

Abstract
γ-Secretase inhibitors (GSIs) reduce amyloid-β (Aβ) peptides but inevitably increase the β-C-terminal fragment (β-CTF) of amyloid precursor protein (APP), potentially having undesirable effects on synapses. In contrast, γ-secretase modulators (GSMs) reduce Aβ42 without increasing β-CTF. Although the Aβ-lowering effects of these compounds have been extensively studied, little effort has been made to investigate their effects on cognition. Here, we compared the effects of two GSIs—(2S)-2-hydroxy-3-methyl-N-[(2S)-1-{[(1S)-3-methyl-2-oxo-2,3,4,5-tetrahydro-1H-3-benzazepin-1-yl]amino}-1-oxopropan-2-yl]butanamide (LY450139, semagacestat) and (2R)-2-[[(4-chlorophenyl)sulfonyl][[2-fluoro-4-(1,2,4-oxazol-3-yl)phenyl]methyl]amino-5,5,5-trifluoropentanamide (BMS-708163)—and a second-generation GSM [{(2S,4R)-1-[(4R)-1,1,1-trifluoro-7-methyloctan-4-yl]-2-[4-(trifluoromethyl)phenyl]piperidin-4-yl}acetic acid (GSM-2)] on spatial working memory in APP-transgenic (Tg2576) and nontransgenic mice using the Y-maze task. While acute dosing with either GSI ameliorated memory deficits in 5.5-month-old Tg2576 mice, these effects disappeared after 8 d subchronic dosing. Subchronic dosing with either GSI rather impaired normal cognition in 3-month-old Tg2576 mice, with no inhibition on the processing of other γ-secretase substrates, such as Notch, N-cadherin, or EphA4, in the brain. LY450139 also impaired normal cognition in wild-type mice; however, the potency was 10-fold lower than that in Tg2576 mice, indicating an APP-dependent mechanism likely with β-CTF accumulation. Immunofluorescence studies revealed that the β-CTF accumulation was localized in the presynaptic terminals of the hippocampal stratum lucidum and dentate hilus, implying an effect on presynaptic function in the mossy fibers. In contrast, both acute and subchronic dosing with GSM-2 significantly ameliorated memory deficits in Tg2576 mice and did not affect normal cognition in wild-type mice. We demonstrated a clear difference between GSI and GSM in effects on functional consequences, providing new insights into strategies for developing these drugs against Alzheimer's disease.
Introduction
Although brain amyloid-β (Aβ) plaque is a pathological hallmark of Alzheimer's disease (AD), actual correlation with memory dysfunction is quite weak. Instead, soluble Aβ level has been found to correlate with disease severity or decline in synaptic density in AD patients (Lue et al., 1999; McLean et al., 1999). Indeed, mutated amyloid precursor protein (APP) transgenic mice have shown cognitive deficits before plaque appearance (Holcomb et al., 1999; Van Dam et al., 2003). Aβ is produced by the sequential proteolytic cleavage of APP. The first cleavage by β-secretase generates the β-C-terminal fragment of APP (β-CTF) (otherwise known as C99) (Sinha et al., 1999; Vassar et al., 1999; Yan et al., 1999), and the second cleavage is induced by γ-secretase (De Strooper et al., 1998; Wolfe et al., 1999). Extensive research efforts over the past decade have identified a number of γ-secretase inhibitors (GSIs) and modulators (GSMs), and their Aβ-lowering effects have been well investigated (Kreft et al., 2009; Oehlrich et al., 2011). However, whether or not these compounds have any effects on improving cognitive function remains poorly understood.
γ-Secretase also cleaves Notch protein and this cleavage is indispensable to activation of Notch signaling (De Strooper et al., 1999; Struhl and Greenwald, 1999). For clinical application of GSIs, researchers have made considerable efforts to dissociate the inhibitory potency of compounds against Notch signaling, which causes risk of developing gastrointestinal and immune disorders (Milano et al., 2004; Wong et al., 2004), from their Aβ-lowering potency. While these compounds also have a tendency to increase β-CTF, this effect has not been regarded as a crucial issue, although several research groups have pointed out its detrimental effects on neurons (Oster-Granite et al., 1996; Nalbantoglu et al., 1997; Suh et al., 2000). A recent report demonstrated that GSIs reduce the dendritic spine density in normal mice but not in APP-knock-out mice, suggesting that the accumulation of APP-CTF (either or both β- and α-CTF) may be synaptotoxic (Bittner et al., 2009).
GSMs, first identified from a subset of nonsteroidal antiinflammatory drugs (Weggen et al., 2001; Eriksen et al., 2003), inhibit the production of Aβ42 without affecting Aβ40 or total Aβ and without increasing β-CTF (Weggen et al., 2003; Kounnas et al., 2010). Although Aβ42 is believed to be a culprit in neuropathology and synaptotoxicity with AD (Jarrett et al., 1993; Iwatsubo et al., 1994; Younkin, 1998; Mucke et al., 2000; McGowan et al., 2005; Lacor et al., 2007; Kuperstein et al., 2010), it remains to be confirmed that a decrease in only Aβ42 but not in total Aβ should actually result in behavioral improvement.
Here, we examined the effects of GSI and GSM on cognitive function in an animal model using the Y-maze task. We directly compared two structurally different GSIs—(2S)-2-hydroxy-3-methyl-N-[(2S)-1-{[(1S)-3-methyl-2-oxo-2,3,4,5-tetrahydro-1H-3-benzazepin-1-yl]amino}-1-oxopropan-2-yl]butanamide (LY450139, semagacestat) (Henley et al., 2009) and (2R)-2-[[(4-chlorophenyl)sulfonyl][[2-fluoro-4-(1,2,4-oxazol-3-yl)phenyl]methyl]amino-5,5,5-trifluoropentanamide (BMS-708163, avagacestat; known as a Notch-sparing GSI) (Gillman et al., 2010)—and a second-generation GSM, {(2S,4R)-1-[(4R)-1,1,1-trifluoro-7-methyloctan-4-yl]-2-[4-(trifluoromethyl)phenyl]piperidin-4-yl}acetic acid (GSM-2) [Garcia Y, Hannam JC, Harrison T, Hamblett CL, Hubbs JL, Kulagowski JJ, Madin A, Ridgill MP, Seward E (2007) International Patent Application WO2007/125364, pending], with respect to their behavioral and neurochemical effects in both APP-transgenic (Tg2576) and wild-type (WT) mice.
Materials and Methods
Drugs.
LY450139, BMS-708163, and GSM-2 were synthesized at Astellas Pharma, Inc. (Tsukuba, Japan), according to the literature (Pu et al., 2008; Gillman et al., 2010) or patent application (Garcia et al., 2007). The chemical structures of the drugs are shown in Figure 1.
Figure 1.

Chemical structures of LY450139, BMS-708163, and GSM-2.
Cellular APP processing assay.
H4 human glioma cells stably overexpressing human wild-type APP695 were maintained in DMEM supplemented with 10% fetal bovine serum and penicillin/streptomycin. Cells were cultured in 96- or 6-well plates overnight, and then treated with each drug at various concentrations for 24 h. Levels of Aβ1-42, Aβ1-40, and Aβ1-38 in the media were measured using separate ELISA kits (Aβ1-42; Wako; Aβ1-40 and Aβ1-38; IBL).
To quantify β-CTF, cells were lysed with RIPA buffer (25 mm Tris, 150 mm NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS; pH 7.6) containing Complete protease inhibitor mixture (Roche Diagnostics) and applied to a human β-CTF ELISA kit (IBL) at 1:20 dilution. Aliquots of the cell lysate were also used for CellTiter-Glo Luminescent Cell Viability Assay (Promega). The cell lysate from the six-well plate was subjected to Western blot analysis.
Cellular Notch signaling assay.
The expression vector of the constitutively active form of Notch (NotchΔE), encoding bases 1–60 and 5193–6657 of the human Notch1 coding region (NM_017617), was constructed into a pcDNA3.1 vector as described previously, with a sequence modification from mouse to human (Kopan et al., 1996). Notch signaling activity was evaluated using Cignal RBP-Jk Reporter Assay kit (SA Biosciences). RBP-Jk protein [CSL/CBF1/Su(H)/Lag1] is a transcription factor activated with Notch intracellular domain produced by γ-secretase. H4 cells were transiently transfected with the human NotchΔE expression vector and the RBP-Jk-responsive luciferase construct using Lipofectamine 2000 (Invitrogen), and then exposed to various concentrations of each drug for 16 h. Notch signaling was measured based on luciferase activity in the cell lysate using the Dual-Glo Luciferase Assay System (Promega).
Animals.
Female Tg2576 mice expressing human APP695 with the Swedish mutation (K670N/M671L) were used (Hsiao et al., 1996). Male transgenic mice were procured through Mayo Foundation for Medical Education and Research (Rochester, MN) and crossbred with female B6SJLF1/J mice at Charles River Laboratories. The offspring of this breeding were then transferred to Astellas Pharma, Inc., and housed in an American Association for Accreditation of Laboratory Animal Care (AAALAC)-approved facility in a temperature- and humidity-controlled colony room (maintained at 23 ± 3°C and 55 ± 15%, respectively) under a 12 h light/dark cycle with water and laboratory chow supplied ad libitum. All in vivo experimental procedures were performed during the light cycle.
Procedures involving animals and their care were conducted in accordance with AAALAC guidelines and Astellas Pharma, Inc., guidelines for the care and use of animals under approved protocols from the Institutional Animal Care and Use Committee of Astellas Pharma, Inc.
Y-maze test.
Spatial working memory in mice was evaluated by recording spontaneous alternation behavior in the Y-maze task (Hsiao et al., 1996). The maze was made of gray polyvinylchloride, with three arms converging at equal angles. Arm dimensions were 40 cm in length, 13 cm in height, and 3 and 10 cm in width at the bottom and top, respectively. Each drug was dissolved or suspended in 0.5% methyl cellulose and orally administered once daily for 8 d. Three hours after administration on either or both days 1 and 8, each animal was placed at the end of one arm and allowed to freely explore the apparatus for 8 min. The sequence and number of all arm entries were recorded for each animal throughout the period. The sequence triads, in which all three arms were represented (ABC, ACB, BAC, BCA, CAB, and CBA), were calculated as successful alternations to evaluate the normal cognition and working memory of the last arm entered. Alternation rate was defined as entries into all three arms on consecutive occasions using the following formula: Alternation rate (%) = Number of alternations/(Number of total arm entries − 2) × 100. Data were eliminated in cases in which the number of total arm entries was <10.
Quantitation of hippocampal Aβ and β-CTF levels.
Immediately after the Y-maze test, animals were decapitated, and the hippocampus was isolated on ice followed by snap freezing with liquid nitrogen and storage at −80°C until use. Frozen samples were homogenized in an ultrasonic homogenizer with a 10-fold volume of TBS (25 mm Tris, 137 mm NaCl, 2.68 mm KCl; pH 7.4) containing Complete protease inhibitor mixture (Roche Diagnostics) on ice followed by ultracentrifugation (100,000 × g, 1 h, 4°C). Levels of Aβx-42 and Aβx-40 in the supernatant were measured using ELISA kits (Wako). To quantify human β-CTF in Tg2576 mice, aliquots of the homogenate were solubilized with 2% SDS and applied to a human β-CTF ELISA kit (IBL) at 1:730 dilution. Mouse endogenous β-CTF in WT mice was also quantified using the human β-CTF ELISA kit at 1:5 dilution. Recombinant mouse β-CTF provided by IBL was used to generate a standard curve instead of the recombinant human β-CTF attached to the kit. SDS was added to the standard samples so that the final SDS concentration (0.33%) was the same as that in the diluted WT mouse samples. Although this concentration of SDS reduced assay performance by ∼30%, quantification was successfully achieved with a log-log linear-fitting standard curve (r = 0.99). The basal levels of Aβx-42, Aβx-40, and β-CTF measured with the procedure described above were 1.9 ± 0.1, 11.6 ± 0.6, and 21.2 ± 2.5 pmol/g tissue, respectively, in Tg2576 mice (mean ± SEM for nine independent experiments), and 0.12 ± 0.00, 0.72 ± 0.02, and 0.48 ± 0.03 pmol/g tissue, respectively, in WT mice (mean ± SEM for six independent experiments). Aliquots of the homogenate from Tg2576 mice were subjected to Western blot analysis.
Quantitation of Notch-target gene expression in vivo.
Three hours after orally administering each drug to Tg2576 mice, the hippocampus and thymus were isolated and stored at −80°C until use. Tissues were homogenized in lysis buffer and processed for total RNA extraction using the QIAGEN RNeasy 96-well plate technology with on-column DNase I treatment. Total RNA from each sample was used to generate cDNA using a SuperScript III (Invitrogen) with random primers. Hes1 and Hey1 mRNA levels were quantified via SYBR Green quantitative PCR assay, and results were normalized using an endogenous reference gene, β-actin. The sequences of the primers used in the SYBR Green assay were as follows: Hes1, forward primer: CCAGCCAGTGTCAACACGA; Hes1, reverse primer: AATGCCGGGAGCTATCTTTCT; Hey1, forward primer: GCGCGGACGAGAATGGAAA; Hey1, reverse primer: TCAGGTGATCCACAGTCATCTG; β-actin, forward primer: CGTGAAAAGATGACCCAGATC; β-actin, reverse primer: GCCTGGATGGCTACGTACATG. The oligonucleotides and Power SYBR Green PCR Master Mix were purchased from Invitrogen and Applied Biosystems, respectively.
Western blot analysis.
The following antibodies were used for immunoblotting: anti-Aβ N-terminal-specific mouse monoclonal antibody (diluted 1:100; clone 82E1; IBL), anti-Aβ1-16 mouse monoclonal antibody (diluted 1:1000; clone 6E10; Covance), anti-APP C-terminal rabbit polyclonal antibody (diluted 1:1000 or 5000; A8717; Sigma-Aldrich), anti-N-cadherin mouse monoclonal antibody (diluted 1:200; 610920; BD Biosciences), anti-EphA4 mouse monoclonal antibody (diluted 1:200; clone 6H7; Merck Chemicals), and anti-β-tubulin mouse monoclonal antibody (1:1000 or 5000; clone AA2; Millipore).
Either the cell lysate or brain homogenate described above was mixed with Laemmli buffer and boiled for 5 min, and then separated by SDS-PAGE on 15% gels. Proteins were transferred to PVDF membranes and labeled with the antibodies listed above, followed by incubation with HRP-conjugated secondary antibodies. Bands were visualized using ECL and quantitated densitometrically with Quantity One 4.6.5 software (Bio-Rad).
Immunofluorescence study.
The following antibodies were used for immunostaining: anti-Aβ N-terminal-specific mouse monoclonal antibody (diluted 1:200; clone 82E1; IBL), anti-APP C-terminal rabbit polyclonal antibody (diluted 1:2000; A8717; Sigma-Aldrich), anti-synaptophysin rabbit monoclonal antibody (diluted 1:500; clone YE269; Millipore), and anti-drebrin A rabbit polyclonal antibody (diluted 1:200; 28023; IBL).
While under isoflurane anesthesia, 10 mm PBS, pH 7.4, was perfused into the left ventricle of each animal. The brain was quickly hemisected and immersion-fixed in 4% paraformaldehyde/phosphate buffer for 2 d at 4°C, and then placed in 16% sucrose/phosphate buffer for 2 d at 4°C. The brain was frozen and cut into 20-μm-thick coronal sections with a cryostat. One section per mouse between −1.70 and −1.94 mm from the bregma was selected and incubated with the antibodies listed above free-floating for 16 h at room temperature. Antibodies were detected with Alexa Fluor 488-conjugated anti-mouse IgG or Alexa Fluor 546-conjugated anti-rabbit IgG (Invitrogen) using an ECLIPSE 80i microscope (Nikon) equipped with an ORCA-ER digital camera (Hamamatsu). Double-stained sections were observed using an LSM5Pascal confocal laser scan microscope (Carl Zeiss). Immunofluorescence intensity in the following areas (16 × 750 μm each, except dentate hilus) was quantified using NIH ImageJ 1.44p: the stratum pyramidale, radiatum, and lacunosum-moleculare of the CA1b region; the distal side of the stratum oriens of the CA1c region; the stratum pyramidale and lucidum of the CA3a to 3b region; the stratum granulare and moleculare; and the whole area of dentate hilus. Immunofluorescence intensity in each area was normalized with that in the CA3 stratum pyramidale of the same section as an internal control to minimize nonspecific variations in signal intensity (Palop et al., 2011). Absolute intensity in the CA3 stratum pyramidale was not altered by drug treatment.
Plasma and brain concentration of LY450139.
Plasma and brain samples were collected at 3 h after drug administration and stored at −80°C until use. LY450139 concentrations in plasma and brain were quantified via HPLC/mass spectrometry assay. Briefly, aliquots of plasma samples and brain homogenates were treated by protein precipitation with acetonitrile containing appropriate internal standard followed by centrifugation. The supernatant was injected onto a LaChrom Elite HPLC system coupled to a NanoFlontier LD linear ion trap time-of-flight mass spectrometer (Hitachi High-Technologies). The HPLC column was an ODS-3 column (2.0 × 50 mm, 5 μm; GL Sciences). The mobile phase was a gradient mixture of 0.1% formic acid and acetonitrile.
Statistical analysis.
Data are presented as mean ± SEM, and all statistical analyses were conducted using SAS software (SAS Institute). Dunnett's multiple-comparison test was used to compare multiple doses in the drug-treated groups with the vehicle-treated group. An unpaired Student's t test was used to compare the single-dose drug-treated group with the vehicle-treated group, and the Tg2576 group with the WT group. For all tests, a value of p < 0.05 was considered statistically significant.
Results
Drug effects on APP processing and Notch signaling in cultured cells
To characterize drug effects on APP processing in vitro, we used H4 cells stably overexpressing human wild-type APP. LY450139 and BMS-708163 reduced the secretion of Aβ42, Aβ40, and Aβ38 in 96-well-cultured media and increased β-CTF in cell lysates as expected, although this increase was unexpectedly attenuated at high concentrations (Fig. 2A). In contrast, GSM-2 only decreased Aβ42, while inversely increasing Aβ38 and having no effect on β-CTF levels. Aβ40 levels were only slightly decreased at high concentrations. No drug affected cell viability at any of the tested concentrations (data not shown). IC50, EC50, and ECmax values are shown in Table 1.
Figure 2.

Drug effects on APP processing and Notch signaling in cells. A, APP-overexpressing H4 cells were exposed to various concentrations of each drug for 24 h in 96-well plates. Aβ42, Aβ40, and Aβ38 secreted in the culture medium (left axis) and β-CTF in the cell lysate (right axis) were measured via ELISA. B, The cells were exposed to 3.2, 16, 80, 2000, and 10,000 nm LY450139 for 24 h in a six-well plate and analyzed by Western blotting with 82E1, 6E10, and anti-APP C-terminal antibody (APP-C). APP-FL, Full-length APP. C, H4 cells transfected with NotchΔE and RBP-Jk-responsive luciferase were exposed to various concentrations of each drug for 16 h. Activity in the Notch signal transduction pathway was measured as luminescence in the cell lysate. Data in A and C are presented as mean ± SEM of three independent experiments. IC50, EC50, or ECmax values are presented in Table 1.
Table 1.
Drug effects on APP processing and Notch signaling in cells
| LY450139 (nm) | BMS-708163 (nm) | GSM-2 (nm) | |
|---|---|---|---|
| Aβ42 | |||
| IC50 | 10.9 ± 0.6 | 0.79 ± 0.15 | 65.2 ± 2.3 |
| Aβ40 | |||
| IC50 | 12.1 ± 0.5 | 0.78 ± 0.09 | >2000 |
| Aβ38 | |||
| IC50 | 12.0 ± 0.3 | 0.66 ± 0.02 | — |
| EC50 | — | — | 81.3 ± 6.0 |
| β-CTF | |||
| ECmaxa | 16.0 ± 0.0 | 0.80 ± 0.00 | No change |
| Notch | |||
| IC50 | 14.1 ± 2.5 | 20.6 ± 1.3 | >3000 |
| Notch/Aβ42 | 1.3 | 26 | >46 |
Values are shown as mean ± SEM for three independent experiments.
aConcentration at which maximum increases in β-CTF were observed.
The biphasic β-CTF response to LY450139 was also found in Western blot analysis of six-well-cultured cells using two antibodies, 82E1 and 6E10, which recognize the N-terminal region of Aβ. In contrast, α-CTF detected with an anti-APP C-terminal antibody showed a simple upward sigmoid response (Fig. 2B). We were unable to conduct β-CTF analysis with this antibody due to interference by the strong α-CTF band.
Drug effects on Notch signaling were evaluated using the Notch intracellular domain reporter gene assay system with human NotchΔE-transfected H4 cells. LY450139 and BMS-708163 inhibited Notch signaling at similar potencies, while GSM-2 had no effect (Fig. 2C). While LY450139 showed minimal Notch-sparing selectivity (Notch IC50/Aβ42 IC50), BMS-708163 and GSM-2 demonstrated 26- and >46-fold selectivity, respectively (Table 1).
Acute and subchronic drug effects on cognitive deficits in Tg2576 mice (Experiment 1)
To identify drug effects on cognitive function, we conducted four different experiments. The objective of Experiment 1 was to elucidate acute and subchronic drug effects on cognitive deficits in Tg2576 mice. Each drug was orally administered to 5.5-month-old Tg2576 mice for 8 d. Y-maze tests were conducted to evaluate spatial working memory 3 h after administration on days 1 and 8. Vehicle-treated Tg2576 mice demonstrated significantly lower spontaneous alternation rates than WT mice in the Y-maze test, suggesting deficits in spatial working memory. On day 1, 1 mg/kg LY450139, 1 mg/kg BMS-708163, and 0.1–0.3 mg/kg GSM-2 significantly ameliorated these cognitive deficits (acute effects). On day 8, however, the GSI effects disappeared, whereas GSM-2 retained its significant effects (subchronic effects) (Fig. 3A).
Figure 3.

Acute and subchronic drug effects on cognitive deficits in Tg2576 mice (Experiment 1). A, Each drug was orally administered to Tg2576 mice (aged 5.5 months) for 8 d, and Y-maze tests were conducted on days 1 and 8. LY450139 and BMS-708163 significantly ameliorated cognitive deficits in Tg2576 mice on day 1, but not on day 8. In contrast, GSM-2 caused significant improvements on both days 1 and 8. Dotted lines, Chance level of alternation behavior. B, Hippocampal Aβ and β-CTF on days 1 and 8 were measured via ELISA. LY450139 and BMS-708163 significantly increased β-CTF, whereas GSM-2 did not. The effects of each drug on Aβ and β-CTF levels were similar between days 1 and 8. ##p < 0.01 by Student's t test. *p < 0.05; **p < 0.01 compared with vehicle group by Dunnett's test. All data are presented as mean ± SEM (n = 7 or 8).
Mice were killed immediately after the Y-maze test on day 8, when hippocampal levels of Aβ42, Aβ40, and β-CTF were determined by ELISA. LY450139 was found to decrease both Aβ42 and Aβ40 at 10 mg/kg (22–23% reduction; p < 0.01) and increase β-CTF at 0.3–10 mg/kg in a dose-dependent manner (15–162% elevation; p < 0.01 at 1–10 mg/kg). BMS-708163 decreased neither Aβ42 nor Aβ40 up to 10 mg/kg but increased β-CTF at 1–10 mg/kg (11–50% elevation; p < 0.01 at 3–10 mg/kg). GSM-2 decreased Aβ42 at 0.1–3 mg/kg (5–18% reduction; p < 0.05 at 0.3 mg/kg, p < 0.01 at 1–3 mg/kg) without affecting Aβ40 or β-CTF levels (Fig. 3B). Given the distinct difference in behavioral effects between acute and subchronic GSI dosing, the three drugs were acutely administered into another set of Tg2576 mice that were killed on day 1, and Aβ and β-CTF levels in these animals were compared with those in the subchronic study. No apparent differences were observed between the two days.
Subchronic drug effects on cognitive deficits in Tg2576 mice at doses that robustly reduce hippocampal Aβ (Experiment 2)
To confirm that subchronic GSI dosing was not effective against cognitive deficits in Tg2576 mice, Y-maze tests were conducted again, this time using a dose range that robustly reduces hippocampal Aβ levels. LY450139 and BMS-708163 were administered to 5.5-month-old Tg2576 mice for 8 d at 3–30 and 6–60 mg/kg, respectively. On day 8, Y-maze tests were conducted 3 h after administration, followed by killing and neurochemical measurements. Hippocampal Aβ42 and Aβ40 levels were markedly decreased, with maximum reductions of 36 and 41% with LY450139, and 43 and 56% with BMS-708163, respectively. Even then, both GSIs failed to restore cognitive deficits in the Y-maze test (Fig. 4). It should be noted that this lack of efficacy was not due to potential worsening of general body condition after multiple dosing with high doses of the GSIs, as we noted no marked differences in the number of total arm entries, body weights, or apparent general condition between the vehicle- and drug-treated groups (data not shown).
Figure 4.

Subchronic drug effects on cognitive deficits in Tg2576 mice at high doses that robustly reduce hippocampal Aβ levels (Experiment 2). A, Each drug was orally administered to Tg2576 mice (aged 5.5 months) for 8 d, and Y-maze tests were conducted on day 8. Only GSM-2 ameliorated the cognitive deficit in Tg2576 mice. B, All three drugs robustly reduced hippocampal Aβ42 levels. GSM-2 did not affect β-CTF levels even at the highest dose. ##p < 0.01 by Student's t test. *p < 0.05; **p < 0.01 compared with vehicle group by Dunnett's test. All data are presented as mean ± SEM (n = 7 or 8).
High doses of GSM-2 were also tested. GSM-2 was administered to Tg2576 mice at 0.03–30 mg/kg in 10-fold increments for 8 d. The broad dose range was chosen because of the shallow slope of the Aβ-lowering dose–efficacy curve compared with that of GSIs. A robust reduction in hippocampal Aβ42 with a maximum decrease of 51% was observed, but not so for Aβ40. GSM-2 significantly ameliorated cognitive deficits in the Y-maze test at 0.3–30 mg/kg (Fig. 4).
Finally, hippocampal β-CTF levels were increased dose-dependently with LY450139 and BMS-708163 administration, with maximum increases of 194 and 149%, respectively; in contrast, values were unchanged with GSM-2 dosing.
Acute and subchronic effects of GSIs on normal cognitive function in younger Tg2576 mice (Experiment 3)
To explore why subchronic treatment with the GSIs failed to restore cognitive deficits, we examined the effect of the GSIs on normal cognitive function in younger Tg2576 mice that had yet to show cognitive deficits. LY450139 or BMS-708163 was administered to 3-month-old Tg2576 mice for 8 d, with Y-maze tests conducted on days 1 and 8. No significant differences were found in the alternation rate between the vehicle-treated Tg2576 mice and WT mice, suggesting that spatial working memory was not impaired at this age. While neither GSI affected the normal alternation rate on day 1, both GSIs significantly and dose-dependently reduced the rate on day 8 (p < 0.01 at 3 mg/kg LY450139; p < 0.05 at 10 mg/kg BMS-708163), indicating that subchronic treatment with GSIs impairs spatial working memory (Fig. 5A). This result may explain why subchronic treatment with the GSIs failed to restore cognitive deficits in 5.5-month-old Tg2576 mice.
Figure 5.

Acute and subchronic effects of GSIs on normal cognitive function in younger Tg2576 mice (Experiment 3). A, Each drug was orally administered to Tg2576 mice (aged 3 months) for 8 d, and Y-maze tests were conducted on days 1 and 8. Both GSIs significantly and dose-dependently impaired normal cognitive function on day 8 but not on day 1. B, GSI-treated mice showed significant and dose-dependent increases in hippocampal β-CTF levels on day 8. *p < 0.05; **p < 0.01 compared with vehicle group by Dunnett's test. All data are presented as mean ± SEM (n = 7 or 8).
Hippocampal Aβ levels in young Tg2576 mice were not affected at 3 mg/kg LY450139 but were reduced at 10 mg/kg BMS-708163. Hippocampal β-CTF levels were dose-dependently increased at 1–3 mg/kg LY450139 (21–62% elevation) and 3–10 mg/kg BMS-708163 (18–63% elevation), appearing to correlate with the decrease in alternation rate (Fig. 5B). The β-CTF-increasing efficacy of the GSIs was almost the same as that observed in 5.5-month-old mice in Experiment 1.
Effects of GSIs on Notch signaling and processing of other γ-secretase substrates in vivo
To address the relationship between GSI-induced cognitive impairment and Notch signaling inhibition, the effects of the GSIs on Notch-target gene expression were examined in vivo. Acute dosing with 30 mg/kg LY450139 to Tg2576 mice (aged 4 months) significantly decreased Hes1 and Hey1 mRNA levels in the thymus, indicating significant Notch signaling inhibition in the thymus. However, we noted no change in mRNA levels of either protein in the hippocampus (Fig. 6). Furthermore, the same dosage of BMS-708163 had no effect on mRNA levels in either the thymus or hippocampus. These findings suggest that the observed GSI-induced cognitive impairment was not due to the inhibition of Notch signaling.
Figure 6.

Effects of GSIs on Notch-target gene expression in vivo. Hippocampus and thymus samples were collected from Tg2576 mice treated with each drug (30 mg/kg), and Hes1 and Hey1 mRNA levels were quantified by quantitative RT-PCR. LY450139 significantly reduced mRNA levels of both proteins in the thymus but not in the hippocampus. BMS-708163 did not affect either mRNA level in the thymus or hippocampus. *p < 0.05; **p < 0.01 compared with vehicle group by Student's t test. Data are presented as mean ± SEM (n = 6 or 7).
In addition to Notch, the effects of LY450139 on the processing of two other γ-secretase substrates were examined by Western blotting. The CTF levels of N-cadherin and EphA4 were clearly increased in the LY450139-treated APP-H4 cells, confirming these proteins were γ-secretase substrates. However, no increase in either CTF was noted in hippocampus samples from Tg2576 mice treated with 30 mg/kg LY450139 for 8 d (the same animals as in Experiment 2), although basal levels of both CTFs were much higher than in the cell samples (Fig. 7). This finding differed distinctly from findings for APP-CTFs, for which we noted a significant increase in LY450139-treated mice. Together, these results suggest that the GSI-induced cognitive impairment is also unlikely due to the inhibition of processing other substrates, or at the very least not N-cadherin or EphA4.
Figure 7.

Effects of LY450139 on the processing of N-cadherin and EphA4 in vivo. Hippocampus samples from Tg2576 mice treated with LY450139 (30 mg/kg) for 8 d were analyzed by Western blotting with antibodies for the C-terminal region of N-cadherin, EphA4, or APP. LY450139 significantly increased the CTF levels of APP but not N-cadherin or EphA4. LY450139 (2 μm)-treated APP-H4 cell lysate was loaded as a positive control. FL, Full length; CTF, C-terminal fragment. **p < 0.01 compared with vehicle group by Student's t test. Data are presented as mean ± SEM (n = 7 or 8).
Acute and subchronic drug effects on normal cognitive function in WT mice (Experiment 4)
We next examined whether or not LY450139 could impair cognitive function in nontransgenic mice. Just as in 3-month-old Tg2576 mice, subchronic but not acute treatment with LY450139 significantly and dose-dependently decreased the alternation rates in WT mice (p < 0.01 at 30 mg/kg); however, this effect required a 10-fold higher dose than that administered to Tg2576 mice (Fig. 8A). This difference in potency was not due to differing pharmacokinetics, since the brain concentration at 10 mg/kg LY450139 was the same between the two strains (5.8 ± 0.7 and 5.9 ± 2.2 ng/g tissue in Tg and WT, respectively; mean ± SEM; n = 4). These findings therefore suggest that LY450139 impairs cognitive function through an APP-dependent pathway, likely involving β-CTF accumulation. In contrast, neither acute nor subchronic treatment with GSM-2 affected normal cognitive function in WT mice (Fig. 8A). We noted no marked differences in the number of total arm entries, body weights, or apparent general condition between the vehicle- and drug-treated groups (data not shown).
Figure 8.

Acute and subchronic drug effects on normal cognitive function in WT mice (Experiment 4). A, Each drug was orally administered to WT mice for 8 d, and Y-maze tests were conducted on days 1 or 8. LY450139 significantly impaired normal cognitive function in WT mice on day 8 but not on day 1. This effect required a 10-fold higher dose than was required in Tg2576 mice. GSM-2 did not affect normal cognitive function. B, LY450139 significantly increased hippocampal mouse endogenous β-CTF levels on day 8, whereas GSM-2 did not despite a robust reduction in mouse endogenous Aβ42. *p < 0.05; **p < 0.01 compared with vehicle group by Dunnett's test. All data are presented as mean ± SEM (n = 7 or 8).
While LY450139 increased hippocampal endogenous Aβ40 in WT mice at 10 mg/kg (15% elevation; p < 0.05), it decreased endogenous Aβ40 and Aβ42 at 30 mg/kg (35 and 34% reduction; p < 0.01). Furthermore, LY450139 increased hippocampal endogenous β-CTF at 3–30 mg/kg in a dose-dependent manner (18–176% elevation; p < 0.01 at 10–30 mg/kg), appearing to correlate with the decrease in alternation rate (Fig. 8B). In contrast, GSM-2 had no effect on endogenous β-CTF levels despite a robust reduction in endogenous Aβ42 (56% reduction at 30 mg/kg; p < 0.01).
Localized accumulation of β-CTF by LY450139 in presynaptic terminals of hippocampal mossy fibers
To determine the sites of β-CTF accumulation in response to LY450139, immunofluorescence of anti-human Aβ/β-CTF N-terminal-specific antibody (82E1) was applied to hippocampal slices from Tg2576 mice. Three-month-old Tg2576 mice were administered 3 mg/kg LY450139 for 8 d, with the brain fixed 3 h after the last administration. We noted that 82E1 immunoreactivities were localized to the stratum lucidum and dentate hilus in both vehicle- and LY450139-treated mice (Fig. 9A). When immunofluorescence intensities in these regions were normalized with that in the CA3 stratum pyramidale of the same slice, LY450139-treated mice showed significantly higher relative immunoreactivity compared with the vehicle-treated mice (Fig. 9B). We also noted an increased relative immunoreactivity by LY450139 in the distal side of the stratum oriens in the CA1c region. These immunoreactivities are suspected to be due to β-CTF and not Aβ, since we detected no immunofluorescence of anti-Aβ40 C-terminal specific antibody (1A10) in these regions (data not shown).
Figure 9.

Localized accumulation of human β-CTF in Tg2576 mouse hippocampus following GSI treatment. Brain slices from Tg2576 mice (aged 3 months) were prepared after subchronic treatment with LY450139 (3 mg/kg) and immunostained with 82E1 antibody. A, Representative microscopic images of the hippocampal slices. LY450139 increased immunofluorescence in the stratum lucidum, dentate hilus, and the distal side of the stratum oriens (arrows). Scale bars: Top, 200 μm; bottom, 30 μm. B, Quantitative analysis of human β-CTF immunoreactivity in each hippocampal region. Data are presented as relative immunofluorescence intensity normalized with the intensity of the CA3 pyramidale in the same slice. *p < 0.05; **p < 0.01 compared with vehicle group by Student's t test. Data are presented as mean ± SEM (n = 7 or 8).
To clarify whether or not β-CTF accumulation occurs in synaptic compartments, hippocampal slices of LY450139-treated mice were coimmunostained with 82E1 and synaptic marker antibodies and observed using a confocal microscope. The 82E1 immunofluorescence in the stratum lucidum showed abundant colocalization with immunofluorescence of synaptophysin (a presynaptic marker) but limited colocalization with that of drebrin A (a postsynaptic marker) (Fig. 10A). The abundant colocalization with synaptophysin was also observed in the dentate hilus (Fig. 10B). These findings suggest that the β-CTF accumulation caused by LY450139 mainly occurred on the large presynaptic terminals of the mossy fibers. No clear colocalization with either synaptophysin or drebrin A was detected in the distal side of the stratum oriens (data not shown).
Figure 10.

Presynaptic accumulation of human β-CTF in the stratum lucidum and dentate hilus in Tg2576 mouse hippocampus following GSI treatment. Hippocampal slices prepared from the same brain samples as in Figure 9 were coimmunostained with 82E1 and synaptic marker antibodies. A, Confocal microscopic images in the stratum lucidum showed abundant colocalization of β-CTF with synaptophysin (syn) (presynaptic marker) but limited colocalization with drebrin A (postsynaptic marker). B, Confocal microscopic images in the dentate hilus also showed abundant colocalization of β-CTF with synaptophysin. Scale bars, 30 μm.
To confirm the localization of the β-CTF accumulation, hippocampal slices were also immunostained with an anti-APP C-terminal antibody. In contrast to 82E1 staining, strong immunoreactivity was observed in pyramidal cells in both vehicle- and LY450139-treated mice, reflecting the expression of full-length APP. While immunoreactivities in the stratum lucidum and dentate hilus were moderate, relative intensities were significantly higher in LY450139-treated mice than in the vehicle-treated mice (Fig. 11). Increased relative intensity by LY450139 was also found in the distal side of the stratum oriens in the CA1c region, all results which are consistent with those for 82E1 staining.
Figure 11.
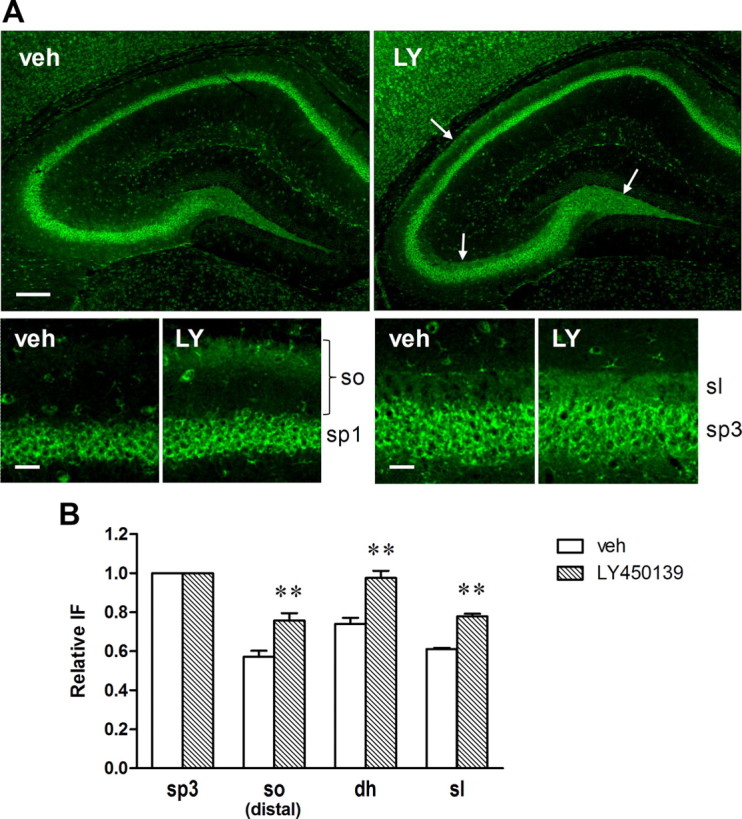
Localized accumulation of APP-CTFs in Tg2576 mouse hippocampus following GSI treatment. Hippocampal slices prepared from the same brain samples as in Figure 9 were immunostained with anti-APP C-terminal antibody. A, Representative microscopic images of the hippocampal slices. LY450139 increased immunofluorescence in the stratum lucidum, dentate hilus, and the distal side of the stratum oriens (arrows). Scale bars: Top, 200 μm; bottom, 30 μm. B, Quantitative analysis of APP-CTF immunoreactivity in each hippocampal region. Data are presented as relative immunofluorescence intensity normalized with the intensity of the CA3 pyramidale in the same slice. **p < 0.01 compared with vehicle group by Student's t test. Data are presented as mean ± SEM (n = 7 or 8).
Anti-APP C-terminal immunostaining was also applied to hippocampal slices from WT mice. The immunoreactivity distribution pattern was similar to that observed in Tg2576 mice, although a longer exposure time was needed because of lower endogenous APP/β-CTF expression. Relative immunoreactivities in the stratum lucidum, dentate hilus, and distal side of the stratum oriens were significantly increased in LY450139 (30 mg/kg)-treated mice but not in GSM-2 (30 mg/kg)-treated mice (Fig. 12).
Figure 12.

Localized accumulation of mouse endogenous APP-CTFs in WT mouse hippocampus following GSI treatment. Brain slices of WT mice were prepared after subchronic treatment with LY450139 (30 mg/kg) or GSM-2 (30 mg/kg) and immunostained with an anti-APP C-terminal antibody. A, Representative microscopic images of the hippocampal slices. LY450139 increased immunofluorescence in the stratum lucidum, dentate hilus, and the distal side of the stratum oriens (arrows), whereas GSM-2 did not. Scale bars: Top, 200 μm; bottom, 30 μm. B, Quantitative analysis of APP-CTF immunoreactivity in each hippocampal region. Data are presented as relative immunofluorescence intensity normalized with the intensity of the CA3 pyramidale in the same slice. *p < 0.05; **p < 0.01 compared with vehicle group by Student's t test. Data are presented as mean ± SEM (n = 7 or 8).
Discussion
Here, we investigated the effects of GSIs and a GSM on cognitive function in mice using the Y-maze task, demonstrating following novel findings: subchronic treatment with the GSIs induced cognitive impairment, this impairment was due not to Notch signaling inhibition but more likely to β-CTF accumulation localized in the presynaptic terminals of hippocampal mossy fibers, and acute and subchronic Aβ42 reduction without Aβ40 reduction by the GSM did ameliorate cognitive deficits in APP-transgenic mice.
Before conducting the behavioral studies, we examined the cellular activity of these drugs in APP-overexpressing H4 cells. LY450139 and BMS-708163 showed potent activities with reducing Aβ levels and increasing α- and β-CTF levels, as expected, although the elevation of β- but not α-CTF was unexpectedly attenuated at higher concentrations (Fig. 2). This attenuation may be due to a shift from β-cleavage to α-cleavage of APP, as the concurrent increase in sAPPα and decrease in sAPPβ in the media were seen by ELISA (data not shown). Alternatively, enhanced cleavage of β-CTF by α-secretase could also be a possible explanation (Beher et al., 2002; Cook et al., 2010; Portelius et al., 2011).
While numerous reports have shown that GSIs reduce Aβ levels in vivo, few have studied the behavioral effects of these compounds. Acute administration with DAPT or GSI-953 (5-chloro-N-[(1S)-3,3,3-trifluoro-1-(hydroxymethyl)-2-(trifluoromethyl)propyl]thiophene-2-sulfonamide) was found to improve cognitive deficits in Tg2576 mice in the contextual fear-conditioning task (Comery et al., 2005; Martone et al., 2009), but neither the chronic nor subchronic effects were examined. In the present study, we investigated both acute and subchronic GSI effects, observing acute cognition improvements with LY450139 and BMS-708163 in the Y-maze task (Fig. 3). Even though hippocampal Aβ reduction was observed only at higher doses, the cognition improvements may be due to a local Aβ reduction in the synaptic cleft or lipid raft (Cirrito et al., 2003; Kawarabayashi et al., 2004; Rushworth and Hooper, 2010), which was difficult to detect with the total homogenate samples. More importantly, the acute effects disappeared after subchronic administration for 8 d (Fig. 3). This finding may be explained by our observation that subchronic treatment with either GSI significantly impaired normal cognition in 3-month-old Tg2576 mice (Fig. 5). In other words, the beneficial effects of Aβ reduction may be cancelled out by the detrimental effects of GSIs.
Notch signaling has been reported to play a key role in memory function (Costa et al., 2003; Wang et al., 2004; Alberi et al., 2011). However, we may conclude that GSI-induced cognitive impairment does not involve Notch signaling inhibition, since either GSI exerted no inhibition on Notch-target gene expression in the hippocampus (Fig. 6). Differing effects of LY450139 between the hippocampus and the thymus could be due to low brain penetration of the compound (brain/plasma ratio, 0.07) or less contribution of γ-secretase to Notch signaling in the adult brain.
γ-Secretase is known to have many type 1 transmembrane protein substrates in addition to APP and Notch (Kopan and Ilagan, 2004; McCarthy et al., 2009), and in some cases, the processing of these substrates, including N-cadherin and EphA4, is assumed to be relevant to memory function (Marambaud et al., 2003; Inoue et al., 2009; Allison et al., 2011; Bot et al., 2011). However, on Western blot analysis, LY450139 appeared to inhibit neither N-cadherin nor EphA4 processing in vivo (Fig. 7), although we cannot exclude the possibility that the reduction in functional intracellular domain released from CTFs could not be detected with the methods used here.
APP β-CTF accumulation as a result of suppressing γ-secretase activity may also be a potential cause of memory impairment (Dewachter et al., 2002; Saura et al., 2005; Bittner et al., 2009). If GSI-induced cognitive impairment was due to β-CTF accumulation, we hypothesized that this effect should be attenuated in non-APP transgenic mice. This was in fact the case, as we were only able to achieve a similar cognition-impairing effect in WT mice administered LY45013 by delivering a dose 10 times that needed in Tg2576 mice (Fig. 8). Furthermore, an apparent correlation was observed between the reduction in Y-maze alternation rate and elevation in hippocampal β-CTF in both Tg2576 and WT mice. These findings indicate that β-CTF accumulation is likely the cause of GSI-induced cognitive impairment, although the causality has not been conclusively proven. Future studies such as usage of APP-knock-out mice or coadministration with a β-secretase inhibitor that reduces β-CTF would be useful for obtaining such proof.
GSI cognition-impairing effects have also been observed in humans. Interim results of phase III trials recently showed that, rather than improving cognition scores in AD patients, LY450139 actually worsens them (http://newsroom.lilly.com/releasedetail.cfm?releaseid=499794). According to a phase II report, LY450139 plasma concentration reached 1000 ng/ml (Fleisher et al., 2008), exceeding the 336 ng/ml that induced significant cognitive impairment in WT mice at 30 mg/kg in the present study. More recently, phase III follow-up study has revealed that the worsened cognition due to LY450139 administration did not return to placebo group levels even 32 weeks after stopping drug treatment (Siemers et al., 2011), indicating presence of irreversible neuronal damage and providing implications for future studies whether longitudinal β-CTF accumulation ultimately results in such damage.
Immunofluorescence studies revealed that β-CTF was localized to the stratum lucidum and dentate hilus in the hippocampus (Figs. 9–12), which well agrees with the localization of β-secretase (Laird et al., 2005). These regions have also been reported to show strong immunoreactivity of presenilin 1 (PS1) as well as GSI-binding (Lee et al., 1996; Yan et al., 2004). GSI-generated β-CTF accumulation in the present study was focally detected in large presynaptic terminals of these regions (Fig. 10), implying that presynaptic function in the mossy fiber pathway but not in the Schaeffer collateral pathway may be affected by GSI activity. This may subsequently explain the apparent discrepancy between our behavioral findings and the electrophysiological findings of a previous report, which showed that 7 d administration with MRK-560 (N-[cis-4-[(4-chlorophenyl)sulfonyl]-4-(2,5-difluorophenyl)cyclohexyl]-1,1,1-trifluoromethanesulfonamide), another GSI, improved LTP impairment in the Schaeffer collateral pathway of Tg2576 mice (Townsend et al., 2010). The presynaptic accumulation of APP-CTFs has also been reported in conditional PS1-knock-out APP-transgenic mice at the hippocampal stratum radiatum (Saura et al., 2005), where no accumulation was found in our GSI-treated mice. This discordance may be due to differences between the long-term genetic inactivation of PS1 and the short-term pharmacological inactivation of γ-secretase as well as due to our using a different strain of APP-transgenic mice. While another intriguing finding in the present study is that β-CTF accumulation was also observed in the distal side of the stratum oriens, further studies are needed to clarify the implications of this finding.
How the accumulated β-CTF exerts synaptotoxicity remains to be elucidated. One possible mechanism is its physical propensity to form nonselective cation channels (Kim and Suh, 1996; Kim et al., 1999), as its subchronic intrahippocampal injection induces working memory impairment and focal apoptosis in the mossy fiber pathway in rats (Jinno et al., 2009). More recently, the elevation of β- but not α-CTF was shown to induce morphological and functional endocytic abnormalities in fibroblasts, similar to those found in patients with AD or Down syndrome, suggesting relevance to neuronal dysfunction (Jiang et al., 2010).
In contrast to the GSIs, GSM-2 showed a potent ability to reduce Aβ42 levels without significant effects on Aβ40 or β-CTF levels, both in vitro and in vivo. Furthermore, both acute and subchronic administration with GSM-2 significantly ameliorated the cognitive deficits in Tg2576 mice (Figs. 3, 4) and had no effect on normal cognition in WT mice (Fig. 8). Although previous studies demonstrated that chronic treatment with first-generation GSMs for 4 or 6 months improved cognitive deficits in APP mice, the relationship to soluble Aβ42 has been unclear, as reductions in Aβ deposits were not accompanied by any changes in soluble Aβ42 levels (Kukar et al., 2007; Imbimbo et al., 2009). To our knowledge, the present study is the first to demonstrate that an acute decrease in soluble Aβ42 without any change in Aβ40 level is sufficient to improve cognitive deficits in APP mice. This effect is likely due to a reduction in synaptic Aβ42 that is secreted in a neuronal activity-dependent manner and depresses synaptic function (Kamenetz et al., 2003; Cirrito et al., 2005; Wei et al., 2010). Another observation of note is that the cognition improvement caused by GSM-2 showed a bell-shaped dose–response in the acute study, a finding similar to that noted with the GSIs, but not in the subchronic study. This is likely because the acute overreduction of synaptic Aβ42 caused an imbalance in neuronal activities, thereby counteracting cognition-improving effects, which was restored under subchronic conditions by synaptic reconstitution.
In conclusion, we demonstrated a clear difference between GSI and GSM in effects on functional consequences. Subchronic treatment with the GSIs caused cognitive impairment, likely due to presynaptic accumulation of β-CTF in the hippocampal mossy fibers, an effect that offsets any benefit to reducing Aβ levels. In contrast, GSM-2 successfully improved cognitive deficits in APP-transgenic mice, suggesting the potential utility of a GSM as a candidate for AD therapeutic regimens.
Footnotes
We thank Dr. Shingo Yamasaki for his kind assistance throughout the present study and his valuable input in the preparation of this manuscript.
The authors declare no competing financial interests.
References
- Alberi L, Liu S, Wang Y, Badie R, Smith-Hicks C, Wu J, Pierfelice TJ, Abazyan B, Mattson MP, Kuhl D, Pletnikov M, Worley PF, Gaiano N. Activity-induced Notch signaling in neurons requires Arc/Arg3.1 and is essential for synaptic plasticity in hippocampal networks. Neuron. 2011;69:437–444. doi: 10.1016/j.neuron.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison JG, Das PM, Ma J, Inglis FM, Jones FE. The ERBB4 intracellular domain (4ICD) regulates NRG1-induced gene expression in hippocampal neurons. Neurosci Res. 2011;70:155–163. doi: 10.1016/j.neures.2011.02.009. [DOI] [PubMed] [Google Scholar]
- Beher D, Wrigley JD, Owens AP, Shearman MS. Generation of C-terminally truncated amyloid-beta peptides is dependent on gamma-secretase activity. J Neurochem. 2002;82:563–575. doi: 10.1046/j.1471-4159.2002.00985.x. [DOI] [PubMed] [Google Scholar]
- Bittner T, Fuhrmann M, Burgold S, Jung CK, Volbracht C, Steiner H, Mitteregger G, Kretzschmar HA, Haass C, Herms J. γ-Secretase inhibition reduces spine density in vivo via an amyloid precursor protein-dependent pathway. J Neurosci. 2009;29:10405–10409. doi: 10.1523/JNEUROSCI.2288-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bot N, Schweizer C, Ben Halima S, Fraering PC. Processing of the synaptic cell adhesion molecule Neurexin-3β by Alzheimer disease α- and γ-secretases. J Biol Chem. 2011;286:2762–2773. doi: 10.1074/jbc.M110.142521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, May PC, O'Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, DeMattos RB, Holtzman DM. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-β metabolism and half-life. J Neurosci. 2003;23:8844–8853. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Comery TA, Martone RL, Aschmies S, Atchison KP, Diamantidis G, Gong X, Zhou H, Kreft AF, Pangalos MN, Sonnenberg-Reines J, Jacobsen JS, Marquis KL. Acute γ-secretase inhibition improves contextual fear conditioning in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2005;25:8898–8902. doi: 10.1523/JNEUROSCI.2693-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook JJ, Wildsmith KR, Gilberto DB, Holahan MA, Kinney GG, Mathers PD, Michener MS, Price EA, Shearman MS, Simon AJ, Wang JX, Wu G, Yarasheski KE, Bateman RJ. Acute γ-secretase inhibition of nonhuman primate CNS shifts amyloid precursor protein (APP) metabolism from amyloid-β production to alternative APP fragments without amyloid-β rebound. J Neurosci. 2010;30:6743–6750. doi: 10.1523/JNEUROSCI.1381-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RM, Honjo T, Silva AJ. Learning and memory deficits in Notch mutant mice. Curr Biol. 2003;13:1348–1354. doi: 10.1016/s0960-9822(03)00492-5. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- Dewachter I, Reversé D, Caluwaerts N, Ris L, Kuipéri C, Van den Haute C, Spittaels K, Umans L, Serneels L, Thiry E, Moechars D, Mercken M, Godaux E, Van Leuven F. Neuronal deficiency of presenilin 1 inhibits amyloid plaque formation and corrects hippocampal long-term potentiation but not a cognitive defect of amyloid precursor protein [V717I] transgenic mice. J Neurosci. 2002;22:3445–3453. doi: 10.1523/JNEUROSCI.22-09-03445.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, Ozols VV, Jessing KW, Zavitz KH, Koo EH, Golde TE. NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J Clin Invest. 2003;112:440–449. doi: 10.1172/JCI18162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisher AS, Raman R, Siemers ER, Becerra L, Clark CM, Dean RA, Farlow MR, Galvin JE, Peskind ER, Quinn JF, Sherzai A, Sowell BB, Aisen PS, Thal LJ. Phase 2 safety trial targeting amyloid beta production with a gamma-secretase inhibitor in Alzheimer disease. Arch Neurol. 2008;65:1031–1038. doi: 10.1001/archneur.65.8.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillman KW, Starrett JE, Parker MF, Xie K, Bronson JJ, Marcin LR, McElhone KE, Bergstrom CP, Mate RA, Williams R, Meredith JE, Burton CR, Barten DM, Toyn JH, Roberts SB, Lentz KA, Houston JG, Zaczek R, Albright CF, Decicco CP, et al. Discovery and evaluation of BMS-708163, a potent, selective and orally bioavailable γ-secretase inhibitor. ACS Med Chem Lett. 2010;1:120–124. doi: 10.1021/ml1000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley DB, May PC, Dean RA, Siemers ER. Development of semagacestat (LY450139), a functional γ-secretase inhibitor, for the treatment of Alzheimer's disease. Expert Opin Pharmacother. 2009;10:1657–1664. doi: 10.1517/14656560903044982. [DOI] [PubMed] [Google Scholar]
- Holcomb LA, Gordon MN, Jantzen P, Hsiao K, Duff K, Morgan D. Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav Genet. 1999;29:177–185. doi: 10.1023/a:1021691918517. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Imbimbo BP, Hutter-Paier B, Villetti G, Facchinetti F, Cenacchi V, Volta R, Lanzillotta A, Pizzi M, Windisch M. CHF5074, a novel γ-secretase modulator, attenuates brain β-amyloid pathology and learning deficit in a mouse model of Alzheimer's disease. Br J Pharmacol. 2009;156:982–993. doi: 10.1111/j.1476-5381.2008.00097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue E, Deguchi-Tawarada M, Togawa A, Matsui C, Arita K, Katahira-Tayama S, Sato T, Yamauchi E, Oda Y, Takai Y. Synaptic activity prompts gamma-secretase-mediated cleavage of EphA4 and dendritic spine formation. J Cell Biol. 2009;185:551–564. doi: 10.1083/jcb.200809151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43) Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- Jarrett JT, Berger EP, Lansbury PT., Jr The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Mullaney KA, Peterhoff CM, Che S, Schmidt SD, Boyer-Boiteau A, Ginsberg SD, Cataldo AM, Mathews PM, Nixon RA. Alzheimer's-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc Natl Acad Sci U S A. 2010;107:1630–1635. doi: 10.1073/pnas.0908953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinno S, Araki K, Matsumoto Y, Suh YH, Yamamoto T. Selective apoptosis induction in the hippocampal mossy fiber pathway by exposure to CT105, the C-terminal fragment of Alzheimer's amyloid precursor protein. Brain Res. 2009;1249:68–78. doi: 10.1016/j.brainres.2008.10.052. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Shoji M, Younkin LH, Wen-Lang L, Dickson DW, Murakami T, Matsubara E, Abe K, Ashe KH, Younkin SG. Dimeric amyloid β protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2004;24:3801–3809. doi: 10.1523/JNEUROSCI.5543-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Suh YH, Lee MH, Ryu PD. Cation selective channels formed by a C-terminal fragment of beta-amyloid precursor protein. Neuroreport. 1999;10:1427–1431. doi: 10.1097/00001756-199905140-00008. [DOI] [PubMed] [Google Scholar]
- Kim SH, Suh YH. Neurotoxicity of a carboxyl-terminal fragment of the Alzheimer's amyloid precursor protein. J Neurochem. 1996;67:1172–1182. doi: 10.1046/j.1471-4159.1996.67031172.x. [DOI] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX. Gamma-secretase: proteasome of the membrane? Nat Rev Mol Cell Biol. 2004;5:499–504. doi: 10.1038/nrm1406. [DOI] [PubMed] [Google Scholar]
- Kopan R, Schroeter EH, Weintraub H, Nye JS. Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proc Natl Acad Sci U S A. 1996;93:1683–1688. doi: 10.1073/pnas.93.4.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kounnas MZ, Danks AM, Cheng S, Tyree C, Ackerman E, Zhang X, Ahn K, Nguyen P, Comer D, Mao L, Yu C, Pleynet D, Digregorio PJ, Velicelebi G, Stauderman KA, Comer WT, Mobley WC, Li YM, Sisodia SS, Tanzi RE, et al. Modulation of γ-secretase reduces β-amyloid deposition in a transgenic mouse model of Alzheimer's disease. Neuron. 2010;67:769–780. doi: 10.1016/j.neuron.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreft AF, Martone R, Porte A. Recent advances in the identification of γ-secretase inhibitors to clinically test the Aβ oligomer hypothesis of Alzheimer's disease. J Med Chem. 2009;52:6169–6188. doi: 10.1021/jm900188z. [DOI] [PubMed] [Google Scholar]
- Kukar T, Prescott S, Eriksen JL, Holloway V, Murphy MP, Koo EH, Golde TE, Nicolle MM. Chronic administration of R-flurbiprofen attenuates learning impairments in transgenic amyloid precursor protein mice. BMC Neurosci. 2007;8:54. doi: 10.1186/1471-2202-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers-Nolten I, Van Der Werf K, Subramaniam V, Braeken D, Callewaert G, Bartic C, D'Hooge R, Martins IC, Rousseau F, Schymkowitz J, De Strooper B. Neurotoxicity of Alzheimer's disease Abeta peptides is induced by small changes in the Abeta42 to Abeta40 ratio. EMBO J. 2010;29:3408–3420. doi: 10.1038/emboj.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC. BACE1, a major determinant of selective vulnerability of the brain to amyloid-β amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci. 2005;25:11693–11709. doi: 10.1523/JNEUROSCI.2766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MK, Slunt HH, Martin LJ, Thinakaran G, Kim G, Gandy SE, Seeger M, Koo E, Price DL, Sisodia SS. Expression of presenilin 1 and 2 (PS1 and PS2) in human and murine tissues. J Neurosci. 1996;16:7513–7525. doi: 10.1523/JNEUROSCI.16-23-07513.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK. A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell. 2003;114:635–645. doi: 10.1016/j.cell.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Martone RL, Zhou H, Atchison K, Comery T, Xu JZ, Huang X, Gong X, Jin M, Kreft A, Harrison B, Mayer SC, Aschmies S, Gonzales C, Zaleska MM, Riddell DR, Wagner E, Lu P, Sun SC, Sonnenberg-Reines J, Oganesian A, et al. Begacestat (GSI-953): a novel, selective thiophene sulfonamide inhibitor of amyloid precursor protein gamma-secretase for the treatment of Alzheimer's disease. J Pharmacol Exp Ther. 2009;331:598–608. doi: 10.1124/jpet.109.152975. [DOI] [PubMed] [Google Scholar]
- McCarthy JV, Twomey C, Wujek P. Presenilin-dependent regulated intramembrane proteolysis and gamma-secretase activity. Cell Mol Life Sci. 2009;66:1534–1555. doi: 10.1007/s00018-009-8435-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, Delucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Milano J, McKay J, Dagenais C, Foster-Brown L, Pognan F, Gadient R, Jacobs RT, Zacco A, Greenberg B, Ciaccio PJ. Modulation of Notch processing by γ-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol Sci. 2004;82:341–358. doi: 10.1093/toxsci/kfh254. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalbantoglu J, Tirado-Santiago G, Lahsaïni A, Poirier J, Goncalves O, Verge G, Momoli F, Welner SA, Massicotte G, Julien JP, Shapiro ML. Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature. 1997;387:500–505. doi: 10.1038/387500a0. [DOI] [PubMed] [Google Scholar]
- Oehlrich D, Berthelot DJC, Gijsen HJ. γ-Secretase modulators as potential disease modifying anti-Alzheimer's drugs. J Med Chem. 2011;54:669–698. doi: 10.1021/jm101168r. [DOI] [PubMed] [Google Scholar]
- Oster-Granite ML, McPhie DL, Greenan J, Neve RL. Age-dependent neuronal and synaptic degeneration in mice transgenic for the C terminus of the amyloid precursor protein. J Neurosci. 1996;16:6732–6741. doi: 10.1523/JNEUROSCI.16-21-06732.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L, Roberson ED. Quantifying biomarkers of cognitive dysfunction and neuronal network hyperexcitability in mouse models of Alzheimer's disease: depletion of calcium-dependent proteins and inhibitory hippocampal remodeling. Methods Mol Biol. 2011;670:245–262. doi: 10.1007/978-1-60761-744-0_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portelius E, Price E, Brinkmalm G, Stiteler M, Olsson M, Persson R, Westman-Brinkmalm A, Zetterberg H, Simon AJ, Blennow K. A novel pathway for amyloid precursor protein processing. Neurobiol Aging. 2011;32:1090–1098. doi: 10.1016/j.neurobiolaging.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Pu YJ, Vaid RK, Boini SK, Towsley RW, Doecke CW, Mitchell D. A practical method for functionalized peptide or amide bond formation in aqueous-ethanol media with EDC as activator. Org Process Res Dev. 2008;13:310–314. [Google Scholar]
- Rushworth JV, Hooper NM. Lipid rafts: linking Alzheimer's amyloid-beta production, aggregation, and toxicity at neuronal membranes. Int J Alzheimers Dis. 2010;2011:603052. doi: 10.4061/2011/603052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura CA, Chen G, Malkani S, Choi SY, Takahashi RH, Zhang D, Gouras GK, Kirkwood A, Morris RG, Shen J. Conditional inactivation of presenilin 1 prevents amyloid accumulation and temporarily rescues contextual and spatial working memory impairments in amyloid precursor protein transgenic mice. J Neurosci. 2005;25:6755–6764. doi: 10.1523/JNEUROSCI.1247-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemers E, Henley D, Sundell K, Sethuraman G, Dean R, Wrobleski K, Mohs R. Evaluating semagacestat, a gamma-secretase inhibitor, in a phase III trial. Alzheimers Dement. 2011;7:S484–S485. [Google Scholar]
- Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999;402:537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- Struhl G, Greenwald I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature. 1999;398:522–525. doi: 10.1038/19091. [DOI] [PubMed] [Google Scholar]
- Suh YH, Kim HS, Lee JP, Park CH, Jeong SJ, Kim SS, Rah JC, Seo JH, Kim SS. Roles of A beta and carboxyl terminal peptide fragments of amyloid precursor protein in Alzheimer disease. J Neural Transm Suppl. 2000;2000:65–82. doi: 10.1007/978-3-7091-6284-2_6. [DOI] [PubMed] [Google Scholar]
- Townsend M, Qu Y, Gray A, Wu Z, Seto T, Hutton M, Shearman MS, Middleton RE. Oral treatment with a gamma-secretase inhibitor improves long-term potentiation in a mouse model of Alzheimer's disease. J Pharmacol Exp Ther. 2010;333:110–119. doi: 10.1124/jpet.109.163691. [DOI] [PubMed] [Google Scholar]
- Van Dam D, D'Hooge R, Staufenbiel M, Van Ginneken C, Van Meir F, De Deyn PP. Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur J Neurosci. 2003;17:388–396. doi: 10.1046/j.1460-9568.2003.02444.x. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chan SL, Miele L, Yao PJ, Mackes J, Ingram DK, Mattson MP, Furukawa K. Involvement of Notch signaling in hippocampal synaptic plasticity. Proc Natl Acad Sci U S A. 2004;101:9458–9462. doi: 10.1073/pnas.0308126101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature. 2001;414:212–216. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Golde TE, Koo EH. Aβ42-lowering nonsteroidal anti-inflammatory drugs preserve intramembrane cleavage of the amyloid precursor protein (APP) and ErbB-4 receptor and signaling through the APP intracellular domain. J Biol Chem. 2003;278:30748–30754. doi: 10.1074/jbc.M304824200. [DOI] [PubMed] [Google Scholar]
- Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, Malinow R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat Neurosci. 2010;13:190–196. doi: 10.1038/nn.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- Wong GT, Manfra D, Poulet FM, Zhang Q, Josien H, Bara T, Engstrom L, Pinzon-Ortiz M, Fine JS, Lee HJ, Zhang L, Higgins GA, Parker EM. Chronic treatment with the γ-secretase inhibitor LY-411,575 inhibits β-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J Biol Chem. 2004;279:12876–12882. doi: 10.1074/jbc.M311652200. [DOI] [PubMed] [Google Scholar]
- Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME. Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature. 1999;402:533–537. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- Yan XX, Li T, Rominger CM, Prakash SR, Wong PC, Olson RE, Zaczek R, Li YW. Binding sites of γ-secretase inhibitors in rodent brain: distribution, postnatal development, and effect of deafferentation. J Neurosci. 2004;24:2942–2952. doi: 10.1523/JNEUROSCI.0092-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younkin SG. The role of A beta 42 in Alzheimer's disease. J Physiol Paris. 1998;92:289–292. doi: 10.1016/s0928-4257(98)80035-1. [DOI] [PubMed] [Google Scholar]