

Abstract
Tar DNA binding protein 43 (TDP-43) mislocalization and aggregation is a hallmark of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar dementia. Moreover, TDP-43 mRNA was found to be upregulated by ∼2.5-fold in the spinal cord of sporadic ALS subjects. Here we have examined the effects of nerve injury in new transgenic mouse models overexpressing by approximately threefold wild-type or mutant (G348C) TDP-43 species. Four weeks after axonal crush of sciatic nerve, TDP-43 transgenic mice remained paralyzed at the injured limb unlike control mice, which had regained most of their normal mobility. In contrast to normal mice, TDP-43 transgenic mice exhibited sustained elevation of TDP-43 cytoplasmic levels in motor neurons after nerve crush, and the relocalization of TDP-43 to the nucleus was delayed by several weeks. After crush, peripherin and ubiquitin levels remained also significantly elevated in TDP-43 transgenic mice compared with control mice. Analysis of the sciatic nerve at 11 d after nerve crush showed that the number of regenerating axons in the distal portion of the lesion was considerably reduced in TDP-43 transgenic mice, especially in TDP-43G348C mice, which exhibited a reduction of ∼40%. In addition, markers of neuroinflammation were detected at much higher levels in TDP-43 transgenic mice. These results suggest that a deregulation of TDP-43 expression in ALS is a phenomenon that can affect the regenerative responses to neuronal injury and regrowth potential of axons.
Introduction
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disorder characterized by the progressive degeneration of motor neurons in the brain and spinal cord. Tar DNA binding protein 43 (TDP-43), a DNA/RNA-binding 43 kDa protein, has been implicated in ALS (Arai et al., 2006; Neumann et al., 2006), and dominant mutations in TARDBP, which codes for TDP-43, were reported by several groups as a primary cause of ALS (Gitcho et al., 2008; Kabashi et al., 2008; Sreedharan et al., 2008; Van Deerlin et al., 2008) and may account for ∼3% of familial ALS cases and ∼1.5% of sporadic cases. TDP-43, normally observed in the nucleus, is detected in pathological inclusions in the cytoplasm and nucleus of both neurons and glial cells of ALS and frontotemporal lobar degeneration with ubiquitin inclusions cases (Arai et al., 2006; Neumann et al., 2006). The inclusions consist prominently of TDP-43 C-terminal fragments of ∼25 kDa. The physiological role of TDP-43 and the pathogenic pathways of TDP-43 abnormalities are not well understood. TDP-43 is essential for embryogenesis (Sephton et al., 2010), and postnatal deletion of the TDP-43 gene in mice caused downregulation of Tbc1d1, a gene that alters body fat metabolism (Chiang et al., 2010). Neuronal overexpression at high levels of wild-type (WT) or mutant TDP-43 in transgenic mice caused a dose-dependent degeneration of cortical and spinal motor neurons but with no cytoplasmic TDP-43 aggregates (Wegorzewska et al., 2009; Stallings et al., 2010; Wils et al., 2010; Xu et al., 2010), raising the possibility that an upregulation of TDP-43 in the nucleus rather than TDP-43 cytoplasmic aggregates may contribute to neurodegeneration. In fact, TDP-43 has been reported to be upregulated in the CSF of patients with ALS (Kasai et al., 2009) and in peripheral blood lymphocytes from ALS patients (Mougeot et al., 2011; Nardo et al., 2011). Recently, we reported that TDP-43 is upregulated in the spinal cord of sporadic ALS cases at both the mRNA and protein levels (Swarup et al. 2011b). An upregulation of TDP-43 has also been observed after axotomy of spinal motor neurons (Moisse et al., 2009a). The fact that TDP-43 is a low-molecular-weight neurofilament mRNA binding protein capable of stabilizing the mRNA transcript (Strong et al., 2007) suggests that TDP-43 has a critical role in mediating the response of the neuronal cytoskeleton to neuronal injury.
To determine the effects of increased levels of TDP-43 on neuronal response to injury, we performed nerve crush experiments in transgenic mice bearing TDP-43 genomic fragments overexpressing at moderate levels (approximately threefold) human WT or mutant TDP-43G348C (Swarup et al., 2011a). Here, we examined the responses to nerve crush injury before the development of clinical symptoms and pathological features in these mice. Our data indicate that a deregulation of TDP-43 in ALS may cause abnormal regenerative responses and reduced axonal outgrowth after neuronal injury.
Materials and Methods
Generation of TDP-43 transgenic mice.
TARDBP (National Center for Biotechnology Information Sequence NM_007375) was amplified by PCR from a human bacterial artificial chromosome clone (clone RPCI-11, clone number 829B14) along with the endogenous promoter (∼4 kb). A315T and G348C mutations in TDP-43 were inserted using site-directed mutagenesis. The full-length genomic TARDBP (TDP-43WT and TDP-43G348C) was linearized by Swa-1 restriction enzyme and 18 kb DNA fragment microinjected in 1 d mouse embryos (having a background of C3H × C57BL/6). The embryos were implanted in pseudo-pregnant mothers (having ICR CD1 background). Founders were bred with nontransgenic C57BL/6 mice to establish stable transgenic lines (Swarup et al., 2011a). The use and maintenance of the mice described here were performed in accordance to the Guide of Care and Use of Experimental Animals of the Canadian Council on Animal Care.
Axonal crush studies.
Nontransgenic, TDP-43WT, TDP-43G348C, GFAP–luciferase (luc), TDP-43WT/GFAP–luc and TDP-43G348C/GFAP–luc mice 3–4 months old of either sex have been used for these studies. The sciatic nerve was crushed at the level of obturator tendon three times for 20 s with number 5 Dumont forceps either without (n = 6, either sex) or with (n = 7, either sex) prechilling in liquid nitrogen. Spinal cord tissue from all strains was examined on non-axotomized controls and at post-injury days 1, 3, 7, 14, and 28. For neurobehavioral assessment (NBA) scores, mice were also examined 42 d after injury. At 11 d after crush injury, animals were killed, and sciatic nerves were dissected and processed for light microscopy as described (Zhu et al., 1997). The sections were stained with toluidine blue. At least six sections were examined for each experiment, and the number of newly myelinated axons at 3 mm distal to the crush site was plotted. Similarly, the number of myelinated axons was counted at 3 mm proximal to the crush injury and at the corresponding level in the contralateral non-axotomized sciatic nerve.
Immunohistochemistry/immunofluorescence microscopy.
Four percent paraformaldehyde fixed spinal cord and brain sections of mice were sectioned and fixed on slides. For immunohistochemistry, tissues were treated with hydrogen-peroxide solution before permeabilization. After blocking with 5% normal goat serum for 1 h at room temperature, primary antibody incubations with anti-phospho-p65 (1:500; Cell Signaling Technology) were performed in 1% normal goat serum in PBS with Tween 20 overnight, followed by an appropriate Alexa Fluor 488 or 594 secondary antibody (1:500; Invitrogen) for 1 h at room temperature. For immunohistochemistry, tissues were incubated in biotinylated secondary antibodies (1:500; Vector Laboratories), incubated in avidin–biotin complex, and developed using Dab Kit (Vector Laboratories). Z-stacked sections were viewed using a 40× or 60× oil-immersion objectives on an Olympus Fluoview Confocal System.
Evaluation of immunostaining.
Immunoreactivity (IR) scores were assigned as described previously (Ravizza et al., 2006; Iyer et al., 2010) by a blinded investigator. Assigned scores represent averages for all cells in the sciatic motor neuron pool. This method was validated first by comparing scores assigned by a second blinded investigator and finding no significant difference and then by using NIH Image J analysis system as described here (Carmona et al., 2007). The overall concordance was >90%, and the overall κ value ranged from 0.86 to 0.95. In case of disagreement, independent reevaluation was performed by both observers to define the final score. Briefly, day 28 post-axonal crush images containing TDP-43-immunostained cells were threshold weighted as described previously (Moisse et al., 2009). The percentage of immunostained area was measured, and the area occupied by Harris's hematoxylin-counterstained nuclei was subtracted to give a quantitative cytosolic TDP-43 IR value. A minimum of four mice per transgenic line per time point was used for analysis (n = 4–6). Two spinal cord sections, >50 μm apart, were analyzed in two levels of lumbar cord between L3 and L5 (four sections per mouse). For peripherin and ubiquitin staining, the percentage of immunostained area was measured as described previously using NIH Image J imaging software (Carmona et al., 2007).
Behavioral assessment.
Neurobehavioural symptoms were assessed on post-injury days 1, 3, 7, 14, and 28 using a rating scale published previously (Marsala and Yaksh, 1994; Moisse et al., 2009b) consisting of two criteria: (A) walking with hindlimbs: 0, normal; 1, toes flat under body when walking but ataxia is present; 2, knuckle walking; 3, movements in hindlimbs but unable to walk; and 4, no hindlimb movement/drags hindlimbs; and (B) placing/stepping reflex: 0, normal; 1, weak; and 2, no stepping. The final score was obtained by adding the score from A with that from B for a total out of 6.
Subcellular fractionation/Western blot assay.
Spinal cord tissues were fractionated using subcellular protein fractionation kit (Thermo Fisher Scientific) following the instructions of the manufacturer. For Western blot assays, blots were incubated with primary antibodies against human monoclonal transactive response DNA-binding protein antibody (1:1000; clone E2-D3; Abnova). Immunoreactive proteins were then visualized by chemiluminescence (PerkinElmer Life and Analytical Sciences) as described previously (Swarup et al., 2011b). Actin (1:10,000; Millipore Bioscience Research Reagents) was used as a cytoplasmic control, and histone (1:1000; Abcam) was used as nuclear control.
ELISA.
The levels of TNF-α, IL-1β, IL-6, MCP-1, and IFN-γ were assayed by multi-analyte ELISA and MIX–MATCH ELISAarray kits (mouse inflammatory cytokine array; SABiosciences) and were performed according to the instructions of the manufacturer.
In vivo bioluminescence imaging.
As described previously (Keller et al., 2009, 2011), the bioluminescence images were gathered using IVIS 200 Imaging System (CaliperLS-Xenogen). Twenty-five minutes before imaging session, the mice received intraperitoneal injection of the luciferase substrate d-luciferine (150 mg/kg for mice between 20 and 25 g, 150–187.5 ml of a solution of 20 mg/ml d-luciferine dissolved in 0.9% saline was injected) (CaliperLS-Xenogen).
Statistical analyses.
For statistical analyses, the data obtained from independent experiments were presented as the mean ± SEM. Two-way ANOVA followed by Bonferroni's multiple comparison tests were used for all IR score, Mac-2-positive (Mac-2+) count, NBA data, axonal caliber distribution, and ELISArray. One-way ANOVA followed by Tukey's multiple comparison tests were used for analysis of axon counts in sciatic nerve and root ganglia. All analyses were executed with Prism 5 software (GraphPad Software). Statistical significance was set at p < 0.05.
Results
Sustained behavioral deficits in TDP-43 transgenic mice after nerve crush
We used presymptomatic 3-month-old mice in all these experiments. The clinical evaluation of the mice paralysis was performed by NBA score. Mice subjected to nerve crush injury from all strains exhibited extreme left hindlimb paralysis immediately after crush such that they dragged the limb while walking. They only extended toes slightly, if at all, when prompted to place the foot of the lesioned side on a bar and grasp. Although all the mice's scores were comparable 1 d after nerve crush, the recovery scores of nontransgenic and TDP-43WT mice differed significantly after 3 d (Fig. 1B). In contrast, the NBA score of TDP-43G348C mice remained high 3 d after nerve crush. Behavioral scores for TDP-43G348C mice improved at post-injury day 42, but they were still significantly different from nontransgenic mice (p < 0.05). The TDP-43G348C mice had a severely impaired mobility even at post-injury day 28 (p < 0.001, n = 5), unlike nontransgenic mice. Similarly, TDP-43WT mice recovered more slowly than nontransgenic mice as shown at post-injury days 14 and 28 (p < 0.001, n = 5) (Fig. 1B).
Figure 1.
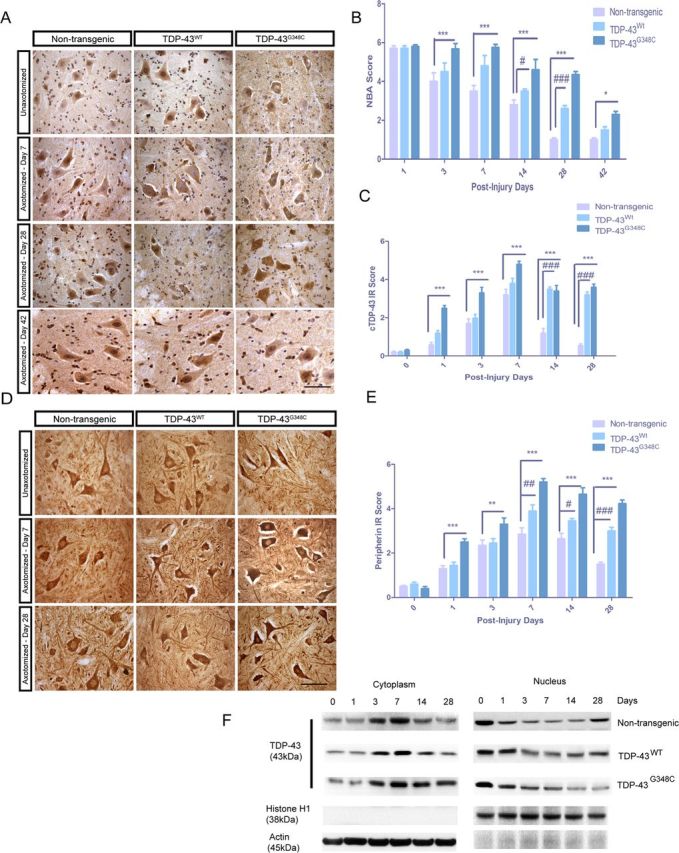
NBA and quantification by IR score of cytoplasmic TDP-43 and peripherin in spinal motor neurons of mice after nerve crush. A, Representative images of immunohistochemistry on non-axotomized and axotomized (days 7, 28, and 42) spinal cord tissues using polyclonal TDP-43 antibody are shown. C, Cytoplasmic TDP-43 immunoreactivity score reveals that, in nontransgenic mice, TDP-43 was translocated in the cytosol with a peak at 7 d after nerve crush and mainly returned in the nucleus 28 d after injury. In TDP-43WT mice, the peak is also at 7 d, but cytosolic TDP-43 was still much higher than nontransgenic mice at 14 and 28 d after injury (###p < 0.001, compared with nontransgenic mice). In TDP-43G348C mice, cytosolic TDP-43 was higher on all days after nerve crush, and its mislocalization was substantially maintained at 28 d (***p < 0.001, compared with nontransgenic mice) (B). At 1 d after nerve crush, NBA scores of all the mice were comparable. TDP-43WT mice recovered slightly more slowly than nontransgenic mice, and their NBA score was significantly higher 14 d (#p < 0.05) and 28 d (###p < 0.001, n = 5, compared with nontransgenic mice) after surgery. NBA score of TDP-43G348C mice was significantly higher from 3 d after nerve crush (*p < 0.05), and they still had a severely impaired mobility at 28 d (***p < 0.001, n = 5, compared with nontransgenic mice) and 42 d (*p < 0.05). Error bar represents mean ± SEM. D, E, Representative images of immunohistochemistry on non-axotomized and axotomized (days 7 and 28) spinal cord tissues using polyclonal peripherin antibody are shown. IR scores reveal that peripherin was upregulated 7 d after injury in nontransgenic mice and decreased rapidly on 14 and 28 d after injury. In both TDP-43WT and TDP-43G348C transgenic mice, the peak IR was at 7 d but the peripherin levels were significantly upregulated 14 and 28 d after injury (**p < 0.01, ***p < 0.001, #p < 0.05, ##p < 0. 01, ###p < 0.001, compared to nontransgenic mice). Scale bars, 50 μm. n = 5 for all quantifications. Error bar represents mean ± SEM. F, Western blot analysis of the cytoplasmic and nuclear fractions from spinal cord tissues of post-axotomized nontransgenic and TDP-43 transgenic mice. In nontransgenic mice, the TDP-43 levels came back to normal levels at 28 d after axotomy. In contrast, at day 28 after axotomy, the cytoplasmic and nuclear TDP-43 levels did not return to normal levels in TDP-43G348C and TDP-43WT samples.
Sustained increases in cytoplasmic TDP-43 and peripherin IR after nerve crush
Cytoplasmic localization of TDP-43 after injury has been reported previously (Moisse et al., 2009a,b). We wanted to determine whether overexpression of WT or mutant TDP-43 in transgenic mice had any effects on the localization of TDP-43. We used TDP-43 polyclonal antibody that recognizes both endogenous mouse and human TDP-43. In nontransgenic control mice, TDP-43 was translocated in the cytoplasm ipsilateral to nerve crush with peak IR at 7 d after nerve crush and sharply returned to the nucleus 14 and 28 d after the injury (Fig. 1A,C). In the spinal cord sections, cytoplasmic TDP-43 was seen mostly in neurons as determined by their shape and size in the staining. In TDP-43WT mice, the peak of TDP-43 IR in the cytoplasm was also seen at 7 d, but it remained much higher than nontransgenic mice at 14 and 28 d after injury (p < 0.001). Moreover, in TDP-43G348C mice, cytosolic TDP-43 IR was significantly higher at 7 d after nerve crush, and its mislocalization was substantially maintained at 28 d (p < 0.001) compared with nontransgenic mice (Fig. 1A,C). Nonetheless, in many neurons ipsilateral to the nerve-crush site, TDP-43 was found relocalized to the nucleus at 42 d after injury in both TDP-43WT and TDP-43G348C mutant mice (Fig. 1A). TDP-43 levels in axotomized spinal cord tissues were also analyzed by subcellular fractionation. Western blot analysis of the cytoplasmic fraction in post-axotomized nontransgenic spinal cord tissues revealed an increase in cytoplasmic TDP-43 levels, reaching highest cytoplasmic TDP-43 levels at 7 d after axotomy. Cytoplasmic levels came down to basal levels at 28 d after axotomy. Nuclear TDP-43 levels decreased sharply in axotomized samples, reaching lowest TDP-43 levels on day 7 and then gradually returning to normal levels at day 28 (Fig. 1F). In contrast, at day 28 after axotomy, the cytoplasmic TDP-43 levels did not return to normal levels in TDP-43G348C and TDP-43WT samples (Fig. 1F). In agreement with these results, nuclear TDP-43 levels remained low in 28 d TDP-43G348C and TDP-43WT samples.
We then investigated the IR and the distribution of peripherin, a type III neuronal intermediate filament (IF) protein, after axotomy. We chose to study peripherin IR because peripherin inclusions are hallmarks of ALS patients (Corbo and Hays, 1992; Migheli et al., 1993) and peripherin expression levels are increased by up to 300% in spinal motor neurons after injury of the sciatic nerve (Troy et al., 1990). In the spinal cord sections, peripherin IR was highest in the cytoplasm 7 d after injury in nontransgenic mice and gradually decreased in IR at 14 and 28 d after injury (Fig. 1D,E). In both TDP-43WT and TDP-43G348C transgenic mice, the peak IR of peripherin was at 7 d after injury. However, TDP-43G348C mice had significantly higher peripherin IR at post-injury days 14 and 28 (p < 0.001), and higher peripherin levels were also seen in TDP-43WT mice (p < 0.001).
Increased ubiquitin expression and microglial proliferation in TDP-43 transgenic mice after nerve crush
The role of increased ubiquitin expression in motor neurons after axotomy have been reported previously (Savedia and Kiernan, 1994). We thus analyzed IR score for ubiquitin using commercially available polyclonal anti-ubiquitin antibody. In nontransgenic control mice, ubiquitin IR in the spinal cord showed peak levels at 7 d after injury and gradually decreased at 14 and 28 d after injury (Fig. 2A,B). Ubiquitin IR also reached its peak at 7 d after injury in TDP-43WT mice and TDP-43G348C mice, but significantly higher ubiquitin IR remained at 14 and 28 d (p < 0.001) compared with nontransgenic mice (Fig. 2A,B).
Figure 2.
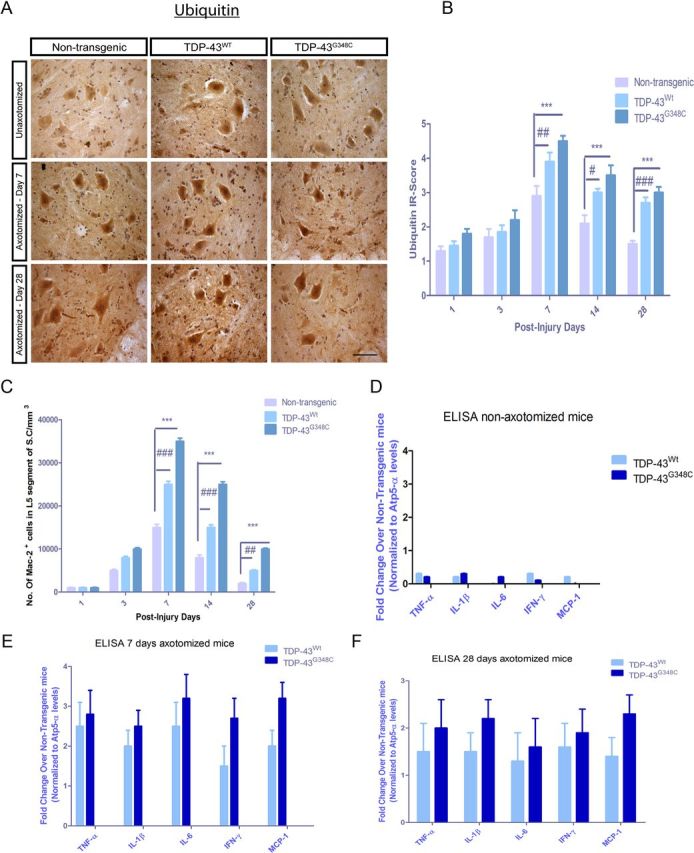
Quantification of ubiquitin and microgliosis in spinal cord of mice after nerve crush. A, B, Nerve-crush-induced upregulation of ubiquitin was at its highest level 7 d after injury in nontransgenic mice and in TDP-43WT mice. Nevertheless, TDP-43WT mice maintained more ubiquitin expression at 28 d (#p < 0.05, ##p < 0.01, ###p < 0.001, compared with nontransgenic mice). The peak level of ubiquitin was also at 7 d in TDP-43G348C mice and stayed higher at 14 and 28 d (***p < 0.001, compared with nontransgenic mice). C, Nerve crush provoked a progressive microgliosis as seen by Mac-2+ cell count with a peak at 7 d in nontransgenic, TDP-43WT, and TDP-43G348C mice. In TDP-43WT mice, Mac-2+ cell counts were significantly higher (25,000 ± 600/mm3) than nontransgenic mice (###p < 0.001). At post-injury days 14 and 28, Mac-2+ counts were significantly higher in TDP-43WT mice compared with nontransgenic mice (##p < 0.01, ###p < 0.001). Mac-2+ cells counts were higher at 3, 7, 14, and 28 d in TDP-43G348C mice compared with nontransgenic mice. TDP-43WT mice had its maximum number of Mac-2+ cells 7 d after injury, and it was sustained until the end of the observations (***p < 0.001, compared with nontransgenic mice). D–F, Mouse inflammatory multiplex ELISA was performed on non-axotomized (D) and 7 d (E) and 42 d (F) after axotomy. In non-axotomized tissues, there was no change in the levels of proinflammatory cytokines and chemokines in TDP-43WT and TDP-43G348C samples compared with nontransgenic samples. At 7 d after injury, the levels of TNF-α (2.5-fold in TDP-43WT and 2.8-fold in TDP-43G348C), IL-6 (2.5-fold in TDP-43WT and 3.2-fold in TDP-43G348C), IL-1β (2-fold in TDP-43WT and 2.5-fold in TDP-43G348C), IFNγ (1.5-fold in TDP-43WT and 2.5-fold in TDP-43G348C), and MCP-1 (2-fold in TDP-43WT and 3.2-fold in TDP-43G348C) were all upregulated compared with nontransgenic control. Although most of the cytokine and chemokine levels had reduced 28 d after axonal crush in nontransgenic mice, the levels of TNF-α (1.5-fold in TDP-43WT and 2.0-fold in TDP-43G348C), IL-6 (1.3-fold in TDP-43WT and 1.6-fold in TDP-43G348C), and IL-1β (1.5-fold in TDP-43WT and 2.2-fold in TDP-43G348C) were significantly upregulated compared with nontransgenic control. Error bar represents mean ± SEM. Scale bars, 50 μm. n = 5 for all quantifications.
We studied the inflammatory response to nerve crush in TDP-43 transgenic mice using Mac-2+ microglial cell counts and mouse inflammatory multianalyte ELISArray (SABiosciences). Nerve crush provoked a progressive microgliosis in the spinal cord of nontransgenic mice, with highest number of Mac-2+ cells (15,000 ± 750/mm3) at 7 d after injury, and it gradually declined at 14 and 28 d (2000 ± 200/mm3) after injury (Fig. 2C). In TDP-43WT mice, Mac-2+ cells were also highest at day 7, but their numbers were significantly higher (25,000 ± 600/mm3) than nontransgenic mice. At post-injury days 14 and 28, Mac-2+ counts were significantly higher in TDP-43WT mice than in nontransgenic mice. The highest Mac-2+ cells counts at 3, 7, 14, and 28 d after crush were detected in TDP-43G348C mice (Fig. 2C). To further evaluate the inflammatory response to nerve crush, we measured the protein levels of proinflammatory cytokines using mouse multianalyte ELISA system. We dissected spinal cords and compared the ipsilateral side with nerve crush with the contralateral side for nontransgenic, TDP-43WT, and TDP-43G348C transgenic mice. The protein levels of all studied proinflammatory cytokines and chemokines were not significantly altered in non-axotomized TDP-43WT and TDP-43G348C mice when compared with their nontransgenic littermates (Fig. 2D). However, the levels of proinflammatory cytokines and chemokines were significantly upregulated in the ipsilateral side of TDP-43WT and TDP-43G348C mice at 7 d after injury when compared with their nontransgenic littermates. At 7 d after injury, the levels of TNF-α (2.5-fold), IL-6 (2.5-fold), IL-1β (2-fold), IFNγ (1.5 fold), and MCP-1(2-fold) were all upregulated in TDP-43WT mice compared with nontransgenic control. Moreover, the upregulation in levels of TNF-α (2.8-fold), IL-6 (3.2-fold), IL-1β (2.5-fold), IFNγ (2.5-fold), and MCP-1(3.2-fold) was more pronounced in TDP-43G348C mice (Fig. 2E). Although the cytokine and chemokine levels had reduced 28 d after axonal crush in nontransgenic mice, the levels of TNF-α (1.5-fold in TDP-43WT and 2.0-fold in TDP-43G348C), IL-6 (1.3-fold in TDP-43WT and 1.6-fold in TDP-43G348C), and IL-1β (1.5-fold in TDP-43WT and 2.2-fold in TDP-43G348C) remained significantly elevated (Fig. 2F).
Altered proinflammatory repertoire of cytokines and chemokines led us to investigate the expression pattern of phosphorylated p65, a subunit of NF-κB. Immunofluorescence of horizontal sections of sciatic nerve and subsequent quantification of phospho-p65+ cells revealed significantly increased p65+ cells in distal sections of TDP-43WT and TDP-43G348C sciatic nerve when compared with nontransgenic control (Fig. 3A,B).
Figure 3.
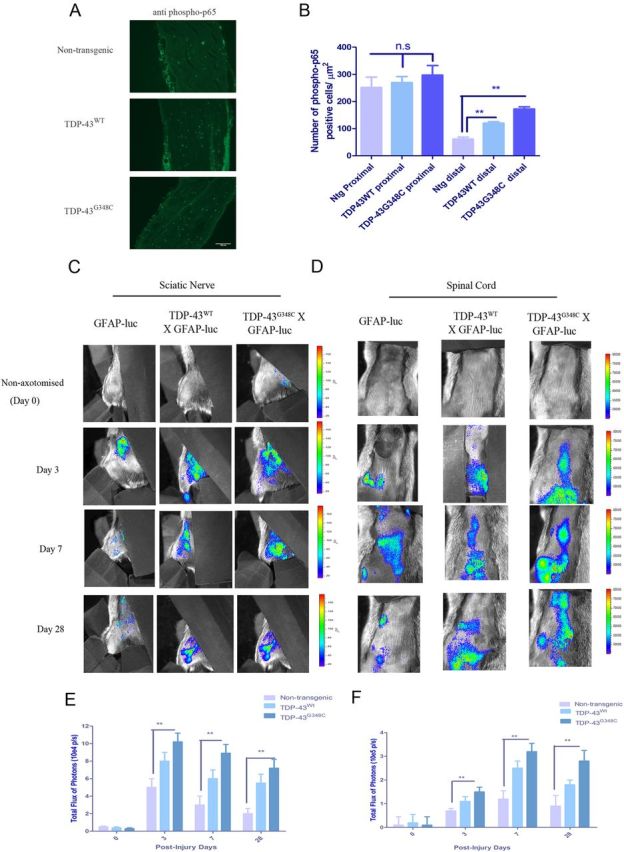
In vivo imaging revealed altered GFAP activation in double transgenic TDP-43;GFAP–luc mice after axotomy. A, Horizontal sections of sciatic nerve from nontransgenic, TDP-43WT, and TDP-43G348C mice were subjected to immunofluorescence using anti-phospho-p65 antibody. Shown are representative images distal to the axotomized site. B, Quantification showing number of phospho-p65+ cells/μm2 of distal and proximal sections of nontransgenic, TDP-43WT, and TDP-43G348C mice. C, D, GFAP–luc, TDP-43WT;GFAP–luc, and TDP-43G348C;GFAP–luc mice were imaged for GFAP promoter luciferase activity after nerve crush in sciatic nerve (C) and spinal cord (D). Representative images from in vivo imaging of sciatic nerve in non-axotomized and 3, 7, and 28 d post-axotomized mice are shown. E, F, Quantitative analysis of in vivo imaging of GFAP signal/bioluminescence (total flux of photon per second) reveals that TDP-43WT;GFAP–luc and TDP-43G348C;GFAP–luc mice have significantly higher counts in sciatic nerve (E) and spinal cord (F) than GFAP–luc controls at 3, 7, and 28 d after axotomy. Error bars represent mean ± SEM. n = 5 per group. **p < 0.01.
Considering an involvement of GFAP upregulation in Schwann cells and astrocytes in nerve regeneration after injury (Keller et al., 2009), we generated and analyzed double transgenic mice carrying a TDP-43 transgene and a GFAP–luc transgene consisting of the reporter luciferase driven by the murine GFAP promoter (Keller et al., 2009; Swarup et al., 2011a). To analyze the spatial and temporal dynamics of GFAP induction in TDP-43 mouse model after axotomy, we performed series of live imaging experiments, starting at 3 d after axotomy until 28 d, and compared it with non-axotomized littermates. Quantitative analysis of the imaging signals in the sciatic nerve revealed an upregulation of GFAP promoter activity (Fig. 3C,D) at 3 d after axotomy in control GFAP–luc mice, with gradual decline at 7 and 28 d after axotomy. In TDP-43WT/GFAP–luc and TDP-43G348C/GFAP–luc mice, there was a significant upregulation in GFAP promoter activity at 3, 7, and 28 d after axotomy compared with nontransgenic GFAP–luc mice (Fig. 3E,F). In the spinal cord, GFAP activity reached a peak at 7 d after axotomy, and it declined 28 d after axotomy. In both TDP-43WT/GFAP–luc and TDP-43G348C/GFAP–luc mice, the level of GFAP promoter activity was higher than nontransgenic controls.
Delayed regeneration of myelinated axons in TDP-43 transgenic mice after nerve crush
We wanted to examine how myelinated axons of peripheral nerves would regenerate in the presence of WT or mutant TDP-43 after axonal crush. We examined the sciatic nerve in the regions proximal and distal to the initial crush 11 d after injury as illustrated in Fig. 4A. Axonal responses in the dorsal root ganglia and in the distal sciatic nerve 11 d after nerve crush were evaluated. Electron microscopy of transversal sections of the distal nerve showed massive degeneration in ipsilateral sections to injury compared with contralateral control nerves (Fig. 4B). No sign of degeneration of myelinated axons was evident in the nerve region 3 mm proximal to crush site (data not shown). Quantifications of the total number of regenerating axons (Fig. 4C) surrounded by myelin (arrows in electron microscopy) revealed a significant difference between nontransgenic (mean of 571 ± 27) and TDP-43G348C transgenic (mean of 335 ± 63) mice. We conclude from these results that TDP-43G348C mice had lower regeneration capacity of myelinated axons in the peripheral nerves compared with nontransgenic controls. TDP-43WT mice also had lower regeneration capacity than nontransgenic mice but to a lesser extent than TDP-43G348C mice.
Figure 4.

Delayed regeneration of axons in the distal sciatic nerve 11 d after nerve crush. A, Schematic representation of the mice indicating the nerve crush site (forceps) and showing where the nerve analysis has been made. DRG, Dorsal root ganglion; D.N., distal nerve sections, 3 mm distal to the nerve crush site. B, Electron microscopy of transversal sections of the distal nerve revealed massive degeneration in ipsilateral (axotomized) sections compared with contralateral (non-axotomized) control nerves (left in B). C, Quantifications of the total number of regenerating axons surrounded by myelin (arrows in B) revealed a significant difference (p = 0.0264, n = 3) between nontransgenic (mean of 571 ± 27) and TDP-43G348C transgenic mice (mean of 335 ± 63) (top right in B). D, Toluidine blue staining of thin sections of L5 ventral root axons from nontransgenic, TDP-43WT, and TDP-43G348C mice showed no significant differences in the axonal count. E, The axonal calibers were measured at 11 d after injury in the sciatic nerve of nontransgenic and transgenic mice. An analysis of the plotted distribution confirms a significant reduction of the axon calibers in TDP-43G348C (p = 0.0261, n = 3, compared with nontransgenic mice). F, G, Axonal counts performed at 11 d after injury in the ventral root (F) and dorsal root (G) of the dorsal root ganglia were not significantly different, neither between the contralateral and the ipsilateral sections nor between any of the axotomized groups. Error bar represents mean ± SEM. Scale bars: B, 5 μm; D, 50 μm.
We then investigated whether nerve crush in TDP-43 transgenic mice changed the number and distribution of axon caliber of the sciatic nerve. The axonal calibers were slightly lower in TDP-43G348C and TDP-43WT mice when compared with nontransgenic (Fig. 4D,E). An analysis of the plotted distribution of axonal caliber confirms a significant shift of TDP-43G348C axon caliber (p = 0.0261, n = 3, compared with nontransgenic mice) (Fig. 4E). However, the numbers of axons in the ventral and dorsal root of the dorsal root ganglia were not different (Fig. 4F,G) between the contralateral and the ipsilateral sections or between any of the transgenic or nontransgenic mice.
Discussion
The recent finding of an upregulation of TDP-43 mRNA in spinal cord of sporadic ALS patients (Swarup et al. 2011b) prompted us to investigate whether overexpression of TDP-43 can affect the regenerative responses after neuronal injury. Here, we took advantage of new transgenic mouse models made with TDP-43 genomic fragments (Swarup et al. 2011a). The results show that an upregulation of TDP-43 WT or mutant altered the nervous tissue responses to peripheral nerve injury, and it delayed axonal outgrowth after nerve crush damage.
The NBA score of TDP-43G348C mice was significantly higher than control mice 3 d after nerve crush, and they had a severely impaired mobility even at post-injury day 28. The behavioral score of TDP-43WT mice was also significantly higher at 14 and 28 d after injury compared with nontransgenic mice. Moreover, TDP-43G348C mice did not recover from nerve crush after 28 d, in contrast to nontransgenic mice that recovered almost completely, whereas TDP-43WT mice recovered partially. Thus, the abnormal NBA scores of TDP-43WT mice and TDP-43G348C mice after nerve injury suggest that changes in TDP-43 homeostasis might affect recovery process after neuronal injury. In response to axonal injury, TDP-43 is relocalized to the cytosol in a time-dependent manner (Moisse et al., 2009b). In agreement with this report, immunohistochemistry of the spinal cord sections revealed cytoplasmic relocalization of TDP-43 after sciatic nerve crush in TDP-43WT and TDP-43G348C transgenic mice. However, this phenomenon was abnormally long lasting in TDP-43 transgenic mice. After nerve crush injury in nontransgenic mice, TDP-43 redistributed to the cytoplasm, reaching a peak of cytoplasmic distribution at 7 d after injury, with normal expression pattern being restored at 28 d after injury (Fig. 1C). In TDP-43WT and TDP-43G348C mice, the peak of TDP-43 IR in the cytoplasm also occurred at 7 d after injury, but the cytoplasmic TDP-43 levels remained at high levels at 14 and 28 d after injury. The TDP-43 expression patterns were only restored at 42 d after injury.
Perikaryal and axonal aggregates of IFs is a hallmark of degenerating motor neurons in ALS (Carpenter, 1968; Corbo and Hays, 1992; Migheli et al., 1993). Peripherin is a type III IF whose expression is known to be upregulated in neuronal injury or stroke (Beaulieu et al., 2002). Using immunohistochemistry, we found that peripherin was upregulated in a time-dependent manner, reaching peak levels in cytoplasm 7 d after injury in nontransgenic mice. Interestingly, peripherin levels in TDP-43G348C mice were significantly higher at all days after injury. Whereas peripherin levels returned back to normal levels at 28 d after injury in nontransgenic mice, peripherin remained significantly upregulated at 28 d after injury in TDP-43WT and TDP-43G348C mice. Ubiquitin, another protein involved in injury (Yamauchi et al., 2008), was also examined for its expression levels. In normal mice, ubiquitin levels in motor neurons were upregulated after injury and returned to the baseline levels at 28 d after injury. In contrast, ubiquitin levels remained significantly upregulated at 28 d after injury in both TDP-43WT and TDP-43G348C mice.
We then examined inflammatory response to sciatic nerve crush in transgenic mice and compared them with nontransgenic mice (Fig. 2C,E). Mac-2+ microglial cell counts were highest at 7 d after injury and gradually declined at 14 and 28 d after injury in nontransgenic mice. In contrast, Mac-2+ cells were significantly higher in both TDP-43WT and TDP-43G348C mice. Higher number of Mac-2+ microglial cells prompted us to evaluate the cytokine and chemokine profiles in the spinal cord tissue samples of 7 and 28 d post-injury mice. Consistent with Mac-2+ cell count, many inflammatory cytokines, such as TNF-α and IL-1β, and chemokines, such as MCP-1, were upregulated 7 d after injury in nontransgenic mice and gradually declined to normal levels 28 d after injury. However, TDP-43WT and TDP-43G348C mice had higher levels of cytokine levels at both 7 and 28 d after injury compared with nontransgenic mice.
The delayed functional recovery of TDP-43 transgenic mice after nerve crush prompted us to investigate the regeneration of myelinating axons. We examined the sciatic nerve in the region distal to the initial crush 11 d after injury (Fig. 4A). Electron microscopy of transversal sections of the distal nerve revealed massive axonal degeneration in nerve sections ipsilateral to injury. Moreover, much less regenerating axons were detected in TDP-43 transgenic mice than in normal mice. For instance, at 11 d after crush, the number of regenerating axons was diminished by 40% in TDP-43G348C transgenic mice compared with normal mice.
It can be concluded that TDP-43G348C mice and to a lesser extent TDP-43WT mice had lower regeneration capacity of myelinated axons in the peripheral nerves compared with nontransgenic controls. How TDP-43 deregulation can affect regenerative responses and delayed axonal outgrowth after nerve injury? We recently reported that overexpression of TDP-43 in either WT or mutant form can cause exaggerated innate immune responses (Swarup et al., 2011b). TDP-43 acts as coactivator of nuclear factor-κB (NF-κB) via interaction of its RNA recognition motif 1 with the NF-κB p65 subunit (Swarup et al., 2011b). Thus, it seems plausible that an exaggerated inflammation in TDP- 43 transgenic mice after nerve crush (Fig. 2) may slow down the repair process. For instance, there is evidence that the inflammatory response through NF-κB activation in Schwann cells at the site of nerve injury may delay the onset of axonal sprouting and regeneration (Smith et al., 2009). Thus, impairment of axonal regeneration in TDP-43WT and TDP-43G348C mice may result in part from the increased number of activated p65+ cells in distal sciatic nerve after crush (Fig. 3A,B). In the future, it would be of relevance to the ALS problem to test pharmacological inhibitors of NF-κB activation in TDP-43 transgenic mice for their capacity to restore regenerative responses and to enhance axonal outgrowth after neuronal injury.
Footnotes
This work was supported by the Canadian Institutes of Health Research and by the Neuromuscular Research Partnership. J.-P.J. holds a Canada Research Chair Tier 1 in mechanisms of neurodegeneration. V.S. was the recipient of the Merit Scholarship for foreign students (Funds of Research of Quebec for Nature and Technologies). We thank Christine Bareil and Genevieve Soucy for technical assistance.
References
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Kriz J, Julien JP. Induction of peripherin expression in subsets of brain neurons after lesion injury or cerebral ischemia. Brain Res. 2002;946:153–161. doi: 10.1016/s0006-8993(02)02830-5. [DOI] [PubMed] [Google Scholar]
- Carmona R, Macías D, Guadix JA, Portillo V, Pérez-Pomares JM, Muñoz-Chápuli R. A simple technique of image analysis for specific nuclear immunolocalization of proteins. J Microsc. 2007;225:96–99. doi: 10.1111/j.1365-2818.2007.01719.x. [DOI] [PubMed] [Google Scholar]
- Carpenter S. Proximal axonal enlargement in motor neuron disease. Neurology. 1968;18:841–851. doi: 10.1212/wnl.18.9.841. [DOI] [PubMed] [Google Scholar]
- Chiang PM, Ling J, Jeong YH, Price DL, Aja SM, Wong PC. Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc Natl Acad Sci U S A. 2010;107:16320–16324. doi: 10.1073/pnas.1002176107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbo M, Hays AP. Peripherin and neurofilament protein coexist in spinal spheroids of motor neuron disease. J Neuropathol Exp Neurol. 1992;51:531–537. doi: 10.1097/00005072-199209000-00008. [DOI] [PubMed] [Google Scholar]
- Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ, White CL, 3rd, Bigio EH, Caselli R, Baker M, Al-Lozi MT, Morris JC, Pestronk A, Rademakers R, Goate AM, Cairns NJ. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63:535–538. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer AM, Zurolo E, Boer K, Baayen JC, Giangaspero F, Arcella A, Di Gennaro GC, Esposito V, Spliet WG, van Rijen PC, Troost D, Gorter JA, Aronica E. Tissue plasminogen activator and urokinase plasminogen activator in human epileptogenic pathologies. Neuroscience. 2010;167:929–945. doi: 10.1016/j.neuroscience.2010.02.047. [DOI] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Kasai T, Tokuda T, Ishigami N, Sasayama H, Foulds P, Mitchell DJ, Mann DM, Allsop D, Nakagawa M. Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2009;117:55–62. doi: 10.1007/s00401-008-0456-1. [DOI] [PubMed] [Google Scholar]
- Keller AF, Gravel M, Kriz J. Live imaging of amyotrophic lateral sclerosis pathogenesis: disease onset is characterized by marked induction of GFAP in Schwann cells. Glia. 2009;57:1130–1142. doi: 10.1002/glia.20836. [DOI] [PubMed] [Google Scholar]
- Keller AF, Gravel M, Kriz J. Treatment with minocycline after disease onset alters astrocyte reactivity and increases microgliosis in SOD1 mutant mice. Exp Neurol. 2011;228:69–79. doi: 10.1016/j.expneurol.2010.12.010. [DOI] [PubMed] [Google Scholar]
- Marsala M, Yaksh TL. Transient spinal ischemia in the rat: characterization of behavioral and histopathological consequences as a function of the duration of aortic occlusion. J Cereb Blood Flow Metab. 1994;14:526–535. doi: 10.1038/jcbfm.1994.65. [DOI] [PubMed] [Google Scholar]
- Migheli A, Pezzulo T, Attanasio A, Schiffer D. Peripherin immunoreactive structures in amyotrophic lateral sclerosis. Lab Invest. 1993;68:185–191. [PubMed] [Google Scholar]
- Moisse K, Volkening K, Leystra-Lantz C, Welch I, Hill T, Strong MJ. Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: implications for TDP-43 in the physiological response to neuronal injury. Brain Res. 2009a;1249:202–211. doi: 10.1016/j.brainres.2008.10.021. [DOI] [PubMed] [Google Scholar]
- Moisse K, Mepham J, Volkening K, Welch I, Hill T, Strong MJ. Cytosolic TDP-43 expression following axotomy is associated with caspase 3 activation in NFL−/− mice: support for a role for TDP-43 in the physiological response to neuronal injury. Brain Res. 2009b;1296:176–186. doi: 10.1016/j.brainres.2009.07.023. [DOI] [PubMed] [Google Scholar]
- Mougeot JL, Li Z, Price AE, Wright FA, Brooks BR. Microarray analysis of peripheral blood lymphocytes from ALS patients and the SAFE detection of the KEGG ALS pathway. BMC Med Genomics. 2011;4:74. doi: 10.1186/1755-8794-4-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardo G, Pozzi S, Pignataro M, Lauranzano E, Spano G, Garbelli S, Mantovani S, Marinou K, Papetti L, Monteforte M, Torri V, Paris L, Bazzoni G, Lunetta C, Corbo M, Mora G, Bendotti C, Bonetto V. Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS One. 2011;6:e25545. doi: 10.1371/journal.pone.0025545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Ravizza T, Boer K, Redeker S, Spliet WG, van Rijen PC, Troost D, Vezzani A, Aronica E. The IL-1beta system in epilepsy-associated malformations of cortical development. Neurobiol Dis. 2006;24:128–143. doi: 10.1016/j.nbd.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Savedia S, Kiernan JA. Increased production of ubiquitin mRNA in motor neurons after axotomy. Neuropathol Appl Neurobiol. 1994;20:577–586. doi: 10.1111/j.1365-2990.1994.tb01012.x. [DOI] [PubMed] [Google Scholar]
- Sephton CF, Good SK, Atkin S, Dewey CM, Mayer P, 3rd, Herz J, Yu G. TDP-43 is a developmentally regulated protein essential for early embryonic development. J Biol Chem. 2010;285:6826–6834. doi: 10.1074/jbc.M109.061846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D, Tweed C, Fernyhough P, Glazner GW. Nuclear factor-kappaB activation in axons and Schwann cells in experimental sciatic nerve injury and its role in modulating axon regeneration: studies with etanercept. J Neuropathol Exp Neurol. 2009;68:691–700. doi: 10.1097/NEN.0b013e3181a7c14e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallings NR, Puttaparthi K, Luther CM, Burns DK, Elliott JL. Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiol Dis. 2010;40:404–414. doi: 10.1016/j.nbd.2010.06.017. [DOI] [PubMed] [Google Scholar]
- Strong MJ, Volkening K, Hammond R, Yang W, Strong W, Leystra-Lantz C, Shoesmith C. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol Cell Neurosci. 2007;35:320–327. doi: 10.1016/j.mcn.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Swarup V, Phaneuf D, Bareil C, Robertson J, Rouleau GA, Kriz J, Julien JP. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain. 2011a;134:2610–2626. doi: 10.1093/brain/awr159. [DOI] [PubMed] [Google Scholar]
- Swarup V, Phaneuf D, Dupr é N, Petri S, Strong M, Kriz J, Julien JP. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor kappaB-mediated pathogenic pathways. J Exp Med. 2011b;208:2429–2447. doi: 10.1084/jem.20111313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy CM, Muma NA, Greene LA, Price DL, Shelanski ML. Regulation of peripherin and neurofilament expression in regenerating rat motor neurons. Brain Res. 1990;529:232–238. doi: 10.1016/0006-8993(90)90832-v. [DOI] [PubMed] [Google Scholar]
- Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen-Plotkin AS, Martinez-Lage M, Steinbart E, McCluskey L, Grossman M, Neumann M, Wu IL, Yang WS, Kalb R, Galasko DR, Montine TJ, Trojanowski JQ, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2009;106:18809–18814. doi: 10.1073/pnas.0908767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, Smits V, Ceuterick-de Groote C, Van Broeckhoven C, Kumar-Singh S. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2010;107:3858–3863. doi: 10.1073/pnas.0912417107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YF, Gendron TF, Zhang YJ, Lin WL, D'Alton S, Sheng H, Casey MC, Tong J, Knight J, Yu X, Rademakers R, Boylan K, Hutton M, McGowan E, Dickson DW, Lewis J, Petrucelli L. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci. 2010;30:10851–10859. doi: 10.1523/JNEUROSCI.1630-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi T, Sakurai M, Abe K, Matsumiya G, Sawa Y. Ubiquitin-mediated stress response in the spinal cord after transient ischemia. Stroke. 2008;39:1883–1889. doi: 10.1161/STROKEAHA.106.455832. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Couillard-Després S, Julien JP. Delayed maturation of regenerating myelinated axons in mice lacking neurofilaments. Exp Neurol. 1997;148:299–316. doi: 10.1006/exnr.1997.6654. [DOI] [PubMed] [Google Scholar]