

Abstract
In the adult mammalian subventricular zone (SVZ), GFAP-positive neural stem cells (NSCs) generate neuroblasts that migrate tangentially along the rostral migratory stream (RMS) toward the olfactory bulb (OB). In the mouse brain, we found that the plasticity inhibitors Nogo-A and Nogo receptor 1 (NgR1) are differentially expressed in the SVZ–OB system, in which Nogo-A identifies immature neuroblasts and NgR1 germinal astrocytes. We therefore examined the role of Nogo-A and NgR1 in the regulation of neurogenesis. Pharmacological experiments show that Nogo-66/NgR1 interaction reduces the proliferation of NSCs. This is consistent with a negative-feedback loop, in which newly generated neurons modulate cell division of SVZ stem cells. Moreover, the Nogo-A–Δ20 domain promotes neuroblast migration toward the OB through activation of the Rho/ROCK (Rho-associated, coiled-coil containing protein kinase) pathway, without the participation of NgR1. Our findings reveal a new unprecedented function for Nogo-A and NgR1 in the homeostatic regulation of the pace of neurogenesis in the adult mouse SVZ and in the migration of neuroblasts along the RMS.
Introduction
Two decades of research have established a clear role for Nogo-A and its receptor NgR1 in neuritic growth control in the mature CNS, including the restriction of structural and synaptic plasticity and the stabilization of connections (Buffo et al., 2000; Lee et al., 2008; Raiker et al., 2010; Schwab 2010; Zagrebelsky et al., 2010; Delekate et al., 2011). Although the bulk of Nogo-A expression is found in oligodendrocytes, its receptor NgR1 is primarily expressed by different subpopulations of mature neurons (Wang et al., 2002; Fischer et al., 2004).
However, both Nogo-A and NgR1 have been found in other neural cell types, including progenitors during development (Mingorance et al., 2004; Mathis et al., 2010), neurosphere cells (Mathis et al., 2010), astrocytes (Wang et al., 2002; Mingorance-Le Meur et al., 2007), and human astrocytoma and glioma cells (Cheung et al., 2009; Xiong et al., 2012), suggesting additional functions for this pathway. For instance, Nogo-A and NgR1 have been implicated in neurosphere cell proliferation (Li et al., 2011), differentiation (Wang and Zhu, 2008; Lööv et al., 2012), and control of tumor malignancy (Liao et al., 2004; Cheung et al., 2009; Xiong et al., 2012). The latter findings point to the participation of Nogo-A/NgR1 signaling in the regulation of other aspects of growth, such as tissue expansion or turnover by cell proliferation.
To address this hypothesis, we studied the expression and function of Nogo-A and NgR1 in the adult germinal zone residing in the lateral wall (LW) of the lateral ventricles [subventricular zone (SVZ)]. Here, neural stem cells (NSCs) with astrocytic features generate intermediate progenitors giving rise to neuroblasts that migrate to the olfactory bulb (OB) along the rostral migratory stream (RMS) (Kriegstein and Alvarez-Buylla, 2009; Ihrie and Alvarez-Buylla, 2011). Using pharmacological approaches along with in vitro and in vivo experiments, we found that Nogo-A/NgR1 signaling regulates the pace of neuronal neogeneration by reducing NSC proliferation. We also provide evidence that neuroblast migration to the OB is supported by the activity of the Nogo-A–Δ20 domain, independently of NgR1.
Materials and Methods
Animals, surgical procedures, and in vivo treatments.
Experiments were performed on different mouse lines, including C57BL/6 mice, GLAST::CreErt2;R26R (Mori et al., 2006), and hGFAP::GFP (Zhuo et al., 1997) animals. Adult male mice (2–4 months of age) were used unless differently stated. Surgical procedures and perfusions were performed under deep general anesthesia [100 mg/kg ketamine (Ketavet; Bayern) and 5 mg/kg xylazine (Rompun; Bayer)]. The experimental plan was designed according to the guidelines of the National Institutes of Health, the European Communities Council (86/609/EEC), and the Italian law for care and use of experimental animals (DL116/92). It was also approved by the Italian Ministry of Health and the Bioethical Committee of the University of Turin.
Mice (GLAST::CreErt2;R26R) received tamoxifen dissolved in corn oil to induce Cre activity and β-galactosidase (β-gal) reporter expression (one administration of 5 mg each by oral gavage for 2 d) before treatment. For acute injections, neutralizing peptide 1–40 (NEP1–40) antagonist (500 μm NEP1–40 in 97.5% PBS plus 2.5% DMSO; Sigma-Aldrich) (GrandPré et al., 2002) or vehicle solution (97.5% PBS plus 2.5% DMSO) were pressure injected into the left cerebral ventricle (coordinates relative to bregma: anterior, 0; lateral, 1 mm; depth, 1.8 mm). Over a period of 20 min, 1 μl was injected. Alternatively, osmotic minipumps (Alzet osmotic pumps 1007D) were implanted at the same coordinates to deliver NEP1–40 peptide, vehicle solution, Nogo-A–Δ20 [11C7, 0.5 mg/ml in saline; raised against an 18-aa peptide in the most active region (Δ20) of Nogo-A; kindly supplied by Novartis Pharma; Oertle et al., 2003] or control (mouse anti-human IgG; Jackson ImmunoResearch) antisera. Choroid plexi (CPs) were dissected from the LWs of postnatal wild-type mice and grafted according to Sawamoto et al. (2006) to inhibit cell migration toward the OB. Animals were killed at 3 d after injection or at 7 or 10 d after minipump implantation.
Proliferation, fate analysis, and immunohistochemistry.
To examine cell proliferation, we used the thymidine analog 5-bromo-2-deoxyuridine (BrdU; Sigma-Aldrich), which is incorporated in the DNA during the S phase of the cell cycle and remains in the DNA even when the cell has exited the active phases of the cells cycle. Depending on treatment, BrdU tagging is suitable for detecting active mitosis, cells that have proliferated over a certain time window, or cells that have exited the cell cycle (Boda et al., 2011). In some experiments, BrdU was administered as a single pulse (50 mg/kg, i.p., in saline) at either the moment of minipump implantation or antagonist injection. In some cases, multiple BrdU injections were required to increase the probability for detection of NSC divisions. Active proliferation was revealed by expression of the M-phase marker phospho-histone H3 (Ph3) or Ki67 (an antigen present in the S-to-M phases of the cell cycle; Boda et al., 2011).
For histological analysis, animals were anesthetized (as above) and transcardially perfused with 4% paraformaldehyde (PFA) in phosphate buffer (PB). Brains were postfixed with 4% PFA overnight, washed in phosphate buffer, cryoprotected in a 30% sucrose solution, and, after cutting, were stained according to a standard immunofluorescence procedure in 0.1 m PB for 48 h (Buffo et al., 2008; Boda et al., 2011). Adult brains were cut into 30-μm-thick coronal sections and collected in PBS. Brain slices of postnatal pups (P5) were placed directly onto glass slides. The sections were stained to detect the expression of different antigens: Nogo-A (11C7, 1:10,000; Novartis Pharma), NgR1 (1:1000, Alomone; 1:400, R&D Systems), glial fibrillary acidic protein (GFAP) (1:1000; Dako), doublecortin (DCX) (1:400; Santa Cruz Biotechnology), Vimentin (1:200; Developmental Studies Hybridoma Bank), βIII-tubulin (1:500; Sigma), brain lipid binding protein (BLBP) (1:500; Millipore), β-gal (1:10,000; Cappel), green fluorescent protein (GFP) (1:700; Invitrogen), BrdU (1:250; Abcam), Ki67 (1:1000; Novacastra), Ph3 (1:100; Millipore Biotechnology), mammalian achaete-schute homolog 1 (Mash1) (1:200; BD Pharmingen), and activated caspase 3 (1:150; Cell Signaling Technology). Sections were incubated overnight with primary antibodies was made overnight at 4°C in PBS with 1.5% donkey or goat serum and 0.5% Triton X-100. Sections were then exposed for 2 h at room temperature to secondary species-specific antibodies conjugated with Alexa Fluor 488 or Alexa Fluor 546 (1:500; Invitrogen), Cy3 (1:500; Jackson ImmunoResearch), or Alexa Fluor 649 (1:500; Invitrogen). Cell nuclei were visualized using 4′,6-diamidino-2-phenylindole (DAPI; Fluka). After processing, sections were mounted on microscope slides with Tris-glycerol supplemented with 10% Mowiol (Calbiochem).
For detection of β-gal, the high-sensitivity tyramide signal amplification kit (PerkinElmer Life and Analytical Sciences) was used (Buffo et al., 2008). This procedure was also applied for anti-NgR1 staining in the case of double labeling with antibodies produced in the same species. To facilitate BrdU recognition, slices were treated with 2N HCl for 20 min at 37°C, followed by 10 min in borate buffer.
Neurosphere assay and explants.
Adult SVZs were dissected and dissociated, and cells (20,000 cells/ml) were cultivated in a standard neurosphere assay (Pastrana et al., 2009). Treatments applied to the culture medium included the following: anti-NgR1 (10 μm; mNogo receptor affinity-purified goat IgG; R & D Systems; Petrinovic et al., 2010; Delekate et al., 2011) or control antibodies (goat anti-mouse IgG; Jackson ImmunoResearch), 11C7 or control antibodies, NEP1–40 (1 μm; Oertle et al., 2003) or vehicle solution, and Nogo-p4 peptide (4 μm in PBS; ADI Co.) (Wang and Zhu, 2008), or vehicle solution. In some cases, primary spheres were dissociated and plated to generate secondary neurospheres, which were then also treated with agonists or antagonists. In other cases, only primary neurosphere were treated. Rate of neurosphere generation was determined as the number of primary neurospheres/number of viable seeded cells. Self-renewal was determined as the number of secondary neurospheres/number of viable seeded cells derived from primary spheres. For assessment of differentiation, neurospheres as a whole or as single-cell suspensions were plated in differentiation medium for 7 d (Buffo et al., 2008). The number of viable cells was determined by trypan blue exclusion. Diameters of living neurospheres were measured using NIS-Element software (Nikon).
Postnatal explants were prepared according to Wichterle et al. (1997). Tissues from the SVZ, RMS, and subependymal layer of the OB (SEL–OB) were embedded in 75% Matrigel growth factor reduced (BD Biosciences) and maintained for 1 d in vitro in Neurobasal medium (Invitrogen) supplemented with B27 (1×; Miltenyi), penicillin/streptomycin (20 U/ml; Sigma), and 0.5 mm glutamine (Invitrogen). Antibodies and compounds were mixed with Matrigel together with the Rho-associated, coiled-coil containing protein kinase (ROCK) inhibitor Y27632 (4-[(1R)-1-aminoethyl]-N-pyridin-4-yl-cyclohexane-1-carboxamide) (10 μg/ml; Sigma-Aldrich). Only vital explants with cells moving out of the tissue core (80–90% of the total) were analyzed, with virtually all of these cells emigrating in chains. In some experiments, explants were grown on a monolayer of RMS-derived astrocytes according to García-Marqués et al. (2010). For analysis, explants were fixed in 4% PFA for 40 min and stained. Vital staining to visualize surface Nogo-A was performed as described previously (Mathis et al., 2010).
To test whether the highly inhibitory Nogo-A region Nogo-A–Δ20 (Oertle et al., 2003) affects SVZ-derived cell adhesion, glass coverslips (1 cm2) were coated with poly-d-lysine (PDL) (5 μg/ml), washed three times, and subsequently coated with Nogo-A–Δ20 (100 pmol, diluted in PBS) for 1 h at 37°C. Unbound Nogo-A–Δ20 was removed by three washes with PBS. Adult SVZs were dissociated, and cells were either plated on PDL only or Nogo-A–Δ20-coated coverslips (12,000 cells/cm2 in DMEM/F-12 supplemented with B27). After 1 h, cells were fixed, stained, and scored. The average number of adhered cells was determined by counting in five randomly chosen fields of view of the coverslips.
Cell isolation and RT-PCR.
Adult mouse SVZs were dissected from 10 to 20 animals and dissociated using Neural Tissue Dissociation kit (Miltenyi). Polysialic acid–neural cell adhesion molecule-positive (PSA-NCAM+) neuroblasts or glutamate aspartate transporter (GLAST)-expressing astrocytes were enriched by positive selection using antibody-conjugated magnetic beads, according to the instructions of the manufacturers (Miltenyi). A double round of selection was applied to isolate GLAST+ cells (94% purity, anti-GLAST antibodies conjugated to allophycocyanin and detected by flow cytometry, FACSCanto; BD Biosciences), whereas a single round yielded ∼90% purity for PSA-NCAM+ cells (anti-PSA-CAM conjugated to phycoerythrin). Total RNA was extracted from the cell fractions with the RNeasy micro kit (Qiagen), and reverse transcribed to single-stranded cDNA with the High-Capacity cDNA Archive kit (Applied Biosystems). cDNAs were amplified in each PCR assay with Go TaqDNA Polymerase (2.5 U/sample; Promega) in a 25 μl reaction mixture containing 1 μm forward and reverse primers, 0.2 mm dNTPs, and 1.5 mm MgCl2. Adult brain cDNA was used as a positive control and negative controls lacking cDNA or Taq polymerase were processed in parallel. Primers used were the following: Nogo-A, forward, 5′-TGAGGGAAGTAGGGATGTGC-3′; reverse, 5′-CAGGTGATGTACGCTCTGGA-3′ (183 bp); NgR, forward, 5′-ATCTTCCTGCATGGCAACCGAAT-3′; reverse, 5′-AGAGGTTGTTGGCAAACAGGTAG-3′ (531 bp); βIII-tubulin, forward, 5′-ATCCACCTTCATTGGCAACAGCAC-3′; reverse, 5′-ACTCGGACACCAGGTCATTCATGT-3′ (173 bp); BLBP, forward, 5′-TGAGTACATGAAAGCTCTGGGCGT-3′; and reverse, 5′-TGAGCTTGTCTCCATCCAACCGAA-3′ (224 bp). Amplifications followed this protocol: 35 cycles, each cycle with 95°C for 30 s, 62°C for 1 min, 72°C for 30 s, after an initial denaturation at 95°C for 3 min.
Image processing and data analyses.
Histological specimens were examined using an E-800 Nikon microscope connected to a color CCD camera and a Leica TCS SP5 confocal microscope. All images were collected with the confocal microscope. Adobe Photoshop 6.0 (Adobe Systems) was used to adjust image contrast and assemble the final plates. Most quantitations were performed by confocal analysis. However, in some instances, the Neurolucida software (MicroBrightField) was used. For evaluation of cell densities or the number of cells per area per section or the extent of labeling after in vivo injections and pump implantations, comparisons were made between hemispheres ipsilateral to the injection/infusion site of control (ctrl) and treated animals (unless differently stated). Measurements were derived from at least three sections per animal. Three to five animals were analyzed for each time point or experimental condition. Number of animals (n) in text includes both treated and control animals. For neurosphere experiments, data are derived from at least three experiments. For explants analyses, ∼30 individual explants were pulled for each experimental condition. For in vitro data, n refers to a single experiment. Isolated or pyknotic cells were identified on the basis of DAPI staining. Staining intensity and percentage of stained areas were quantified using NIH ImageJ software (http://rsb.info.nih.gov/ij/). Statistical analyses were performed by SigmaStat software package (Jandel Scientific) and included unpaired and paired Student's t test, one-way ANOVA, and χ2 test. In all instances, p < 0.05 was considered as statistically significant. Data are expressed as averages ± SEM.
Results
Nogo-A and NgR1 expression pattern in the adult and postnatal SVZ–OB system
The expression patterns of Nogo-A and NgR1 in the SVZ–OB system were assessed in coronal telencephalic sections of adult and postnatal (P5) mice. In the adult brain, anti-Nogo-A and anti-NgR1 immunostaining displayed a reciprocal expression pattern and a clear segregation to distinct cell types (Fig. 1A, SVZ, and A′, RMS). Nogo-A antiserum labeled DCX+ neuroblasts (Fig. 1B–D), whereas anti-NgR1 antibodies stained GFAP+ cells (Fig. 1F–G′). Quantitative analyses of confocal images throughout the SVZ–OB system confirmed that the vast majority of neuroblasts expressed Nogo-A and that all Nogo-A+ cells displayed immature neuronal traits (Fig. 1E). Conversely, NgR1 was expressed by the vast majority of GFAP+ astroglial cells and NgR1+ elements consistently displayed an astroglial phenotype (Fig. 1H). As additional evidence for the segregation of Nogo-A and NgR1 to distinct cell types, no significant overlap of Nogo-A and GFAP or NgR1 and DCX was detected by confocal and automated analyses (Figs. 1I–L, 2A,B). No evidence was found for NgR1 expression in Mash1+ transit amplifying cells (Fig. 2C).
Figure 1.

Expression patterns of Nogo-A and NgR1 in the adult SVZ–OB system. A, A′, Anti-Nogo-A and anti-NgR1 labeling appear complementary in both the DH–SVZ (A) and the RMS (A′). B–D, Nogo-A labeling identifies DCX-expressing neuroblasts in the DH–SVZ (B), RMS (C), and LW (D). E, Quantifications of Nogo-A and DCX coexpression in the SVZ–OB system (n = 5). F–G′, The astrocytic marker GFAP and NgR1 colocalize in germinal astrocytes. H, Quantification of NgR1 and GFAP coexpression in the SVZ–OB system (n = 5). I–L, Absence of costaining for Nogo-A and GFAP, or NgR1 and DCX. Error bars indicate SEM. Scale bars: A, A′, F, F′, I, J, 30 μm; B–D, K, I, 20 μm.
Figure 2.

A, Quantification of anti-GFAP and anti-Nogo-A immunostaining colocalization in the adult SVZ–OB system by automatic pixel analysis. B, Anti-NgR1 and anti-DCX immunostaining overlap coefficient in the SVZ–OB system. C, NgR1-expressing cells do not coexpress Mash1. D, RT-PCR analysis on MACS-sorted PSA-NCAM+ or GLAST+ cells reveals distinct expression levels for Nogo-A and NgR1 in these cell types. E–H, Expression patterns of Nogo-A and NgR1 in the postnatal SVZ. Nogo-A+ cells express DCX (E) but are negative for the astrocytic marker BLBP (F). Conversely, NgR1+ cells do not costain for the neuronal protein βIII-tubulin (G) but express the astroglial marker vimentin (H). Error bars indicate SEM. Scale bars: C, 50 μm; E, 40 μm; F, G, 20 μm; H, 30 μm.
To further confirm our immunohistochemical observations, RT-PCR was performed on adult SVZ cells selected by magnetic-activated cell sorting (MACS) on the basis of neuroblast (Peretto et al., 2005) or astrocyte lineage (Ninkovic et al., 2007) surface markers (PSA-NCAM and GLAST, respectively). Consistent with immunohistochemical data, Nogo-A or NgR1 transcripts are highly enriched in the correspondent neuronal or astroglial fractions (Fig. 2D). Moreover, postnatal (P5) brains displayed an expression pattern fully consistent with that of the adult SVZ–OB system (Fig. 2E–H). Thus, Nogo-A and NgR1 are reciprocally expressed by neuroblasts and astrocytes in the adult and postnatal SVZ–OB system.
Pharmacological antagonization of NgR1 signaling increases the rate of neurosphere formation and stimulates proliferation
To shed light on the role(s) of the Nogo-A/NgR1 signaling in the SVZ, we first evaluated the effects of Nogo-A/NgR1 pharmacological antagonization/activation on the capability of adult SVZ progenitors to generate neurospheres in response to mitogens (neurosphere assay; Reynolds and Weiss, 1992), an index of the progenitor activation state (Pastrana et al., 2009).
To neutralize NgR1 activation attributable to binding of the Nogo-66 domain, we applied the NEP1–40 peptide (Nogo-A extracellular peptide residues 1–40 amino acids of Nogo-66) (GrandPré et al., 2002) or the NgR1 function blocking antibody (Petrinovic et al., 2010; Delekate et al., 2011). Nogo-A–Δ20 activity was blocked by the specific 11C7 neutralizing antibody (Oertle et al., 2003). In parallel experiments, NgR1 was activated by treatments with Nogo-p4, the active segment of Nogo-66 (Wang and Zhu, 2008). NgR1 antagonization by both peptide and antibody treatments increased the rate of primary neurosphere generation by 1.6-fold compared with control cultures at 7 d in vitro (Fig. 3A,B,D). Furthermore, NgR1 antagonization significantly augmented the size of primary neurospheres with respect to controls, indicating enhanced proliferation (Fig. 3A,B,E). Conversely, when NgR1 was activated by Nogo-p4, we observed a remarkable decrease in both the number and the diameter of neurospheres (Fig. 3A,C,D,E). Notably, 11C7 antibody treatment did not faithfully reproduce the effects of NgR1 neutralization (rate of neurosphere generation, ctrl, 100 ± 13.3%; 11C7, 111.1 ± 40.1%; p = 0.81; diameter, ctrl, 78.9 ± 8.9 μm; 11C7, 79.3 ± 22.1 μm; p = 0.98), suggesting that the Δ20 domain is much less effective than the Nogo-66 loop in regulating the function of SVZ progenitors. When the outcomes of NgR1 antagonization or activation were tested on secondary neurosphere generation, the effects of both treatments were remarkably amplified (Fig. 3F). Furthermore, overall the effects were maintained over three passages even when only primary neurospheres were treated (data not shown), indicating that NgR1 is potently acting on mechanisms that control self-renewal.
Figure 3.

Neurosphere assay during NgR1 antagonization or activation. A–C, Micrographs depict neurospheres produced in control condition (A) and after NEP1–40 (B) or Nogo-p4 exposure (C). D, Quantification of the rate of primary neurosphere generation at 7 d of NgR1 blockade (by either NEP1–40 or anti-α-NgR1 treatment; ctrl vs NEP1–40, p = 0.005, n = 4; ctrl vs anti-NgR, p = 0.021, n = 3, paired t test) or of activation by Nogo-p4 (ctrl vs p4, p = 0.015, n = 3, paired t test). Because no differences were detected among distinct controls (p = 0.29, n = 4), data were pooled together. E, Primary neurosphere size increases after NgR1 antagonization, whereas it is reduced when NgR1 is activated (ctrl vs NEP1–40, p = 0.011; ctrl vs anti-NgR1, p = 0.013; ctrl vs Nogo-p4, p = 0.027, n = 3). F, The rate of secondary sphere formation is greatly enhanced when NgR1 is antagonized, although it is strongly reduced by the Nogo-p4 NgR1 activator. NS, Neurospheres. Paired t test: *p < 0.05, **p < 0.01. Error bars indicate SEM. Scale bars, 100 μm.
Neurosphere cells always maintained the capability to differentiate along all the three neural lineages (data not shown), revealing unaltered multipotency. Altogether, these in vitro results point to a role for NgR1-mediated signals in restricting activation and proliferation of adult SVZ progenitors.
NgR1 antagonization expands the SVZ NSC pool by cell proliferation in vivo
Starting from these in vitro findings, we investigated the role of Nogo-66-to-NgR1 signaling in the SVZ in vivo. To visualize NSCs, we used adult hGFAP::GFP mice (Zhuo et al., 1997), in which SVZ NSCs are highlighted by GFP expression driven by the human GFAP (hGFAP) promoter (Doetsch et al., 1999a). We injected the NEP1–40 peptide or vehicle solution in the lateral ventricles and investigated whether NgR1 inhibition affected NSC number and proliferation at 3 d after treatment (Fig. 4A). BrdU intraperitoneal injection was used to monitor NSC proliferation. In the peptide-treated hemispheres, the number of GFP+ cells was increased compared with controls in both the dorsal horn (DH)–SVZ (Fig. 4B–D, 1.6-fold increase) and the LW (Fig. 4E–G, twofold increase). Moreover, the high-density cellular layer underneath the ventricular surface displayed a 1.6-fold thickening (Fig. 4H). Notably, although in controls we hardly detected any GFP+/BrdU+ cells, they were much more frequent during NEP1–40 administration (2.8-fold increase; Fig. 4I–M) and included numerous mitotic figures (Fig. 4J).
Figure 4.
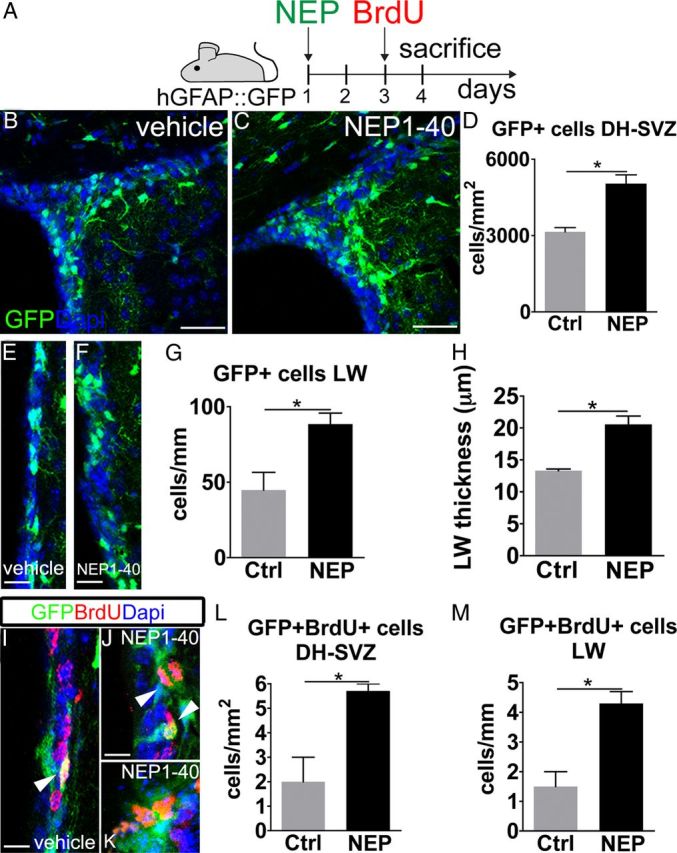
NgR1 antagonization in adult hGFAP::GFP mice. A, Experimental design. B, C, E, F, During NgR1 antagonization (C, F), the number of GFP+ cells increases compared with vehicle administration (B, E) in both the DH–SVZ and LW. D, G, GFP+ cells after NEP1–40 treatment in the DH–SVZ (D) and LW (G) (DH–SVZ, p = 0.027; LW, p = 0.042, unpaired t test, n = 6). H, Increase in the thickness of the high-density layer underneath the ventricular surface in NEP1–40-treated mice (p = 0.024, unpaired t test, n = 6). J, K, Examples of GFP/BrdU double-labeled cells (arrowheads) in vehicle-treated (I) and NEP1–40-treated (J, K) animals. L, M, Quantification of GFP/BrdU double-expressing cells in the DH–SVZ (L) and LW (M) (DH–SVZ, p = 0.023; LW, p = 0.015, unpaired t test, n = 6). Unpaired t test: *p < 0.05. Error bars indicate SEM. Scale bars: B, C, 50 μm; I–K, 10 μm; E, F, 20 μm.
To examine the progeny of NSCs at this time point, we repeated the same experiment using GLAST::CreErt2;R26R mice (Mori et al., 2006), in which a cohort of NSCs expresses β-gal during tamoxifen administration (Fig. 5A) and inherits reporter gene expression in its derivatives (Ninkovic et al., 2007). Consistent with the results obtained with the hGFAP::GFP line, in NEP1–40-treated animals, we observed a significant thickening of the LW (ctrl, 13.3 ± 0.2 μm; NEP1–40, 20.5 ± 1.3 μm; p = 0.01; n = 6) and a marked increase in the number of β-gal+ cells that were mostly positive for GFAP (Fig. 5B–G). In addition, β-gal+/Mash1+ transit amplifying cells were moderately increased (∼1.5-fold), suggesting that the immediate NSC progeny was just starting to expand (Fig. 5H,I). NEP1–40 treatment had no effect on DCX+ derivatives (Fig. 5J–M). These data are consistent with the early phases of a primary effect of NgR1 antagonization on the activation and proliferation of SVZ NSCs. They further suggest that, via interaction with NgR1, Nogo-A expressed by neuroblasts exerts a negative feedback on NSC proliferation, thereby limiting the pace of neurogenesis. To test this hypothesis, we induced a massive accumulation of neuroblasts at the SVZ by altering CP signaling (Sawamoto et al., 2006) over 2 weeks (Fig. 5N,O) and found a remarkable reduction of DH–SVZ β-gal+/BrdU+/DCX-negative cells (Fig. 5P,R), consistent with a negative control exerted by new neurons on NSC proliferation. Importantly, this decrease was reverted to control values by NEP1–40-mediated disruption of Nogo-A/NgR1 interaction (Fig. 5P,Q,S), showing an inhibitory action of NgR1 on NSCs. Similar effects were observed at the LW (ctrl, 1.6 ± 0.3 β-gal+/BrdU+/DCX-negative cells/mm; CP no cells, CP + NEP1–40, 1.7 ± 0.3 cells/mm; n = 10; one-way ANOVA, F(2,7) = 11.07, p = 0.07, post hoc Tukey's test; ctrl vs CP + NEP1–40, p = 1.00; ctrl vs CP, p = 0.013, CP vs CP + NEP1–40, p = 0.09).
Figure 5.

NgR1 neutralization in adult GLAST::CreErt2;R26R mice. A, Experimental design. Tam, Tamoxifen. B–E, β-gal+/GFAP+ cells in the DH-SVZ (B, C) and LW (D, E) of vehicle-treated (B, B′, D) and NEP1–40-threated (C, C′, E) animals. F, G, Quantifications of β-gal+ and β-gal+/DCX+ cells per DH–SVZ (F) or LW (G) per section during NgR1 antagonization (DH–SVZ, β-gal+ cells, p = 0.001; β-gal+/DCX+ cells, p = 0.25; LW, β-gal+ cells, p = 0.048; β-gal+/DCX+ cells, p = 0.11, unpaired t test, n = 6). H, I, Images illustrate the increase in β-gal+/Mash1+ cells (arrowheads) during NgR1 antagonization in the LW. J–M, Pictures illustrate the increase in β-gal-expressing cells after NEP1–40 administration in the DH–SVZ (J, K) and LW (L, M). Most of these cells do not express DCX. N, Grafting of CP and NEP1–40 injection: experimental design. O–S, When CP is grafted rostrally along the RMS (O, arrowheads), neuroblasts accumulate at the SVZ (compare R, ctrl intact, and Q, CP grafted) and proliferation of β-gal+/DCX-negative cells (yellow arrowheads, see insets) decreases at the DH–SVZ (P). However, NEP1–40 injection reverts proliferation to control values (P; one-way ANOVA, F(2,7) = 8.9, p = 0.016, n = 10; post hoc Tukey's test, ctrl vs CP, p = 0.019; ctrl vs CP + NEP1–40, p = 0.88; CP vs CP + NEP1–40, p = 0.03), despite persisting neuroblast amassing (S). Unpaired t test: *p < 0.05, **p < 0.01. Error bars indicate SEM. Scale bars: B–E, 20 μm; J, K, Q–S, 30 μm; L, M, 15 μm; H, I, 10 μm; O, 100 μm.
NgR1 antagonization increases neurogenesis in vivo
To further understand whether interfering with NgR1 signaling modifies the neurogenic capability of NSCs, NEP1–40 or vehicle solutions were chronically infused in the lateral ventricles for 1 week via osmotic minipumps (Fig. 6A). In NEP1–40-treated hemispheres, GFAP-expressing cells were packed along both the LW and DH–SVZ (Fig. 6B,D,D′), and the SVZ layers appeared thickened (Fig. 6C,D), consistent with a persistent activation of NSCs. At this time point, the number of β-gal+/DCX+ cells was also greatly expanded in the DH–SVZ (Fig. 6E–G, 1.98-fold increase), suggesting that the protracted treatment enhanced neurogenic activity of the enlarged pool of SVZ NSCs. These results could also be attributable to the reduced emigration of β-gal+/DCX+ cells from the SVZ. However, this possibility was excluded because the number of β-gal+/DCX+ cells in the RMS was unchanged (NEP1–40, 1680.1 ± 150.8 cells/mm2 vs ctrl, 1405.1 ± 180 cells/mm2, p = 0.81, n = 8), and the RMS maintained its normal organization and size (Fig. 6H,I). To further substantiate this point, we treated wild-type mice and injected a single pulse of BrdU intraperitoneally at the moment of minipump implantation to tag cells either exiting the cell cycle at the beginning of treatment (thereby maintaining high levels of BrdU) or continuing to cycle over the week (thereby diluting the BrdU). The density of BrdU+ nuclei was markedly increased in the DH–SVZ (Fig. 6L–N), and most of these cells were DCX+ neuroblasts (Fig. 6L′,M′). By selecting strongly BrdU-immunolabeled nuclei, we quantified elements that became postmitotic at the beginning of the treatment and should have moved toward the OB. However, no differences were found in the ratio between strongly BrdU-labeled neuroblasts (sBrdU+/DCX+ cells) in the RMS and DH–SVZ of NEP1–40- and vehicle-infused mice (ratio NEP1–40 RMS/SVZ, 1.9 ± 0.5 vs ctrl, 1.3 ± 0.3, p = 0.36; sBrdU+/DCX+ cells, DH–SVZ, NEP1–40, 532.9 ± 70.3 DCX+/sBrdU+ cells/mm2 vs ctrl, 377.1 ± 37.8 DCX+/sBrdU+ cells/mm2; RMS, NEP1–40, 680.5 ± 81.7 DCX+/sBrdU+ cells/mm2 vs ctrl, 505.4 ± 81.1 DCX+/sBrdU+ cells/mm2, n = 8), showing that NgR1 inhibition did not impair neuroblast progression to the OB. This result was further corroborated by experiments on SVZ explants (see below).
Figure 6.

Chronic NgR1 inhibition in adult GLAST::CreErt2;R26R and wild-type mice. A, Experimental design. B, Expansion of the area along the LW labeled for GFAP after NgR1 antagonization (p = 0.022, unpaired t test, n = 8). C–D′, Anti-GFAP staining increases in the LW (D) and DH–SVZ (D′) of NEP1–40-treated animals. E, F, H, I, Micrographs depict β-gal/DCX-expressing cells in control (E, H) and NEP1–40-treated (F, I) animals at 7 d of survival. J, K, Images illustrate the RMS of control (J) and NEP1–40-treated (K) animals 10 d after starting the treatment. G, After NEP1–40 administration, the number of these cells augments in the DH–SVZ (p = 0.006, unpaired t test, n = 6). L, M, Pictures illustrate cells with strong (arrowheads) or low (arrows) BrdU staining in the DH–SVZ of vehicle-treated (L) and NEP1–40-treated (M) mice. L′, M′, BrdU+/DCX+ cells in the DH–SVZ in control (L′) and NgR1 antagonized mice (M′). N, Quantification of BrdU+ cell increase in the DH–SVZ after NgR1 antagonization (p = 0.012, unpaired t test, n = 8). O, P, Representative images of Ki67- and DCX-expressing cells in the DH–SVZ of vehicle-treated (O) and peptide-treated (P) animals. Unpaired t test: *p < 0.05, **p < 0.01. Error bars indicate SEM. Scale bars: C, C′, O, P, 20 μm; H, I, 30 μm; D, D′, 35 μm; J, K, 45 μm; E, F, L, M, 50 μm.
Parallel analysis of neuroblasts displaying low levels of BrdU, indicative of more recent generation, yielded increased numbers in the NEP1–40-treated DH–SVZ (NEP1–40, 2571 ± 233.2 BrdU+ cells/mm2 vs ctrl, 1400.8 ± 62.8 BrdU+ cells/mm2, p = 0.008, n = 6). Among such neuroblasts, Ki67+ actively dividing cells were also expanded during NgR1 inhibition (Fig. 6O,P, 1.9-fold increase of Ki67+/DCX+ cells; NEP1–40, 2328.2 ± 259.9 cells/mm2 vs ctrl, 1226.5 ± 143.5 cells/mm2). This phenomenon was proportional to a global enhancement of active proliferation (Fig. 6O,P, 1.9-fold increase of Ki67+ cells; 3910 ± 303 cells/mm2 in the DH–SVZ in NEP1–40-treated animals vs 2018.5 ± 420.3 cells/mm2 in controls, p < 0.05), consistent with an incremented neurogenesis sustained by general stimulation of SVZ functioning.
The finding that such an incremented neurogenesis is not reflected by an enlargement of the RMS size raises the possibility that part of the newly generated neuroblasts undergo apoptosis while progressing rostrally. However, we found no evidence of enhanced apoptosis, as detected by anti-active caspase staining (ctrl, 0.5 ± 0.1 cells/SVZ–LW, NEP1–40, 0.6 ± 0.2 cells/SVZ-LW, p = 0.29; RMS, ctrl, 0.3 ± 0.2 cells/RMS, NEP1–40, 0.2 ± 0.1 cells/RMS, p = 0.32) and inspection of pyknotic nuclei (ctrl, 0.9 ± 0.1 cells/SVZ-LW, NEP1–40, 0.7 ± 0.01 cells/SVZ-LW, p = 0.78; RMS, ctrl, 0.7 ± 0.03 cells/RMS, NEP1–40, 0.6 ± 0.2 cells/RMS, p = 0.81). Furthermore, at longer survival times, we did observe an increased cellularity and an expansion of the RMS (Fig. 6J,K; ctrl, 10,178.4 ± 867.3 μm2 vs NEP1–40, 14,825.3 ± 1226.8 μm2, p = 0.03, n = 6), indicating that, after 1 week of treatment, the bulk of newly produced cells had not yet reached more rostral sites. Together, these results reveal a role for NgR1-mediated signals in the regulation of SVZ NSC neurogenesis.
Nogo-A–Δ20 supports the migration of SVZ–RMS neuroblasts independently of NgR1 in vitro
To unveil additional roles of Nogo-A/NgR1 signaling in SVZ–OB neurogenesis, we used postnatal explants of the SVZ, RMS, and SEL–OB as an in vitro model to investigate neuroblast migration toward the OB. Nogo-A+ neuroblasts move out of the explant core by chain migration, as occurs in the RMS in vivo (Fig. 7A,B) (Wichterle et al., 1997). Treatment with 11C7 to inhibit Nogo-A–Δ20 (Fig. 7C–E) induced a reduction of the areas of neuroblast migration that was more pronounced with increasing antibody concentrations (Fig. 7F). Both SVZ and RMS explants displayed smaller migration areas compared with controls (Fig. 7C–E), whereas no reduction was observed in SEL–OB explants, indicating different effects of Nogo-A signaling on tangential (RMS) or radial (OB) migration. Importantly, reduction of migratory areas was not attributable to cell death (number of pyknotic nuclei was unaltered; ctrl, 0.3 ± 0.1% vs 11C7, 0.4 ± 0.04%, p = 0.6) or to decreased production of neuroblasts by diminished proliferation (Fig. 7G). Visual inspection and quantification of the number of isolated cells (ctrl, 0.2 ± 0.1% vs 11C7, 0.3 ± 0.2%, p = 0.7, n = 4) revealed that cell chains were preserved, although 11C7 treatment increased the frequency of shorter chains (Fig. 7H; χ132 = 115.5, p < 0.0001, n = 455). These results suggest that Nogo-A–Δ20-driven signals support the tangential migratory capacity of neuroblasts but are not implicated in chain formation.
Figure 7.

Nogo-A and NgR1 inhibition in SVZ-OB explants. A, B, Micrographs show Nogo-A+ cells (A) that express DCX in SVZ-OB explants at 1 d in vitro. C, Quantification of the decrease in the migration areas of 11C7 exposed explants versus controls (SVZ, ctrl vs 11C7, p = 0.012, n = 5; RMS, ctrl vs 11C7, p = 0.008, n = 4; OB, ctrl vs 11C7, p = 0.218, n = 4, paired t test). D, E, Examples of reduced neuroblast migration after 11C7 treatment. F, Analysis of neuroblast migration areas at increasing concentrations of 11C7 (50 ng/ml, p = 0.59; 100 ng/ml, p = 0.036; 0.5 μg/ml, p = 0.030; n = 3). G, Number of Ph3+ cells in control and 11C7-treated explants (SVZ, RMS, p > 0.05, n = 3). H, Quantification of the distance from the explant core of neuroblasts moving in chains in 11C7-treated or control explants. I, J, Representative images of control (I) and NgR1 antagonized (J) explants at 1 d in vitro. K, Quantification shows no changes in the migration areas in NgR1-antagonized explants compared with controls (anti-α-NgR vs ctrl, SVZ, p = 0.89; RMS, p = 0.79; OB, p = 0.52, n = 5; NEP1–40 vs ctrl, SVZ, p = 0.91; RMS, p = 0.67; OB, p = 0.61; n = 3). L, M, NgR1 antagonization does not change neuroblast migration areas when explants are plated on a feeder layer of RMS astrocytes (NEP1–40 vs ctrl, SVZ, 96.6 ± 7.2%, 100 ± 18.2%, p = 0.87; RMS, 117.2 ± 15.2%, 100 ± 10.7%, p = 0.52; n = 3). N, O, Quantification of the decrease in the migration areas during ROCK inhibition (J, SVZ, ctrl vs Y27632, p = 0.003; ctrl vs 11C7 + Y27632, p = 0.004; SVZ, Y27632 + 11C7 vs Y27632, p = 0.31, unpaired t test, n = 4; K, RMS, ctrl vs Y27632, p = 0.009; ctrl vs 11C7 + Y27632, p = 0.03, unpaired t test; RMS, Y27632 + 11C7 vs Y27632, p = 0.42; n = 4). P, Q, Neuroblast emigration from explants during ROCK blockade. Unpaired t test: *p < 0.05, **p < 0.01, ***p < 0.0001. Error bars indicate SEM. Scale bars: A, B, 50 μm; D, E, G–J, 100 μm; P, Q, 40 μm.
Some astrocytes can also move from the explant to the matrix, in which they might promote neuroblast migration through paracrine and/or contact-mediated mechanisms, including Nogo-A/NgR1 interactions. To test this hypothesis, we exposed explants to NgR1 antagonists (anti-NgR1 antiserum and NEP1–40), but no changes in neuroblast migration were observed (Fig. 7I–K), even when the explants were plated on RMS astrocyte monolayers (García-Marqués et al., 2010) to better mimic in vivo conditions (Fig. 7L–M; NEP1–40 vs ctrl, SVZ, 96.6 ± 7.2%, 100 ± 18.2%, p = 0.87; RMS, 117.2 ± 15.2%, 100 ± 10.7%, p = 0.52, n = 3). Thus, consistent with in vivo observations, NgR1 signaling is not involved in neuroblast migration.
Nogo-A migratory function is mediated by ROCK signaling and anti-adhesive effects
Whereas the Nogo-66 sequence binding to NgR1 is present in all the three major Nogo isoforms (Nogo-A–Nogo-C), Nogo-A interacts with an additional receptor (subunit) that is not yet characterized (Schwab, 2010). Our data indicate that this Nogo-A-specific receptor, and not NgR1, influences neuroblast migration. Notably, the effects of Nogo-A–Δ20 as well as those of Nogo-66 are known to operate through the activation of the GTPase RhoA and its effector protein ROCK (Petrinovic et al., 2010; Schwab, 2010). To assess whether this pathway mediates Nogo-A action in neuroblast migration, we applied the ROCK pharmacological inhibitor Y27632 (Petrinovic et al., 2010) on SVZ and RMS explants. After treatment, we observed a dramatic decrease in cell migration (Fig. 7N–Q).
To clarify whether Nogo-A could operate through intracellular pathways other than ROCK activation, we compared the effects of ROCK inhibition with those of simultaneous application of both ROCK inhibitor and 11C7. The latter experimental condition resulted in a significant decrease in migratory areas, but this effect was not different from Y27632 alone (Fig. 7N,O). These data show that Nogo-A action on neuroblast migration is exclusively mediated by the classical ROCK signaling cascade. In addition, our observations indicate that the Nogo-A–Δ20 effect on neuroblast migration is NgR1 independent and likely causes the activation of an unidentified receptor, which operates through the ROCK pathway. This intracellular effector could promote cell migration by regulating the balance between cell adhesion and repulsion. We therefore assessed the adhesiveness of adult SVZ-derived cells to a Nogo-A–Δ20-coated substrate. In this condition, βIII-tubulin+ cells were less able to adhere to coverslips compared with when they were plated on PDL-coated glass (βIII-tubulin cells per field; PDL, 13.2 ± 0.42; Nogo-A–Δ20, 5.6 ± 0.76, p < 0.001, n = 3). Together, our data suggest that Nogo-A provides anti-adhesive signals, thus promoting neuroblast progression and sustaining their migration.
Nogo-A–Δ20 domain mediates neuroblast migration in vivo
To confirm in vivo the role of Nogo-A–Δ20 in neuroblast migration, 11C7 neutralizing antiserum or control antibodies were infused intraventricularly by osmotic minipumps for 7 d in adult mice (Fig. 8A). A pulse of BrdU was administered at the time of minipump insertion. As for Nogo-66-NgR1 antagonization, these experiments were performed in both GLAST::CreERT2; R26R and wild-type mice. In agreement with an effect distinct from that of direct NgR1 inhibition (see above), but in line with in vitro data on neurosphere formation, no significant changes were observed in GFAP expression (Fig. 8B,C; ctrl, 1.17 ± 0.45%; 11C7, 1.13 ± 0.16%, p = 0.91, n = 6), SVZ thickness (ctrl, 8.61 ± 1.16 μm; 11C7, 10.9 ± 2.92 μm, p = 0.43, n = 6), or mitotic activity (Ki67+ cells; 11C7, 2144.5 ± 267 cells/mm2 vs ctrl, 1987.5 ± 135.3 cells/mm2, p = 0.87, n = 6). However, 11C7 application triggered an approximate threefold reduction in the ratio between β-gal+/DCX+ cells in the RMS or SVZ (Fig. 8D–F; ratio RMS/SVZ, 11C7 vs ctrl β-gal+/DCX+ cells, DH–SVZ, 11C7, 2005.3 ± 41.5 cells/mm2; ctrl, 1075.9 ± 158.2 cells/mm2; RMS, 11C7, 1055.7 ± 143.6 cells/mm2; ctrl, 1412.2 ± 248.5 cells/mm2). These data are consistent with an amassing of neuroblasts in the SVZ determined by reduced migration to the OB. To further corroborate this conclusion, DCX+ cells with strong BrdU labeling, i.e., those born at the beginning of the treatment (see above), were quantified in wild-type animals. Nogo-A–Δ20 neutralization markedly reduced the ratio between DCX+/BrdU+ cells that had reached the RMS or remained the SVZ (Fig. 8G,H,K; DH–SVZ, 11C7, 435 ± 49.9 vs ctrl, 252.3 ± 71.8 BrdU+/DCX+/mm2; RMS, 11C7, 320.2 ± 50.1 vs ctrl, 574 ± 9.9 BrdU+/DCX+/mm2), showing an inhibition of neuroblast progression toward the OB. No cell spreading out of the SVZ/RMS territories indicative of altered chain formation or disruption of glial tubes was observed (Fig. 8G–J).
Figure 8.

Chronic Nogo-A inhibition in adult GLAST::CreErt2;R26R and wild-type mice. A, Experimental design. B, C, Anti-GFAP immunostaining of the LW in control conditions and after 11C7 treatment. D, E, β-gal+/DCX+ cells in the DH–SVZ of control and 11C7 animals. F, Ratio of β-gal+/DCX+ cell densities in the RMS and DH–SVZ of control and treated mice (p = 0.038, unpaired t test, n = 7). G–J, Micrographs show accumulation of BrdU+/DCX+ cells in the DH–SVZ of 11C7-treated animals and their decrease in number in the RMS. K, Ratio of BrdU+/DCX+ cell densities in the RMS and DH–SVZ of control and treated mice (p = 0.002, unpaired t test, n = 9). Unpaired t test: *p < 0.05, **p < 0.01. Error bars indicate SEM. Scale bars: B, C, 10 μm; D, E, 40 μm; I, J, 30 μm.
Finally, to rule out the possibility that increased proliferation could contribute to cell accumulation in the SVZ, we quantified DCX+/Ki67+ cells and found no change in neuroblast proliferation (DH–SVZ, 11C7 DCX+/Ki67+, 1506.5 ± 200.5 cells/mm2 vs ctrl, 1477.9 ± 110.9 cells/mm2, p = 0.91, n = 9). Although these data clearly show that Nogo-A promotes neuroblast mobilization via its Δ20 domain, they also indicate that DCX+ cell accumulation attributable to 7 d of 11C7 treatment does not reduce SVZ activity, as could be expected by the Nogo-A-dependent inhibition found during CP grafting. This suggests that Nogo-A mediated reduction of NSC activity builds up slowly and requires >7 d to become overtly detectable.
Discussion
Nogo-A and its receptor NgR1 are master regulators of CNS plasticity that physiologically restrict structural remodeling and hamper axon regeneration during injury. Here we provide the first evidence that Nogo-A/NgR1 signaling is implicated in another form of neuroplasticity, namely neurogenesis in the adult SVZ. We find that neuroblast-mediated Nogo-A signaling restricts NSC proliferation through the activation of NgR1. In this way, the rate of neuronal neogeneration can be dynamically adjusted according to the quantity of actually produced neurons. In addition, we show that Nogo-A promotes the tangential migration of neuroblasts toward the OB, independent of NgR1. Altogether, our observations point to a dual mechanism wherein newly generated neuroblasts use Nogo-A signaling to modulate neurogenic processes in the SVZ, while promoting their own emigration toward the OB.
Nogo-A/NgR1 signaling regulates NSC function
Many intrinsic mechanisms and environmental factors regulate neurogenic processes in the SVZ–OB. Based on the evidence that depletion of NSC derivatives stimulate mitotic activity of NSCs, it was hypothesized that neuroblasts inhibit NSC proliferation in physiological conditions (Doetsch et al., 1999b), but the underlying molecular mediators are essentially unknown. Here, we provide insights into this mechanism by showing that the Nogo-66 domain of Nogo-A expressed by neuroblasts negatively feeds back to NgR1+ NSCs to limit their activation, proliferation, and self-renewal. A negative action on primary progenitors has also been proposed previously for the neurotransmitter GABA released by neuroblasts, whose levels in the extracellular space can modulate NSC proliferation according to the amount of neogenerated neurons (Liu et al., 2005). In addition to soluble signals, however, regulation of NSC function may also exploit direct cell-to-cell interactions. Eph/ephrin signaling could mediate such a contact effect, because interfering with this pathway modifies NSC proliferation (Conover et al., 2000; Ricard et al., 2006). Nonetheless, the cellular distribution of these molecules in the SVZ is unclear, and their positive or negative effect on NSC proliferation is still uncertain. Therefore, it is difficult to unequivocally identify Eph/ephrin as molecular mediators of a negative feedback loop exerted by neuroblasts on NSCs. Direct interactions between neural precursor cells and NSCs have instead been implicated in the Notch-dependent reduction of NSC number and self-renewal triggered by epidermal growth factor receptor-mediated expansion of the NSC progeny (Aguirre et al., 2010). However, the molecular interactors acting in this cell-to cell interplay remain to be fully understood. In our study, a clear contact inhibition operating through Nogo-66/NgR1 interactions emerges. Grafts of CPs along the RMS arrest neuroblast migration and induce the accumulation of neogenerated neurons in the SVZ (Sawamoto et al., 2006), which is associated with a significant decrease of NSC proliferation. Most importantly, disruption of Nogo-66/NgR1 interaction completely reverses this effect, highlighting the prominent role of cell-to-cell contact.
Nogo-A/NgR1-mediated inhibition on NSCs would be released as neuroblasts emigrate away or die, thus allowing reactivation of neurogenesis. Notably, neuroblast emigration toward the OB is promoted by Nogo-A itself via the interaction of the Nogo-A–Δ20 domain with an uncharacterized receptor expressed by neogenerated neurons (see below). Hence, our results indicate a dual role for Nogo-A. On the one hand, through distinct transduction machineries, Nogo-A inhibits progenitor amplification, but on the other hand favors the release of its own inhibition by promoting neuroblast emigration from the SVZ. To our knowledge, such a mechanism of action is unique among known modulators of germinal niche activity and appears most suited to regulate the rhythm of neurogenesis in the SVZ according to the number of actually produced neurons.
The molecular pathways associated with NgR1 activation in SVZ NSCs are still undefined. Notch signaling appears to be a promising candidate. Known as a key regulator of NSC proliferation and self-renewal, it is inhibited by expansion of NSC derivatives (Hitoshi et al., 2002; Alexson et al., 2006; Aguirre et al., 2010) and regulates embryonic neurosphere formation after neutralization of Nogo-66–NgR1 interactions (Lööv et al., 2012). In addition, the gap junction components Connexins sustain progenitor proliferation (Miragall et al., 1997) and may be inhibited by NgR1 activation (Wang et al., 2011). Finally, the possible contribution of additional ligands of NgR1, such as Nogo-B and Nogo-C (Schwab, 2010), also remains to be explored.
Nogo-A supports neuroblast migration through the activation of Rho/ROCK pathway
The reciprocal expression pattern of Nogo-A in neuroblasts migrating along the RMS and NgR1 in the surrounding astrocytes of the glial tubes, prima facie, suggests a possible role for their interaction in the migratory process. However, this possibility is excluded by the lack of effects on cell migration, chain formation, and RMS organization after NgR1 antagonization in vivo or in vitro. Conversely, blockade of the Nogo-A–Δ20 domain reduces tangential migration, indicating that this process is modulated by homotypic interactions between neuroblasts and disclosing distinct functions for the amino-Nogo and Nogo-66 domains.
Nogo-A–Δ20 neutralization does not overtly impair chain formation or disrupt the RMS structure, indicating that mechanisms underlying reciprocal recognition and adhesion among neuroblasts are preserved. Rather, the observation that Nogo-A–Δ20 coating repels neuroblasts suggests that the Δ20 domain promotes cell-to-cell detachment to facilitate the effective sliding of neuroblasts onto each other. We also show that the Nogo-A–Δ20 effects on migration involve the intracellular Rho/ROCK pathway, a potent regulator of cytoskeleton dynamics and a common downstream effector of Nogo receptor-mediated functions (Schwab, 2010). Collectively, these data suggest that Nogo-A–Δ20 supports tangential migration by modulating cell-to-cell adhesion possibly through cytoskeleton reorganization. Furthermore, they show that an uncharacterized receptor (subunit), distinct from NgR1, mediates Nogo-A–Δ20 functions in SVZ neuroblasts.
These findings on neuroblast progression to the OB are in general agreement with previous studies showing that Nogo-A regulates cell motility, adhesion, and migration of neuronal precursors in the developing cortex (Oertle et al., 2003; Mingorance-Le Meur et al., 2007; Mathis et al., 2010). However, although our observations are consistent with the positive effect of Nogo-A on the tangential migration of early-born interneurons from the medial ganglionic eminence (Mingorance-Le Meur et al., 2007), they do not match the enhanced migration/motility observed in cells from Nogo-A knock-out mice (Mathis et al., 2010) and during Nogo-A/NgR1 neutralization (Oertle et al., 2003). These discrepancies are likely related to different migratory systems, each of which may be endowed with specific regulatory mechanisms. We are in fact comparing homotypic sliding of SVZ neuroblasts onto each other (chain migration), interneuron movements along corticofugal axons (Mingorance-Le Meur et al., 2007), migration along radial glia scaffolds (Mathis et al., 2010), and in vitro movements of single cells on substrates (Oertle et al., 2003; Mathis et al., 2010). For instance, NgR1 activation does not promote migration in the RMS, but it effectively reduces motility of forebrain-derived precursors (Mathis et al., 2010). Interestingly, Nogo-A–Δ20 loses its promoting function when neuroblasts reach the SEL–OB and switch from tangential chain migration to radial movement along vascular scaffolds (Bovetti et al., 2007). Thus, Nogo-A function in cell migration varies according to the molecular and cellular substrates interacting in distinct migratory systems.
In conclusion, we show here that Nogo-A contributes to the control of SVZ homeostasis by a dual mechanism (Fig. 9), involving distinct signaling domains. Heterotypic contact interaction between neuroblasts and NSCs negatively regulates mitotic rates to adjust the rhythm of neuronal neogeneration. Coincidentally, homotypic interaction among neuroblasts favors tangential migration and releases NSCs from feedback inhibition.
Figure 9.

Adult SVZ neurogenic niche and its modulation by the Nogo-A/NgR1 interaction. A, At the level of the LW of the lateral ventricle, NSCs divide to generate transit amplifying progenitors (TAPs), which, in turn, give rise to neuroblasts. B, The Nogo-66 domain on neuroblast membranes interacts with NgR1+ NSCs and limits both their proliferation and neurogenic activity. Conversely, the N terminal of Nogo-A, containing the Δ20 domain, regulates neuroblast migration through the interaction with an uncharacterized mediator that leads to the activation of the Rho/ROCK pathway.
Footnotes
This work and C.R.'s fellowship were supported by Compagnia di San Paolo (Grant 2008.2269 to A.B.). We thank Giovanna Ponti for fruitful discussions, Elena Parmigiani and Ornella Plicato for technical support, Miltenyi Biotec for help with cell separation, and Ian Martin Williams for comments on this manuscript. We are also grateful to Gwendolyn Behrendt, Silvia DeMarchis, and Donatella Garzotto for help in setting up the explant experiments.
The authors declare no competing financial interests.
References
- Aguirre A, Rubio ME, Gallo V. Notch and EGFR pathway interaction regulates neural stem cell number and self-renewal. Nature. 2010;467:323–327. doi: 10.1038/nature09347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexson TO, Hitoshi S, Coles BL, Bernstein A, van der Kooy D. Notch signaling is required to maintain all neural stem cell populations—irrespective of spatial or temporal niche. Dev Neurosci. 2006;28:34–48. doi: 10.1159/000090751. [DOI] [PubMed] [Google Scholar]
- Boda E, Viganò F, Rosa P, Fumagalli M, Labat-Gest V, Tempia F, Abbracchio MP, Dimou L, Buffo A. The GPR17 receptor in NG2 expressing cells: focus on in vivo cell maturation and participation in acute trauma and chronic damage. Glia. 2011;59:1958–1973. doi: 10.1002/glia.21237. [DOI] [PubMed] [Google Scholar]
- Bovetti S, Hsieh YC, Bovolin P, Perroteau I, Kazunori T, Puche AC. Blood vessels form a scaffold for neuroblast migration in the adult olfactory bulb. J Neurosci. 2007;27:5976–5980. doi: 10.1523/JNEUROSCI.0678-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffo A, Zagrebelsky M, Huber AB, Skerra A, Schwab ME, Strata P, Rossi F. Application of neutralizing antibodies against NI-35/250 myelin-associated neurite growth inhibitory proteins to the adult rat cerebellum induces sprouting of uninjured Purkinje cell axons. J Neurosci. 2000;20:2275–2286. doi: 10.1523/JNEUROSCI.20-06-02275.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP, Mori T, Götz M. Origin and progeny of reactive gliosis: a source of multipotent cells in the injured brain. Proc Natl Acad Sci U S A. 2008;105:3581–3586. doi: 10.1073/pnas.0709002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung HC, Hai T, Zhu W, Baggerly KA, Tsavachidis S, Krahe R, Cote GJ. Splicing factors PTBP1 and PTBP2 promote proliferation and migration of glioma cell lines. Brain. 2009;132:2277–2288. doi: 10.1093/brain/awp153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover JC, Doetsch F, Garcia-Verdugo JM, Gale NW, Yancopoulos GD, Alvarez-Buylla A. Disruption of Eph/ephrin signaling affects migration and proliferation in the adult subventricular zone. Nat Neurosci. 2000;3:1091–1097. doi: 10.1038/80606. [DOI] [PubMed] [Google Scholar]
- Delekate A, Zagrebelsky M, Kramer S, Schwab ME, Korte M. NogoA restricts synaptic plasticity in the adult hippocampus on a fast time scale. Proc Natl Acad Sci U S A. 2011;108:2569–2574. doi: 10.1073/pnas.1013322108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, Caillé I, Lim DA, García-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999a;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Doetsch F, García-Verdugo JM, Alvarez-Buylla A. Regeneration of a germinal layer in the adult mammalian brain. Proc Natl Acad Sci U S A. 1999b;96:11619–11624. doi: 10.1073/pnas.96.20.11619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer D, He Z, Benowitz LI. Counteracting the Nogo receptor enhances optic nerve regeneration if retinal ganglion cells are in an active growth state. J Neurosci. 2004;24:1646–1651. doi: 10.1523/JNEUROSCI.5119-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Marqués J, De Carlos JA, Greer CA, López-Mascaraque L. Different astroglia permissivity controls the migration of olfactory bulb interneuron precursors. Glia. 2010;58:218–230. doi: 10.1002/glia.20918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GrandPré T, Li S, Strittmatter SM. Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature. 2002;417:547–551. doi: 10.1038/417547a. [DOI] [PubMed] [Google Scholar]
- Hitoshi S, Alexson T, Tropepe V, Donoviel D, Elia AJ, Nye JS, Conlon RA, Mak TW, Bernstein A, van der Kooy D. Notch pathway molecules are essential for the maintenance, but not the generation, of mammalian neural stem cells. Genes Dev. 2002;16:846–858. doi: 10.1101/gad.975202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihrie RA, Alvarez-Buylla A. Lake-front property: a unique germinal niche by the lateral ventricles of the adult brain. Neuron. 2011;70:674–686. doi: 10.1016/j.neuron.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009;32:149–184. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Raiker SJ, Venkatesh K, Geary R, Robak LA, Zhang Y, Yeh HH, Shrager P, Giger RJ. Synaptic function for the Nogo-66 receptor NgR1: regulation of dendritic spine morphology and activity-dependent synaptic strength. J Neurosci. 2008;28:2753–2765. doi: 10.1523/JNEUROSCI.5586-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao H, Duka T, Teng FY, Sun L, Bu WY, Ahmed S, Tang BL, Xiao ZC. Nogo-66 and myelin-associated glycoprotein (MAG) inhibit the adhesion and migration of Nogo-66 receptor expressing human glioma cells. J Neurochem. 2004;90:1156–1162. doi: 10.1111/j.1471-4159.2004.02573.x. [DOI] [PubMed] [Google Scholar]
- Li X, Su H, Fu QL, Guo J, Lee DH, So KF, Wu W. Soluble NgR fusion protein modulates the proliferation of neural progenitor cells via the notch pathway. Neurochem Res. 2011;36:2363–2372. doi: 10.1007/s11064-011-0562-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang Q, Haydar TF, Bordey A. Nonsynaptic GABA signaling in postnatal subventricular zone controls proliferation of GFAP-expressing progenitors. Nat Neurosci. 2005;8:1179–1187. doi: 10.1038/nn1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lööv C, Fernqvist M, Walmsley A, Marklund N, Erlandsson A. Neutralization of Lingo-1 during in vitro differentiation of neural stem cells results in proliferation of immature neurons. PLoS One. 2012;7:e29771. doi: 10.1371/journal.pone.0029771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathis C, Schröter A, Thallmair M, Schwab ME. Nogo-A regulates neural precursor migration in the embryonic mouse cortex. Cereb Cortex. 2010;20:2380–2390. doi: 10.1093/cercor/bhp307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingorance A, Fontana X, Solé M, Burgaya F, Ureña JM, Teng FY, Tang BL, Hunt D, Anderson PN, Bethea JR, Schwab ME, Soriano E, del Río JA. Regulation of Nogo and Nogo receptor during the development of the entorhino-hippocampal pathway and after adult hippocampal lesions. Mol Cell Neurosci. 2004;26:34–49. doi: 10.1016/j.mcn.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Mingorance-Le Meur A, Zheng B, Soriano E, del Río JA. Involvement of the myelin-associated inhibitor nogo-a in early cortical development and neuronal maturation. Cereb Cortex. 2007;17:2375–2386. doi: 10.1093/cercor/bhl146. [DOI] [PubMed] [Google Scholar]
- Miragall F, Albiez P, Bartels H, de Vries U, Dermietzel R. Expression of the gap junction protein connexin43 in the subependymal layer and the rostral migratory stream of the mouse: evidence for an inverse correlation between intensity of connexin43 expression and cell proliferation activity. Cell Tissue Res. 1997;287:243–253. doi: 10.1007/s004410050749. [DOI] [PubMed] [Google Scholar]
- Mori T, Tanaka K, Buffo A, Wurst W, Kühn R, Götz M. Inducible gene deletion in astroglia and radial glia—a valuable tool for functional and lineage analysis. Glia. 2006;54:21–34. doi: 10.1002/glia.20350. [DOI] [PubMed] [Google Scholar]
- Ninkovic J, Mori T, Götz M. Distinct modes of neuron addition in adult mouse neurogenesis. J Neurosci. 2007;27:10906–10911. doi: 10.1523/JNEUROSCI.2572-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertle T, van der Haar ME, Bandtlow CE, Robeva A, Burfeind P, Buss A, Huber AB, Simonen M, Schnell L, Brösamle C, Kaupmann K, Vallon R, Schwab ME. Nogo-A inhibits neurite outgrowth and cell spreading with three discrete regions. J Neurosci. 2003;23:5393–5406. doi: 10.1523/JNEUROSCI.23-13-05393.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastrana E, Cheng LC, Doetsch F. Simultaneous prospective purification of adult subventricular zone neural stem cells and their progeny. Proc Natl Acad Sci U S A. 2009;106:6387–6392. doi: 10.1073/pnas.0810407106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretto P, Giachino C, Aimar P, Fasolo A, Bonfanti L. Chain formation and glial tube assembly in the shift from neonatal to adult subventricular zone of the rodent forebrain. J Comp Neurol. 2005;487:407–427. doi: 10.1002/cne.20576. [DOI] [PubMed] [Google Scholar]
- Petrinovic MM, Duncan CS, Bourikas D, Weinman O, Montani L, Schroeter A, Maerki D, Sommer L, Stoeckli ET, Schwab ME. Neuronal Nogo-A regulates neurite fasciculation, branching and extension in the developing nervous system. Development. 2010;137:2539–2550. doi: 10.1242/dev.048371. [DOI] [PubMed] [Google Scholar]
- Raiker SJ, Lee H, Baldwin KT, Duan Y, Shrager P, Giger RJ. Oligodendrocyte-myelin glycoprotein and Nogo negatively regulate activity-dependent synaptic plasticity. J Neurosci. 2010;30:12432–12445. doi: 10.1523/JNEUROSCI.0895-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- Ricard J, Salinas J, Garcia L, Liebl DJ. EphrinB3 regulates cell proliferation and survival in adult neurogenesis. Mol Cell Neurosci. 2006;31:713–722. doi: 10.1016/j.mcn.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Sawamoto K, Wichterle H, Gonzalez-Perez O, Cholfin JA, Yamada M, Spassky N, Murcia NS, Garcia-Verdugo JM, Marin O, Rubenstein JL, Tessier-Lavigne M, Okano H, Alvarez-Buylla A. New neurons follow the flow of cerebrospinal fluid in the adult brain. Science. 2006;311:629–632. doi: 10.1126/science.1119133. [DOI] [PubMed] [Google Scholar]
- Schwab ME. Functions of Nogo proteins and their receptors in the nervous system. Nat Rev Neurosci. 2010;11:799–811. doi: 10.1038/nrn2936. [DOI] [PubMed] [Google Scholar]
- Wang F, Zhu Y. The interaction of Nogo-66 receptor with Nogo-p4 inhibits the neuronal differentiation of neural stem cells. Neuroscience. 2008;151:74–81. doi: 10.1016/j.neuroscience.2007.10.034. [DOI] [PubMed] [Google Scholar]
- Wang X, Chun SJ, Treloar H, Vartanian T, Greer CA, Strittmatter SM. Localization of Nogo-A and Nogo-66 Receptor proteins at sites of axon-myelin and synaptic contact. J Neurosci. 2002;22:5505–5515. doi: 10.1523/JNEUROSCI.22-13-05505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wu Y, Liu M, Wang J, Ju G. Nogo-66 inhibits the dye-coupling of astrocytic gap junctions in vitro. Neurochem Res. 2011;36:1129–1134. doi: 10.1007/s11064-011-0460-z. [DOI] [PubMed] [Google Scholar]
- Wichterle H, Garcia-Verdugo JM, Alvarez-Buylla A. Direct evidence for homotypic, glia-independent neuronal migration. Neuron. 1997;18:779–791. doi: 10.1016/s0896-6273(00)80317-7. [DOI] [PubMed] [Google Scholar]
- Xiong N, Shen J, Li S, Li J, Zhao H. Expression of Nogo-66 receptor in human astrocytoma is correlated with tumor malignancy. Mol Biol Rep. 2012;39:2625–2632. doi: 10.1007/s11033-011-1015-8. [DOI] [PubMed] [Google Scholar]
- Zagrebelsky M, Schweigreiter R, Bandtlow CE, Schwab ME, Korte M. Nogo-A stabilizes the architecture of hippocampal neurons. J Neurosci. 2010;30:13220–13234. doi: 10.1523/JNEUROSCI.1044-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo L, Sun B, Zhang CL, Fine A, Chiu SY, Messing A. Live astrocytes visualized by green fluorescent protein in transgenic mice. Dev Biol. 1997;187:36–42. doi: 10.1006/dbio.1997.8601. [DOI] [PubMed] [Google Scholar]