

Abstract
Memory stabilization following encoding (synaptic consolidation) or memory reactivation (reconsolidation) requires gene expression and protein synthesis (Dudai and Eisenberg, 2004; Tronson and Taylor, 2007; Nader and Einarsson, 2010; Alberini, 2011). Although consolidation and reconsolidation may be mediated by distinct molecular mechanisms (Lee et al., 2004), disrupting the function of the transcription factor CREB impairs both processes (Kida et al., 2002; Mamiya et al., 2009). Phosphorylation of CREB at Ser133 recruits CREB binding protein (CBP)/p300 coactivators to activate transcription (Chrivia et al., 1993; Parker et al., 1996). In addition to this well known mechanism, CREB regulated transcription coactivators (CRTCs), previously called transducers of regulated CREB (TORC) activity, stimulate CREB-mediated transcription, even in the absence of CREB phosphorylation. Recently, CRTC1 has been shown to undergo activity-dependent trafficking from synapses and dendrites to the nucleus in excitatory hippocampal neurons (Ch'ng et al., 2012). Despite being a powerful and specific coactivator of CREB, the role of CRTC in memory is virtually unexplored. To examine the effects of increasing CRTC levels, we used viral vectors to locally and acutely increase CRTC1 in the dorsal hippocampus dentate gyrus region of mice before training or memory reactivation in context fear conditioning. Overexpressing CRTC1 enhanced both memory consolidation and reconsolidation; CRTC1-mediated memory facilitation was context specific (did not generalize to nontrained context) and long lasting (observed after virally expressed CRTC1 dissipated). CREB overexpression produced strikingly similar effects. Therefore, increasing CRTC1 or CREB function is sufficient to enhance the strength of new, as well as established reactivated, memories without compromising memory quality.
Introduction
The stabilization of long-term memory following encoding (consolidation) or memory reactivation (reconsolidation) requires gene expression and de novo protein synthesis (Dudai and Eisenberg, 2004; Tronson and Taylor, 2007; Nader and Einarsson, 2010; Alberini, 2011). Although the precise molecular mechanisms critically mediating these processes may differ (Lee et al., 2004; Alberini, 2005), we, and others, showed disrupting cAMP/Ca2+ responsive element binding protein (CREB) function impairs both synaptic consolidation (Bourtchuladze et al., 1994; Yin et al., 1994; Guzowski and McGaugh, 1997; Bartsch et al., 1998; Kida et al., 2002; Pittenger et al., 2002; Frankland et al., 2004) and reconsolidation (Kida et al., 2002; Mamiya et al., 2009; Yang et al., 2011). In contrast, increasing CREB promotes memory consolidation (Josselyn et al., 2001; Wallace et al., 2004; Han et al., 2007; Restivo et al., 2009; Zhou et al., 2009; Sekeres et al., 2010). Whether increasing CREB function similarly promotes memory reconsolidation is unknown.
CREB modulates the transcription of genes with cAMP responsive elements (CREs) (Shaywitz and Greenberg, 1999; De Cesare and Sassone-Corsi, 2000; Mayr and Montminy, 2001; Lonze and Ginty, 2002). Phosphorylation of CREB at Ser133 promotes recruitment of CREB binding protein (CBP) and p300 to stimulate transcription (Sheng et al., 1991; Radhakrishnan et al., 1997; Chawla et al., 1998; Shaywitz and Greenberg, 1999; Kornhauser et al., 2002). Interestingly, phosphorylation of CREB at Ser133 is not always sufficient to stimulate transcription, suggesting additional transcriptional modulators are involved. Indeed, CREB regulated transcription coactivators (CRTCs) were identified as potent modulators of CREB-mediated transcription (Iourgenko et al., 2003; Conkright et al., 2003b). Although CRTCs may potentiate the ability of CREB to recruit CBP/p300 (Xu et al., 2007), CRTCs stimulate CRE-dependent transcription via a phosphorylation-independent interaction with the basic leucine zipper domain of CREB (Iourgenko et al., 2003; Conkright et al., 2003b). Therefore, CRTCs may provide a powerful mechanism for specifically enhancing CREB function.
In mice, CRTCs are encoded by three genes (CRTC1–3) (Iourgenko et al., 2003; Conkright et al., 2003b) with CRTC1 showing highest brain expression (Wu et al., 2006; Zhou et al., 2006; Kovács et al., 2007; Altarejos et al., 2008; Watts et al., 2011). Under basal conditions, CRTC1 is sequestered in the cytoplasm; dephosphorylation of CRTC1 by calcium promotes nuclear translocation (Screaton et al., 2004) while elevated cAMP increases the nuclear persistence of CRTC1 (Ch'ng et al., 2012). Nuclear accumulation of CRTC1 is a sensitive readout of synaptic activity in hippocampal neurons (Ch'ng et al., 2012). Intact CRTC1 function is necessary for CRE-mediated transcription; decreasing CRTC1 levels or blocking the interaction between CRTC1 and CREB disrupts, while overexpressing CRTC1 increases, CRE-mediated transcription in culture (Zhou et al., 2006; Kovács et al., 2007; Ch'ng et al., 2012). Although the role of CRTC1 in memory is unexplored, previous results show that blocking CRTC1 function disrupts, while increasing CRTC1 levels enhances, L-LTP in hippocampal slices (Zhou et al., 2006; Kovács et al., 2007).
Here we examined the effects of increasing CRTC1 or CREB function in a regionally and temporally specific manner on memory consolidation and reconsolidation.
Materials and Methods
HSV vectors
Wild-type full-length CRTC1 or CREB cDNAs (kindly provided by Dr. Satoshi Kida, Tokyo University of Agriculture) were subcloned into the bicistronic herpes simplex virus (HSV) vectors that coexpress green fluorescent protein (GFP) as a fluorescent reporter (HSV-p1005; Russo et al., 2009). In this vector, GFP expression is driven by a CMV promoter whereas CRTC1 or CREB expression is driven by the constitutive promoter for the HSV immediate-early gene IE 4/5. Transgene expression using this viral system typically peaks 3 d, and dissipates within 10–14 d, following microinjection (Josselyn et al., 2001; Barrot et al., 2002; Vetere et al., 2011) (see Fig. 2b). As a control, we used HSV-expressing GFP alone. HSV virus was packaged using a replication-defective helper virus, purified on a sucrose gradient, and pelleted and resuspended in 10% sucrose, as previously described (Carlezon et al., 1998; Han et al., 2007, 2008, 2009). The average titer of the virus stocks was typically 4.0 × 107 infectious units/ml.
Figure 2.

Microinjection of CRTC1 vector induces robust expression of CRTC1 in the DG. a, Vector microinjection induces robust localized transgene expression (GFP, green) in DG of dorsal hippocampus. Left, Coronal brain images (adapted from Paxinos and Franklin, 2001) depicting the AP extent of typical viral vector infection (−1.46 to −3.08 mm posterior to bregma). Right, Corresponding image showing transgene expression (GFP, green) following vector microinjection (assessed 4 d postmicroinjection; counterstained with DAPI, blue). Scale bar, 200 μm. b, HSV preferentially infects excitatory neurons (DG granule cells) (assessed 4 d postmicroinjection, top; counterstained with DAPI, blue). Scale bar, 50 μm. HSV-driven transgene expression dissipates by 10 d postmicroinjection (bottom). c, Microinjection of CRTC1 vector increases CRTC1 protein levels. Immunohistochemical staining for CRTC1 protein (red) in the DG 4 d following microinjection of GFP vector (top) or CRTC1 vector (bottom). Mice microinjected with CRTC1 vector show higher levels of CRTC1 protein levels than mice microinjected with GFP vector, in infected neurons (green). Scale bars: 200 μm (top); 50 μm (bottom).
Preparation of primary hippocampal neurons
Primary hippocampal neurons were prepared from E18–E19 mice (see below). Briefly, hippocampi were collected in cold PBS and dissociated using trypsin (0.25%, 12 min at 37°C) and a glass Pasteur pipette. Neurons were plated onto poly-l-lysine-treated glass coverslips (immunostaining) or culture plates (luciferase assay) in minimum essential medium with 10% horse serum, 0.6% glucose, 1 mm glutamax, 50 μg/ml streptomycin, and 50 U/ml penicillin (Gibco-Invitrogen). Media was replaced with Neurobasal medium (Invitrogen) containing B27 supplement (2%; Invitrogen), penicillin-streptomycin (50 μg/ml penicillin, 50 U/ml streptomycin), and glutamine (1 mm; Sigma) 4–5 h later.
Immunostaining of primary hippocampal neurons
To visualize plasmid-induced CRTC1 protein expression and localization, hippocampal neurons 5 d in vitro (DIV) were transfected with plasmids expressing GFP-CRTC1 or GFP alone. Twenty-four hours later, neurons were treated with KCl (50 mm)/FSK (20 μm) or vehicle for 4 h. Neurons were washed with PBS, fixed with 4% paraformaldehyde (PFA) in PBS, permeabilized and blocked [0.3% Triton X-100, 2% normal goat serum (NGS; Jackson ImmunoResearch Laboratories), 0.5% bovine serum albumin (BSA; Bioshop Canada) in PBS] then incubated at 4°C overnight with rabbit anti-CRTC1 polyclonal antibody (1:1000; Cell Signaling Technology). After washing in PBS, neurons were incubated with goat anti-rabbit Alexa 568 antibody (1:500; Invitrogen) for 1 h at room temperature, washed with PBS, counterstained with Hoechst, and mounted with PermaFluor Mounting medium (Thermo Scientific). Images were obtained using a confocal laser scanning microscope (LSM 710; Zeiss).
Luciferase assays
DIV 5 neurons were transfected (using Lipofectamine 2000) with the CRE reporter plasmid [500 ng; generated by replacing d2eGFP transgene in pCRE-d2eGFP (Clontech) with the luciferase coding region from the MRE reporter plasmid, pGL3-TATA-DesMEF, with HindIII and XbaI (Vetere et al., 2011)]. TK-pRL vector-expressing Renilla luciferase (250 ng; Promega) was used as internal control. Twenty-four hours later, neurons were infected with GFP or CRTC1 vector and medium replaced 6 h later. Twenty-four hours later, neurons were treated with KCl (50 mm)/FSK (20 μm) or vehicle for 4 h. Neurons were lysed and luciferase assays conducted using a Dual Luciferase Assay kit (Promega). Firefly and Renilla luciferase activity levels were quantified by a luminometer (Berthold Microlumat LB 96V; Fisher Scientific) and CRE-luciferase activity was normalized to Renilla-luciferase activity. Data represent means from four independent experiments, with internal duplicates or triplicates for each condition.
Mice
Adult female F1 hybrid (C57 BL/6NTac × 129S6/SvEvTac) mice were used for all behavioral experiments. Mice were bred at the Hospital for Sick Children and group housed (3–5 mice per cage) on a 12 h light/dark cycle with food and water available ad libitum. Behavioral experiments were conducted during the light phase of the cycle. All procedures were conducted in accordance with the policies of the Hospital for Sick Children Animal Care and Use Committee and conformed to both the Canadian Council on Animal Care and National Institutes of Health Guidelines on the Care and Use of Laboratory Animals.
Surgery
Mice were pretreated with atropine sulfate (0.1 mg/kg, i.p.), anesthetized with chloral hydrate (400 mg/kg, i.p.) and placed in a stereotaxic frame. Skin was retracted and holes drilled in the skull bilaterally above the dorsal hippocampus (anteroposterior (AP) = −2.3, mediolateral = ±1.5, ventral = −1.8 mm from bregma) according to Paxinos and Franklin (2001). Viral vector (2.0 μl/side) was microinjected through glass micropipettes connected via polyethylene tubing to a microsyringe (Hamilton) at a rate of 0.1 μl/min. Micropipettes were left in place an additional 5 min following microinjection to ensure diffusion of vector. Micropipettes were slowly retracted, the incision closed, and mice treated with analgesic (ketoprofen, 5 mg/kg, s.c.).
Verifying location of vector microinjection and extent of viral infection
Four days following vector microinjection (except for Fig. 3d), mice were transcardially perfused with 0.1 m PBS followed by 4% PFA. Brains were fixed overnight (4°C) and transferred to a 30% sucrose solution. Coronal brain slices (50 μm) across the entire AP extent of the hippocampus were collected using a cryostat (Leica CM1850). Every second section was mounted on a gel-coated glass slide and coverslipped with Vectashield fluorescence mounting medium containing DAPI (Vector Laboratories). Mice with extensive cortical or hippocampal damage were excluded from subsequent statistical analyses. Consistent with previous reports from several labs (Carlezon et al., 1998; Josselyn et al., 2001; Wallace et al., 2004; Brightwell et al., 2005; Han et al., 2007; Vetere et al., 2011) microinjection of HSV vectors produces robust localized transgene expression with minimal tissue damage around the site of microinjection (Fig. 2a,c). GFP-immunofluorescence (which did not differ between vectors) was used to determine placement and extent of the viral infection for each mouse. Based on this, each mouse was classified as a “hit” or “miss” by an examiner unaware of the treatment condition and behavioral results. Mice were defined as “hits” if robust bilateral GFP expression was observed in the dentate gyrus (DG) of dorsal hippocampus in at least five consecutive brain sections (across the AP plane). All other mice were classified as miss (including those with unilateral, weak, or no transgene expression in the target region). Only mice determined to be a bilateral hit were included in subsequent data analysis.
Figure 3.
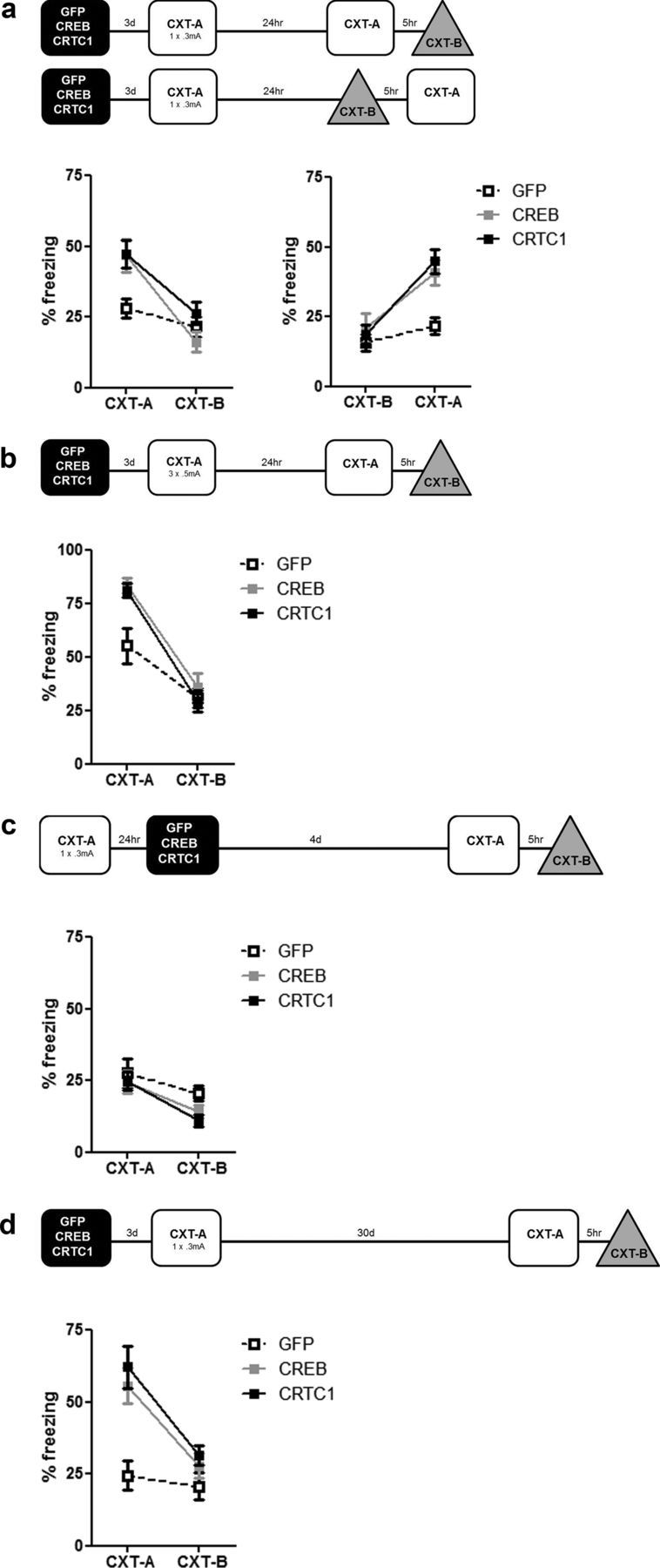
Locally and acutely increasing CRTC1 or CREB levels in DG during training enhance consolidation of contextual fear memory; this memory enhancement is context specific, not due to an effect on memory expression, and long lasting. a, Microinjection of CRTC1 or CREB vector in DG before weak training (1 × 0.3 mA shock) enhances contextual fear memory (CRTC1 vector, n = 29; CREB, n = 24; GFP, n = 27). This memory enhancement is specific for the training context (CXT-A), and does not generalize to a similar, nonshocked context (CXT-B), regardless of context testing order (CXT-A then CXT-B, left, or CXT-B then CXT-A, right). Mean ± SEM. b, Microinjection of CRTC1 or CREB vector in DG before strong training (3 × 0.5 mA shocks) enhances memory for contextual fear; this memory is specific to the training context (CXT-A) (CRTC1 vector, n = 10; CREB, n = 9; GFP, n = 8). c, Microinjection of CRTC1 or CREB vector after training does not facilitate memory expression (in either context) indicating that the enhancement of context memory by these vectors is not due to effects on memory expression/retrieval (CRTC1, n = 12; CREB, n = 11; GFP, n = 11). d, Memory enhancement produced by microinjection of CRTC1 or CREB vector is long lasting and maintains precision. Microinjection of CRTC1 or CREB vector before training (1 × 0.3 mA shock) enhances memory for contextual fear even when tested 30 d later (after transgene expression has dissipated) (CRTC1 vector, n = 12; CREB, n = 16; GFP, n = 9). This memory enhancement is context specific (only observed in CXT-A).
Typically, we observed numerous GFP+ neurons in a circular region (diameter ∼1.6 mm) centered at the site of microinjection. To determine the percentage area of target region infected by vector, we first traced the target region (−1.46 to −3.08 mm AP, corresponding to plates 43–56 in Paxinos and Franklin, 2001) across 15 serial sections in random brains classified as hit (CRTC1 vector, n = 14; CREB vector, n = 14; GFP vector, n = 11) then examined the number and extent of GFP+ neurons within this target region (Stereo Investigator 8 software; MBF Bioscience). We observed GFP+ cells in ∼62–72% of the total target area (CRTC1 vector = 63.00 ± 7.98%, CREB vector = 72.11 ± 4.67%; GFP vector = 62.54 ± 8.58%). Importantly, there was no difference between the percentage dentate gyrus (DG) area infected by the different vectors (F(2,36) = 0.59, p > 0.05). To estimate the number of infected cells, we stereologically counted GFP+ cells in the target region in a subset of brains (4–5 brains per vector). The number of cells infected by CRTC1 vector was 120,825 ± 19,184, for CREB vector 114,066 ± 11,174 and for GFP vector 138,531 ± 47,302. (The overall number of DAPI+ cells in this target region was 527,027 ± 22,108.) Therefore, the overall number of DG cells we infected with our viral vectors was ∼20–23%. The number of cells infected did not vary between vectors (F(2,11) = 0.21, p > 0.05) or correlate with any behavioral measure.
Immunohistochemistry
To examine the type of cell in which CRTC1 protein is endogenously expressed, we examined the overlap of antibodies against CRTC1 protein with cell markers specific for excitatory neurons (α calcium calmodulin II, α-CaMKII), glial cells (GFAP), or interneurons (GAD67, parvalbumin). Images were obtained using a confocal laser scanning microscope (LSM 710; Zeiss).
GFAP, GAD67, and parvalbumin staining.
Brain sections (35 μm) from (homecage wild-type) mice were incubated with blocking solution (0.1% BSA, 2% NGS, 0.3% Triton-X) for 2 h (room temperature, RT) then incubated with rabbit anti-CRTC1 polyclonal (1:500; Cell Signaling Technology) and one of the following primary antibodies: mouse monoclonal anti-GFAP (1:500; Cell Signaling Technology), mouse monoclonal anti-GAD67 (1:500; Millipore), or mouse monoclonal anti-parvalbumin (1:500; Sigma-Aldrich) at 4°C for 24 h. Sections were washed with PBS 0.1 m, then incubated with goat-anti-rabbit Alexa 568 (1:500;,Invitrogen) and goat-anti-mouse Alexa 633 (1:500; Invitrogen) for 2 h at RT.
α-CaMKII staining.
Staining for α-CaMKII was similar except brain tissue was incubated with blocking solution (anti-mouse IgG blocking in 1% H2O2) for 1 h at RT then incubated with mouse monoclonal anti-αCaMKII antibody (1:1000; Millipore) at 4°C for 24 h. Sections were washed with PBS, incubated with donkey-anti-mouse horseradish peroxidase (1:500) for 1 h at RT and signal amplified with TSA-FCM (30 min).
CRTC1 staining.
To verify that microinjection of CRTC1 vector increased expression of CRTC1 protein, we used an antibody specific for CRTC1. Coronal brain sections (50 μm) from mice microinjected with GFP or CRTC1 vector were incubated with blocking solution (0.1% BSA, 2% NGS, 0.3% Triton X-100) for 2 h RT, then incubated with rabbit anti-CRTC1 polyclonal antibody (1:500) at 4°C for 24 h. Sections were washed with PBS, then incubated with goat-anti-rabbit Alexa 568 (1:500) for 2 h at RT. Sections were washed with PBS, counterstained with Hoechst, mounted on slides, and coverslipped using PermaFluor mounting medium.
c-Fos staining.
To examine whether CRTC1 increased neuronal activity, we quantified c-Fos levels in mice microinjected with CRTC1 or GFP vector (as above) and either maintained in the homecage or 90 min following strong training in context fear conditioning. Mice were perfused as above and coronal brain sections (35 μm) were incubated with blocking solution (0.1% BSA, 2% NGS, 0.3% Triton X-100) for 2 h RT, then incubated with rabbit anti-c-Fos polyclonal antibody (1:1000; Calbiochem, PC38) at 4°C for 12–16 h. Sections were washed with PBS, then incubated with goat-anti-rabbit Alexa 568 (1:500) for 2 h at RT, counterstained, mounted, and coverslipped as above. To quantify c-Fos levels in the infected area, we used GFP expression to outline the infected region of the DG across 12–15 serial sections per mouse, then used stereological counting (optical fractionator method) to quantify the number of c-Fos+ neurons within this infected region (Stereo Investigator 8 software; MBF Bioscience). The average number of c-Fos+ cells per infected region was calculated (using values generated by Stereo Investigator) with the following formula: estimated c-Fos+ population using mean section thickness/total area (μm2) = c-Fos+ cells per um2. Values were averaged per mouse and averaged across mice per group (see Fig. 1d).
Figure 1.
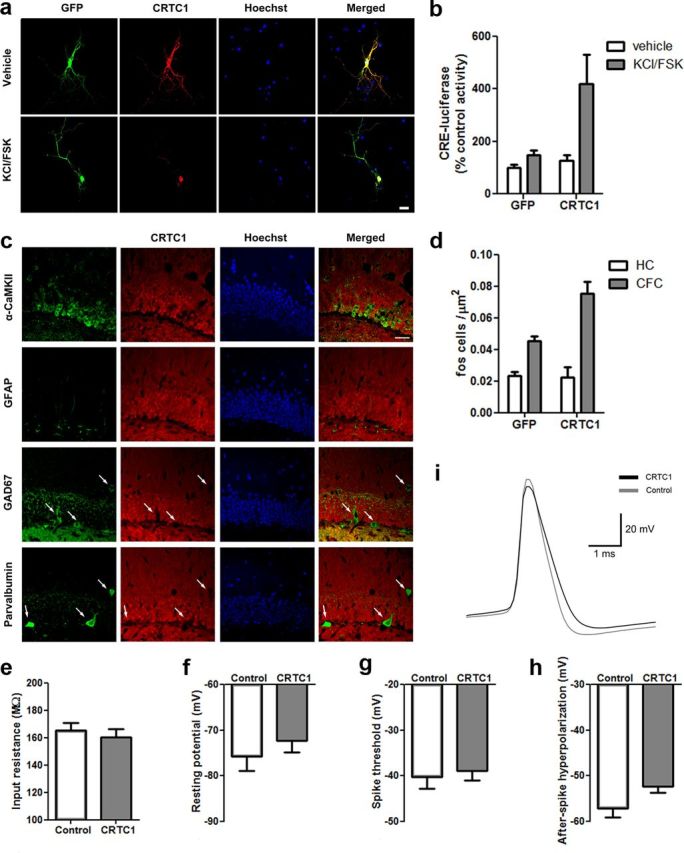
CRTC1 in the DG of the dorsal hippocampus. a, Similar to endogenous CRTC1 protein, plasmid-derived CRTC1 undergoes nuclear translocation following stimulation (KCl/FSK for 4 h) in primary hippocampal neurons. Scale bar, 20 μm. b, CRTC1 increased CRE-dependent transcription following stimulation in primary hippocampal neurons. (CRTC1, n = 4; GFP, n = 4). c, In the DG, CRTC1 is endogenously expressed exclusively in excitatory neurons (not interneurons or glia). Overlap of immunohistochemical staining for endogenous CRTC1 protein (red) and markers of different cell types: green, excitatory neurons (α-CaMKII); glia (GFAP); or interneurons (GAD67 or parvalbumin). Hoechst (blue) identifies DG granule cell layer. Merged images shows that endogenous CRTC1 protein is colocalized in cells positive for α-CaMKII (but not GFAP, GAD67, or parvalbumin). White arrows show lack of CRTC1 staining overlap in GAD67+ or parvalbumin+ cells. Scale bar, 50 μm. d, Context fear conditioning (CFC) increases c-Fos levels in mice microinjected with GFP vector and this effect is potentiated in mice microinjected with CRTC1 vector. CRTC1 overexpression had no effect on c-Fos levels homecage (HC) control mice (CFC–CRTC1, n = 4; CFC–GFP, n = 4; HC–CRTC1, n = 4, HC–GFP, n = 4). e, Input resistance, (f) resting potential, and (g) spike threshold (mV) did not differ between cells infected with CRTC1 vector or control cells whereas AHP (h, i) was decreased in cells infected with CRTC1 vector relative to control cells (CRTC1, n = 7; Control, n = 7). Means ± SEM.
Slice electrophysiology
One day following microinjection of CRTC1 vector into the DG, mice were perfused with cold modified artificial CSF (mACSF) containing the following (in mm): 180 sucrose, 25 sodium bicarbonate, 25 glucose, 2.5 KCl, 1.25 sodium phosphate, 2 MgCl2, 1 CaCl2, 0.4 sodium ascorbate, and 3 sodium pyruvate, saturated with 95% O2/5% CO2 and with pH and osmolarity adjusted to 7.4 and ∼295 mOsm, respectively. Brains were quickly removed and placed for 30 s in a chilled mACSF slurry. The cerebellum and anterior forebrain were removed and the posterior portion of the brain was glued to a slicing stage with an agarose block placed behind it. The stage was then placed in a slicing chamber filled with mACSF chilled to a slurry and continuously oxygenated with 95% O2/5% CO2 during slicing. Brain slices (350 μm) were prepared on a Vibratome 1000 and slices recovered for 1 h at RT in 50 ml of continuously oxygenated Earle's Balanced Salt Solution with 50 μl of 3 m CaCl2 and 150 μl of 3 m MgCl2 added.
During recording, slices were placed in a recording chamber perfused with continuously oxygenated ACSF containing the following (in mm): 125 NaCl, 25 sodium bicarbonate, 25 glucose, 2.5 KCl, 1.25 sodium phosphate, 1 MgCl2, 2 CaCl2, 0.4 sodium ascorbate, and 3 sodium pyruvate, saturated with 95% O2/5% CO2 and with pH and osmolarity adjusted to 7.4 and ∼295 mOsm, respectively, maintained at 36°C with a TC-344B temperature controller and SH-27B in-line solution heater (Warner Instruments). Whole-cell recording pipettes with tip resistances of 4–7 MΩ were pulled from thin-walled borosilicate glass (World Precision Instruments, TW-150F) using a Sutter Instruments P-87. Pipettes were filled with a potassium gluconate-based internal solution containing the following (in mm): 130 potassium gluconate, 10 KCl, 10 HEPES, 0.2 EGTA, 4 ATP, 0.3 GTP, and 10 phosphocreatine, with pH and osmolarity adjusted to 7.4 and ∼295 mOsm, respectively. Whole-cell recordings (CRTC1-infected, n = 7, Control noninfected, n = 7) were performed using a Multiclamp 700B amplifier and digitized using an Axon Digidata 1440A (Molecular Devices). Recordings were made from both fluorescent and nonfluorescent granule cells in the DG, visualized with an Olympus BX51WI equipped with infrared differential interference contrast and GFP epifluorescence. Recordings were performed in current-clamp and current steps from −100 to 200 pA were injected for 1 s in 20 pA steps. To estimate spike threshold and after-spike hyperpolarization (AHP) levels, elicited spike waveforms were averaged for each cell. The threshold was defined as the voltage at the time of the peak in the average waveform's second derivative, while the AHP was defined as the minimum voltage in a 3 ms window following the peak of the average waveform. Reported voltage levels were not corrected for any liquid junction potential. All reagents used for electrophysiology were obtained from Sigma.
Contextual fear-conditioning apparatus
Fear-conditioning chambers (Context-A, CXT-A; 31 × 24 ×21 cm; MED Associates) consisted of two stainless steel and two clear acrylic walls, with a stainless steel shock-grid floor (bars 3.2 mm diameter, spaced 7.9 mm apart). A stainless steel drop-pan containing a 70% ethanol solution was placed below the grid floor. A fan provided low-level white noise during training and testing in CXT-A. To examine the specificity of context fear memory, mice were also tested in a no-shock context, Context-B (CXT-B). CXT-B was a modified version of CXT-A with an opaque white acrylic triangular wall insert placed inside the chamber and the shock-grid floor covered by a smooth opaque white acrylic floor. The door of the chamber was covered with an opaque sheet with horizontal black and white stripes. During testing in CXT-B, neither the ethanol nor fan was used (Wang et al., 2009).
Behavior was monitored by overhead cameras, which digitized video images at 4 Hz. An automated frame-by-frame analysis of movement was used to generate freezing scores (Freezeframe software; Actimetrics). Reactivity to shock was assessed by comparing distance traveled in 2 s before shock onset (pre-US), to distance traveled during the 2 s shock (US). Reactivity Index = (US − pre-US)/(US + pre-US). Importantly, we observed no difference in shock reactivity between vectors for any experiment (see Results).
General behavioral procedures
Context fear training
Weak training.
Mice were placed in a conditioning chamber (CXT-A) and 2 min later received an unsignaled shock (0.3 mA, 2 s). Mice remained in the chamber for an additional 60 s before being returned to their homecage.
Strong training.
Strong training was similar to above except that mice received three unsignaled shocks (0.5 mA, 2 s) spaced 60 s apart.
Context fear testing
Mice were placed in the context in which they were previously shocked (CXT-A), returned to the homecage, and 5 h later, placed in the no-shock context (CXT-B). For Experiment 1 (see Fig. 3a), the order of context test was counterbalanced. The percentage of time spent freezing during each 5 min test session was used as an index of memory. Freezing was defined as an immobilized, crouched position, with an absence of any movement except respiration (Blanchard and Blanchard, 1969; Bolles and Fanselow, 1982).
Specific behavioral procedures
Experiment 1: Effects of increasing CRTC1 or CREB on consolidation of a weak context fear memory
Mice were microinjected with vector (CRTC1 vector, n = 29; CREB, n = 24; GFP, n = 27) and 3 d later given weak fear training. Twenty-four hours later mice were placed in either CXT-A or CXT-B (for 5 min), and 5 h later they were placed in the alternate context. The order of context test was counterbalanced in this experiment only.
Experiment 2: Effects of increasing CRTC1or CREB on consolidation of a strong context fear memory
Mice were microinjected with vector (CRTC1, n = 10; CREB, n = 9; GFP, n = 8) and 3 d later given strong fear training. Twenty-four hours later, mice were tested in CXT-A and 5 h later, tested in CXT-B.
Experiment 3: Effects of increasing CRTC1 or CREB on expression of a weak context fear memory
Mice were fear conditioned using the weak training protocol and 24 h later, microinjected with vector (CRTC1, n = 12; CREB, n = 11; GFP, n = 11). Four days later, mice were tested in CXT-A and 5 h later, tested in CXT-B.
Experiment 4: Examining the enduring effects of increasing CRTC1 or CREB on consolidation of a weak context fear memory
Mice were microinjected with vector (CRTC1, n = 12; CREB, n = 16; GFP, n = 9) and 3 d later given weak fear training. Thirty days later, at a time when transgene expression driven by HSV vector had long dissipated, mice were tested in CXT-A and 5 h later, tested in CXT-B.
Experiment 5: Effects of increasing CRTC1 or CREB on reconsolidation of a weak context fear memory
Mice were fear conditioned using the weak training protocol and, 26 d later (after the memory was consolidated), microinjected with vector (CRTC1, n = 10; CREB, n = 10; GFP, n = 10). Three days following microinjection, mice were replaced in CXT-A in the absence of the shock (for 45 s) to reactivate the context fear memory. Mice were removed from CXT-A and returned to the homecage. Twenty-four hours later, mice were tested as above in CXT-A then CXT-B. As a control, a “no memory reactivation” condition was included (no-reactivation groups: CRTC1, n = 6, CREB, n = 6; GFP, n = 6). Mice were treated identically except that the reactivation procedure was omitted (mice remained in the homecage).
Data analyses
To analyze CRE-luciferase activity, we used an ANOVA with between-group factor Plasmid (CRTC1, GFP) and within-group factor Stimulation (Stim, KCl/FSK; No Stim, unstimulated vehicle control). Context fear memory data were analyzed using an ANOVA with between-group factor Vector (CRTC1, CREB, GFP) and within-group factor Context (CXT-A, shock context; CXT-B, no-shock context). Significant interactions or main effects were further analyzed using post hoc Tukey's HSD tests. c-Fos expression data were analyzed using a 2 × 2 ANOVA with Vector (CRTC1, GFP) and Treatment (homecage, HC; context fear condition; CFC) as factors. Significant interactions or main effects were further analyzed using post hoc Fisher's LSD tests. Data obtained from the electrophysiological experiments were analyzed using the Mann–Whitney U test.
Results
Microinjection of CRTC1 or CREB vector increases CRTC1 or CREB function in the DG of dorsal hippocampus
We first examined endogenous expression of CRTC1 in the brain. Consistent with previous findings (Zhou et al., 2006; Watts et al., 2011), we observed high levels of CRTC1 in the hippocampus. Importantly, we found that in the DG, CRTC1 is expressed exclusively in excitatory dentate granule cells, not in glia or interneurons (Fig. 1c). Therefore, to examine the effects of locally and acutely increasing CRTC1 levels in the DG region of dorsal hippocampus on memory, we used replication-defective HSV vectors. HSV is neurotropic (Fink et al., 1996; Barrot et al., 2002) and, following microinjection into the DG, selectively infects excitatory neurons (Fig. 1c). In this way, our viral vectors increase CRTC1 levels only in neurons in which CRTC1 is endogenously expressed. To increase CRTC1 levels, we used HSV encoding wild-type CRTC1; this vector also expressed GFP, allowing visualization of infected neurons (CRTC1 vector). As a control, we used a vector expressing GFP alone (GFP vector). Importantly, we observed no evidence of toxicity associated with these vectors either in cultured neurons or following microinjection in vivo.
Previous findings indicate that a similar vector-expressing CREB (CREB vector) increases CREB levels and function (CRE-mediated transcription) both in vitro and in vivo (Barrot et al., 2002; Han et al., 2007, 2009; Sekeres et al., 2010; Larson et al., 2011). We verified that microinjection of CRTC1 vector into the DG similarly increased CRTC1 protein above endogenous levels (Fig. 2c). Similar to endogenous CRTC1 protein (Zhou et al., 2006; Li et al., 2009; Ch'ng et al., 2012), transgenic CRTC1 protein is normally sequestered in the cytoplasm, but translocates to the nucleus following stimulation by cAMP and calcium (Fig. 1a). Furthermore, we found that increasing CRTC1 levels in primary hippocampal neurons increased CRE-dependent transcription under unstimulated (basal) and stimulated (KCl/FSK) for 4 h conditions (Fig. 1b). This observation was supported by the results of an ANOVA, which revealed a significant Construct × Stimulation interaction (F(1,12) = 4.75; p < 0.05), as well as significant main effects of Construct (F(1,12) = 7.10, p <0.05) and Stimulation (F(1,12) = 9.10, p < 0.05). The finding that CRTC1 increases CRE-luciferase reporter activity in primary hippocampal neurons is consistent with previous results (Zhou et al., 2006; Altarejos et al., 2008). Together, these data show that CRTC1 vector increases both CRTC1 levels and CRE-mediated transcription.
CRTC1 vector increases excitability of cells in the DG
To examine whether CRTC1 influences neuronal excitability, we used in vitro recording techniques. The electrophysiological characteristics of DG cells infected with CRTC1 vector versus noninfected control cells from the same slice were compared using whole-cell patch-clamp recordings in current-clamp mode. We observed that increasing CRTC1 levels decreased the AHP of cells following stimulation (Fig. 1h,i). This observation was supported by the results of the Mann–Whitney U test, which revealed that the distributions between CRTC1-infected and noninfected cells differed significantly (Mann–Whitney U = 10, n1 = n2 = 7, p < 0.05). Importantly, mean resting potential (mV) (Fig. 1f; Mann–Whitney U = 18, n1 = n2 = 7, p > 0.05), mean input resistance (mΩ) (Fig. 1e; Mann–Whitney U = 18, n1 = n2 = 7, p > 0.05), and mean spike threshold (Fig. 1g; Mann–Whitney U = 21, n1 = n2 = 7, p > 0.05) did not differ between the groups. Together, these data are consistent with the interpretation that increasing CRTC1 increased neuronal excitability, perhaps by mediating active K+ currents.
Microinjection of CRTC1 vector increases c-Fos expression in the DG of dorsal hippocampus
To further examine the effects of CRTC1 on neuronal excitability, we used c-Fos as a marker of neuronal activity. We microinjected mice with CRTC1 or GFP vector and 3 d later examined c-Fos in the infected region of the DG either in mice maintained in the homecage or 90 min following context fear conditioning. We found that increasing CRTC1 levels increased c-Fos expression in the infected region of the DG in mice trained for context fear conditioning (Fig. 1d). This observation was supported by the results of an ANOVA, which revealed a significant Vector × Treatment interaction (F(1,12) = 8.42; p <0.05), as well as significant main effects of Vector (F(1,12) = 7.30, p <0.05) and Treatment (F(1,12) = 49.17, p <0.01). Post hoc analyses revealed that mice microinjected with either GFP or CTRC1 vector had higher levels of c-Fos following fear conditioning compared with mice maintained in their homecage. Moreover, fear-conditioned mice with CRTC1 vector had higher c-Fos activation than fear-conditioned mice with GFP vector (p <0.05). There was no difference between c-Fos expressions in homecage control mice (p >0.05). Together, these results suggest that neurons with increased CRTC1 expression are more excitable. These data are consistent with previous findings that overexpression of CREB increases intrinsic excitability (Dong et al., 2006; Lopez de Armentia et al., 2007; Zhou et al., 2009).
Experiment 1: Increasing CRTC1 or CREB in DG at the time of training facilitates consolidation of a weak contextual fear memory
To examine the effects of increasing CRTC1 or CREB function in DG on consolidation of a contextual fear memory, we microinjected CRTC1, CREB, or GFP vector 3 d before contextual fear training, which normally induces weak memory (mice received single 0.3 mA shock in CXT-A) (Fig. 3a). One day following training, contextual fear memory was assessed by returning mice to the training context (CXT-A). As expected, mice microinjected with GFP vector showed low levels of freezing when replaced in CXT-A; however, mice with CRTC1 or CREB vector showed enhanced freezing (Fig. 3a). To examine the specificity of this increased freezing, we retested mice in a novel, alternate context not previously paired with shock (Context B, CXT-B). All groups, regardless of vector, showed similarly low freezing in CXT-B (Fig. 3a). The results of a Vector (CRTC1, CREB, GFP) by Context (CXT-A, CXT-B) ANOVA supported this observation and revealed a significant Vector × Context interaction (F(2,45) = 8.19, p < 0.001) as well as a significant main effect of Context (F(1,45) = 65.81, p < 0.001), but not Vector (F(2,45) = 3.02, p > 0.05). Post hoc analyses conducted on the significant interaction indicated that mice with either CRTC1 or CREB vector froze more in CXT-A than mice with GFP vector, but that all groups showed low freezing in CXT-B. Interestingly, there was no difference between freezing levels in mice with CRTC1 or CREB vector (in either context). Testing mice in CXT-B and then CXT-A produced similar results: significant Vector × Context interaction (F(2,29) = 4.32, p < 0.05), significant main effects of Context (F(1,29) = 37.62, p < 0.001), and Vector (F(2,29) = 6.55, p < 0.001 (Fig. 3a). Importantly, the increase in freezing observed in mice with CRTC1 or CREB vector cannot be attributed to differences in shock sensitivity during training (F(2,45) = 0.33, p > 0.05). Therefore, increasing CRTC1 or CREB levels before training increased freezing in the context previously paired with shock, but did not enhance generalization of freezing to a nonshock context. These findings suggest that overexpressing CRTC1 or CREB increased memory strength, without compromising memory specificity.
Experiment 2: Increasing CRTC1 or CREB in DG at the time of training further facilitates consolidation of a strong contextual fear memory
We next examined the effects of similarly increasing CRTC1 or CREB function on context fear memory induced by a stronger training protocol (three 0.5 mA shocks in CXT-A) (Fig. 3b). When subsequently tested in CTX-A, mice with CRTC1 or CREB vector showed greater freezing than mice with GFP vector, but similar to above, this memory enhancement was context specific, as all groups showed low freezing in the no-shock context (Fig. 3b) [(significant Vector × Context interaction (F(2,24) = 4.90, p < 0.05), as well as significant main effects of Vector (F(2,24) = 3.77, p < 0.05) and Context (F(1,24) = 124.62, p < 0.001)]. Post hoc analyses revealed that mice with CRTC1 or CREB vector froze more in CXT-A than mice with GFP vector, but that all groups froze at equally low levels in CXT-B (p > 0.05). Together, these results indicate that increasing CRTC1 or CREB function in the DG before training enhances context fear memory induced by either weak or strong training. Furthermore, the enhanced memory produced by CRTC1 or CREB overexpression is specific and does not generalize to a novel context.
Experiment 3: Increasing CRTC1 or CREB in DG does not enhance expression of a previously acquired contextual fear memory
In the above experiments, we microinjected vectors before training such that mice were both trained and tested with high CRTC1 or CREB levels in the DG. To examine whether the enhancement in freezing produced by CRTC1 or CREB vectors was due to facilitated memory expression/memory retrieval, we performed a similar experiment but microinjected CRTC1, CREB, or GFP vectors 24 h after weak training, at a time when cellular/synaptic consolidation is thought to be complete (for review, see Dudai, 2004). When tested 4 d after microinjection, all groups froze at low levels in the shock context (CXT-A) and still lower levels in the no-shock context (CXT-B) [no significant effect of Vector × Context (F(2,31) = 0.69, p > 0.05), Vector (F(2,31) = 1.45, p > 0.05), but a significant effect of Context (F(1,31) = 0.19, p < 0.001), in which all groups froze more in CXT-A than CXT-B] (Fig. 3c). Therefore, increasing CRTC1 or CREB levels does not affect the expression or context specificity of a previously acquired fear memory. Together, these results indicate that increasing CRTC1 or CREB function in the DG before, but not after, training enhances memory.
Experiment 4: The memory facilitation produced by increasing CRTC1 or CREB in DG at the time of training is long lasting
The above finding suggests that increasing CRTC1 or CREB at the time of training enhances memory consolidation. Importantly, using this vector system, transgene expression peaks ∼3 d following microinjection, and dissipates within 10–14 d after microinjection (Barrot et al., 2002; Vetere et al., 2011) (Fig. 2b). Therefore, to investigate whether the memory enhancement produced by microinjecting CRTC1 or CREB vector persists beyond transgene expression, we trained mice as before (3 d following microinjection, at a time of high transgenic expression of CRTC1 or CREB) but tested mice long after transgene expression had dissipated (33 d following microinjection). When tested at this later time, mice previously microinjected with CRTC1 or CREB vector froze more in CXT-A than mice with GFP vector. This enhancement was context-specific as all groups froze at equally low levels in CXT-B [significant Vector × Context (F(2,34) = 5.44, p < 0.05), Vector (F(2,34) = 5.90, p < 0.01), and Context (F(1,34) = 37.35, p < 0.001) (Fig. 3d]. Therefore, increasing CRTC1 or CREB in the DG before training facilitates the consolidation of context fear memory; this enhancement is both precise (observed only in the training context) and enduring (observed even after transgene expression dissipated). Together, these results are consistent with the interpretation that increasing CRE-mediated transcription in the DG around the time of training enhances memory consolidation. Once this memory has been consolidated, elevated CRE-mediated transcription is no longer necessary to maintain this memory in terms of strength and precision.
Experiment 5: Increasing CRTC1 or CREB in DG facilitates reconsolidation of fear memory
We found that increasing CRTC1 or CREB in the DG before training facilitates memory consolidation. Memory reactivation (by exposure to cues present during initial memory encoding) may trigger a second wave of consolidation (reconsolidation). Similar to initial consolidation, reconsolidation also requires protein synthesis (Nader et al., 2000; Sara, 2000; Debiec et al., 2002) and intact CREB function (Kida et al., 2002; Mamiya et al., 2009; Kim et al., 2011). Therefore, we next asked whether increasing CRTC1 or CREB before memory reactivation would similarly enhance memory reconsolidation. We trained unoperated mice using the weak training protocol and 26 d later (when memory was consolidated) mice were microinjected with CRTC1, CREB, or GFP vectors 3 d before memory reactivation (placement in CXT-A for 45 s without shock). In this way, the time between vector microinjection and training (in this experiment, reactivation) remained constant. To examine the stability of the reactivated memory (memory reconsolidation), mice were tested 24 h later in CXT-A. To examine the specificity of this memory, mice were tested the no-shock context (CXT-B) 5 h later. As expected following weak fear training, all groups showed low levels of freezing during the memory reactivation session (F(2,19) = 0.72, p > 0.05) (Fig. 4a). In contrast, when tested 24 h after memory reactivation, mice microinjected with CRTC1 or CREB vector showed higher freezing than mice with GFP vector in CXT-A (Fig. 4a) but equally low levels in CXT-B [significant effects of Vector × Context (F(2,27) = 9.54, p < 0.001), Context (F(1,27) = 39.06, p < 0.001), but not Vector (F(2,27) = 2.89, p > 0.05)]. This finding is consistent with the interpretation that increasing CRTC1 or CREB before memory reactivation enhanced memory reconsolidation.
Figure 4.

Increasing CRTC1 or CREB levels in DG enhances reconsolidation of an established contextual fear memory. a, Microinjection of CRTC1 or CREB vector before reactivation of an established weak contextual fear memory enhances subsequent memory expression in a context-specific manner. Naive mice were trained with a weak protocol (1 × 0.3 mA shock), and 26 d later were microinjected with vector (CRTC1-R, n = 10; CREB-R, n = 10; GFP-R, n = 10). Three days following vector microinjection, all groups showed similar low levels of freezing when initially re-exposed to the training context (for 45 s) to reactivate the memory (left graph). In subsequent test session (24 h later), mice with GFP vector showed low levels of freezing (in both contexts). However, mice with CRTC1 or CREB vector showed enhanced memory, which was context specific (right graph). b, Memory reactivation is necessary for the enhancement of an established memory by CRTC1 or CREB vectors (reconsolidation). Mice were trained as above, similarly microinjected with vectors, but not re-exposed to the training context (no reactivation, NR) after vector microinjection (CRTC1-NR, n = 6; CREB-NR, n = 6; GFP-NR, n = 6). During the subsequent test, all groups showed equally low levels of freezing. Means ± SEM.
To confirm that this memory enhancement was critically dependent on reactivation of the context fear memory at a time of high CRTC1 or CREB levels, we conducted a similar experiment except that mice were maintained in the homecage following microinjection of vector (no memory reactivation). When subsequently tested in both CXT-A and CXT-B, all mice showed equally low levels of freezing, regardless of vector or context [no significant Vector × Context interaction (F(2,15) = 0.47, p > 0.05), main effect of Context (F(1,15) = 4.32, p > 0.05), or Vector (F(2,15) = 0.04, p > 0.05)] (Fig. 4b). Therefore, the enhancement of memory produced by microinjecting CRTC1 or CREB vector was critically dependent on memory reactivation.
Discussion
Extensive evidence implicates CREB-dependent gene transcription in memory (Bourtchuladze et al., 1994; Yin et al., 1994; Guzowski and McGaugh, 1997; Bartsch et al., 1998; Kida et al., 2002; Pittenger et al., 2002; Frankland et al., 2004). Multiple signaling pathways phosphorylate CREB at Ser133 (Shaywitz and Greenberg, 1999; Mayr and Montminy, 2001; Lonze and Ginty, 2002), which stimulates the recruitment of coactivators CBP/p300 (Chrivia et al., 1993; Parker et al., 1996). However, this phosphorylation event is not always sufficient to activate transcription (Impey et al., 1996; Mayr and Montminy, 2001) suggesting that CREB-mediated transcription is regulated by additional mechanisms.
In 2003, two laboratories identified a new family of CREB-specific coactivators, now referred to as CRTCs (Iourgenko et al., 2003; Conkright et al., 2003b). CRTC is thought to enhance transcription by facilitating the interaction of CREB with the RNA polymerase II pre-initiation complex (Conkright et al., 2003b; Xu et al., 2007; Altarejos and Montminy, 2011) or perhaps increasing the occupancy of CREB on DNA binding sites of some promoters (Wang et al., 2010; Altarejos and Montminy, 2011). Although CRTC1 and CBP/p300 activate CRE-mediated transcription through different mechanisms, their effects can be synergistic (Ravnskjaer et al., 2007; Xu et al., 2007). In addition to CREB, CBP/p300 binds to ∼400 protein partners, thereby making CBP/p300 among the most heavily connected nodes in the known mammalian protein–protein interactome (Goodman and Smolik, 2000; Bedford et al., 2010). In contrast, CRTC seems to be a dedicated modulator of CRE-containing genes (Conkright et al., 2003b). Despite being a powerful and specific coactivator of CREB, the role of CRTC in memory has been virtually unexplored.
Our studies identify a critical role for CRTC1 in both memory consolidation and reconsolidation. We show that increasing CRTC1 function enhances memory strength without compromising memory quality. To examine the effects of increasing CRTC1 levels on different memory phases, we used context fear conditioning. Context fear conditioning is well suited for investigating the molecular basis of consolidation and reconsolidation as infusions of the protein synthesis inhibitor anisomycin directly into dorsal hippocampus around the time of training (synaptic consolidation) or memory reactivation (reconsolidation) disrupt subsequent memory expression (Quevedo et al., 1999; Taubenfeld et al., 2001; Debiec et al., 2002; Frankland et al., 2006; Suzuki et al., 2008; Mamiya et al., 2009). Moreover, by examining fear in both a trained and nontrained context, this task allowed us to assess memory specificity or quality. We found that mice with increased CRTC1 show increased freezing, but only in the context previously paired with shock. This indicates that CRTC1 promotes the formation of a precise context fear memory (rather than a nonspecific increase in fear or anxiety). We targeted our manipulation to the DG, the primary relay station for inputs to the hippocampus (Treves et al., 2008), because previous studies show this region is particularly important in context fear acquisition (Lee and Kesner, 2004; Hernández-Rabaza et al., 2008).
We found that increasing CRTC1 levels in the DG region of dorsal hippocampus before training facilitates memory consolidation of a weak fear memory without affecting memory quality. That is, we observed an increase in freezing only in the context previously paired with shock; this increase in freezing did not generalize to a similar no-shock context. Similarly increasing CRTC1 levels also enhanced contextual fear memory produced by stronger training, in a context-specific manner. This CRTC1-induced enhancement in memory consolidation is not due to an effect on memory retrieval/expression as similarly increasing CRTC1 levels after training did not affect the expression or specificity of a weak fear memory. Interestingly, increasing CREB levels produced strikingly similar effects in all experiments. Therefore, increasing CRTC1 or CREB levels around the time of memory encoding enhances memory consolidation without compromising memory quality. The increase in memory produced by increasing CRTC1 or CREB levels at the time of training was long lasting and was observed even after viral expression of CRTC1 or CREB dissipated, suggesting that once consolidated, this strong and specific context fear memory no longer required elevated CRTC1 or CREB function.
Memory retrieval is thought to be an active constructive process (Schacter et al., 1998) that functions to modify previously acquired memories (Sara, 2000; Dudai, 2006; Lee, 2009). Here we showed that an established conditioned fear memory was strengthened (without further training) if reactivated at a time of high CRTC1 or CREB levels in the DG. Specifically, we trained naive mice using a weak protocol, and 26 d later, microinjected vector. Three days following vector microinjection, mice were re-exposed to the shock context (but did not receive a shock). When subsequently tested, mice with GFP vector showed low conditioned fear memory (as expected). However, mice trained with a weak protocol but microinjected with CRTC1 or CREB vector before context re-exposure, showed enhanced memory. This finding indicates that the strength of an established memory can be increased even relatively long after acquisition. Moreover, the quality of the memory was not affected, as conditioned fear did not generalize to the no-shock context. The enhanced memory was dependent on re-exposure to the training context, and not observed in similarly trained mice maintained in the homecage (no context re-exposure). In this way, the memory enhancement produced by CRTC1 or CREB differs from that produced by overexpressing the atypical protein kinase C isoform, protein kinase Mζ (PKMζ). For instance, Shema et al. (2011) found that virally increasing PKMζ expression in insular cortex 6 d after conditioned taste aversion training (at a time when the memory trace was consolidated) enhanced subsequent memory, even though rats were not re-exposed to the taste previously paired with illness.
Memory retrieval likely engages the population of neurons involved in the original memory trace and memory coding is thought to be especially sparse in the DG (Leutgeb et al., 2007a,b). Therefore, the present findings that increasing CRTC1 or CREB in ∼20% of DG cells in the target region enhanced reconsolidation is perhaps surprising. These data suggest that either the population of neurons involved in the memory trace overlapped with the population of neurons infected or that increasing CRTC1 or CREB in DG enhanced overall circuit function (rather than producing a cell autonomous effect).
The majority of experiments investigating the molecular basis of memory consolidation, and especially reconsolidation, have examined whether a given molecule (or protein synthesis in general) is necessary for these processes by inferring normal function from loss-of-function studies. Although the results from these types of experiments have greatly increased our understanding of the mechanisms underlying memory, alternative interpretations to the observed behavioral deficits in memory reconsolidation experiments have been offered (e.g., observed decrease in memory may be due to a temporary inability to access the memory trace; Lattal et al., 2004). In contrast to loss-of function studies, we assessed whether increasing CRTC1 or CREB function enhances memory consolidation and reconsolidation. One advantage of this approach is that any memory enhancement is unlikely to be easily attributable to performance and/or memory retrieval effects. Here we show that increasing CRTC1 or CREB in DG enhances both memory consolidation and reconsolidation. Our findings are consistent with results from Tronson et al. (2006) who showed that infusions of a protein kinase A (PKA) agonist into the amygdala enhanced reconsolidation of a tone fear memory. As PKA may both phosphorylate CREB at Ser133 (Impey et al., 1998; West et al., 2002; Cohen and Greenberg, 2008) and promote nuclear translocation of CRTC1 (Bittinger et al., 2004), the present findings suggest a potential molecular mechanism for this enhancement of memory reconsolidation.
Since its characterization >20 years ago, CREB has been implicated in diverse brain processes, including neural development, addiction, circadian rhythms, and memory (Bourtchuladze et al., 1994; Yin et al., 1994; Ding et al., 1997; Carlezon et al., 1998; von Gall et al., 1998; Gau et al., 2002; Lonze and Ginty, 2002; McClung and Nestler, 2003; Cohen and Greenberg, 2008; Eckel-Mahan et al., 2008; Briand and Blendy, 2010). CREB may modulate up to one-quarter of the mammalian genome (Conkright et al., 2003a; Impey et al., 2004; Zhang et al., 2005), suggesting that different subsets of CREB-target genes may be involved in different processes. CREB-dependent gene transcription is modulated by several mechanisms, including post-translational modifications of CREB (phosphorylation, acetylation, ubiquitination, sumoylation, and glycosylation) (Taylor et al., 2000; Kornhauser et al., 2002; Comerford et al., 2003; Lamarre-Vincent and Hsieh-Wilson, 2003; Lu et al., 2003; Altarejos and Montminy, 2011; Rexach et al., 2012) and interaction with different cofactors, including CBP/p300 and CRTC (Goodman and Smolik, 2000; Mayr and Montminy, 2001; Vo and Goodman, 2001; Conkright et al., 2003b; Screaton et al., 2004). The combination of different CREB regulatory mechanisms might be one way to orchestrate the transcription of specific CREB target genes under different conditions (the CREB regulon; Impey et al., 2004).
Footnotes
This work was supported by from the Canadian Institutes of Health Research (CHIR) Grants MOP-74650 (S.A.J.) and MOP-86762 (P.W.F.), EJLB Foundation (S.A.J.), and Natural Science and Engineering Research Council (S.A.J.). M.J.S. received support from a Restracomp Fellowship (Hospital for Sick Children), a CIHR Frederick Banting and Charles Best Canada Graduate Scholarships Doctoral Award, and the Faculty of Medicine at the University of Toronto. D.S. was supported by the Alzheimer's Society of Canada. B.R. was supported by a Banting Postdoctoral Fellowship (CIHR). We thank Mika Yamamoto, Russell Braybon, and Antonietta (Toni) DeCristofaro for excellent technical assistance.
The authors declare no competing financial interests.
References
- Alberini CM. Mechanisms of memory stabilization: are consolidation and reconsolidation similar or distinct processes? Trends Neurosci. 2005;28:51–56. doi: 10.1016/j.tins.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Alberini CM. The role of reconsolidation and the dynamic process of long-term memory formation and storage. Front Behav Neurosci. 2011;5:12. doi: 10.3389/fnbeh.2011.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. 2011;12:141–151. doi: 10.1038/nrm3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altarejos JY, Goebel N, Conkright MD, Inoue H, Xie J, Arias CM, Sawchenko PE, Montminy M. The Creb1 coactivator Crtc1 is required for energy balance and fertility. Nat Med. 2008;14:1112–1117. doi: 10.1038/nm.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrot M, Olivier JD, Perrotti LI, DiLeone RJ, Berton O, Eisch AJ, Impey S, Storm DR, Neve RL, Yin JC, Zachariou V, Nestler EJ. CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc Natl Acad Sci U S A. 2002;99:11435–11440. doi: 10.1073/pnas.172091899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch D, Casadio A, Karl KA, Serodio P, Kandel ER. CREB1 encodes a nuclear activator, a repressor, and a cytoplasmic modulator that form a regulatory unit critical for long-term facilitation. Cell. 1998;95:211–223. doi: 10.1016/s0092-8674(00)81752-3. [DOI] [PubMed] [Google Scholar]
- Bedford DC, Kasper LH, Fukuyama T, Brindle PK. Target gene context influences the transcriptional requirement for the KAT3 family of CBP and p300 histone acetyltransferases. Epigenetics. 2010;5:9–15. doi: 10.4161/epi.5.1.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittinger MA, McWhinnie E, Meltzer J, Iourgenko V, Latario B, Liu X, Chen CH, Song C, Garza D, Labow M. Activation of cAMP response element-mediated gene expression by regulated nuclear transport of TORC proteins. Curr Biol. 2004;14:2156–2161. doi: 10.1016/j.cub.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Blanchard RJ, Blanchard DC. Crouching as an index of fear. J Comp Physiol Psychol. 1969;67:370–375. doi: 10.1037/h0026779. [DOI] [PubMed] [Google Scholar]
- Bolles RC, Fanselow MS. Endorphins and behavior. Annu Rev Psychol. 1982;33:87–101. doi: 10.1146/annurev.ps.33.020182.000511. [DOI] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- Briand LA, Blendy JA. Molecular and genetic substrates linking stress and addiction. Brain Res. 2010;1314:219–234. doi: 10.1016/j.brainres.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brightwell JJ, Smith CA, Countryman RA, Neve RL, Colombo PJ. Hippocampal overexpression of mutant creb blocks long-term, but not short-term memory for a socially transmitted food preference. Learn Mem. 2005;12:12–17. doi: 10.1101/lm.85005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, Duman RS, Neve RL, Nestler EJ. Regulation of cocaine reward by CREB. Science. 1998;282:2272–2275. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- Chawla S, Hardingham GE, Quinn DR, Bading H. CBP: a signal-regulated transcriptional coactivator controlled by nuclear calcium and CaM kinase IV. Science. 1998;281:1505–1509. doi: 10.1126/science.281.5382.1505. [DOI] [PubMed] [Google Scholar]
- Ch'ng TH, Uzgil B, Lin P, Avliyakulov NK, O'Dell TJ, Martin KC. Activity-dependent transport of the transcriptional coactivators CRTC1 from synapse to nucleus. Cell. 2012;150:207–221. doi: 10.1016/j.cell.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- Cohen S, Greenberg ME. Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu Rev Cell Dev Biol. 2008;24:183–209. doi: 10.1146/annurev.cellbio.24.110707.175235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comerford KM, Leonard MO, Karhausen J, Carey R, Colgan SP, Taylor CT. Small ubiquitin-related modifier-1 modification mediates resolution of CREB-dependent responses to hypoxia. Proc Natl Acad Sci U S A. 2003;100:986–991. doi: 10.1073/pnas.0337412100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conkright MD, Guzmán E, Flechner L, Su AI, Hogenesch JB, Montminy M. Genome-wide analysis of CREB target genes reveals a core promoter requirement for cAMP responsiveness. Mol Cell. 2003a;11:1101–1108. doi: 10.1016/s1097-2765(03)00134-5. [DOI] [PubMed] [Google Scholar]
- Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB, Montminy M. TORCs: transducers of regulated CREB activity. Mol Cell. 2003b;12:413–423. doi: 10.1016/j.molcel.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Debiec J, LeDoux JE, Nader K. Cellular and systems reconsolidation in the hippocampus. Neuron. 2002;36:527–538. doi: 10.1016/s0896-6273(02)01001-2. [DOI] [PubMed] [Google Scholar]
- De Cesare D, Sassone-Corsi P. Transcriptional regulation by cyclic AMP-responsive factors. Prog Nucleic Acid Res Mol Biol. 2000;64:343–369. doi: 10.1016/s0079-6603(00)64009-6. [DOI] [PubMed] [Google Scholar]
- Ding JM, Faiman LE, Hurst WJ, Kuriashkina LR, Gillette MU. Resetting the biological clock: mediation of nocturnal CREB phosphorylation via light, glutamate, and nitric oxide. J Neurosci. 1997;17:667–675. doi: 10.1523/JNEUROSCI.17-02-00667.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Green T, Saal D, Marie H, Neve R, Nestler EJ, Malenka RC. CREB modulates excitability of nucleus accumbens neurons. Nat Neurosci. 2006;9:475–477. doi: 10.1038/nn1661. [DOI] [PubMed] [Google Scholar]
- Dudai Y. The neurobiology of consolidations, or, how stable is the engram? Annu Rev Psychol. 2004;55:51–86. doi: 10.1146/annurev.psych.55.090902.142050. [DOI] [PubMed] [Google Scholar]
- Dudai Y. Reconsolidation: the advantage of being refocused. Curr Opin Neurobiol. 2006;16:174–178. doi: 10.1016/j.conb.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Dudai Y, Eisenberg M. Rites of passage of the engram: reconsolidation and the lingering consolidation hypothesis. Neuron. 2004;44:93–100. doi: 10.1016/j.neuron.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Eckel-Mahan KL, Phan T, Han S, Wang H, Chan GC, Scheiner ZS, Storm DR. Circadian oscillation of hippocampal MAPK activity and cAmp: implications for memory persistence. Nat Neurosci. 2008;11:1074–1082. doi: 10.1038/nn.2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink DJ, DeLuca NA, Goins WF, Glorioso JC. Gene transfer to neurons using herpes simplex virus-based vectors. Annu Rev Neurosci. 1996;19:265–287. doi: 10.1146/annurev.ne.19.030196.001405. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Josselyn SA, Anagnostaras SG, Kogan JH, Takahashi E, Silva AJ. Consolidation of CS and US representations in associative fear conditioning. Hippocampus. 2004;14:557–569. doi: 10.1002/hipo.10208. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Ding HK, Takahashi E, Suzuki A, Kida S, Silva AJ. Stability of recent and remote contextual fear memory. Learn Mem. 2006;13:451–457. doi: 10.1101/lm.183406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gau D, Lemberger T, von Gall C, Kretz O, Le Minh N, Gass P, Schmid W, Schibler U, Korf HW, Schütz G. Phosphorylation of CREB Ser142 regulates light-induced phase shifts of the circadian clock. Neuron. 2002;34:245–253. doi: 10.1016/s0896-6273(02)00656-6. [DOI] [PubMed] [Google Scholar]
- Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- Guzowski JF, McGaugh JL. Antisense oligodeoxynucleotide-mediated disruption of hippocampal cAMP response element binding protein levels impairs consolidation of memory for water maze training. Proc Natl Acad Sci U S A. 1997;94:2693–2698. doi: 10.1073/pnas.94.6.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JH, Kushner SA, Yiu AP, Cole CJ, Matynia A, Brown RA, Neve RL, Guzowski JF, Silva AJ, Josselyn SA. Neuronal competition and selection during memory formation. Science. 2007;316:457–460. doi: 10.1126/science.1139438. [DOI] [PubMed] [Google Scholar]
- Han JH, Yiu AP, Cole CJ, Hsiang HL, Neve RL, Josselyn SA. Increasing CREB in the auditory thalamus enhances memory and generalization of auditory conditioned fear. Learn Mem. 2008;15:443–453. doi: 10.1101/lm.993608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JH, Kushner SA, Yiu AP, Hsiang HL, Buch T, Waisman A, Bontempi B, Neve RL, Frankland PW, Josselyn SA. Selective erasure of a fear memory. Science. 2009;323:1492–1496. doi: 10.1126/science.1164139. [DOI] [PubMed] [Google Scholar]
- Hernández-Rabaza V, Hontecillas-Prieto L, Velázquez-Sanchez C, Ferragud A, Pérez-Villaba A, Arcusa A, Barcia JA, Trejo JL, Canales JJ. The hippocampal dentate gyrus is essential for generating contextual memories of fear and drug-induced reward. Neurobiol Learn Mem. 2008;90:553–559. doi: 10.1016/j.nlm.2008.06.008. [DOI] [PubMed] [Google Scholar]
- Impey S, Mark M, Villacres EC, Poser S, Chavkin C, Storm DR. Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron. 1996;16:973–982. doi: 10.1016/s0896-6273(00)80120-8. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- Impey S, McCorkle SR, Cha-Molstad H, Dwyer JM, Yochum GS, Boss JM, McWeeney S, Dunn JJ, Mandel G, Goodman RH. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell. 2004;119:1041–1054. doi: 10.1016/j.cell.2004.10.032. [DOI] [PubMed] [Google Scholar]
- Iourgenko V, et al. Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc Natl Acad Sci U S A. 2003;100:12147–12152. doi: 10.1073/pnas.1932773100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josselyn SA, Shi C, Carlezon WA, Jr, Neve RL, Nestler EJ, Davis M. Long-term memory is facilitated by cAMP response element-binding protein overexpression in the amygdala. J Neurosci. 2001;21:2404–2412. doi: 10.1523/JNEUROSCI.21-07-02404.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida S, Josselyn SA, Peña de Ortiz S, Kogan JH, Chevere I, Masushige S, Silva AJ. CREB required for the stability of new and reactivated fear memories. Nat Neurosci. 2002;5:348–355. doi: 10.1038/nn819. [DOI] [PubMed] [Google Scholar]
- Kim R, Moki R, Kida S. Molecular mechanisms for the destabilization and restabilization of reactivated spatial memory in the Morris water maze. Mol Brain. 2011;4:9. doi: 10.1186/1756-6606-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornhauser JM, Cowan CW, Shaywitz AJ, Dolmetsch RE, Griffith EC, Hu LS, Haddad C, Xia Z, Greenberg ME. CREB transcriptional activity in neurons is regulated by multiple, calcium-specific phosphorylation events. Neuron. 2002;34:221–233. doi: 10.1016/s0896-6273(02)00655-4. [DOI] [PubMed] [Google Scholar]
- Kovács KA, Steullet P, Steinmann M, Do KQ, Magistretti PJ, Halfon O, Cardinaux JR. TORC1 is a calcium- and cAMP-sensitive coincidence detector involved in hippocampal long-term synaptic plasticity. Proc Natl Acad Sci U S A. 2007;104:4700–4705. doi: 10.1073/pnas.0607524104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamarre-Vincent N, Hsieh-Wilson LC. Dynamic glycosylation of the transcription factor CREB: a potential role in gene regulation. J Am Chem Soc. 2003;125:6612–6613. doi: 10.1021/ja028200t. [DOI] [PubMed] [Google Scholar]
- Larson EB, Graham DL, Arzaga RR, Buzin N, Webb J, Green TA, Bass CE, Neve RL, Terwilliger EF, Nestler EJ, Self DW. Overexpression of CREB in the nucleus accumbens shell increases cocaine reinforcement in self-administering rats. J Neurosci. 2011;31:16447–16457. doi: 10.1523/JNEUROSCI.3070-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattal KM, Honarvar S, Abel T. Effects of post-session injections of anisomycin on the extinction of a spatial preference and on the acquisition of a spatial reversal preference. Behav Brain Res. 2004;153:327–339. doi: 10.1016/j.bbr.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Lee I, Kesner RP. Differential contributions of dorsal hippocampal subregions to memory acquisition and retrieval in contextual fear-conditioning. Hippocampus. 2004;14:301–310. doi: 10.1002/hipo.10177. [DOI] [PubMed] [Google Scholar]
- Lee JL. Reconsolidation: maintaining memory relevance. Trends Neurosci. 2009;32:413–420. doi: 10.1016/j.tins.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JL, Everitt BJ, Thomas KL. Independent cellular processes for hippocampal memory consolidation and reconsolidation. Science. 2004;304:839–843. doi: 10.1126/science.1095760. [DOI] [PubMed] [Google Scholar]
- Leutgeb JK, Moser EI. Enigmas of the dentate gyrus. Neuron. 2007a;55:176–178. doi: 10.1016/j.neuron.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Leutgeb JK, Leutgeb S, Moser MB, Moser EI. Pattern separation in the dentate gyrus and CA3 of the hippocampus. Science. 2007b;315:961–966. doi: 10.1126/science.1135801. [DOI] [PubMed] [Google Scholar]
- Li S, Zhang C, Takemori H, Zhou Y, Xiong ZQ. TORC1 regulates activity-dependent CREB-target gene transcription and dendritic growth of developing cortical neurons. J Neurosci. 2009;29:2334–2343. doi: 10.1523/JNEUROSCI.2296-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Lopez de Armentia M, Jancic D, Olivares R, Alarcon JM, Kandel ER, Barco A. CREB-mediated gene expression increases the intrinsic excitability of CA1 pyramidal neurons. J Neurosci. 2007;27:13909–13918. doi: 10.1523/JNEUROSCI.3850-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Hutchins AE, Doyle CM, Lundblad JR, Kwok RP. Acetylation of cAMP-responsive element-binding protein (CREB) by CREB-binding protein enhances CREB-dependent transcription. J Biol Chem. 2003;278:15727–15734. doi: 10.1074/jbc.M300546200. [DOI] [PubMed] [Google Scholar]
- Mamiya N, Fukushima H, Suzuki A, Matsuyama Z, Homma S, Frankland PW, Kida S. Brain region-specific gene expression activation required for reconsolidation and extinction of contextual fear memory. J Neurosci. 2009;29:402–413. doi: 10.1523/JNEUROSCI.4639-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- McClung CA, Nestler EJ. Regulation of gene expression and cocaine reward by CREB and DeltaFosB. Nat Neurosci. 2003;6:1208–1215. doi: 10.1038/nn1143. [DOI] [PubMed] [Google Scholar]
- Nader K, Einarsson EO. Memory reconsolidation: an update. Ann N Y Acad Sci. 2010;1191:27–41. doi: 10.1111/j.1749-6632.2010.05443.x. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- Parker D, Ferreri K, Nakajima T, LaMorte VJ, Evans R, Koerber SC, Hoeger C, Montminy MR. Phosphorylation of CREB at Ser-133 induces complex formation with CREB-binding protein via a direct mechanism. Mol Cell Biol. 1996;16:694–703. doi: 10.1128/mcb.16.2.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Ed. 2. San Diego: Academic; 2001. [Google Scholar]
- Pittenger C, Huang YY, Paletzki RF, Bourtchuladze R, Scanlin H, Vronskaya S, Kandel ER. Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus-dependent spatial memory. Neuron. 2002;34:447–462. doi: 10.1016/s0896-6273(02)00684-0. [DOI] [PubMed] [Google Scholar]
- Quevedo J, Vianna MR, Roesler R, de-Paris F, Izquierdo I, Rose SP. Two time windows of anisomycin-induced amnesia for inhibitory avoidance training in rats: protection from amnesia by pretraining but not pre-exposure to the task apparatus. Learn Mem. 1999;6:600–607. doi: 10.1101/lm.6.6.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan I, Pérez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: a model for activator:coactivator interactions. Cell. 1997;91:741–752. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
- Ravnskjaer K, Kester H, Liu Y, Zhang X, Lee D, Yates JR, 3rd, Montminy M. Cooperative interactions between CBP and TORC2 confer selectivity to CREB target gene expression. EMBO J. 2007;26:2880–2889. doi: 10.1038/sj.emboj.7601715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restivo L, Tafi E, Ammassari-Teule M, Marie H. Viral-mediated expression of a constitutively active form of CREB in hippocampal neurons increases memory. Hippocampus. 2009;19:228–234. doi: 10.1002/hipo.20527. [DOI] [PubMed] [Google Scholar]
- Rexach JE, Clark PM, Mason DE, Neve RL, Peters EC, Hsieh-Wilson LC. Dynamic O-GlcNAc modification regulates CREB-mediated gene expression and memory formation. Nat Chem Biol. 2012;8:253–261. doi: 10.1038/nchembio.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SJ, et al. Nuclear factor kappa B signaling regulates neuronal morphology and cocaine reward. J Neurosci. 2009;29:3529–3537. doi: 10.1523/JNEUROSCI.6173-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sara SJ. Retrieval and reconsolidation: toward a neurobiology of remembering. Learn Mem. 2000;7:73–84. doi: 10.1101/lm.7.2.73. [DOI] [PubMed] [Google Scholar]
- Schacter DL, Norman KA, Koutstaal W. The cognitive neuroscience of constructive memory. Annu Rev Psychol. 1998;49:289–318. doi: 10.1146/annurev.psych.49.1.289. [DOI] [PubMed] [Google Scholar]
- Screaton RA, Conkright MD, Katoh Y, Best JL, Canettieri G, Jeffries S, Guzman E, Niessen S, Yates JR, 3rd, Takemori H, Okamoto M, Montminy M. The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell. 2004;119:61–74. doi: 10.1016/j.cell.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Sekeres MJ, Neve RL, Frankland PW, Josselyn SA. Dorsal hippocampal CREB is both necessary and sufficient for spatial memory. Learn Mem. 2010;17:280–283. doi: 10.1101/lm.1785510. [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- Shema R, Haramati S, Ron S, Hazvi S, Chen A, Sacktor TC, Dudai Y. Enhancement of consolidated long-term memory by overexpression of protein kinase Mzeta in the neocortex. Science. 2011;331:1207–1210. doi: 10.1126/science.1200215. [DOI] [PubMed] [Google Scholar]
- Sheng M, Thompson MA, Greenberg ME. CREB: a Ca(2+)-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science. 1991;252:1427–1430. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Mukawa T, Tsukagoshi A, Frankland PW, Kida S. Activation of LVGCCs and CB1 receptors required for destabilization of reactivated contextual fear memories. Learn Mem. 2008;15:426–433. doi: 10.1101/lm.888808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenfeld SM, Milekic MH, Monti B, Alberini CM. The consolidation of new but not reactivated memory requires hippocampal C/EBPbeta. Nat Neurosci. 2001;4:813–818. doi: 10.1038/90520. [DOI] [PubMed] [Google Scholar]
- Taylor CT, Furuta GT, Synnestvedt K, Colgan SP. Phosphorylation-dependent targeting of cAMP response element binding protein to the ubiquitin/proteasome pathway in hypoxia. Proc Natl Acad Sci U S A. 2000;97:12091–12096. doi: 10.1073/pnas.220211797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treves A, Tashiro A, Witter MP, Moser EI. What is the mammalian dentate gyrus good for? Neuroscience. 2008;154:1155–1172. doi: 10.1016/j.neuroscience.2008.04.073. [DOI] [PubMed] [Google Scholar]
- Tronson NC, Taylor JR. Molecular mechanisms of memory reconsolidation. Nat Rev Neurosci. 2007;8:262–275. doi: 10.1038/nrn2090. [DOI] [PubMed] [Google Scholar]
- Tronson NC, Wiseman SL, Olausson P, Taylor JR. Bidirectional behavioral plasticity of memory reconsolidation depends on amygdalar protein kinase A. Nat Neurosci. 2006;9:167–169. doi: 10.1038/nn1628. [DOI] [PubMed] [Google Scholar]
- Vetere G, Restivo L, Cole CJ, Ross PJ, Ammassari-Teule M, Josselyn SA, Frankland PW. Spine growth in the anterior cingulate cortex is necessary for the consolidation of contextual fear memory. Proc Natl Acad Sci U S A. 2011;108:8456–8460. doi: 10.1073/pnas.1016275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. J Biol Chem. 2001;276:13505–13508. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]
- von Gall C, Duffield GE, Hastings MH, Kopp MD, Dehghani F, Korf HW, Stehle JH. CREB in the mouse SCN: a molecular interface coding the phase-adjusting stimuli light, glutamate, PACAP, and melatonin for clockwork access. J Neurosci. 1998;18:10389–10397. doi: 10.1523/JNEUROSCI.18-24-10389.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace TL, Stellitano KE, Neve RL, Duman RS. Effects of cyclic adenosine monophosphate response element binding protein overexpression in the basolateral amygdala on behavioral models of depression and anxiety. Biol Psychiatry. 2004;56:151–160. doi: 10.1016/j.biopsych.2004.04.010. [DOI] [PubMed] [Google Scholar]
- Wang SH, Teixeira CM, Wheeler AL, Frankland PW. The precision of remote context memories does not require the hippocampus. Nat Neurosci. 2009;12:253–255. doi: 10.1038/nn.2263. [DOI] [PubMed] [Google Scholar]
- Wang Y, Inoue H, Ravnskjaer K, Viste K, Miller N, Liu Y, Hedrick S, Vera L, Montminy M. Targeted disruption of the CREB coactivator Crtc2 increases insulin sensitivity. Proc Natl Acad Sci U S A. 2010;107:3087–3092. doi: 10.1073/pnas.0914897107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts AG, Sanchez-Watts G, Liu Y, Aguilera G. The distribution of messenger RNAs encoding the three isoforms of the transducer of regulated cAMP responsive element binding protein activity in the rat forebrain. J Neuroendocrinol. 2011;23:754–766. doi: 10.1111/j.1365-2826.2011.02178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Griffith EC, Greenberg ME. Regulation of transcription factors by neuronal activity. Nat Rev Neurosci. 2002;3:921–931. doi: 10.1038/nrn987. [DOI] [PubMed] [Google Scholar]
- Wu Z, Huang X, Feng Y, Handschin C, Gullicksen PS, Bare O, Labow M, Spiegelman B, Stevenson SC. Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1alpha transcription and mitochondrial biogenesis in muscle cells. Proc Natl Acad Sci U S A. 2006;103:14379–14384. doi: 10.1073/pnas.0606714103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Kasper LH, Lerach S, Jeevan T, Brindle PK. Individual CREB-target genes dictate usage of distinct cAMP-responsive coactivation mechanisms. EMBO J. 2007;26:2890–2903. doi: 10.1038/sj.emboj.7601734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CH, Huang CC, Hsu KS. Generalization of fear inhibition by disrupting hippocampal protein synthesis-dependent reconsolidation process. Neuropsychopharmacology. 2011;36:1992–2008. doi: 10.1038/npp.2011.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JC, Wallach JS, Del Vecchio M, Wilder EL, Zhou H, Quinn WG, Tully T. Induction of a dominant negative CREB transgene specifically blocks long-term memory in Drosophila. Cell. 1994;79:49–58. doi: 10.1016/0092-8674(94)90399-9. [DOI] [PubMed] [Google Scholar]
- Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, Chen H, Jenner R, Herbolsheimer E, Jacobsen E, Kadam S, Ecker JR, Emerson B, Hogenesch JB, Unterman T, Young RA, Montminy M. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A. 2005;102:4459–4464. doi: 10.1073/pnas.0501076102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wu H, Li S, Chen Q, Cheng XW, Zheng J, Takemori H, Xiong ZQ. Requirement of TORC1 for late-phase long-term potentiation in the hippocampus. PLoS One. 2006;1:e16. doi: 10.1371/journal.pone.0000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Won J, Karlsson MG, Zhou M, Rogerson T, Balaji J, Neve R, Poirazi P, Silva AJ. CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nat Neurosci. 2009;12:1438–1443. doi: 10.1038/nn.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]