

Abstract
Neuroinflammation plays a fundamental role in the pathogenesis of Alzheimer's disease (AD), resulting in the extensive activation of microglial and astroglial cells. Here we describe the role of myeloid-related protein Mrp14, a recently described amplifier of inflammation, in Alzheimer's disease and in the related amyloid precursor protein/presenilin1 (APP/PS1) mouse model. Detection of Mrp14 in control, mildly cognitive impaired, and AD patients revealed a strong induction of Mrp14 in protein extracts as well as in the cerebrospinal fluid, but not in blood plasma. In APP/PS1 mice, Mrp14 and its heterodimeric partner Mrp8 was found to be upregulated in microglial cells surrounding amyloid plaques. Functionally, loss of Mrp14 led to increased phagocytosis of fibrillar amyloid β (Aβ) in microglia cells in vitro and in vivo. Generating APP/PS1-transgenic mice deficient for Mrp14, we observed a decrease of key cytokines involved in APP processing, a reduction of BACE1 expression and activity, and consequently overall Aβ deposition. We therefore conclude that Mrp14 promotes APP processing and Aβ accumulation under neuroinflammatory conditions.
Introduction
Alzheimer's disease (AD) harbors an extensive microgliosis and astrogliosis in areas of plaque formation and neurofibrillary tangle formation (Heneka et al., 2010). While some aspects of this neuroinflammatory component may exert neuroprotective action, there is evidence that inflammation drives disease progression, e.g., through the inflammation-induced upregulation of BACE1 (beta-secretase 1) expression (Sastre et al., 2003, 2006) and inflammation-induced aggregation of amyloid β (Aβ) (Kummer et al., 2011).
Microglia represent the most important part of the innate immune system of the brain, defending it against invading pathogens and other harmful external agents. For that, they are equipped with a set of pattern recognition receptors (PRRs) including the Toll-like receptors (TLRs) (Beutler, 2004; Mariathasan and Monack, 2007). Activation of these receptors induces the secretion of inflammatory cytokines, thereby spreading the inflammatory reaction. Besides pathogens, there are endogenous PRR ligands called damage-associated molecular pattern (DAMP) molecules that have been described as amplifying the inflammatory reaction (Oppenheim and Yang, 2005; Zhang and Mosser, 2008).
Two members of the S100 protein family, myeloid-related proteins Mrp14 (S100A9) and Mrp8 (S100A8), have been identified as extracellular DAMPs next to their function as modulators of the tubulin cytoskeleton during cell migration (Vogl et al., 2004; Ehrchen et al., 2009). These molecules form a heterodimeric complex (called calprotectin) that amplifies inflammation via autocrine and paracrine mechanisms after release by stimulated phagocytes via unconventional secretion (Ehrchen et al., 2009). It has been reported that the Mrp14/Mrp8 (S110A9/S100A8) complex is able to induce TLR4 responses independently of lipopolysaccharide (Vogl et al., 2007). In addition, correlation of inflammatory processes in Alzheimer's disease brains and Mrp14 has been observed in the past (Shepherd et al., 2006). siRNA-mediated knock-down of Mrp14 in the Tg2576 mouse model resulted in a reduction of amyloid β burden and improved memory formation (Ha et al., 2010).
Here we investigated the Mrp14 expression in cerebrospinal fluid, blood plasma, and brain tissue from age-matched healthy controls, mildly cognitive impaired (MCI), and AD patients. Further, the effect of Mrp14 gene deficiency in amyloid precursor protein/presenilin1 (APP/PS1) transgenic mice on amyloid pathology, APP processing, Aβ degradation, and phagocytosis of microglia was assessed.
Materials and Methods
Animals.
APP/PS1 transgenic animals were obtained from The Jackson Laboratory (catalog no. 005864) (Jankowsky et al., 2001). Mrp14(−/−) mice were generated by targeted gene disruption of the Mrp14 gene as described (Manitz et al., 2003). Mice were housed in groups of four under standard conditions at 22°C and a 12 h light-dark cycle with free access to food and water. For experiments, only female mice were used. Animals were deeply anesthetized, transcardially perfused with PBS, and brains were removed. Animal care and handling was performed according to the declaration of Helsinki and approved by the local ethical committees.
Primary microglial cell culture.
Primary microglial cell cultures were prepared asdescribed previously (Terwel et al., 2011). Briefly, mixed glial cultures were prepared from P1-P3 mice and cultured in DMEM supplemented with 10% FCS and 100 U ml−1 penicillin/streptomycin.
Brain protein extraction.
Snap-frozen brain hemispheres were extracted as described previously (Kummer et al., 2011). Briefly, tissue was homogenized in PBS with protease inhibitor mixture (Sigma). The homogenate was extracted in 25 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.5% sodium deoxycholate, and 1% NP-40 for 30 min on ice. After centrifugation at 100000 × g for 30 min at 4°C, the resulting supernatant containing the soluble proteins was saved and the pellet was sonified in 25 mm Tris-HCl, pH 7.5, and 2% SDS, resulting in the solubilization of insoluble proteins. Protein concentration in the soluble fraction was determined using the BCA protein assay (Thermo Scientific).
BACE mRNA determination by qPCR.
Total RNA was isolated from brain homogenates using TRIzol (Sigma-Aldrich). RNA was quantified spectrophotometrically using a NanoDrop 1000 (Peqlab). Three micrograms of total RNA were reverse-transcribed into cDNA by using SuperScript III Reverse Transcriptase (Invitrogen). Real-time quantitative PCR assays were performed on a StepOnePlus Real-Time PCR System (Applied Biosystems) using Power SYBR Green (Applied Biosystems). The composition of the reaction mixture was as follows: 1 μl of cDNA corresponding to 40 ng of total RNA, 100 nm of each primer, and 2× Power SYBR Green PCR Master Mix (Applied Biosystems) in a total volume of 25 μl. The primer sequences were the following: GAPDH, 5′-TCACCAGGGCTGCCATTTGC-3′ (forward) and 5′-GACTCCACGACATACTCAGC-3′ (reverse); BACE1, 5′-CTGCCACAGCGAAGTTACTG-3′ (forward) and 5′-CCGCGGCTACTACCTAGAGA-3′ (reverse); and BACE2 5′-TACTACTGCCCGTGTCCACC-3′ (forward) and 5′-ACAACCTGAGGGGAAAGTCC-3′ (reverse).
Samples were analyzed simultaneously for GAPDH mRNA as the internal control. Each sample was assayed in triplicate and normalized to GAPDH.
Western blotting.
Protein samples (25 μg) were separated by 4–12% NuPAGE (Invitrogen) using MES or MOPS buffer and transferred to nitrocellulose membranes. For detection of Aβ, blots were boiled for 5 min in water. Aβ was detected using antibody 6E10 (1:2000; Covance), APP and C-terminal fragments (CTF) using antibody 140 (1:2500; (Wahle et al., 2006), insulin-degrading enzyme (IDE) using antibody PC730 (1:5000; Merck), GRP75 using antibody N52A/42 (University of California Davis/NIH NeuroMab Facility, Davis, CA), Mrp14 using antibody sc-8114 (Santa Cruz Biotechnology), glial fibrillary acidic protein (GFAP) using anti-GFAP serum (1:2000; Dako), and tubulin using antibody E7 (1:5000; Developmental Studies Hybridoma Bank, Iowa City, IA) as a loading control, followed by incubation with appropriate horseradish peroxidase-conjugated secondary antibodies. Immunoreactivity was detected by enhanced chemiluminescence reaction (Millipore), and luminescence intensities were analyzed using ChemiDoc XRS documentation system (Bio-Rad).
ELISA measurements.
Quantitative determination of Aβ40, Aβ42, interferon γ (IFN-γ), interleukin 6 (IL-6), interleukin 12 heterodimer (p70) (IL-12 p70), and tumor necrosis factor α (TNFα) was performed using a electrochemoluminescence ELISA (Meso Scale) according to the manufacturer's protocol. SDS-containing samples were diluted so that the final SDS concentration was 0.01% or lower. Mrp8-Mrp14 concentrations in plasma, lysates, and cerebrospinal fluid were determined by ELISA using the ELISA protocol for human Mrp8-Mrp14 (Vogl et al., 2007). Antibodies to human MRP14 were replaced by the mouse counterparts, and purified mouse Mrp8-Mrp14 complex was used as standard.
Phagocytosis of Aβ.
Microglial phagocytosis of fibrillar FAM-labeled Aβ1–42 (Anaspec) was measured by plate-based assay as described previously (Terwel et al., 2011). For determination of Aβ phagocytosis in vivo, mice were intraperitoneally injected 3 h before sacrification with 10 mg/kg methoxy-XO4 (Klunk et al., 2002) (kindly provided by Dr. Alfons Verbruggen, Katholieke Universiteit Leuven, Leuven, Belgium) in 50% DMSO/50% NaCl (0.9%), pH 12. Mice were perfused with ice-cold PBS and the brains were removed, chopped into pieces using scalpels and incubated in HBSS and 10% FCS containing 0.144 mg/ml collagenase type IV (Sigma-Aldrich) for 1 h at 37°C. Homogenization was achieved by pipetting through a 19 G needle. The homogenate was filtered through a cell strainer (70 μm) and centrifuged at 155 × g at 4°C for 10 min without brake. The pellet was resuspended in 9 ml of 70% Percoll in PBS, underlayered with 10 ml of ice-cold 37% Percoll in PBS, and overlayered with 6 ml of ice-cold PBS. The gradient was centrifuged at 800 × g at 4°C for 25 min without braking. Microglial cells were recovered from the 37/70% Percoll interphase, diluted with 3 vol PBS and centrifuged at 880 × g at 4°C for 25 min (Beckman Allegra) without a break. The pellet containing the microglial cells was resuspended in 200 μl PBS. For flow cytometry analysis, 50 μl of cells were diluted with 0.5 ml HBSS and centrifuged at 250 × g for 5 min at 4°C. Binding of antibodies to Fc-receptors was prevented by adding 1 μg of Fc block (BD Bioscience) and incubating for 10 min on ice. Cells were taken up in 50 μl of primary antibody mix (CD11b-APC (1:100, BioLegend, catalog no. 101212), CD45-FITC (1:100, eBioscience, catalog no. 11-0451), CD36-PE, (1:100, eBioscience, catalog no. 12-0361) and incubated for 30 min on ice. Cells were centrifuged at 250 × g for 5 min at 4°C and resuspended in 200 μl HBSS. For control and compensation, corresponding isotype control antibodies were used. Cells were measured on a FACSCanto II flow cytometer (BD Bioscience). For analysis, the CD11b+ CD45+ population was gated. Wild-type mice injected with methoxy-XO4 were used to determine the methoxy-XO4-threshold for non-phagocytosing cells, and unstained wild-type cells were used to determine background fluorescence.
Histology.
Brains were fixed in 4% paraformaldehyde for 18 h, washed in 70% ethanol for 2 h, and stored in PBS afterward. Brain hemispheres were sectioned saggitally using a vibratome (Leica). Antigen retrieval was performed by heating sections at 80°C in 100 mm citric acid, pH 6. Unspecific binding was prevented by blocking with 20% goat serum in TBS and 0.1% Triton X-100. Sections were stained free floating using the antibodies IC16 against Aβ, polyclonal serum murMRP14(VII) and anti-murMRP14 (III) against Mrp14, polyclonal serum murMRP8(V) against Mrp8 (both generated by J.R.), Iba1-antiserum (Wako), and antibody MCA711 against CD11b (AbSerotec). Sections were dried for 10 min and mounted in ImmoMount (Thermo Scientific). Sections were analyzed using a BX61 microscope equipped with an disk-scanning unit to achieve confocality (Olympus) or on a A1-MP laser-scanning microscope (Nikon). Pictures were deconvoluted using Cell∧P software (Olympus).
Human brain samples.
Frozen postmortem brain material from parietal cortex of histologically confirmed AD cases (Braak stages V–VI) as well as age-matched controls who had died from non-neurological disease, was derived from the Neurological Tissue Bank of the Biobank-Hospital Clínic-IDIBAPS (Barcelona, Spain). Postmortem times were similar between controls and AD cases. Postmortem times across all cases varied from 3,5–5 h. Age of patients was 75 ± 6 years. Non-fixed tissue samples were extracted as described previously (Kummer et al., 2011), with the exception that 25 mm Tris-HCl pH 7.5, 150 mm NaCl, and 1% Triton X-100 was used instead of RIPA buffer. We used the ubiquitously expressed chaperone GRP75 as a loading control. Cerebrospinal fluid (CSF) and plasma samples were from 10 control (age, 65.8 ± 6.710 SD; mini-mental state examination (MMSE), 29.8 ± 0.42 SD), mildly cognitive impaired (age, 72.7 ± 4.76 SD; MMSE, 26 ± 2.49 SD), and 10 diagnosed AD patients (age, 72.8 ± 6.41 SD; MMSE, 20.4 ± 2.76 SD).
Statistical analysis.
Results were expressed as mean ± SEM of at least three experiments. For normally distributed samples, Student‘s t test was used for single comparisons of means, and one-way ANOVA was used for multiple comparisons of means combined with Tukey's post hoc test to evaluate statistical significance. The levels for statistical significance were *p < 0.05, **p < 0.01, and ***p < 0.001. All evaluations were done using GraphPad Prism 5.03 software.
Results
Increased expression of Mrp14 in Alzheimer's disease
Since inflammation is one of the key features of AD, we tested whether the inflammatory molecule Mrp14 is changed under disease conditions. We observed a strongly increased expression of Mrp14 in brain lysates of AD patients compared to healthy, age-matched controls by Western blot (Fig. 1A). Quantification revealed a >9-fold increase compared to the control group (Fig. 1B). Since the analysis of cerebrospinal fluid is a common procedure for establishing the diagnosis of AD, we also tested CSF of control, mildly cognitive impaired, and AD patients using a Mrp14/Mrp8 sandwich ELISA (Fig. 1C). Mrp14/Mrp8 was only slightly increased in MCI patients but strongly upregulated in AD patients, demonstrating its release from brain parenchyma. In contrast, analysis of plasma samples derived from age-matched controls, MCI, and AD patients did not reveal any changes in the Mrp14/Mrp8 concentration (Fig. 1D).
Figure 1.
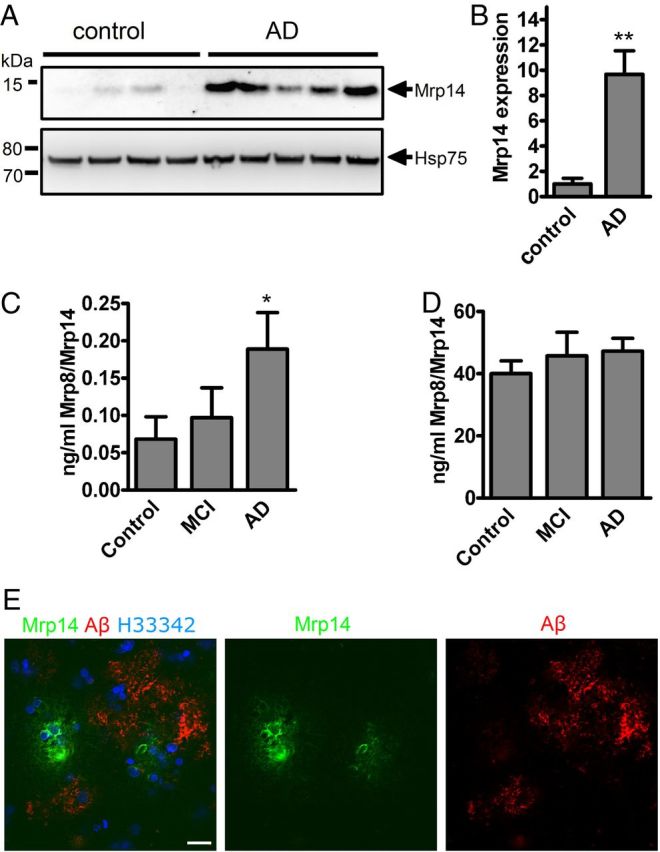
A, Brain lysates from control and AD patients were separated by NuPAGE and immunoblotted using antibody sc-8114 against Mrp14. B, Densitometric analysis of A (mean of n = 4 control and n = 5 AD patients ± SEM, Student's t test, *p < 0.05). C, The Mrp14/Mrp8 complex was determined in the cerebrospinal fluid of control, MCI, and Alzheimer's disease patients by sandwich ELISA (n = 10, mean ± SEM, one-way ANOVA, Tukey post hoc test, *p < 0.05). D, Determination of the Mrp14/Mrp8 complex in the plasma of control, MCI, and AD patients (n = 10, mean ± SEM). E, AD brain section immunostained using antibodies IC16 against Aβ and polyclonal antiserum against Mrp14. Nuclei were stained using H33342 (bar = 10 μm).
Finally, immunohistochemical analysis of AD brain sections (Braak stage V–VI) using antibodies against Mrp14 and Aβ revealed Mrp14-positive cells in the vicinity of amyloid plaques (Fig. 1E). We therefore conclude that Mrp14 is a neuroinflammatory marker for AD.
Mrp14/Mrp18 are expressed in activated microglia surrounding amyloid plaques
Expression of Mrp14 has been demonstrated in microglia under a variety of pathological conditions in humans, including AD (Roth et al., 2003; Shepherd et al., 2006). Testing the expression of Mrp14 in the APP/PS1 mouse model, we observed intracellular expression of Mrp14 and its binding partner Mrp8 in the vicinity of amyloid plaque (Fig. 2A,B). In addition, these cells coexpressed the microglial marker CD11b (Fig. 2C) and Iba1 (data not shown), identifying these cells are microglia.
Figure 2.
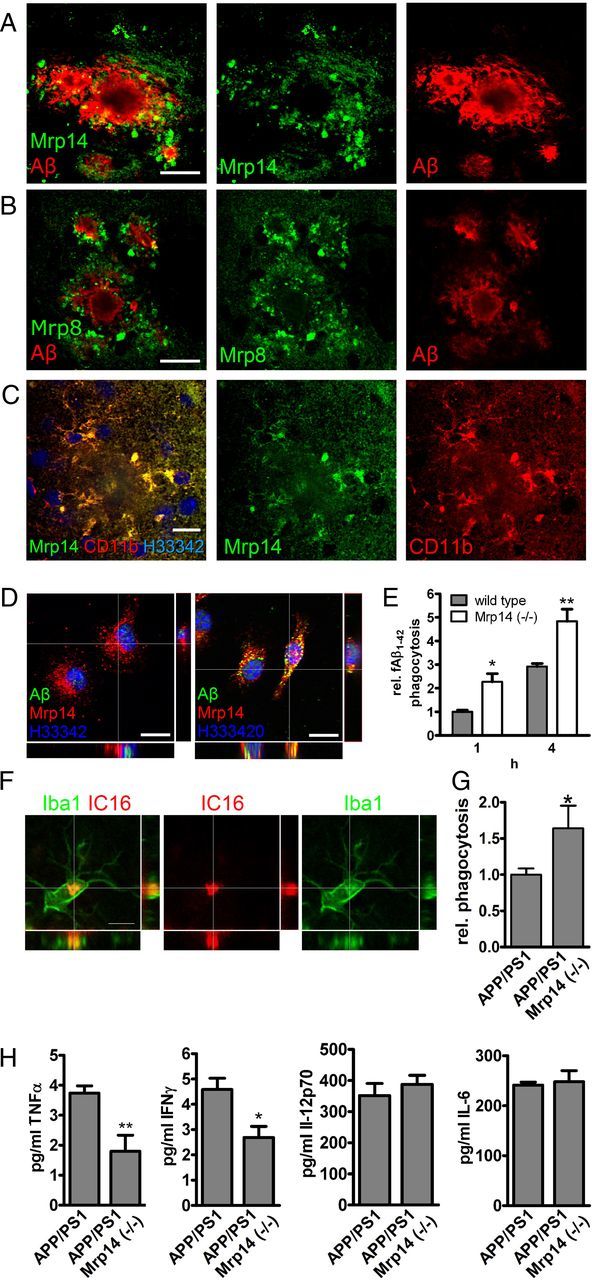
A–C, Brain section of 9-month-old APP/PS1 mice were double-stained with antibody IC16 against Aβ and a polyclonal serum against Mrp14 (scale bar, 20 μm) (A), antibody IC16 against Aβ and a polyclonal serum against Mrp8 (scale bar, 20 μm) (B), and a polyclonal serum against Mrp14 and antibody MCA711 against CD11b (scale bar, 10 μm) (C). D, Immunocytochemical detection of Mrp14 in primary mouse microglia in the absence (left) or presence (right) of 0.5 μm FAM-labeled Aβ1–42 for 1 h (scale bars, 10 μm). E, Quantification of FAM-labeled Aβ1–42 uptake in primary mouse microglia from wild-type and Mrp14(−/−) mice using an plate-based assay (representative experiment performed in pentaduplicate ± SEM, Student's t test, *p < 0.05, **p < 0.01). F, Immunostaining of 9-month-old APP/PS1 mouse using antibody IC16 against Aβ and an anti-Iba1 antiserum (scale bar, 5 μm). G, Quantification of the in vivo phagocytosis assay of 16-month-old APP/PS1 and APP/PS1 Mrp14(−/−) mice (mean of n = 4 for APP/PS1 and n = 3 for APP/PS1 Mrp14(−/−) ± SEM, Student's t test, *p < 0.05). H, Determination of the inflammatory cytokines TNFα, INFγ, IL-6, and IL12 p70 in the lysate of APP/PS1 and APP/PS1 Mrp14(−/−) mice by multiplex ELISA (mean of n = 7 ± SEM, Student's t test, *p < 0.05, **p < 0.01).
Aβ phagocytosis is increased by Mrp14 deficiency
Phagocytosis of extracellular debris is one of the most important functions of microglial cells. It has been shown that microgliaare indeed capable of phagocytosing Aβ peptides. We therefore tested the ability of Mrp14(−/−) microglia to take up fibrillar FAM-labeled Aβ1–42 in vitro. Mrp14 is expressed intracellularly in primary mouse microglia (Fig. 2D). Incubation of these cells with Aβ1–42 resulted in the intracellular accumulation that colocalized with Mrp14 only to a minor extent (Fig. 2D). Comparison of the phagocytic capabilities of wild-type and Mrp14(−/−) microglia showed that absence of Mrp14 strongly induced phagocytosis compared to wild-type microglia after 1 and 4 h (Fig. 2E).
In APP/PS1 mice we observed microglial uptake of Aβ, in particular in the vicinity of amyloid plaques (Fig. 2F). To determine the influence of Mrp14 deficiency on the uptake of Aβ in vivo, we injected wild-type, APP/PS1, and APP/PS1 Mrp14(−/−) mice with the amyloid dye methoxy-XO4 following the analysis of microglia by flow cytometry, a new method recently established in our laboratory. For this, mice were injected with methoxy-X04 3 h before scarification, and microglia were prepared by density gradient fractionation. Methoxy-X04-positive CD11b+/CD45+ cells were detected in APP/PS1, but were almost absent in wild-type mice. More importantly we observed increased signal for methoxy-XO4 in APP/PS1 Mrp14(−/−) mice, suggesting increased uptake of Aβ (Fig. 2G). Therefore, loss of Mrp14 may release microglia from their phagocytic arrest during AD.
Since Mrp14(−/−) has been implicated in the production of inflammatory cytokines (Vogl et al., 2007), we compared the amount of TNFα, IL-6, IL-12 p70, and INF-γ in lysates of APP/PS1 and APP/PS1 Mrp14(−/−) at 9 month of age using a multiplex ELISA. Levels of TNFα and INTγ were reduced by 50% in APP/PS1 Mrp14(−/−) mice whereas IL-6 and Il12p70 remained unaffected, suggesting a selective regulation of cytokine production by Mrp14 (Fig. 2H).
APP/PS1 Mrp14(−/−) mice show reduced amyloid deposition
To test whether knock-out of Mrp14 has an impact on amyloid deposition, we crossbred the APP/PS1 model with Mrp14(−/−) mice. Mice were analyzed at 9 months of age. Even so the level of the APP-transgene remained unaffected, and the steady-statelevel of APP CTF, as well as that of RIPA soluble Aβ, decreased (Fig. 3A,B). Determination of Aβ levels in the RIPA- and SDS-soluble fraction by ELISA revealed a robust reduction of Aβ40 and Aβ42 in the SDS-soluble fraction (Fig. 3C). We could confirm the observation from the immunoblot analysis for Aβ40 in the RIPA-soluble fraction, but we failed to detect Aβ42 (Fig. 3C). In addition, we observed reduced plaque pathology in APP/PS1 Mrp14(−/−) mice using thioflavin S histochemistry in hippocampal (Fig. 3D) and cortical (Fig. 3E) sections. Changes in the CTF of APP and the reduction of Aβ in the RIPA-soluble fraction suggest that the amyloidogenic processing of APP is affected in response to MRP14 deficiency. Testing the expression of BACE1 and 2 by quantitative PCR, we observed a reduction in the mRNA levels of both enzymes (Fig. 3F), offering one explanation for the Aβ-lowering effect of Mrp14 gene deletion. BACE1 expression was found in neurons and astrocytes using NeuN and GFAP as marker proteins, respectively (Fig. 3G,H), suggesting that Mrp14(−/−) influences BACE1 mRNA levels by an paracrine mechanism.
Figure 3.
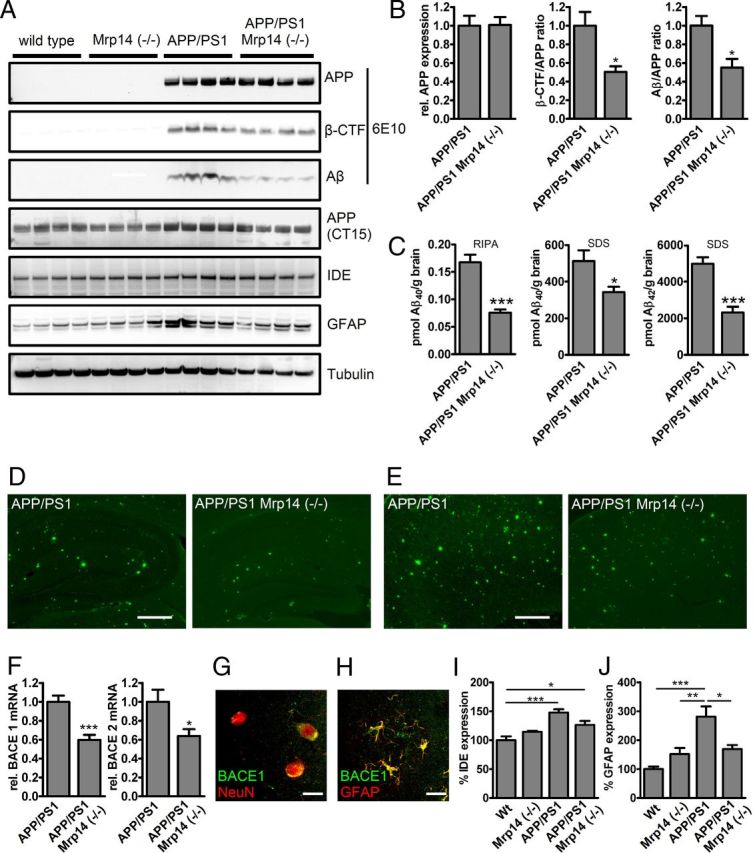
A, Western blot analysis of RIPA-soluble fractions of 9-month-old APP/PS1 and APP/PS1 Mrp14(−/−) mice for APP, CTFs, Aβ, IDE, GFAP, and tubulin. B, Densitometric analysis of A for APP expression, β-CTF/APP ratio, and Aβ/APP ratio in APP/PS1 and APP/PS1 Mrp14(−/−) mice (mean of n = 4 ± SEM, Student's t test, *p < 0,05, ***p < 0.001). C, ELISA analysis of RIPA-soluble and SDS-soluble fractions for Aβ40 and Aβ42 in APP/PS1 and APP/PS1 Mrp14(−/−) mice (mean of n = 7 ± SEM, Student's t test, *p < 0.05, ***p < 0,001). D, Representative images of thioflavin S-stained hippocampi from APP/PS1 and APP/PS1 Mrp14(−/−) mice (scale bar, 10 μm). E, Representative images of thioflavin S-stained cortices from APP/PS1 and APP/PS1 Mrp14(−/−) mice (scale bar, 10 μm) F, Determination of BACE1 and BACE2 mRNA expression (rel., relative) by qPCR in APP/PS1 and APP/PS1 Mrp14(−/−) mice (mean of n = 8 ± SEM, Student's t test, *p < 0,05, ***p < 0,001). G–I, Densitometric analysis of A for IDE expression in APP/PS1 and APP/PS1 Mrp14(−/−) mice (mean of n = 4 ± SEM, Student's t test, *p < 0.05, **p < 0.01). Wt, Wild type. J, Densitometric analysis of A for GFAP expression in APP/PS1 and APP/PS1 Mrp14(−/−) mice (mean of n = 4 ± SEM, Student's t test, *p < 0.05, **p < 0.01).
In addition, we observed that expression of insulin-degrading enzyme was increased in the APP/PS1 mouse model compared to wild-type and Mrp14(−/−) mice and that expression was lower in APP/PS1 Mrp14(−/−) mice without showing statistical significance (Fig. 3E), whereas when immunoblotting for the astrocytic marker GFAP, APP/PS1 Mrp14(−/−) mice showed reduced expression of this protein, demonstrating that loss of Mrp14(−/−) ameliorates inflammation in this model (Fig. 3G).
Discussion
Neuroinflammation is a key component of Alzheimer disease with diverse functions and consequences during disease progression. Microglia represent key players of the inflammatory response in AD by releasing a variety of proinflammatory molecules. It has been reported that Mrp14 and its binding partner Mrp8 are secreted by activated phagocytes and strongly upregulated in numerous inflammatory diseases, including sepsis, rheumatoid arthritis, and vasculitis (Roth et al., 2003).
Here we report a characteristic upregulation of Mrp14 in brain lysates as well as in cerebrospinal fluid of AD patients that was not observed in blood plasma of these patients, pointing toward a role of this inflammatory mediator in AD. Transferring this finding into an AD mouse model, we observed that lack of Mrp14 results in decreased Aβ burden. Similar results have been observed in a model of local Mrp14 suppression in Tg2576 mice (Ha et al., 2010).
Here we show that Aβ reduction was due to altered processing of APP caused by decreased transcription of BACE1 and 2, which lowers the formation of β-CTFs, the direct precursors of Aβ. The impact of neuroinflammation on the expression of BACE1 has been recently elucidated in vitro (Sastre et al., 2003) and in vivo (Heneka et al., 2005; Sastre et al., 2006). Under the influence of the proinflammatory molecules TNFα and IFNγ, the activity of the BACE1 promoter is induced, resulting in increased generation of β-CTF (β-secretase-cleaved C-terminal fragment of APP) and subsequently to more Aβ generation. This induction can be suppressed by binding of the anti-inflammatory transcription factor PPARγ to a specific response element within the BACE1 promoter (Sastre et al., 2006).
A mixture of classical activation, acquired deactivation, and increasing alternative activation is observed in AD (Town et al., 2005; Colton et al., 2006) and may ultimately further our understanding of the complex roles that microglia may play in neurodegenerative diseases, as well as how to manipulate them therapeutically.
In addition, we observed that deletion of Mrp14 stimulates the phagocytosis of microglia. Since Mrp14 is released by microglia upon exposure to Aβ (Ha et al., 2010), this result points toward an autocrine mechanism, which has been suggested previously (Ehrchen et al., 2009). We therefore speculate that Mrp14 drives microglia into a proinflammatory state thereby compromising microglial key functions such as phagocytosis. The observed detrimental impact of long lasting Aβ-induced inflammatory stimulation of microglia and the resulting deficits in Aβ clearance is in agreement with this hypothesis (Hickman et al., 2008).
Potential pathways for the amplification of phagocyte activation by Mrp14 have been suggested. There is a direct interaction of Mrp8 with the TLR4/MD2 complex with subsequent induction of TNFα (Vogl et al., 2007). In addition, the receptor for advanced glycation end products (RAGE) is a receptor for Mrp14 (Gebhardt et al., 2008; Turovskaya et al., 2008). This is of particular interest, since overexpression of RAGE in microglia results in increased amyloid plaque load and worsens behavioral performance in APP-transgenic mice via a p38/Erk1/2-mediated pathway leading to enhanced TNFα production (Fang et al., 2010).
These findings are corroborated by another molecule from the S100 family, S100B, which is plays a role in AD as well (Mrak and Griffin, 2001). Overexpression of this glial activation marker in astrocytes using the Tg2576 mouse model of AD resulted in the increase of Aβ deposits, BACE1 expression accompanied by β-CTF accumulation, activation of microglia and astrocytes, and production of proinflammatory cytokines (Mori et al., 2010).
In conclusion, lack of Mrp14 reduces the production of proinflammatory cytokines in an AD mouse model with dual consequences. First, reduced inflammation decreases the transcription of BACE1 and BACE2, thereby limiting the generation of Aβ and its subsequent deposition in senile plaques. Secondly, prevention of excessive microglial activation favors amyloid degradation by microglial phagocytosis, thereby acting to protect against Alzheimer pathology. We therefore suggest that reduction of Mrp14 in the chronic inflammatory phase of AD may be a treatment opportunity.
Footnotes
This study was supported by a grant from the Deutsche Forschungsgemeinschaft (HE 3350/4-1 und HE 3350 4-2; KFO177, TP4) to M.T.H. We are indebted to the Neurological Tissue Bank of the IDIBAPS Biobank, Barcelona, Spain, for procuring human brain samples, and we thank Dr. Ellen Gelpi for helpful suggestions. We are grateful to Drs. Sascha Weggen and C. Piertzik for providing antibody IC16. The E7 antibody developed by M. Klymkowsky and the Abl-93 antibody developed by J. T. August were obtained from the Developmental Studies Hybridoma Bank and developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242.
References
- Beutler B. Toll-like receptors and their place in immunology. Where does the immune response to infection begin? Nat Rev Immunol. 2004;4:498. doi: 10.1038/nri1401. [DOI] [PubMed] [Google Scholar]
- Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrchen JM, Sunderkötter C, Foell D, Vogl T, Roth J. The endogenous Toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J Leukoc Biol. 2009;86:557–566. doi: 10.1189/jlb.1008647. [DOI] [PubMed] [Google Scholar]
- Fang F, Lue L-F, Yan S, Xu H, Luddy JS, Chen D, Walker DG, Stern DM, Yan S, Schmidt AM, Chen JX, Yan SS. RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer's disease. FASEB J. 2010;24:1043–1055. doi: 10.1096/fj.09-139634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhardt C, Riehl A, Durchdewald M, Németh J, Fürstenberger G, Müller-Decker K, Enk A, Arnold B, Bierhaus A, Nawroth PP, Hess J, Angel P. RAGE signaling sustains inflammation and promotes tumor development. J Exp Med. 2008;205:275–285. doi: 10.1084/jem.20070679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha T-Y, Chang K-A, Kim J, Kim H-S, Kim S, Chong YH, Suh Y-H. S100a9 knockdown decreases the memory impairment and the neuropathology in Tg2576 mice, AD animal model. PLoS ONE. 2010;5:e8840. doi: 10.1371/journal.pone.0008840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Sastre M, Dumitrescu-Ozimek L, Dewachter I, Walter J, Klockgether T, Van Leuven F. Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. J Neuroinflammation. 2005;2:22. doi: 10.1186/1742-2094-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, O'Banion MK, Terwel D, Kummer MP. Neuroinflammatory processes in Alzheimer's disease. J Neural Transm. 2010;117:919–947. doi: 10.1007/s00702-010-0438-z. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Bacskai BJ, Mathis CA, Kajdasz ST, McLellan ME, Frosch MP, Debnath ML, Holt DP, Wang Y, Hyman BT. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol. 2002;61:797–805. doi: 10.1093/jnen/61.9.797. [DOI] [PubMed] [Google Scholar]
- Kummer MP, Hermes M, Delekate A, Hammerschmidt T, Kumar S, Terwel D, Walter J, Pape H-C, König S, Roeber S, Jessen F, Klockgether T, Korte M, Heneka MT. Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron. 2011;71:833–844. doi: 10.1016/j.neuron.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Manitz M-P, Horst B, Seeliger S, Strey A, Skryabin BV, Gunzer M, Frings W, Schönlau F, Roth J, Sorg C, Nacken W. Loss of S100A9 (MRP14) results in reduced interleukin-8-induced CD11b surface expression, a polarized microfilament system, and diminished responsiveness to chemoattractants in vitro. Mol Cell Biol. 2003;23:1034–1043. doi: 10.1128/MCB.23.3.1034-1043.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- Mori T, Koyama N, Arendash GW, Horikoshi-Sakuraba Y, Tan J, Town T. Overexpression of human S100B exacerbates cerebral amyloidosis and gliosis in the Tg2576 mouse model of Alzheimer's disease. Glia. 2010;58:300–314. doi: 10.1002/glia.20924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrak RE, Griffin WS. Interleukin-1, neuroinflammation, and Alzheimer's disease. Neurobiol Aging. 2001;22:903–908. doi: 10.1016/s0197-4580(01)00287-1. [DOI] [PubMed] [Google Scholar]
- Oppenheim JJ, Yang D. Alarmins: chemotactic activators of immune responses. Curr Opin Immunol. 2005;17:359–365. doi: 10.1016/j.coi.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Roth J, Vogl T, Sorg C, Sunderkötter C. Phagocyte-specific S100 proteins: a novel group of proinflammatory molecules. Trends Immunol. 2003;24:155–158. doi: 10.1016/s1471-4906(03)00062-0. [DOI] [PubMed] [Google Scholar]
- Sastre M, Dewachter I, Landreth GE, Willson TM, Klockgether T, van Leuven F, Heneka MT. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J Neurosci. 2003;23:9796–9804. doi: 10.1523/JNEUROSCI.23-30-09796.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre M, Dewachter I, Rossner S, Bogdanovic N, Rosen E, Borghgraef P, Evert BO, Dumitrescu-Ozimek L, Thal DR, Landreth G, Walter J, Klockgether T, van Leuven F, Heneka MT. Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc Natl Acad Sci U S A. 2006;103:443–448. doi: 10.1073/pnas.0503839103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd CE, Goyette J, Utter V, Rahimi F, Yang Z, Geczy CL, Halliday GM. Inflammatory S100A9 and S100A12 proteins in Alzheimer's disease. Neurobiol Aging. 2006;27:1554–1563. doi: 10.1016/j.neurobiolaging.2005.09.033. [DOI] [PubMed] [Google Scholar]
- Terwel D, Steffensen KR, Verghese PB, Kummer MP, Gustafsson J-Å, Holtzman DM, Heneka MT. Critical role of astroglial apolipoprotein E and liver X receptor-α expression for microglial Aβ phagocytosis. J Neurosci. 2011;31:7049–7059. doi: 10.1523/JNEUROSCI.6546-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turovskaya O, Foell D, Sinha P, Vogl T, Newlin R, Nayak J, Nguyen M, Olsson A, Nawroth PP, Bierhaus A, Varki N, Kronenberg M, Freeze HH, Srikrishna G. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis. 2008;29:2035–2043. doi: 10.1093/carcin/bgn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl T, Ludwig S, Goebeler M, Strey A, Thorey IS, Reichelt R, Foell D, Gerke V, Manitz MP, Nacken W, Werner S, Sorg C, Roth J. MRP8 and MRP14 control microtubule reorganization during transendothelial migration of phagocytes. Blood. 2004;104:4260–4268. doi: 10.1182/blood-2004-02-0446. [DOI] [PubMed] [Google Scholar]
- Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MAD, Nacken W, Foell D, van der Poll T, Sorg C, Roth J. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13:1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- Wahle T, Thal DR, Sastre M, Rentmeister A, Bogdanovic N, Famulok M, Heneka MT, Walter J. GGA1 is expressed in the human brain and affects the generation of amyloid beta-peptide. J Neurosci. 2006;26:12838–12846. doi: 10.1523/JNEUROSCI.1982-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Mosser DM. Macrophage activation by endogenous danger signals. J Pathol. 2008;214:161–178. doi: 10.1002/path.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]