced increases in PKMζ expression in the synaptoneurosomes. To confirm the involvement of OXTR-mediated PLC activation, we examined the effect of OXT in the presence of OXTR antagonist L-371257 or PLC blocker U73122. We found that pretreatment of the synaptoneurosomes with L-371257 (3 μm) or U73122 (10 μm) completely blocked OXT (1 μm)-induced increases in PKMζ protein synthesis (Fig. 6A,B). Neither L-371257 nor U73122 treatment alone had an effect on basal levels of PKMζ protein (Fig. 6A,B).
Figure 6.

Involvement of OXTR/PLC and EGFR transactivation in OXT-induced increases in PKMξ synthesis. A, B, Representative immunoblot and summary histogram showing pretreatment of the synaptoneurosomes with OXTR antagonist L-371257 (3 μm) or PLC blocker U73122 (10 μm) blocked OXT (1 μm)-induced increases in PKMξ synthesis. C, Representative immunoblot and summary histogram showing OXT (1 μm)-induced increases in PKMξ expression in the synaptoneurosomes was blocked by pretreatment with EGFR inhibitor AG1478 (1 μm), PI3K inhibitor LY294002 (20 μm), or MAPK kinase inhibitor U0126 (10 μm). D, Summary of experiments showing pretreatment of hippocampal slices with AG1478 (1 μm) blocked OXT (1 μm)-induced enhancement of LTP. Representative traces of fEPSPs were taken at the time indicated by number. Dash lines show level of baseline. E, Representative immunoblot and summary histogram showing OXT (1 μm)-induced increases in PKMξ expression were not affected by pretreatment of the synaptoneurosomes with Trk family of receptor tyrosine kinase inhibitor K252a (0.2 μm). The total number of experiments is indicated by n in parenthesis. Data are represented as mean ± SEM. *p < 0.05 compared with vehicle-treated group.
Because OXT has been shown to transactivate EGFR tyrosine kinase in both PHM1 immortalized human myometrial cells (Zhong et al., 2003) and bovine endometrial epithelial cells (Krishnaswamy et al., 2010), we therefore examined whether activation of OXTR could transactivate EGFR and subsequently stimulate PKMζ protein synthesis. As expected, we found that pretreatment of the synaptoneurosomes with a tyrophostin AG1478 (1 μm), a selective inhibitor of EGFR (Levitzki and Gazit, 1995), completely blocked OXT (1 μm)-induced increases in PKMζ protein synthesis (Fig. 6C). However, K252a (0.2 μm), a potent inhibitor of the Trk family of receptor tyrosine kinases (Tapley et al., 1992), did not alter the effect of OXT (Fig. 6D), suggesting some specificity of OXTR-induced transactivation of EGFR. Because PI3K and ERK1/2 are two major downstream effectors activated by EGFR, we next examined the possible role of PI3K and ERK1/2 in OXT-induced increases in PKMζ protein synthesis. Accordingly, we pretreated the synaptoneurosomes with PI3K inhibitor LY294002 (20 μm) or MAPK kinase inhibitor U0126 (10 μm) and found that both of them effectively blocked OXT (1 μm)-induced increases in PKMζ protein synthesis (Fig. 6C). Moreover, consistent with its ability to inhibit OXT-induced increases in PKMζ protein synthesis in the synaptoneurosomes, AG1478 (1 μm) also blocked OXT-induced enhancement of LTP (110–120 min after HFS: rapamycin, 106.8 ± 5.6% of baseline, n = 5; Fig. 6E). AG1478 treatment alone had no effect on synaptic transmission before or after TS (data not shown). These results indicate that EGFR serves as an important effector in a signaling pathway connecting OXTR to PKMζ protein synthesis.
To further characterize the transactivation of EGFR by OXTR, we measured the tyrosine phosphorylation of EGFR by using a phospho-specific antibody that recognizes the autophosphorylation of EGFR at Tyr1068 in the synaptoneurosomes. We found that OXT (1 μm) stimulated a rapid increase in the tyrosine phosphorylation of EGFR, reaching a maximum within 10 min and remaining elevated for at least 25 min (Fig. 7A). The increase in tyrosine phosphorylation was blocked by pretreatment with L-371257 (3 μm), U73122 (10 μm), or AG1478 (1 μm) (Fig. 7B–D), supporting a signaling model that OXTR transactivates EGFR through a PLC-dependent mechanism.
Figure 7.

OXT stimulates EGFR-dependent PI3K and ERK1/2 activation in the synaptoneurosomes. A, Representative immunoblot and summary histogram showing OXT (1 μm) induces increases in tyrosine phosphorylation of EGFR in a time-dependent manner. B–D, Representative immunoblot and summary histogram showing pretreatment of the synaptoneurosomes with L-371257 (3 μm), U73122 (10 μm), or AG1478 (1 μm) blocked OXT (1 μm)-induced increases in EGFR tyrosine phosphorylation. E, Representative immunoblot and summary histogram showing pretreatment of the synaptoneurosomes with AG1478 (1 μm) blocked OXT (1 μm)-induced increases in Akt phosphorylation at both Thr308 and Ser473 sites. F, Representative immunoblot and summary histogram showing pretreatment of the synaptoneurosomes with AG1478 (1 μm) blocked OXT (1 μm)-induced increases in ERK1/2 phosphorylation. G, Representative immunoblot and summary histogram showing pretreatment of the synaptoneurosomes with LY294002 (20 μm) significantly inhibited OXT (1 μm)-induced increases in ERK1/2 phosphorylation. H, Representative immunoblot and summary histogram showing pretreatment of the synaptoneurosomes with U0126 (10 μm) had no effect on OXT (1 μm)-induced increases in Akt phosphorylation. Inhibitors were applied 30 min before the addition of OXT for 30 min. The total number of experiments is indicated by n in parenthesis. Data are represented as mean ± SEM. *p < 0.05 compared with vehicle-treated group.
To investigate whether OXT stimulates PI3K activity, we indirectly monitored the activation of PI3K by measuring the phosphorylation of its downstream target Akt at both Thr308 and Ser473 sites (Franke et al., 1995). We found that OXT (1 μm) treatment significantly increased the level of phosphorylated Akt, which was blocked by AG1478 pretreatment (1 μm) (Fig. 7E). Likewise, OXT (1 μm) also induced a significant increase in the phosphorylation of ERK1/2, and this increase was also blocked by AG1478 (1 μm) pretreatment (Fig. 7F). These results indicate that activation of OXTR may lead to transactivation of EGFR and its PI3K and ERK1/2 downstream signaling pathways. Because a close interdependence between PI3K and ERK1/2 signaling pathways have been reported in a wide variety of cell types including neurons (Chandler et al., 2001; Perkinton et al., 2002; Yang et al., 2008), we therefore asked whether there exists a crosstalk between the PI3K and ERK1/2 signaling pathways in mediating OXT-induced increases in PKMζ protein synthesis. We found that pretreatment with PI3K inhibitor LY294002 (20 μm) caused a significant reduction of OXT-induced ERK1/2 phosphorylation (Fig. 7G). In contrast, OXT-induced Akt phosphorylation was not significantly altered by MAPK kinase inhibitor U0126 (10 μm; Fig. 7H), indicating that PI3K may act upstream of ERK1/2 to mediate OXT regulation.
To further delineate whether the crosstalk between OXTR and EGFR is mediated by extracellular release of EGFR ligands, we used three different approaches. First, we pretreated the synaptoneurosomes with the functional anti-EGF antibody at 10 μg/ml, a concentration sufficient to inhibit EGF (20 ng/ml)-induced tyrosine phosphorylation of EGFR, for 30 min before OXT (1 μm) application, and found that OXT-induced EGFR tyrosine phosphorylation remained normal (Fig. 8A). Because exogenously applied heparin has been shown to effectively inhibit EGFR transactivation by competing with cell surface-associated heparin sulfate proteoglycans for HB-EGF binding (Raab and Klagsbrun, 1997; Kalmes et al., 2000), we therefore tested whether HB-EGF is required for OXT-induced EGFR transactivation by pretreatment of the synaptoneurosomes with heparin (50 μg/ml), a concentration that has been shown to effectively block angiotensin-II or HB-EGF-induced EGFR tyrosine phosphorylation (Chiu et al., 2005). We found no changes in OXT-induced EGFR tyrosine phosphorylation in the presence of heparin (Fig. 8B). Because the activity of matrix metalloproteinase (MMP) has been shown to be required to mediate extracellular release of EGFR ligands (Prenzel et al., 1999; Chiu et al., 2005), we examined the influence of a broad-spectrum MMP inhibitor GM6001 (10 μm), a concentration that has been shown to block angiotensin-II-induced EGFR tyrosine phosphorylation (Chiu et al., 2005), on OXT-induced EGFR tyrosine phosphorylation. As shown in Figure 8C, OXT-induced tyrosine phosphorylation of EGFR was not significantly altered by pretreatment with GM6001. Together, these findings suggest that OXT-induced EGFR transactivation does not require extracellular release of EGFR ligands.
Figure 8.
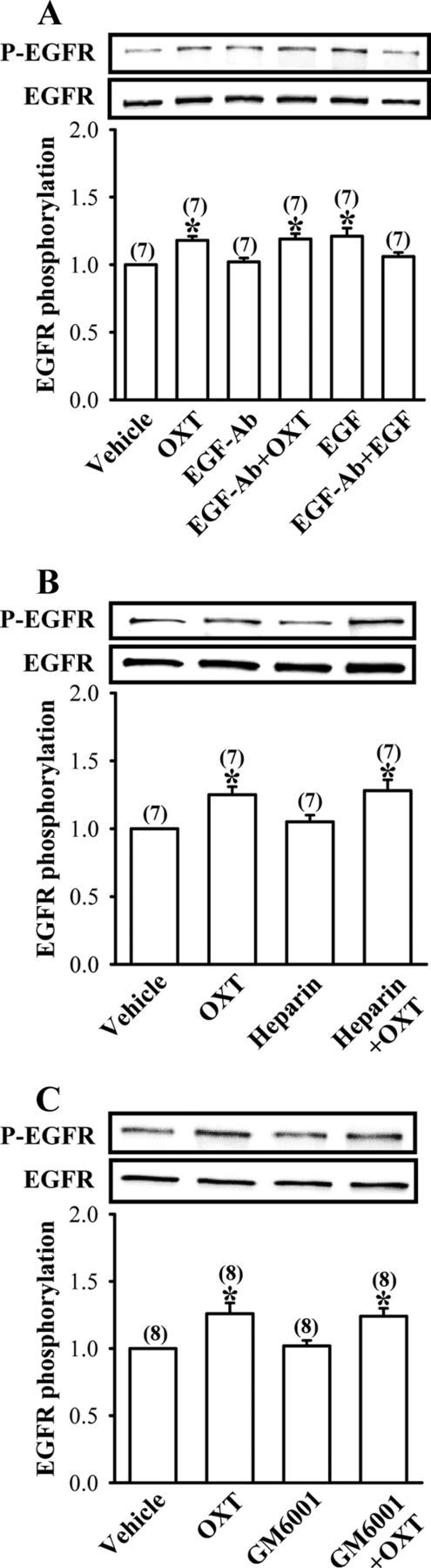
OXT-induced EGFR transactivation does not require extracellular release of EGFR ligands or MMP activity. A, Representative immunoblot and summary histogram showing OXT (1 μm)-induced increases in EGFR tyrosine phosphorylation in the synaptoneurosomes was not affected by pretreatment with a functional anti-EGF antibody (EGF-Ab) at a concentration of 10 μg/ml, which was sufficient to completely block EGF (20 ng/ml)-induced tyrosine phosphorylation of EGFR. B, Representative immunoblot and summary histogram showing pretreatment of the synaptoneurosomes with heparin (50 μg/ml) had no effect on OXT (1 μm)-induced increases in EGFR tyrosine phosphorylation. C, Representative immunoblot and summary histogram showing OXT (1 μm)-induced increases in EGFR tyrosine phosphorylation in the synaptoneurosomes was not affected by pretreatment with MMP inhibitor GM6001 (10 μm). Inhibitors were applied 30 min before the addition of OXT for 30 min. The total number of experiments is indicated by n in parenthesis. Data are represented as mean ± SEM. *p < 0.05 compared with vehicle-treated group.
We next investigated whether OXT-induced increases in PKMζ protein synthesis also occurred in the cultured hippocampal neurons. We first confirmed that the cultured hippocampal neurons faithfully reproduced the increased expression of PKMζ protein to OXT treatment that was observed in acute slice and synaptoneurosomal preparations (Fig. 9A). Consistently, pretreatment of the cultured preparations with L-371257 (3 μm) or AG1478 (1 μm) completely blocked OXT (1 μm)-induced increases in PKMζ protein synthesis. In addition, the effect of OXT on PKMζ protein synthesis was blocked by pretreatment of the cultured hippocampal neurons with rapamycin (200 nm), LY294002 (20 μm), or U0126 (10 μm), respectively (Fig. 9B). These results strengthen our hypothesis that PKMζ synthesis is upregulated by OXT through a mechanism that involves EGFR transactivation and the PI3K/ERK1/2/mTOR signaling pathway.
Figure 9.
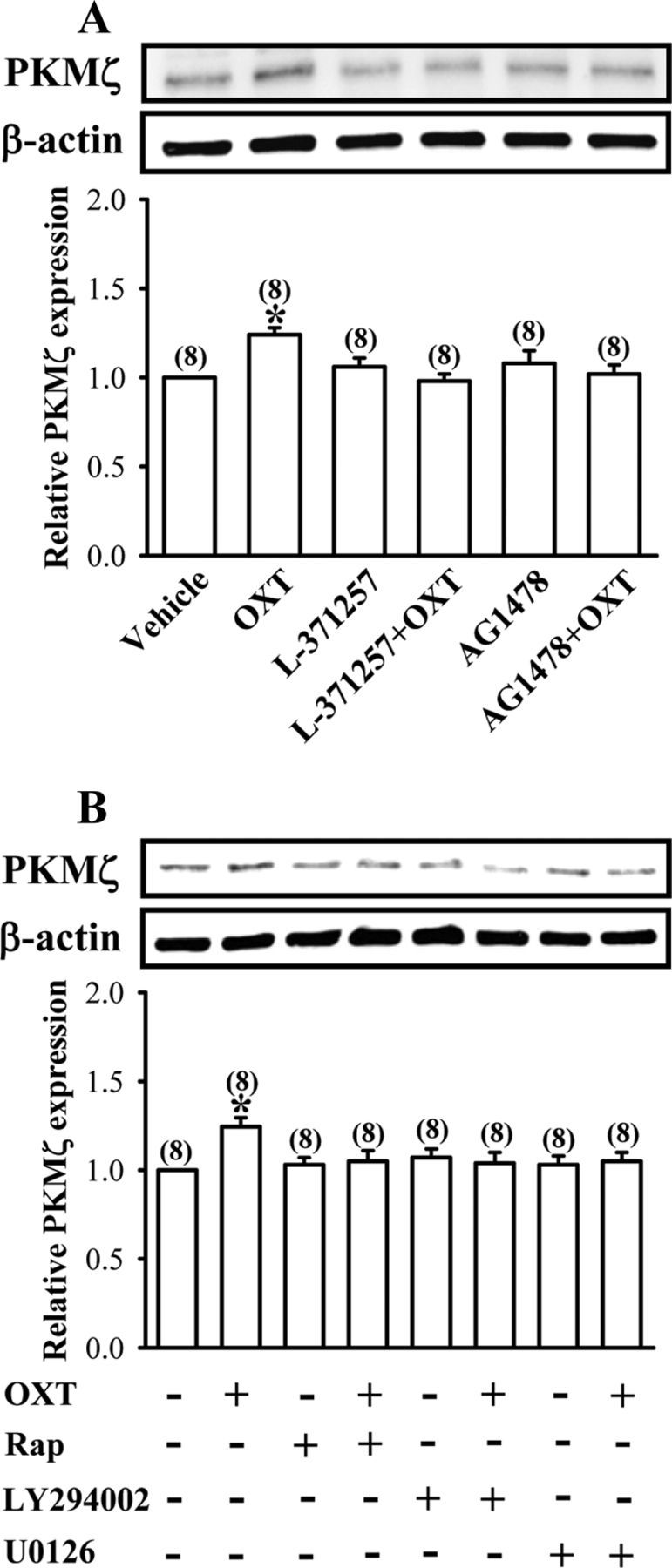
OXT stimulates PKMξ synthesis in cultured hippocampal neurons. A, Representative immunoblot and summary histogram showing pretreatment of the cultured hippocampal neurons with either L-371257 (3 μm) or AG1478 (1 μm) blocked OXT (1 μm)-induced increases in PKMξ expression. B, Representative immunoblot and summary histogram showing OXT (1 μm)-induced increases in PKMξ expression was blocked by pretreatment of the cultured hippocampal neurons with rapamycin (Rap, 200 nm), LY294002 (20 μm), or U0126 (10 μm), respectively. Inhibitors were applied 30 min before the addition of OXT for 30 min. The total number of experiments is indicated by n in parenthesis. Data are represented as mean ± SEM. *p < 0.05 compared with vehicle-treated group.
Endogenous OXT contributes to the maintenance of the late-phase but not early-phase of LTP
We finally examined the possible contribution of endogenous OXT in the maintenance of LTP. To this end, late-phase LTP was induced by three 1 s trains of TS at 100 Hz separated by 5 min in the presence of OXTR antagonist L-371257 (1 μm). In the control slice, TS produced synaptic potentiation that was stable for >2 h (110–120 min after last TS: 136.8 ± 6.7% of baseline, n = 5). In the slices incubated with L-371257, however, TS produced synaptic potentiation that was stable for ∼1 h (50–60 min after last TS: 132.6 ± 6.8% of baseline, n = 6) and then gradually declined to baseline by 2 h after TS (110–120 min after last TS: 110.2 ± 6.2% of baseline, n = 6; Fig. 10), suggesting a crucial role for endogenous OXT in the maintenance of late-phase LTP.
Figure 10.

Blockade of OXTR significantly inhibits the maintenance of late-phase LTP. A, Summary of experiments showing pretreatment of hippocampal slices with OXTR antagonist L-371257 (3 μm) significantly inhibited late-phase LTP induced by three trains of TS at 100 Hz separated by an intertrain interval of 5 min. B, Summary histogram depicting the effect of L-371257 on late-phase LTP in slices from female dorsal hippocampus. The magnitudes of LTP were measured 1 h (50–60 min) or 2 h (110–120 min) after TS. Representative traces of fEPSPs were taken at the time indicated by number. Dash lines show level of baseline. The total number of animals examined is indicated by n in parenthesis. Data are represented as mean ± SEM. *p < 0.05 compared with vehicle-treated group.
Discussion
The present study demonstrates that OXT enhances the maintenance of LTP in hippocampal CA1 region of both sexes via activation of the OXTR-associated conventional PLC signaling pathway. This enhancement requires a rapid and persistent increase in dendritic PKMζ protein synthesis via an mTOR-mediated mechanism. More importantly, OXT-induced increases in PKMζ expression relies on the transactivation of EGFR and subsequent activation of its downstream PI3K and ERK1/2 signaling pathways. Furthermore, our results reveal a functional contribution of endogenous OXT in the maintenance of late-phase LTP induced by multiple trains of TS.
OXT has previously been shown to affect either the establishment or the maintenance of LTP at excitatory central synapses (Dubrovsky et al., 2002; Tomizawa et al., 2003; Fang et al., 2008), but the exact underlying mechanisms still remain obscure. Dubrovsky et al. (2002) have shown that intracerebroventricular injection of OXT can elicit synaptic depression of previously established LTP in the dentate gyrus of anesthetized rats via its inhibitory effect on ATPase and ectoprotein kinase activity. On the contrary, Tomizawa et al. (2003) showed that OXT enhances hippocampal CA1 LTP in vitro and cognitive function in vivo during motherhood in mice through the activation of MAPK cascade and consequent cAMP-responsive element binding protein phosphorylation. In accordance with findings by Tomizawa et al. (2003), the current results show that activation of OXTR enhances the ability of subthreshold synaptic stimulation to induce late-phase LTP. We have, in addition, extended these findings by demonstrating that OXT exerts its LTP-enhancing effect via the activation of conventional OXTR-associated PLC and the signaling pathways downstream of EGFR.
An important finding in this study is the identification of PKMζ that contributes to OXT-induced enhancement of LTP. PKMζ is expressed at relatively high levels in the hippocampus (Naik et al., 2000) and de novo protein synthesis of PKMζ has been widely implicated in the maintenance of late-phase LTP (Sacktor et al., 1993; Ling et al., 2002; Pastalkova et al., 2006). The observation that inhibiting PKMζ with ZIP effectively reversed established LTP induced by conjoint activation of OXTR and subthreshold stimulation supports a pivotal role for PKMζ in the maintenance of OXT-induced LTP (Fig. 4). Furthermore, we propose that OXTR activation regulates the increased translation of PKMζ from preexisting mRNAs, rather than increased transcription, because incubation of slices with the transcription inhibitor actinomycin D does not affect OXT-induced increases in PKMζ expression (Fig. 5C) and PKMζ protein increases within minutes after OXT treatment, too rapid for new transcription of PKMζ mRNAs (Kelleher et al., 2004). More importantly, our data indicate that OXT stimulated PKMζ protein synthesis in isolated synaptoneurosomes, raising the possibility that the new PKMζ protein synthesis occurs locally in the synaptodendritic compartment. These findings confirm previous reports showing that PKMζ mRNAs are locally available at synapses for site-specific translation (Muslimov et al., 2004; Kelly et al., 2007). Due to the specificity of OXT-induced synthesis of PKMζ to enhance the synaptic responses of the tetanized pathway without affecting baseline synaptic transmission, our results raise the possibility that newly generated PKMζ may act as a plasticity-related protein (PRP) to interact with recently potentiated “tagged” synapses and then prolong potentiation. In support of this, Sajikumar et al. (2005) indicated that PKMζ is an LTP-specific PRP and is crucial for the transformation of early- into late-LTP during both synaptic tagging and cross-tagging. Furthermore, our observation that OXT-induced increases in PKMζ protein synthesis were blocked by PI3K, MEK, and mTOR inhibitors supports a mechanism involving the PI3K/Akt/mTOR and ERK1/2 signaling pathways. This is in line with earlier reports indicating that multiple signaling kinases important for LTP induction, in particular the Ca2+/calmodulin-dependent protein kinase II, MAPK, PI3K, and mTOR, can release the translational block on PKMζ synthesis in dendrites (Kelly et al., 2007; Sacktor, 2011).
A pressing question that follows these observations is how OXT activates PI3K and ERK1/2 signaling pathways. Our finding that the OXT-induced increases in PI3K and ERK1/2 activation were blocked by OXTR, PLC, and EGFR inhibitors supports pathways involving OXTR-associated conventional PLC and EGFR transactivation. This study provides, to our knowledge, the first evidence for the transactivation of EGFR by OXTR in brain. This regulation has been demonstrated in myometrial and epithelial cells (Zhong et al., 2003; Krishnaswamy et al., 2010), where the EGFR tyrosine kinase may serve as an important effector in a signaling pathway connecting OXTR to PI3K and ERK1/2 to regulate gene expression. Interestingly, it was noted that the inhibition of PLC with U73122 completely prevented OXT-induced increases in PI3K and ERK1/2 activation, suggesting that OXT-induced PLC activation mediates the transactivation of EGFR and its downstream PI3K and ERK1/2 signaling. Although several previous studies have indicated that extracellular release EGFR ligands and MMP activity are required for GPCR-induced EGFR transactivation (Prenzel et al., 1999; Chiu et al., 2005), our results do not support a role for EGFR ligands or MMP activity in mediating the crosstalk between OXTR and EGFR in hippocampal neurons (Fig. 8). This discrepancy in findings can be partially reconciled by accounting for differences in GPCR types and cell types, resulting in activation of different cellular processes that vary in their mode of action. Although the mechanism by which OXTR/PLC transactivates EGFR remains to be established, activation of proline-rich tyrosine kinase 2 (Pyk2) and its association with Src kinase may be involved in this event. Indeed, it has been reported that OXT can stimulate angiogenesis via a PLC-dependent activation of Pyk2 and Src mechanism (Cattaneo et al., 2009) and that Pyk2 is involved in Ca2+-stimulated EGFR transactivation in PC12 cells (Zwick et al., 1999).
How does PKMζ maintain OXT-induced LTP? Although we did not address this issue in the present study, several candidate mechanisms have been recently proposed. Yao et al. (2008) demonstrated that PKMζ maintains late-phase LTP by persistently upregulating N-ethylmaleimide-sensitive factor/glutamate receptor subunit 2-dependent AMPA receptor trafficking pathway to increase postsynaptic AMPA receptors. Interestingly, it has been suggested that immediately after translation, PKMζ exhibits a low level of activity until it binds to phosphoinositide-dependent protein kinase 1 (PDK1), which phosphorylates PKMζ at Thr410 and converts it into a conformation with high constitutive activity (Kelly et al., 2007). As such, it is plausible that the phosphorylation of PKMζ by PDK1 may ensure optimal PKMζ activity to maintain OXT-induced LTP. However, we were unable to assess levels of phosphorylated PKMζ in the synaptoneurosomes, since available phosphorylation site-specific antibodies did not yield a reliable signal. Further investigation is needed to examine this possibility.
What could be the functional consequence of OXT-induced enhancement of LTP? Given LTP, in hippocampal CA1 region, is generally assumed as a synaptic mechanism underlying the formation of spatial and episodic memories (Bliss and Collingridge, 1993; Martin et al., 2000), this enhancement of LTP by OXT may therefore promote long-term memory formation. While we did not directly test the effects of OXT on behavioral performance in hippocampus-dependent tasks in this study, a prior study has shown that intracerebroventricular injection of OXT into virgin female mice significantly improved long-term spatial memory in the radial arm maze, whereas an injection of OXTR antagonist in multiparous mice inhibited the improved spatial memory (Tomizawa et al., 2003). It is worth noting, however, that de Oliveira et al. (2007) showed that systemically administered OXT before testing impaired memory retrieval for the step-down inhibitory avoidance, a hippocampus-dependent associative learning task (Gold, 1986). Such amnesic effect of OXT on long-term memory might be caused by a decrease in glucocorticoid release from the adrenal gland, which is critically involved in enhancing memory of inhibitory avoidance training (Borrell et al., 1983). These results let us speculate that the effects of OXT on long-term memory formation could be polymodal, depending on the dose, route of administration, and types of memory tasks. Furthermore, the present study extend previous work (Tomizawa et al., 2003) by showing that OXT enhances the maintenance of LTP in the CA1 region of hippocampal slices from both nulliparous female and male rats, contradicting the idea that OXT exerts its effects in a sex-dependent manner (Ferguson et al., 2000; Monks et al., 2003).
Although our data show that OXT (1 μm) can consistently enhance the maintenance of LTP, it is unclear whether the endogenous OXT level could trigger this enhancing action. Because OXTR antagonist effectively prevented the maintenance of late-phase LTP induced by three trains of TS, we suggest that OXT release in response to strong conditioning stimuli is important for late-phase LTP maintenance.
In conclusion, we provide compelling evidence that OXTR activation facilitates induction of a protein-synthesis-dependent form of late-phase LTP in hippocampal CA1 region. Our data support a sequential signaling model, in which OXTR activation stimulates PLC, leading to the transactivation of EGFR, which in turn activates PI3K and ERK1/2 to increase dendritic PKMζ protein synthesis via mTOR signaling, thereby contributing to the maintenance of LTP.
Footnotes
This work was supported by research grants from the National Health Research Institute (NHRI-EX100-9618NI) and from the National Science Council (NSC101-2321-B-006-024), Taiwan.
References
- Balkowiec A, Katz DM. Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J Neurosci. 2002;22:10399–10407. doi: 10.1523/JNEUROSCI.22-23-10399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Borrell J, De Kloet ER, Versteeg DH, Bohus B. Inhibitory avoidance deficit following short-term adrenalectomy in the rat: the role of adrenal catecholamines. Behav Neural Biol. 1983;39:241–258. doi: 10.1016/s0163-1047(83)90910-x. [DOI] [PubMed] [Google Scholar]
- Brown EJ, Schreiber SL. A signaling pathway to translational control. Cell. 1996;86:517–520. doi: 10.1016/s0092-8674(00)80125-7. [DOI] [PubMed] [Google Scholar]
- Buijs RM, Swaab DF. Immuno-electron microscopical demonstration of vasopressin and oxytocin synapses in the limbic system of the rat. Cell Tissue Res. 1979;204:355–365. doi: 10.1007/BF00233648. [DOI] [PubMed] [Google Scholar]
- Cattaneo MG, Lucci G, Vicentini LM. Oxytocin stimulates in vitro angiogenesis via a Pyk-2/Src-dependent mechanism. Exp Cell Res. 2009;315:3210–3219. doi: 10.1016/j.yexcr.2009.06.022. [DOI] [PubMed] [Google Scholar]
- Chandler LJ, Sutton G, Dorairaj NR, Norwood D. N-methyl-d-aspartate receptor-mediated bidirectional control of extracellular signal-regulated kinase activity in cortical neuronal cultures. J Biol Chem. 2001;276:2627–2636. doi: 10.1074/jbc.M003390200. [DOI] [PubMed] [Google Scholar]
- Chiu T, Santiskulvong C, Rozengurt E. EGF receptor transactivation mediates ANG II-stimulated mitogenesis in intestinal epithelial cells through the PI3-kinase/Akt/mTOR/p70S6K1 signaling pathway. Am J Physiol Gastrointest Liver Physiol. 2005;288:G182–G194. doi: 10.1152/ajpgi.00200.2004. [DOI] [PubMed] [Google Scholar]
- Cohen H, Kaplan Z, Kozlovsky N, Gidron Y, Matar MA, Zohar J. Hippocampal microinfusion of oxytocin attenuates the behavioural response to stress by means of dynamic interplay with the glucocorticoid-catecholamine responses. J Neuroendocrinol. 2010;22:889–904. doi: 10.1111/j.1365-2826.2010.02003.x. [DOI] [PubMed] [Google Scholar]
- Deadwyler SA, Dunwiddie T, Lynch G. A critical level of protein synthesis is required for long-term potentiation. Synapse. 1987;1:90–95. doi: 10.1002/syn.890010112. [DOI] [PubMed] [Google Scholar]
- de Oliveira LF, Camboim C, Diehl F, Consiglio AR, Quillfeldt JA. Glucocorticoid-mediated effects of systemic oxytocin upon memory retrieval. Neurobiol Learn Mem. 2007;87:67–71. doi: 10.1016/j.nlm.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Dubrovsky B, Harris J, Gijsbers K, Tatarinov A. Oxytocin induces long-term depression on the rat dentate gyrus: possible ATPase and ectoprotein kinase mediation. Brain Res Bull. 2002;58:141–147. doi: 10.1016/s0361-9230(01)00748-1. [DOI] [PubMed] [Google Scholar]
- Fang LY, Quan RD, Kaba H. OxytocDubrovskyin facilitates the induction of long-term potentiation in the accessory olfactory bulb. Neurosci Lett. 2008;438:133–137. doi: 10.1016/j.neulet.2007.12.070. [DOI] [PubMed] [Google Scholar]
- Ferguson JN, Young LJ, Hearn EF, Matzuk MM, Insel TR, Winslow JT. Social amnesia in mice lacking the oxytocin gene. Nat Genet. 2000;25:284–288. doi: 10.1038/77040. [DOI] [PubMed] [Google Scholar]
- Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res. 1988;452:57–65. doi: 10.1016/0006-8993(88)90008-x. [DOI] [PubMed] [Google Scholar]
- Gainer H, Wray S. Oxytocin and vasopressin. From genes to peptides. Ann N Y Acad Sci. 1992;652:14–28. doi: 10.1111/j.1749-6632.1992.tb34342.x. [DOI] [PubMed] [Google Scholar]
- Gimpl G, Fahrenholz F. The oxytocin receptor system: structure, function, and regulation. Physiol Rev. 2001;81:629–683. doi: 10.1152/physrev.2001.81.2.629. [DOI] [PubMed] [Google Scholar]
- Gold PE. The use of avoidance training in studies of modulation of memory storage. Behav Neural Biol. 1986;46:87–98. doi: 10.1016/s0163-1047(86)90927-1. [DOI] [PubMed] [Google Scholar]
- Hardt O, Migues PV, Hastings M, Wong J, Nader K. PKMζ maintains 1-day- and 6-day-old long-term object location but not object identity memory in dorsal hippocampus. Hippocampus. 2010;20:691–695. doi: 10.1002/hipo.20708. [DOI] [PubMed] [Google Scholar]
- Hollingsworth EB, McNeal ET, Burton JL, Williams RJ, Daly JW, Creveling CR. Biochemical characterization of a filtered synaptoneurosome preparation from guinea pig cerebral cortex: cyclic adenosine 3′:5′-monophosphate-generating systems, receptors, and enzymes. J Neurosci. 1985;5:2240–2253. doi: 10.1523/JNEUROSCI.05-08-02240.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Hsu KS. Sustained activation of metabotropic glutamate receptor 5 and protein tyrosine phosphatases mediate the expression of (S)-3,5-dihydroxyphenylglycine-induced long-term depression in the hippocampal CA1 region. J Neurochem. 2006;96:179–194. doi: 10.1111/j.1471-4159.2005.03527.x. [DOI] [PubMed] [Google Scholar]
- Huang YF, Yang CH, Huang CC, Tai MH, Hsu KS. Pharmacological and genetic accumulation of hypoxia-inducible factor-1α enhances excitatory synaptic transmission in hippocampal neurons through the production of vascular endothelial growth factor. J Neurosci. 2010;30:6080–6093. doi: 10.1523/JNEUROSCI.5493-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttner WB, Schiebler W, Greengard P, De Camilli P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol. 1983;96:1374–1388. doi: 10.1083/jcb.96.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel TR, Gelhard R, Shapiro LE. The comparative distribution of forebrain receptors for neurohypophyseal peptides in monogamous and polygamous mice. Neuroscience. 1991;43:623–630. doi: 10.1016/0306-4522(91)90321-e. [DOI] [PubMed] [Google Scholar]
- Kalmes A, Vesti BR, Daum G, Abraham JA, Clowes AW. Heparin blockade of thrombin-induced smooth muscle cell migration involves inhibition of epidermal growth factor (EGF) receptor transactivation by heparin-binding EGF-like growth factor. Circ Res. 2000;87:92–98. doi: 10.1161/01.res.87.2.92. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Govindarajan A, Tonegawa S. Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron. 2004;44:59–73. doi: 10.1016/j.neuron.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Kelly MT, Crary JF, Sacktor TC. Regulation of protein kinase Mζ synthesis by multiple kinases in long-term potentiation. J Neurosci. 2007;27:3439–3444. doi: 10.1523/JNEUROSCI.5612-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnaswamy N, Lacroix-Pepin N, Chapdelaine P, Taniguchi H, Kauffenstein G, Chakravarti A, Danyod G, Fortier MA. Epidermal growth factor receptor is an obligatory intermediate for oxytocin-induced cyclooxygenase 2 expression and prostaglandin F2 alpha production in bovine endometrial epithelial cells. Endocrinology. 2010;151:1367–1374. doi: 10.1210/en.2009-1304. [DOI] [PubMed] [Google Scholar]
- Laudanna C, Mochly-Rosen D, Liron T, Constantin G, Butcher EC. Evidence of ζ protein kinase C involvement in polymorphonuclear neutrophil integrin-dependent adhesion and chemotaxis. J Biol Chem. 1998;273:30306–30315. doi: 10.1074/jbc.273.46.30306. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Macbeth AH, Pagani JH, Young WS., 3rd Oxytocin: the great facilitator of life. Prog Neurobiol. 2009;88:127–151. doi: 10.1016/j.pneurobio.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuner B, Caponiti JM, Gould E. Oxytocin stimulates adult neurogenesis even under conditions of stress and elevated glucocorticoids. Hippocampus. 2012;22:861–868. doi: 10.1002/hipo.20947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267:1782–1788. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- Lin HJ, Huang CC, Hsu KS. Effects of neonatal dexamethasone treatment on hippocampal synaptic function. Ann Neurol. 2006;59:939–951. doi: 10.1002/ana.20885. [DOI] [PubMed] [Google Scholar]
- Ling DS, Benardo LS, Serrano PA, Blace N, Kelly MT, Crary JF, Sacktor TC. Protein kinase Mζ is necessary and sufficient for LTP maintenance. Nat Neurosci. 2002;5:295–296. doi: 10.1038/nn829. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Domes G, Kirsch P, Heinrichs M. Oxytocin and vasopressin in the human brain: social neuropeptides for translational medicine. Nat Rev Neurosci. 2011;12:524–538. doi: 10.1038/nrn3044. [DOI] [PubMed] [Google Scholar]
- Monks DA, Lonstein JS, Breedlove SM. Got milk? Oxytocin triggers hippocampal plasticity. Nat Neurosci. 2003;6:327–328. doi: 10.1038/nn0403-327. [DOI] [PubMed] [Google Scholar]
- Muslimov IA, Nimmrich V, Hernandez AI, Tcherepanov A, Sacktor TC, Tiedge H. Dendritic transport and localization of protein kinase Mζ mRNA: implications for molecular memory consolidation. J Biol Chem. 2004;279:52613–52622. doi: 10.1074/jbc.M409240200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik MU, Benedikz E, Hernandez I, Libien J, Hrabe J, Valsamis M, Dow-Edwards D, Osman M, Sacktor TC. Distribution of protein kinase Mζ and the complete protein kinase C isoform family in rat brain. J Comp Neurol. 2000;426:243–258. doi: 10.1002/1096-9861(20001016)426:2<243::aid-cne6>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Neumann ID. Brain oxytocin: a key regulator of emotional and social behaviors in both females and males. J Neuroendocrinol. 2008;20:858–865. doi: 10.1111/j.1365-2826.2008.01726.x. [DOI] [PubMed] [Google Scholar]
- Neumann ID, Landgraf R. Advances in vasopressin and oxytocin-from genes to behavior to disease. Prog Brain Res. 2008;170:xi–xiii. doi: 10.1016/S0079-6123(08)00448-2. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Abel T, Kandel ER. Requirement of a critical period of transcription for induction of a late phase of LTP. Science. 1994;265:1104–1107. doi: 10.1126/science.8066450. [DOI] [PubMed] [Google Scholar]
- Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton AA, Sacktor TC. Storage of spatial information by the maintenance mechanism of LTP. Science. 2006;313:1141–1144. doi: 10.1126/science.1128657. [DOI] [PubMed] [Google Scholar]
- Pedersen CA, Prange AJ., Jr Induction of maternal behavior in virgin rats after intracerebroventricular administration of oxytocin. Proc Natl Acad Sci U S A. 1979;76:6661–6665. doi: 10.1073/pnas.76.12.6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkinton MS, Ip JK, Wood GL, Crossthwaite AJ, Williams RJ. Phosphatidylinositol 3-kinase is a central mediator of NMDA receptor signalling to MAP kinase (Erk1/2), Akt/PKB and CREB in striatal neurones. J Neurochem. 2002;80:239–254. doi: 10.1046/j.0022-3042.2001.00699.x. [DOI] [PubMed] [Google Scholar]
- Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- Raab G, Klagsbrun M. Heparin-binding EGF-like growth factor. Biochim Biophys Acta. 1997;1333:F179–F199. doi: 10.1016/s0304-419x(97)00024-3. [DOI] [PubMed] [Google Scholar]
- Rao A, Steward O. Evidence that protein constituents of postsynaptic membrane specializations are locally synthesized: analysis of proteins synthesized within synaptosomes. J Neurosci. 1991;11:2881–2895. doi: 10.1523/JNEUROSCI.11-09-02881.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacktor TC. How does PKMζ maintain long-term memory? Nat Rev Neurosci. 2011;12:9–15. doi: 10.1038/nrn2949. [DOI] [PubMed] [Google Scholar]
- Sacktor TC, Osten P, Valsamis H, Jiang X, Naik MU, Sublette E. Persistent activation of the ζ isoform of protein kinase C in the maintenance of long-term potentiation. Proc Natl Acad Sci U S A. 1993;90:8342–8346. doi: 10.1073/pnas.90.18.8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajikumar S, Navakkode S, Sacktor TC, Frey JU. Synaptic tagging and cross-tagging: the role of protein kinase Mζ in maintaining long-term potentiation but not long-term depression. J Neurosci. 2005;25:5750–5756. doi: 10.1523/JNEUROSCI.1104-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano P, Yao Y, Sacktor TC. Persistent phosphorylation by protein kinase Mζ maintains late-phase long-term potentiation. J Neurosci. 2005;25:1979–1984. doi: 10.1523/JNEUROSCI.5132-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano P, Friedman EL, Kenney J, Taubenfeld SM, Zimmerman JM, Hanna J, Alberini C, Kelley AE, Maren S, Rudy JW, Yin JC, Sacktor TC, Fenton AA. PKMζ maintains spatial, instrumental, and classically conditioned long-term memories. PLoS Biol. 2008;6:2698–2706. doi: 10.1371/journal.pbio.0060318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Morphology of vasopressin and oxytocin neurons and their central and vascular projections. Prog Brain Res. 1983;60:101–114. doi: 10.1016/S0079-6123(08)64378-2. [DOI] [PubMed] [Google Scholar]
- Stanton PK, Sarvey JM. Blockade of long-term potentiation in rat hippocampal CA1 region by inhibitors of protein synthesis. J Neurosci. 1984;4:3080–3088. doi: 10.1523/JNEUROSCI.04-12-03080.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica L, Zhu PJ, Huang W, Zhou H, Kozma SC, Costa-Mattioli M. Selective pharmacogenetic inhibition of mammalian target of Rapamycin complex I (mTORC1) blocks long-term synaptic plasticity and memory storage. Proc Natl Acad Sci U S A. 2011;108:3791–3796. doi: 10.1073/pnas.1014715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapley P, Lamballe F, Barbacid M. K252a is a selective inhibitor of the tyrosine protein kinase activity of the trk family of oncogenes and neurotrophin receptors. Oncogene. 1992;7:371–381. [PubMed] [Google Scholar]
- Tomizawa K, Iga N, Lu YF, Moriwaki A, Matsushita M, Li ST, Miyamoto O, Itano T, Matsui H. Oxytocin improves long-lasting spatial memory during motherhood through MAP kinase cascade. Nat Neurosci. 2003;6:384–390. doi: 10.1038/nn1023. [DOI] [PubMed] [Google Scholar]
- Tsokas P, Grace EA, Chan P, Ma T, Sealfon SC, Iyengar R, Landau EM, Blitzer RD. Local protein synthesis mediates a rapid increase in dendritic elongation factor 1A after induction of late long-term potentiation. J Neurosci. 2005;25:5833–5843. doi: 10.1523/JNEUROSCI.0599-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyzio R, Cossart R, Khalilov I, Minlebaev M, Hübner CA, Represa A, Ben-Ari Y, Khazipov R. Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science. 2006;314:1788–1792. doi: 10.1126/science.1133212. [DOI] [PubMed] [Google Scholar]
- Veinante P, Freund-Mercier MJ. Distribution of oxytocin- and vasopressin-binding sites in the rat extended amygdala: a histoautoradiographic study. J Comp Neurol. 1997;383:305–325. [PubMed] [Google Scholar]
- Wigström H, Gustafsson B. Facilitated induction of hippocampal long-lasting potentiation during blockade of inhibition. Nature. 1983;301:603–604. doi: 10.1038/301603a0. [DOI] [PubMed] [Google Scholar]
- Yang PC, Yang CH, Huang CC, Hsu KS. Phosphatidylinositol 3-kinase activation is required for stress protocol-induced modification of hippocampal synaptic plasticity. J Biol Chem. 2008;283:2631–2643. doi: 10.1074/jbc.M706954200. [DOI] [PubMed] [Google Scholar]
- Yao Y, Kelly MT, Sajikumar S, Serrano P, Tian D, Bergold PJ, Frey JU, Sacktor TC. PKMζ maintains late long-term potentiation by N-ethylmaleimide-sensitive factor/GluR2-dependent trafficking of postsynaptic AMPA receptors. J Neurosci. 2008;28:7820–7827. doi: 10.1523/JNEUROSCI.0223-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young WS, 3rd, Gainer H. Transgenesis and the study of expression, cellular targeting and function of oxytocin, vasopressin and their receptors. Neuroendocrinology. 2003;78:185–203. doi: 10.1159/000073702. [DOI] [PubMed] [Google Scholar]
- Zaninetti M, Raggenbass M. Oxytocin receptor agonists enhance inhibitory synaptic transmission in the rat hippocampus by activating interneurons in stratum pyramidale. Eur J Neurosci. 2000;12:3975–3984. doi: 10.1046/j.1460-9568.2000.00290.x. [DOI] [PubMed] [Google Scholar]
- Zhong M, Yang M, Sanborn BM. Extracellular signal-regulated kinase 1/2 activation by myometrial oxytocin receptor involves GαqGβγ and epidermal growth factor receptor tyrosine kinase activation. Endocrinology. 2003;144:2947–2956. doi: 10.1210/en.2002-221039. [DOI] [PubMed] [Google Scholar]
- Zwick E, Wallasch C, Daub H, Ullrich A. Distinct calcium-dependent pathways of epidermal growth factor receptor transactivation and Pyk2 tyrosine phosphorylation in PC12 cells. J Biol Chem. 1999;274:20989–20996. doi: 10.1074/jbc.274.30.20989. [DOI] [PubMed] [Google Scholar]