

Abstract
Multiple sclerosis (MS) is a degenerative autoimmune disease of the CNS. Experimental autoimmune encephalomyelitis (EAE) is a commonly used murine model for MS. Here we report that CD137 ligand (CD137L, 4-1BB ligand, TNFS9), a member of the TNF superfamily, is critical for the development of EAE. EAE symptoms were significantly ameliorated in CD137L−/− mice. In the absence of CD137L, myelin oligodendrocyte glycoprotein (MOG)-specific T-cells secreted lower levels of Th1/Th17 cell-associated cytokines. MOG-specific T-cells also trafficked less efficiently to the CNS in CD137L−/− mice, possibly as a consequence of reduced expression of vascular cell adhesion molecule-1 (VCAM-1), which regulates leukocyte extravasation. Thus, CD137L regulates many functions of MOG-specific T-cells that contribute to EAE and may represent a novel therapeutic target for the treatment of MS.
Introduction
Multiple sclerosis (MS) is a T-cell-mediated autoimmune disease that affects the CNS (Goverman, 2009). The murine model of experimental autoimmune encephalomyelitis (EAE) has been used to elucidate many aspects of MS pathogenesis (Baxter, 2007). In EAE, priming of antigen-specific T-cells occurs in the periphery after immunization with a myelin-derived peptide. The encephalitogenic T-cells subsequently migrate to the CNS where they are reactivated by antigen-presenting cells (APCs) presenting self-antigen (Bailey et al., 2007). After the reactivation of effector T-cells in the CNS, a second wave of myeloid cells enters the CNS and causes demyelination resulting in the clinical manifestations of EAE, such as limb weakness and paralysis (Juedes and Ruddle, 2001; King et al., 2009; Mildner et al., 2009).
Optimal priming of T-cells by APCs requires engagement of the T-cell receptor (TCR) and costimulatory molecules expressed on T-cells (Racke et al., 2000). A number of studies have shown important roles for different costimulatory molecules in the development of EAE (Croxford et al., 1998a). The costimulatory molecule CD28 and its ligands B7-1 and B7-2 are critical for both the induction and effector phase of EAE (Chang et al., 1999). Several costimulatory molecules belonging to the TNFR/TNF superfamily have been implicated in EAE (Racke et al., 2000). For instance, CD137 (4-1BB, TNFRSF9) and CD137 ligand (CD137L, 4-1BB ligand, TNFS9) are increased in serum and CSF of MS patients (Sharief, 2002; Liu et al., 2006, 2008). CD137 is mainly expressed by activated T-cells but can also be expressed by dendritic cells, natural killer (NK) cells, neutrophils, and endothelial cells. It delivers potent costimulatory signals to T-cells that enhance cytokine secretion, proliferation, and survival. CD137L is constitutively expressed by APCs and is upregulated upon cell activation. Interestingly, in addition to acting as a ligand by cross-linking CD137, CD137L can receive signals and transmit them into the cells on which it is expressed, a process referred to as reverse signal transduction (Shao and Schwarz, 2011).
Here we show that CD137L is critical for EAE development. Priming, cytokine secretion, and trafficking of myelin oligodendrocyte glycoprotein (MOG)-specific T-cells and inflammatory monocytes were deregulated in CD137L−/− mice leading to a significant amelioration of EAE symptoms. Decreased leukocyte infiltration to the CNS correlated with a lower expression of vascular cell adhesion molecule-1 (VCAM-1), which is essential for the ability of T-cells to cross the blood–brain barrier.
Materials and Methods
Mice.
C57BL/6 mice were obtained from the Centre for Animal Resources of the National University of Singapore. CD137L−/− mice were a gift of Amgen and bred in-house under pathogen-free conditions. Institutional guidelines for animal care and use were followed throughout the experiments.
Induction of EAE and clinical evaluation.
Six- to eight-week-old male C57BL/6 or CD137L−/− mice were injected subcutaneously with 100 μg of myelin oligodendrocyte glycoprotein peptide, MOG35–55 (Sigma-Aldrich) and 1 mg of heat-killed Mycobacterium tuberculosis H37RA (Difco) emulsified in complete Freund's adjuvant (CFA). Pertussis toxin (200 ng in PBS; List Biological Laboratories) was injected intraperitoneally on days 0 and 2 after immunization. EAE clinical symptoms were scored daily as follows: 0, no clinical signs; 1, loss of tail tonicity; 2, impaired righting reflex; 3, partial hindlimb paralysis; 4, total hindlimb paralysis (Croxford et al., 1998b).
Immunohistochemistry.
Mice were killed by an overdose of CO2. Spinal cords were removed from perfused mice by intrathecal hydrostatic pressure using PBS, embedded in OCT compound (Tissue-Tek, Electron Microscopy Science) and frozen at −80°C. Cryosections (6 μm) from the lumbar region of the spinal cord were fixed with cold acetone for 5 min, blocked with 0.2% BSA for 10 min, and then stained with antibodies against CD68 (AbD Serotec), TCR-β (eBioscience), CD31 (Millipore), or VCAM-1 (Abcam). Fluorescently conjugated Cy3, Cy2, Dy649, or Dy549 secondary antibodies (Jackson ImmunoResearch Laboratories) were used for detection. Sections were counterstained with DAPI for cell nuclei visualization and mounted for analysis. To detect demyelination, sections were incubated with Luxol fast blue to stain myelin and with cresyl violet for counterstaining. Specimens were viewed by fluorescence microscopy (Axio Imager.Z1, Axiocam HRM camera; Carl Zeiss Micro Imaging). MetaMorph software (Molecular Devices) was used to analyze the images.
Cell proliferation assay and cytokine quantification.
Lymph node cells (5 × 105 cells/200 μl) obtained from MOG-immunized mice at day 10 post-immunization (p.i.) were stimulated with different concentrations of MOG35–55 peptide or media alone for 4 d in 96-well round bottom plates. During the last 18 h, cultures were pulsed with 0.5 μCi of [3H]thymidine (PerkinElmer) and incorporated radioactivity was measured using a TopCount Microplate Scintillation Counter (Packard Instruments). From a duplicate plate, supernatants were collected after 72 h of culture and frozen at −80°C until analyzed by ELISA (R&D Systems or eBioscience). Serum cytokines were analyzed using BioPlex (Bio-Rad).
Preparation of CNS-infiltrating mononuclear cells for flow cytometry.
Mice were perfused through the left cardiac ventricle with 50 ml of PBS. The spinal cords of 3–8 mice per group were removed by intrathecal hydrostatic pressure using PBS, pooled, and homogenized through a 70 μm cell strainer. After centrifugation, the cells were resuspended in 30% isotonic Percoll (Sigma-Aldrich) and overlaid onto a 70% isotonic Percoll solution. The gradient was centrifuged at 500 × g for 20 min at room temperature. The mononuclear cells were collected from the interphase and washed with PBS. Infiltrating CNS cells were characterized by fluorescence-activated cell sorting (FACS). T-cells were stained with the following antibodies: anti-CD45-V500 (BD PharMingen), anti-CD3-A700, anti-CD4-PE-Cy7, anti-CD25-APC, anti-CD137-PE, anti-Foxp3-PerCP-Cy5.5, anti-CD8-PB (BD PharMingen). For staining of myeloid cells, anti-CD45-V500, anti-CD11b-PE, anti-CD11c-PerCP-Cy5.5, anti-Ly6C-APC, anti-Ly6G-PE-Cy7, and anti-B220-FITC were used. For intracellular cytokine staining, cells were restimulated ex vivo for 5 h with 50 nm Phorbol 12,13-dibutyrate (PdBu) and 500 ng/ml ionomycin calcium salt (Sigma-Aldrich) or media alone, in the presence of brefeldin-A (eBioscience), then surface-stained for anti-CD45-V500, anti-CD3-A700, and anti-CD4-FITC. After fixation/permeabilization with the BD Fix/Perm kit, cells were stained with anti-IFN-γ-APC and anti-IL-17A-PE (BD PharMingen) antibodies. Mononuclear cells from the spleens were obtained by mechanical disruption through a 70 μm nylon cell strainer. For blood samples, ∼500 μl of blood were collected in 0.5 m EDTA-containing tubes. After centrifugation, red blood cells from single-cell suspensions of either spleens or blood were lysed, and then washed and stained for surface markers as described above. Analysis was performed with an LSR Fortessa flow cytometer (BD Biosciences) and FlowJo software (Version 9.3.1, TreeStar). Unless otherwise stated antibodies were from eBioscience. All stainings were incubated for 30 min at 4°C and contained a live/dead fixable dead stain kit in APC-Cy7 (Invitrogen). Total cell numbers were determined by flow cytometry using CountBright Absolute Counting Beads (Invitrogen).
Statistical analysis.
Prism software (Version 5, GraphPad) was used to analyze the data using the unpaired Student's t test. p < 0.05 was considered statistically significant.
Results
Decreased EAE susceptibility of CD137L−/− mice
Previous studies indicated a role for CD137 and its ligand in MS (Sharief, 2002; Liu et al., 2006, 2008). To gain further insight into the role of CD137-CD137L in EAE, we immunized wild-type (WT) and CD137L−/− mice with MOG35–55 peptide. All WT mice (8 of 8) developed EAE-related symptoms whereas only 60% of CD137L−/− mice (6 of 10) developed clinical signs of EAE (Fig. 1A). The mean day of disease onset was similar for WT and CD137L−/− mice (13 ± 0.9 vs 14.7 ± 1.6, respectively). However, the mean clinical score at the peak of disease, day 18 p.i., was significantly reduced in CD137L−/− mice (0.6 ± 0.3 compared with 3.2 ± 0.4 in the WT mice). The mean EAE score was also reduced when disease-free mice were excluded from the analysis (1.6 ± 0.7 in CD137L−/− mice compared with 3.2 ± 0.4 in WT mice (p < 0.05)). In agreement with a milder disease progression, CD137L−/− mice transiently lost weight, but recovered their initial weight by day 20 p.i. (Fig. 1B). MS is a demyelinating inflammatory disorder (Goverman, 2009). To assess CNS demyelination in CD137L-deficient and WT mice, spinal cord sections were stained with Luxol fast blue to stain myelin. Histological examination of the spinal cord revealed that the CNS of CD137L−/− mice was protected from demyelination in contrast to WT mice (Fig. 1C). In summary, our data demonstrate that the initiation and progression of EAE critically depends on CD137L expression.
Figure 1.
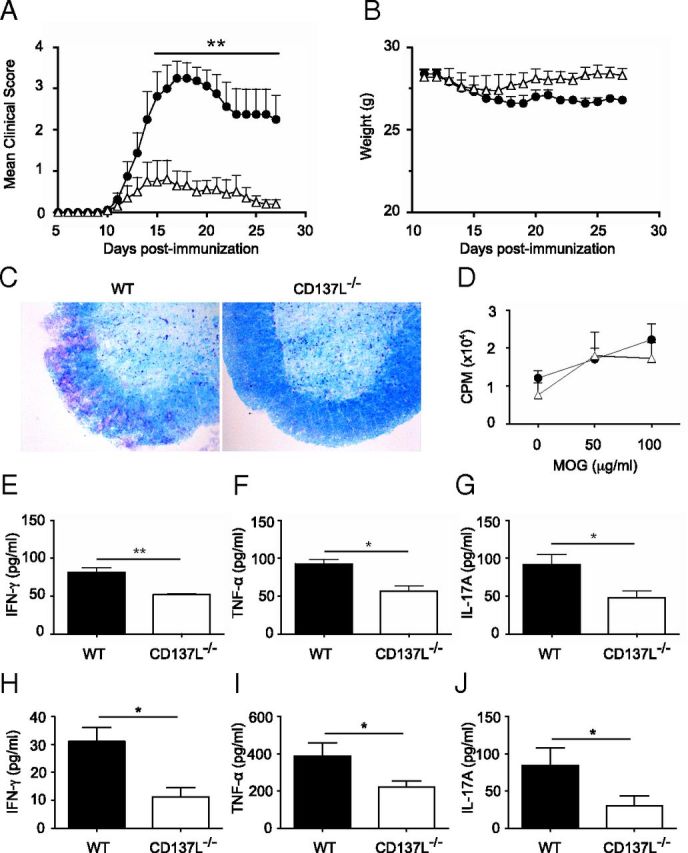
CD137L−/− mice are partially resistant to EAE. A, Clinical score of WT (filled circles) and CD137L−/− (empty triangles) mice immunized with MOG35–55 in CFA and PTx. B, Weight of mice during the course of the disease. Results represent means ± SEM (n = 8–10/group). **p < 0.01. C, Spinal cord sections from EAE mice at day 18 p.i. were evaluated for demyelination using Luxol fast blue staining and cresyl violet for counterstaining. Images are representative staining of 4 mice per group from two independent experiments. D, Lymph node cells from MOG-immunized mice were harvested 10 d p.i. and cultured with indicated concentrations of MOG35–55 peptide for 4 d. Cells were pulsed with [3H]thymidine for the last 18 h of culture and [3H]thymidine incorporation in WT (filled circles) and CD137L−/− (empty triangles) lymph node cells was analyzed. E–G, Levels of IFN-γ (E), TNF-α (F), and IL-17A (G) in the culture supernatants of WT (black bars) and CD137L−/− (white bars) DLN cells cultured in presence of 100 μg/ml MOG peptide were determined by ELISA after 72 h of culture. Results are means ± SEM (n = 3/group). *p < 0.05. H–J, The levels of IFN-γ (H), TNF-α (I), and IL-17A (J) in the sera of WT (black bars) and CD137L−/− (white bars) mice at day 18 post-MOG immunization were determined by BioPlex-ELISA. Results show means ± SEM (n = 3–4/group). Results are representative of three independent experiments.
Defective T-cell activation in CD137L−/− EAE mice
T-cells play a key role in the development of EAE (Goverman, 2009). CD137L was shown to deliver costimulatory signals that can augment T-cell activation and survival (Wang et al., 2009). To determine whether the resistance of CD137L−/− mice to EAE was due to a defect in the priming of autoreactive T-cells, we restimulated draining lymph node (DLN) cells from day 10 p.i. EAE mice with MOG peptide in vitro. No difference in the incorporation of thymidine between groups was observed suggesting that the ability of DLN cells to proliferate was not reduced in CD137L−/− EAE mice (Fig. 1D). To test whether effector functions of T-cells were impaired in CD137L−/− EAE mice, we measured the level of secreted cytokines that are important for the pathogenesis of EAE (Murphy et al., 2010). Restimulation of DLN cells derived from CD137L−/− EAE mice induced lower levels of IFN-γ, TNF-α and IL-17A, compared with DLN cells of WT mice (Fig. 1E–G). In agreement with this observation, IFN-γ, TNF-α and IL-17A serum levels of CD137L−/− EAE mice were reduced when compared with WT mice (Fig. 1H–J). These data suggest that impaired secretion of cytokines by T cells contributes to the reduced susceptibility of CD137L−/− mice to EAE.
Decreased infiltration of leukocytes in the spinal cord of CD137L−/− EAE mice
EAE immunopathology is characterized by perivascular infiltration of the CNS by inflammatory cells (Goverman, 2009). To test whether the absence of CD137L leads to fewer infiltrating immune cells in the CNS, sections of the spinal cord were analyzed by fluorescence microscopy for the presence of immune cells at day 18 p.i. A similar distribution of immune cells, mainly consisting of T cells and monocytes was found in the lateral and ventral white matter spinal cord in both mouse strains (Fig. 2A). No infiltrating cells were observed in the CNS of naive mice from either genotype (data not shown). Moreover, the number of T cells and monocytes in the blood of naive mice was similar in both strains (Fig. 2B). To quantify and characterize the T-cell and monocyte subsets, mononuclear cells from pooled spinal cords were isolated at the peak of disease and analyzed for expression of different immune cell markers by FACS. In agreement with the immunohistological analysis, the number of infiltrating T-cells and monocytes in the spinal cord of CD137L−/− mice was significantly decreased when compared with WT mice (Fig. 2C,D). In contrast, similar numbers of leukocytes were present in the spleen (Fig. 2E). Regulatory T-cells (Tregs) (CD4+CD25+Foxp3+) can suppress the development of EAE (Zhang et al., 2010). To test whether CD137L−/− mice were protected from EAE due to elevated Tregs numbers, we measured CD25 and Foxp3 expression in the infiltrating CD4+ T-cells of the CNS and in the spleen of EAE mice. The percentage and absolute numbers of Tregs were decreased in the spinal cords, but not spleen, of CD137L−/− mice when compared with WT mice (Fig. 2F,G). To test the activation status of CD4+ T-cells in the CNS and spleen of EAE mice, we stained T-cells for CD137 expression. Similar numbers of activated CD4+ T-cells were present in the spleen of WT and CD137L−/− EAE mice (Fig. 2F). However, the number of CD4+CD137+ T cells was decreased in the CNS of CD137L−/− when compared with WT mice (Fig. 2G). CNS infiltrating inflammatory monocytes are important in the effector phase of EAE (Mildner et al., 2009), whereas neutrophils have been shown to play a role during the early developmental phase of EAE (McColl et al., 1998). In fact, fewer inflammatory monocytes (Ly6ChiLy6Gint) and neutrophils (Ly6CintLy6Ghi) were found in the spinal cords of CD137L−/− mice. Hence, the reduced infiltration of the CNS by immune cells may contribute to the lower susceptibility of CD137L−/− mice to EAE.
Figure 2.
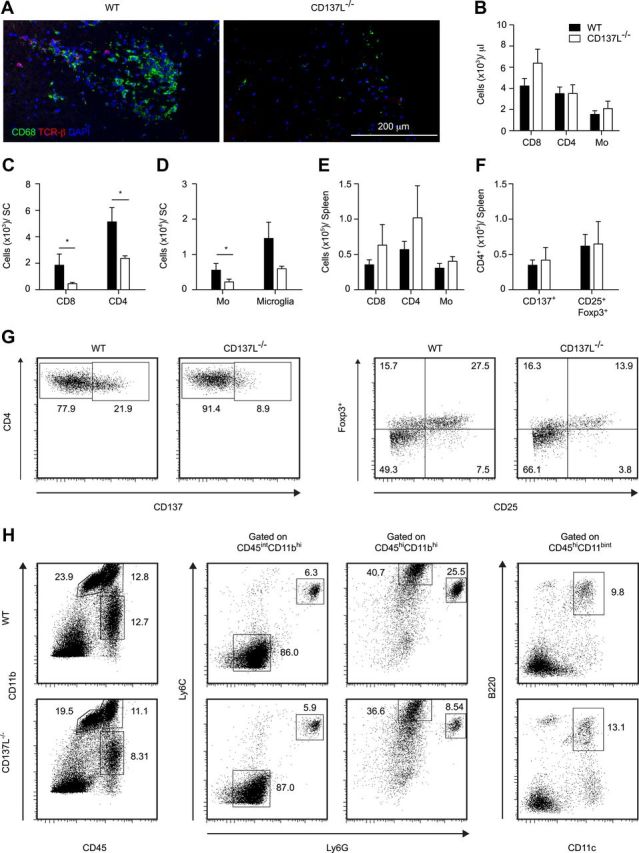
Reduced infiltration of immune cells in the spinal cord of CD137L−/− EAE mice. A, Histological analysis of spinal cord sections from WT and CD137L−/− MOG-immunized mice at day 18 p.i. stained with anti-CD68 (green), anti-TCR-β (red) and DAPI (blue). Data are representative of two independent experiments. B, Cells isolated from the blood of naive mice were stained for CD3, CD4, CD8, CD45, CD11b, CD11c, Ly6C and Ly6G and analyzed by FACS. C–F, Cells isolated at day 18 p.i. from pooled spinal cords (C, D) or spleens from individual mice (E, F) of MOG-immunized mice were stained for CD3, CD4, CD8, CD25, CD137, Foxp3, CD45, CD11b, CD11c, B220, Ly6C and Ly6G and analyzed by FACS. C, D, Graphs represent 4 independent experiments with 3–8 spinal cords pooled per group and were combined for statistical analysis. G, Dot-plots gated on CD4+ T-cells represent activated CD4+CD137+ T-cells and CD4+CD25+Foxp3+ Tregs cells in the spinal cord. H, Phenotypic analysis of CNS infiltrating myeloid cells. Depicted dot-plots are gated on live cells. Microglia were differentiated from monocytes (Mo) (CD45hiCD11bhi) by their intermediate CD45 expression CD45intCD11bhi. Ly6G and Ly6C were used to identify inflammatory monocytes (Ly6ChiLy6Gint) and neutrophils (Ly6CintLy6Ghi). Inflammatory dendritic cells were identified as CD45hiCD11bintB220+CD11c+ cells.
Reduced cytokine secretion by CNS infiltrating T-cells in CD137L−/− EAE mice
IFN-γ secreted by Th1 cells and IL-17 produced by Th17 cells play an important role in the pathology of EAE (Murphy et al., 2010). To investigate whether the reduced T-cell numbers in the CNS of CD137L-deficient mice result in lower numbers of IFN-γ- and IL-17-producing T-cells, spinal cord mononuclear cells of WT and CD137L−/− EAE mice were isolated at day 18 p.i., restimulated ex vivo and stained for intracellular IFN-γ and IL-17A. The percentage and absolute number of CNS infiltrating CD4+ T-cells that secrete IFN-γ and IL-17A were reduced in CD137L−/− EAE mice when compared with WT mice (Fig. 3A,B). In summary, our data suggest CD137L is critical for the recruitment and/or reactivation of Th1 and Th17 cells in the spinal cord of EAE mice.
Figure 3.

Decreased numbers of effector CD4+ T-cells in CNS of MOG-immunized CD137L−/− mice. Isolated spinal cord cells at day 18 p.i. were restimulated ex vivo with PdBu and ionomycin or media alone for 5 h in the presence of brefeldin-A. Cells were stained for CD45, CD3, CD4 and intracellular IFN-γ and IL-17A expression and analyzed by FACS. A, Dot-plots depict CD4 and intracellular IFN-γ (left) or IL-17A (right) expression derived from WT (top row) and CD137L−/− (bottom row) mice. B, Absolute numbers of CD4+ T-cells producing IFN-γ or IL-17A in spinal cord are shown in the graph. Data represent means ± SEM of 3 independent experiments (n = 3–8) combined for statistical analysis.
Impaired upregulation of VCAM-1 in the CNS of CD137L−/− EAE mice
Infiltration of immune cells into the CNS follows the breakdown of the blood–brain barrier. The extravasation of leukocytes into the CNS is mediated by adhesion molecules upregulated on inflamed vascular endothelium, such as VCAM-1 (Engelhardt and Ransohoff, 2005). Since we observed impaired T-cell and monocyte infiltration in the CNS of CD137L−/− EAE mice, we compared the expression of VCAM-1 on the vascular endothelium of CD137L−/− and WT EAE mice. VCAM-1 was highly expressed on vascular endothelial cells in the CNS of WT but not CD137L−/− EAE mice (Fig. 4A,B). These data indicate that CD137L deficiency is associated with decreased endothelial VCAM-1 expression, which likely contributes to the reduced leukocyte infiltration to the CNS and a reduced EAE pathogenesis.
Figure 4.

Reduced VCAM-1 expression in spinal cord of CD137L−/− mice at the peak of EAE. A, Spinal cord sections from WT (top) and CD137L−/− (bottom) MOG-immunized mice at day 18 p.i. were stained for CD31 (red), VCAM-1 (green) and DAPI (blue). Staining was analyzed by fluorescence microscopy. B, Quantification of the VCAM-1+ endothelial cells in the spinal cord. Results show average area of double-positive cells ± SEM (n = 6). Images are representative of two independent experiments.
Discussion
In this study, we examined the role of CD137L costimulation and its importance in the development of EAE. Our results show that CD137L−/− mice are highly resistant to EAE induction and the associated demyelination. Their resistance correlated with reduced cytokine secretion by lymph node cells, which suggests a critical role for CD137L in the priming of T-cells during EAE. Furthermore, fewer immune cells were present in the CNS of CD137L−/− mice indicating that CD137L is important for trafficking and/or stimulation of immune cells in the CNS, a key event in the pathogenesis of EAE (Goverman, 2009). CD137L was found to play an important role for the recruitment and/or activation of IFN-γ and IL-17A secreting CD4+ T-cells in the CNS. The reduced cytokine production and CD137 cell surface expression of CD4+ T-cells in the CNS of CD137L−/− mice could lead to impaired activation of microglia cells. IFN-γ and IL-17A upregulate MHC-class II and costimulatory molecules on microglia (Murphy et al., 2010) and CD137+CD4+ T-cells may stimulate CD137L-dependent killing of myelin-producing oligodendrocytes by microglia (Yeo et al., 2011). These results suggest that CD137L could regulate several important steps required for EAE development.
A key event in the pathogenesis of EAE is the migration of leukocytes from blood vessels into the CNS (Engelhardt and Ransohoff, 2005). Several studies have shown the involvement of CD137-CD137L signaling in leukocyte recruitment at sites of inflammation (Drenkard et al., 2007; Jeon et al., 2010). Here, we observed reduced cellular infiltrates in the CNS of CD137L-deficient mice, suggesting that CD137-CD137L interactions may affect inflammatory cell trafficking to the CNS. The adhesion molecule VCAM-1 binds to VLA-4 expressed by leukocytes and is an essential factor for leukocyte extravasation to the CNS (Cruz-Orengo et al., 2011). CD137L might be critical for VCAM-1 expression as engagement of CD137 on the vascular endothelial cells by an agonistic CD137 antibody was shown to increase the expression of VCAM-1, ICAM-1 and E-selectin, leading to enhanced recruitment of T-cells in a tumor model (Palazón et al., 2011). In line with these studies, we observed decreased VCAM-1 expression in the CNS of CD137L−/− EAE mice. In addition, VCAM-1 upregulation is induced by TNF-α (Gimenez et al., 2004), which we found reduced in CD137L-deficient mice. The decreased VCAM-1 expression in the CD137L−/− mice correlated with reduced leukocyte infiltration to the CNS.
In contrast to our findings, two studies observed that stimulation of CD137 using an agonistic anti-CD137 antibody reduced the severity of EAE (Sun et al., 2002; Kim et al., 2011). Stimulating CD137 in vivo using an agonistic antibody overactivated T cells, which lead to deletion of pathogenic T cells. Moreover, anti-CD137 antibody treatment inhibited the generation of Th17 cells and instead skewed T-cell differentiation toward Tregs. Recent studies found that engagement of CD137 on Tregs enhanced their immune suppressive capacity (Elpek et al., 2007; Zhang et al., 2007). These results support the possibility that agonistic anti-CD137 antibody treatment may lead to reduce EAE pathogenesis by enhancing the activity of Tregs. Contrary to the enhancement of Tregs numbers observed in the previous studies we found similar numbers of Tregs in the spleens of WT and CD137L−/− EAE mice, but reduced numbers of Tregs in the spinal cord of CD137L−/− mice. These results suggest that Tregs do not contribute to the protection of CD137L−/− mice from EAE. Another explanation for the differences observed by Sun et al. (2002) and Kim et al. (2011) and our study may be due to the bidirectional signaling in CD137-CD137L system. The use of agonistic anti-CD137 antibody is expected to maintain or even enhance signaling through CD137 while signaling through CD137L would be reduced as CD137 is bound to the antibody. However, in CD137L-deficient mice both signaling of CD137 as well as that of CD137L are abrogated. The CD137L reverse-signal transduction pathway is thought to amplify inflammatory reactions. A recent study showed that triggering the CD137L signaling pathway in epithelial cells was sufficient to initiate acute tissue inflammation (Kim et al., 2012). This can explain why in our system the lack of CD137L signaling leads to a reduced inflammation of the CNS and therefore reduced EAE susceptibility of CD137L−/− mice.
In summary, our data show that CD137L−/− mice are largely resistant to EAE. We present evidence that CD137L is important for the activation of encephalitogenic T-cells effector function and trafficking to the CNS. The reduced infiltration of T-cells and inflammatory monocytes into the CNS correlated with impaired VCAM-1 upregulation. The demonstration of a key role of CD137L costimulation in EAE may have important implications for MS, and our data suggest that CD137L could be a promising target for intervention in MS and possibly other autoimmune diseases.
Footnotes
This study was supported by an A*STAR-NKTH Joint Grant (10/1/21/24/637) to H.S. and the National Research Foundation Grant HUJ-CREATE-Cellular and Molecular Mechanisms of Inflammation to S.G.
References
- Bailey SL, Schreiner B, McMahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4+ T(H)-17 cells in relapsing EAE. Nat Immunol. 2007;8:172–180. doi: 10.1038/ni1430. [DOI] [PubMed] [Google Scholar]
- Baxter AG. The origin and application of experimental autoimmune encephalomyelitis. Nat Rev Immunol. 2007;7:904–912. doi: 10.1038/nri2190. [DOI] [PubMed] [Google Scholar]
- Chang TT, Jabs C, Sobel RA, Kuchroo VK, Sharpe AH. Studies in B7-deficient mice reveal a critical role for B7 costimulation in both induction and effector phases of experimental autoimmune encephalomyelitis. J Exp Med. 1999;190:733–740. doi: 10.1084/jem.190.5.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croxford JL, O'Neill JK, Ali RR, Browne K, Byrnes AP, Dallman MJ, Wood MJ, Feldmann M, Baker D. Local gene therapy with CTLA4-immunoglobulin fusion protein in experimental allergic encephalomyelitis. Eur J Immunol. 1998a;28:3904–3916. doi: 10.1002/(SICI)1521-4141(199812)28:12<3904::AID-IMMU3904>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Croxford JL, Triantaphyllopoulos K, Podhajcer OL, Feldmann M, Baker D, Chernajovsky Y. Cytokine gene therapy in experimental allergic encephalomyelitis by injection of plasmid DNA-cationic liposome complex into the central nervous system. J Immunol. 1998b;160:5181–5187. [PubMed] [Google Scholar]
- Cruz-Orengo L, Holman DW, Dorsey D, Zhou L, Zhang P, Wright M, McCandless EE, Patel JR, Luker GD, Littman DR, Russell JH, Klein RS. CXCR7 influences leukocyte entry into the CNS parenchyma by controlling abluminal CXCL12 abundance during autoimmunity. J Exp Med. 2011;208:327–339. doi: 10.1084/jem.20102010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenkard D, Becke FM, Langstein J, Spruss T, Kunz-Schughart LA, Tan TE, Lim YC, Schwarz H. CD137 is expressed on blood vessel walls at sites of inflammation and enhances monocyte migratory activity. FASEB J. 2007;21:456–463. doi: 10.1096/fj.05-4739com. [DOI] [PubMed] [Google Scholar]
- Elpek KG, Yolcu ES, Franke DDH, Lacelle C, Schabowsky RH, Shirwan H. Ex-vivo expansion of CD4(+)CD25(+)FoxP3(+) T regulatory cells based on synergy between IL-2 and 4–1BB signaling. J Immunol. 2007;179:7295–7304. doi: 10.4049/jimmunol.179.11.7295. [DOI] [PubMed] [Google Scholar]
- Engelhardt B, Ransohoff RM. The ins and outs of T-lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol. 2005;26:485–495. doi: 10.1016/j.it.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Gimenez MAT, Sim JE, Russell JH. TNFR1-dependent VCAM-1 expression by astrocytes exposes the CNS to destructive inflammation. J Neuroimmunol. 2004;151:116–125. doi: 10.1016/j.jneuroim.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol. 2009;9:393–407. doi: 10.1038/nri2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon HJ, Choi JH, Jung IH, Park JG, Lee MR, Lee MN, Kim B, Yoo JY, Jeong SJ, Kim DY, Park JE, Park HY, Kwack K, Choi BK, Kwon BS, Oh GT. CD137 (4–1BB) Deficiency reduces atherosclerosis in hyperlipidemic mice. Circulation. 2010;121:1124–1133. doi: 10.1161/CIRCULATIONAHA.109.882704. [DOI] [PubMed] [Google Scholar]
- Juedes AE, Ruddle NH. Resident and infiltrating central nervous system APCs regulate the emergence and resolution of experimental autoimmune encephalomyelitis. J Immunol. 2001;166:5168–5175. doi: 10.4049/jimmunol.166.8.5168. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Lee JS, Kim JD, Cha HJ, Kim A, Lee SK, Lee SC, Kwon BS, Mittler RS, Cho HR, Kwon B. Reverse signaling through the costimulatory ligand CD137L in epithelial cells is essential for natural killer cell-mediated acute tissue inflammation. Proc Natl Acad Sci U S A. 2012;109:E13–E22. doi: 10.1073/pnas.1112256109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Choi BK, Shin SM, Kim CH, Oh HS, Park SH, Lee DG, Lee MJ, Kim KH, Vinay DS, Kwon BS. 4–1BB triggering ameliorates experimental autoimmune encephalomyelitis by modulating the balance between Th17 and regulatory T cells. J Immunol. 2011;187:1120–1128. doi: 10.4049/jimmunol.1002681. [DOI] [PubMed] [Google Scholar]
- King IL, Dickendesher TL, Segal BM. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood. 2009;113:3190–3197. doi: 10.1182/blood-2008-07-168575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GZ, Gomes AC, Putheti P, Karrenbauer V, Kostulas K, Press R, Hillert J, Hjelmström P, Gao XG. Increased soluble 4–1BB ligand (4–1BBL) levels in peripheral blood of patients with multiple sclerosis. Scand J Immunol. 2006;64:412–419. doi: 10.1111/j.1365-3083.2006.01796.x. [DOI] [PubMed] [Google Scholar]
- Liu GZ, Gomes AC, Fang LB, Gao XG, Hjelmström P. Decreased 4–1BB expression on CD4+CD25high regulatory T cells in peripheral blood of patients with multiple sclerosis. Clin Exp Immunol. 2008;154:22–29. doi: 10.1111/j.1365-2249.2008.03730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl SR, Staykova MA, Wozniak A, Fordham S, Bruce J, Willenborg DO. Treatment with anti-granulocyte antibodies inhibits the effector phase of experimental autoimmune encephalomyelitis. J Immunol. 1998;161:6421–6426. [PubMed] [Google Scholar]
- Mildner A, Mack M, Schmidt H, Brück W, Djukic M, Zabel MD, Hille A, Priller J, Prinz M. CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain. 2009;132:2487–2500. doi: 10.1093/brain/awp144. [DOI] [PubMed] [Google Scholar]
- Murphy ÁC, Lalor SJ, Lynch MA, Mills KHG. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav Immun. 2010;24:641–651. doi: 10.1016/j.bbi.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Palazón A, Teijeira A, Martínez-Forero I, Hervás-Stubbs S, Roncal C, Peñuelas I, Dubrot J, Morales-Kastresana A, Pérez-Gracia JL, Ochoa MC, Ochoa-Callejero L, Martínez A, Luque A, Dinchuk J, Rouzaut A, Jure-Kunkel M, Melero I. Agonist anti-CD137 mAb act on tumor endothelial cells to enhance recruitment of activated T lymphocytes. Cancer Res. 2011;71:801–811. doi: 10.1158/0008-5472.CAN-10-1733. [DOI] [PubMed] [Google Scholar]
- Racke MK, Ratts RB, Arredondo L, Perrin PJ, Lovett-Racke A. The role of costimulation in autoimmune demyelination. J Neuroimmunol. 2000;107:205–215. doi: 10.1016/s0165-5728(00)00230-7. [DOI] [PubMed] [Google Scholar]
- Shao Z, Schwarz H. CD137 ligand, a member of the tumor necrosis factor family, regulates immune responses via reverse signal transduction. J Leukoc Biol. 2011;89:21–29. doi: 10.1189/jlb.0510315. [DOI] [PubMed] [Google Scholar]
- Sharief MK. Heightened intrathecal release of soluble CD137 in patients with multiple sclerosis. Eur J Neurol. 2002;9:49–54. doi: 10.1046/j.1468-1331.2002.00323.x. [DOI] [PubMed] [Google Scholar]
- Sun Y, Lin X, Chen HM, Wu Q, Subudhi SK, Chen L, Fu YX. Administration of agonistic anti-4–1BB monoclonal antibody leads to the amelioration of experimental autoimmune encephalomyelitis. J Immunol. 2002;168:1457–1465. doi: 10.4049/jimmunol.168.3.1457. [DOI] [PubMed] [Google Scholar]
- Wang C, Lin GHY, McPherson AJ, Watts TH. Immune regulation by 4–1BB and 4–1BBL: complexities and challenges. Immunol Rev. 2009;229:192–215. doi: 10.1111/j.1600-065X.2009.00765.x. [DOI] [PubMed] [Google Scholar]
- Yeo AY, Martínez Gómez JM, Croxford JL, Gasser S, Ling EA, Schwarz H. CD137 ligand activated microglia induces oligodendrocyte apoptosis via reactive oxygen species. J Neuroinflamm. 2011;9:173. doi: 10.1186/1742-2094-9-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Podojil JR, Chang J, Luo X, Miller SD. TGF-β-induced myelin peptide-specific regulatory T cells mediate antigen-specific suppression of induction of experimental autoimmune encephalomyelitis. J Immunol. 2010;184:6629–6636. doi: 10.4049/jimmunol.0904044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Gao F, Wang Q, Wang X, Zhu F, Ma C, Sun W, Zhang L. Agonistic anti-4–1BB antibody promotes the expansion of natural regulatory T cells while maintaining Foxp3 expression. Scand J Immunol. 2007;66:435–440. doi: 10.1111/j.1365-3083.2007.01994.x. [DOI] [PubMed] [Google Scholar]