

Abstract
The correct balance between excitation and inhibition is crucial for brain function and disrupted in several pathological conditions. Excitatory neuronal circuits in the primary somatosensory cortex (S1) are modulated by local inhibitory neurons with the balance of this excitatory and inhibitory activity important for function. The activity of excitatory layer 2/3 neurons (L2/3) in the S1 cortex is increased in chronic pain, but it is not known how the local interneurons, nor the balance between excitation and inhibition, may change in chronic pain. Using in vivo two-photon calcium imaging and electrophysiology, we report here that the response of L2/3 local inhibitory neurons to both sensory stimulation and to layer 4 electrical stimulation increases in inflammatory chronic pain. Local application into L2/3 of a GABAA receptor blocker further enhanced the activity of S1 excitatory neurons and reduced pain thresholds, whereas local application of the GABAA receptor modulators (muscimol and diazepam) transiently alleviated the allodynia. This illustrates the importance of the local inhibitory pathways in chronic pain sensation. A reduction in the expression and function of the potassium–chloride cotransporter 2 occurred during chronic pain, which reduces the efficacy of the inhibitory inputs to L2/3 excitatory neurons. In summary, both excitatory and inhibitory neuronal activities in the S1 are enhanced in the chronic pain model, but the increased inhibition is insufficient to completely counterbalance the increased excitation and alleviate the symptoms of chronic pain.
Introduction
Inhibitory neurons in cortical areas play key roles in the maintenance of brain function. An imbalance between excitation and inhibition, attributable to abnormal inhibitory neuron activity, contributes to several diseases, including epilepsy, depression, and schizophrenia (Croarkin et al., 2011; Lewis et al., 2005). The primary somatosensory cortex (S1) contributes to nociception, specifically coding the location, intensity, and duration of pain sensation (Bushnell et al., 1999). Under chronic pain conditions, structural remodeling of the synapses onto S1 excitatory neurons occurs (Kim and Nabekura, 2011), and an increase in the activity of layer 2/3 (L2/3) excitatory neurons in the S1 contributes to the allodynia that characterizes the chronic pain behavior (Eto et al., 2011). L2/3 GABAergic inhibitory neurons in the S1 innervate the L2/3 excitatory neurons, forming feedforward and feedback inhibition that can attenuate excessive activity of the excitatory neurons (Zhu et al., 2004; Xu and Callaway, 2009). Hence, the transmission of the sensory and pain information by excitatory L2/3 pyramidal neurons to other cortical areas (Ferezou et al., 2007; Kamatani et al., 2007) depends on the integration of these excitatory and inhibitory inputs.
In the present study, we therefore investigated changes in inhibition in S1 in chronic pain and report that the activity of L2/3 inhibitory neurons is increased. Furthermore, pharmacological manipulation of the S1 inhibition markedly affects allodynia. There is also, however, disruption of Cl− homeostasis in the L2/3 excitatory neurons resulting in inhibition being less efficacious. Although there is net increase in inhibition within the cortical circuit, this is insufficient to balance the enhanced excitatory activity and attenuate chronic pain behavior.
Materials and Methods
Animal preparation.
We used 4- to 5-week-old male C57BL/6 and VGAT (vesicular GABA transporter)–Venus mice, with the latter specifically expressing Venus in inhibitory neurons (Wang et al., 2009). Mice were maintained on a 12 h light/dark cycle. Food and water were provided ad libitum. To induce chronic inflammatory pain, 40 μl of complete Freund's adjuvant (CFA) (Sigma) was injected subcutaneously into the hindpaw at 4 weeks of age, and experiments were performed from 5 weeks. Control mice were injected with 40 μl of saline. All relevant experimental protocols were approved by the Animal Research Committee of the National Institutes of Natural Sciences, Japan.
Surgery and optical imaging of intrinsic neuronal activity.
Mice were anesthetized with urethane (1.7 g/kg body weight, i.p.) and atropine (0.4 mg/kg body weight, i.p.) and implanted with a small cranial window above S1 as reported previously (Eto et al., 2011). Drugs [or artificial CSF (aCSF)] were applied to S1 via a gap in the coverslip that covered this cranial window. An intrinsic optical signal in response to mechanical stimulation of the hindpaw was used to identify S1. This involved a change in intensity of reflected blue light (490 nm) (Shibuki et al., 2003) that was evoked in S1 by 1 s application of a 4 g/cm2 metal plate to the hindpaw using a custom-made manipulator.
In vivo Ca2+ imaging.
Multicell bolus loading of L2/3 neurons with the Ca2+ indicator fura-2 AM (Invitrogen) was performed as reported previously (Kameyama et al., 2010). Fura-2 AM was dissolved in dimethylsulfoxide and then mixed with 20% Pluonic F-127 (Invitrogen) to yield a stock concentration of 10 mm that was then diluted 1:10 with aCSF that also included sulforhodamine 101 (SR101; 0.1 mm). Fura-2 was excited at 800 nm, and both Venus and SR101 were excited at 950 nm, using a two-photon laser-scanning microscope based on a mode-locked titanium–sapphire laser. Fura-2-stained cells were all distinguished as inhibitory and excitatory neurons by the presence or absence of Venus, respectively, whereas astrocytes were identified by SR101 fluorescence. Electrical excitation used a bipolar electrode inserted into S1 at a depth of 500–550 μm, as reported previously (Eto et al., 2011). Electrical pulses were generated using a stimulator (SEN-7203; Nihon Kohden). Each trial consisted of 10 square 50 μΑ current pulses of 200 μs in duration applied at a frequency of 20 Hz. Peripheral sensory stimulation (see Fig. 1) used mechanical pressure applied to the hindpaw as described above.
Figure 1.
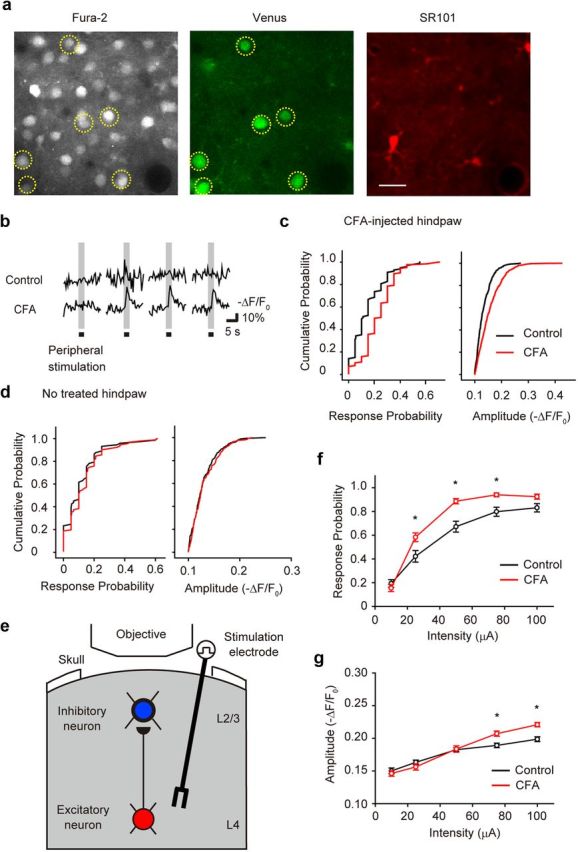
Intracortical plasticity between L4 and the L2/3 inhibitory neurons in the S1 contribute to the enhanced L2/3 interneuron response to sensory stimulation under chronic pain conditions. a, Ca2+ fluorescence (left) in L2/3 inhibitory neurons, identified as fluorescence in VGAT–Venus mice (middle, yellow dotted circles). Astrocytes were identified by SR101 fluorescence (right). Scale bar, 20 μm. b, Typical traces of sensory-evoked Ca2+ transients of L2/3 inhibitory neurons in control (top traces) and CFA-injected (bottom traces) mice. c, d, Cumulative probability histograms of response probability per cell and amplitude of Ca2+ transients in the S1 region contralateral to a CFA-injected hindpaw (c) or to a non-injected normal hindpaw (d); in c, a significant difference between control and CFA-injected mice was observed (Kolmogorov–Smirnov test, p < 0.05). e, Schematic drawing of in vivo Ca2+ imaging of L2/3 inhibitory neurons in response electrical stimulation of L4. f, g, The relationship between stimulus intensity and response probability per cell (f), and amplitude of Ca2+ transients (g) in response to L4 stimulation at the indicated intensities, in control and CFA-injected mice. *p < 0.05, significantly different from the control group by ANOVA, followed by a Bonferroni's test; error bars represent ± SEM.
Fluorescence data acquisition and analyses.
Images (256 × 256 pixels, 0.428 s/frame) were analyzed with Olympus Fluoview software. The baseline intensity F0 was obtained by averaging values during the prestimulus period (5 s). Cells were considered responsive if the change in fluorescence intensity was >10% of the baseline value, the detection threshold of our system. Because an increase in intracellular calcium concentration induces reduction of fura-2 signal, the response was expressed as −ΔF/F0.
Slice preparation and patch-clamp recordings.
Slices were prepared as reported previously (Eto et al., 2011). For measuring IPSCs in L2/3 pyramidal neurons, patch pipettes were filled with an internal solution containing the following (in mm): 130 cesium methanesulfonate, 4 CsCl, 2 EGTA, 4 Mg-ATP, and 10 HEPES, and 5 QX-314, pH 7.3. Neurons were voltage clamped at a holding potential of 0 mV (Multiclamp 700B; Molecular Devices). Blockers of NMDA [dl-(−)-2-amino-5-phosphonopentanoic acid (AP-5; 50 μm)] and non-NMDA [6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 10 μm)] receptors were included in the solution. To measure the somatodendritic chloride gradient, the composition of the patch pipette solution was as follows (in mm): 18 KCl, 111 K-gluconate, 0.5 CaCl2, 2 NaOH, 10 glucose, 10 HEPES, 2 Mg-ATP, and 5 BAPTA, with pH adjusted to 7.3 with KOH. Slices were perfused with an aCSF solution containing 50 μm AP-5, 10 μm CNQX, 1 μm tetrodotoxin (to block voltage-dependent Na+ channels), and 10 μm bumetanide (to selectively block the sodium–potassium–chloride cotransporter 1). To quantify the GABA reversal potential, a 13.5 mV liquid junction potential was accounted for in the data shown in Figure 4b–e. GABA (1 mm) was applied by pressure ejection (50 ms in duration) from a glass pipette. The GABA solution contained 0.1 μm SR101 so as to monitor the drug diffusion area, which was ∼4 μm in diameter. For gramicidin-perforated patch-clamp recordings, the patch pipette solution contained 150 mm KCl, 10 mm HEPES, pH 7.3 with Tris-OH, and 50–80 μg/ml gramicidin.
Figure 4.

Decrease of KCC2 expression and function in the S1 cortex in chronic pain. a–c, The somatodendritic chloride gradient in L2/3 pyramidal neurons was abolished in chronic pain conditions. a, Schematic diagram of the experimental arrangement: a standard whole-cell patch pipette imposing a Cl− load into the cell stimulates KCC2-mediated Cl− efflux along the dendrites. b, c, Typical current–voltage relationship of the currents in response to local application of GABA at the soma (green line) and dendrites (black line), in control (b) and CFA-injected (c) mice. Insets show typical GABA response current traces at different voltages. d, EGABA for the dendritic application (white columns) and soma application (green columns) in control and CFA-injected mice, with and without furosemide (Furo). *p < 0.05, significantly different from the data of dendrites in control mice by ANOVA, followed by a Bonferroni's test. e, Difference between EGABA of dendrites and soma in control mice (white columns) and CFA-injected mice (red columns). *p < 0.05, significantly different from the data of ΔEGABA in control mice without furosemide by ANOVA, followed by a Bonferroni's test. Error bars represent ± SEM. f, GABAergic IPSCs recorded in L2/3 pyramidal neurons using gramicidin-perforated patch-clamp recordings to maintain intracellular Cl− intact. Top traces show typical traces in neurons from control (left) and CFA (right) mice, at holding potentials between −110 and −40 mV (10 mV increments). Bottom graph shows averaged reversal potentials of these IPSCs (EGABA) in neurons from control and CFA-injected mice. g, Western blots showing reduced KCC2 expression in S1 cortex from CFA-injected mice. Top shows a sample blot, which used β-actin as a control. Bottom graphs show averaged relative expression in controls (n = 7) and chronic pain conditions (n = 7). *p < 0.05, Student's t test.
Mechanical allodynia tests and drug application.
The mechanical pain thresholds were measured using von Frey hairs (Chaplan et al., 1994), in the same mice, before and 7 d after injection of saline or CFA into the hindpaw. To evaluate drug effects on these thresholds, a small injection hole (1 mm in diameter) was made in the skull above S1. Surgery and pressure injection of drugs into L2/3 was conducted under brief anesthesia using isoflurane (1–3% as needed, with 30% oxygen balanced with nitrogen).
Western blotting.
Brain slices containing S1 L2/3 were collected and homogenized in ice-cold SDS buffer (2% SDS, 10% glycerol, 2% 2-mercaptoethanol, 75 mm Tris-HCl). Samples were separated using 7% SDS-PAGE. Membranes were probed with anti-potassium-chloride cotransporter 2 (KCC2) antibody (1:500) and anti-β-actin antibody (1:1000).
Statistical analyses.
All values were expressed as mean ± SEM. Statistical comparisons were performed using ANOVA, followed by a Bonferroni's test, or using unpaired Student's t test. The difference between cumulative distributions was assessed using the Kolmogorov–Smirnov test. A difference was considered significant at p < 0.05.
Results
Increase in activity of inhibitory neurons in chronic pain
Inflammatory chronic pain was induced by subcutaneous injection of CFA into mice hindpaws. The intrinsic optical signal arising from neuronal activity was used to identify the S1 contralateral to the injected hindpaw. In vivo Ca2+ responses in S1 neurons were imaged with fura-2 AM and GABAergic interneurons identified as Venus positive in VGAT–Venus mice (Fig. 1a). Mechanical stimulation of the hindpaw evoked larger and more frequent Ca2+ transients in L2/3 inhibitory neurons in CFA-injected mice (Fig. 1b,c). The relative fluorescence change and probability of a Ca2+ response in these L2/3 inhibitory neurons was 0.156 ± 0.002 (578 responses in 4 animals) and 0.232 ± 0.012 (126 trials in 4 animals), respectively, in CFA-injected mice. These values were significantly different from the amplitude and response probability in control mice, which were 0.133 ± 0.002 (300 responses in 3 animals) and 0.152 ± 0.013 (99 trials in 3 animals), respectively (Fig. 1b,c). Ca2+ responses in the opposite hemisphere S1 L2/3 inhibitory neurons in response to stimulation of the non-injected contralateral hindpaw were not different between control (three animals) and CFA-injected mice (three animals; Fig. 1d). These results demonstrate that the activity of the L2/3 inhibitory neurons that specifically respond to mechanical stimulation of the CFA-injected hindpaw shows a chronic increase in activity associated with chronic inflammatory pain. In addition, both the response probability and amplitude of Ca2+ transients in response to in vivo electrical stimulation of the layer 4 (L4) region in S1 (Fig. 1e) increased in CFA-injected mice compared with those in control mice (Fig. 1f,g). This indicates that either the intrinsic excitability of the L2/3 interneurons or the excitatory input to these L2/3 neurons increases in chronic pain, similar to the enhanced excitatory input from L4 to L2/3 excitatory neurons that we have reported recently (Eto et al., 2011).
Increase in the net GABA inhibition of excitatory neurons is insufficient to offset mechanical allodynia
To examine the functional consequences of this increased inhibitory neuron activity, we investigated the in vivo effects of GABAA receptor modulating drugs on activity of their targets: the L2/3 excitatory neurons. The amplitude of Ca2+ transients in the cortex in response to sensory stimulation has been reported to linearly correlate to the number of action potentials (Kerr et al., 2005). The amplitude of Ca2+ responses in L2/3 excitatory neurons evoked by L4 stimulation was greater in CFA-injected mice (346 cells, 4 animals, p < 0.05) than that in control mice (454 cells, 5 animals; Fig. 2b,c), as we reported previously (Eto et al., 2011). Application of 0.6 and 3 μm SR95531 [2-(3-carboxypropyl)-3-amino-6-(4-methoxyphenyl)pyridazinium bromide], a GABAA receptor blocker, concentration dependently increased the amplitude of Ca2+ responses in both control and CFA-injected mice (control, five animals; CFA, three animals; Fig. 2b,c). The extent of this increase for both SR95531 concentrations was greater in CFA-injected mice than that in controls (Fig. 2d). This result suggests that net effect of GABA inhibition on L2/3 excitatory neurons in the S1 is greater in the chronic pain model than that in control mice. However, this enhanced inhibition to the excitatory neurons is insufficient to offset the enhanced activity of the excitatory neurons during chronic pain. We also examined the effects of GABAA receptor modulators injected into S1 on the mechanical pain thresholds using the von Frey filament. Microinjection of a low dose of 0.6 μm SR95531 (0.5 μl) into the S1 did not affect mechanical sensitivity in control mice (five animals) but further reduced the mechanical pain thresholds in CFA-injected mice (five animals; Fig. 2e). Hence, the increased inhibitory neuronal activity is modestly alleviating the mechanical allodynia but not able to prevent it. Microinjection of a GABAA receptor agonist (muscimol, 50 μm, 0.5 μl) or a positive GABAA receptor allosteric modulator (diazepam, 10 μm, 0.5 μl) could conversely increase the mechanical thresholds in CFA-injected mice, whereas the vehicle had no effect (Fig. 2f,g). Hence, additional activation of GABAA receptors in the L2/3 of the S1 could completely (although transiently) attenuate chronic pain behavior, with important possible clinical relevance. Together, these in vivo data indicate that, although the activity of L2/3 inhibitory neurons is significantly enhanced, the net effect of this increased GABA inhibition is insufficient to offset the increased excitation and alleviate pain behavior. Additional enhancement of GABAA-mediated inhibition can, however, counteract the excessive excitation and eliminate allodynia.
Figure 2.
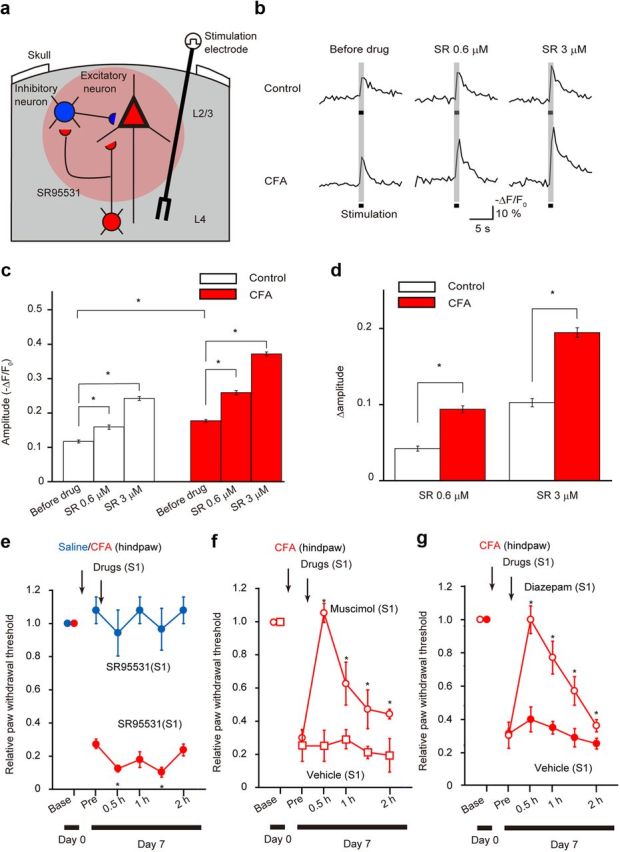
Effects of GABAA receptor modulators on the hyperactivity of S1 excitatory neurons and on pain behavior. a, Schematic drawing of in vivo Ca2+ imaging of L2/3 excitatory neurons in response to L4 electrical stimulation in the presence of SR95531 (SR) (red circle) to block the output from the hyperactive inhibitory neurons. b, Averaged traces of L4 stimulation-evoked Ca2+ transients in L2/3 excitatory neurons in control (top traces) and CFA-injected (bottom traces) mice before (left) and after the local application of 0.6 μm (middle) and 3 μm (right) SR95531. c, Amplitude of Ca2+ transients of L2/3 excitatory neurons in control (white columns) and CFA-injected (red columns) mice, with and without SR95531. d, Comparison of the effects of SR95531 between control (white columns) and CFA-injected (red columns) mice. The y-axis (Δamplitude) shows the difference in the amplitude of the Ca2+ transients before and after application of SR95531. *p < 0.05, significantly different from the control group by ANOVA, followed by a Bonferroni's test; error bars represent ± SEM. e–g, Effects of microinjection of GABAA receptor drugs into the L2/3 region of S1 cortex on paw-withdrawal thresholds tested with von Frey hairs. e, SR95531 (0.6 μm), a GABAA receptor antagonist, reduced withdrawal thresholds beyond the reduction already present as a result of chronic allodyniain CFA-injected mice (red filled circles). This dose of SR95531 had no effect in control mice (blue filled circles). f, Muscimol (50 μm) transiently alleviated mechanical allodynia in CFA-injected mice (red open circles), whereas vehicle injection had no effect (red open squares). g, Diazepam (10 μm) transiently alleviated mechanical allodynia in CFA-injected mice (open circles), whereas vehicle injection had no effect (filled circles). *p < 0.05, significantly different from the value at just before drug application by ANOVA, followed by a Bonferroni's test; error bars represent ± SEM.
Locus of the enhanced neuronal activity
We next examined the synaptic output of the L2/3 inhibitory interneurons, conducting whole-cell patch-clamp recording of IPSCs in L2/3 excitatory pyramidal neurons in brain slices containing the S1 hindlimb area, in the presence of CNQX and AP-5 (Fig. 3). The frequency and amplitude of spontaneous IPSCs were not different between control and CFA-injected mice (frequency: 4.62 ± 0.86 Hz, nine cells in controls; 5.80 ± 0.74 Hz, seven cells in CFA; amplitude: 23.9 ± 3.00 pA, nine cells in controls; 26.1 ± 3.38 pA, seven cells in CFA; Fig. 3a–c). Evoked IPSCs (eIPSCs) were induced by local paired electrical stimulation of the L2/3 inhibitory neurons using a range of stimulus intensities (Fig. 3d). The stimulus intensity–eIPSC amplitude relationship and the eIPSCs paired-pulse ratios (second IPSC amplitude/first IPSC amplitude) were also not significantly different between control and CFA-injected mice (controls, eight cells; CFA, nine cells) (Fig. 3e–g). The results suggest that the transmission from inhibitory synapses to L2/3 excitatory neurons were not altered during chronic inflammatory pain. We next examined IPSCs in L2/3 pyramidal neurons when the interneurons were activated by feedforward inputs from activated L4 neurons, simultaneously recording L4 activated EPSCs in the same L2/3 pyramidal neurons. EPSCs were recorded at the IPSC reversal potential (from −55 to −60 mV), whereas IPSCs were recorded at the EPSC reversal potential (0 mV). CNQX was used to isolate the feedforward IPSCs from the IPSCs evoked by direct activation of any inputs. (feedforward IPSC = IPSCs without CNQX − IPSCs with CNQX). Both the feedforward IPSCs and the direct L4 evoked EPSCs increased in CFA-injected mice (n = 8) compared with those in normal mice (n = 7), but the ratio of EPSC amplitude to IPSC amplitude did not significantly change (control, 0.50 ± 0.09; CFA, 0.35 ± 0.06; p > 0.05). Together, these results strongly suggest that the increased feedforward activation of L2/3 interneurons by L4 inputs contributes greatly to their enhanced activity. However, the L4 inputs also results in enhanced activity of excitatory L2/3 pyramidal neurons in chronic pain, with the balance of excitation and inhibition in S1 L2/3 being less affected.
Figure 3.

Inhibitory synaptic transmission to L2/3 pyramidal neurons in the S1 was not altered in chronic pain. a, Typical traces of spontaneous IPSCs in control (top) and CFA-injected (bottom) mice. b, c, Mean frequency of spontaneous IPSCs (b) and IPSC amplitudes (c) in control (white columns) and CFA-injected (red columns) mice. d, Schematic drawing of whole-cell patch-clamp recording of IPSCs from L2/3 pyramidal neurons in response to L2/3 electrical stimulation. e, Averaged traces of IPSCs evoked by paired-pulse stimulation at an intensity of 30 μA in slices from control (left) and CFA-injected (right) mice. f, g, Stimulus intensity–IPSC amplitude curves (f) and stimulus intensity–paired-pulse ratio relationships (g) in neurons from control (black line) and CFA-injected (red line) mice. Error bars represent ± SEM.
Reduction of KCC2 expression in chronic pain
The above results also suggest that the postsynaptic GABAA receptor responses were not altered during chronic pain. However, the postsynaptic GABA response also depends on the electrochemical Cl− gradient, which was artificially maintained in the above standard whole-cell patch-clamp experiments. Neuronal Cl− homeostasis is disrupted in a number of pathological conditions, primarily attributable to loss of function of the potassium–chloride cotransporter KCC2 (Nabekura et al., 2002; Coull et al., 2005). To examine whether Cl− homeostasis is altered in L2/3 pyramidal neurons in the chronic pain model, initially examined efficacy of Cl− extrusion between soma and dendrites (somatodendritic gradient) by loading the cell soma with 19 mm Cl− (18 mm KCl plus 0.5 mm CaCl2) through the whole-cell patch-clamp pipette and recorded the reversal potential of the response to GABA (EGABA) locally applied to the soma and to the dendrites 60 μm away (Jarolimek et al., 1999; Khirug et al., 2010; Fig. 4a). For control mice, the EGABA obtained from local dendritic GABA application was more negative than that obtained for the somatic GABA response (dendrites, −64.05 ± 3.50 mV; soma, −55.86 ± 1.92 mV; n = 7, p < 0.05) (Fig. 4b,d). This reflects the active efflux of Cl− from dendrites primarily via KCC2. In CFA-injected mice, there was no difference between EGABA of the dendritic and soma GABA response (dendrites, −52.4 ± 3.93 mV; soma, −54.6 ± 2.58 mV; n = 7, p < 0.05) (Fig. 4c,d). Hence, there was no somatodendritic ΔEGABA (EGABA at dendrite − EGABA at soma) in CFA-injected mice compared with the negative value obtained in control mice (control, −8.16 ± 2.05 mV, n = 7; CFA, 2.07 ± 2.60 mV, n = 7; p < 0.05). Application of 1 mm furosemide, a KCC2 inhibitor, reduced the somatodendritic ΔEGABA in control mice (n = 7) but did not affect the value in CFA-injected mice (Fig. 4d,e). We next more directly examined these suggested changes to intracellular Cl− by measuring the EGABA of the synaptic GABA response to local electrical stimulation using the gramicidin-perforated patch-clamp method (Kitamura et al., 2008), which prevents dialysis of intracellular Cl− into the recording patch pipette and allows electrical measurements while maintaining intracellular chloride intact. EGABA in CFA-injected mice (−59.4 ± 2.82 mV, n = 6) was significantly more positive than that in control mice (−69.9 ± 1.83 mV, n = 8, p < 0.05; Fig. 4f). Finally, we measured KCC2 expression using Western blots of S1 cortex and found that KCC2 expression levels was significantly decreased in chronic pain (p < 0.05) (Fig. 4g). Thus, reduction of KCC2 expression results in a less strongly hyperpolarizing GABAergic response, which would be expected to reduce the efficacy of GABAergic inhibition in S1.
Discussion
In the present study, we demonstrate hyperactivity of L2/3 inhibitory neurons in the S1 in response to mechanical stimulation in the inflammatory chronic pain model. The inhibitory neurons also showed enhanced activity when excited by local electrical stimulation, indicating that plasticity in the S1 cortex itself contributes to the enhanced response to sensory stimulation. These L2/3 inhibitory neurons are activated by sensory stimulation (Zhu et al., 2004) via a neuronal pathway that includes excitatory inputs from L4 (Xu and Callaway, 2009). Excitatory synapses onto interneurons can undergo synaptic plasticity, such as long-term potentiation in the interneurons of the hippocampus and amygdale (Mahanty and Sah, 1998; Kullmann and Lamsa, 2007) and spike-timing-dependent plasticity of cortical interneurons (Lu et al., 2007). Chronic inflammatory pain in response to CFA in the hindpaw is associated with enhanced afferent activity in peripheral sensory nerves (Ji et al., 2003), resulting in increased excitation of L4 neurons and subsequently L2/3 excitatory and inhibitory neurons. Increased activation of these cortical areas is also likely to result from hyperexcitability of responses in the spinal cord during chronic pain. We have shown recently that the activity of L2/3 excitatory neurons are also increased in the same chronic pain model (Eto et al., 2011), and L2/3 inhibitory neurons also receive feedback input from these L2/3 excitatory neurons (Kaiser et al., 2004). In short, we suggest that increased peripheral and central afferent activity in the mechanosensation and pain pathways induces local synaptic plasticity within the S1 cortex that results in enhanced excitatory inputs to both L2/3 excitatory and inhibitory neurons and resultant chronic potentiation of responses of these neurons.
The L2/3 inhibitory neurons project locally to the excitatory L2/3 neurons in which they can function to reduce excessive excitation of the excitatory neurons evoked by sensory inputs (Zhu et al., 2004; Xu and Callaway, 2009). We show here that, although the activity of these inhibitory interneurons is increased in chronic pain, this is clearly insufficient to prevent the hyperactivity of the L2/3 excitatory neurons and the allodynia that characterize chronic inflammatory pain. The inhibitory inputs were having some effect, because the GABAA receptor antagonist SR95531 increased Ca2+ transients in L2/3 excitatory neurons and reduced mechanical thresholds in the chronic pain mice even further. The effects of SR95531 in CFA-injected mice were greater than in control, consistent with the elevated activity of the L2/3 inhibitory neurons. Although the elevated inhibitory activity could only attenuate and not offset allodynia, local injection of the GABAA receptor activators muscimol or diazepam could completely alleviate allodynia, indicating additional scope to enhance the local inhibition in S1 that could be used clinically.
The increased inhibition may be an adaptive response to try to counteract the effects of the enhanced excitation and thereby reduce chronic pain behavior, but such an adaptation appears only partially effective. The efficacy of synaptic transmission between these inhibitory neurons and the excitatory neurons is also important for determining the net inhibition in S1. Our patch-clamp results in S1 brain slices indicate that the presynaptic GABA release probability and the postsynaptic receptor number are unchanged in chronic pain. Hence, “shunting” inhibition should be increased in chronic pain. However, the postsynaptic response also depends on the intracellular Cl− concentration, which is primarily regulated by the KCC2 chloride transporter (Kakazu et al., 1999; Rivera et al., 1999). In CFA-injected mice, chronic pain is associated with a decrease in KCC2 expression and a decrease in Cl− efflux in L2/3 pyramidal excitatory neurons. This results in increased intracellular Cl− concentration and a less hyperpolarizing GABAA receptor response. A similar downregulation of KCC2 expression and function is seen in numerous pathological conditions, including in the dorsal horn of the spinal cord in which it plays a key role in chronic pain (Coull et al., 2005). Increased neuronal activity can reduce KCC2 function by dephosphorylation (Lee et al., 2011) or reduced expression (Fiumelli et al., 2005; Kitamura et al., 2008), and a similar increased activity in S1 may trigger KCC2 downregulation. In other brain areas, release of trophic factors, such as brain-derived neurotrophic factor, from neurons or glia may mediate activity-dependent downregulation of KCC2 function (Kohara et al., 2001; Coull et al., 2005; Wake et al., 2007). The loss of KCC2 function would reduce the effectiveness of inhibitory inputs from L2/3 interneurons onto L2/3 excitatory neurons. Although inhibitory neuron activity is increased, the reduced efficacy of their synaptic outputs may contribute to the inability of the enhanced inhibitory activity to alleviate chronic pain.
In summary, chronic inflammatory pain is associated with increased activity of both the L2/3 excitatory neurons and L2/3 inhibitory neurons. However, the balance between excitation and inhibition is the critical functional outcome, and this balance is weighed toward excessive excitation in chronic pain. A loss of KCC2 and the resultant reduced efficacy of GABAergic inhibition may contribute to this excitation–inhibition imbalance in chronic pain. The enhanced inhibition does alleviate the pain behavior somewhat but is not able to prevent the enhanced L2/3 excitation or the allodynia. However, the inhibitory neuronal circuits in S1 and their mechanism remain as powerful components of the pain behavior and valid targets for new drugs to treat chronic pain.
Footnotes
This study was supported by Core Research for Evolutional Science and Technology at the Japan Science and Technology Agency and Grants-in-Aid for Scientific Research (A) (J.N.) and Grants-in-Aid for Young Scientists (B) (Grant 23790653) from the Ministry of Education, Culture, Sports, Science, and Technology (K.E.). We are grateful to Dr. A. Miyawaki for providing pCS2–Venus and Y. Yanagawa for providing VGAT–Venus mice. We thank Dr. H. Wake and Dr. S. K. Kim for discussions about this manuscript.
The authors declare no competing financial interests.
References
- Bushnell MC, Duncan GH, Hofbauer RK, Ha B, Chen JI, Carrier B. Pain perception: is there a role for primary somatosensory cortex? Proc Natl Acad Sci U S A. 1999;96:7705–7709. doi: 10.1073/pnas.96.14.7705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Croarkin PE, Levinson AJ, Daskalakis ZJ. Evidence for GABAergic inhibitory deficits in major depressive disorder. Neurosci Biobehav Rev. 2011;35:818–825. doi: 10.1016/j.neubiorev.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Eto K, Wake H, Watanabe M, Ishibashi H, Noda M, Yanagawa Y, Nabekura J. Inter-regional contribution of enhanced activity of the primary somatosensory cortex to the anterior cingulate cortex accelerates chronic pain behavior. J Neurosci. 2011;31:7631–7636. doi: 10.1523/JNEUROSCI.0946-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferezou I, Haiss F, Gentet LJ, Aronoff R, Weber B, Petersen CC. Spatiotemporal dynamics of cortical sensorimotor integration in behaving mice. Neuron. 2007;56:907–923. doi: 10.1016/j.neuron.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Fiumelli H, Cancedda L, Poo MM. Modulation of GABAergic transmission by activity via postsynaptic Ca2+-dependent regulation of KCC2 function. Neuron. 2005;48:773–786. doi: 10.1016/j.neuron.2005.10.025. [DOI] [PubMed] [Google Scholar]
- Jarolimek W, Lewen A, Misgeld U. A furosemide-sensitive K+–Cl− cotransporter counteracts intracellular Cl− accumulation and depletion in cultured rat midbrain neurons. J Neurosci. 1999;19:4695–4704. doi: 10.1523/JNEUROSCI.19-12-04695.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Kaiser KM, Lübke J, Zilberter Y, Sakmann B. Postsynaptic calcium influx at single synaptic contacts between pyramidal neurons and bitufted interneurons in layer 2/3 of rat neocortex is enhanced by backpropagating action potentials. J Neurosci. 2004;24:1319–1329. doi: 10.1523/JNEUROSCI.2852-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakazu Y, Akaike N, Komiyama S, Nabekura J. Regulation of intracellular chloride by cotransporters in developing lateral superior olive neurons. J Neurosci. 1999;19:2843–2851. doi: 10.1523/JNEUROSCI.19-08-02843.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamatani D, Hishida R, Kudoh M, Shibuki K. Experience-dependent formation of activity propagation patterns at the somatosensory S1 and S2 boundary in rat cortical slices. Neuroimage. 2007;35:47–57. doi: 10.1016/j.neuroimage.2006.08.049. [DOI] [PubMed] [Google Scholar]
- Kameyama K, Sohya K, Ebina T, Fukuda A, Yanagawa Y, Tsumoto T. Difference in binocularity and ocular dominance plasticity between GABAergic and excitatory cortical neurons. J Neurosci. 2010;30:1551–1559. doi: 10.1523/JNEUROSCI.5025-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JN, Greenberg D, Helmchen F. Imaging input and output of neocortical networks in vivo. Proc Natl Acad Sci U S A. 2005;102:14063–14068. doi: 10.1073/pnas.0506029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khirug S, Ahmad F, Puskarjov M, Afzalov R, Kaila K, Blaesse P. A single seizure episode leads to rapid functional activation of KCC2 in the neonatal rat hippocampus. J Neurosci. 2010;30:12028–12035. doi: 10.1523/JNEUROSCI.3154-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Nabekura J. Rapid synaptic remodeling in the adult somatosensory cortex following peripheral nerve injury and its association with neuropathic pain. J Neurosci. 2011;31:5477–5482. doi: 10.1523/JNEUROSCI.0328-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura A, Ishibashi H, Watanabe M, Takatsuru Y, Brodwick M, Nabekura J. Sustained depolarizing shift of the GABA reversal potential by glutamate receptor activation in hippocampal neurons. Neurosci Res. 2008;62:270–277. doi: 10.1016/j.neures.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Kohara K, Kitamura A, Morishima M, Tsumoto T. Activity-dependent transfer of brain-derived neurotrophic factor to postsynaptic neurons. Science. 2001;291:2419–2423. doi: 10.1126/science.1057415. [DOI] [PubMed] [Google Scholar]
- Kullmann DM, Lamsa KP. Long-term synaptic plasticity in hippocampal interneurons. Nat Rev Neurosci. 2007;8:687–699. doi: 10.1038/nrn2207. [DOI] [PubMed] [Google Scholar]
- Lee HH, Deeb TZ, Walker JA, Davies PA, Moss SJ. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat Neurosci. 2011;14:736–743. doi: 10.1038/nn.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Lu JT, Li CY, Zhao JP, Poo MM, Zhang XH. Spike-timing-dependent plasticity of neocortical excitatory synapses on inhibitory interneurons depends on target cell type. J Neurosci. 2007;27:9711–9720. doi: 10.1523/JNEUROSCI.2513-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanty NK, Sah P. Calcium-permeable AMPA receptors mediate long-term potentiation in interneurons in the amygdala. Nature. 1998;394:683–687. doi: 10.1038/29312. [DOI] [PubMed] [Google Scholar]
- Nabekura J, Ueno T, Okabe A, Furuta A, Iwaki T, Shimizu-Okabe C, Fukuda A, Akaike N. Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury. J Neurosci. 2002;22:4412–4417. doi: 10.1523/JNEUROSCI.22-11-04412.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Shibuki K, Hishida R, Murakami H, Kudoh M, Kawaguchi T, Watanabe M, Watanabe S, Kouuchi T, Tanaka R. Dynamic imaging of somatosensory cortical activity in the rat visualized by flavoprotein autofluorescence. J Physiol. 2003;549:919–927. doi: 10.1113/jphysiol.2003.040709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Watanabe M, Moorhouse AJ, Kanematsu T, Horibe S, Matsukawa N, Asai K, Ojika K, Hirata M, Nabekura J. Early changes in KCC2 phosphorylation in response to neuronal stress result in functional downregulation. J Neurosci. 2007;27:1642–1650. doi: 10.1523/JNEUROSCI.3104-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kakizaki T, Sakagami H, Saito K, Ebihara S, Kato M, Hirabayashi M, Saito Y, Furuya N, Yanagawa Y. Fluorescent labeling of both GABAergic and glycinergic neurons in vesicular GABA transporter (VGAT)-Venus transgenic mouse. Neuroscience. 2009;164:1031–1043. doi: 10.1016/j.neuroscience.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Xu X, Callaway EM. Laminar specificity of functional input to distinct types of inhibitory cortical neurons. J Neurosci. 2009;29:70–85. doi: 10.1523/JNEUROSCI.4104-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Stornetta RL, Zhu JJ. Chandelier cells control excessive cortical excitation: characteristics of whisker-evoked synaptic responses of layer 2/3 nonpyramidal and pyramidal neurons. J Neurosci. 2004;24:5101–5108. doi: 10.1523/JNEUROSCI.0544-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]