

Abstract
Dopamine replacement with levodopa (l-DOPA) represents the mainstay of Parkinson's disease (PD) therapy. Nevertheless, this well established therapeutic intervention loses efficacy with the progression of the disease and patients develop invalidating side effects, known in their complex as l-DOPA-induced dyskinesia (LID). Unfortunately, existing therapies fail to prevent LID and very few drugs are available to lessen its severity, thus representing a major clinical problem in PD treatment. D2-like receptor (D2R) agonists are a powerful clinical option as an alternative to l-DOPA, especially in the early stages of the disease, being associated to a reduced risk of dyskinesia development. D2R agonists also find considerable application in the advanced stages of PD, in conjunction with l-DOPA, which is used in this context at lower dosages, to delay the appearance and the extent of the motor complications. In advanced stages of PD, D2R agonists are often effective in delaying the appearance and the extent of motor complications. Despite the great attention paid to the family of D2R agonists, the main reasons underlying the reduced risk of dyskinesia have not yet been fully characterized. Here we show that the striatal NMDA/AMPA receptor ratio and the AMPA receptor subunit composition are altered in experimental parkinsonism in rats. Surprisingly, while l-DOPA fails to restore these critical synaptic alterations, chronic treatment with pramipexole is associated not only with a reduced risk of dyskinesia development but is also able to rebalance, in a dose-dependent fashion, the physiological synaptic parameters, thus providing new insights into the mechanisms of dyskinesia.
Introduction
The treatment of Parkinson's disease (PD) is a major clinical challenge. Despite therapeutic advances, PD still leads to severe disability and reduced quality of life (Lees et al., 2009; Jenkinson et al., 2012).
The central pillar in PD clinical practice remains, after almost half century since its introduction, levodopa [l-3,4-dihydroxyphenylalanine (l-DOPA)], a dopamine (DA) precursor (Birkmayer and Hornykiewicz, 1998; Olanow et al., 2006). However, over time, l-DOPA treatment leads to the emergence of invalidating motor side effects, such as l-DOPA-induced dyskinesia (LID) (Obeso et al., 2000; Ahlskog and Muenter, 2001). The mechanisms underlying LID appear complex and not fully understood (Calabresi et al., 2008; Fisone and Bezard, 2011). Thus, the prevention of LID is still a major clinical problem (Gottwald and Aminoff, 2011; Blandini and Armentero, 2012). Among the numerous players involved in the progressive appearance of LID, abnormal striatal glutamatergic transmission exerts a critical role (Calabresi et al., 2010). This has been demonstrated in experiments showing that targeting of striatal glutamatergic transmission can influence LID, which is associated with altered trafficking of NMDA receptor (NMDAR) subunits (Gardoni et al., 2006, 2012; Varanese et al., 2010; Ahmed et al., 2011). In addition to glutamatergic dysfunction, abnormal activation of D1-like receptors (D1Rs) is crucial for LID induction (Feyder et al., 2011). Increased localization of D1Rs at the cell surface, caused by impaired receptor internalization and trafficking, is associated with LID (Guigoni et al., 2007; Berthet et al., 2009). Pharmacological or genetic inactivation of D1R reduces LID and decreases the associated molecular alterations (Jenner, 2008; Darmopil et al., 2009; Voon et al., 2009; Cenci and Konradi, 2010; Lebel et al., 2010). However, at present, drugs targeting glutamatergic transmission and D1R signal transduction either produce unsatisfactory results or are not clinically tolerated.
Great attention has been paid to the development of D2-like receptor (D2R) agonists as a possible alternative to l-DOPA. These agonists constitute a powerful clinical option, showing several advantages, such as direct receptor stimulation without metabolic modifications (required for l-DOPA) and prolonged pharmacological action (Rascol et al., 2011). They can significantly alleviate motor symptoms in de novo PD, be used as monotherapy, particularly in young patients, and can reduce the risk for developing dyskinesia thanks to longer half-lives compared with l-DOPA's pulsatile effects (Hauser et al., 2007; Watts et al., 2010; Jenner et al., 2011; Calandrella and Antonini, 2012). Additionally, D2 agonists, when administered as adjunctive therapy to l-DOPA in the advanced stages of PD, delay the appearance and the extent of the serious motor complications induced by l-DOPA (Rinne et al., 1998; Rascol et al., 2000).
However, the synaptic and molecular mechanisms underlying the action of D2 agonists have never been fully understood, despite their widespread clinical use. Here, using a combined behavioral and cellular approach, we show that in experimental parkinsonism the NMDAR/AMPA receptor (AMPAR) ratio and the composition of both NMDAR and AMPAR subunits are altered in striatal spiny neurons. Surprisingly, while l-DOPA fails to restore these critical synaptic alterations, chronic treatment with the D2R agonist pramipexole rebalances, in a dose-dependent fashion, these physiological parameters, thus providing new insights into the mechanisms of dyskinesia.
Materials and Methods
Animals.
Adult male Wistar rats (150–250 g) were used for the study (n = 280). All experiments were approved by the European Communities Council Directive of November 1986 (86/609/ECC), and by the Italian Health Ministry.
Nigrostriatal lesion.
Male Wistar rats were deeply anesthetized with chloral hydrate (400 mg/ml/kg; n = 260) and were injected with 6-hydroxydopamine (6-OHDA, 12 μg/4 μl of saline containing 0.1% ascorbic acid) via a Hamilton syringe into the medial forebrain bundle, at a rate of 0.38 μl/min [anteroposterior = −4.4; lateral = +1.2; ventrodorsal = −7.8] (Fig. 1). Control rats (n = 10) were injected with vehicle at the same coordinates. Fifteen days later, the rats were tested with a 0.05 mg/kg subcutaneous injection of apomorphine, and the contralateral turns were counted with automatic rotometers for 40 min. Only those rats consistently making at least 200 contralateral turns (Schwarting and Huston, 1996) were used for the electrophysiological recordings performed 45 d later (2 months after the lesion), to avoid possible interference of apomorphine treatment with the plastic changes occurring in DA D2 receptor function following chronic dopaminergic denervation. The severity of the lesion was also quantified afterward by striatal and nigral TH immunohistochemistry (data not shown).
Figure 1.

Timeline of the experimental plan. Two weeks after the lesion with 6-OHDA, rats were tested with the DA agonist apomorphine (APO) and then selected for successful lesions, based on the number of contraversive turns, counted with automatic rotometers for 40 min as previously reported. Two months after the lesion, the animals were randomly allotted in five different experimentally treated groups: Control group (saline, i.p.); I Pramipexole group (0.1 mg/kg, i.p., twice a day); II Pramipexole group (0.3 mg/kg, i.p., twice a day); III Pramipexole group (1 mg/kg, i.p., once a day); and l-DOPA group (10 mg/kg, twice a day). At the end of chronic pharmacological treatment, animals were killed by cervical dislocation and coronal corticostriatal slices were prepared. After ≥30 min, the slices were used for electrophysiological recordings.
Behavioral testing of motor deficits, stepping test.
Each animal was systematically handled on a regular basis (i.e., taken by hand from its cage, put on a table, gently touched, moved from one place to another, and then, after several minutes of continuous contact with the human hand, returned to its home cage) for several days before the first assessment. Forelimb akinesia (Fig. 1) was assessed using a modified version of the stepping test (Paillé et al., 2007). Briefly, the experimenter firmly suspended the rat's hindquarters while it supported its weight on its forelimbs. Then, the experimenter moved the rat backwards along the table (0.9 m in 5 s) three times consecutively per session. All the sessions were video recorded to allow the number of adjusting steps to be counted afterward by an investigator blinded to the state of the rat (i.e., 6-OHDA-lesioned or sham-operated). For each session, the total score calculated was the sum of the number of adjusting steps observed in the three tests (for the contralateral paw). The sessions took place between 2:30 and 4:30 P.M. 1 week before surgery and 2 weeks after surgery (just before the apomorphine-induced rotation test and during the pharmacological treatments).
l-DOPA and DA agonist treatment.
Two months after the lesion, 6-OHDA full-lesioned rats (n = 205) were allotted into three experimental groups. The first group was treated with l-DOPA (n = 50, 10 mg/kg l-DOPA plus 6 mg/kg benserazide, twice daily, i.p.). The second group of 6-OHDA rats (n = 155) received the non-ergot D2-like agonist pramipexole [0.1 (n = 35) and 0.3 (n = 41) mg/kg twice daily, i.p., and 1 mg/kg (n = 60) injected once daily, i.p.]. The last group of 6-OHDA-lesioned rats (n = 19) was used for control electrophysiological recordings. Since we have noted, in a pilot experiment, that treatment with the higher dose of pramipexole 1 mg/kg injected once daily was able to induce in rats a prolonged turning behavior, with severe loss of weight, we chose to treat the animals chronically only once daily with this dosage.
Drug-induced abnormal involuntary movements (AIMs, or dyskinesia) were recorded on alternate days three times a week using a validated AIM scale in rat, and were classified into three subtypes: axial, limb, and orolingual AIMs, as previously described (Cenci et al., 1998; Lundblad et al., 2002; Picconi et al., 2003; Gardoni et al., 2006). Each of these subtypes was scored on a severity scale from 0 to 4. Total AIMs score for each session was obtained by summing the monitoring period scores. Two-way ANOVA and Tukey's HSD post hoc test were used for statistical comparison when appropriate.
Electrophysiology.
The preparation and maintenance of coronal corticostriatal slices have been described previously (Calabresi et al., 1992; Picconi et al., 2003). Corticostriatal coronal slices, 240 μm thickness, were prepared. A single slice was transferred to a recording chamber and submerged in a continuously flowing artificial CSF (ACSF) solution (room temperature, 3 ml/min) gassed with 95% O2/5% CO2. The composition of the ACSF solution was as follows (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 11 glucose, and 25 NaHCO3.
Whole-cell patch-clamp recordings.
Whole-cell patch-clamp recordings were performed from medium spiny neurons (MSNs) visualized using infrared differential interference contrast microscopy in the dorsal striatum (Eclipse FN1, Nikon) (Bagetta et al., 2011). Recordings were made with a Multiclamp 700B amplifier (Molecular Devices), using borosilicate glass pipettes (outer diameter, 1.5 mm; inner diameter, 0.86 mm) pulled on a P-97 Puller (Sutter Instruments). Pipette resistances ranged from 3.5 to 5 MΩ. Membrane currents were continuously monitored and access resistance measured in voltage clamp was in the range of 5–30 MΩ before electronic compensation (60–80% routinely used). Current–voltage relationships were obtained by applying steps of current of Δ50 pA in both depolarizing and hyperpolarizing direction (from −400 to 400 pA, 500 ms). For spontaneous EPSCs (sEPSCs), pipettes were filled with an internal solution containing the following (in mm): 120 CsMeSO3, 15 CsCl, 8 NaCl, 0.2 EGTA, 10 HEPES, 2 Mg-ATP, 0.3 Na-GTP, 10 tetraethylammonium, and 5 QX-314 [N-(2,6-dimethylphenylcarbamoylmethyl) triethylammonium bromide], pH 7.2, with CsOH. Picrotoxin (50 μm) was added to block GABAA currents. MSNs were clamped at the holding potential (Vh) of −70 mV. Data were acquired with pClamp 10.2 (Molecular Devices); current was filtered at 0.1 kHz, digitized at 200 μs using Clampex 10.2 (gap-free mode), analyzed offline using the automatic detection, and subsequently checked manually for accuracy. The threshold amplitude for the detection of an event (5 pA) was adjusted above root mean square noise level (2–3 pA at Vh = −70 mV).
A bipolar stimulating electrode was located intrastriatally, close to the recording electrode (50–150 μm), to evoke glutamate-mediated synaptic currents [evoked EPSCs, (eEPSCs)] in MSNs, in the continuous presence of picrotoxin (50 μm). Stimuli were delivered at 0.1 Hz. In this condition, the evoked potentials were fully blocked by MK-801 or AP-5 (30 μm) and CNQX (10 μm). Baseline current level was detected as mean current value recorded between 10 and 50 ms before electrical stimulation. The maximal current value recorded between 10 and 150 ms after stimulation was considered as eEPSC peak. eEPSC amplitude was obtained by subtracting the baseline current value from the peak current value. Only cells that had stable baseline and peak current levels for at least 10 min before drug application were used for statistical analysis. All drugs were from Sigma-Aldrich and were applied by switching the control perfusion to drug-containing solution. Experiments were discarded if the access resistance varied by >20%.
For the NMDAR/AMPAR ratio experiments, MSNs were voltage-clamped at −70 and +40 mV to record, respectively, AMPAR-mediated and NMDAR-mediated EPSCs. The NMDA EPSC component was individuated by using the kinetic method, considering the peak amplitude at 50 ms after the beginning of the event. The NMDAR/AMPAR ratio was calculated by dividing the NMDAR peak by the AMPAR peak (Paillé et al., 2010). In the experiments with SCH23390, slices were incubated with the antagonist (10 μm) for at least 2 h and then the NMDAR/AMPAR ratio was recorded.
The rectification pattern of the AMPAR was calculated using voltage-clamp method (holding level was between −70 mV and +40 mV). The rectification index (IR) was calculated by the current–voltage relationship as the ratio of EPSC amplitudes between −70 and +40 mV.
Data are expressed as means ± SEM, and Student's t test and one-way ANOVA were used for statistical analysis, unless otherwise stated. Differences between groups were considered significant when Bonferroni post hoc test resulted of p < 0.05.
Western blot analysis.
Western blot analysis was performed in total homogenate or in Triton-insoluble postsynaptic fractions (TIFs, see below). To check for specificity, three different D3 DA receptor antibodies were tested and used. The following antibodies were used: polyclonal D3 DA receptor antibodies purchased from Abcam and Santa Cruz Biotechnology (H-50 and H-14); polyclonal GluN2A antibody purchased from Sigma-Aldrich; monoclonal GluN2B antibody purchased from NeuroMab; polyclonal GluA1 antibody purchased from Merck Millipore; monoclonal GluA2 antibody purchased from NeuroMab; monoclonal tubulin antibody purchased from Sigma-Aldrich.
Subcellular fractionation.
Subcellular fractionation of striatal tissue was performed as previously reported with minor modifications (Gardoni et al., 2006). Striata were homogenized in ice-cold sucrose 0.32 m containing 1 mm HEPES, 1 mm MgCl2, 1 mm EDTA, 1 mm NaHCO3, 0.1 mm PMSF, pH 7.4. The homogenized tissue was centrifuged at 1000 × g for 5 min. The resulting supernatant (S1) was centrifuged at 13,000 × g for 15 min to obtain a crude membrane fraction (P2 fraction). The pellet was resuspended in buffer containing 75 mm KCl and 1% Triton X-100 and centrifuged at 100,000 × g for 1 h. The final pellet, referred to as TIF, was homogenized in a glass-glass potter in 20 mm HEPES and stored at −80°C until processing. TIF was used instead of the classical postsynaptic density because the amount of the starting material was very limited. All purifications were performed in presence of a complete set of protease (Complete) and phosphatase (PhosSTOP) inhibitors (Roche Diagnostics).
Results
Forelimb akinesia after chronic treatment with dopaminergic drugs: pramipexole vs l-DOPA treatment
Three different doses of pramipexole (0.1 mg/kg or 0.3 mg/kg, given twice daily and 1 mg/kg once daily) and one dose of l-DOPA (10 mg/kg plus benserazide 6 mg/kg, twice daily) administered for 3 weeks, were challenged to assess the improvement of motor performance in hemiparkinsonian rats and also to study cellular physiology in vitro (Fig. 1). From a behavioral point of view, the stepping test, a well known motor task, was used to assess motor function. The scores were collected at the beginning of every week of treatment. As showed in Figure 2A, all the rats taking part in the study progressively recovered the use of the right paw (contralateral to the lesion) (within group main factor: F(3,108) = 9.55; p < 0.001). Comparisons between baseline and post-treatment scores across the groups (Fig. 2A) (group–session interaction: F(3,36) = 2.95; p < 0.05) showed similar forelimb akinesia results for the baselines of the four groups. Only the low dose of pramipexole failed to show any antiparkinsonian effect in the behavioral paradigm used (pramipexole 0.1 mg/kg, p > 0.05; pramipexole 0.3 mg/kg, p < 0.001; pramipexole 1 mg/kg, p < 0.001; l-DOPA, p < 0.001). The forelimb akinesia measured prior the beginning of the treatment (Fig. 2B, Baseline) was comparable among the four groups. Interestingly, the higher dose of pramipexole (1 mg/kg) was more effective than chronic l-DOPA treatment (°p < 0.05) in recovering the use of the right paw, but not statistically different from the 0.3 mg/kg pramipexole dose (p > 0.05).
Figure 2.
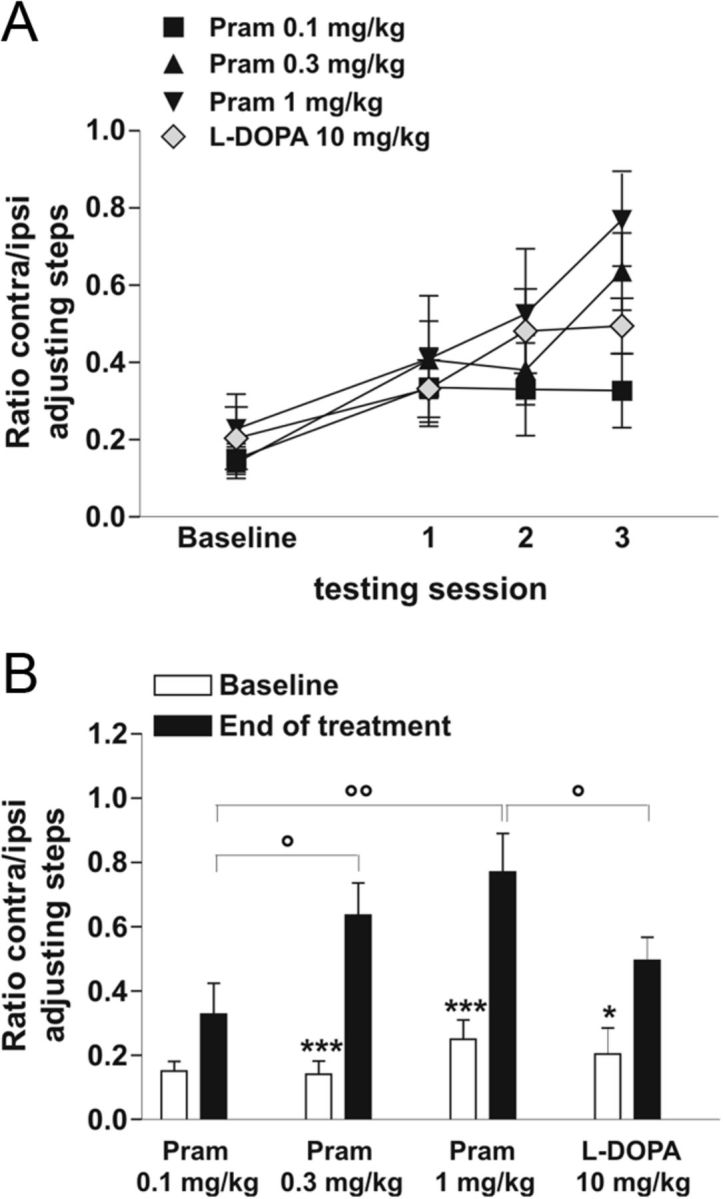
Restoration of forelimb akinesia after chronic treatment with dopaminergic drugs: pramipexole versus l-DOPA treatment. A, Graph shows the motor effect of the drugs at the beginning of every week of treatment. All treatments increased the performance in stepping test, without any overall statistically significant differences. B, Graph shows the therapeutic effect of pramipexole versus l-DOPA on the forelimb akinesia, measured prior the beginning of the treatment (Baseline), compared with the last session test (End of treatment). Interestingly, 1 mg/kg doses of pramipexole were more effective than chronic l-DOPA treatment (°p < 0.05), but not statistically different from 0.3 mg/kg pramipexole dose (*p < 0.05, ***p < 0.001; Baseline vs End of treatment) (°p < 0.05, °°p < 0.01 between groups).
AIMs induction after chronic treatment with pramipexole versus l-DOPA
AIMs were recorded on alternate days, three times a week using a validated rat AIMs scale, as previously described (Cenci et al., 1998; Lundblad et al., 2002; Picconi et al., 2003; Gardoni et al., 2006). During the chronic drug treatment period, l-DOPA, but not pramipexole at the dosages of 0.1 mg/kg and 0.3 mg/kg, induced a gradual development of AIMs affecting the trunk, limb, and orolingual region (Fig. 3A; group–session interaction: F(24,280) = 3.93, p < 0.001, post hoc test p < 0.05; #l-DOPA vs pramipexole 0.1 mg/kg; *l-DOPA vs pramipexole 0.3 mg/kg). Only the experimental group of 6-OHDA-lesioned rats treated with the high dosage of pramipexole (1 mg/kg) showed abnormal movements (Fig. 3A). Interestingly, the dyskinetic movements induced by the higher dosage of pramipexole were significantly lower than AIMs induced by l-DOPA (l-DOPA vs pramipexole 1 mg/kg, §p < 0.05).
Figure 3.
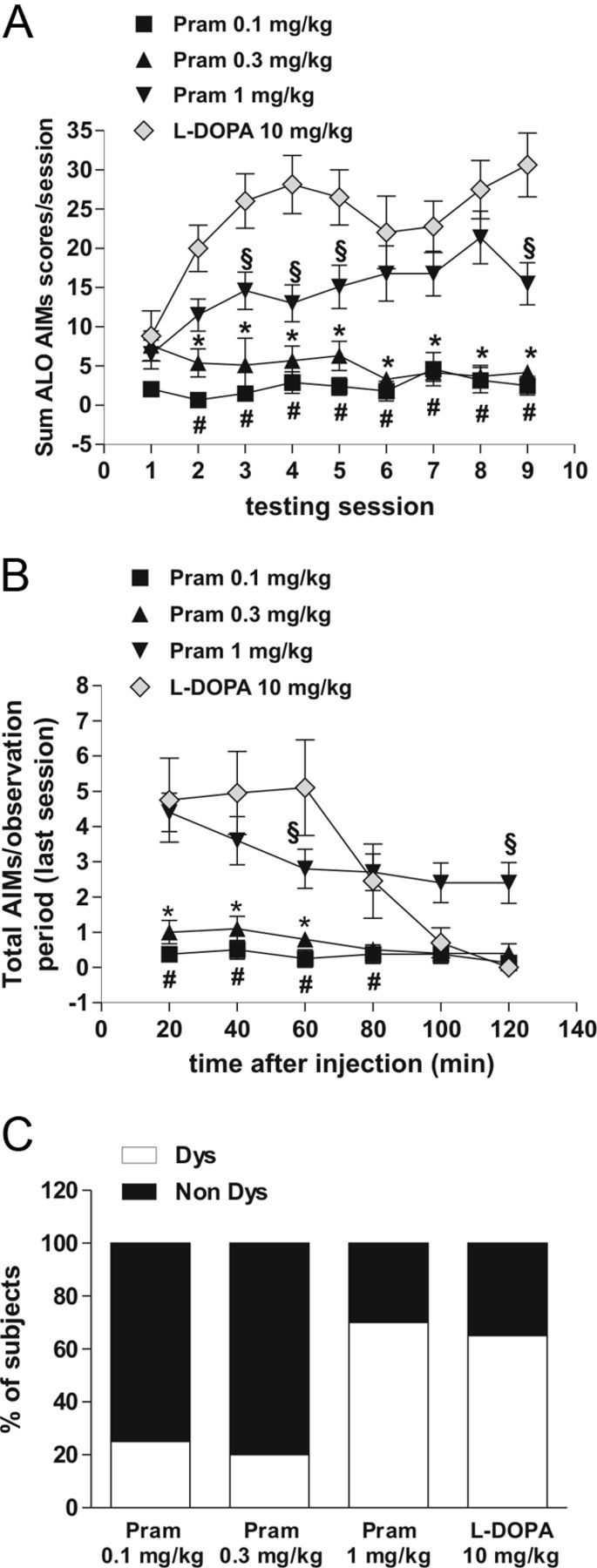
Therapeutic doses of pramipexole induced fewer dyskinetic movements than l-DOPA. A, l-DOPA but not 0.1 mg/kg and 0.3 mg/kg pramipexole induced the progressive development of AIMs (post hoc test, p < 0.05; #l-DOPA vs pramipexole 0.1 mg/kg; *l-DOPA vs pramipexole 0.3 mg/kg). Pramipexole at the dose of 1 mg/kg induced AIMs. However, AIMs induced by this high dose of pramipexole were significantly fewer than those triggered by l-DOPA (p < 0.05; §l-DOPA vs pramipexole 1 mg/kg). B, Graph shows the development of AIMs measured at the end of the treatment during the last session test. C, Administration of 0.1 mg/kg and 0.3 mg/kg pramipexole induced AIMs in a lower percentage of animals (25% Dys rats for the 0.1 mg/kg group; 20% Dys rats for the 0.3 mg/kg group) than after administration of 1 mg/kg pramipexole (70% Dys rats for the 1 mg/kg group) and l-DOPA (65% Dys).
The analysis of the percentage of PD animals developing AIMs, measured during every session test (Fig. 3A) and at the end of the treatment (last session test, Fig. 3B), showed that the administration of the low doses of pramipexole was associated with a lower incidence of AIMs [25% dyskinetic (Dys) rats for the 0.1 mg/kg group; 20% Dys rats for the 0.3 mg/kg group] when compared with the pramipexole high-dose group (70% Dys rats for the 1 mg/kg group) and the l-DOPA group (65% Dys) (Fig. 3C). None of the tested drugs induced dyskinetic movements or motor abnormalities in control sham-operated rats (data not shown).
Spontaneous activity alterations induced by 6-OHDA lesion recovered after l-DOPA or pramipexole treatment
It has been previously shown that the intrinsic properties of striatal MSNs, such as current-evoked firing discharge and current–voltage relationship, are not significantly altered by 6-OHDA lesion and l-DOPA treatment (Picconi et al., 2003). In the present study, comparable results were obtained in MSNs recorded from pramipexole-treated animals (data not shown).
Conversely, compelling evidence suggest that glutamate transmission is dramatically increased after 6-OHDA lesion (Calabresi et al., 1993; Schwarting and Huston, 1996; Tang et al., 2001; Picconi et al., 2004; VanLeeuwen et al., 2010) especially in terms of frequency and amplitude of sEPSCs. Notably, these alterations are overcome by the restoration of DA levels through “subchronic” l-DOPA treatment (Picconi et al., 2004).
In the present study, through the administration of two doses of the D2-like agonist pramipexole (1 mg/kg once daily or 0.3 mg/kg twice daily, for 3 weeks), we further characterized the effect exerted by DA replacement following chronic l-DOPA treatment on spontaneous excitatory synaptic transmission and the consequence of the DAergic transmission stimulation. The lowest dose of pramipexole (0.1 mg/kg twice daily) was discarded from all the electrophysiological studies due to lack of any therapeutic effect. As reported in Figure 4A,B, both frequency (one-way ANOVA, F(4,42) = 4.184, p < 0.01) and amplitude (F(4,42) = 6.046, p < 0.001) of the glutamatergic sEPSCs were significantly increased compared with control levels after 6-OHDA lesion [n = 6 6-OHDA vs n = 8 control (CTRL), *p < 0.05, frequency, **p < 0.01, amplitude]. Interestingly, these parameters were successfully rescued to control levels by chronic l-DOPA administration (n = 14 l-DOPA vs CTRL p > 0.05) as well as pramipexole therapy, with the same efficacy, regardless of the two different doses used (Fig. 4A–C; n = 9 pramipexole 0.3 mg/kg vs CTRL, p > 0.05; n = 6 pramipexole 1 mg/kg vs CTRL, p > 0.05).
Figure 4.
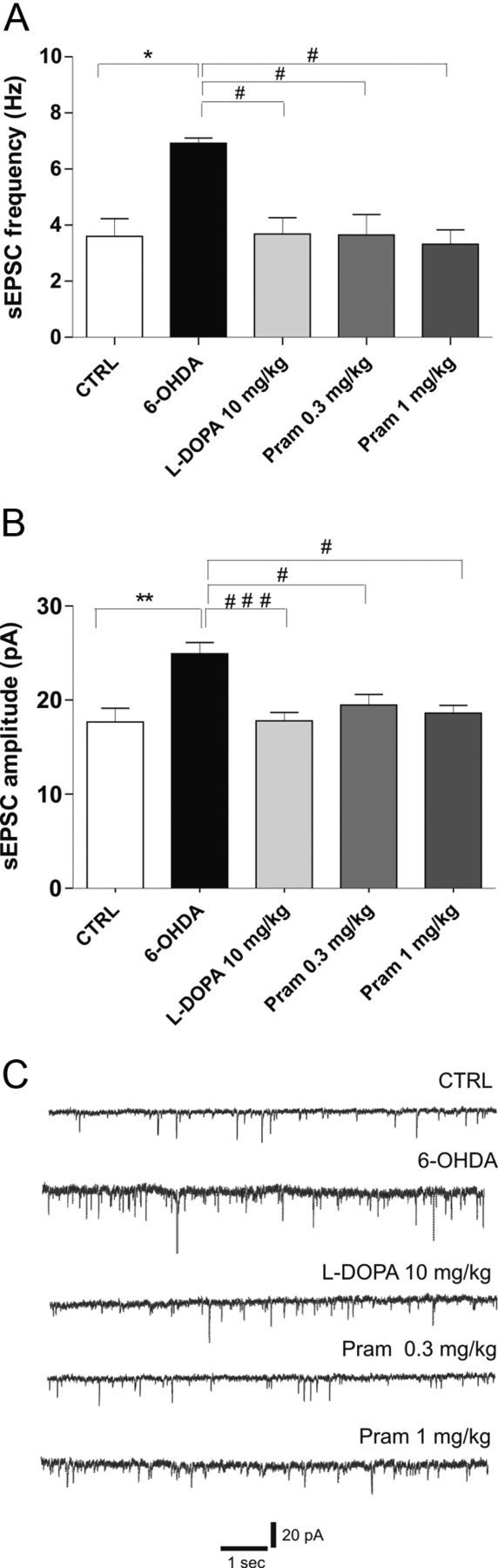
6-OHDA-lesion-related alterations in spontaneous activity of MSNs are rescued by either chronic l-DOPA or pramipexole treatment. A, B, Bar graphs show the averaged values of the frequency (A) and amplitude (B) of spontaneous events recorded in MSNs from the different experimental groups: control (CTRL), 6-OHDA-lesioned rats (6-OHDA), l-DOPA-treated rats, 0.3 mg/kg pramipexole-treated rats, and 1 mg/kg pramipexole-treated rats (n = 8 CTRL vs n = 6 6-OHDA, *p < 0.05 frequency, **p < 0.01 amplitude; n = 14 l-DOPA vs 6-OHDA, #p < 0.05 frequency, ###p < 0.001 amplitude; n = 9 pramipexole 0.3 mg/kg vs 6-OHDA, #p < 0.05 frequency, #p < 0.05 amplitude, n = 6 pramipexole 1 mg/kg vs 6-OHDA, #p < 0.05 frequency, #p < 0.05 amplitude; one-way ANOVA with Bonferroni comparison). C, Representative traces from whole-cell patch-clamp experiments showing glutamatergic sEPSCs recorded from MSNs in control condition (upper trace) and in all the other groups of animals studied.
NMDAR/AMPAR ratio alteration after 6-OHDA lesion and DAergic therapy
To measure postsynaptic responsiveness between the different groups of animals, AMPAR-evoked eEPSCs (Vh, −70 mV) were compared with evoked NMDAR currents from the same cell (and stimulation). This enabled us to calculate an NMDAR/AMPAR current ratio for each individual neuron. This parameter provides a measure of NMDAR activity relative to that one of AMPAR, regardless of the number of presynaptic afferents recruited and stimulating electrode positioning.
As reported in Figure 5A, direct comparison with MSNs recorded from control rats demonstrated a significant effect of the lesion in reducing the NMDAR/AMPAR ratio at the postsynaptic site (one-way ANOVA, F(4,44) = 8.88, p < 0.0001, n = 10 6-OHDA vs n = 10 CTRL, post hoc test ***p < 0.001). Neither l-DOPA nor the high dose of pramipexole rescued the physiological ratio of these receptors (n = 10 l-DOPA vs 6-OHDA, p > 0.05; n = 6 pramipexole 1 mg/kg vs 6-OHDA, p > 0.05). Conversely, MSNs recorded from animals treated with the low therapeutic dose of pramipexole (0.3 mg/kg) showed a NMDAR/AMPAR ratio comparable to the one obtained in control condition (n = 9 pramipexole 0.3 mg/kg vs CTRL, p > 0.05).
Figure 5.
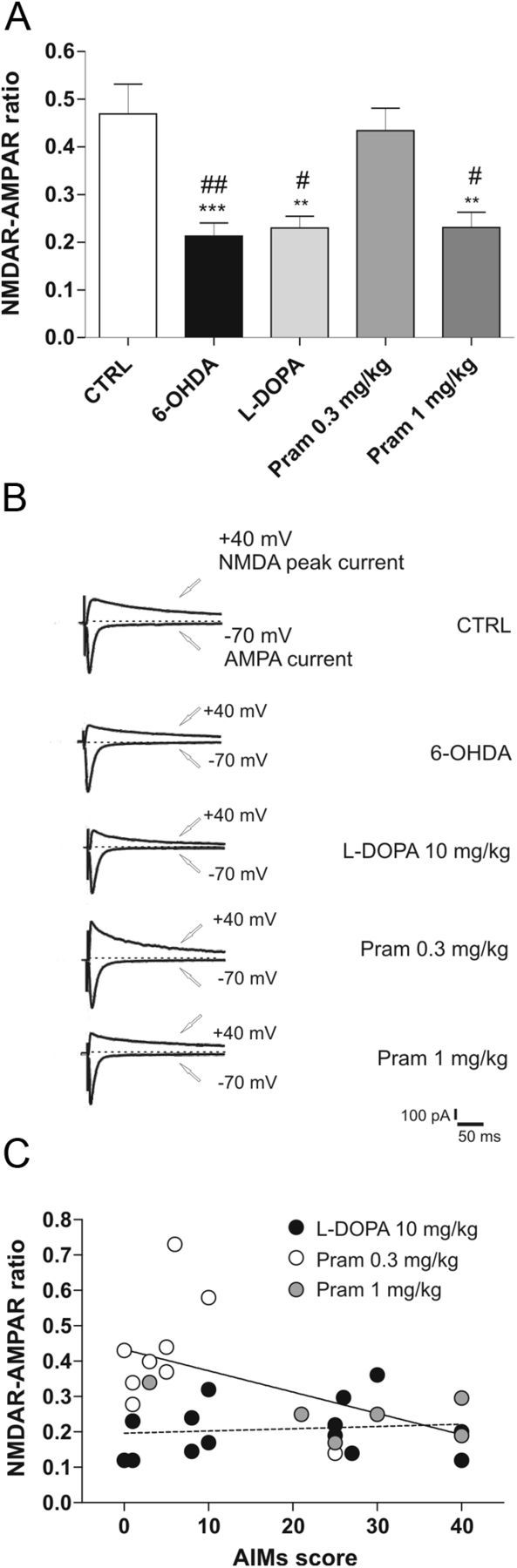
NMDAR/AMPAR ratio reduction after 6-OHDA-lesion and DAergic treatment. A, Summary plot reporting the significant reduction of NMDAR/AMPAR ratio induced by 6-OHDA lesion; neither l-DOPA treatment nor the higher dose of pramipexole rescues this parameter to control levels (n = 10 6-OHDA vs n = 10 CTRL, ***p < 0.001; n = 10 l-DOPA vs CTRL, **p < 0.01; n = 9; pramipexole 0.3 mg/kg vs CTRL, p > 0.05; n = 6, pramipexole 1 mg/kg vs CTRL, **p < 0.01; 6-OHDA vs pramipexole 0.3 mg/kg, ##p < 0.01; l-DOPA vs pramipexole 0.3 mg/kg, #p < 0.05; pramipexole 1 mg/kg vs pramipexole 0.3 mg/kg, #p < 0.05; one-way ANOVA with Bonferroni comparison). B, Representative traces from whole-cell patch-clamp experiments showing AMPAR-evoked and NMDAR-evoked currents, recorded at −70 mV and +40 mV respectively, in the presence of 50 μm picrotoxin. NMDAR component was measured at +40 mV considering the peak amplitude after 50 ms from the beginning of the event. C, Plotting of individual mean AIMs score during the treatment with l-DOPA (filled circles) and pramipexole (open circles) against the individual NMDAR/AMPAR ratio. Significant correlation between the two variables in the group treated with pramipexole (p < 0.05) but not in that treated with l-DOPA is revealed by the linear regression (n = 15 l-DOPA, Spearman's r = 0.1355, p > 0.05; n = 15 pramipexole, Spearman's r = −0.5189, p < 0.05).
Then, in looking for a possible relationship between the alteration of NMDAR/AMPAR ratio in MSNs and the behavioral motor abnormalities, we analyzed the correlation between individual AIM scores during the 21 d of treatment with l-DOPA or pramipexole and the respective ratio of the two receptors. Interestingly, the plotting of individual mean AIMs score during the treatment against the individual NMDAR/AMPAR ratio (Fig. 5C) revealed a highly significant correlation between the two variables in the group treated with pramipexole (white circles, pramipexole 0.3 mg/kg; gray circles, pramipexole 1 mg/kg) (n = 15, Spearman's r = −0.5189, p < 0.05) but not in the group treated with l-DOPA (n = 15, Spearman's r = 0.1355, p > 0.05).
Extent of NR2A/NRB subunit ratio in animals treated with l-DOPA or pramipexole
To further comprehend the mechanisms underlying the alterations in NMDAR/AMPAR ratio found, we performed a molecular analysis of the TIF to obtain the relative abundance of NMDAR and AMPAR subunits at the postsynaptic site. It is known that AIMs are associated with consistent synaptic rearrangement of NMDAR NR2-type subunits, leading to an increase of NR2A/NR2B subunit ratio in the postsynaptic compartment (Dunah and Standaert, 2003; Gardoni et al., 2006). In this respect, we analyzed the effects of the two doses of the D2-like agonist pramipexole (1 mg/kg once daily or 0.3 mg/kg twice daily) or l-DOPA (10 mg/kg plus benserazide 6 mg/kg, twice daily) (n = 3 rats for each group) administered for 3 weeks, on the relative abundance of these two regulatory subunits of the NMDAR in the TIF. As shown in Figure 6A, we found a significant increase in the NR2A/NR2B ratio only in dyskinetic 1 mg/kg pramipexole-treated animals (Student's t test, 6-OHDA vs pramipexole 1 mg/kg Dys, *p < 0.05; pramipexole 1 mg/kg Dys vs pramipexole 1 mg/kg Non Dys, #p < 0.05). Interestingly, no alteration of NMDAR regulatory subunits was observed between 0.3 mg/kg and 1 mg/kg pramipexole-treated animals not showing a dyskinetic behavior (Student's t test, pramipexole 1 mg/kg Non Dys vs pramipexole 0.3 mg/kg Non Dys, p > 0.05), thus confirming the presence of a correlation between induction of AIMs and increased NR2A/NR2B ratio. Similarly, and in agreement with previous studies (Gardoni et al., 2006), alteration of NR2A/NR2B ratio correlates with onset of dyskinesia also following treatment with l-DOPA (Fig. 6B, Student's t test, l-DOPA Non Dys vs l-DOPA Dys, #p < 0.05; 6-OHDA vs l-DOPA Dys, *p < 0.05).
Figure 6.

Alteration of NR2A/NRB subunit ratio in dyskinetic animals treated with l-DOPA or pramipexole. A, B, 6-OHDA-lesioned animals were treated chronically with 0.3 mg/kg pramipexole, with 1 mg/kg pramipexole (A), or with l-DOPA (B) and divided into two groups based on the presence (Dys) or absence (Non Dys) of dyskinesia. Striatal TIFs from the above-indicated experimental groups were analyzed by Western blot (wb) analysis with NR2A, NR2B, and tubulin antibodies (see Materials and Methods). The same amount of proteins was loaded per lane. Histogram in A shows the quantification of Western blotting experiments as NR2A/NR2B ratio after normalization on tubulin levels (Student's t test, 6-OHDA vs pramipexole 1 mg/kg, *p < 0.05; pramipexole 0.3 mg/kg Non Dys vs pramipexole 1 mg/kg Dys, #p < 0.05; pramipexole 1 mg/kg Non Dys vs pramipexole 1 mg/kg Dys, #p < 0.05). B, Histogram shows the NR2A/NR2B distribution between parkinsonian and l-DOPA-treated rats (6-OHDA vs l-DOPA Dys, *p < 0.05; l-DOPA Non Dys vs l-DOPA Dys, #p < 0.05).
However, significant modifications of the AMPAR amount were not found at the postsynaptic compartment (data not shown). Nevertheless, we are aware that this latter result does not exclude a possible modification of the receptor trafficking to the plasma membrane within the postsynaptic compartment or changes in its activity, which may contribute to NMDAR/AMPAR ratio alterations.
AMPAR subunit composition is differently affected by l-DOPA or pramipexole treatment
We then investigated whether, in addition to the different extent in NMDAR/AMPAR ratio found following DAergic denervation and replacement, AMPAR subunit modifications might also take place at glutamatergic synapses. In fact, synapses in MSNs contain a mixed AMPAR population that can undergo a rapid activity-dependent switch in subtypes. The GluR2 subunit in particular is a key determinant of biophysical properties of AMPARs. In fact, an arginine residue in the pore-lining region makes GluR2-containing AMPARs impermeable to calcium ions (Ca2+) which thus display a characteristic linear current–voltage relationship (Pellegrini-Giampietro et al., 1997; Cull-Candy et al., 2006). In contrast, AMPARs lacking GluR2 subunits are permeable to Ca2+ and are characterized electrophysiologically by an inward-rectifying current–voltage relationship. For this purpose, we measured the IR, calculated as the ratio between the AMPAR-evoked current at −70 mV and at +40 mV, to test the hypothesis that an exchange in subunit composition and then Ca2+ permeability might occur after DAergic denervation, as observed in the MPTP model of PD (VanLeeuwen et al., 2010). It is noteworthy that the IR value considerably increased after 6-OHDA lesion and it reached a level of significant difference compared with control level (Fig. 7A; one-way ANOVA F(4,38) = 10.86, ***p < 0.001, n = 7 6-OHDA vs n = 8 CTRL, post hoc test **p < 0.01), suggesting a stronger inward-rectifying current–voltage relationship, further evident after either l-DOPA treatment or the higher dose of pramipexole (Fig. 7A, n = 11 l-DOPA vs n = 8 CTRL, ***p < 0.001; n = 7 pramipexole 1 mg/kg vs CTRL, **p < 0.01). Interestingly, 0.3 mg/kg pramipexole administered twice daily, representing the therapeutic dose free from dyskinetic side effects, was able to bring the IR back to control level (Fig. 7A, n = 7 pramipexole 0.3 mg/kg vs CTRL, p > 0.05; pramipexole 0.3 mg/kg vs 6-OHDA, ##p < 0.01; pramipexole 0.3 mg/kg vs l-DOPA, ###p < 0.001).
Figure 7.
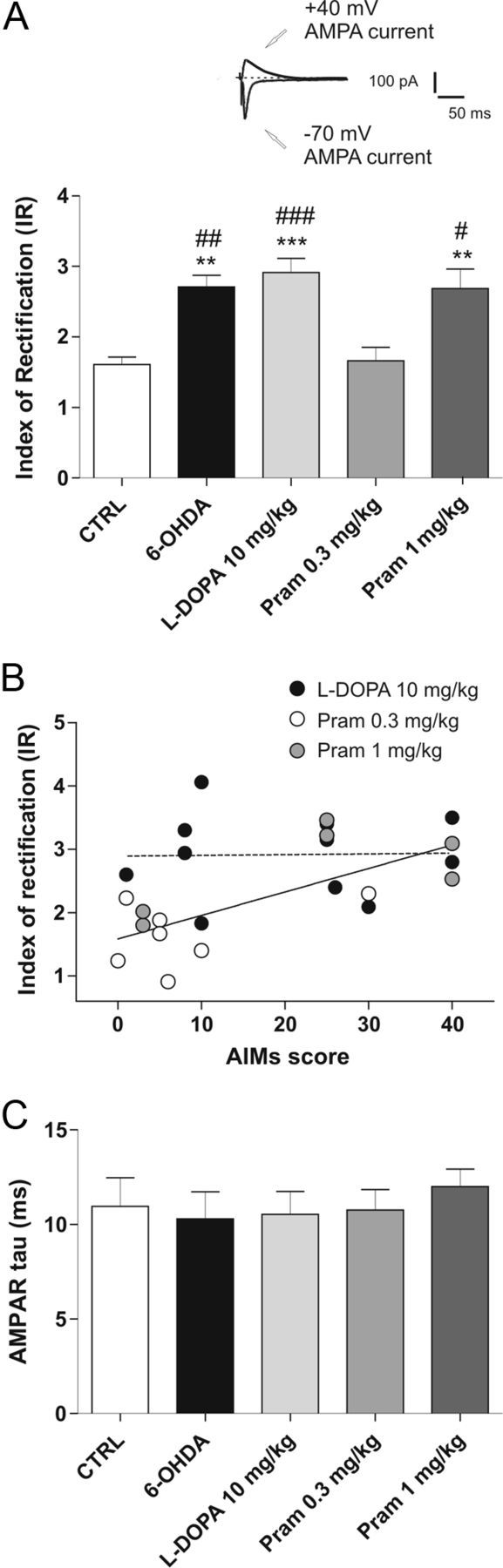
Modifications of AMPAR subunit complex after 6-OHDA lesion and pharmacological treatments. A, Top of the graph shows example of an individual experiment obtained in control condition to calculate the IR of AMPAR (scale bars, 50 ms and 100 pA). In the bottom, the bar graph shows the IR in the different groups, calculated as the ratio of the AMPAR-mediated EPSCs recorded at −70 mV and +40 mV (n = 7 6-OHDA vs n = 8 CTRL, **p < 0.01; n = 11 l-DOPA vs CTRL, ***p < 0.001; n = 7 pramipexole 1 mg/kg vs CTRL, **p < 0.01; n = 7 6-OHDA vs pramipexole 0.3 mg/kg, ## p < 0.01; l-DOPA vs pramipexole 0.3 mg/kg, ###p < 0.001; pramipexole 1 mg/kg vs pramipexole 0.3 mg/kg, #p < 0.05; one-way ANOVA with Bonferroni comparison). B, IR values obtained from pramipexole-treated and l-DOPA-treated rats were correlated with AIM score. The linear regression revealed a significant correlation between these two variables among the pramipexole-treated group (n = 11 l-DOPA, Pearson's r = 0.0238, p > 0.05; n = 13 pramipexole, Pearson's r = 0.7126, p < 0.01). C, Decay time constants of AMPAR, calculated by fitting with a single exponential, are summarized in the bar graph.
To evaluate the predictive value of IR on dyskinesia development, we investigated the eventual correlation between IR and the AIMs score as reported in Figure 7B, where a correlation between IR and AIMs score in the rats treated with either pramipexole (white circles, pramipexole 0.3 mg/kg; gray circles, pramipexole 1 mg/kg) or l-DOPA is shown (n = 11 l-DOPA, Pearson's r = 0.0238, p > 0.05; n = 13 pramipexole, Pearson's r = 0.7126, p < 0.01).
Having found an increased proportion of GluR2-lacking AMPARs at synaptic level after either DAergic lesion and l-DOPA or high dose of pramipexole treatment, we examined whether the modification of AMPAR assemblage affected the kinetics of the recorded currents at these synapses. The decay phase of the AMPAR EPSC was best fitted by a single exponential curve at −70 mV (Ozawa et al., 1998). In all the groups recorded, no significant difference was observed in the kinetic properties, as shown in Figure 7C. This result suggests that EPSCs displaying different rectification properties may have a similar decay time course, at least in MSNs.
Alteration of D3R levels in dyskinetic animals treated with l-DOPA or pramipexole
We hypothesized that the appearance of dyskinetic behavior and the related synaptic abnormalities after chronic treatment with l-DOPA or the high dose of pramipexole, might be dependent on the convergence of these two drugs on D3R-mediated signaling. The importance of D3R in the emergence of l-DOPA-induced dyskinesia is still matter of debate (Berthet et al., 2009; Visanji et al., 2009). However, evidence from several experiments suggests that a significant D3R overexpression takes place in this condition (Bordet et al., 2000) and contributes to the overactivity of D1R pathway.
Interestingly, in dyskinetic animals obtained after chronic treatment with the high dose of pramipexole as well as in l-DOPA-treated dyskinetic rats (Fig. 8A), a significant increase of D3R quantity is detectable (Student's t test, n = 3 rats for each group; pramipexole 1 mg/kg Non Dys vs pramipexole 1 mg/kg Dys, ##p < 0.01; pramipexole 0.3 mg/kg Non Dys vs pramipexole 1 mg/kg Dys, ##p < 0.01; l-DOPA Non Dys vs l-DOPA Dys, #p < 0.05). It is noteworthy that, even the chronic treatment with a low dose of pramipexole can induce the overexpression of D3R compared with control (pramipexole 0.3 mg/kg Non Dys vs CTRL, **p < 0.01). However, this increase is “less severe” when compared with the levels of D3R found in dyskinetic animals obtained with the high dosage. These results further support a central role for D3R in the emergence of dyskinesia, likely due to its ability of boosting the D1R pathway.
Figure 8.
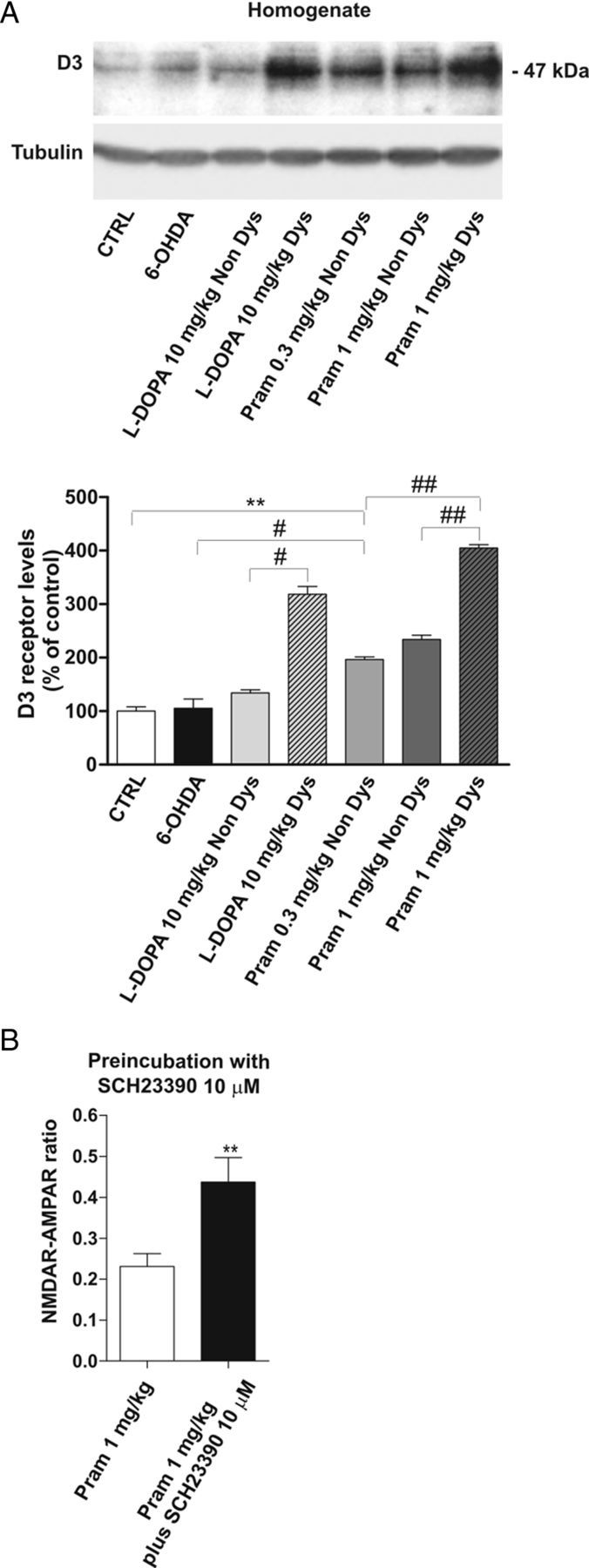
Alteration of D3R levels in dyskinetic animals treated with l-DOPA or pramipexole. A, 6-OHDA-lesioned animals were treated chronically with 10 mg/kg l-DOPA, 0.3 mg/kg pramipexole, or 1 mg/kg pramipexole and divided into two groups based on the presence (Dys) or absence (Non Dys) of dyskinesia. Striatal lysates from the above-indicated experimental groups were analyzed by Western blot (wb) analysis with D3 antibodies (see Materials and Methods). The same amount of proteins was loaded per lane. Histograms show the quantification of wb experiments after normalization on tubulin levels (CTRL vs pramipexole 0.3 mg/kg Non Dys, **p < 0.01; 6-OHDA vs pramipexole 0.3 mg/kg Non Dys, #p < 0.05; pramipexole 1 mg/kg Non Dys vs pramipexole 1 mg/kg Dys ##p < 0.01; pramipexole 0.3 mg/kg Non Dys vs pramipexole 1 mg/kg Dys ##p < 0.01; l-DOPA Non Dys vs l-DOPA Dys, #p < 0.05). B, Bar graph compares the NMDAR/AMPAR ratio recorded in vitro from dyskinetic animals after chronic treatment with the high dose of pramipexole, with or without a prolonged incubation of the D1 antagonist SCH23390 (n = 6 pramipexole 1 mg/kg vs n = 5 pramipexole 1 mg/kg plus SCH23390, **p < 0.01).
To further prove this concept, we incubated corticostriatal slices obtained from dyskinetic animals treated with 1 mg/kg pramipexole, with the D1R antagonist SCH23390 (10 μm), for at least 2 h. The NMDAR/AMPAR ratio was measured in this condition. As expected, this treatment was capable of bringing the synaptic parameter back to control level (Fig. 8B) (Student's t test, n = 6 pramipexole 1 mg/kg vs n = 5 pramipexole 1 mg/kg plus SCH23390, **p < 0.01). Similar results were obtained with the IR (data not shown). The time window of 2 h incubation, chosen as a shorter incubation period, did not affect the NMDAR/AMPAR ratio (n = 4, data not shown). This observation might suggest that the modification of the synaptic parameter is not simply a result of an acute D1R antagonism, but rather involves a more complex downstream machinery. In fact, a recent experimental work revealed the profound impact of D1 receptor prolonged activaction/inactivation in the control of NMDAR and AMPAR subunit arrangement (Vastagh et al., 2012).
Discussion
The current study provides the first evidence that in an experimental model of PD, a chronic and “mild level” of D2-like receptor stimulation, in addition to its therapeutic action and the reduced level of dyskinetic movements compared with l-DOPA, overcomes profound synaptic alterations occurring at striatal glutamatergic synapses (Calabresi et al., 1993; Picconi et al., 2004; VanLeeuwen et al., 2010). In fact, chronic treatment with an intermediate dose (0.3 mg/kg twice daily) of pramipexole retunes the NMDAR/AMPAR ratio imbalance and seems to restore the correct assemblage of AMPAR subunits at the postsynaptic site as shown by the normalization of the IR of AMPAR currents.
Our study also provides relevant pathophysiological information concerning the role of the increased spontaneous glutamatergic activity in the parkinsonian striatum. In fact, while we confirmed in this study that both the amplitude and the frequency of sEPSCs are increased in this PD model, we also observed as a new finding that both l-DOPA and pramipexole treatments are able to reverse these synaptic abnormalities regardless the motor side effects produced. This latter observation indicates that, although changes in the features of sEPSCs represent a consistent synaptic alteration in PD models, they do not seem to play a critical role in the control of the dyskinetic behavior.
Interestingly, changes in glutamate-mediated and DA-dependent striatal synaptic plasticity have been already implicated in the pathophysiology of LID. In particular, in dyskinetic animals we have reported lack of depotentiation (reversal of NMDA-dependent long-term potentiation) (Picconi et al., 2003) and loss of long-term depression (Picconi et al., 2011), indicating that chronic l-DOPA treatment inducing dyskinesia impairs the scaling down of glutamatergic synapses. Here we integrate this view and propose an additional mechanism further linking abnormal involuntary movements triggered by DAergic stimulation to an abnormal glutamatergic transmission.
In fact, we found that chronic l-DOPA, as well as the high dose of pramipexole, fails to rescue the physiological NMDAR/AMPAR ratio and both these treatments induce a similar degree of dyskinesia. Surprisingly, however, we observed that the abnormal NMDAR/AMPAR ratio was correlated to high AIM score in the pramipexole-treated group but not in the l-DOPA-treated group. In fact, the low dose of pramipexole (0.3 mg/kg), free from dyskinetic side effects, was associated with a NMDAR/AMPAR ratio similar to that seen in control animals. In contrast, the high dose of the drug, inducing elevated AIMs scores, resulted in abnormal values of this parameter in the neurons recorded from dyskinetic animals. The strict relationship between high AIMs score and abnormal NMDAR/AMPAR ratio was completely lost in the l-DOPA-treated group where, even in those subjects not showing dyskinesia, an “abnormal” change in NMDAR/AMPAR ratio was observed. One possible mechanism explaining this apparently contrasting result might be the absence of selectivity that l-DOPA has for D1 and D2 receptors. Furthermore, one could also speculate that, because almost all subjects chronically treated with l-DOPA presumably will develop dyskinesia over time (similarly to PD patients), the alteration of the NMDAR/AMPAR ratio simply precedes the appearance of abnormal behavior in non-dyskinetic animals.
Among the increasing number of D2-like receptor agonists, pramipexole is currently the most prescribed for PD treatment, showing a preferential affinity also for the DA-D3 receptor subtype (D3R) and lacking affinity for D1 and D5 receptors (Gerlach et al., 2003). In the striatum, D3Rs, once activated, exert a synergistic effect on D1R-mediated transmission through direct intramembrane interaction (Fiorentini et al., 2008; Marcellino et al., 2008). Pramipexole binding to D3R boosts D1R function, which explains, in part, the antiparkinsonian efficacy of this compound. On the other hand, the activation of D2R by pramipexole reduces indirect pathway neuron excitability, which physiologically acts in the opposite direction to the direct pathway in movement control. The synergy of these two actions may account for the efficacy of this compound in the treatment of PD symptoms but also for the side effects induced by the high dose of this agonist.
D3 receptors comprise <1% of DA receptors in the caudate putamen in physiological condition, suggesting that the abundance of the D1R–D3R heteromer is also low. However, in pathological condition, such as in l-DOPA-induced dyskinesia, this fine regulation is impaired. Thus, D3R overexpression has been observed in striatal neurons of the direct pathway associated with defect in D1R internalization, which was counteracted by a D3R antagonist, suggesting a role of the D3R in immobilizing the D1R at the plasma membrane (Fiorentini et al., 2008; Marcellino et al., 2008).
In line with these results, we believe that the alteration of the NMDAR/AMPAR ratio is likely dependent on dysregulation of D1R activation. Pramipexole, in fact, might indirectly enable the activity of D1R through D1R–D3R heteromerization and, at high dosage, it could determine sensitization of D1R-mediated pathway, as observed following l-DOPA-treatment. This concept is supported by evidence showing that a high dose of pramipexole massively increases the expression of D3R in dyskinetic animals.
Our results suggest that a moderate dose of pramipexole, free from dyskinetic side effects, restores the NMDAR/AMPAR ratio, possibly allowing a finely balanced activation of D1R, as suggested by the intermediate increase of D3R. However, sustained activation of D3R obtained with a high dose of pramipexole, could have detrimental effects on both motor performance and synaptic circuitry via the abnormal indirect activation of the D1-dependent transmission.
It is well known that alterations in NMDAR/AMPAR ratio can be related to changes in activity and/or abundance of NMDAR or AMPAR, or to both events. This latter condition seems to be more probable in the present scenario. In fact, modifications of the abundance of NMDAR subunits (Gardoni and Di Luca, 2006; Gardoni et al., 2012), as well as abnormal phosphorylation of striatal NMDAR and AMPAR subunits, have been pointed out in several experimental studies in animal models of LID (Dunah et al., 2004; Santini et al., 2007).
Interestingly, by showing changes in the NR2A/NR2B subunit ratio, we provide for the first time evidence that the dyskinetic phenotype emerging after chronic treatment with a D2R agonist (i.e., pramipexole) is also accompanied by a perturbed NMDAR assemblage at the postsynaptic compartment, and that this action is a function of the dose used for the treatment.
In addition, here we show that both parkinsonism and dyskinesia are associated with a switch of AMPAR subunit composition within glutamatergic synapses. In particular, we demonstrated an increased IR in MSNs, suggesting a possible abnormal insertion of GluR2-lacking AMPARs at corticostriatal synapses. In fact, this rise in IR indicates that, in these conditions, a switch toward a nonlinear voltage–current relationship occurs within glutamatergic synapses and stems from a major availability of Ca2+-permeable receptors (i.e., GluR2-lacking AMPARs). This particular AMPAR subtype is widely expressed in the CNS, where it contributes to synaptic transmission and changes in synaptic efficacy (Isaac et al., 2007). Nevertheless, compelling evidence indicates that several neurological disorders, such as cerebral ischemia, amyotrophic lateral sclerosis, pain, and epilepsy, are associated with alterations in GluR2 function (Cull-Candy et al., 2006). This result is in agreement with previous data demonstrating that dopaminergic lesion as well as dyskinetic condition are associated with increased activity of calcium-permeable AMPAR due to hyperphosphorylation of GluR1 subunit (Santini et al., 2007).
Here we show that, in striking contrast to l-DOPA, the intermediate dose of pramipexole, which has antiparkinsonian effects and is associated with reduced AIMs score, restores the AMPAR subunit composition, in addition to rescuing the physiological ratio in the two glutamatergic receptors. In fact, this moderate dose of pramipexole is able to correct the IR that was altered after 6-OHDA lesion. However, the higher dose of pramipexole is unable to rescue this parameter to control levels.
Among the possible mechanisms implicated in the increased availability of GluR2-lacking receptors at the postsynaptic site, those that might be essential include the abnormal NMDAR activity and Ca2+ influx in l-DOPA-induced dyskinesia. In fact, several lines of evidence show that NMDAR signaling is essential in GluR2 subunit internalization in physiological condition (Hayashi and Huganir, 2004).
Thus, because Ca2+ permeability of AMPARs depends on the absence of GluR2 subunits (Mishina et al., 1991), increased insertion of GluR2-lacking receptors obviously triggers “abnormal” cellular Ca2+ dynamics, related downstream pathways, and eventually “pathological” synaptic plasticity in a vicious cycle that leads to the amplification of NMDAR dysfunction.
In accordance with our results, a recent paper from Kobylecki et al. (2010), using a selective GluR2-lacking AMPAR antagonist (IEM-1460), which was able to significantly reduce the induction and expression of dyskinesia, revealed the pivotal role of these receptors in LID induction and expression.
In conclusion, the findings of the present work cast further light on the cellular mechanisms that subtend the development of dyskinesia during treatment with l-DOPA and D2-like receptor agonists. Moreover, our results confer further emphasis to the role of therapeutic strategies alternative to l-DOPA, such as those represented by D2R/D3R agonists family.
Footnotes
This work was supported by grants from European Community contract number 222918 (REPLACES [Restorative Plasticity at Corticostriatal Excitatory Synapses)] FP7-Thematic priority HEALTH (P.C.), Programmi di Ricerca Scientifica di Rilevante Interesse Nazionale-PRIN2008 (PC), Progetto Giovani Ricercatori Ministero Sanità 2008 (B.P.), and Cariplo Foundation project number 2010-0661 (B.P. and F.G.), and by support from Boehringer-Ingelheim to the Santa Lucia Foundation (2007–2009). We are indebted to Boehringer-Ingelheim also for the kind gift of pramipexole. We also thank Dr. Bernd Sommer and Dr. Bastian Hengerer their helpful comments.
P.C. serves as an editorial board member of Lancet Neurology, The Journal of Neuroscience, Movement Disorders, and Synapse; and receives research support from Bayer Schering, Biogen, Boehringer Ingelheim, Eisai, Merck Sharp & Dohme, Novartis, Lundbeck, Sanofi-Aventis, Sigma-Tau, and UCB Pharma, Ricerca Corrente Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Ricerca Finalizzata IRCCS [European Community Grants SYNSCAFF (Synaptic Scaffolding Proteins Orchestrating Cortical Synapse Organisation During Development) and REPLACES (Restorative Plasticity at Corticostriatal Excitatory Synapses)], the Italian Ministry of Health, and Agenzia Italiana del Farmaco. All other authors declare no competing financial interests.
References
- Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord. 2001;16:448–458. doi: 10.1002/mds.1090. [DOI] [PubMed] [Google Scholar]
- Ahmed I, Bose SK, Pavese N, Ramlackhansingh A, Turkheimer F, Hotton G, Hammers A, Brooks DJ. Glutamate NMDA receptor dysregulation in Parkinson's disease with dyskinesias. Brain. 2011;134:979–986. doi: 10.1093/brain/awr028. [DOI] [PubMed] [Google Scholar]
- Bagetta V, Picconi B, Marinucci S, Sgobio C, Pendolino V, Ghiglieri V, Fusco FR, Giampà C, Calabresi P. Dopamine-dependent long-term depression is expressed in striatal spiny neurons of both direct and indirect pathways: implications for Parkinson's disease. J Neurosci. 2011;31:12513–12522. doi: 10.1523/JNEUROSCI.2236-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthet A, Porras G, Doudnikoff E, Stark H, Cador M, Bezard E, Bloch B. Pharmacological analysis demonstrates dramatic alteration of D1 dopamine receptor neuronal distribution in the rat analog of l-DOPA-induced dyskinesia. J Neurosci. 2009;29:4829–4835. doi: 10.1523/JNEUROSCI.5884-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkmayer W, Hornykiewicz O. The effect of l-3,4-dihydroxy phenylalanine (=DOPA) on akinesia in parkinsonism. Parkinsonism Relat Disord. 1998;4:59–60. doi: 10.1016/s1353-8020(98)00013-3. [DOI] [PubMed] [Google Scholar]
- Blandini F, Armentero MT. New pharmacological avenues for the treatment of L-DOPA-induced dyskinesias in Parkinson's disease: targeting glutamate and adenosine receptors. Expert Opin Investig Drugs. 2012;21:153–168. doi: 10.1517/13543784.2012.651457. [DOI] [PubMed] [Google Scholar]
- Bordet R, Ridray S, Schwartz JC, Sokoloff P. Involvement of the direct striatonigral pathway in levodopa-induced sensitization in 6-hydroxydopamine-lesioned rats. Eur J Neurosci. 2000;12:2117–2123. doi: 10.1046/j.1460-9568.2000.00089.x. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci. 1992;12:4224–4233. doi: 10.1523/JNEUROSCI.12-11-04224.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Mercuri NB, Sancesario G, Bernardi G. Electrophysiology of dopamine-denervated striatal neurons. Implications for Parkinson's disease. Brain. 1993;116:433–452. [PubMed] [Google Scholar]
- Calabresi P, Di Filippo M, Ghiglieri V, Picconi B. Molecular mechanisms underlying levodopa-induced dyskinesia. Mov Disord. 2008;23(Suppl 3):S570–S579. doi: 10.1002/mds.22019. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Di Filippo M, Ghiglieri V, Tambasco N, Picconi B. Levodopa-induced dyskinesias in patients with Parkinson's disease: filling the bench-to-bedside gap. Lancet Neurol. 2010;9:1106–1117. doi: 10.1016/S1474-4422(10)70218-0. [DOI] [PubMed] [Google Scholar]
- Calandrella D, Antonini A. Pulsatile or continuous dopaminomimetic strategies in Parkinson's disease. Parkinsonism Relat Disord. 2012;18(Suppl 1):S120–S122. doi: 10.1016/S1353-8020(11)70037-2. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Konradi C. Maladaptive striatal plasticity in L-DOPA-induced dyskinesia. Prog Brain Res. 2010;183:209–233. doi: 10.1016/S0079-6123(10)83011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci MA, Lee CS, Björklund A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. Eur J Neurosci. 1998;10:2694–2706. [PubMed] [Google Scholar]
- Cull-Candy S, Kelly L, Farrant M. Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr Opin Neurobiol. 2006;16:288–297. doi: 10.1016/j.conb.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Darmopil S, Martín AB, De Diego IR, Ares S, Moratalla R. Genetic inactivation of dopamine D1 but not D2 receptors inhibits L-DOPA-induced dyskinesia and histone activation. Biol Psychiatry. 2009;66:603–613. doi: 10.1016/j.biopsych.2009.04.025. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Standaert DG. Subcellular segregation of distinct heteromeric NMDA glutamate receptors in the striatum. J Neurochem. 2003;85:935–943. doi: 10.1046/j.1471-4159.2003.01744.x. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Sirianni AC, Fienberg AA, Bastia E, Schwarzschild MA, Standaert DG. Dopamine D1-dependent trafficking of striatal N-methyl-D-aspartate glutamate receptors requires Fyn protein tyrosine kinase but not DARPP-32. Mol Pharmacol. 2004;65:121–129. doi: 10.1124/mol.65.1.121. [DOI] [PubMed] [Google Scholar]
- Feyder M, Bonito-Oliva A, Fisone G. L-DOPA-induced dyskinesia and abnormal signaling in striatal medium spiny neurons: focus on dopamine D1 receptor-mediated transmission. Front Behav Neurosci. 2011;5:71. doi: 10.3389/fnbeh.2011.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentini C, Busi C, Gorruso E, Gotti C, Spano P, Missale C. Reciprocal regulation of dopamine D1 and D3 receptor function and trafficking by heterodimerization. Mol Pharmacol. 2008;74:59–69. doi: 10.1124/mol.107.043885. [DOI] [PubMed] [Google Scholar]
- Fisone G, Bezard E. Molecular mechanisms of l-DOPA-induced dyskinesia. Int Rev Neurobiol. 2011;98:95–122. doi: 10.1016/B978-0-12-381328-2.00004-3. [DOI] [PubMed] [Google Scholar]
- Gardoni F, Di Luca M. New targets for pharmacological intervention in the glutamatergic synapse. Eur J Pharmacol. 2006;545:2–10. doi: 10.1016/j.ejphar.2006.06.022. [DOI] [PubMed] [Google Scholar]
- Gardoni F, Picconi B, Ghiglieri V, Polli F, Bagetta V, Bernardi G, Cattabeni F, Di Luca M, Calabresi P. A critical interaction between NR2B and MAGUK in l-DOPA induced dyskinesia. J Neurosci. 2006;26:2914–2922. doi: 10.1523/JNEUROSCI.5326-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardoni F, Sgobio C, Pendolino V, Calabresi P, Di Luca M, Picconi B. Targeting NR2A-containing NMDA receptors reduces L-DOPA-induced dyskinesias. Neurobiol Aging. 2012;33:2138–2144. doi: 10.1016/j.neurobiolaging.2011.06.019. [DOI] [PubMed] [Google Scholar]
- Gerlach M, Double K, Arzberger T, Leblhuber F, Tatschner T, Riederer P. Dopamine receptor agonists in current clinical use: comparative dopamine receptor binding profiles defined in the human striatum. J Neural Transm. 2003;110:1119–1127. doi: 10.1007/s00702-003-0027-5. [DOI] [PubMed] [Google Scholar]
- Gottwald MD, Aminoff MJ. Therapies for dopaminergic-induced dyskinesias in Parkinson disease. Ann Neurol. 2011;69:919–927. doi: 10.1002/ana.22423. [DOI] [PubMed] [Google Scholar]
- Guigoni C, Doudnikoff E, Li Q, Bloch B, Bezard E. Altered D(1) dopamine receptor trafficking in parkinsonian and dyskinetic non-human primates. Neurobiol Dis. 2007;26:452–463. doi: 10.1016/j.nbd.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Hauser RA, Rascol O, Korczyn AD, Jon Stoessl A, Watts RL, Poewe W, De Deyn PP, Lang AE. Ten-year follow-up of Parkinson's disease patients randomized to initial therapy with ropinirole or levodopa. Mov Disord. 2007;22:2409–2417. doi: 10.1002/mds.21743. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Huganir RL. Tyrosine phosphorylation and regulation of the AMPA receptor by SRC family tyrosine kinases. J Neurosci. 2004;24:6152–6160. doi: 10.1523/JNEUROSCI.0799-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Ashby MC, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Jenkinson C, Dummett S, Kelly L, Peters M, Dawson J, Morley D, Fitzpatrick R. The development and validation of a quality of life measure for the carers of people with Parkinson's disease (the PDQ-Carer) Parkinsonism Relat Disorders. 2012;18:483–487. doi: 10.1016/j.parkreldis.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Jenner P. Molecular mechanisms of L-DOPA-induced dyskinesia. Nat Rev Neurosci. 2008;9:665–677. doi: 10.1038/nrn2471. [DOI] [PubMed] [Google Scholar]
- Jenner P, McCreary AC, Scheller DK. Continuous drug delivery in early- and late-stage Parkinson's disease as a strategy for avoiding dyskinesia induction and expression. J Neural Transm. 2011;118:1691–1702. doi: 10.1007/s00702-011-0703-9. [DOI] [PubMed] [Google Scholar]
- Kobylecki C, Cenci MA, Crossman AR, Ravenscroft P. Calcium-permeable AMPA receptors are involved in the induction and expression of l-DOPA-induced dyskinesia in Parkinson's disease. J Neurochem. 2010;114:499–511. doi: 10.1111/j.1471-4159.2010.06776.x. [DOI] [PubMed] [Google Scholar]
- Lebel M, Chagniel L, Bureau G, Cyr M. Striatal inhibition of PKA prevents levodopa-induced behavioural and molecular changes in the hemiparkinsonian rat. Neurobiol Dis. 2010;38:59–67. doi: 10.1016/j.nbd.2009.12.027. [DOI] [PubMed] [Google Scholar]
- Lees AJ, Hardy J, Revesz T. Parkinson's disease. Lancet. 2009;373:2055–2066. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- Lundblad M, Andersson M, Winkler C, Kirik D, Wierup N, Cenci MA. Pharmacological validation of behavioural measures of akinesia and dyskinesia in a rat model of Parkinson's disease. Eur J Neurosci. 2002;15:120–132. doi: 10.1046/j.0953-816x.2001.01843.x. [DOI] [PubMed] [Google Scholar]
- Marcellino D, Ferré S, Casadó V, Cortés A, Le Foll B, Mazzola C, Drago F, Saur O, Stark H, Soriano A, Barnes C, Goldberg SR, Lluis C, Fuxe K, Franco R. Identification of dopamine D1–D3 receptor heteromers. Indications for a role of synergistic D1–D3 receptor interactions in the striatum. J Biol Chem. 2008;283:26016–26025. doi: 10.1074/jbc.M710349200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishina M, Sakimura K, Mori H, Kushiya E, Harabayashi M, Uchino S, Nagahari K. A single amino acid residue determines the Ca2+ permeability of AMPA-selective glutamate receptor channels. Biochem Biophys Res Commun. 1991;180:813–821. doi: 10.1016/s0006-291x(05)81137-4. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Olanow CW, Nutt JG. Levodopa motor complications in Parkinson's disease. Trends Neurosci. 2000;23:S2–S7. doi: 10.1016/s1471-1931(00)00031-8. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Obeso JA, Stocchi F. Continuous dopamine-receptor treatment of Parkinson's disease: scientific rationale and clinical implications. Lancet Neurol. 2006;5:677–687. doi: 10.1016/S1474-4422(06)70521-X. [DOI] [PubMed] [Google Scholar]
- Ozawa S, Kamiya H, Tsuzuki K. Glutamate receptors in the mammalian central nervous system. Prog Neurobiol. 1998;54:581–618. doi: 10.1016/s0301-0082(97)00085-3. [DOI] [PubMed] [Google Scholar]
- Paillé V, Henry V, Lescaudron L, Brachet P, Damier P. Rat model of Parkinson's disease with bilateral motor abnormalities, reversible with levodopa, and dyskinesias. Mov Disord. 2007;22:533–539. doi: 10.1002/mds.21308. [DOI] [PubMed] [Google Scholar]
- Paillé V, Picconi B, Bagetta V, Ghiglieri V, Sgobio C, Di Filippo M, Viscomi MT, Giampà C, Fusco FR, Gardoni F, Bernardi G, Greengard P, Di Luca M, Calabresi P. Distinct levels of dopamine denervation differentially alter striatal synaptic plasticity and NMDA receptor subunit composition. J Neurosci. 2010;30:14182–14193. doi: 10.1523/JNEUROSCI.2149-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini-Giampietro DE, Gorter JA, Bennett MV, Zukin RS. The GluR2 (GluR-B) hypothesis: Ca(2+)-permeable AMPA receptors in neurological disorders. Trends Neurosci. 1997;20:464–470. doi: 10.1016/s0166-2236(97)01100-4. [DOI] [PubMed] [Google Scholar]
- Picconi B, Centonze D, Håkansson K, Bernardi G, Greengard P, Fisone G, Cenci MA, Calabresi P. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat Neurosci. 2003;6:501–506. doi: 10.1038/nn1040. [DOI] [PubMed] [Google Scholar]
- Picconi B, Centonze D, Rossi S, Bernardi G, Calabresi P. Therapeutic doses of L-dopa reverse hypersensitivity of corticostriatal D2-dopamine receptors and glutamatergic overactivity in experimental parkinsonism. Brain. 2004;127:1661–1669. doi: 10.1093/brain/awh190. [DOI] [PubMed] [Google Scholar]
- Picconi B, Bagetta V, Ghiglieri V, Paillè V, Di Filippo M, Pendolino V, Tozzi A, Giampà C, Fusco FR, Sgobio C, Calabresi P. Inhibition of phosphodiesterases rescues striatal long-term depression and reduces levodopa-induced dyskinesia. Brain. 2011;134:375–387. doi: 10.1093/brain/awq342. [DOI] [PubMed] [Google Scholar]
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson's disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med. 2000;342:1484–1491. doi: 10.1056/NEJM200005183422004. [DOI] [PubMed] [Google Scholar]
- Rascol O, Lozano A, Stern M, Poewe W. Milestones in Parkinson's disease therapeutics. Mov Disord. 2011;26:1072–1082. doi: 10.1002/mds.23714. [DOI] [PubMed] [Google Scholar]
- Rinne UK, Bracco F, Chouza C, Dupont E, Gershanik O, Marti Masso JF, Montastruc JL, Marsden CD. Early treatment of Parkinson's disease with cabergoline delays the onset of motor complications. Results of a double-blind levodopa controlled trial. The PKDS009 Study Group. Drugs. 1998;55(Suppl 1):23–30. doi: 10.2165/00003495-199855001-00004. [DOI] [PubMed] [Google Scholar]
- Santini E, Valjent E, Usiello A, Carta M, Borgkvist A, Girault JA, Hervé D, Greengard P, Fisone G. Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in l-DOPA-induced dyskinesia. J Neurosci. 2007;27:6995–7005. doi: 10.1523/JNEUROSCI.0852-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarting RK, Huston JP. The unilateral 6-hydroxydopamine lesion model in behavioral brain research. Analysis of functional deficits, recovery and treatments. Prog Neurobiol. 1996;50:275–331. doi: 10.1016/s0301-0082(96)00040-8. [DOI] [PubMed] [Google Scholar]
- Tang K, Low MJ, Grandy DK, Lovinger DM. Dopamine-dependent synaptic plasticity in striatum during in vivo development. Proc Natl Acad Sci U S A. 2001;98:1255–1260. doi: 10.1073/pnas.031374698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanLeeuwen JE, Petzinger GM, Walsh JP, Akopian GK, Vuckovic M, Jakowec MW. Altered AMPA receptor expression with treadmill exercise in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned mouse model of basal ganglia injury. J Neurosci Res. 2010;88:650–668. doi: 10.1002/jnr.22216. [DOI] [PubMed] [Google Scholar]
- Varanese S, Howard J, Di Rocco A. NMDA antagonist memantine improves levodopa-induced dyskinesias and “on-off” phenomena in Parkinson's disease. Mov Disord. 2010;25:508–510. doi: 10.1002/mds.22917. [DOI] [PubMed] [Google Scholar]
- Vastagh C, Gardoni F, Bagetta V, Stanic J, Zianni E, Giampà C, Picconi B, Calabresi P, Di Luca M. NMDA receptor composition modulates dendritic spine morphology in striatal medium spiny neurons. J Biol Chem. 2012;287:18103–18114. doi: 10.1074/jbc.M112.347427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visanji NP, Fox SH, Johnston T, Reyes G, Millan MJ, Brotchie JM. Dopamine D3 receptor stimulation underlies the development of L-DOPA-induced dyskinesia in animal models of Parkinson's disease. Neurobiol Dis. 2009;35:184–192. doi: 10.1016/j.nbd.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Voon V, Fernagut PO, Wickens J, Baunez C, Rodriguez M, Pavon N, Juncos JL, Obeso JA, Bezard E. Chronic dopaminergic stimulation in Parkinson's disease: from dyskinesias to impulse control disorders. Lancet Neurol. 2009;8:1140–1149. doi: 10.1016/S1474-4422(09)70287-X. [DOI] [PubMed] [Google Scholar]
- Watts RL, Lyons KE, Pahwa R, Sethi K, Stern M, Hauser RA, Olanow W, Gray AM, Adams B, Earl NL, Earl NL. Onset of dyskinesia with adjunct ropinirole prolonged-release or additional levodopa in early Parkinson's disease. Mov Disord. 2010;25:858–866. doi: 10.1002/mds.22890. [DOI] [PubMed] [Google Scholar]