

Abstract
The grik2 gene, coding for the kainate receptor subunit GluK2 (formerly GluR6), is associated with autism spectrum disorders and intellectual disability. Here, we tested the hypothesis that GluK2 could play a role in the appropriate maturation of synaptic circuits involved in learning and memory. We show that both the functional and morphological maturation of hippocampal mossy fiber to CA3 pyramidal cell (mf-CA3) synapses is delayed in mice deficient for the GluK2 subunit (GluK2−/−). In GluK2−/− mice this deficit is manifested by a transient reduction in the amplitude of AMPA-EPSCs at a critical time point of postnatal development, whereas the NMDA component is spared. By combining multiple probability peak fluctuation analysis and immunohistochemistry, we have provided evidence that the decreased amplitude reflects a decrease in the quantal size per mf-CA3 synapse and in the number of active synaptic sites. Furthermore, we analyzed the time course of structural maturation of CA3 synapses by confocal imaging of YFP-expressing cells followed by tridimensional (3D) anatomical reconstruction of thorny excrescences and presynaptic boutons. We show that major changes in synaptic structures occur subsequently to the sharp increase in synaptic transmission, and more importantly that the course of structural maturation of synaptic elements is impaired in GluK2−/− mice. This study highlights how a mutation in a gene linked to intellectual disability in the human may lead to a transient reduction of synaptic strength during postnatal development, impacting on the proper formation of neural circuits linked to memory.
Introduction
Precise control of the development of synapses and neuronal connectivity is critical for accurate neural network activity and normal brain function at the adult stage. Improper function of synapses is a common feature associated with intellectual disability (ID) and autism spectrum disorders (ASDs) (Toro et al., 2010). Gain and loss of function approaches in mouse models of ID have revealed alterations in synaptic function and plasticity, as well as in spine morphology and in the balance between excitation and inhibition (Humeau et al., 2009; Boda et al., 2010).Together, these data strongly suggest that abnormal synaptic homeostasis represents an underlying mechanism to ID and ASD in the adult. Here, we take a further and complementary step to understand how mutation of a gene associated with ID and ASD in humans impairs the course of synaptic maturation in vivo.
Loss of function mutations in the grik2 gene, encoding the GluK2 (formerly GluR6) subunit of kainate receptors (KARs), cosegregate with nonsyndromic autosomal recessive ID (Motazacker et al., 2007). In the adult, KARs play an important role in the regulation of network activity by acting at both presynaptic and postsynaptic sites to regulate neuronal excitability and neurotransmitter release (Contractor et al., 2011). Thus, the lack of a functional GluK2 may directly impact on the activity of cortical synaptic circuits (Sachidhanandam et al., 2009), hence on higher brain function (Ko et al., 2005). Because of its association with neurodevelopmental disorders, GluK2 may also serve in the activity-dependent refinement of synaptic connections during development, leading to improper maturation of neuronal networks.
KARs and especially those comprising the GluK2 subunit are particularly abundant at presynaptic and postsynaptic levels at hippocampal mossy fiber-CA3 synapses (Mulle et al., 1998; Contractor et al., 2001). Hippocampal mossy fibers (mfs) are the axons of dentate granule cells that make characteristic synaptic connections with large spines on CA3 pyramidal neurons (Henze et al., 2000). These glutamatergic synapses display a number of unique functional features that have been well studied in the mature rodent brain (Nicoll and Schmitz, 2005) and they play a major role in learning and memory (Kesner, 2007). Mf-CA3 synapses constitute an attractive model to investigate patterns of synaptic maturation and to establish direct links between the different steps of morphological and functional development of synaptic properties. Mf-CA3 synapses undergo several stages of functional maturation during the first 3 weeks of postnatal development, with a “critical” phase at the beginning of the second week (postnatal day 6–9) during which the composition of postsynaptic glutamate receptors and the strength of synaptic connections abruptly change (Marchal and Mulle, 2004).
We have previously proposed a role for KARs in the functional maturation of CA3 synapses (Marchal and Mulle, 2004). Here we have taken a further step by examining which quantal parameters account for the marked developmental maturation of presynaptic and postsynaptic elements at mf-CA3 synapses during the first 3 weeks after birth. Furthermore, we have combined a detailed analysis of synaptic properties with a morphometric analysis of thorny excrescences and presynaptic mossy fiber terminals. These analyses were performed in parallel in control mice and in mice deficient for the GluK2 subunit, to test the hypothesis of a defective synaptic maturation in vivo, in a mouse model of autosomal nonsyndromic ID.
Materials and Methods
Experimental procedures followed the recommendations of the CNRS ethics committee and the French Ministry of Agriculture and Forestry concerning animal care (authorization number, A33093). GluK2 knock-out mice (GluK2−/−) were generated on a hybrid C57BL6/SV129 background (Mulle et al., 1998) and backcrossed for 12 generations on a FVB background. Wild-type (GluK2+/+) and GluK2−/− mice used were from heterozygous breeding pairs.
Slice preparation and electrophysiology.
Pups and young mice of either sex were killed by cervical dislocation. Parasagittal brain slices (320–350 μm thick) were prepared from P6 to P21 mice (P0 being the day of birth) in a solution containing the following (in mm): 80 NaCl, 2.5 KCl, 25 NaHCO3, 1.5 NaH2PO4, 7 MgCl2, 0.5 CaCl2, 10 glucose, and 75 sucrose (equilibrated with 95% O2/5% CO2). Slices were incubated at 33°C for 30 min and subsequently stored at room temperature in an extracellular solution composed of (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, 11 glucose (equilibrated with 95% O2/5% CO2). Whole-cell voltage-clamp recordings (3–4 MΩ electrodes, −70 mV holding potential) were made from hippocampal CA3 pyramidal cells visualized by infrared video-microscopy. Experiments were performed in extracellular solution near physiological temperature (33°C). Intracellular solution was composed of the following (in mm): 122 Cs methanesulfonate, 2 NaCl, 10 HEPES, 10 EGTA, 2 MgCl2, 4 ATP-Na, pH 7.3. Bicuculline (10 μm) was added to the bath to inhibit GABAA receptors and d-AP5 (25 μm) or NBQX (10 μm) were used respectively to record AMPA/kainate-EPSCs (mf-EPSCs) at −70 mV or NMDA-EPSCs at +30 mV in isolation. The series resistance of the cell was monitored during the recording using a −10 mV hyperpolarizing voltage step of 50 ms length occurring at the beginning of each recording. The average series resistance was 15.6 ± 0.3 MΩ (n = 293 cells) and cells were discarded if it changed by >20%. Recordings were made using an EPC 9.0 amplifier (HEKA Elektronik), filtered at 2.9 kHz with a Bessel filter, digitized at 10 kHz, and stored on a personal computer for additional analysis (IGOR PRO 5.0; Wave-Metrics).
Mf-CA3 EPSCs were evoked by minimal intensity stimulation (Marchal and Mulle, 2004; Sachidhanandam et al., 2009). A glass microelectrode was placed in the hilus of the dentate gyrus to stimulate mf. While recording from a CA3 pyramidal cell, the stimulating electrode was moved to a position where a sharp EPSC with fixed latency was evoked. Stimulation intensity (200 μs pulse; <20 μA) was adjusted just above the sharp threshold for activation of a synaptic response. Using such low minimal stimulations, no prominent polysynaptic activation was observed. On average, the rise time (10–90%) of EPSCs recorded at 0.1 Hz (n = 78) was 1.3 ± 0.04 ms and the tau weighted was 6 ± 0.3 ms. Mf stimulation was assessed by its short-term facilitation properties (Salin et al., 1996) (40 ms paired pulse ratio or 1 Hz/0.1 Hz ratio >3) except for some P6 or P7 recordings where the facilitation ratio could be smaller because of their immature state. The group II mGluR agonist LCCG-1 (10 μm) was applied to confirm that mfs were stimulated (Lanore et al., 2010). Low-frequency facilitation was evoked by increasing stimulation frequency from 0.1 to 1 Hz.
For strontium (Sr2+) experiments, slices were bathed in an extracellular solution containing SrCl2 (4 mm) and no Ca2+. Asynchronous EPSCs (aEPSCs) were evoked by trains of five stimuli at 50 Hz repeated 100 times at 30 s intervals. Only events occurring within a 400 ms time window beginning 50 ms after the last stimulus in each train were considered for statistical analysis. aEPSCs were detected using an amplitude criterion set at 7.5 pA (three times the basal noise level; SD, 2–3 pA; leading to <1.5% false positive events). Events with a rise time (20–80%) and a decay time (fitted with a single-exponential) between 0.2–2 ms and 1–20 ms, respectively, were kept for further analysis. The peak amplitude of the individual aEPSC was calculated on a 0.4 ms window centered on the maximum of the peak amplitude. Individual events were baseline subtracted using a 2 ms window before the onset of the aEPSC.
For multiple probability peak fluctuation analysis (MPFA), mf-CA3 EPSCs were recorded in solutions containing either 2 or 8 mm Ca2+ concentrations and evoked at various frequencies (0.1, 1, and 3 Hz) to yield a wide range of release probabilities. For each condition, a single mf was stimulated 60 times and the amplitude of the last 50 mf-CA3 EPSCs were measured (Fig. 1d) in a 0.4 ms window around the maximum of the peak of the EPSC. The time stability of the last 50 CA3 EPSCs was assessed using Spearman rank order correlation analysis (in Igor pro running under NeuroMatic, http://www.neuromatic.thinkrandom.com/) for each condition (Silver, 2003). The baseline noise variance, determined from a 0.4 ms window before the stimulation artifact, was subtracted from the synaptic variance and the synaptic variance was then plotted against the mean at each release probability condition. MPFA was performed using a multinomial quantal model (Silver et al., 1998; Silver, 2003). The mean amplitude of the synaptic response is I = npq, where I is the mean current, n the number of functional release site, p the probability of release, and q the quantal size. The variance (σ2) can be expressed as a function of I using the following relationship: σ2 = (qI − I2/n) (1 + CVqII2) + qICVqI2, where CVqII and CVqI are type II (intersite) and type I (intrasite) quantal coefficients of variation (CV), respectively. The variance–mean relationship plotted over a range of release probabilities is approximately parabolic. The equation for the parabola is as follows: y = Ax − Bx2 where y is the variance and x the mean amplitude of mf-CA3 EPSCs. The total quantal variability is (CVq2 = CVqI2 + CVqII2). However, it is not practical to determine the intrasite quantal variability at the mf-CA3 synapse because individual sites cannot be stimulated in isolation. We thus measured CVq2 by determining the CV2 of aEPSCs elicited in Sr2+ (Foster and Regehr, 2004) (GluK2+/+: P6–P7: 1.09; P8–P9: 0.84, P14–P16: 0.69, P19–P21: 0.61; GluK2−/−: P8–P9: 0.65, P19–P21: 0.87). We assumed that CVqI2 = CVqII2, which is likely to introduce relatively little error (Clements, 2003; Silver, 2003). We also assumed that each active zone contained a fixed number of vesicles that can be released by a presynaptic action potential with a uniform probability of release p. Vesicles at a given active zone were considered to release transmitter onto a common pool of postsynaptic receptors. n denotes the number of independent “release sites” at which quantal release events can take place.
Figure 1.
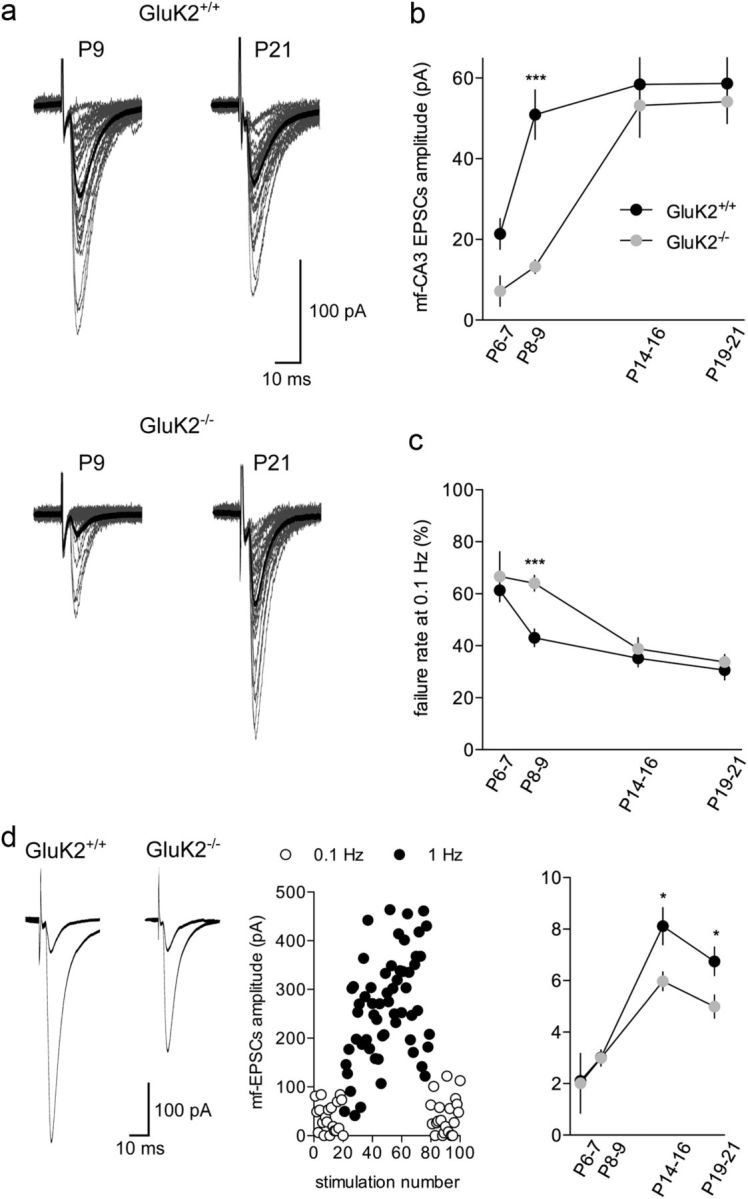
The time course of functional synaptic maturation is delayed in GluK2−/− mice. a, Representative traces (30 superimposed traces in gray) recorded at 0.1 Hz in GluK2+/+ and GluK2−/− mice at P9 (left) and P21 (right). The average mf-CA3 EPSC is represented in black. b, Summary graph of the average mf-CA3 EPSCs recorded at 0.1 Hz between P6 and P21 for GluK2+/+ and GluK2−/− mice. c, Summary graph of failures at 0.1 Hz between P6 and P21 for GluK2+/+ and GluK2−/− mice. d, Average of 50 traces (left) illustrating low-frequency facilitation of mf-CA3 EPSCs when shifting frequency stimulation from 0.1 to 1 Hz for GluK2+/+ and GluK2−/− mice. Graph illustrating the time course of frequency facilitation when shifting the stimulation frequency from 0.1 to 1 Hz. Each dot represents the amplitude of a single EPSC (white dots: 0.1 Hz, black dots: 1 Hz). This example has been recorded in a GluK2+/+ mouse at P19. Summary graph of the mean frequency facilitation ratio from 0.1 to 1 Hz for GluK2+/+ and GluK2−/− mice. Mann–Whitney test: *p < 0.05; ***p < 0.001.
All drugs were obtained from Tocris Cookson, Sigma, or Ascent Scientific.
Generation of lentiviral vectors.
The myristoylated-palmitoylated YFP-expressing lentiviral vector (mYFP) is based on the FUW plasmid (generously provided by C. Lois, California Institute of Technology, Pasadena, CA). FUW vector carries a CMV enhancer, a substituted 5′ LTR for viral RNA expression, an ubiquitin promoter, a polylinker, and the woodchuck hepatitis virus posttranscriptional regulatory element (WRE) followed by a self-inactivating 3′ LTR. The membrane-bound YFP is based on the plasmid MyrPalm-mYFP in pcDNA3, originally from Dr. R.Y. Tsien's laboratory, and was a gift from Dr. Helge Ewers (Institute of Biochemistry, ETH Zurich, Zurich, Switzerland). It contains three amino acid substitution/insertions (19K, E209D, L240K) and two silent mutations compared with the published sequence. The BamHI-EcoRI-digested MyrPalm-mYFP fragment carrying the entire coding sequence was subcloned into the vector pGEM-7zf(+) (Promega) at identical positions. The resulting clone was digested by BamHI and EcoRI, ligated into identical positions of the polylinker of FUW, and transformed into Stbl3 chemically competent E. coli bacteria (Invitrogen). Sequence integrity was verified by restriction enzyme cleavages and partial DNA sequencing.
Viral gene transfer and stereotaxic delivery.
Mouse pups (P1–P2) were anesthetized by isoflurane inhalation. The virus solution was injected with a brief pulse of air delivered by a picospritzer (Intracell). Approximately 100 nl of virus solution was distributed in the CA3 layer (Y: +0.3 mm from lambda; X: ±2.3 mm; Z: −1.5 mm) or in the dentate gyrus (Y: +0.7 mm from lambda; X: ±1.3 mm; Z: −1 mm) to label CA3 pyramidal cells and granule cells respectively using a binocular microscope (Nikon SMZ800; zoom used: 45×) equipped with a scaled crosshair reticle. After surgery, animals recovered within minutes and were returned to their mother. The injection capillaries were pulled on a puller (model P-97, Sutter Instruments) using micropipettes (5 μl, ringcaps). The specific settings resulted in elongated tips that were trimmed to obtain a sharply edged tip with a diameter of ∼30 μm.
After in vivo protein expression for a minimum of 4 d, mice were deeply anesthetized using 0.7% chlorohydrate and perfused transcardially with 0.9% NaCl for 1 min followed by 4% formaldehyde in 0.1 mm PBS for 10 min. Brains were removed, postfixed in the same fixative for 4 h at 4°C, and conserved at 4°C in PBS azide (0.05%). Coronal sections of the hippocampus (60 μm) were cut on a vibratome (Leica VT1000S) and examined with confocal microscopy.
Confocal laser-scanning fluorescence microscopy and image analysis.
Confocal image stacks of PFA-fixed brain slices were acquired with a Leica TCS SP2 laser-scanning microscope with 488 nm (YFP) excitation wavelength using 63× Leica oil objectives or a confocal spinning-disk (Leica DMI6000) equipped with a confocal Scanner Unit CSU-X1 (Yokogawa), an Evolve EMCCD camera (Roper Scientific), and a galvanometric stage (Leica Microsystems), using the objective HCX PL Apo CS 63×oil NA 1.4 and the 491 nm diode laser. High-resolution 3D stacks were generated using Imaris 7.1.1 software (Bitplane) to characterize and analyze presynaptic terminals and thorny excrescences. The analysis of filopodia was made using ImageJ software on 2D image projections of 3D acquired confocal stacks. Illustrations correspond to 3D reconstructions smoothened by the median filter.
Immunohistochemistry.
Pups and young mice were killed by cervical dislocation. Brains were quickly removed, snap frozen in liquid nitrogen, and stored at −80°C before sectioning. Coronal sections (14 μm) of hippocampus were permeabilized with Clark liquid (1 volume acetic acid/6 volume ethanol) for 15 min. Nonspecific binding was blocked with PBS containing 3% BSA. Synaptic structures were labeled with mouse anti-SAP102 and rabbit anti-Munc13.1 (1:500, Synaptic Systems) or anti-GluA1 (1:500, Abcam) on coated slides followed by overnight incubation with Alexa 488-conjugated goat anti-rabbit antibody and Alexa 568-conjugated goat anti-mouse antibody (2 mg/ml, Invitrogen).
Immunostainings were visualized with a confocal spinning disk microscope equipped with a 100× oil/1.4 NA objective and appropriate diode lasers. Images of the mossy fiber termination zone in the CA3b region of hippocampus were captured with an interval of 0.2 μm in the z-axis. Camera aperture, magnification, light power, and exposure time were fixed for all images. Ten stacks from a minimum of five animals of each genotype and age were acquired randomly. Images were later processed as described below. To compute the density of colocalization between presynaptic elements (Munc13.1) and postsynaptic clusters (SAP102), analysis was performed using the “Colocalization” module of Imaris 7.1.1 software. Two stacks, one for each type of staining, were analyzed, allowing 3D identification of synaptic structures as individual objects, which were then automatically counted and characterized. These objects are used to determine the number of postsynaptic clusters that are colocalized to a presynapse per μm3.
To count the number of GluA1 clusters per surface area in the CA3b stratum lucidum region and to measure their integrated intensity, images were treated by a segmentation program allowing clusters to be detected as single objects, which were then automatically counted. An intensity threshold was applied to define the stratum lucidum region contour (excluding cell bodies) and calculate its area. For each cluster detected as an object, a region mask was created and transferred onto the original image to measure the integrated intensity after background correction.
Statistical analysis.
Values are presented as mean ± SEM of n experiments unless otherwise stated. For statistical analysis, nonparametric tests were used. Mann–Whitney test was used for comparison of two groups and Kruskal–Wallis test followed by a Dunn's multiple-comparison test for comparison between more than two groups. Statistical differences were considered as significant when p < 0.05.
Results
Functional maturation of mf-CA3 synapses is delayed in GluK2−/− mice
We investigated the time course of maturation of mf-CA3 synaptic transmission at different stages of postnatal development in GluK2+/+ and GluK2−/− mice between P6 and P21. For this purpose, we recorded EPSCs evoked in CA3 pyramidal cells by stimulation of mfs (mf-CA3 EPSCs) in hippocampal slices (Fig. 1a). Consistent with previous observations (Marchal and Mulle, 2004; Lanore et al., 2010), the average amplitude of mf-CA3 EPSCs recorded at low stimulation rate (0.1 Hz) abruptly increased between P6 (P6–P7) and P9 (P8–P9) (P6: 21 ± 4 pA, n = 28, P9: 51 ± 6 pA, n = 46; p < 0.01) (Fig. 1b). This abrupt change in mf-CA3 EPSCs amplitude was delayed in GluK2−/− mice (P9: 13 ± 2 pA, n = 35, p < 0.0001) (Fig. 1b) pointing to an impairment in the course of functional maturation of synaptic properties in these mice.
We attempted to describe which synaptic parameters account for the sharp developmental increase of mf-CA3 EPSCs amplitude in control mice, hence providing an explanation for the differences observed in GluK2−/− mice. The developmental increase of mf-CA3 EPSCs amplitude was paralleled by a decrease in the rate of synaptic failures in control mice (P6: 61 ± 4%, n = 28 and P9: 43 ± 3%, n = 46) (Fig. 1c). The increase of mf-CA3 EPSCs amplitude and the reduction in failure rate between P6 and P9 in control mice may be attributed to an increase in release probability. However, the extent of mf-CA3 EPSC facilitation observed when switching the rate of stimulation from 0.1 to 1 Hz (“low-frequency facilitation,” LFF) (Fig. 1d) did not change significantly between P6 and P9, arguing against a major change in release probability (Fig. 1d). In addition, no significant difference in LFF was observed between the two genotypes at P6 (GluK2+/+: 2.1 ± 0.3, n = 11; GluK2−/−: 2.0 ± 1.0, n = 4) and P9 (GluK2+/+: 3.0 ± 0.3, n = 32; GluK2−/−: 2.9 ± 0.3, n = 27) (Fig. 1d). The extent of LFF was, however, significantly lower in GluK2−/− mice as compared with GluK2+/+ at P14–P16 (GluK2+/+: 8.1 ± 0.7, n = 16 and GluK2−/−: 5.9 ± 0.3, n = 12, p < 0.05) and P19–P21 (GluK2+/+: 7.8 ± 0.7, n = 12 and GluK2−/−: 5.5 ± 0.5, n = 9, p < 0.05). At these stages, decreased LFF is likely caused by the absence of facilitatory presynaptic KARs containing GluK2 subunits (Contractor et al., 2001; Pinheiro et al., 2007).
The analysis of failure rates and LFF led to apparent contradictions regarding the basis for the abrupt increase in synaptic strength between P6 and P9, and the cause of the delayed maturation of mf-CA3 synaptic transmission in GluK2−/− mice. However, failure rates may also depend on the number of release sites (i.e., the larger number of release sites, the lower the failure rates), on failures of action potential propagation between granule cell soma and presynaptic terminals, or on the presence of postsynaptically silent sites (devoid of AMPA receptors, AMPARs).
We used multiple-probability fluctuation analysis (MPFA) to estimate the quantal parameters that describe synaptic efficacy (Silver et al., 1998): the number of release sites (n), the mean probability of release (p), and the amplitude of the response to a single vesicle release (quantum, q). Mf-CA3 synaptic transmission was investigated over a wide range of experimentally imposed release probabilities by increasing stimulation rate (from 0.1 to 3 Hz) (Fig. 2a) and modifying the extracellular Ca2+ concentration. In 8 mm Ca2+, mf-CA3 EPSCs at 0.1 Hz were potentiated by 319 ± 42% at P6, 373 ± 68% at P9, 344 ± 69% at P14–P16 and 868 ± 311% at P19–P21 (data not shown). The mean variance of the EPSCs was plotted against the mean EPSC amplitude and the resulting variance-mean relationship was fitted with a parabola (Fig. 2b). In control mice, the estimated n increased progressively and significantly between P6 and P21 (P6: 7.9 ± 1.9, n = 6; P9: 11.0 ± 1.2, n = 17; P14–P16: 15.5 ± 2.4, n = 12: P19–P21: 31.3 ± 5.6, n = 10) (Fig. 2c), while the estimated p significantly dropped between P9 and P21 (P6: 0.09 ± 0.01, n = 6; P9: 0.16 ± 0.02, n = 17; P14–P16: 0.07 ± 0.01, n = 12: P19–P21: 0.04 ± 0.007, n = 10) (Fig. 2d). The estimated q increased sharply between P6 and P9 and remained almost stable afterward (P6: 27 ± 3 pA, n = 6; P9: 49 ± 6 pA, n = 17; P14–P16: 58 ± 3 pA, n = 12; P19–P21: 49 ± 7 pA, n = 10) (Fig. 2e). As an additional estimation of the quantal amplitude, we measured the amplitude of aEPSCs (Fig. 2f), assumed to correspond to the quantal release of neurotransmitter (Bekkers and Clements, 1999). For each age group in control mice, the quantal amplitude estimated by MPFA and by aEPSC amplitude was in the same range; thus, the average amplitude of aEPSCs increased sharply between P6 and P9 (P6: 29 ± 1 pA, 6 cells, n = 1496 events; P9: 47 ± 1 pA, 12 cells, n = 4704 events; P14–P16: 58 ± 1 pA, 14 cells, n = 6155 events; P19–P21: 49 ± 1 pA, 14 cells, n = 7775 events) (Fig. 2f).
Figure 2.

Multiple peak fluctuation analysis and asynchronous recordings in GluK2+/+ mice between P6 and P21. a, Representatives traces (30 superimposed traces in gray) recorded at different frequencies in 2 and 8 mm extracellular Ca2+ in a GluK2+/+ mouse at P19. The average mf-CA3 EPSCs are represented in black. Note the decrease in the percentage of failures and in the amplitude variance (gray traces) when increasing release probability. b, Relationship between variance and mf-CA3 EPSCs mean amplitude for the recordings shown in a. The variance–mean plot was then fitted with a simple parabola (continuous line). c, Summary graph of the estimated number of release sites between P6 and P21 in GluK2+/+ mice. d, Summary graph of the estimated release probability at 0.1 Hz between P6 and P21 in GluK2+/+ mice. e, Summary graph of the estimated quantal amplitude between P6 and P21 in GluK2+/+ mice. f, Representative recordings of mf-CA3 EPSCs in response to a train of 5 mf stimuli (50 Hz, every 30 s) in control conditions (top) or in an extracellular medium where Ca2+ has been replaced by 4 mm Sr2+ (bottom) in GluK2+/+ at P21. Asterisks display asynchronous EPSCs. Summary graph of the aEPSCs amplitude between P6 and P21 in GluK2+/+mice (right).
We then analyzed these quantal parameters in GluK2−/− mice at P9, the time point where the major difference in mf-CA3 EPSCs amplitude was observed. No significant difference in p was observed between GluK2+/+ and GluK2−/− mice (Fig. 3a). On the contrary, the mean estimated n was smaller in GluK2−/− mice as compared with GluK2+/+, probably because of a low proportion of synapses with an estimated n larger than 7 (GluK2−/−: 6.9 ± 1.6, n = 9; p < 0.05) (Fig. 3b, c). Strikingly, q measured by MPFA at P9 (GluK2−/−: 29 ± 5 pA, n = 9; p < 0.0001) (Fig. 3d) and the mean aEPSC amplitude (GluK2−/−: 38 ± 1 pA, 16 cells, n = 4796 events; p < 0.0001) (Fig. 3e) were significantly smaller in GluK2−/− compared with GluK2+/+. At a later maturation stage (P21) when basal synaptic transmission is normalized in GluK2−/− mice, neither quantal parameters appeared significantly different between genotypes (Fig. 3a–e).
Figure 3.

The delay in functional maturation in GluK2−/− mice at P8–P9 is due to a decrease in n and q. a, Summary graph of the estimated probability of release at 0.1 Hz in GluK2+/+ and GluK2−/− mice at P8–P9 and P19–P21. b, Summary graph of the estimated number of release sites in GluK2+/+ and GluK2−/− mice at P8–P9 and P19–P21 (left). c, Frequency distribution (%) of the estimated number of release sites in GluK2+/+ at P6–P7 and P8–P9 and in GluK2−/− mice at P8–P9. d, Summary graph of the estimated quantal amplitude between in GluK2+/+ and GluK2−/− mice at P8–P9 and P19–P21. e, Representative recordings of mf-CA3 EPSCs in response to a train of 5 mf stimuli (50 Hz) in an extracellular medium where Ca2+ has been replaced by 4 mm Sr2+ in GluK2+/+ and GluK2−/− mice at P8–P9. Asterisks display asynchronous EPSCs. Summary graph of the aEPSC amplitude in GluK2+/+and GluK2−/−mice at P8–P9 and P19–P21. Mann–Whitney test: *p < 0.05, ***p < 0.001.
To get further insight into the parameters explaining the delayed functional maturation of mf-CA3 synapses in GluK2−/− mice, we performed immunohistological experiments on fixed brain slices with the postsynaptic marker SAP102 and the presynaptic marker Munc13-1. We quantified colocalized Munc13.1 and SAP102 clusters per μm3 in the stratum lucidum, which likely represent multiple neighboring synaptic sites. A change in the number (and/or volume) of colocalized Munc/SAP 102 clusters is likely to reflect a change in the number of synaptic sites (Fig. 4a). The number of colocalized Munc13.1/SAP102 clusters per μm3 was decreased in GluK2−/− as compared with GluK2+/+ mice at P6, P9, and P14 (P6, GluK2+/+: 0.04 ± 0.01, n = 12 mice and GluK2−/−: 0.01 ± 0.01, n = 8 mice; P9, GluK2+/+: 0.04 ± 0.01, n = 14 mice and GluK2−/−: 0.01 ± 0.01, n = 16 mice; P14, GluK2+/+: 0.07 ± 0.01, n = 10 mice and GluK2−/−: 0.04 ± 0.01, n = 14 mice; P21, GluK2+/+: 0.04 ± 0.01, n = 11 mice and GluK2−/−: 0.05 ± 0.01, n = 11 mice) (Fig. 4b). In addition, the density of immunolabeled clusters of GluA1, a subunit of AMPARs, as well as the integrated intensity of GluA1 immunofluorescence were significantly decreased in GluK2−/− mice at P9 (density: GluK2+/+: 0.44 ± 0.03, n = 6 and GluK2−/−: 0.31 ± 0.01 n = 7, p < 0.01; integrated intensity: GluK2+/+: 331 ± 12, n = 6 and GluK2−/−: 233 ± 22, n = 7, p < 0.01) (Fig. 4c–e) consistent with a decrease in q. Altogether, the delay in functional synaptic maturation in GluK2−/− mice between P6 and P9 is best explained by a reduction of q and of the number of active synaptic sites per mf-CA3 synapse.
Figure 4.

Synaptic markers in the stratum lucidum are decreased in GluK2−/− mice at P9. a, Staining of the stratum lucidum at P9 with Munc13-1, SAP102 antibodies, and the merge images in CA3b in GluK2+/+ and GluK2−/− mice. b, Summary graph of colocalized voxels between Munc13-1/SAP102 for GluK2+/+and GluK2−/− mice between P6 and P21. c, Staining of the stratum lucidum at P9 with GluA1 antibody in CA3b in GluK2+/+ and GluK2−/− mice. d, Summary graphs of the integrated intensity (left) and the density (right) of GluA1 clusters at P9 for GluK2+/+ and GluK2−/−mice. Mann–Whitney test: *p < 0.05, **p < 0.01.
We investigated the relationships between the AMPA and NMDA components, in particular at P9, the stage of major differences between GluK2+/+ and GluK2−/− mice. The amplitude of NMDA-EPSCs at 0.1 Hz did not change significantly between P9 and P21 in GluK2+/+ mice (P9: 20 ± 4 pA, n = 16 and P21: 33 ± 5 pA, n = 15) (Fig. 5a,b). Consistent with the stable mf-CA3 EPSCs amplitude between P9 and P21, the NMDA/AMPA ratio did not change significantly (Fig. 5c). Additionally, we noticed that NMDA-EPSCs progressively displayed faster decay kinetics (Fig. 5d) and lower sensitivity to Ro25–6981 (10 μm) during the course of maturation (% of inhibition: P9: 50 ± 6%, n = 10, P14: 25 ± 7%, n = 11, and P21: − 3 ± 10%, n = 11; data not shown), indicating a progressive removal of GluN2B subunit from NMDARs as described at other synapses. Contrary to AMPA-EPSCs, the amplitude of NMDA-EPSCs was not significantly different in GluK2−/− mice as compared with GluK2+/+ mice at P9 (Fig. 5b). Consequently, the NMDA/AMPA ratio at P9 was larger in GluK2−/− mice than in GluK2+/+ mice (GluK2−/−: 1.1 ± 0.2, n = 8; GluK2+/+: 0.4 ± 0.1, n = 12; p < 0.05) (Fig. 5c) and similar to the ratio in P6 GluK2+/+ mice (1.4 ± 0.3, n = 17, data not shown), arguing again in favor of a delayed synapse maturation in GluK2−/− mice. At P21, neither NMDA-EPSCs amplitude and decay kinetics, nor the NMDA/AMPA ratio were different between both genotypes (Fig. 5a–d). Thus the GluK2−/− mutation selectively impacts the time course of development of the AMPA component, rather than the NMDA component of mf-CA3 synapses.
Figure 5.

NMDA-EPSCs at hippocampal mf-CA3 synapses during maturation are not affected in GluK2−/− mice. a, Average of 30 traces illustrating the NMDA/AMPA ratio at P8–P9 and P19–P21 for GluK2+/+ and GluK2−/− mice. b, Summary graph of the NMDA-EPSCs amplitude at 0.1 Hz between P6 and P21 for GluK2+/+ and GluK2−/− mice. c, Summary graph of the NMDA/AMPA ratio between P6 and P21 for GluK2+/+ and GluK2−/− mice. d, Average traces of NMDA-EPSCs at P6 and P21 illustrating the change in decay with maturation. Summary graph (right) of the tau weighted of NMDA-EPSCs between P6 and P21 for GluK2+/+ and GluK2−/− mice. Mann–Whitney test: *p < 0.05.
Correlated functional and morphological maturation is impaired in GluK2−/− mice
We then took a morphometric approach to examine the coordinated functional and morphological changes of presynaptic and postsynaptic elements during the course of maturation in GluK2+/+ and in GluK2−/− mice. We first investigated the maturation of thorny excrescences (TEs), the postsynaptic elements of mf-CA3 synapses, by confocal microscopy of YFP-expressing CA3 pyramidal cells (Fig. 6a,b), followed by 3D reconstruction (Fig. 6c). In GluK2+/+ mice, the volume of TEs was small at P6 and P9, but significantly increased between P9 and P14 (P6: 2.8 ± 0.8 μm3, n = 16; P9: 1.4 ± 0.4 μm3, n = 19; P14: 11.3 ± 1.1 μm3, n = 85; P21: 14.8 ± 2.2 μm3, n = 107; p < 0.001) (Fig. 6d,e). This contrasts with the abrupt increase in the strength of synaptic transmission observed earlier during development, between P6 and P9. The same analysis performed in GluK2−/− mice revealed a marked delay in the appearance of TEs in CA3 pyramidal cells, with no identified TEs at P6 and only 29% of the cells (2 cells out of 7) displaying TEs at P9 (as compared with 50% in GluK2+/+ mice) (Fig. 6d). Furthermore, the volume of TEs in GluK2−/− at P14 was smaller as compared with GluK2+/+ mice (GluK2−/−: 3.4 ± 0.5 μm3, n = 19, p < 0.01) (Fig. 6e,f). These observations indicate a progressive maturation of the number and volume of the postsynaptic elements, in a manner that does not directly relate to the amplitude of mf-CA3 EPSCs. In addition to the delay in functional maturation, GluK2−/− mice displayed a marked delay in the appearance and maturation of the postsynaptic elements of mf-CA3 synapses.
Figure 6.

The maturation of postsynaptic mossy fiber elements is delayed in GluK2−/− mice. a, Schematic representation of the experimental procedure to infect CA3 pyramidal cells in pups (P1–P2). A micropipette is used to inject the mYFP-expressing lentivirus. After minimum 4 d of expression, tissues were prepared for imaging (DG, dentate gyrus). Pictures of a hippocampus labeled with mYFP. b, Projection of 3D-reconstructed confocal image stacks of a CA3 pyramidal cell labeled by lentivirus infection with mYFP at P21 in GluK2+/+ mouse. c, Example images of a z series acquisition of a thorny excrescence and an example of a thorny excrescence reconstructed with surface rendering (below, in white) with Imaris software. d, Examples of cells at P9 in GluK2+/+ and GluK2−/− mice with thorny excrescences indicated by the white arrows. e, Graph of the thorny excrescence volume between P9 and P21 for GluK2+/+ and GluK2−/− mice. f, Reconstructed volumes of thorny excrescences at P14 for GluK2+/+ and GluK2−/− mice. Mann–Whitney test: ***p < 0.001.
Presynaptic boutons of mf-CA3 terminals develop after birth, with the first mf-CA3 synaptic contacts being visible by electron microscopy at P0–P3 (Amaral and Dent, 1981). We quantified the morphological characteristics of presynaptic terminals during the course of maturation using the same lentiviral injection approach but labeling dentate gyrus granule cells (Fig. 7a). In GluK2+/+ mice, the presynaptic terminal structure was homogeneous at P6 and became more complex around P9 (Fig. 7b). Between P9 and P14, the appearance of filopodia on the presynaptic boutons caused a dramatic complexification of the structure (Fig. 7b) (see quantification below). The volume of presynaptic boutons was small at P6 and P9 (P6: 3.3 ± 0.4 μm3, n = 8 mice; P9: 5.2 ± 1.1 μm3, n = 11 mice), but abruptly increased between P9 and P14 (P14: 28.9 ± 3.0 μm3, n = 5 mice; p < 0.01) and remained stable later on (P21: 16.9 ± 2.6 μm3, n = 8 mice), in accordance with the maturation of TEs (Fig. 7c). The index of complexity, calculated by dividing bouton volume by its area, rapidly increased between P6 and P14 (P6: 0.06 ± 0.01 μm, n = 8 mice, P9: 0.24 ± 0.02 μm, n = 11 mice, P14: 0.42 ± 0.02 μm, n = 5 mice, P21: 0.39 ± 0.01 μm, n = 5 mice) (Fig. 7d). In GluK2−/− mice, a parallel developmental profile was found for both parameters although it was markedly delayed. The volume of presynaptic elements were decreased at P9 and P14 in GluK2−/− (P9: GluK2−/−: 1.4 ± 0.1 μm3, n = 9 mice, p < 0.001; P14: GluK2−/−: 20.5 ± 2.7 μm3, n = 8 mice, p < 0.05) as well as their complexity at P9 (GluK2−/−: 0.05 ± 0.01 μm, n = 9 mice, p < 0.001) (Fig. 7c,d), suggesting an impaired morphological maturation of the presynaptic compartment which parallel defects observed for the postsynaptic compartment. We also investigated how filopodial extensions develop on mf presynaptic boutons during maturation. Filopodia emerge from mf presynaptic terminals and contact interneurons (Acsády et al., 1998). In GluK2+/+ mice, filopodial extensions were absent at P6 (GluK2+/+: n = 18 mice) and the percentage of boutons with filopodia markedly increased from P9 to P21 (P9: 5 ± 1%, n = 16 mice; P14: 27 ± 4%, n = 4 mice; P21: 62 ± 2%, n = 16 mice) (Fig. 7e). In parallel, the number of filopodia per bouton increased between P9 and P14 (P9: 1 ± 0.2, n = 26 boutons; P14: 3 ± 0.2, n = 34 boutons; P21: 3 ± 0.07, n = 196 boutons) (Fig. 7f), and their length slightly increased until P21 (P9: 2 ± 0.2 μm, n = 42 filopodia; P14: 3 ± 0.2 μm, n = 104 filopodia; P21: 3 ± 0.09 μm, n = 739 filopodia) (Fig. 7g). Consistent with a delayed morphological maturation in GluK2−/− mice, the percentage of boutons showing filopodia was decreased as compared with GluK2+/+ mice at P9 (GluK2−/−: 1 ± 0.7%, n = 9 mice, p < 0.05) (Fig. 7e), at a time when only a few boutons display filopodia in wild-type mice. The number of boutons with filopodia was not different between the two genotypes at P14 and P21, although a modest increase in the number of filopodia was found in GluK2−/− mice at P14 (3.8 ± 0.1, n = 17 boutons, p < 0.01) (Fig. 7f), and these filopodia were shorter (2 ± 0.05 μm, n = 485 filopodia, p < 0.001) (Fig. 7g).
Figure 7.
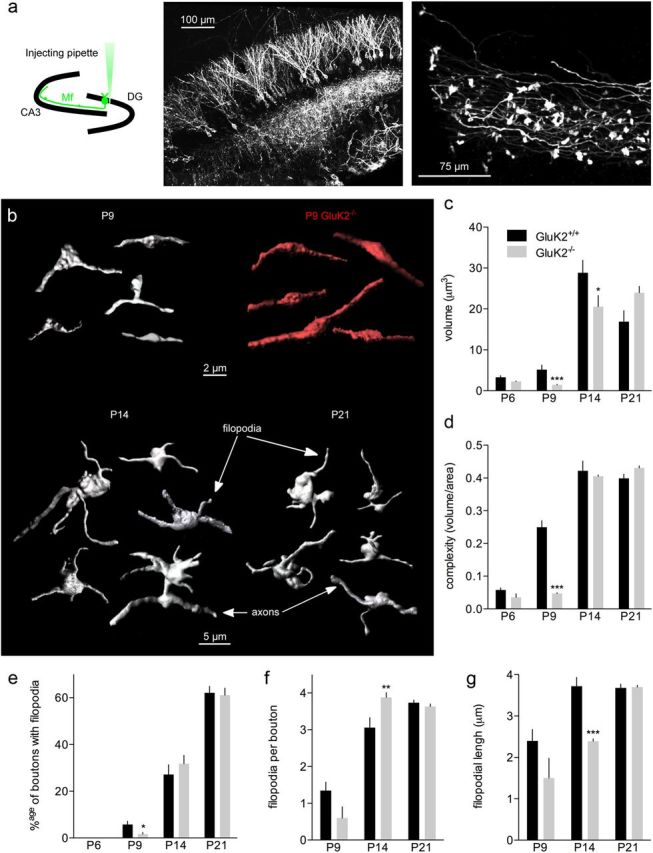
The maturation of presynaptic boutons is impaired in GluK2−/− mice. a, Schematic representation of the experimental procedure to infect granule cells in pups (P1–P2). A micropipette is used to inject the mYFP-expressing lentivirus (DG, dentate gyrus). Pictures of granule cells and mossy fibers labeled with mYFP in the stratum lucidum. b, Examples of presynaptic terminals reconstructed with surface rendering for GluK2+/+ and GluK2−/− mice between P9 and P21. c, Summary graph of the presynaptic terminal volume between P6 and P21 for GluK2+/+ and GluK2−/− mice. d, Summary graph of the presynaptic terminal complexity between P6 and P21 for GluK2+/+ and GluK2−/− mice. The index of complexity is calculated by dividing the volume by the area. (Number of animals: P6–P7: GluK2+/+: n = 8, GluK2−/−: n = 4; P8–P9: GluK2+/+: n = 11, GluK2−/−: n = 9; P14–P16: GluK2+/+: n = 5, GluK2−/−: n = 8; P19–P21: GluK2+/+: n = 6, GluK2−/−: n = 6.) e–g, Summary graphs of the percentage of boutons with filopodia (e), the number of filopodia per bouton (f), and the filopodial length (g) between P6 and P21 for GluK2+/+ and GluK2−/− mice. Mann–Whitney test: *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
Here, we analyzed in parallel the functional and morphological maturation of Mf-CA3 synapses during postnatal development in control mice and in mice deficient for the KAR subunit gene grik2. We show that the precise timing of functional and structural maturation of hippocampal mf-CA3 synapses is impaired in these mice. This suggests that a transient defect in synaptic transmission during postnatal development may be a critical aspect of neurodevelopmental cognitive disorders in humans with mutations of the grik2 gene.
Quantal analysis at developing hippocampal mossy fiber-CA3 synapses
We have used the MPFA method to estimate the developmental changes in quantal parameters at mf-CA3 synapse. At P21, the estimated quantal amplitude (49 ± 7 pA) differs from previous results obtained at rat mossy fiber synapses (Jonas et al., 1993; Lawrence et al., 2004). This difference may arise from experimental conditions (species, voltage, temperature), or from the analysis. We found nevertheless that the estimated quantal size is consistent with q measured by the amplitude of asynchronous release events at all ages tested in both genotypes. The average release probability of individual sites is estimated to be <10%, hence much lower than the estimate for mf-interneurons synapses (range 0.34–0.51) (Lawrence et al., 2004). Importantly, our estimate of n with the MPFA method at P21 (range 7–59; 31.3 ± 5.6) is well within the range of the number of release sites measured by ultrastructural serial reconstruction analysis (range 7–45; average 25) (Rollenhagen et al., 2007).
Strikingly, the number of release sites per mf-bouton, estimated by the MPFA method, is relatively stable between P6 and P9 and displays a robust increase between P9 and P21, consistent with changes in the density of synaptic sites estimated by the colocalized presynaptic and postsynaptic markers Munc13-1 and SAP102. This increase in n is in great part counterbalanced by the decrease in the release probability. Between P6 and P9, the abrupt increase of mf-CA3 EPSC amplitude is thus best explained by a change in the quantal size and by an increase in the number of functional synaptic sites. Several parameters may account for the increase in q. At a presynaptic level, the vesicular glutamate concentration may in principle affect quantal size at glutamatergic synapses (Wojcik et al., 2004; Wilson et al., 2005) over development, possibly linked to changes in VGLUT1-2 expression (Herzog et al., 2006). Most likely, the increase in q between P6 and P9 is linked to a change in the density, clustering, and properties of postsynaptic AMPARs although we cannot rule out an effect of cable filtering due to changes in dendritic properties. PSD-95 expression increases rapidly after the first week of postnatal development in rat (Sans et al., 2000), and increased expression of PSD-95 has been associated with an increase of AMPA-EPSCs amplitude and more generally to glutamatergic synapse maturation (El-Husseini et al., 2000). The robust developmental changes of mf-CA3 synapses may rely on the insertion of new AMPARs at preexisting sites and/or on a switch in receptor composition from Ca2+-permeable to Ca2+ impermeable receptors (Ho et al., 2007). Synaptic NMDA-EPSCs remained stable at the critical P6–P9 stage. Consequently, the NMDA/AMPA ratio at hippocampal mf-CA3 synapses markedly decreased between P6 and P9, suggesting that the increase in mf-CA3 synaptic transmission is due to an insertion of AMPARs at preexisting sites. This is consistent with an increase in the quantal size and/or with the existence of “silent” synaptic sites, which are unsilenced during maturation.
Morphological maturation of hippocampal mossy fiber synapses
Using 3D reconstruction analysis following targeted expression of membrane-bound YFP, we observed major morphometric changes at mf-CA3 synapses within a short period of development, between P9 and P14, reminiscent of electron microscopy data in the rat (Amaral and Dent, 1981). Up to P9, only very few postsynaptic spines (of small volume) could be observed in CA3 pyramidal cells in the stratum lucidum, although we cannot exclude that emergent structures could not be detected with the resolution of confocal microscopy. In contrast, at P14, CA3 pyramidal cells displayed large and complex TEs similar to those reported at mature stages (Qin et al., 2001; Rollenhagen et al., 2007). Presynaptic terminals increased in volume and became more complex starting at P6 and up to P14, preceding the appearance of TEs. The complexification of presynaptic elements was associated with the appearance of filopodia extensions. Filopodial extensions arising from mf presynaptic terminals and contacting CA3 interneurons (Acsády et al., 1998), progressively increased in number between P9 and P14. It should be mentioned that only 62% of the boutons contained filopodia at P21, indicating that the developmental maturation of mf boutons may not be complete at this stage, or that some mf-CA3 synapses may lack feedforward connectivity. Interestingly, the short-term temporal dynamics of inhibitory circuit recruitment differs between juvenile (P16–P20) and young adult rats (Mori et al., 2004; Torborg et al., 2010). Future studies should extend these analyses to earlier stages of postnatal development at which a massive and sharp increase in the number of filopodia occur. Filopodia on mf boutons appear to be highly plastic processes. Interestingly, one-trial and incremental learning lead to rapid and robust (2.5-fold) increases in the number of filopodia extensions of mf terminals (Ruediger et al., 2011), likely impacting on the activity of CA3 microcircuits through increased feedforward inhibition (Mori et al., 2004; Torborg et al., 2010), although this has not yet been demonstrated. In contrast, the rather mild differences in the number of filopodia observed between wild-type and GluK2−/− mice suggest that the major deficits relate to the robust difference in mf-CA3 EPSC amplitude rather than to defective feedforward inhibitory connectivity.
Maturation of Mf-CA3 synapses thus appear to proceed in two overlapping steps, a step of functional maturation (insertion of AMPARs) followed by a step of structural modifications (addition of new synaptic sites). Our data provide evidence for a delay between the increased amplitude of mf-CA3 EPSCs, which mainly occurs between P6 and P9, and the major structural changes observed after P9, for both the presynaptic and the postsynaptic elements. These two steps may be causally related as it has been shown at other synapses that postsynaptic NMDA-dependent plasticity leads to insertion (or removal) of AMPARs (Collingridge et al., 2004) and concomitant and subsequent structural modifications (Maletic-Savatic et al., 1999). Accordingly, in CA1 pyramidal cells spines can be generated by local ionophoretic application of glutamate nearby the dendritic shaft (Richards et al., 2005). Moreover, dendritic spine changes are associated with long-term potentiation in many brain regions (Engert and Bonhoeffer, 1999; Toni et al., 1999).
Kainate receptors and synaptic maturation
Both presynaptic and postsynaptic KARs contain the GluK2 subunit at mf-CA3 synapses, at least in mature (circa P21) mice (Mulle et al., 1998; Contractor et al., 2001). A major observation of the study is that the morphofunctional maturation of mf-CA3 synapses is altered in GluK2−/− mice. A marked impairment of synaptic function is most clearly visible at P9 and is transitory. Interestingly, the absence of GluK2 during development markedly affects AMPA-EPSCs but not NMDA-EPSCs. We also found a marked delay in the addition of synaptic sites, as indicated by the MPFA analysis, by the decrease of colocalized SAP102/Munc13-1 and by the density and intensity of GluA1 puncta. The morphometric analysis of mf-CA3 synapses confirmed the clear delay in the functional maturation of mf-CA3 synapses in the absence of GluK2. The delayed maturation in GluK2−/− mice fits well with the time window of the appearance of synaptic KARs, reinforcing the importance of KARs for the functional maturation of mf-CA3 synapses (Marchal and Mulle, 2004).
The mechanisms by which KARs can regulate synaptic maturation are not known. The physical presence of GluK2 subunits, independent of their activity, may be sufficient for the proper maturation of the synapse, for instance by interaction with scaffolding proteins such as PSD-95 (Garcia et al., 1998), or with adhesion molecules such as N-cadherin (Coussen et al., 2002). Similarly to the role of NMDARs in the development of synaptic circuits (van Zundert et al., 2004), the influx of Ca2+ through unedited KARs (Seeburg and Hartner, 2003) may also play an important role in the refinement of synaptic transmission. Along these lines, the edition of GluK2-containing KARs was shown to be developmentally regulated (Bernard et al., 1999). Our study reinforces the role of KARs in the regulation of the activity of neural networks at critical stages of development (Bahn et al., 1994; Lauri et al., 2005, 2006; Sallert et al., 2007; Vesikansa et al., 2007; Sallert et al., 2009; Caiati et al., 2010; Juuri et al., 2010; Segerstråle et al., 2010). Whereas GluK1-containing KARs appear to play a role in the regulation of activity of hippocampal networks during early postnatal development (Lauri et al., 2006; Segerstråle et al., 2010), GluK2-containing KARs may be involved in the localized refinement of synaptic contacts.
Synaptic activation of KARs plays a role in information transfer at mf-CA3 synapses in the adult by controlling synaptic integration and spike transmission efficacy through an indirect G-protein-coupled mechanism at the postsynaptic level (Ruiz et al., 2005) and a direct ionotropic action of GluK2 at both presynaptic and postsynaptic level (Sachidhanandam et al., 2009; Pinheiro et al., 2012). In addition, GluK2 is necessary for the proper maturation of mf-CA3 synapses between P6 and P9, by plasticity processes reminiscent of those involving NMDARs, i.e., insertion of AMPARs and correlated structural modifications. Hence, there are two separate consequences for the lack of functional GluK2-containing KARs at mf-CA3 synapses, a delay in morphofunctional synaptic maturation and a long-lasting impairment of synaptic integration and spike transfer. Loss-of-function mutations of GluK2 in human (Motazacker et al., 2007) may similarly impact both on the development of proper hippocampal circuits and on their function, leading to cognitive deficits associated with ID.
Footnotes
This work was supported by the Centre National de la Recherche Scientifique (CNRS), the Ministere de la Recherche of France, the Conseil Regional of Aquitaine, the European Commission (SYNSCAFF program, contract number LSH-2003-511995), the Fondation pour la Recherche Medicale, and the Agence Nationale de la Recherche (contract “ASD/LD Syngene”). We thank Audrey Lacquemant, Noëlle Grosjean, and Pierre Costet for genotyping and technical assistance with mouse breeding. Confocal microscopy and image analysis were performed at the Bordeaux Imaging Center with the help of Philippe Legros, Christel Poujol, Laure Malicieux, Sébastien Marais, and Florian Levet. We thank R. Angus Silver for comments on the MPFA analysis, Thomas Kuner and Darius B. Schwenger for their help with morphological analysis, and Lucie Bard for comments on the manuscript.
The authors declare no competing financial interests.
References
- Acsády L, Kamondi A, Sík A, Freund T, Buzsáki G. GABAergic cells are the major postsynaptic targets of mossy fibers in the rat hippocampus. J Neurosci. 1998;18:3386–3403. doi: 10.1523/JNEUROSCI.18-09-03386.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral DG, Dent JA. Development of the mossy fibers of the dentate gyrus: I. A light and electron microscopic study of the mossy fibers and their expansions. J Comp Neurol. 1981;195:51–86. doi: 10.1002/cne.901950106. [DOI] [PubMed] [Google Scholar]
- Bahn S, Volk B, Wisden W. Kainate receptor gene expression in the developing rat brain. J Neurosci. 1994;14:5525–5547. doi: 10.1523/JNEUROSCI.14-09-05525.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Clements JD. Quantal amplitude and quantal variance of strontium-induced asynchronous EPSCs in rat dentate granule neurons. J Physiol. 1999;516:227–248. doi: 10.1111/j.1469-7793.1999.227aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard A, Ferhat L, Dessi F, Charton G, Represa A, Ben-Ari Y, Khrestchatisky M. Q/R editing of the rat GluR5 and GluR6 kainate receptors in vivo and in vitro: evidence for independent developmental, pathological and cellular regulation. Eur J Neurosci. 1999;11:604–616. doi: 10.1046/j.1460-9568.1999.00479.x. [DOI] [PubMed] [Google Scholar]
- Boda B, Dubos A, Muller D. Signaling mechanisms regulating synapse formation and function in mental retardation. Curr Opin Neurobiol. 2010;20:519–527. doi: 10.1016/j.conb.2010.03.012. [DOI] [PubMed] [Google Scholar]
- Caiati MD, Sivakumaran S, Cherubini E. In the developing rat hippocampus, endogenous activation of presynaptic kainate receptors reduces GABA release from mossy fiber terminals. J Neurosci. 2010;30:1750–1759. doi: 10.1523/JNEUROSCI.4566-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD. Variance-mean analysis: a simple and reliable approach for investigating synaptic transmission and modulation. J Neurosci Methods. 2003;130:115–125. doi: 10.1016/j.jneumeth.2003.09.019. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5:952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- Contractor A, Swanson G, Heinemann SF. Kainate receptors are involved in short- and long-term plasticity at mossy fiber synapses in the hippocampus. Neuron. 2001;29:209–216. doi: 10.1016/s0896-6273(01)00191-x. [DOI] [PubMed] [Google Scholar]
- Contractor A, Mulle C, Swanson GT. Kainate receptors coming of age: milestones of two decades of research. Trends Neurosci. 2011;34:154–163. doi: 10.1016/j.tins.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussen F, Normand E, Marchal C, Costet P, Choquet D, Lambert M, Mège RM, Mulle C. Recruitment of the kainate receptor subunit glutamate receptor 6 by cadherin/catenin complexes. J Neurosci. 2002;22:6426–6436. doi: 10.1523/JNEUROSCI.22-15-06426.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- Foster KA, Regehr WG. Variance-mean analysis in the presence of a rapid antagonist indicates vesicle depletion underlies depression at the climbing fiber synapse. Neuron. 2004;43:119–131. doi: 10.1016/j.neuron.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Garcia EP, Mehta S, Blair LA, Wells DG, Shang J, Fukushima T, Fallon JR, Garner CC, Marshall J. SAP90 binds and clusters kainate receptors causing incomplete desensitization. Neuron. 1998;21:727–739. doi: 10.1016/s0896-6273(00)80590-5. [DOI] [PubMed] [Google Scholar]
- Henze DA, Urban NN, Barrionuevo G. The multifarious hippocampal mossy fiber pathway: a review. Neuroscience. 2000;98:407–427. doi: 10.1016/s0306-4522(00)00146-9. [DOI] [PubMed] [Google Scholar]
- Herzog E, Takamori S, Jahn R, Brose N, Wojcik SM. Synaptic and vesicular co-localization of the glutamate transporters VGLUT1 and VGLUT2 in the mouse hippocampus. J Neurochem. 2006;99:1011–1018. doi: 10.1111/j.1471-4159.2006.04144.x. [DOI] [PubMed] [Google Scholar]
- Ho MT, Pelkey KA, Topolnik L, Petralia RS, Takamiya K, Xia J, Huganir RL, Lacaille JC, McBain CJ. Developmental expression of Ca2+-permeable AMPA receptors underlies depolarization-induced long-term depression at mossy fiber CA3 pyramid synapses. J Neurosci. 2007;27:11651–11662. doi: 10.1523/JNEUROSCI.2671-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Gambino F, Chelly J, Vitale N. X-linked mental retardation: focus on synaptic function and plasticity. J Neurochem. 2009;109:1–14. doi: 10.1111/j.1471-4159.2009.05881.x. [DOI] [PubMed] [Google Scholar]
- Jonas P, Major G, Sakmann B. Quantal components of unitary EPSCs at the mossy fibre synapse on CA3 pyramidal cells of rat hippocampus. J Physiol. 1993;472:615–663. doi: 10.1113/jphysiol.1993.sp019965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juuri J, Clarke VR, Lauri SE, Taira T. Kainate receptor-induced ectopic spiking of CA3 pyramidal neurons initiates network bursts in neonatal hippocampus. J Neurophysiol. 2010;104:1696–1706. doi: 10.1152/jn.00840.2009. [DOI] [PubMed] [Google Scholar]
- Kesner RP. Behavioral functions of the CA3 subregion of the hippocampus. Learn Mem. 2007;14:771–781. doi: 10.1101/lm.688207. [DOI] [PubMed] [Google Scholar]
- Ko S, Zhao MG, Toyoda H, Qiu CS, Zhuo M. Altered behavioral responses to noxious stimuli and fear in glutamate receptor 5 (GluR5)- or GluR6-deficient mice. J Neurosci. 2005;25:977–984. doi: 10.1523/JNEUROSCI.4059-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanore F, Blanchet C, Fejtova A, Pinheiro P, Richter K, Balschun D, Gundelfinger E, Mulle C. Impaired development of hippocampal mossy fibre synapses in mouse mutants for the presynaptic scaffold protein Bassoon. J Physiol. 2010;588:2133–2145. doi: 10.1113/jphysiol.2009.184929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauri SE, Segerstråle M, Vesikansa A, Maingret F, Mulle C, Collingridge GL, Isaac JT, Taira T. Endogenous activation of kainate receptors regulates glutamate release and network activity in the developing hippocampus. J Neurosci. 2005;25:4473–4484. doi: 10.1523/JNEUROSCI.4050-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauri SE, Vesikansa A, Segerstråle M, Collingridge GL, Isaac JT, Taira T. Functional maturation of CA1 synapses involves activity-Dependent Loss of Tonic Kainate Receptor-Mediated Inhibition of Glutamate Release. Neuron. 2006;50:415–429. doi: 10.1016/j.neuron.2006.03.020. [DOI] [PubMed] [Google Scholar]
- Lawrence JJ, Grinspan ZM, McBain CJ. Quantal transmission at mossy fibre targets in the CA3 region of the rat hippocampus. J Physiol. 2004;554:175–193. doi: 10.1113/jphysiol.2003.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- Marchal C, Mulle C. Postnatal maturation of mossy fibre excitatory transmission in mouse CA3 pyramidal cells: a potential role for kainate receptors. J Physiol. 2004;561:27–37. doi: 10.1113/jphysiol.2004.069922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Abegg MH, Gähwiler BH, Gerber U. A frequency-dependent switch from inhibition to excitation in a hippocampal unitary circuit. Nature. 2004;431:453–456. doi: 10.1038/nature02854. [DOI] [PubMed] [Google Scholar]
- Motazacker MM, Rost BR, Hucho T, Garshasbi M, Kahrizi K, Ullmann R, Abedini SS, Nieh SE, Amini SH, Goswami C, Tzschach A, Jensen LR, Schmitz D, Ropers HH, Najmabadi H, Kuss AW. A defect in the ionotropic glutamate receptor 6 gene (GRIK2) is associated with autosomal recessive mental retardation. Am J Hum Genet. 2007;81:792–798. doi: 10.1086/521275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulle C, Sailer A, Pérez-Otaño I, Dickinson-Anson H, Castillo PE, Bureau I, Maron C, Gage FH, Mann JR, Bettler B, Heinemann SF. Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6-deficient mice. Nature. 1998;392:601–605. doi: 10.1038/33408. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci. 2005;6:863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- Pinheiro PS, Perrais D, Coussen F, Barhanin J, Bettler B, Mann JR, Malva JO, Heinemann SF, Mulle C. GluR7 is an essential subunit of presynaptic kainate autoreceptors at hippocampal mossy fiber synapses. Proc Natl Acad Sci U S A. 2007;104:12181–12186. doi: 10.1073/pnas.0608891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro PS, Lanore F, Veran J, Artinian J, Blanchet C, Crépel V, Perrais D, Mulle C. Selective block of postsynaptic kainate receptors reveals their function at hippocampal mossy fiber synapses. Cereb Cortex. 2012 doi: 10.1093/cercor/bhs022. Advance online publication. Retrieved February 17, 2012. [DOI] [PubMed] [Google Scholar]
- Qin L, Marrs GS, McKim R, Dailey ME. Hippocampal mossy fibers induce assembly and clustering of PSD95-containing postsynaptic densities independent of glutamate receptor activation. J Comp Neurol. 2001;440:284–298. doi: 10.1002/cne.1386. [DOI] [PubMed] [Google Scholar]
- Richards DA, Mateos JM, Hugel S, de Paola V, Caroni P, Gähwiler BH, McKinney RA. Glutamate induces the rapid formation of spine head protrusions in hippocampal slice cultures. Proc Natl Acad Sci U S A. 2005;102:6166–6171. doi: 10.1073/pnas.0501881102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollenhagen A, Sätzler K, Rodriguez EP, Jonas P, Frotscher M, Lübke JHR. Structural Determinants of Transmission at Large Hippocampal Mossy Fiber Synapses. J Neurosci. 2007;27:10434–10444. doi: 10.1523/JNEUROSCI.1946-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruediger S, Vittori C, Bednarek E, Genoud C, Strata P, Sacchetti B, Caroni P. Learning-related feedforward inhibitory connectivity growth required for memory precision. Nature. 2011;473:514–518. doi: 10.1038/nature09946. [DOI] [PubMed] [Google Scholar]
- Ruiz A, Sachidhanandam S, Utvik JK, Coussen F, Mulle C. Distinct subunits in heteromeric kainate receptors mediate ionotropic and metabotropic function at hippocampal mossy fiber synapses. J Neurosci. 2005;25:11710–11718. doi: 10.1523/JNEUROSCI.4041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachidhanandam S, Blanchet C, Jeantet Y, Cho YH, Mulle C. Kainate receptors act as conditional amplifiers of spike transmission at hippocampal mossy fiber synapses. J Neurosci. 2009;29:5000–5008. doi: 10.1523/JNEUROSCI.5807-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salin PA, Scanziani M, Malenka RC, Nicoll RA. Distinct short-term plasticity at two excitatory synapses in the hippocampus. Proc Natl Acad Sci U S A. 1996;93:13304–13309. doi: 10.1073/pnas.93.23.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallert M, Malkki H, Segerstråle M, Taira T, Lauri SE. Effects of the kainate receptor agonist ATPA on glutamatergic synaptic transmission and plasticity during early postnatal development. Neuropharmacology. 2007;52:1354–1365. doi: 10.1016/j.neuropharm.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Sallert M, Rantamäki T, Vesikansa A, Anthoni H, Harju K, Yli-Kauhaluoma J, Taira T, Castren E, Lauri SE. Brain-derived neurotrophic factor controls activity-dependent maturation of CA1 synapses by downregulating tonic activation of presynaptic kainate receptors. J Neurosci. 2009;29:11294–11303. doi: 10.1523/JNEUROSCI.0560-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans N, Petralia RS, Wang YX, Blahos J, 2nd, Hell JW, Wenthold RJ. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci. 2000;20:1260–1271. doi: 10.1523/JNEUROSCI.20-03-01260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg PH, Hartner J. Regulation of ion channel/neurotransmitter receptor function by RNA editing. Curr Opin Neurobiol. 2003;13:279–283. doi: 10.1016/s0959-4388(03)00062-x. [DOI] [PubMed] [Google Scholar]
- Segerstråle M, Juuri J, Lanore F, Piepponen P, Lauri SE, Mulle C, Taira T. High firing rate of neonatal hippocampal interneurons is caused by attenuation of afterhyperpolarizing potassium currents by tonically active kainate receptors. J Neurosci. 2010;30:6507–6514. doi: 10.1523/JNEUROSCI.4856-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver RA. Estimation of nonuniform quantal parameters with multiple-probability fluctuation analysis: theory, application and limitations. J Neurosci Methods. 2003;130:127–141. doi: 10.1016/j.jneumeth.2003.09.030. [DOI] [PubMed] [Google Scholar]
- Silver RA, Momiyama A, Cull-Candy SG. Locus of frequency-dependent depression identified with multiple-probability fluctuation analysis at rat climbing fibre-Purkinje cell synapses. J Physiol. 1998;510:881–902. doi: 10.1111/j.1469-7793.1998.881bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toni N, Buchs PA, Nikonenko I, Bron CR, Muller D. LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature. 1999;402:421–425. doi: 10.1038/46574. [DOI] [PubMed] [Google Scholar]
- Torborg CL, Nakashiba T, Tonegawa S, McBain CJ. Control of CA3 output by feedforward inhibition despite developmental changes in the excitation-inhibition balance. J Neurosci. 2010;30:15628–15637. doi: 10.1523/JNEUROSCI.3099-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toro R, Konyukh M, Delorme R, Leblond C, Chaste P, Fauchereau F, Coleman M, Leboyer M, Gillberg C, Bourgeron T. Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet. 2010;26:363–372. doi: 10.1016/j.tig.2010.05.007. [DOI] [PubMed] [Google Scholar]
- van Zundert B, Yoshii A, Constantine-Paton M. Receptor compartmentalization and trafficking at glutamate synapses: a developmental proposal. Trends Neurosci. 2004;27:428–437. doi: 10.1016/j.tins.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Vesikansa A, Sallert M, Taira T, Lauri SE. Activation of kainate receptors controls the number of functional glutamatergic synapses in the area CA1 of rat hippocampus. J Physiol. 2007;583:145–157. doi: 10.1113/jphysiol.2007.133975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NR, Kang J, Hueske EV, Leung T, Varoqui H, Murnick JG, Erickson JD, Liu G. Presynaptic regulation of quantal size by the vesicular glutamate transporter VGLUT1. J Neurosci. 2005;25:6221–6234. doi: 10.1523/JNEUROSCI.3003-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik SM, Rhee JS, Herzog E, Sigler A, Jahn R, Takamori S, Brose N, Rosenmund C. An essential role for vesicular glutamate transporter 1 (VGLUT1) in postnatal development and control of quantal size. Proc Natl Acad Sci U S A. 2004;101:7158–7163. doi: 10.1073/pnas.0401764101. [DOI] [PMC free article] [PubMed] [Google Scholar]