

Abstract
The molecular and cellular bases of motor neuron diseases (MNDs) are still poorly understood. The diseases are mostly sporadic, with ∼10% of cases being familial. In most cases of familial motor neuronopathy, the disease is caused by either gain-of-adverse-effect mutations or partial loss-of-function mutations in ubiquitously expressed genes that serve essential cellular functions. Here we show that deletion of Scyl1, an evolutionarily conserved and ubiquitously expressed gene encoding the COPI-associated protein pseudokinase SCYL1, causes an early onset progressive MND with characteristic features of amyotrophic lateral sclerosis (ALS). Skeletal muscles of Scyl1−/− mice displayed neurogenic atrophy, fiber type switching, and disuse atrophy. Peripheral nerves showed axonal degeneration. Loss of lower motor neurons (LMNs) and large-caliber axons was conspicuous in Scyl1−/− animals. Signs of neuroinflammation were seen throughout the CNS, most notably in the ventral horn of the spinal cord. Neural-specific, but not skeletal muscle-specific, deletion of Scyl1 was sufficient to cause motor dysfunction, indicating that SCYL1 acts in a neural cell-autonomous manner to prevent LMN degeneration and motor functions. Remarkably, deletion of Scyl1 resulted in the mislocalization and accumulation of TDP-43 (TAR DNA-binding protein of 43 kDa) and ubiquilin 2 into cytoplasmic inclusions within LMNs, features characteristic of most familial and sporadic forms of ALS. Together, our results identify SCYL1 as a key regulator of motor neuron survival, and Scyl1−/− mice share pathological features with many human neurodegenerative conditions.
Introduction
Motor neuron diseases (MNDs) are a group of progressive neurological disorders characterized by the degeneration and death of upper and/or LMNs, resulting in denervation and atrophy of muscles, paralysis, and, in most cases, premature death. In humans, the diseases are predominantly sporadic with ∼10% of cases being familial, most of which are dominantly inherited and originate from the expression of mutant genes. Only rare cases of MNDs are recessively inherited and are the result of partial loss-of-function mutations (Lorson et al., 2010; Ferraiuolo et al., 2011). Similarly, inherited forms of MNDs in mice, including mouse models of amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy, are the result of either gain-of-adverse-effect or partial loss-of-function mutations (Burgess et al., 2010). In all instances, loss-of-function mutations in mice involving orthologs of genes associated with MNDs do not result in motor neuron degeneration. Instead, targeted deletion of causative genes in mice most often results in early embryonic lethality, likely because of their wide, often ubiquitous, expression and the fundamental functions they perform within cells (Hicks et al., 2000; Sephton et al., 2009; Wu et al., 2009; Burgess et al., 2010). The causes for the majority of sporadic forms of MNDs are still unknown, but multiple lines of evidence now suggest that redistribution of TDP-43 (TAR DNA-binding protein of 43 kDa) from its normal predominantly nuclear location to cytoplasmic inclusions drives motor neuron degeneration (Lagier-Tourenne and Cleveland, 2009; Lee et al., 2011).
The mouse autosomal recessive mutation muscle deficient (mdf) is characterized by an early onset progressive motor neuron disorder (Blot et al., 1995) and is the result of a frame-shift mutation in the gene encoding SCYL1 (Schmidt et al., 2007). SCYL1 is an evolutionarily conserved and ubiquitously expressed protein (Liu et al., 2000) thought to regulate essential cellular functions, such as the nucleocytoplasmic shuttling of transfer RNA, COPI-mediated Golgi-to-endoplasmic reticulum (ER) trafficking, and maintenance of the Golgi apparatus morphology (Burman et al., 2008, 2010; Chafe and Mangroo, 2010). SCYL1 is characterized by an N-terminal kinase domain, HEAT repeats, and a C-terminal segment of unknown structure. In the mdf mice, the thymidine insertion causes a frame shift and creates a premature STOP codon, which reduces SCYL1 protein expression (Schmidt et al., 2007). The reduced level of SCYL1 in these mice suggests that the mdf mutation may act as a loss-of-function mutation. However, given that SCYL1 is ubiquitously expressed and highly conserved throughout evolution and is thought to regulate essential cellular processes, it remains unclear whether the mdf mutation causes a complete rather than a partial loss of function.
In this study, we used genetically engineered mice to show that loss of SCYL1 function in mice causes growth abnormalities and an early onset progressive MND with characteristic features of ALS, including mislocalization of TDP-43 and ubiquilin 2 from their normal locations into cytoplasmic inclusions. We further demonstrate that SCYL1 acts in a neural cell-autonomous manner to protect motor neurons from degeneration.
Materials and Methods
Antibodies.
The following antibodies were used in this study: anti-AP50 clone 31 (611351; BD Biosciences), anti-FLAG M2 (F3165; Sigma-Aldrich), rabbit polyclonal anti-FLAG antibody (F7425; Sigma-Aldrich), anti-rabbit IgG, HRP-linked antibody (7074; Cell Signaling Technologies), anti-mouse IgG, HRP-linked antibody (7076; Cell Signaling Technologies), anti-ubiquitin (MK-11-3), anti-Iba1 (CP290A; BioCore Medical), anti-GFAP (Z0334; Dako), anti-TDP-43 (10782-2-AP; Protein Tech Group), anti-FUS/TLS (IHC00074; Bethyl Laboratories), anti-ubiquilin 2 (clone 5F5, H00029978; Novus Biologicals), and anti-phospho(409/410)-TDP43 antibody (22309-1-AP; Protein Tech Group).
Generation of SCYL1 antibody.
A peptide corresponding to the amino acids 706–726 (SSVEPPPEGTRLASEYNWGGA) of SCYL1 was synthesized by the Hartwell Center for Bioinformatics and Biotechnology (St. Jude Children's Research Hospital) and conjugated to glutaraldehyde-activated keyhole limpet hemocyanin (KLH). The KLH-conjugated peptide was then used to immunize rabbits (Rockland Immunochemicals). The antibody (serum 7645) was enriched by affinity chromatography over the corresponding peptide. Selectivity of the antibody was tested by Western blot analyses of brain extracts obtained from Scyl1+/+, Scyl1+/−, Scyl1−/−, Scyl1+/fl, and Scyl1fl/fl mice. The antiserum was also tested for cross-reactivity against other SCYL family members by RNAi-mediated knockdowns. No cross-reactivity with other SCYL proteins was found. In immunostaining applications, the antiserum against SCYL1 reacted with an unidentified nuclear protein.
Western blot.
Mouse tissues were lysed in RIPA buffer [50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% NP-40 (Igepal-CA-630), 0.5% sodium deoxycholate, 0.1% SDS, and complete protease inhibitors (Roche)]. Western blot analyses were performed as described previously (Pelletier et al., 2006).
DNA constructs.
All oligonucleotides used in this study were produced by the Hartwell Center for Bioinformatics and Biotechnology. The plasmid pBR322–DTA (diphtheria toxin A) was created by subcloning the cDNA encoding DTA followed by a splice donor (Yanagawa et al., 1999) into pBR322. PL452 and PL451 plasmids were obtained from Dr. Neil A. Copeland (National Cancer Institute, Frederick, MD) (Liu et al., 2003). PL451–TK plasmid was generated by subcloning the DNA fragment containing the TK cassette into the BstBI sites of PL451. Plasmids encoding the Flp and Cre recombinases (pMC–Cre) were obtained from Klaus Rajewski (Immune Disease Institute, Boston, MA). The Scyl1 targeting construct was engineered using gap-repair technology as described previously (Liu et al., 2003). Briefly, a 31 kb fragment containing genomic sequences of the Scyl1 gene was subcloned by gap repair into pBR322–DTA. A first cassette containing the neomycin-resistance gene flanked by two loxP sites (i.e., PL452) was inserted in intron 1. After excision of the neomycin cassette by the Cre recombinase, a second cassette, containing the FLAG epitope followed by a STOP codon, the neomycin-resistance, and thymidine kinase cDNAs flanked by two Frt sites and a second loxP site (PL451-TK), was inserted before the stop codon in exon 17. The FLAG epitope (NH2-DYKDDDDK-COOH) was inserted, in-frame, with the coding sequence of SCYL1 to obtain the final targeting construct (Fig. 1A). Locations of the loxP sites were chosen such that all coding exons, with the exception of exon 1, are deleted during Cre recombination, ensuring the generation of a null allele. The conditional allele also encoded a C-terminally FLAG-tagged version of SCYL1.
Figure 1.

SCYL1 deficiency causes growth defects and early onset progressive motor deficits. A, Schematic representation of the Scyl1 locus, the targeting vector, and predicted Scyl1 mutated locus. After Cre- or Flp-mediated recombination in ES cells, Scyl1− and Scyl1fl loci were generated. Following Cre-mediated recombination, only exon 1 remained. Exons are indicated with gray bars. Black bars represent XhoI sites. Black triangles indicate loxP and Frt sites. Gray box indicates the Neo–TK cassette. Diphtheria toxin cassette is illustrated by a black arrow. The black circle labeled with an f indicates the position of the FLAG epitope. B, C, SCYL1 and FLAG-tagged SCYL1 expression in the cerebrum of Scyl1+/+, Scyl1+/, Scyl1+/fl, Scyl1fl/fl, and Scyl1−/− mice. Cerebrum extracts obtained from adult Scyl1+/+, Scyl1+/−, Scyl1+/fl, Scyl1fl/fl, and Scyl1−/− mice were resolved by SDS-PAGE and analyzed by Western blotting, using antibodies against Scyl1 (α-SCYL1) (B) or the FLAG epitope (C). Note the absence of SCYL1 (S1) in Scyl1-deficient mice. Also note the expression of FLAG-tagged SCYL1 (S1-F) in Scyl1+/fl and Scyl1fl/fl mice. Nonspecific (ns) bands served as loading control. D, Representative photograph of 6-week-old Scyl1+/+ and Scyl1−/− male littermates illustrating the size differences between Scyl1+/+ and Scyl1−/− mice. E, Growth curve of male (circle) and female (square) Scyl1+/+ (white) and Scyl1−/− (black) mice from 3 to 9 weeks of age. Scyl1−/− mice did not recover and stayed smaller than their wild-type littermates. Values are expressed as the mean ± SEM. Males: Scyl1+/+, n = 9; Scyl1−/−, n = 5; females: Scyl1+/+, n = 6; Scyl1−/−, n = 13. F, Disease progression in Scyl1-deficient mice. The clinical progression of defects in mice was assessed by an objective grading system (for details, see Materials and Methods). Values are expressed as the mean ± SEM. Scyl1+/+, n = 13; Scyl1−/−, n = 13. G, Representative photograph of hindlimbs obtained from 12-week-old Scyl1+/+ and Scyl1−/− males showing massive muscle wasting in Scyl1−/− mice. H, Representative photograph of an end-stage Scyl1−/− mouse. The hindlimbs of the Scyl1−/− mouse are atrophied and dorsally contracted. In some mice, flexion of the hindlimb digits is still possible. Also note the flattening of the pelvis. At this stage of the disease, the affected mice move by the power of their front legs. I, Motor defects in Scyl1-deficient mice. The inverted grid test was performed on 4-week-old and 8- to 12-week-old Scyl1+/+ and Scyl1−/− mice to assess their motor functions for up to 120 s as described in Materials and Methods. Four-week-old Scyl1+/+, n = 22; 4-week-old Scyl1−/−, n = 11; 8- to 12-week-old Scyl1+/+, n = 10; 8- to 12-week-old Scyl1−/−, n = 10.
Generation of mice.
The Scyl1 targeting construct was used to target the Scyl1 gene in EmbryoMax mouse embryonic stem (ES) cell line, strain 129/SVEV (CMTI-1 cells; Millipore). Positive clones were identified by Southern blot analyses of XhoI-digested genomic DNA using 5′ and 3′ external probes. To identify single recombination events, XhoI-digested genomic DNA obtained from positive clones were also analyzed by Southern blot using a neomycin probe. ES cell clones bearing the null (Scyl1−) or the conditional (Scyl1fl) allele were generated in vitro by electroporating ES cell clones with plasmids encoding CRE or FLP recombinases. 1-(2-Deoxy-2-fluoro-β-d-arabinofuranosyl)-5-iodouracil (0.5 μm) was used as negative selection during this process. Properly rearranged clones were identified by Southern blot analysis using the 5′ and 3′ external probes and PCR. Two clones of each allele were then injected into blastocysts. Blastocysts were implanted into BALB/c recipient females to produce chimeras. Chimeras were crossed to C57BL/6 mice (The Jackson Laboratory) to obtain heterozygous mice. Heterozygous mice were bred once on C57BL/6, and their progeny were used for heterozygous intercross. CkmmCre [B6.FVB(129S4)-Tg(Ckmm-cre)5Khn/J #006475] and NesCre [B6.Cg-Tg(Nes-cre)1Kln/J #003771] lines were purchased from The Jackson Laboratory.
PCR genotyping.
Mouse genotypes were routinely determined by PCR on tail DNA. By using the following primers: S1F01 (5′-GCTGCTCCGAAGGCCGCGGCCGA-3′), S1R51 (5′-GATTATGTACACTAGATGTGCCTGA-3′), and S1R02 (5′-GAGGAGAGTAAGATGGGTAGA-3′), bands of 521 bp, 251 bp, and 625 bp corresponding to the wild-type (Scyl1+), null (Scyl1−), and conditional alleles (Scyl1fl), respectively, are obtained. Mice bearing the Nestin–Cre locus were genotyped by PCR according to The Jackson Laboratory protocol (http://jaxmice.jax.org/strain/003771.html). Mice bearing the CkmmCre locus were genotyped by PCR according to The Jackson Laboratory protocol (http://jaxmice.jax.org/strain/006475.html). Wild-type and mdf alleles were detected in two distinct PCR reactions. The wild-type allele of Scyl1 was detected using primers mdf-wt-F01 (5′-ACAGTCAACACGCAGATTTTC-3′) and mdf-R01 (5′-TAGCTGCCAGTGTGAGCTGTGTCCTAG-3′). The mdf allele was detected using the primers mdf-F01 (5′-ACAGTCAACACGCAGATTTTT-3′) and mdf-R01.
Mouse husbandry.
All animals were housed in an Association for Assessment and Accreditation of Laboratory Animal Care International-accredited facility and maintained in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All experiments involving animals were reviewed and approved by the St. Jude Children's Research Hospital Institutional Animal Care and Use Committee.
Scoring system.
Disease progression in Scyl1−/− mice was evaluated using a scoring system in which a score of 0 was given to any animal showing no abnormal behaviors. A score of 1 was given to animals with growth defects or retardation. A score of 2 was given to animals showing growth defects and posterior waddle. A score of 3 was given to animals showing growth defects, posterior waddle, pronounced abnormal gait. A score of 4 was given to animals showing growth defects, abnormal gait, and exhibiting tremor when suspended by their tails. A score of 5 was given to animals showing growth defects, abnormal gait, tremor, and partial paralysis of the hindlimb. A score of 6 was given to animals with growth defects and complete paralysis of the hindlimbs. A score of 7 was given to animals with growth defects, paralysis of the hindlimbs, and flattening of the pelvis. A score of 7 constituted the endpoint.
Grip test.
The inverted-grid test was used to assess neuromuscular function and muscular strength. Mice were placed on top of an elevated cage grid. The cage grid was inverted, and the time until release of the animals was measured. A maximum score of 120 s was given to the animals that did not fall.
Histology and immunohistochemistry.
Immediately after the animals were killed, mice were perfused with 10% buffered Formalin (Thermo Fisher Scientific) through the left cardiac ventricle. The brain and spinal cords of perfused mice were left in place within the skull and vertebral columns, respectively, and postfixed by immersion in 10% buffered Formalin for at least 24 h before being decalcified in formic acid (TBD-2 Decalcifier; Thermo Fisher Scientific). Tissues were embedded in paraffin, sectioned at 4 μm, mounted on positively charged glass slides (Superfrost Plus; Thermo Fisher Scientific), and dried in a 60°C oven for 20 min. The polymeric biotin-free horseradish peroxidase method on a BOND-MAX stainer (Leica Microsystems) was used for immunohistochemistry. Tissue sections were placed in the automated BOND-MAX stainer and pretreated with enzyme-induced epitope retrieval for 8 min, followed by incubation with the primary antibodies. Binding of primary antibody to tissue sections was visualized by the Bond Polymer Refine Detection kit (Leica Microsystems), with 3,3′-diaminobenzidine as the chromogenic substrate and a light hematoxylin counterstain.
Ventral horn motor neurons in the lumbar region of the spinal cord were quantified using a light microscope and the NIH Image J software version 1.42q (http://rsbweb.nih.gov/ij/) on hematoxylin and eosin (H&E)-stained sections. With H&E staining, the motor neurons were considered healthy if the cells demonstrated clearly defined Nissl substance, a large nucleus, and clearly defined nucleoli.
Myelinated axons in the sciatic nerve were quantified using a light microscope and NIH Image J on toluidine blue-stained cross-sections. The number of myelinated axons in 3.5- and 8-week-old Scyl1+/+ and Scyl1−/− mice (three for each genotype and age group) was determined and plotted.
Axonal diameters in the sciatic nerve were quantified by using a light microscope and NIH Image J on toluidine blue-stained cross-sections. A total of 5882 axons from three 8-week-old Scyl1+/+ mice, 4911 axons from three 8-week-old Scyl1−/− mice, 4050 axons from three 4-week-old Scyl1+/+ mice, and 2820 axons from three 4-week-old Scyl1−/− were evaluated. g ratios were determined for 100 fibers per sciatic nerve from three mice per genotype and age group.
Purkinje cell numbers in the cerebellum were quantified by using a light microscope and the NIH Image J on H&E-stained sections as described previously (Schmidt et al., 2007). A total of 25 segments from three 17-week-old Scyl1+/+ males, 21 segments from three 17-week-old Scyl1−/− males, and three 17-week-old Scyl1mdf/mdf males were evaluated. The number of Purkinje cells for an arbitrarily defined length of a segment was determined and plotted.
Cell density in the granular and molecular layers was determined by using a light microscope and NIH Image J software on H&E-stained sections. A total of six images from three 8-week-old Scyl1+/+ and three 8-week-old Scyl1−/− mice were taken. The number of cells per 10,000 μm2 was determined.
The number of lumbar ventral horn motor neurons showing rarefaction of cytosolic organelles (including motor neurons with enlarged vacuoles) and chromatolysis was quantified under blinded conditions by using a light microscope and NIH Image J on toluidine blue-stained semithin sections. A total of 23 images obtained from three 8-week-old Scyl1+/+, 30 images (90,000 μm2) from three 8-week-old Scyl1−/−, 30 images from three 4-week-old Scyl1+/+, and 30 images from three 4-week-old Scyl1−/− mice were analyzed, and the number of motor neurons with rarefaction of cytosolic organelles or central chromatolysis per 90,000 μm2 was determined.
The number of ventral horn motor neurons showing cytoplasmic TDP-43 was determined under blinded conditions. A total of 48 images of spinal ventral horn (from the cervical to the lumbar region) obtained from three 8-week-old Scyl1+/+ mice and 52 images obtained from three 8-week-old Scyl1−/− mice were analyzed.
Transmission electron microscopy.
Immediately after the animals were killed, mice were perfused with 4% glutaraldehyde in 0.1 m sodium cacodylate buffer, pH 7.4, and postfixed in 2% osmium tetroxide in 0.1 m sodium cacodylate buffer with 0.3% potassium ferrocyanide for 2 h. After rinsing in the same buffer, the tissue was dehydrated through a series of graded ethanol to propylene oxide, infiltrated and embedded in epoxy resin, and polymerized at 70°C overnight. Semithin sections (0.5 μm) were stained with toluidine blue for light microscope examination. Ultrathin sections (80 nm) were cut and imaged using a JEOL 1200 electron microscope with an AMT XR 111 camera.
Mitochondrial swelling was quantified in randomly selected ventral horn motor neurons of the lumbar region of the spinal cord of 8-week-old Scyl1+/+ and Scyl1−/− mice. The number of healthy and swollen (those with disrupted cristae) mitochondria was determined based on morphology using transmission electron microscopy and scored. Twenty-three randomly selected motor neurons from three Scyl1+/+ and 29 motor neurons from three Scyl1−/− mice were analyzed.
Biochemical fractionation of spinal cord extracts.
Frozen spinal cord tissues from Scyl1+/+ and Scyl1−/− mice were used for sequential extractions of proteins. Lumbar spinal cord tissues (∼0.05 g) were homogenized in RIPA buffer at 4°C for 30 min. The lysates were cleared by centrifugation, and the RIPA-insoluble fractions were then extracted with 500 μl of urea buffer [7 m urea, 40 mm Tris, pH 8.8, 4% NP-40, and 10 mm DTT containing complete protease inhibitors and phosSTOP (Roche)]. Proteins (20 μg) were loaded on SDS-PAGE and analyzed by Western blotting as described previously (Pelletier et al., 2006).
Statistical analyses.
Statistical analyses were performed using GraphPad Prism software, version 5.00. Except when indicated, Student's t test was used, and P values <0.05 were considered significant.
Results
Growth defect and motor dysfunction in Scyl1-deficient mice
To gain insights into SCYL1 protein function, mice bearing a conditional or a null allele of Scyl1 were generated as detailed in Materials and Methods and illustrated in Figure 1A. Locations of the loxP sites were chosen such that, during Cre-dependent recombination, most of the coding sequence was deleted. The conditional allele was also engineered to encode for a C-terminally FLAG-tagged version of SCYL1. Cre- or Flp-mediated recombination of properly targeted ES cell clones generated ES cells bearing a null allele (Scyl1−) or a conditional allele (Scyl1fl), respectively (Fig. 1A). Two ES cell clones (for each allele) transmitted the mutant alleles. Mice obtained from either clone showed the same phenotypes.
Scyl1+/− and Scyl1+/fl mice were viable, fertile, and exhibited no overt abnormalities. Mating of Scyl1+/− mice yielded Scyl1+/+, Scyl1+/−, and Scyl1−/− mice in the expected Mendelian ratio. Similarly, mating of Scyl1+/fl mice yielded Scyl1+/+, Scyl1+/fl, and Scyl1fl/fl mice according to the normal Mendelian ratio. Western blot analyses, using an SCYL1-specific antibody generated in our laboratory (see Materials and Methods) demonstrated the absence of SCYL1 in Scyl1−/− mice (Fig. 1B). Using the M2 anti-FLAG antibody, a band of 100 kDa corresponding to the FLAG-tagged SCYL1 (S1-F) was detected in Scyl1+/fl and Scyl1fl/fl mice (Fig. 1C). Importantly, SCYL1 levels in Scylfl/fl mice were comparable with those in Scyl1+/+ mice (Fig. 1B). Scyl1fl/fl mice were viable, fertile, and otherwise appeared normal, indicating that the FLAG epitope does not interfere with SCYL1 protein function.
At birth, Scyl1−/− mice were indistinguishable from their wild-type littermates. However, by 3 weeks of age, both male and female Scyl1−/− mice exhibited growth retardation, which was maintained through their lives (Fig. 1D,E). At 8 weeks of age, Scyl1-deficient males had 35% lower body weight than control littermates (Scyl1+/+, 23.1 ± 1.1 g, n = 8; Scyl1−/− mice, 15.0 ± 0.8 g, n = 5). Similarly, 8-week-old females weighed 25% less than control littermates (Scyl1+/+, 17.5 ± 1.2 g, n = 4; Scyl1−/−, 13.4 ± 0.8 g, n = 8). More strikingly, Scyl1-deficient mice exhibited progressive motor dysfunction, the progression of which was monitored using a scoring system (Fig. 1F; for details, see Materials and Methods). By 4 weeks of age, Scyl1-deficient mice developed a posterior waddle and abnormal gait. By 6 weeks of age, they displayed tremor when suspended by their tails. By 6–8 weeks of age, muscle wasting was evident in the rear legs (Fig. 1G). By 8–20 weeks of age, their rear legs were paralyzed, and they dragged themselves with their front legs. At this stage, most Scyl1-deficient mice exhibited dorsoventral flattening of the pelvis (Fig. 1H). Animals that attained a score of 7 had to be killed. Therefore, the lifespan of Scyl1-deficient mice was not determined.
Motor functions in Scyl1+/+ and Scyl1−/− were assessed by the mesh grip test. By 4 weeks of age, control animals held on to the inverted cage grid for up to 111 ± 6 s, whereas mutant animals held on for only 31 s ± 10 s (Fig. 1I). By 8 weeks of age, Scyl1−/− mice could not hold on to the inverted cage grid for more than 1 s (1 ± 1 s), whereas 8-week-old control animals remained suspended for >2 min (Fig. 1I). Together, these observations indicate that loss of SCYL1 function(s) in mice causes growth retardation, muscle wasting, and motor dysfunction.
Neurogenic myopathy in Scyl1-deficient mice
To gain insights into the motor dysfunction caused by the loss of Scyl1, histopathological examinations were performed of all four legs of 4- to 20-week-old Scyl1-deficient animals. As early as 4 weeks of age, evidence of neurogenic atrophy was present in skeletal muscles, with particularly severe involvement of the rectus femoris and triceps brachii muscles. In 8- and 20-week-old mice, evidence of neurogenic atrophy was more widespread and conspicuous (Fig. 2A). Characteristic changes in affected muscles included diffuse centralization of myocyte nuclei associated with the marked variations in muscle fiber diameter, with groups of angulated (atrophied) fibers often adjacent to less severely affected rounded myocytes (Fig. 2B). Notably, the severity of neurogenic atrophy differed markedly between specific muscles. For example, in the rear leg, the quadriceps muscle was severely affected, whereas most posterior muscles of the rear leg lacked the typical lesions of neurogenic atrophy, such as centralization of nuclei and clusters of shrunken angular fibers. Muscle fibers in the more normal muscles were noticeably rounder and smaller than those of control mice, but the normal peripheral localization of nuclei and generally uniform diameter of the rounded myocytes in these fascicles were most suggestive of disuse atrophy, secondary to the neurogenic atrophy affecting the anterior muscles. Similarly, in the forelegs, neurogenic atrophy of the anteriorly located biceps brachii muscle was accompanied by diffuse disuse atrophy of the posteriorly located triceps brachii muscle. In muscles affected by neurogenic atrophy, immunohistochemical detection of fast and slow myosin revealed the occurrence of fiber-type switching: there were increased numbers of atrophic type I fibers in affected muscles of Scyl1-deficient mice (Fig. 2C). None of these abnormalities was found in control animals. Together, these results indicate that loss of Scyl1 in mice caused a bilaterally symmetrical progressive neurogenic myopathy, with particularly severe involvement of the rectus femoris and biceps brachii muscles.
Figure 2.
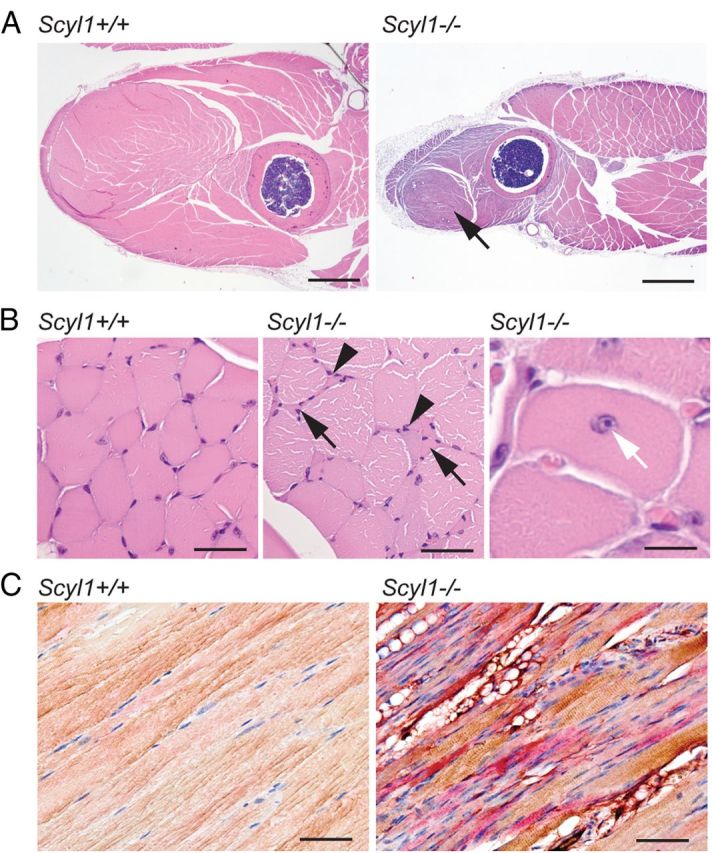
Myopathological abnormalities in Scyl1-deficient mice. A, Representative micrographs of H&E-stained sections of the anterior muscles in the rear legs of 12-week-old Scyl1+/+ and Scyl1−/− mice. Note the massive muscle wasting in the quadriceps femoris of Scyl1−/− mice (arrow). Scale bar, 1 mm. B, Higher magnification of H&E-stained sections of quadriceps femoris muscle fibers obtained from Scyl1+/+ and Scyl1−/− mice. Lesions in Scyl1−/− muscles include fibers of different sizes, angulated (atrophied) fibers surrounded by rounded fibers, group atrophy (arrows), nuclear clumps (arrowheads), and centrally localized nuclei (white arrow). Scale bars: left, middle, 50 μm; right, 25 μm. C, Representative micrographs of longitudinal sections of quadriceps femoris from 12-week-old Scyl1+/+ and Scyl1−/− mice illustrating fiber type switching from type II (fast, brown) to type I (slow, red) in Scyl1−/− muscles. Scale bar, 50 μm.
Neuroinflammation and motor neuron degeneration in Scyl1-deficient mice
Because skeletal muscles of Scyl1-deficient mice had features suggestive of neurogenic atrophy, we examined these mice for abnormalities in peripheral nerves and spinal cord. Specifically, axon numbers, axonal diameter, and myelination (g ratio) were analyzed in the sciatic nerves of 3.5-week-old (before onset of disease) and 8-week-old Scyl1+/+ and Scyl1−/− animals.
In 3.5-week-old animals, at onset of the disease, the sciatic nerves of Scyl1+/+ and Scyl1−/− mice were histologically indistinguishable (Fig. 3A). Axonal caliber distribution (Fig. 3C) and the total number of myelinated axons in the sciatic nerve (Fig. 3E) of Scyl1−/− mice were similar to those found in Scyl1+/+ mice. Similarly, myelination was comparable in young Scyl1+/+ and Scyl1−/− mice; g ratio of sciatic nerve fibers in Scyl1+/+ mice was 0.55 ± 0.03 versus 0.56 ± 0.04 in Scyl1−/− mice.
Figure 3.

Loss of large-diameter axons in peripheral nerves of Scyl1-deficient mice. A, B, Representative micrograph of toluidine blue-stained semithin sections of the sciatic nerve obtained from 3.5-week-old (A) and 8-week-old (B) Scyl1+/+ and Scyl1−/− mice. Scale bar, 20 μm. C, D, Histogram of the frequency of myelinated axons of the sciatic nerve in function of their axonal diameter in 3.5-week-old (C) or 8-week-old (D) mice. A total of 5882 axons from three 8-week-old Scyl1+/+ mice, 4911 axons from three 8-week-old Scyl1−/− mice, 4050 axons from three 3.5-week-old Scyl1+/+, and 2820 axons from three 3.5-week-old Scyl1−/− mice were evaluated. Note the significant reduction in large-caliber axons (≥5 μm) and increased frequency of small-caliber axons (<1–2 μm) in 8-week-old Scyl1-deficient mice. Values are expressed as the mean ± SEM. *p < 0.05. E, Myelinated axon counts in the sciatic nerve of 3.5- and 8-week-old Scyl1+/+ and Scyl1−/− mice. Values are expressed as the mean ± SEM; n = 3 each. F, Segmental demyelination in the sciatic nerve of Scyl1−/− mice. H&E-stained longitudinal sections of sciatic nerves obtained from 4- and 8-week-old Scyl1+/+ (left) and Scyl1−/− male mice. Arrows indicates myelin ovoids with myelin debris. Scale bar, 50 μm.
In 8-week-old animals, the total number of myelinated axons was significantly lower in Scyl1−/− than in Scyl1+/+ animals (Fig. 3E). Also, the distribution in axonal caliber was significantly different between Scyl1+/+ and Scyl1−/− animals (Fig. 3B). In the sciatic nerve of 8-week-old Scyl1−/− mice, large-caliber axons (>5 μm in diameter) were almost completely absent and the frequency of small-caliber axons was increased (Fig. 3D). The g ratios in the sciatic nerve of 8-week-old Scyl1+/+ and Scyl1−/− animals were similar (0.62 ± 0.01 vs 0.58 ± 0.02, respectively), suggesting the absence of major myelination defects in Scyl1−/− mice. Longitudinal sections of the sciatic nerve of Scyl1-deficient mice, however, revealed segmental degeneration of the myelin sheath (Fig. 3F). Signs of segmental demyelination in peripheral nerves were visible by 4 weeks of age and became conspicuous by 8 weeks of age, with large vacuoles being clearly visible (Fig. 3F).
The reduced number of myelinated axons combined with the shift in axonal diameter in sciatic nerves of 8-week-old but not 3.5-week-old mutant animals indicated that large-caliber axons were lost in Scyl1−/− mice. Consistent with this, we noted a reduction in the number of spinal ventral horn motor neurons in 8-week-old animals (6.6 ± 0.5 motor neurons per field in control animals, n = 42 fields from three different mice vs 2.9 ± 0.3 in mutant animals, n = 50 from three different animals; p < 0.01). Histological examination of toluidine blue-stained sections of the spinal ventral horn also revealed an increase in the percentage of motor neurons with rarefaction of cytosolic organelles (Fig. 4A,B) and central chromatolysis (Fig. 4A,C). Transmission electron microscopy also revealed the presence of swollen mitochondria in degenerating neurons of Scyl1-deficient mice (Fig. 4D). The number of swollen mitochondria in randomly selected lumbar ventral horn motor neurons of 8-week-old Scyl1−/− mice was threefold that in 8-week-old Scyl1+/+ mice. Mitochondrial swelling was seen in 75 (4.9%) of the 1548 mitochondria analyzed in motor neurons of Scyl1+/+ mice versus 247 (16.7%) of the 1479 mitochondria analyzed in Scyl1−/− mice.
Figure 4.

Degenerating motor neurons in the ventral horn of Scyl1−/− mice. A, Representative micrographs of toluidine blue-stained lumbar ventral horn motor neurons of Scyl1+/+ (top left) and Scyl1−/− (top right and bottom panels) mice. Morphological changes in motor neurons of the lumbar spinal ventral horn included rarefaction of cytosolic organelles, enlarged cytoplasmic vacuoles, and central chromatolysis. White arrows indicate healthy motor neurons. Black arrows point to motor neurons with rarefaction of cytosolic organelles. Black arrowhead points to a motor neuron with enlarged vacuoles. White arrowhead points to a motor neuron showing central chromatolysis. Scale bar, 20 μm. B, Quantification of lumbar ventral horn motor neurons showing rarefaction of cytosolic organelles (including motor neurons with enlarged vacuoles) in 4- and 8-week-old Scyl1+/+ and Scyl1−/− mice. Values are expressed as the mean ± SEM. A minimum of 23 images from three different mice per genotype and age group were analyzed (see Materials and Methods). C, Quantification of chromatolytic lumbar ventral horn motor neurons in 4- and 8-week-old Scyl1+/+ and Scyl1−/− mice. Values are expressed as the mean ± SEM. A minimum of 23 images from three different mice per genotype and age group were analyzed (see Materials and Methods). D, Mitochondrial swelling in motor neurons of Scyl1-deficient mice. In motor neurons of Scyl1+/+ mice, mitochondria of the perikaryon appear normal (top panel and inset). In select LMNs of Scyl1−/− mice, mitochondria are swollen, with disrupted cristae (bottom panel and inset). Mitochondrial swelling was observed in 75 (4.9%) of the 1549 mitochondria analyzed from Scyl1+/+ motor neurons versus 247 (16.7%) of the 1479 mitochondria analyzed in Scyl1−/− mice. Scale bar, 1 μm. E, Neuroinflammation in the spinal ventral horn of Scyl1-deficient mice. Immunohistochemistry using antibodies against Iba1 and GFAP on spinal ventral horn sections obtained from 8-week-old Scyl1+/+ and Scyl1−/− mice. Note the increased Iba1 and GFAP staining in the ventral horn of Scyl1-deficient mice. Scale bars: top row, 100 μm; bottom row, 50 μm.
Consistent with the loss of large motor neurons, there was evidence of inflammation associated with neurodegeneration, including mild-to-moderate astrogliosis and microgliosis, in the spinal cord of Scyl1-deficient mice. In spinal cord sections from control mice, immunoreactivity for both GFAP and Iba1 demonstrated the fine cytoplasmic processes of normal astrocytes and microglia, respectively (Fig. 4E). In contrast, in spinal cords of Scyl1−/− mice, there was an increase in GFAP immunoreactivity throughout the spinal gray matter, indicating astrogliosis. There was also an increase in the number and intensity of Iba1-positive microglia in ventral horn areas of the spinal cord, which indicates microglial activation (Fig. 4E).
Because upper motor neurons may be affected in MNDs, histopathological and immunohistochemical evaluations of the brain, in particular the motor cortex and cerebellum, were performed in 4- to 20-week-old Scyl1+/+ and Scyl1−/− mice. Despite a small reduction in the size of Scyl1−/− brains (males: Scyl1+/+, 0.45 ± 0.01 g, n = 6; Scyl1−/−, 0.38 ± 0.01 g, n = 3, p = 0.0041; females: Scyl1+/+, 0.46 ± 0.01 g, n = 7; Scyl1−/− 0.40 ± 0.01 g, n = 8, p = 0.0115), there was no brain region-specific atrophy. For example, cerebellum weight/brain weight ratios (0.14 ± 0.01 in Scyl1+/+ vs 0.16 ± 0.01 in Scyl1−/− mice, n = 3 for each genotype, p = 0.3092) were similar in 8-week-old Scyl1+/+ and Scyl1−/− mice. Moreover, there were no notable morphological abnormalities in the architecture of Scyl1−/− cerebrum (data not shown) and cerebellum (Fig. 5A). The layered structures of the cerebellum on Scyl1-deficient mice were normal (Fig. 5B). Purkinje cell numbers (Fig. 5C), molecular layer cell density (Scyl1+/+, 22.0 ± 3.2 cells/10,000 μm2, n = 3; Scyl1−/−, 18.17 ± 1.8 cells/10,000 μm2, n = 3, p = 0.3155) and granular layer cell density (Scyl1+/+, 263.3 ± 11.9 cells/10,000 μm2,n = 3; Scyl1−/−, 276.8 ± 10.5 cells/10,000 μm2, n = 3, p = 0.4147) were similar in Scyl1+/+ and Scyl1−/− mice.
Figure 5.
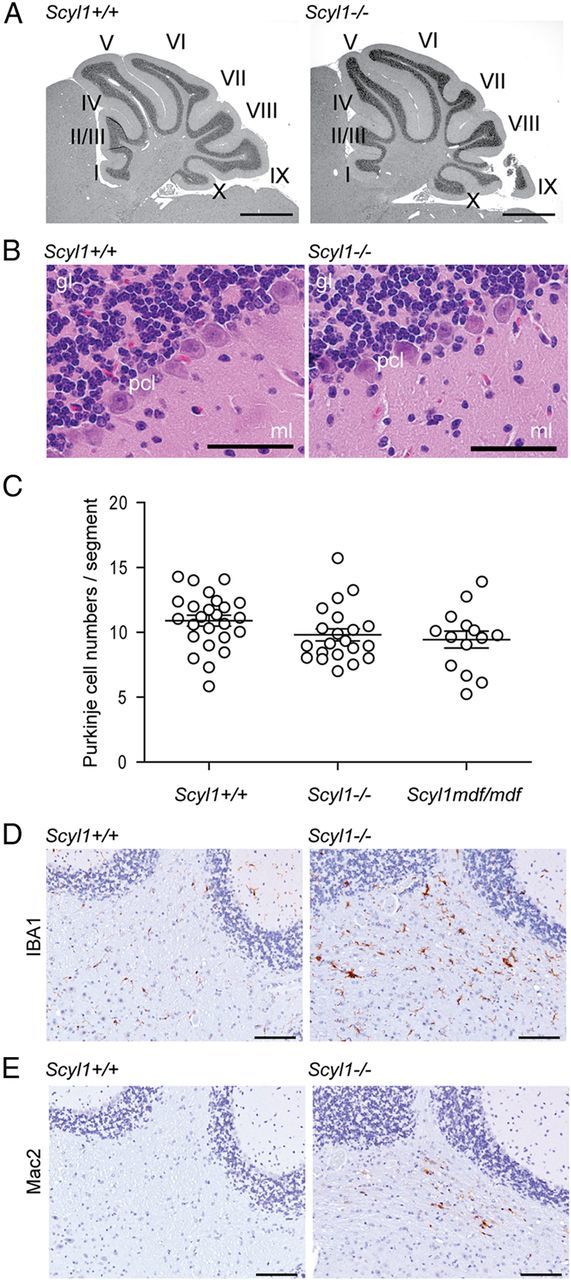
Histological examination of the layered structures of Scyl1+/+ and Scyl1−/− cerebellum. A, Representative micrographs of H&E-stained sagittal sections of cerebellums obtained from 17-week-old Scyl1+/+ and Scyl1−/− mice. No gross deformation in the cerebellar structures was observed in Scyl1−/− mice. I–X, Cerebellar lobules. Scale bar, 1 mm. B, Representative micrographs of H&E-stained sections of 8-week-old Scyl1+/+ and Scyl1−/− mice showing the layered structures of the cerebellum. The layered structures of the cerebellum of Scyl1−/− were undistinguishable from Scyl1+/+ mice. gl, Granular layer; pcl, Purkinje cell layer; ml, molecular layer. Scale bar, 50 μm. C, Purkinje cell numbers in Scyl1+/+ and Scyl1−/− mice. Purkinje cells numbers were determined as described in Materials and Methods. For Purkinje cell counts, a total of 25 segments from three Scyl1+/+, 21 segments from three Scyl1−/−, and 14 segments from three Scyl1mdf/mdf male mice of 17 weeks of age were counted. Values are expressed as mean ± SEM. p > 0.05. D, E, Microglial activation in the white track of the cerebellar peduncle. Immunohistochemistry using antibodies against Iba1 (D) and Mac2 (E) on cerebellar sections of 8-week-old Scyl1+/+ (left) and Scyl1−/− (right) mice. Note the increased Iba1 and Mac2 staining in the cerebellar peduncle of Scyl1-deficient mice. Scale bar, 100 μm.
This was unexpected because Schmidt et al. (2007) reported that 6-week-old and 9-month-old Scyl1mdf/mdf mice have 40% fewer Purkinje cells than Scyl1+/+ mice, a finding that was not reported in the original description of mdf mice (Blot et al., 1995). The mdf mouse is characterized by growth retardation, neurogenic atrophy, and muscle wasting (Blot et al., 1995). Examination of Scyl1−/− mice in parallel with Scyl1mdf/mdf mice revealed that the onset and progression of disease were the same between both strains. Thus, to test whether these changes in the architecture of the cerebellum could reflect differences between Scyl1mdf/mdf and Scyl1−/− mice, Purkinje cell numbers in Scyl1mdf/mdf were also quantified. No significant differences were found in Purkinje cell numbers among 17-week-old Scyl1+/+, Scyl1−/−, and Scyl1mdf/mdf mice (Fig. 5C).
Despite the lack of changes in the cellular composition of the cerebellum, immunohistochemical analysis showed evidence of neuroinflammation in the cerebellar peduncle of Scyl1-deficient mice (Fig. 5D,E). Glial cell activation in this structure may result from degenerating axons.
Together, our results indicate that loss of SCYL1 function in mice causes motor neuron degeneration and neuroinflammation in specific locations in the CNS. Also, among all neuronal cells, LMNs of the lateral motor column are most severely affected by the loss of SCYL1.
Tissue-specific deletion of Scyl1
Because SCYL1 is ubiquitously expressed in mammalian tissues (Liu et al., 2000), we next sought to confirm that the motor dysfunction seen in Scyl1-deficient mice was neurogenic rather than myogenic in origin. To test this, mice bearing the conditional allele of Scyl1 (Scyl1+/fl) were crossed with mice expressing the Cre recombinase under either the muscle creatine kinase promoter (CkmmCre) or the rat nestin promoter (NesCre). The Ckmm promoter drives expression specifically in skeletal muscles by embryonic day 13.5 (E13.5), whereas the rat nestin promoter drives expression in the neural compartment by E10.5 (Zimmerman et al., 1994; Tronche et al., 1999; Wang et al., 1999). Western blot analysis confirmed the tissue specific and complete recombination of the Scyl1 allele in CkmmCre+;Scyl1fl/fl mice, because no SCYL1 protein could be detected in the skeletal muscles of these animals (Fig. 6A). In contrast, SCYL1 was detected in the brain and liver of the same animal and in CkmmCre+;Scyl1+/fl animals (Fig. 6A). Similarly, Western blot analyses showed the selective and complete recombination of the Scyl1 locus in cells of neural origin as SCYL1 protein levels were undetectable in the brains of NesCre+;Scyl1fl/fl mice compared with NesCre+;Scyl1+/fl mice. In contrast, SCYL1 was expressed in the liver and skeletal muscles of the same animal (Fig. 6A). To further substantiate the loss of SCYL1 protein in the spinal cord of NesCre+;Scyl1fl/fl mice, immunohistochemical stainings using an antibody against SCYL1 were also performed and revealed that >90% of neuronal cells of NesCre+;Scyl1fl/fl mice no longer expressed SCYL1, but there were scattered large neurons that still expressed SCYL1 (Fig. 6B). In comparison, immunohistochemical detection of SCYL1 in Scyl1+/+ and NesCre+;Scyl1+/fl mice showed neuronal expression, a pattern that was absent in Scyl1−/− mice (Fig. 6B, bottom row).
Figure 6.
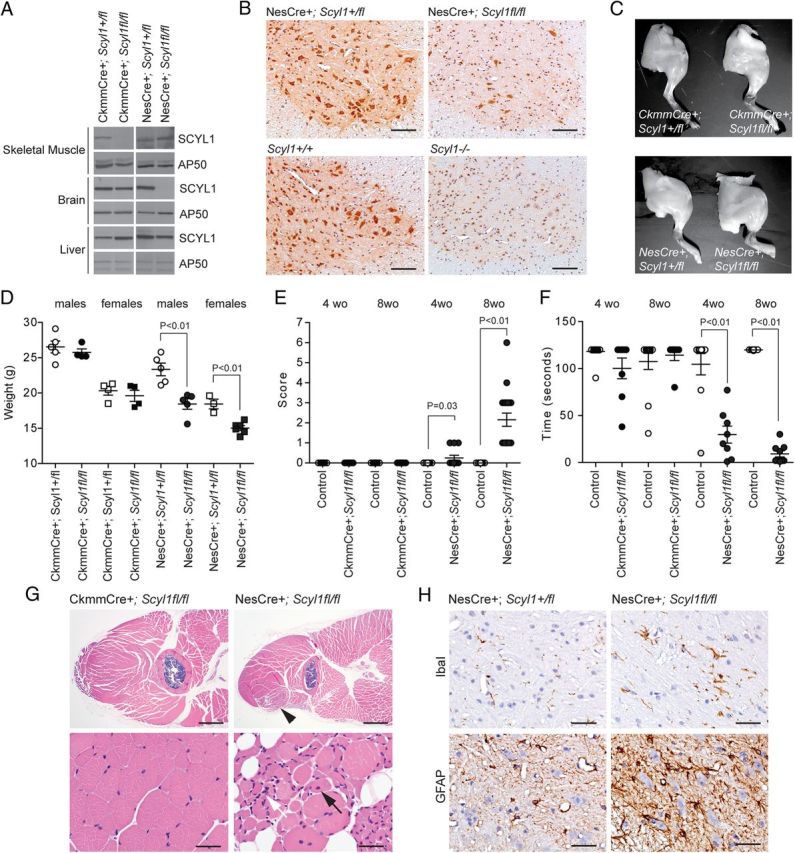
Neural- and skeletal muscle-specific deletion of Scyl1. A, Western blot analysis of brain, liver, and skeletal muscle extracts obtained from CkmmCre+;Scyl1+/fl, CkmmCre+;Scyl1fl/fl, NesCre+;Scyl1+/fl, and NesCre+;Scyl1fl/fl mice using antibodies against SCYL1 or AP50 as loading control. Note the selective absence of SCYL1 in the brain of NesCre+;Scyl1fl/fl mice and in the skeletal muscle of CkmmCre+;Scyl1fl/fl mice. B, Immunohistochemical staining of spinal cord cross-sections from NesCre+;Scyl1+/fl and NesCre+;Scyl1fl/fl, Scyl1+/+ and Scyl1−/− mice, using an anti-SCYL1 antibody. In NesCre+;Scyl1fl/fl sections, >90% of neuronal cells are deficient in SCYL1 expression. Note the absence of SCYL1 in all neurons of spinal sections obtained from Scyl1−/− mice compared with Scyl1+/+ mice. A nonspecific nuclear staining is detected by using this antibody. This nuclear staining was also seen in Scyl1−/− mouse embryonic fibroblasts (data not shown) and in sections obtained from Scyl1−/− mice. Scale bar, 200 μm. C, Hindlimb morphology of neural and skeletal muscle mutants of Scyl1. Representative photographs of hindlimbs obtained from 12-week-old CkmmCre+;Scyl1+/fl, CkmmCre+;Scyl1fl/fl, NesCre+;Scyl1+/fl, and NesCre+;Scyl1fl/fl mice. D, Body weight of 8-week-old male (circles) and female (squares) CkmmCre+;Scyl1+/fl (males, n = 5; females, n = 4), CkmmCre+;Scyl1fl/fl (males, n = 4; females, n = 4), NesCre+;Scyl1+/fl (males, n = 5; females, n = 3), and NesCre+;Scyl1fl/fl (males, n = 5; females, n = 6) mice. Values are expressed as the mean ± SEM. E, Early onset progressive motor deficit in neural-specific but not muscle-specific mutant of Scyl1. Score of 4- and 8-week-old CkmmCre+;Scyl1+/fl [4-week-old (wo) animals, n = 18; 8-week-old animals, n = 8], CkmmCre+;Scyl1fl/fl (4-week-old animals, n = 12; 8-week-old animals, n = 7), NesCre+;Scyl1+/fl (4-week-old animals, n = 10; 8-week-old animals, n = 15), and NesCre+; Scyl1fl/fl (4-week-old animals, n = 12; 8-week-old animals, n = 19) mice. Values are expressed as the mean ± SEM. F, Motor defects in tissue-specific mutants of Scyl1. The inverted grid test was performed on 4- and 8-week-old CkmmCre+;Scyl1+/fl (4-week-old animals, n = 25; 8-week-old animals, n = 12), CkmmCre+;Scyl1fl/fl (4-week-old animals, n = 8; 8-week-old animals, n = 7), NesCre+;Scyl1+/fl (4-week-old animals, n = 10; 8-week-old animals, n = 15), and NesCre+;Scyl1fl/fl (4-week-old animals, n = 12; 8-week-old animals, n = 9) mice to assess their motor functions as described in Materials and Methods. Values are expressed as the mean ± SEM. G, Myopathy in neural-specific but not in muscle-specific mutants of Scyl1. Representative micrographs of skeletal muscles (quadriceps femoris) from CkmmCre+;Scyl1fl/fl (n = 3) mice and NesCre+;Scyl1fl/fl (n = 3) mice. Lesions in NesCre+;Scyl1fl/fl muscles include fibers of different size, angulated (atrophied) fibers surrounded by rounded fibers, group atrophy, nuclear clumps (white arrow), and centrally localized nuclei (black arrow). Scale bars: top row, 1 mm; bottom row, 50 μm. H, Neuroinflammation in the spinal ventral horn of NesCre+;Scyl1fl/fl mice. Representative immunohistochemical staining using antibodies against Iba1 (top row) and GFAP (bottom row) on spinal ventral horn sections obtained from 8-week-old control NesCre+;Scyl1+/fl (n = 3) and NesCre+;Scyl1fl/fl (n = 3) mice. Note the increased Iba1 and GFAP staining in the ventral horn of NesCre+;Scyl1fl/fl mice compared with control animals. Scale bar, 50 μm.
CkmmCre+;Scyl1fl/fl mice were produced according to the normal Mendelian ratio. Unlike Scyl1-deficient mice, CkmmCre+;Scyl1fl/fl mice exhibited neither muscle wasting nor growth defects (Fig. 6C,D). CkmmCre+;Scyl1fl/fl mice did not develop a posterior waddle, abnormal gait, or any of the phenotypes seen in Scyl1-deficient mice (Fig. 6E). Mouse limb strength was shown normal in the mesh grip test in 4- and 8-week-old animals (Fig. 6F). Histological and immunohistochemical analyses did not reveal any significant abnormalities in skeletal muscles from CkmmCre+;Scyl1fl/fl mice. Group atrophy, fiber type switching, and muscle wasting were absent in these mice (Fig. 6G). Because motor neurons critically depend on target-derived growth factors for their development and survival, we also analyzed the spinal cord of CkmmCre+;Scyl1fl/fl mice for signs of neurodegeneration and neuroinflammation. No abnormalities were found in the ventral horn of the spinal cord of CkmmCre+;Scyl1fl/fl animals. These results indicate that the motor defects and myopathy seen in Scyl1-deficient mice are not the result of a muscle-autonomous defect.
NesCre+;Scyl1fl/fl mice were also produced according to the normal Mendelian ratio and, as seen in Scyl1-deficient mice, exhibited growth defects. NesCre+;Scyl1fl/fl mice had a 20% lower body weight than control littermates, and this was maintained throughout their lives (Fig. 6D). More importantly, we found that neural-specific deletion of Scyl1 caused motor dysfunction. However, the progression of the disease was slower and the disease was less severe than that in Scyl1-deficient mice. By 4 weeks of age, NesCre+;Scyl1fl/fl mice had not developed the posterior waddle or abnormal gait characteristic of Scyl1-deficient mice, but all mice exhibited grip strength deficits on the mesh grip test (Fig. 6E,F). By 8 weeks of age, NesCre+;Scyl1fl/fl mice developed an abnormal gait and posterior waddle and clearly showed strength deficits (Fig. 6E,F). In contrast to what was observed in Scyl1−/− mice, NesCre+;Scyl1fl/fl mice did not exhibit tremor when suspended by their tails and the majority did not become paralytic. Flattening of the pelvis was also absent in these mice. Gross morphological analysis of hindlimbs did not reveal any massive muscle wasting as seen in Scyl1-deficient animals (Fig. 6C). Nevertheless, histopathological analyses revealed the presence of lesions in skeletal muscles of NesCre+;Scyl1fl/fl mice. As seen in Scyl1-deficient mice, central nuclei and shrunken myocytes were found within specific muscles of forelimbs and hindlimbs, most notably the rectus femoris and biceps brachii muscles (Fig. 6G). The lesions, however, were milder and more sharply defined from surrounding muscles than those in Scyl1-deficient animals. Histological examination of NesCre+;Scyl1fl/fl mice also revealed the presence of neuroinflammation in similar locations in the CNS, in particular in the ventral horn of the spinal cord (Fig. 6H). Also, there was a slight reduction in the number of large motor neurons of the lateral motor column in NesCre+;Scyl1fl/fl mice compared with those in NesCre+;Scyl1+/fl mice (data not shown). These results indicate that neural-specific loss of Scyl1 recapitulated, albeit to a lesser extent, the phenotypic changes seen in Scyl1-deficient mice.
The results demonstrate that SCYL1 acts, at least in part, in a neuron-autonomous manner to prevent motor neuron degeneration and atrophy of muscle fibers. Moreover, the presence of activated microglia and astrocytes in the spinal ventral horn of NesCre+;Scyl1fl/fl mice also suggests that neuroinflammation is likely triggered by damaged neurons in these mice. Last, the growth retardation seen in these mice is probably related to impaired locomotion but does not rule out a possible role for SCYL1 in regulating feeding behaviors and/or the neuroendocrine system.
TDP-43- and ubiquilin 2-positive cytoplasmic inclusions in Scyl1-deficient motor neurons
Collectively, our results indicate that SCYL1 plays a critical role in the survival of large LMNs. To elucidate the possible mechanisms by which Scyl1 deficiency leads to degeneration of LMNs, we used immunohistochemistry to determine the distribution of several MND-associated proteins, including the familial and sporadic ALS-associated proteins FUS/TLS, TDP-43, and ubiquilin 2, in motor neurons of Scyl1-deficient animals. Although no changes were found in the subcellular localization of FUS/TLS and ubiquitin in the spinal ventral horn of Scyl1−/− mice compared with control littermates, striking differences were observed in the cellular localization of TDP-43 and ubiquilin 2. In the spinal cord of Scyl1+/+ animals, TDP-43 staining was restricted to the nucleus of neuronal cells, although there were some scattered large neurons containing sharply defined small granules positive for TDP-43 (Fig. 7A). In marked contrast, in the spinal cords of Scyl1-deficient mice, scattered ventral horn motor neurons showed reduced to absent nuclear localization of TDP-43 accompanied by intensely positive staining of cytoplasm and granular cytoplasmic aggregates (Fig. 7A). On average, 0.04 ± 0.03 motor neuron with mislocalized TDP-43 per slice was found in Scyl1+/+ mice [two motor neurons with mislocalized TDP-43 (barely discernable) in 48 images analyzed], whereas an average of 2.39 ± 0.3 was found in Scyl1−/− mice (124 motor neurons with mislocalized TDP-43 in 52 images analyzed, p < 0.01). Mislocalization of TDP-43 to cytoplasmic inclusions in motor neurons was sparse in young (4-week-old) animals and intensified with age, which correlated with disease progression in these mice (data not shown). There were also differences in the subcellular localization of ubiquilin 2 (Fig. 7A). In the spinal cord of Scyl1+/+ mice, ubiquilin 2 was evenly distributed within motor neurons, whereas in Scyl1-deficient mice, it accumulated within the cytosol and cytoplasmic inclusions. To determine whether TDP-43 mislocalization was the result of a neural cell-autonomous defect, we also analyzed the spinal cord of NesCre+;Scyl1fl/fl mice for the presence of TDP-43 mislocalization. As shown in Figure 7B, neural-specific deletion of Scyl1 was sufficient to cause TDP-43 mislocalization in some motor neurons.
Figure 7.
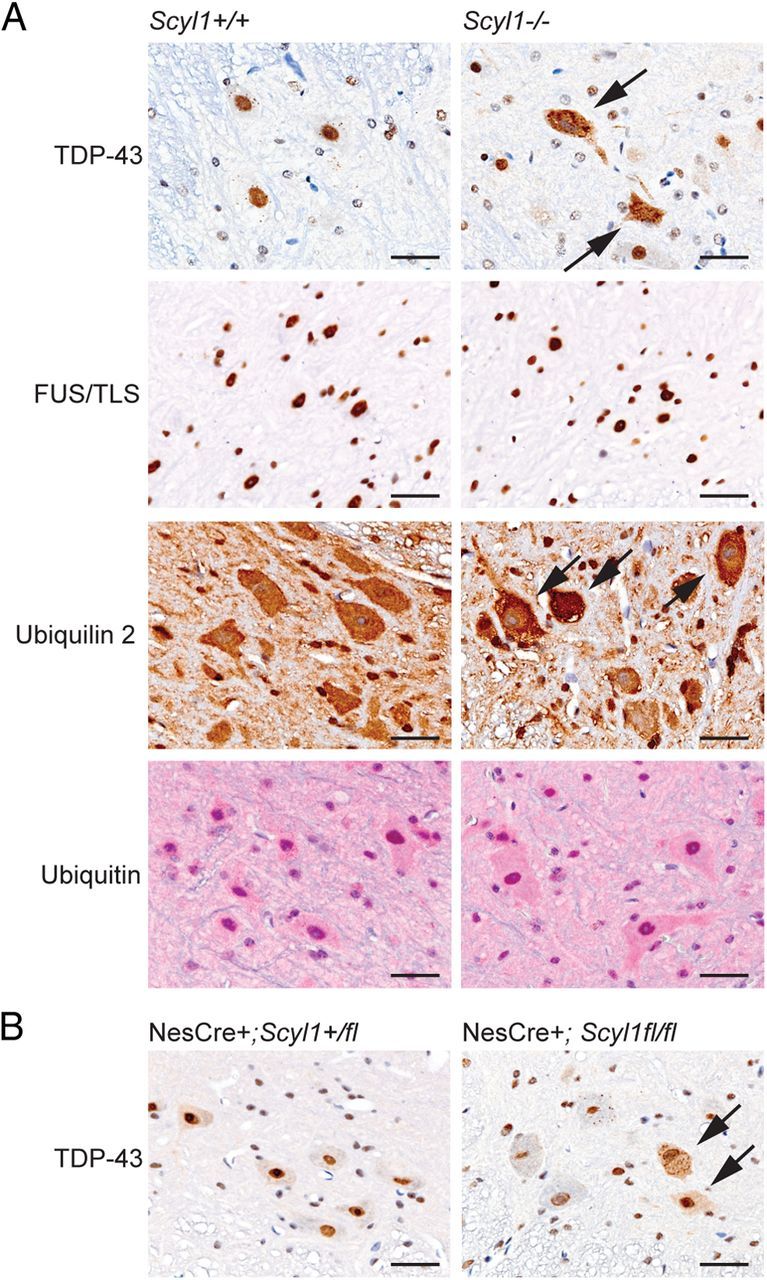
Nuclear to cytoplasmic relocalization of TDP-43 in Scyl1-deficient motor neurons. A, Immunohistochemistry using antibodies against TDP-43, FUS/TLS, ubiquilin 2, and ubiquitin on spinal ventral horn sections obtained from 8- to 12-week-old Scyl1+/+ and Scyl1−/− mice. Note the relocalization of TDP-43 (arrows) but not FUS/TLS from the nucleus to cytoplasmic inclusions in large motor neurons of Scyl1−/− mice. Also note the cytoplasmic accumulation of ubiquilin 2 (arrows) but not ubiquitin in select motor neurons of the spinal ventral horn of Scyl1-deficient mice. Data shown are representative of three mice for each genotype. Scale bars, 25 μm. B, Immunohistochemistry using antibodies against TDP-43 on spinal ventral horn sections obtained from 8- to 12-week-old NesCre+;Scyl1+/fl and NesCre+;Scyl1fl/fl mice. Note the relocalization of TDP-43 (arrows) in large motor neurons of NesCre+;Scyl1fl/fl mice. Data shown are representative of three mice for each genotype. Scale bars, 25 μm.
In addition to the aberrant localization of TDP-43 to cytoplasmic inclusions, abnormal posttranslational modifications, such as hyperphosphorylation, ubiquitylation, cleavage, and formation of Sarkosyl-insoluble aggregates of TDP-43, have been observed in patients with ALS, most of which are restricted to brain tissues (Igaz et al., 2008; Neumann et al., 2009). To test whether similar changes occurred in the spinal cord of Scyl1−/− animals, Western blot analyses using antibodies against TDP-43 and phosphorylated TDP-43 on RIPA and urea fractions isolated from Scyl1+/+ and Scyl1−/− spinal cords were performed. No differences in the migration pattern, solubility, or phosphorylation of TDP-43 were found between extracts from 8-week-old Scyl1+/+ or Scyl1−/− mice (data not shown), a finding that is consistent with what is seen in spinal extracts obtained from patients with ALS (Igaz et al., 2008; Neumann et al., 2009).
Discussion
Our results indicate that Scyl1-deficient mice suffer from an early onset progressive MND with characteristic features of progressive muscular atrophy, a subtype of ALS affecting only LMNs. Phenotypic changes included progressive loss of motor function, decline in limb muscle strength, severe muscle wasting, and paralysis of the rear legs. Morphological changes characteristic of neurogenic atrophy were found in skeletal muscles of Scyl1-deficient animals. In line with this, neuron-specific deletion, but not muscle-specific deletion, of Scyl1 recapitulated, albeit to a lesser extent, the phenotypic changes seen in Scyl1-deficient mice. Loss of large-caliber axons in peripheral nerves and motor neurons of the lateral motor column were conspicuous in Scyl1-deficient animals. Neuroinflammation was found multifocally throughout the CNS and in particular in the spinal ventral horn. Remarkably, redistribution of TDP-43 from the nucleus of large motor neurons to cytoplasmic aggregates and the presence of ubiquilin 2 inclusions, hallmarks of ALS and many neurodegenerative conditions, were present in Scyl1-deficient mice.
The phenotypes of the Scyl1−/− mice are identical to those found in mdf mice (Blot et al., 1995). By 4 weeks of age, both strains exhibited growth defects, loss of strength, and a posterior waddle. Muscle wasting was evident in the rear legs by 6–8 weeks of age, and both strains of mice became paralytic by 8–20 weeks of age. The mdf mutation consists of a single thymidine insertion, creating a premature STOP codon, destabilizating mRNA transcript, and causing a marked reduction in SCYL1 expression (Schmidt et al., 2007). Our data confirm the original assumption that the mdf mutation acts as a loss-of-function mutation (Schmidt et al., 2007). However, unlike what was previously reported in mdf mice (Schmidt et al., 2007), we did not find morphological abnormalities in the cerebellum and optic nerves of Scyl1-deficient mice or reduced numbers of Purkinje cells. However, we found microglial activation in the cerebellar peduncle that could be caused by degeneration of axon tracts in that structure.
In most cases of MNDs, with the exception of SMARD1 (spinal muscular atrophy with respiratory distress type 1) (Grohmann et al., 2001, 2003), ventral horn motor neuron degeneration is the result of gain-of-adverse-effect mutations or partial loss-of-function mutations (Schmalbruch et al., 1991; Rosen, 1993; Wong et al., 1995; Monani et al., 2000; Schmitt-John et al., 2005; Neumann et al., 2006; Lai et al., 2007; Chevalier-Larsen et al., 2008; Kwiatkowski et al., 2009; Vance et al., 2009; Deng et al., 2011; Lee et al., 2011). Null mutations of disease-causing genes are often embryonic lethal as a result of their essential cellular functions (Schrank et al., 1997; Monani et al., 1999; Hicks et al., 2000; LaMonte et al., 2002; Hafezparast et al., 2003; Schmitt-John et al., 2005; Lai et al., 2006, 2007; Ilieva et al., 2009; Sephton et al., 2009; Wu et al., 2009). To our knowledge, Scyl1 represents the first example of a gene whose complete loss of function causes motor neuron degeneration in viable mice. This was unexpected, especially given the proposed roles of SCYL1 in several essential cellular functions and raises the possibility that other members of the SCYL family, which includes SCYL2 and SCYL3, may compensate for the loss of SCYL1 in vivo.
The mechanisms of disease pathogenesis for MNDs are not well understood. A critical question is whether the changes seen in motor neurons, including their death, arise from the neuron itself or from toxic byproducts of neighboring cells. Studies with mice expressing mutant forms of SOD1 showed that the convergence of damages arising from multiple neighboring non-neuronal as well as neuronal cells causes the disease (Ilieva et al., 2009). Here, we show that neural-specific loss of Scyl1 causes motor dysfunctions, neuroinflammation, and TDP-43 mislocalization, indicating that SCYL1 acts in a neuron-autonomous manner to protect motor neurons from degeneration. However, the fact that NesCre+;Scyl1fl/fl mice did not develop full-blown pathology compared with Scyl1−/− mice also suggests that non-neural cells may contribute to the degeneration process.
By virtue of its interaction with the COPI complex, SCYL1 is proposed to regulate Golgi-to-ER retrograde transport of the KDELR, a COPI-dependent process (Burman et al., 2008). Although we confirmed that SCYL1 has the ability to interact with the COPI complex, we could not confirm by using Scyl1−/− primary mouse embryonic fibroblasts a role for SCYL1 in the retrieval of the KDELR from the Golgi apparatus or in the maintenance of Golgi morphology. This indicates that SCYL1 is either dispensable or plays an accessory role in COPI functions. In line with this, loss of SCYL1 function in mice, although debilitating, has milder consequences than the loss of COPI functions in lower organisms. Loss of COPI functions in yeast, Caenorhabditis elegans, Drosophila melanogaster, and zebrafish result in lethality (Faulstich et al., 1996; Coutinho et al., 2004; Hamamichi et al., 2008; Jayaram et al., 2008). Although this argues against a role of SCYL1 in regulating COPI functions, it is important to note that other members of the SCYL family may compensate for the loss of SCYL1. Thus, whether SCYL1 regulates COPI functions, acts as an adaptor protein, or is itself a cargo is still unclear, and additional studies are required to unravel the precise molecular function of SCYL1.
A link between COPI and neurodegeneration has been made recently. The neurological 17 (Nur17) mutant mouse is characterized by a coat color dilution and ataxia with cerebellar involvement (Xu et al., 2010). The mouse has a unique missense mutation in the Arcn1 gene, which encodes the COPD subunit of the heptameric complex COPI. In contrast to the mdf mutation, which affects SCYL1 expression at the mRNA level, Nur17 mutation does not impair expression of COPD. Instead, the mutation, located in the vicinity of its cargo binding site, likely affects recruitment or cargo selection by the COPI complex (Xu et al., 2010). However, there is no report of spinal cord or skeletal muscle lesions in Nur17.
The mechanism by which SCYL1 protects motor neurons from degeneration is unknown, but our findings suggest that SCYL1 may prevent motor neuron degeneration by regulating TDP-43 and ubiquilin 2 homeostasis. In the absence of SCYL1, TDP-43 relocalizes from its predominantly nuclear location to cytoplasmic inclusions in select motor neurons. Similarly, changes in ubiquilin 2 localization were seen in Scyl1-deficient motor neurons. TDP-43 is a ubiquitously expressed DNA/RNA-binding protein involved in RNA metabolism (Buratti and Baralle, 2001; Mercado et al., 2005; Chen-Plotkin et al., 2010). TDP-43 was identified several years ago as the major constituent of cytoplasmic inclusions in ALS and tau-negative FTLD-U (also known as FTLD-TDP) (Neumann et al., 2006). Accumulating evidence suggests that, in these diseases, the mislocalization of TDP-43 from its normal nuclear location to cytoplasmic inclusions negatively impacts its normal nuclear function and leads to neurodegeneration (Banks et al., 2008; Gitcho et al., 2008; Kabashi et al., 2008; Sreedharan et al., 2008; Van Deerlin et al., 2008; Corrado et al., 2009; Daoud et al., 2009; Lagier-Tourenne and Cleveland, 2009; Lee et al., 2011). However, it should be noted that debate still exists as to whether TDP-43 mislocalization is always causally linked to neurodegeneration or is secondary to disease pathology (Rothstein, 2007; Baloh, 2011; Lee et al., 2011). Thus, whether or not TDP-43 mislocalization in Scyl1-deficient mice is causally linked to neurodegeneration or is the result of degenerating neurons is still unclear, and additional studies will be required to address this.
Ubiquilins are thought to function as ubiquitin–proteasome shuttle factors involved in ER-associated protein degradation (Lim et al., 2009). A recent study showed that mutations in UBQLN2, which encodes ubiquilin 2, are associated with dominant X-linked ALS and ALS/dementia and that aberrant accumulation of ubiquilin 2 into cytoplasmic inclusions is a common feature of a large spectrum of ALS cases (Deng et al., 2011). Interestingly, SCYL1 localizes to the ER-to-Golgi-intermediate-compartment (ERGIC) (Burman et al., 2008), a site of protein sorting, conformation-based quality control, and folding along the secretory pathway (Appenzeller-Herzog and Hauri, 2006), raising the possibility of SCYL1 involvement in ERGIC-associated protein degradation. Deregulation of this pathway may trigger a cascade of events leading to, among other effects, accumulation of TDP-43 and ubiquilin 2 in cytoplasmic inclusions.
Collectively, our results indicate that SCYL1 plays a critical role in the preventing of LMN degeneration and identify a link between SCYL1 and the regulation of TDP-43 and ubiquilin 2 homeostasis. The histopathological similarities between the MND caused by the loss of SCYL1 and ALS indicate that Scyl1−/− and Scyl1fl/fl mice represent valuable tools to further our understanding of the pathogenesis of MNDs. Moreover, the early onset and full penetrance of the disease may make the Scyl1−/− mouse a superior mouse model for the rapid and effective screen of pharmacological agents. The phenotype of the Scyl1-deficient mouse also suggests that some of the unmapped recessively inherited and/or sporadic forms of MNDs might result from a loss of SCYL1 function or component of its pathway. Understanding SCYL1 protein functions in motor neurons and its role in non-neuronal cells will be beneficial to understanding the molecular and cellular bases of motor neuron disorders.
Footnotes
This work was supported by Cancer Center Core Grant CA21765, National Institutes of Health Grants R01DK42932 and P01 HL53740 (J.N.I.), and the American Lebanese Syrian Associated Charities. We also thank the Animal Imaging Center, the Veterinary Pathology Core Laboratory, the Histology Laboratory, the Electron Microscopy Facility, the Hartwell Center for Bioinformatics and Biotechnology, and the Genetics/Transgenic Core Unit (all at the St. Jude Children's Research Hospital, Memphis, TN).
The authors declare no competing financial interests.
References
- Appenzeller-Herzog C, Hauri HP. The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J Cell Sci. 2006;119:2173–2183. doi: 10.1242/jcs.03019. [DOI] [PubMed] [Google Scholar]
- Baloh RH. TDP-43: the relationship between protein aggregation and neurodegeneration in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. FEBS J. 2011;278:3539–3549. doi: 10.1111/j.1742-4658.2011.08256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks GT, Kuta A, Isaacs AM, Fisher EM. TDP-43 is a culprit in human neurodegeneration, and not just an innocent bystander. Mamm Genome. 2008;19:299–305. doi: 10.1007/s00335-008-9117-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blot S, Poirier C, Dreyfus PA. The mouse mutation muscle deficient (mdf) is characterized by a progressive motoneuron disease. J Neuropathol Exp Neurol. 1995;54:812–825. [PubMed] [Google Scholar]
- Buratti E, Baralle FE. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem. 2001;276:36337–36343. doi: 10.1074/jbc.M104236200. [DOI] [PubMed] [Google Scholar]
- Burgess RW, Cox GA, Seburn KL. Neuromuscular disease models and analysis. Methods Mol Biol. 2010;602:347–393. doi: 10.1007/978-1-60761-058-8_20. [DOI] [PubMed] [Google Scholar]
- Burman JL, Bourbonniere L, Philie J, Stroh T, Dejgaard SY, Presley JF, McPherson PS. Scyl1, mutated in a recessive form of spinocerebellar neurodegeneration, regulates COPI-mediated retrograde traffic. J Biol Chem. 2008;283:22774–22786. doi: 10.1074/jbc.M801869200. [DOI] [PubMed] [Google Scholar]
- Burman JL, Hamlin JN, McPherson PS. Scyl1 regulates Golgi morphology. PLoS One. 2010;5:e9537. doi: 10.1371/journal.pone.0009537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chafe SC, Mangroo D. Scyl1 facilitates nuclear tRNA export in mammalian cells by acting at the nuclear pore complex. Mol Biol Cell. 2010;21:2483–2499. doi: 10.1091/mbc.E10-03-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol. 2010;6:211–220. doi: 10.1038/nrneurol.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier-Larsen ES, Wallace KE, Pennise CR, Holzbaur EL. Lysosomal proliferation and distal degeneration in motor neurons expressing the G59S mutation in the p150Glued subunit of dynactin. Hum Mol Genet. 2008;17:1946–1955. doi: 10.1093/hmg/ddn092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrado L, Ratti A, Gellera C, Buratti E, Castellotti B, Carlomagno Y, Ticozzi N, Mazzini L, Testa L, Taroni F, Baralle FE, Silani V, D'Alfonso S. High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum Mutat. 2009;30:688–694. doi: 10.1002/humu.20950. [DOI] [PubMed] [Google Scholar]
- Coutinho P, Parsons MJ, Thomas KA, Hirst EM, Saúde L, Campos I, Williams PH, Stemple DL. Differential requirements for COPI transport during vertebrate early development. Dev Cell. 2004;7:547–558. doi: 10.1016/j.devcel.2004.07.020. [DOI] [PubMed] [Google Scholar]
- Daoud H, Valdmanis PN, Kabashi E, Dion P, Dupré N, Camu W, Meininger V, Rouleau GA. Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J Med Genet. 2009;46:112–114. doi: 10.1136/jmg.2008.062463. [DOI] [PubMed] [Google Scholar]
- Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, Jiang H, Hirano M, Rampersaud E, Jansen GH, Donkervoort S, Bigio EH, Brooks BR, Ajroud K, Sufit RL, Haines JL, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulstich D, Auerbach S, Orci L, Ravazzola M, Wegchingel S, Lottspeich F, Stenbeck G, Harter C, Wieland FT, Tschochner H. Architecture of coatomer: molecular characterization of delta-COP and protein interactions within the complex. J Cell Biol. 1996;135:53–61. doi: 10.1083/jcb.135.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;7:616–630. doi: 10.1038/nrneurol.2011.152. [DOI] [PubMed] [Google Scholar]
- Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ, White CL, 3rd, Bigio EH, Caselli R, Baker M, Al-Lozi MT, Morris JC, Pestronk A, Rademakers R, Goate AM, Cairns NJ. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63:535–538. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grohmann K, Varon R, Stolz P, Schuelke M, Janetzki C, Bertini E, Bushby K, Muntoni F, Ouvrier R, Van Maldergem L, Goemans NM, Lochmüller H, Eichholz S, Adams C, Bosch F, Grattan-Smith P, Navarro C, Neitzel H, Polster T, Topaloğlu H, et al. Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1) Ann Neurol. 2003;54:719–724. doi: 10.1002/ana.10755. [DOI] [PubMed] [Google Scholar]
- Grohmann K, Schuelke M, Diers A, Hoffmann K, Lucke B, Adams C, Bertini E, Leonhardt-Horti H, Muntoni F, Ouvrier R, Pfeufer A, Rossi R, Van Maldergem L, Wilmshurst JM, Wienker TF, Sendtner M, Rudnik-Schöneborn S, Zerres K, Hübner C. Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat Genet. 2001;29:75–77. doi: 10.1038/ng703. [DOI] [PubMed] [Google Scholar]
- Hafezparast M, Klocke R, Ruhrberg C, Marquardt A, Ahmad-Annuar A, Bowen S, Lalli G, Witherden AS, Hummerich H, Nicholson S, Morgan PJ, Oozageer R, Priestley JV, Averill S, King VR, Ball S, Peters J, Toda T, Yamamoto A, Hiraoka Y, et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300:808–812. doi: 10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- Hamamichi S, Rivas RN, Knight AL, Cao S, Caldwell KA, Caldwell GA. Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson's disease model. Proc Natl Acad Sci U S A. 2008;105:728–733. doi: 10.1073/pnas.0711018105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks GG, Singh N, Nashabi A, Mai S, Bozek G, Klewes L, Arapovic D, White EK, Koury MJ, Oltz EM, Van Kaer L, Ruley HE. Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat Genet. 2000;24:175–179. doi: 10.1038/72842. [DOI] [PubMed] [Google Scholar]
- Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M, Clark CM, Elman LB, Miller BL, Grossman M, McCluskey LF, Trojanowski JQ, Lee VM. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol. 2008;173:182–194. doi: 10.2353/ajpath.2008.080003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram SA, Senti KA, Tiklová K, Tsarouhas V, Hemphälä J, Samakovlis C. COPI vesicle transport is a common requirement for tube expansion in Drosophila. PLoS One. 2008;3:e1964. doi: 10.1371/journal.pone.0001964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136:1001–1004. doi: 10.1016/j.cell.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C, Xie C, McCormack SG, Chiang HC, Michalak MK, Lin X, Chandran J, Shim H, Shimoji M, Cookson MR, Huganir RL, Rothstein JD, Price DL, Wong PC, Martin LJ, Zhu JJ, Cai H. Amyotrophic lateral sclerosis 2-deficiency leads to neuronal degeneration in amyotrophic lateral sclerosis through altered AMPA receptor trafficking. J Neurosci. 2006;26:11798–11806. doi: 10.1523/JNEUROSCI.2084-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C, Lin X, Chandran J, Shim H, Yang WJ, Cai H. The G59S mutation in p150(glued) causes dysfunction of dynactin in mice. J Neurosci. 2007;27:13982–13990. doi: 10.1523/JNEUROSCI.4226-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascaño J, Tokito M, Van Winkle T, Howland DS, Holzbaur EL. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34:715–727. doi: 10.1016/s0896-6273(02)00696-7. [DOI] [PubMed] [Google Scholar]
- Lee EB, Lee VM, Trojanowski JQ. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci. 2012;13:38–50. doi: 10.1038/nrn3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim PJ, Danner R, Liang J, Doong H, Harman C, Srinivasan D, Rothenberg C, Wang H, Ye Y, Fang S, Monteiro MJ. Ubiquilin and p97/VCP bind erasin, forming a complex involved in ERAD. J Cell Biol. 2009;187:201–217. doi: 10.1083/jcb.200903024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SC, Lane WS, Lienhard GE. Cloning and preliminary characterization of a 105 kDa protein with an N-terminal kinase-like domain. Biochim Biophys Acta. 2000;1517:148–152. doi: 10.1016/s0167-4781(00)00234-7. [DOI] [PubMed] [Google Scholar]
- Lorson CL, Rindt H, Shababi M. Spinal muscular atrophy: mechanisms and therapeutic strategies. Hum Mol Genet. 2010;19:R111–R118. doi: 10.1093/hmg/ddq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado PA, Ayala YM, Romano M, Buratti E, Baralle FE. Depletion of TDP 43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene. Nucleic Acids Res. 2005;33:6000–6010. doi: 10.1093/nar/gki897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, McPherson JD. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, Jablonka S, Schrank B, Rossol W, Prior TW, Morris GE, Burghes AH. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Neumann M, Kwong LK, Lee EB, Kremmer E, Flatley A, Xu Y, Forman MS, Troost D, Kretzschmar HA, Trojanowski JQ, Lee VM. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009;117:137–149. doi: 10.1007/s00401-008-0477-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier S, Gingras S, Funakoshi-Tago M, Howell S, Ihle JN. Two domains of the erythropoietin receptor are sufficient for Jak2 binding/activation and function. Mol Cell Biol. 2006;26:8527–8538. doi: 10.1128/MCB.01035-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD. TDP-43 in amyotrophic lateral sclerosis: pathophysiology or patho-babel? Ann Neurol. 2007;61:382–384. doi: 10.1002/ana.21155. [DOI] [PubMed] [Google Scholar]
- Schmalbruch H, Jensen HJ, Bjaerg M, Kamieniecka Z, Kurland L. A new mouse mutant with progressive motor neuronopathy. J Neuropathol Exp Neurol. 1991;50:192–204. doi: 10.1097/00005072-199105000-00002. [DOI] [PubMed] [Google Scholar]
- Schmidt WM, Kraus C, Höger H, Hochmeister S, Oberndorfer F, Branka M, Bingemann S, Lassmann H, Müller M, Macedo-Souza LI, Vainzof M, Zatz M, Reis A, Bittner RE. Mutation in the Scyl1 gene encoding amino-terminal kinase-like protein causes a recessive form of spinocerebellar neurodegeneration. EMBO Rep. 2007;8:691–697. doi: 10.1038/sj.embor.7401001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt-John T, Drepper C, Mussmann A, Hahn P, Kuhlmann M, Thiel C, Hafner M, Lengeling A, Heimann P, Jones JM, Meisler MH, Jockusch H. Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse. Nat Genet. 2005;37:1213–1215. doi: 10.1038/ng1661. [DOI] [PubMed] [Google Scholar]
- Schrank B, Götz R, Gunnersen JM, Ure JM, Toyka KV, Smith AG, Sendtner M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci U S A. 1997;94:9920–9925. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton CF, Good SK, Atkin S, Dewey CM, Mayer P, 3rd, Herz J, Yu G. TDP-43 is a developmentally regulated protein essential for early embryonic development. J Biol Chem. 2010;285:6826–6834. doi: 10.1074/jbc.M109.061846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen-Plotkin AS, Martinez-Lage M, Steinbart E, McCluskey L, Grossman M, Neumann M, Wu IL, Yang WS, Kalb R, Galasko DR, Montine TJ, Trojanowski JQ, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wilhelmsson H, Graff C, Li H, Oldfors A, Rustin P, Brüning JC, Kahn CR, Clayton DA, Barsh GS, Thorén P, Larsson NG. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat Genet. 1999;21:133–137. doi: 10.1038/5089. [DOI] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Wu LS, Cheng WC, Hou SC, Yan YT, Jiang ST, Shen CK. TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis. 2010;48:56–62. doi: 10.1002/dvg.20584. [DOI] [PubMed] [Google Scholar]
- Xu X, Kedlaya R, Higuchi H, Ikeda S, Justice MJ, Setaluri V, Ikeda A. Mutation in archain 1, a subunit of COPI coatomer complex, causes diluted coat color and Purkinje cell degeneration. PLoS Genet. 2010;6:e1000956. doi: 10.1371/journal.pgen.1000956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagawa Y, Kobayashi T, Ohnishi M, Kobayashi T, Tamura S, Tsuzuki T, Sanbo M, Yagi T, Tashiro F, Miyazaki J. Enrichment and efficient screening of ES cells containing a targeted mutation: the use of DT-A gene with the polyadenylation signal as a negative selection maker. Transgenic Res. 1999;8:215–221. doi: 10.1023/a:1008914020843. [DOI] [PubMed] [Google Scholar]
- Zimmerman L, Parr B, Lendahl U, Cunningham M, McKay R, Gavin B, Mann J, Vassileva G, McMahon A. Independent regulatory elements in the nestin gene direct transgene expression to neural stem cells or muscle precursors. Neuron. 1994;12:11–24. doi: 10.1016/0896-6273(94)90148-1. [DOI] [PubMed] [Google Scholar]