

Abstract
Amyloid-β peptide (Aβ) is believed to play a central role in the pathogenesis of Alzheimer's disease. In view of the side effects associated with inhibiting the secretases that produce Aβ, new molecular targets are required to provide alternative therapeutic options. We used RNA interference (RNAi) to systematically screen the Drosophila genome to identify genes that modulate Aβ production upon knockdown. RNAi of 41 genes in Drosophila cells significantly lowered Aβ without affecting general secretion or viability. After the γ-secretase complex components, the most potent effect was observed for platelet activating factor acetylhydrolase α (Paf-AHα), and, in mammalian cells, the effect was replicated for its ortholog PAFAH1B2. Knockdown of PAFAH1B2 strongly reduced Aβ secretion from human cells, and this effect was confirmed in primary cells derived from PAFAH1B2 knock-out mice. Reduced Aβ production was not attributable to altered β-amyloid precursor protein (APP) ectodomain shedding but was a result of an enhanced degradation of APP C-terminal fragments (CTFs) in the absence of PAFAH1B2 but not its close homolog PAFAH1B3. Enhanced degradation of APP CTFs was selective because no such effects were obtained for Notch or E-/N-cadherin. Thus, we have identified an important protein that can selectively modify Aβ generation via a novel mechanism, namely enhanced degradation of its immediate precursor. In view of the absence of a neurological phenotype in PAFAH1B2 knock-out mice, targeted downregulation of PAFAH1B2 may be a promising new strategy for lowering Aβ.
Introduction
Aggregation and accumulation of amyloid-β (Aβ) peptides in the CNS is thought to underlie most cases of Alzheimer's disease (Haass and Selkoe, 2007). Strategies that can limit the production or release of Aβ are therefore considered to have therapeutic potential. Aβ is derived from amyloid precursor protein (APP), a type I transmembrane protein that traffics through the secretory pathway to the plasma membrane (Haass, 2004; Thinakaran and Koo, 2008). β-Secretase-mediated ectodomain shedding of APP generates a β-C-terminal fragment (βCTF), and subsequent γ-secretase cleavage of βCTF generates Aβ (Steiner et al., 2008). Alternatively, APP can be shed by α-secretase within the Aβ region, thus precluding the generation of the toxic Aβ peptide (Lichtenthaler, 2011). It is unclear how the generation of Aβ is regulated, but potentially there are several steps along the pathway in which other proteins could intervene, such as with the activity of the secretase enzymes or the metabolic fate of Aβ and its immediate precursor the APP βCTF. To find such modulators, we performed a genome-wide RNA interference (RNAi) screen in Drosophila S2 cells that express the human APP βCTF and secrete human Aβ. This allowed a systematic assessment of the contribution of individual genes toward Aβ generation. Drosophila cells were chosen for their several-fold advantages. First, there is less toxicity associated with RNAi as a result of a lack of an interferon response (Boutros and Ahringer, 2008). Second, invertebrates have less genetic redundancy, allowing robust phenotypes to be more readily observed upon single gene knockdowns (Boutros et al., 2004). Third, 60% of known human disease genes are present in Drosophila (Rubin et al., 2000), and ectodomain shedding and γ-secretase activity are well conserved (Fossgreen et al., 1998; Six et al., 2003), making it an ideal model system for RNAi-mediated analysis of Aβ generation.
By screening Drosophila S2 cells expressing the human APP βCTF, we identified platelet activating factor acetylhydrolase α (Paf-AHα) as one of the most potent modifiers of Aβ secretion. RNAi-mediated knockdown of the mammalian ortholog PAFAH1B2 resulted in a highly significant reduction of Aβ secretion in a range of cell types. Additional characterization of cells lacking PAFAH1B2 revealed that the reduced Aβ levels were attributable to a reduction of APP CTFs, whereby an enhanced degradation of APP CTFs occurs in the absence PAFAH1B2. Thus, PAFAH1B2 is essential for efficient generation of Aβ, and it provides a novel target for achieving Aβ reduction in Alzheimer's disease.
Materials and Methods
Reagents and antibodies.
The following reagents were used: Effectene (Qiagen), Lipofectamine2000 and Lipofectamine RNAiMax (Invitrogen), protease inhibitor mixture and leupeptin (Roche), synthetic Aβ peptide standards (Bachem), Steadylite HTS reagent for measuring firefly luciferase (fLuc) activity (PerkinElmer Life and Analytical Sciences), and GAR2 for measuring Gaussia luciferase activity (Targeting Systems). The following antibodies were used: mouse monoclonal 4G8 to Aβ17–24 (Covance), C-terminal-specific anti-Aβ38 (Meso Scale Discovery), C-terminal-specific BAP24 anti-Aβ40 and BAP15 anti-Aβ42 (kind gifts from Dr. Manfred Brockhaus, Roche, Basel, Switzerland), 22C11 mouse monoclonal to the APP N terminus for soluble APP (sAPP) detection (Millipore), 192 rabbit polyclonal to sAPPβ (kind gift from Drs. Dale Schenk and Peter Seubert, Elan Pharmaceuticals, San Francisco, CA), chicken polyclonal to PAFAH1B2 (Abcam), mouse monoclonal to PAFAH1B3 (Abnova), rabbit polyclonal to amyloid precursor protein-like 2 (APLP2) (Calbiochem), rabbit polyclonal N1660 to human nicastrin (NCT) (Sigma), mouse monoclonal to β-actin (Sigma), 9E10 mouse monoclonal to Myc-tagged F-NEXT (FLAG-tagged Notch1 extracellular truncation)/NICD (Notch1 intracellular domain) (Santa Cruz Biotechnology), and mouse monoclonal to E-cadherin and mouse monoclonal to N-cadherin (BD Biosciences). New antibodies, 2C11 (mouse monoclonal) and 5G11 (rat monoclonal), were generated against the APP C terminus and mouse sAPPα, respectively (Colombo et al., 2012). Rabbit polyclonal 6687 to the APP C terminus (Steiner et al., 2000) was used for immunocytochemistry (ICC), together with mouse monoclonal LAMP2 (Hybridoma Bank, National Institutes of Health), and Alexa Fluor-488-conjugated anti-rabbit and Alexa Fluor-555-conjugated anti-mouse antibodies (Invitrogen).
RNAi screen in Drosophila S2 cells.
A library of 14,333 long double-stranded RNAs (dsRNAs) covering ∼91% of all protein coding genes in the Drosophila genome was used for screening (Horn et al., 2010). Two hundred fifty nanograms of dsRNA (5 μl) per well were spotted in duplicate in 384-well plates. Using a Multidrop liquid dispenser, cells were plated at a density of 20,000 per well in 20 μl of serum-free Schneider's media and incubated alone with the individual dsRNAs for 1 h at 25°C before the addition of 30 μl of serum-containing media and additional incubation. The following day, cells were transfected using 1.5 μl of Effectene per well together with 42 ng of pAc5:SP-C99 and 8 ng of an fLuc reporter construct to control for viability. Cells were incubated for an additional 3 d at 25°C before aliquoting of conditioned media into four 96-well daughter plates using a Beckman FX liquid-handling robot. Plates containing conditioned media were immediately frozen and stored at −80°C before being subjected to immunoassay analysis. fLuc activity was determined by adding 25 μl of Steadylite HTS reagent to the cells, incubating for 5 min, and then measuring the luminescence. All signals were analyzed using the high-throughput screening software CellHTS (Boutros et al., 2006). Briefly, measured intensities were normalized per plate (for each of the two replicates) by the respective plate median to account for plate-to-plate differences. We only considered measurements with SDs <30% of the mean of the two replicates. z-scores were then computed as the number of SDs that the actual normalized value was above or below the normalized plate median. Finally, the mean of the z-scores from the two replicates was calculated. Positive and negative z-scores in the immunoassay indicated decreased and increased secretion of Aβ peptides, respectively. Positive and negative z-scores in the fLuc assay indicated decreased and increased viability, respectively. For the secondary screen, dsRNAs were freshly synthesized and spotted in triplicate in 384-well plates and the above procedure was repeated, except that a secretion reporter plasmid encoding Gaussia luciferase was additionally transfected. Gaussia luciferase activity was determined by adding 30 μl of GAR2 reagent containing 8% stabilizer to 30 μl of conditioned media and immediately measuring luminescence.
siRNA/short-hairpin RNA oligonucleotides and transfection.
For screening genes in human embryonic kidney 293 cells expressing APP carrying the Swedish mutation (HEK293–APPsw), non-overlapping siRNA pools from two different sources were used, namely, siGENOME SMARTpool siRNAs (Dharmacon) together with nontargeting control pool 2 (Dharmacon) and FlexiTube GeneSolution siRNAs (Qiagen) together with nontargeting control siRNA (Qiagen). As an additional negative control, H2O was used in a mock transection and was found to behave identically to both nontargeting controls. shRNAs against human PAFAH1B2 were purchased from Sigma as pLKO MISSION clones (#377 and #894) together with a nontargeting control shRNA.
cDNA constructs.
SP-C99 (Lichtenthaler et al., 1999), fLuc, and secretory Gaussia luciferase were cloned with HindIII/NotI restriction sites into individual pAc5 vectors (Invitrogen) for expression in Drosophila S2 cells. Human PAFAH1B2 was purchased from Imagenes, sequence verified, and subcloned by PCR mutagenesis into pcDNA3.1/zeo(+) (Invitrogen) using KpnI/NotI restriction sites. For lentiviral expression, PAFAH1B2 cDNA was cloned into a FUΔZeo viral vector (Kuhn et al., 2010). The S48A mutant was generated by Quikchange mutagenesis (Stratagene) using oligonucleotide primers corresponding to the point mutation.
Mammalian cell culture and treatments.
HEK293 cells and primary mouse embryonic fibroblasts (MEFs) were cultured in DMEM supplemented with 10% FCS and penicillin/streptomycin. SH-SY5Y cells were cultured in DMEM F-12 supplemented with 15% FCS, non-essential amino acids, and penicillin/streptomycin. For RNAi screening of the 30 orthologous candidate genes in HEK293–APPsw cells (Citron et al., 1992), cells were plated without antibiotics at a density of 200,000 cells per 24-well and, using 1 μl of Lipofectamine RNAiMax, were immediately reverse transfected with 10 nm of individual siRNA oligo pools. Two days later, media were replaced, and, after an overnight incubation, the conditioned media and cell lysates were harvested. For lentivirus transduction, cells were plated on 24-well or 6-well plates and, at 70% confluency, were transduced with lentiviruses containing a nontargeting control shRNA or shRNAs against PAFAH1B2. Again, 2 d later, media were replaced, and, after an overnight incubation, the conditioned media and cell lysates were harvested. To inhibit lysosomal proteases, media were replaced with fresh media containing leupeptin at a concentration of 100 μm, and cells were incubated overnight (16 h) before harvesting (Haass et al., 1992).
ICC.
Cells were fixed in 4% paraformaldehyde in PBS for 30 min, followed by washing three times in PBS and simultaneous permeabilization and blocking for 1 h in ICC buffer (0.2% Triton X-100 and 1% BSA in PBS). Cells were stained with the 6687 polyclonal antibody to the APP C terminus together with anti-LAMP2 (both at 1:500 in ICC buffer for 2 h) and washed five times in PBS before incubation with Alexa Fluor-488-conjugated anti-rabbit and Alexa Fluor-555-conjugated anti-mouse antibodies (1:250 in ICC buffer for 1 h). After washing five times in PBS, coverslips were mounted on glass slides. Confocal images were obtained with an inverted laser-scanning confocal microscope (Carl Zeiss Axiovert 200M) with a 100×/1.4 numerical aperture oil-immersion lens, and pictures were taken and analyzed with the LSM 510 confocal software (Carl Zeiss).
Quantification of Aβ.
Secreted Aβ peptides in conditioned medium were quantified by a sandwich immunoassay using the MSD Sector Imager 2400 as described previously (Page et al., 2010). For more sensitive detection of Aβ species and for endogenous Aβ, the MSD Aβ Triplex sandwich immunoassay was used. Here, MSD C-terminal-specific antibodies are pre-spotted into each well. For detection, ruthenylated 6E10 or 4G8 antibodies were used for human or mouse Aβ, respectively. The corresponding concentrations of Aβ peptides were calculated using the MSD Discovery Workbench software. Aβ concentrations were always normalized to cellular protein content, and, in each graph, the bars represent the mean of at least four experiments and error bars indicate the SEM (*p < 0.05, **p < 0.01, ***p < 0.001, unpaired Student's t test).
Protein analysis.
Cells pellets were resuspended in lysis buffer (150 mm NaCl, 10 mm Tris, pH 7.5, 2 mm EDTA, and 1% NP-40 plus protease inhibitors), and lysates were loaded on appropriate percentage SDS-PAGE gels and immunoblotted with the relevant primary antibodies. Secreted Aβ was analyzed from medium conditioned overnight for 16 h by combined immunoprecipitation/immunoblotting using antibodies 3552/2D8 (Page et al., 2010), followed by Tris–bicine urea SDS-PAGE (for individual Aβ species) or 10–20% Tris–tricine SDS-PAGE (for total Aβ).
Results
A genome-wide RNAi screen in cultured Drosophila cells
It has been demonstrated that Drosophila possess a BACE (β-site APP-cleaving enzyme)-like protease (Carmine-Simmen et al., 2009), but it does not generate Aβ at the same site as human BACE1, instead generating a larger Aβ peptide from human APP (Greeve et al., 2004). To establish a system in which authentic human Aβ could be efficiently generated in a Drosophila cell line by γ-secretase activity alone, a construct was used that contains the APP βCTF fused to a signal peptide (SP-C99; Lichtenthaler et al., 1999). Drosophila S2 cells expressing this construct generated and secreted all three major Aβ peptide species, as shown by immunoprecipitation of conditioned media (Fig. 1A). Aβ ratios were similar to that from transfected human cells, i.e., Aβ40 represents >80% of total secreted Aβ, with Aβ38 and Aβ42 representing ∼10% each (Page et al., 2010). SP-C99-transfected S2 cells were then tested for their responsiveness to dsRNA-mediated knockdown of key genes involved in APP processing, with dsRNA against GFP used as a negative control. Knockdown of Drosophila presenilin (Psn), the catalytic subunit of γ-secretase, almost completely abolished Aβ generation (Fig. 1B, left), demonstrating the efficiency of RNAi in Drosophila cells. This was accompanied by an accumulation of βCTF (C99) and its α-secretase cleaved product αCTF (C83). In contrast to Psn, knockdown of Kuzbanian (Kuz), the Drosophila homolog of ADAM10 (Rooke et al., 1996; Kuhn et al., 2010), resulted in a significant increase in Aβ generation. This was presumably because silencing Kuz resulted in decreased turnover of βCTF to αCTF (Fig. 1B, left), therefore increasing the relative amount of βCTF substrate available to γ-secretase. The Aβ effects measured by Western blot were fully replicated by Aβ immunoassay (Fig. 1B, right), thus validating the use of the immunoassay for the high-throughput RNAi screen.
Figure 1.
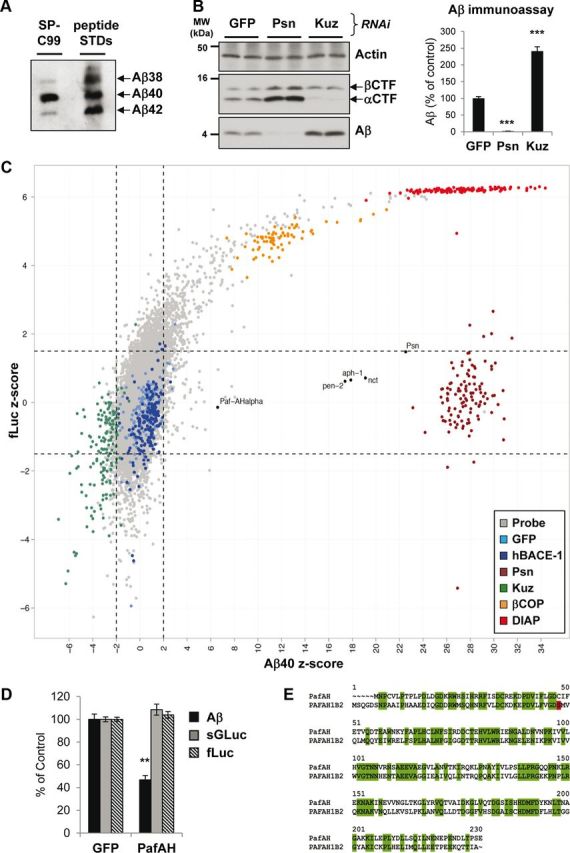
A genome-wide RNAi screen in cultured Drosophila cells for modifiers of Aβ secretion. A, Tris–bicine urea SDS-PAGE immunoblot of conditioned media from Drosophila S2 cells expressing pAc5:SP-C99. Standard peptides confirmed that Aβ species corresponding to Aβ38, Aβ40, and Aβ42 were generated. Aβ species were generated in a similar ratio to human cells, with Aβ40 the predominant species and Aβ38 and Aβ42 minor species. B, S2 cells after treatment with dsRNAs against GFP, Drosophila Psn, and Kuz. SDS-PAGE immunoblot demonstrated that RNAi of Psn abolished Aβ generation, whereas RNAi of Kuz increased it (left). Both CTFs accumulate after Psn knockdown, whereas alphaCTF is specifically reduced after Kuz knockdown. An Aβ immunoassay of conditioned media confirmed the Aβ effects (right). MW, Molecular weight. C, Graph of mean Aβ40 z-scores plotted against mean fLuc z-scores from the genome-wide RNAi screen. Each dot represents a single gene. Multiple controls were included on every plate: negative controls, GFP (light blue) and human BACE1 (dark blue); positive controls, Psn (dark red), Kuz (green), βCOP (β-coatomer protein) (orange), and DIAP (red). βCOP is required for general secretion and viability and DIAP (Drosophila inhibitor of apoptosis protein) is a gene essential for viability. The negative controls GFP and human BACE1 varied around a z-score of 0 for both Aβ40 and fLuc. Psn controls showed very high Aβ40 z-scores, corresponding to strong reductions in Aβ secretion but no effect on fLuc, with a z-score for fLuc varying at ∼0. The dashed lines indicate the significance thresholds: genes were considered as hits if the Aβ40 z-score was >2 or less than −2 and fLuc z-score was between −1.5 and 1.5. The vast majority (∼99%) of Drosophila genes (Probe; gray dots) varied around a z-score of 0 for both Aβ40 and fLuc, in a similar range to the negative controls. However, some showed specific increases in Aβ (in the direction of Kuz) or specific reductions in Aβ (in the direction of Psn). The positions of the four γ-secretase complex components (Psn, Nct, Aph-1, and Pen-2) and Paf-AHα are labeled. D, S2 cells after treatment with dsRNAs against GFP or Paf-AHα. In the media, Aβ was reduced by 50%. This was a specific effect because there was no change in the secretion of a Gaussia luciferase secretion reporter (sGLuc) or in cellular fLuc activity. E, Protein sequence homology between Drosophila Paf-AHα and human PAFAH1B2 demonstrates a highly conserved protein. Green boxes highlight the 46% of amino acids that are identical, and the red box highlights the active serine (48) in the human protein. Bar graphs show mean and SEM of six individual experiments (**p < 0.01, ***p < 0.001, unpaired Student's t test).
Nearly 15,000 dsRNAs designed to target ∼91% of Drosophila protein coding genes (Horn et al., 2010) were screened in duplicate 384-well plates for their ability to modulate Aβ secretion in Drosophila S2 cells. Cellular fLuc was measured as a viability control. The duplicate measurements were highly correlated (Pearson's correlation of 0.82), indicating a robust assay. Aβ40 was measured as the most prominent Aβ species. Aβ and fLuc measurements for each gene were assigned z-scores, which is a measure of their relative deviation from individual plate median values. Positive z-scores indicate a decrease in Aβ or fLuc, whereas negative z-scores indicate an increase in Aβ or fLuc. Negative control dsRNAs performed as expected, because GFP and human BACE1 generated average Aβ40 z-scores of 0.8 and 0.6, respectively (i.e. close to 0), and did not significantly affect fLuc activity (Fig. 1C). Knockdown of the positive controls Psn and Kuz generated average z-scores of 27.8 and −2.9, respectively (Fig. 1C). Knockdown of Psn did not affect fLuc activity, although a tendency for increased fLuc activity was observed for Kuz knockdown (Fig. 1C). Hits were defined as wells with z-scores >2 or less than −2 for Aβ, together with z-scores between −1.5 and 1.5 for fLuc, which indicated normal transfection efficiency and viability. The majority of genes screened did not show any effect on the generation of Aβ, as shown by their clustering around a z-score of 0 (Fig. 1C, gray dots). Candidate genes that significantly modulated Aβ secretion numbered 140 (∼1% of all genes screened), and these were then reassessed in a secondary screen to confirm their phenotypes. To exclude genes that merely affected general secretion, an additional plasmid was transfected in this screen that encoded a secretory Gaussia luciferase reporter and the efficiency of its secretion was assessed by measuring the luminescence of the conditioned media. All potential false positives were removed, leaving 41 genes that produced highly significant and specific changes in Aβ upon knockdown compared with control nontargeting dsRNAs. The gene identities and the magnitude of their effects are given in Table 1. All genes affected Aβ38 and Aβ42 to an equal extent as Aβ40.
Table 1.
A list of the final 41 genes from the Drosophila secondary screen, which upon knockdown significantly modulated Aβ secretion
| Hit gene | Drosophila annotation ID | % of Aβ after KD | Human gene ID | Human symbol | Human hit |
|---|---|---|---|---|---|
| 1 | CG18803 | 3 | 5663 | PSEN1 | * |
| 2 | CG7012 | 6 | 23385 | NCSTN | * |
| 3 | CG2855 | 8 | 51107 | APH1A | * |
| 4 | CG33198 | 8 | 55851 | PSENEN | * |
| 5 | CG8962 | 36 | 5049 | PAFAH1B2 | * |
| 6 | CG33696 | 38 | 7201 | TRHR | * |
| 7 | CG9743 | 42 | 6319 | SCD | * |
| 8 | CG18766 | 43 | 345222 | C4orf44 | * |
| 9 | CG15749 | 44 | 10655 | DMRT2 | |
| 10 | CG11073 | 44 | 200958 | MUC20 | |
| 11 | CG31714 | 46 | 22997 | IGSF9B | |
| 12 | CG7263 | 49 | 9131 | AIFM1 | |
| 13 | CG8253 | 50 | 55093 | WDYHV1 | |
| 14 | CG9357 | 52 | 1116 | CHI3L1 | |
| 15 | CG13071 | 53 | N/A | N/A | |
| 16 | CG18208 | 53 | 150 | ADRA2A | |
| 17 | CG17603 | 53 | 6872 | TAF1 | * |
| 18 | CG3732 | 55 | 9406 | ZRAB2 | * |
| 19 | CG4491 | 56 | 80139 | ZNF703 | |
| 20 | CG12926 | 59 | 79183 | TTPAL | |
| 21 | CG4183 | 64 | 1410 | CRYAB | |
| 22 | CG17212 | 64 | 54933 | RHBDL2 | * |
| 23 | CG2096 | 64 | 5500 | PPP1CB | * |
| 24 | CG5874 | 68 | 7469 | WHSC2 | * |
| 25 | CG14572 | 68 | N/A | N/A | |
| 26 | CG13630 | 69 | 23173 | METAP1 | * |
| 27 | CG7938 | 72 | 126295 | ZNF57 | * |
| 28 | CG1793 | 72 | 9441 | MED26 | |
| 29 | CG11865 | 73 | 7092 | TLL1 | |
| 30 | CG6686 | 155 | 9092 | SART1 | |
| 31 | CG42258 | 158 | N/A | N/A | |
| 32 | CG1249 | 160 | 6633 | SNRPD2 | * |
| 33 | CG12857 | 163 | 84541 | KBTBD8 | |
| 34 | CG32134 | 164 | 2261/2263 | FGFR2/FGFR1 | |
| 35 | CG7757 | 171 | 9129 | PRPF3 | |
| 36 | CG8427 | 175 | 6634 | SNRPD3 | * |
| 37 | CG8749 | 178 | 6625 | SNRP70 | * |
| 38 | CG4528 | 188 | 6629 | SNRPB2 | * |
| 39 | CG6391 | 189 | 11165 | NUDT3 | * |
| 40 | CG6159 | 210 | 10640 | EXOC5 | |
| 41 | CG3160 | 215 | 80055 | PGAP1 |
The Drosophila annotation identification (ID) is given, together with the percentage of Aβ measured after knockdown (KD) in Drosophila cells and the corresponding human gene ID and symbol. The top four genes correspond to the four γ-secretase complex components, and their knockdowns potently reduced Aβ. An additional 25 genes reduced Aβ, and 12 genes increased Aβ upon knockdown. Genes that were confirmed to modulate Aβ in human cells by at least 25% in either direction are indicated with an asterisk. N/A, Not applicable.
The four γ-secretase complex components Psn, Nct, Aph-1 (anterior pharynx-defective protein 1), and Pen-2 (presenilin enhancer protein 2) were identified as the top four genes that lowered Aβ upon knockdown (Fig. 1C; Table 1). Of the 37 novel Drosophila hits, 30 have human orthologs as determined by BLAST analysis of protein sequences. Next, we used HEK293–APPsw cells, a well-established and frequently used cell line for studying Aβ generation (Citron et al., 1992; Haass et al., 1995; Page et al., 2008), to verify the 30 candidate genes in a human cell context. Using non-overlapping siRNA pools from two different sources, it was determined that, upon knockdown, 16 novel genes significantly modified Aβ secretion (indicated with asterisks in Table 1).
PAFAH1B2 knockdown reduces Aβ secretion in human cells
From the Drosophila Aβ-lowering hits, Paf-AHα was identified to have one of the most potent reductions of Aβ secretion upon knockdown (Fig. 1C,D). Paf-AHα has a strong homology (46% sequence identity; Fig. 1E) to its human counterpart PAFAH1B2. Knockdown of PAFAH1B2 in human cells had an equally potent effect on Aβ secretion, thus making it the most attractive target for detailed characterization. RNAi-mediated knockdown of PAFAH1B2 in HEK293–APPsw cells potently reduced Aβ secretion, by 60% for pool 1 and 40% for pool 2 (Fig. 2A). The protein levels of APP and actin were unaffected. Likewise, the level of NCT and the maturation of this protein, a key indicator of a functional γ-secretase complex (Edbauer et al., 2002), were unaffected. As expected from a hit that was identified as modifying Aβ generation from a “pre-shed” C99 stub, total sAPP (sAPPα and sAPPβ) secretion into the medium was, on average, not significantly affected across siRNA pools (Fig. 2A).
Figure 2.

Knockdown of PAFAH1B2 in human cells using different RNAi approaches reduces Aβ. A, HEK293–APPsw cells transfected with non-overlapping control or PAFAH1B2-targeting siRNA pools from two sources. Efficient knockdowns of PAFAH1B2 were achieved for both pools, and this corresponded to strong reductions in Aβ secretion, by 60% for pool 1 and 40% for pool 2. Total sAPP was, on average, not significantly affected across siRNA pools. Mature (m.) and immature (im.) full-length APP and mature and immature NCT and actin were unaffected. B, SH-SY5Y cells transfected with the same non-overlapping control or PAFAH1B2-targeting siRNA pools from two sources. Efficient knockdowns of PAFAH1B2 were achieved for both pools, although not quite as good as in the HEK293 cells. This corresponded to strong reductions in endogenous Aβ secretion, by 40 and 30%, respectively. Total sAPP, full-length APP, and mature and immature NCT and actin were unaffected. C, SH-SY5Y cells transduced with non-overlapping control or PAFAH1B2-targeting shRNAs. Highly efficient knockdowns of PAFAH1B2 were achieved for both shRNAs, and this corresponded to potent reductions in endogenous Aβ secretion, by 40 and 70% respectively. Total sAPP, full-length APP, and mature and immature NCT and actin were unaffected. Con, Control; MW, molecular weight. Bar graphs show mean and SEM of six individual experiments (**p < 0.01, ***p < 0.001, unpaired Student's t test).
To study the effect of PAFAH1B2 on Aβ secretion in a neuronally relevant cell line, siRNA-mediated knockdown experiments were repeated in SH-SY5Y neuroblastoma cells. PAFAH1B2 knockdown correlated with significant reductions in the endogenous Aβ produced by this cell line without affecting total sAPP secretion (Fig. 2B). In another RNAi strategy, we used two validated shRNAs targeting different regions of the PAFAH1B2 mRNA to the siRNA pools. Lentiviral delivery of these shRNAs resulted in potent silencing of PAFAH1B2 expression in SH-SY5Y cells (Fig. 2C). Compared with control nontargeting shRNA, both shRNAs targeting PAFAH1B2 resulted in significant reductions in endogenous Aβ secretion, shRNA-1 by 74% and shRNA-2 by 44% (Fig. 2C). In each case, the levels of total sAPP and cellular APP, actin, and NCT remained unaffected.
PAFAH1B2 is a phospholipase highly expressed in brain (Koizumi et al., 2003). Through its serine esterase activity, PAFAH1B2 is responsible for the hydrolysis of platelet activating factor (Blank et al., 1981; Hattori et al., 1993), although in Drosophila, Paf-AHα is catalytically inactive (Sheffield et al., 2000). Other roles have been attributed to the mammalian protein, such as in microtubule dynamics (Zhang et al., 2007), in the Wnt pathway during development (Livnat et al., 2010), and in subcellular trafficking (Bechler et al., 2011). In mammals, the protein forms part of the tetrameric PAFAH1B enzyme, an intracellular G-protein-like complex consisting of a dimer of catalytic subunits (either PAFAH1B2 or PAFAH1B3) and a regulatory PAFAH1B1 dimer (Tarricone et al., 2004). PAFAH1B3 is the predominant catalytic subunit during development, but PAFAH1B2 predominates in adult brain (Arai et al., 2002). Knockdown of Drosophila PAFAH1B1 (Lis1) had no effect on Aβ in the RNAi screen and knockdown of PAFAH1B1 and PAFAH1B3 in HEK293–APPsw cells also had no effect on Aβ (data not shown).
Primary cells lacking PAFAH1B2 exhibit reduced Aβ secretion
We next sought to check the phenotype of primary MEF cells from mice carrying deletions for PAFAH1B2, PAFAH1B3, or both subunits (Livnat et al., 2010). Compared with wild-type (WT) MEFs, PAFAH1B2−/− MEFs secreted ∼55% less endogenous Aβ into conditioned media (Fig. 3A), confirming the phenotype observed with RNAi knockdown in other cell types. Reduced Aβ secretion by ∼60% was also observed for double-knock-out PAFAH1B2−/−, PAFAH1B3−/− MEFs, whereas MEFs lacking PAFAH1B3 alone showed a mild but not significant reduction of secreted Aβ compared with WT MEFs. Consistent with the RNAi data, MEFs lacking the PAFAH1B catalytic subunits exhibited no alterations in APP ectodomain shedding, with sAPPα and sAPPβ secretion unaltered for all knock-out cell lines (Fig. 3A).
Figure 3.

Primary MEF cells lacking PAFAH1B2 show reduced Aβ. A, Comparison of WT and PAFAH1B2−/− MEFs showed that, when PAFAH1B2 was lacking, endogenous Aβ secretion was reduced by 55%. Loss of PAFAH1B3 caused a mild but insignificant reduction of Aβ, but loss of both subunits caused a slightly enhanced reduction of Aβ to ∼60%. Compared with actin, endogenous full-length APP was slightly elevated for MEFs lacking PAFAH1B2, but the ectodomain cleavage products sAPPα and sAPPβ remained unaffected. B, WT or PAFAH1B2−/− MEFs were transduced with either control lentiviruses (Vec) or lentiviruses containing PAFAH1B2 cDNAs with WT function (B2 wt) or inactive serine esterase function (B2 S48A). Transduction of PAFAH1B2−/− MEFs with both PAFAH1B2 cDNAs reversed the endogenous Aβ deficit, restoring levels to that of WT MEFs. Endogenous full-length APP and actin were unaffected. An increase in PAFAH1B2 level, as seen for the WT cells upon cDNA transduction, did not result in any additional increase in endogenous Aβ. MW, Molecular weight. Bar graphs show mean and SEM of at least four individual experiments (**p < 0.01, ***p < 0.001, unpaired Student's t test).
To confirm the link between PAFAH1B2 and Aβ secretion, a rescue experiment was performed whereby PAFAH1B2 cDNA was reintroduced into PAFAH1B2−/− MEFs via lentiviral transduction. Transduction allowed robust expression of PAFAH1B2 cDNA and rescued the reduced Aβ production (Fig. 3B). This confirmed that PAFAH1B2 is specifically required for Aβ production. Interestingly, reintroduction of the S48A inactive serine esterase mutant (Bechler et al., 2011) also resulted in the same degree of rescue, suggesting that esterase activity is not required for the PAFAH1B2-mediated effect on Aβ. This is consistent with the absence of any reduction of Aβ from cells lacking the other catalytic subunit, PAFAH1B3. An increase of PAFAH1B2 expression above normal levels, as seen when WT MEFs were transduced with the rescue cDNA, did not however lead to an additional increase in Aβ levels (Fig. 3B). This is in agreement with the observation that PAFAH1B2 cDNA overexpression in HEK293–APPsw cells led to only a very mild (5%) but not significant increase in Aβ secretion (data not shown).
Selective degradation of APP and APLP2 CTFs upon loss of PAFAH1B2
Additional analysis of APP processing in HEK293–APPsw cells revealed that, upon potent knockdown of PAFAH1B2 with two different lentivirally delivered shRNAs, both APP CTFs were strongly reduced compared with the control condition, although mature and immature full-length APP levels were unaffected (Fig. 4A). Analysis of endogenous APP processing in MEFs revealed that cells lacking PAFAH1B2 exhibited a slight elevation in APP holoprotein levels, but strikingly, there was a prominent reduction of APP CTFs compared with WT MEFs (Fig. 4B). PAFAH1B3−/− MEFs displayed no such alteration in APP CTFs compared with WT MEFs (Fig. 4B), which is a noteworthy difference in phenotype considering the high degree of homology between the two proteins. These effects correlate with the levels of Aβ measured from these cells (Fig. 3A). We next examined the closest homolog of APP, APLP2, to see whether its processing was also altered in the absence of the PAFAH1B subunits. In exactly the same way, the CTFs of APLP2 were markedly reduced in PAFAH1B2−/− MEFs, although total full-length APLP2 levels were unaffected. PAFAH1B3−/− MEFs behaved like WT MEFs, showing no change in APLP2 CTFs (Fig. 4B). Analysis of two other endogenous type I transmembrane proteins, E-cadherin and N-cadherin, revealed that the CTF reduction was selective. The CTF levels of both cadherins, which are also γ-secretase substrates, were unchanged in the absence of either PAFAH1B2 or PAFAH1B3 (Fig. 4C). In a similar way to APP, full-length levels of E-cadherin were slightly increased in the absence of PAFAH1B2. Another important substrate for γ-secretase is Notch (Steiner et al., 2008). We decided to analyze the effect of PAFAH1B2 on Notch processing in cells expressing APP and F-NEXT (Okochi et al., 2002), a flagged Notch1 extracellular truncation that, like APP CTFs, is a direct substrate of γ-secretase to generate NICD (Schroeter et al., 1998). After potent shRNA-mediated knockdown of PAFAH1B2 in this cell line, APP CTFs were again strongly reduced, but the levels of F-NEXT and its cleavage product NICD were unaltered (Fig. 4D).
Figure 4.

Loss of PAFAH1B2 in HEK293–APPsw and primary MEF cells reduces APP CTFs. A, HEK293–APPsw cells virally transduced with shRNAs against PAFAH1B2 resulted in highly potent knockdowns of PAFAH1B2 compared with nontargeting control. This correlated with a dramatic reduction of both the αCTF and βCTF. A shorter exposure of the immunoblot showed that full-length mature (m.) and immature (im.) APP levels were unaffected. Actin was also unaffected. B, In primary MEF cells, analysis of APP and APLP2 processing by Tris–tricine SDS-PAGE revealed that, in the absence of PAFAH1B2, endogenous APP and APLP2 CTFs were dramatically reduced. No such change was observed for MEFs lacking PAFAH1B3. As before, the level of full-length APP was slightly elevated in PAFAH1B2−/− MEFs. C, Analysis of N-cadherin (NCAD) and E-cadherin (ECAD) processing by 10% SDS-PAGE revealed full-length proteins and CTFs running at ∼45 and 36 kDa, respectively. For E-cadherin, the level of full-length protein was slightly increased in the absence of PAFAH1B2, but for both E- and N-cadherin, the CTF levels were unchanged in the absence of either PAFAH1B2 or PAFAH1B3. D, HEK293–APPsw/F-NEXT cells virally transduced with shRNAs against PAFAH1B2 resulted in potent knockdowns of PAFAH1B2 compared with nontargeting control. In contrast to the reduced levels of APP CTFs, the levels of F-NEXT and its γ-secretase generated product, NICD, were completely unaffected. MW, Molecular weight.
Loss of PAFAH1B2 increases lysosomal degradation of APP CTFs
Considering that APP CTFs were reduced in addition to Aβ secretion, we reasoned that APP CTFs did not meet γ-secretase for the proteolytic processing that would give rise to Aβ. It is known that there are two pools of APP CTFs: one that can meet γ-secretase to give rise to Aβ and one that is instead trafficked to the lysosome for degradation, in which it can no longer give rise to Aβ (Haass et al., 1992, 1993). We therefore hypothesized that there could be an increased APP CTF degradation in the lysosomes in the absence of PAFAH1B2, and we decided to test this possibility by lysosomal protease inhibition. Treatment of WT cells with leupeptin, a classical lysosomal protease inhibitor, increased APP CTFs (Fig. 5A) as reported previously (Haass et al., 1992). Treatment of PAFAH1B2−/− cells with leupeptin recovered the reduced APP CTFs to a similar extent as for WT cells (Fig. 5A,B). This suggests that APP CTFs are initially generated in PAFAH1B2−/− cells but that they are more rapidly degraded by lysosomal proteases. We conclude that the reduction of Aβ generation observed in the absence of PAFAH1B2 can be explained by an increased degradation of the βCTF. Leupeptin treatment did not restore Aβ levels to normal (data not shown), but this was expected because the treatment cannot re-route the APP CTFs to be available for γ-secretase cleavage (Haass et al., 1993). To directly assess a role for increased lysosomal degradation of APP CTFs upon loss of PAFAH1B2, we performed ICC in HEK293 cells overexpressing APP. These cells were chosen because the APP expression in the MEF cells was too low to be detected by ICC. Interestingly, knockdown of PAFAH1B2 revealed an increased LAMP2 staining compared with control cells (Fig. 5C, arrowheads), which is indicative of clustered and/or enlarged lysosome structures. Treatment of control and PAFAH1B2 knockdown cells with leupeptin increased LAMP2 staining, and this increase was more pronounced in PAFAH1B2 knockdown cells (Fig. 5C). After leupeptin treatment, a partial colocalization of APP with LAMP2 was also observed in both cases, as indicated by the yellow staining (arrows) in the enlarged panels, and this colocalization was more pronounced in PAFAH1B2 knockdown cells. Consistent with the recovery of APP CTFs after leupeptin treatment of PAFAH1B2−/− cells (Fig. 5A), the increased APP staining in PAFAH1B2 knockdown cells most likely reflects the increase in APP CTFs accumulating during leupeptin treatment, although additional staining of full-length APP must be considered (Haass et al., 1992).
Figure 5.

Loss of PAFAH1B2 increases lysosomal degradation of APP CTFs. A, WT and PAFAH1B2−/− MEFs were treated in the absence or presence of 100 μm of the lysosomal protease inhibitor leupeptin. Treatment resulted in a moderate increase in APP CTFs for WT cells. Untreated knock-out MEFs showed reduced APP CTFs, as before, but during treatment with leupeptin, they were restored to normal levels. B, Quantification of APP CTF immunoblots for nine independent replicates of the experiment performed in A, with APP CTF levels from untreated WT cells set to 100%. The increase in APP CTFs after leupeptin treatment was highly significant for both WT and PAFAH1B2−/− MEFs (***p < 0.001, unpaired Student's t test), whereas the difference in the extent of APP CTF recovery was not significant between the two cell types. C, Confocal microscopy of HEK293–APP cells treated with a nontargeting siRNA pool or an siRNA pool against PAFAH1B2, in the presence or absence of 100 μm leupeptin (Leu). Cells were stained with 6687 polyclonal antibody to the APP C terminus (green) and LAMP2 antibody (red). Knockdown of PAFAH1B2 revealed an increased LAMP2 staining compared with control cells (arrowheads), with some larger clusters clearly visible. Treatment of control and PAFAH1B2 knockdown cells with leupeptin increased LAMP2 staining in both cases, but this increase was more pronounced in PAFAH1B2 knockdown cells. After leupeptin treatment, a partial colocalization of APP with LAMP2 was also observed in both cases, as indicated by the yellow staining (arrows) in the enlarged panels. This partial colocalization was more pronounced after PAFAH1B2 knockdown and leupeptin treatment. Scale bar, 15 μm. Con, Control.
If lysosomal degradation of APP CTFs is the key underlying mechanism of the observed effects, prevention of lysosomal targeting of APP should eliminate the PAFAH1B2-mediated effect on Aβ. Because the APP C-terminal domain is known to be important for internalization and intracellular trafficking (Haass et al., 1993; Selkoe et al., 1996; Kawasumi et al., 2004), we next asked whether it is required for the PAFAH1B2-mediated effect on Aβ generation. To this end, Aβ secretion from cells stably expressing a C-terminal truncated APP (HEK293–APPΔC) (Haass et al., 1993) was investigated in the context of PAFAH1B2 knockdown. Using potent shRNAs, effective silencing of PAFAH1B2 was achieved in both HEK293–APP and HEK293–APPΔC cells (Fig. 6A). This resulted in a strong reduction of Aβ secretion for HEK293–APP cells, but for the cells expressing the truncated form of APP, there was no effect on Aβ secretion (Fig. 6A). Thus, the APP intracellular domain is necessary for the PAFAH1B2-mediated effect on Aβ. Together with the findings described in Figure 5, we have identified a protein that can reduce the generation of Aβ through an increased degradation of its immediate precursor, the βCTF, in lysosomes. The APP intracellular domain, which is known to influence intracellular trafficking by binding to a variety of adaptor proteins (Selkoe et al., 1996; Kawasumi et al., 2004), is required for this effect (summarized in Fig. 6B).
Figure 6.

The APP intracellular domain is required for the PAFAH1B2-mediated effect on Aβ. A, HEK293 cells stably expressing full-length APP (FL) or C-terminally truncated APP (ΔC) were treated with lentiviruses carrying a nontargeting shRNA or shRNAs against PAFAH1B2. Potent knockdowns were achieved in both cell lines, but although this corresponded to reductions in Aβ for cells expressing full-length APP, there was no change in Aβ for cells expressing the truncated version. Protein levels of APP and actin were unaffected in each case. B, Model depicting how PAFAH1B2 can influence Aβ generation. Under WT conditions, APP is internalized from the plasma membrane into vesicles after which it is cleaved by β-secretase to generate the βCTF. Thereafter, a large portion of the βCTF is trafficked through the early endosomes, in which it meets γ-secretase and is cleaved to generate the Aβ peptide, which is then secreted. Another portion of the βCTF is trafficked directly to the lysosomes in which it cannot give rise to Aβ and is degraded. Some CTFs may be degraded by other, as yet undefined, mechanisms as depicted by the dotted line and question mark. Upon loss of PAFAH1B2, the trafficking of βCTFs to the lysosome may be promoted, which results in their enhanced degradation before they can meet γ-secretase for the generation of Aβ. In the case of APPΔC, internalization of the protein is blocked and so any changes in post-endocytic traffic induced by loss of PAFAH1B2 do not affect the generation of Aβ. β-Secretase and γ-secretase can cleave APPΔC to a lesser extent at the plasma membrane as depicted here, or en route to the plasma membrane, to generate Aβ. MW, Molecular weight. Bar graphs show mean and SEM of four individual experiments (*p < 0.05, **p < 0.01, unpaired Student's t test).
Discussion
A genome-wide RNAi screen in Drosophila S2 cells identified 41 genes capable of specifically modifying Aβ secretion without affecting general secretion or viability. As expected, the four γ-secretase complex components showed the most potent Aβ-lowering effects upon knockdown, with <10% Aβ remaining. This validated the RNAi screen because it demonstrated that the approach could robustly identify relevant Aβ-modulator genes in an unbiased screen. There was a large difference in the magnitude of phenotype between these four genes and the other genes identified, suggesting that no other essential γ-secretase complex protein exists as expected (Edbauer et al., 2003), but rather proteins that serve a modulatory role in Aβ generation. Indeed, another high-throughput RNAi screen found a potent modulator of BACE1 activity but did not describe the identification of any γ-secretase modulators (Majercak et al., 2006). Knockdown of pigeon, the Drosophila homolog of the recently identified GSAP (γ-secretase activating protein) (He et al., 2010), showed no significant change in Aβ secretion (data not shown). GPR3 (G-protein-coupled receptor 3), a protein shown to influence γ-secretase localization and Aβ production in neurons (Thathiah et al., 2009), is not present in Drosophila and could therefore not be assessed. This is one limitation of using an invertebrate system, but our rationale was that the core conserved modulators would be identified in this robust system. Knockdown of Paf-AHα showed one of the strongest phenotypes after the γ-secretase complex components, and knockdown of its homolog, PAFAH1B2, in mammalian cells verified the gene as a potent and relevant modifier of Aβ production.
PAFAH1B2 knockdown reduced Aβ secretion in neuronal and peripheral cell types—in cells expressing both exogenous and endogenous APP. This effect was not influenced by the efficiency of β-secretase cleavage, because Aβ reduction was equally strong in the HEK293–APPsw cells. Indeed, the phenotype was identified in Drosophila independently of APP ectodomain shedding, by screening a pre-shed βCTF. The phenotype of primary MEFs from PAFAH1B2 knock-out mice was consistent with that observed in the other cells, with a ∼60% reduction of Aβ secretion. Importantly, the Aβ deficit could be efficiently rescued by reintroduction of the PAFAH1B2 gene. The inactive serine esterase mutant of PAFAH1B2 could also mediate rescue, suggesting that esterase activity is not required for the effect on Aβ. Indeed, loss of PAFAH1B3, also a serine esterase, had no effect on Aβ. This is consistent with the identification of the effect in Drosophila, because Paf-AHα has been shown to be catalytically inactive (Sheffield et al., 2000). It does not possess the catalytic triad like its mammalian counterpart, and it is catalytically inert for PAF hydrolysis. Indeed, Sheffield et al. proposed that, in addition to its catalytic function, the mammalian protein may also have additional roles that do not require catalysis. Further analysis of APP processing revealed that, in the absence of PAFAH1B2, there was a stark reduction in the levels of the APP CTFs. This phenotype was also observed for the close homolog of APP, APLP2. In both cases, the effect was observed specifically for PAFAH1B2 and not for its close homolog PAFAH1B3, and this correlated with the effects measured for Aβ. Other proteins that give rise to CTFs as substrates for γ-secretase, such as E-cadherin and N-cadherin, did not show any such alteration in their CTF levels. Moreover, PAFAH1B2 knockdown did not affect the levels of NICD, an important finding, because a disturbance in this crucial signaling pathway is an unwanted side effect of γ-secretase inhibition (Citron, 2010). It has been shown previously that Fluvastatin treatment lowers Aβ by modifying the endosomal/lysosomal pathway and promoting the degradation of APP CTFs (Shinohara et al., 2010), but PAFAH1B2 represents to our knowledge the first protein to have such an effect on APP CTFs and Aβ.
The reduction of the APP CTFs observed in PAFAH1B2−/− cells could be explained by an increased lysosomal degradation, because treatment with the lysosomal protease inhibitor leupeptin restored APP CTF levels to the same extent as in WT cells. Aβ levels were not restored after leupeptin treatment, but this is consistent with previous work that showed that such a treatment could not increase Aβ production (Haass et al., 1992, 1993), because only a pre-lysosomal βCTF in earlier compartments can be efficiently cleaved to give rise to the peptide. Confocal microscopy showed that PAFAH1B2 knockdown cells had increased lysosomal staining and leupeptin treatment of these cells increased APP within lysosomes. This suggests a recovery of APP CTFs that would otherwise have been degraded. We cannot exclude that another, as yet undefined, pathway may play an additional role, but the data presented here provide strong evidence for a prominent role of increased lysosome degradation of APP CTFs in the absence of PAFAH1B2. A recent study using antibodies against the APP C terminus demonstrated a rapid and direct transport from the cell surface to lysosomes, in a previously unrecognized selective pathway (Lorenzen et al., 2010). This transport was impaired for APP mutants that produce more Aβ, suggesting that preferential transport to the lysosome favors degradation of APP CTFs, whereas normal endocytosis through the early endosomes favors more efficient APP CTF cleavage and Aβ generation. Loss of PAFAH1B2 may thus favor such an alternative trafficking pathway, whereby APP CTFs are more efficiently transported to the lysosomes for degradation (Fig. 6B). This is in line with our finding that PAFAH1B2 knockdown did not reduce Aβ generation in cells expressing a C-terminal truncated APP, demonstrating that the intracellular domain and presumably internalization is important for the effect that PAFAH1B2 exerts on Aβ. APPΔC can give rise to Aβ on the late secretory pathway or at the plasma membrane (Haass et al., 1993), so any PAFAH1B2-mediated influence on post-endocytic traffic would not affect Aβ in this case (Fig. 6B). Interestingly, the APP, cadherin, and Notch families differ in the fact that the APP family members possess an NPXY internalization motif in their C-terminal tails (Selkoe et al., 1996), whereas the cadherins and Notch do not. Although we have not formally investigated the contribution of the NPXY motif for the PAFAH1B2-dependent APP CTF degradation, this could provide an explanation as to the differential effects observed for the CTFs of these proteins. Thus, one would expect that other NPXY-containing proteins could be affected, but these are relatively few in number (Chen et al., 1990) and perhaps the APP CTF is even functionally dispensable. Indeed, it has been shown that only the secreted sAPPα ectodomain fragment fulfills the essential functions of APP, because it alone rescues the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice (Ring et al., 2007). In support of this, the loss of PAFAH1B2 does not have any effect on the general health of the cells investigated here or in the mouse knock-outs generated. Two independent studies demonstrated that PAFAH1B2−/− and PAFAH1B3−/− mice were developmentally normal and the expected Mendelian ratios were observed; the mice displayed only defects in spermatogenesis (Koizumi et al., 2003; Yan et al., 2003). A subsequent study showed certain neuronal migration defects for the double-knock-out mice (Livnat et al., 2010), although this is in stark contrast to PAFAH1B1 knock-out mice, in which there is a fatal disturbance in neuronal migration. However, no such developmental or neuropathological phenotypes have been described for the single subunit knock-out mice.
Together, we provide evidence that the loss of PAFAH1B2 potently reduces Aβ by promoting the degradation of its immediate precursor, the βCTF. Loss of PAFAH1B2 is well tolerated by the brain; thus, a targeted downregulation of PAFAH1B2 may be a novel and viable therapeutic strategy to lower brain Aβ.
Footnotes
This work was funded by the Ludwig Maximilians University Excellent Program, which supports C.H. with a research professorship, the Federal Ministry of Education and Research (Competence Network of Degenerative Dementias) (H.S., S.F.L., and C.H.), and the German Research Foundation [Collaborative Research Center (SFB596) “Molecular Mechanisms of Neurodegeneration” (H.S., S.F.L., and C.H.)]. The work in the laboratory of M.B. was supported by the Helmholtz Alliance for Systems Biology and the CellNetworks Cluster of Excellence. We thank Gabriele Basset, Katrin Moschke, and Brigitte Nuscher for excellent technical assistance.
The authors declare declare no competing financial interests.
References
- Arai H, Koizumi H, Aoki J, Inoue K. Platelet-activating factor acetylhydrolase (PAF-AH) J Biochem. 2002;131:635–640. doi: 10.1093/oxfordjournals.jbchem.a003145. [DOI] [PubMed] [Google Scholar]
- Bechler ME, Doody AM, Ha KD, Judson BL, Chen I, Brown WJ. The phospholipase A enzyme complex PAFAH Ib mediates endosomal membrane tubule formation and trafficking. Mol Biol Cell. 2011;22:2348–2359. doi: 10.1091/mbc.E09-12-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank ML, Lee T, Fitzgerald V, Snyder F. A specific acetylhydrolase for 1-alkyl-2-acetyl-sn-glycero-3-phosphocholine (a hypotensive and platelet-activating lipid) J Biol Chem. 1981;256:175–178. [PubMed] [Google Scholar]
- Boutros M, Ahringer J. The art and design of genetic screens: RNA interference. Nat Rev Genet. 2008;9:554–566. doi: 10.1038/nrg2364. [DOI] [PubMed] [Google Scholar]
- Boutros M, Kiger AA, Armknecht S, Kerr K, Hild M, Koch B, Haas SA, Paro R, Perrimon N Heidelberg Fly Array Consortium. Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science. 2004;303:832–835. doi: 10.1126/science.1091266. [DOI] [PubMed] [Google Scholar]
- Boutros M, Brás LP, Huber W. Analysis of cell-based RNAi screens. Genome Biol. 2006;7:R66. doi: 10.1186/gb-2006-7-7-r66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmine-Simmen K, Proctor T, Tschäpe J, Poeck B, Triphan T, Strauss R, Kretzschmar D. Neurotoxic effects induced by the Drosophila amyloid-beta peptide suggest a conserved toxic function. Neurobiol Dis. 2009;33:274–281. doi: 10.1016/j.nbd.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WJ, Goldstein JL, Brown MS. NPXY, a sequence often found in cytoplasmic tails, is required for coated pit-mediated internalization of the low density lipoprotein receptor. J Biol Chem. 1990;265:3116–3123. [PubMed] [Google Scholar]
- Citron M. Alzheimer's disease: strategies for disease modification. Nat Rev Drug Discov. 2010;9:387–398. doi: 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- Colombo A, Wang H, Kuhn PH, Page R, Kremmer E, Dempsey PJ, Crawford HC, Lichtenthaler SF. Constitutive alpha- and beta-secretase cleavages of the amyloid precursor protein are partially coupled in neurons, but not in frequently used cell lines. Neurobiol Dis. 2012;49C:137–147. doi: 10.1016/j.nbd.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edbauer D, Winkler E, Haass C, Steiner H. Presenilin and nicastrin regulate each other and determine amyloid beta-peptide production via complex formation. Proc Natl Acad Sci U S A. 2002;99:8666–8671. doi: 10.1073/pnas.132277899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of gamma-secretase activity. Nat Cell Biol. 2003;5:486–488. doi: 10.1038/ncb960. [DOI] [PubMed] [Google Scholar]
- Fossgreen A, Brückner B, Czech C, Masters CL, Beyreuther K, Paro R. Transgenic Drosophila expressing human amyloid precursor protein show gamma-secretase activity and a blistered-wing phenotype. Proc Natl Acad Sci U S A. 1998;95:13703–13708. doi: 10.1073/pnas.95.23.13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greeve I, Kretzschmar D, Tschäpe JA, Beyn A, Brellinger C, Schweizer M, Nitsch RM, Reifegerste R. Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J Neurosci. 2004;24:3899–3906. doi: 10.1523/JNEUROSCI.0283-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C. Take five: BACE and the gamma-secretase quartet conduct Alzheimer's amyloid beta-peptide generation. EMBO J. 2004;23:483–488. doi: 10.1038/sj.emboj.7600061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Haass C, Koo EH, Mellon A, Hung AY, Selkoe DJ. Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature. 1992;357:500–503. doi: 10.1038/357500a0. [DOI] [PubMed] [Google Scholar]
- Haass C, Hung AY, Schlossmacher MG, Teplow DB, Selkoe DJ. beta-Amyloid peptide and a 3-kDa fragment are derived by distinct cellular mechanisms. J Biol Chem. 1993;268:3021–3024. [PubMed] [Google Scholar]
- Haass C, Lemere CA, Capell A, Citron M, Seubert P, Schenk D, Lannfelt L, Selkoe DJ. The Swedish mutation causes early-onset Alzheimer's disease by beta-secretase cleavage within the secretory pathway. Nat Med. 1995;1:1291–1296. doi: 10.1038/nm1295-1291. [DOI] [PubMed] [Google Scholar]
- Hattori M, Arai H, Inoue K. Purification and characterization of bovine brain platelet-activating factor acetylhydrolase. J Biol Chem. 1993;268:18748–18753. [PubMed] [Google Scholar]
- He G, Luo W, Li P, Remmers C, Netzer WJ, Hendrick J, Bettayeb K, Flajolet M, Gorelick F, Wennogle LP, Greengard P. Gamma-secretase activating protein is a therapeutic target for Alzheimer's disease. Nature. 2010;467:95–98. doi: 10.1038/nature09325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn T, Sandmann T, Boutros M. Design and evaluation of genome-wide libraries for RNA interference screens. Genome Biol. 2010;11:R61. doi: 10.1186/gb-2010-11-6-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasumi M, Matsuda S, Matsuoka M, Nishimoto I. Cytoplasmic tail adaptors of Alzheimer's amyloid-beta protein precursor. Mol Neurobiol. 2004;30:185–200. doi: 10.1385/MN:30:2:185. [DOI] [PubMed] [Google Scholar]
- Koizumi H, Yamaguchi N, Hattori M, Ishikawa TO, Aoki J, Taketo MM, Inoue K, Arai H. Targeted disruption of intracellular type I platelet activating factor-acetylhydrolase catalytic subunits causes severe impairment in spermatogenesis. J Biol Chem. 2003;278:12489–12494. doi: 10.1074/jbc.M211836200. [DOI] [PubMed] [Google Scholar]
- Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, Kremmer E, Rossner S, Lichtenthaler SF. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010;29:3020–3032. doi: 10.1038/emboj.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenthaler SF. Alpha-secretase in Alzheimer's disease: molecular identity, regulation and therapeutic potential. J Neurochem. 2011;116:10–21. doi: 10.1111/j.1471-4159.2010.07081.x. [DOI] [PubMed] [Google Scholar]
- Lichtenthaler SF, Multhaup G, Masters CL, Beyreuther K. A novel substrate for analyzing Alzheimer's disease gamma-secretase. FEBS Lett. 1999;453:288–292. doi: 10.1016/s0014-5793(99)00730-9. [DOI] [PubMed] [Google Scholar]
- Livnat I, Finkelshtein D, Ghosh I, Arai H, Reiner O. PAF-AH catalytic subunits modulate the Wnt pathway in developing GABAergic neurons. Front Cell Neurosci. 2010;4.pii:19. doi: 10.3389/fncel.2010.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzen A, Samosh J, Vandewark K, Anborgh PH, Seah C, Magalhaes AC, Cregan SP, Ferguson SS, Pasternak SH. Rapid and direct transport of cell surface APP to the lysosome defines a novel selective pathway. Mol Brain. 2010;3:11. doi: 10.1186/1756-6606-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majercak J, Ray WJ, Espeseth A, Simon A, Shi XP, Wolffe C, Getty K, Marine S, Stec E, Ferrer M, Strulovici B, Bartz S, Gates A, Xu M, Huang Q, Ma L, Shughrue P, Burchard J, Colussi D, Pietrak B, et al. LRRTM3 promotes processing of amyloid-precursor protein by BACE1 and is a positional candidate gene for late-onset Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:17967–17972. doi: 10.1073/pnas.0605461103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okochi M, Steiner H, Fukumori A, Tanii H, Tomita T, Tanaka T, Iwatsubo T, Kudo T, Takeda M, Haass C. Presenilins mediate a dual intramembranous gamma-secretase cleavage of Notch-1. EMBO J. 2002;21:5408–5416. doi: 10.1093/emboj/cdf541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page RM, Baumann K, Tomioka M, Pérez-Revuelta BI, Fukumori A, Jacobsen H, Flohr A, Luebbers T, Ozmen L, Steiner H, Haass C. Generation of Abeta38 and Abeta42 is independently and differentially affected by familial Alzheimer disease-associated presenilin mutations and gamma-secretase modulation. J Biol Chem. 2008;283:677–683. doi: 10.1074/jbc.M708754200. [DOI] [PubMed] [Google Scholar]
- Page RM, Gutsmiedl A, Fukumori A, Winkler E, Haass C, Steiner H. Beta-amyloid precursor protein mutants respond to gamma-secretase modulators. J Biol Chem. 2010;285:17798–17810. doi: 10.1074/jbc.M110.103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring S, Weyer SW, Kilian SB, Waldron E, Pietrzik CU, Filippov MA, Herms J, Buchholz C, Eckman CB, Korte M, Wolfer DP, Müller UC. The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J Neurosci. 2007;27:7817–7826. doi: 10.1523/JNEUROSCI.1026-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooke J, Pan D, Xu T, Rubin GM. KUZ, a conserved metalloprotease-disintegrin protein with two roles in Drosophila neurogenesis. Science. 1996;273:1227–1231. doi: 10.1126/science.273.5279.1227. [DOI] [PubMed] [Google Scholar]
- Rubin GM, Yandell MD, Wortman JR, Gabor Miklos GL, Nelson CR, Hariharan IK, Fortini ME, Li PW, Apweiler R, Fleischmann W, Cherry JM, Henikoff S, Skupski MP, Misra S, Ashburner M, Birney E, Boguski MS, Brody T, Brokstein P, Celniker SE, et al. Comparative genomics of the eukaryotes. Science. 2000;287:2204–2215. doi: 10.1126/science.287.5461.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeter EH, Kisslinger JA, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393:382–386. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Yamazaki T, Citron M, Podlisny MB, Koo EH, Teplow DB, Haass C. The role of APP processing and trafficking pathways in the formation of amyloid beta-protein. Ann N Y Acad Sci. 1996;777:57–64. doi: 10.1111/j.1749-6632.1996.tb34401.x. [DOI] [PubMed] [Google Scholar]
- Sheffield PJ, Garrard S, Caspi M, Aoki J, Arai H, Derewenda U, Inoue K, Suter B, Reiner O, Derewenda ZS. Homologs of the alpha- and beta-subunits of mammalian brain platelet-activating factor acetylhydrolase Ib in the Drosophila melanogaster genome. Proteins. 2000;39:1–8. doi: 10.1002/(sici)1097-0134(20000401)39:1<1::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Shinohara M, Sato N, Kurinami H, Takeuchi D, Takeda S, Shimamura M, Yamashita T, Uchiyama Y, Rakugi H, Morishita R. Reduction of brain beta-amyloid (Abeta) by fluvastatin, a hydroxymethylglutaryl-CoA reductase inhibitor, through increase in degradation of amyloid precursor protein C-terminal fragments (APP-CTFs) and Abeta clearance. J Biol Chem. 2010;285:22091–22102. doi: 10.1074/jbc.M110.102277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Six E, Ndiaye D, Laabi Y, Brou C, Gupta-Rossi N, Israel A, Logeat F. The Notch ligand Delta1 is sequentially cleaved by an ADAM protease and gamma-secretase. Proc Natl Acad Sci U S A. 2003;100:7638–7643. doi: 10.1073/pnas.1230693100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, Kostka M, Romig H, Basset G, Pesold B, Hardy J, Capell A, Meyn L, Grim ML, Baumeister R, Fechteler K, Haass C. Glycine 384 is required for presenilin-1 function and is conserved in bacterial polytopic aspartyl proteases. Nat Cell Biol. 2000;2:848–851. doi: 10.1038/35041097. [DOI] [PubMed] [Google Scholar]
- Steiner H, Fluhrer R, Haass C. Intramembrane proteolysis by gamma-secretase. J Biol Chem. 2008;283:29627–29631. doi: 10.1074/jbc.R800010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarricone C, Perrina F, Monzani S, Massimiliano L, Kim MH, Derewenda ZS, Knapp S, Tsai LH, Musacchio A. Coupling PAF signaling to dynein regulation: structure of LIS1 in complex with PAF-acetylhydrolase. Neuron. 2004;44:809–821. doi: 10.1016/j.neuron.2004.11.019. [DOI] [PubMed] [Google Scholar]
- Thathiah A, Spittaels K, Hoffmann M, Staes M, Cohen A, Horr é K, Vanbrabant M, Coun F, Baekelandt V, Delacourte A, Fischer DF, Pollet D, De Strooper B, Merchiers P. The orphan G protein-coupled receptor 3 modulates amyloid-beta peptide generation in neurons. Science. 2009;323:946–951. doi: 10.1126/science.1160649. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283:29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Assadi AH, Wynshaw-Boris A, Eichele G, Matzuk MM, Clark GD. Previously uncharacterized roles of platelet-activating factor acetylhydrolase 1b complex in mouse spermatogenesis. Proc Natl Acad Sci U S A. 2003;100:7189–7194. doi: 10.1073/pnas.1236145100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Assadi AH, McNeil RS, Beffert U, Wynshaw-Boris A, Herz J, Clark GD, D'Arcangelo G. The Pafah1b complex interacts with the reelin receptor VLDLR. PLoS One. 2007;2:e252. doi: 10.1371/journal.pone.0000252. [DOI] [PMC free article] [PubMed] [Google Scholar]