

Abstract
Correct brain functioning relies on the precise activity of a myriad of synapses assembling neurons in complex networks. In the hippocampus, highly diverse inhibitory circuits differently govern several physiologically relevant network activities. Particularly, perisomatic inhibition provided by specific interneurons was proposed to control emotional states, and could therefore be affected by mood disorders and their therapy. We found that both chronic and acute administration of two major antidepressants, imipramine and fluoxetine, strongly and directly altered GABA-mediated (GABAergic) hippocampal neurotransmission in mice and rats, independently of their effects on amine reuptake systems. These drugs affected GABA release from synapses formed by fast-spiking cells, but not interneurons expressing cannabinoid receptor type 1, resulting in the disruption of γ oscillations. This differential effect, shared by two types of antidepressants, suggests a new mechanism of action of these medications, and a possible role of perisomatic inhibition in depressive disorders.
Introduction
One of the hallmarks of the cerebral cortex, including the hippocampus, is the vast heterogeneity of its locally projecting inhibitory GABAergic interneurons (Freund and Buzsáki, 1996; Klausberger and Somogyi, 2008; Wang, 2010). This rich assortment of inhibitory circuits, together with their ability of performing a highly controlled division of labor during cortical activities, is crucial for elaborate cognitive functions. Remarkably, impairment of specific cortical GABAergic circuits underlies several psychiatric diseases (Benes and Berretta, 2001; Levitt et al., 2004; Lewis et al., 2005; Orekhova et al., 2007).
Synapses impinging upon hippocampal pyramidal neuron cell bodies are almost exclusively GABAergic and originate from two major basket cell subtypes, the parvalbumin (PV)-positive fast-spiking (FS) interneurons and cholecystokinin (CCK)-positive basket cells that express cannabinoid receptor type 1 (CB1R) (Freund and Katona, 2007). PV+ basket cells sustain high-frequency firing, receive strong excitation, release GABA very reliably, and are considered the clockwork of cortical networks, as they synchronize a large population of principal cells (Buzsáki and Draguhn, 2004; Freund and Katona, 2007; Klausberger and Somogyi, 2008). Conversely, basket cells expressing CB1Rs (and CCK) receive less excitation, cannot sustain high-frequency firing, release GABA more asynchronously and unreliably (Hefft and Jonas, 2005; Daw et al., 2009), and are negatively modulated by endocannabinoids (Wilson et al., 2001; Kano et al., 2009). Notably, CCK+ cells are the specific target of subcortical neuromodulators, such as acetylcholine and serotonin (Freund and Katona, 2007; Varga et al., 2009), and this, together with their more “capricious” GABAergic transmission, led to the hypothesis that CCK+ cells exert a fine-tuning of hippocampal activities and might play a key role in the control of mood (Freund and Katona, 2007; Varga et al., 2009).
Mood disorders, and particularly major depressive disorder (MDD), are a cluster of psychiatric diseases proposed to originate from reduced brain levels of monoamines (serotonin, norepinephrine, and dopamine) that influence synaptic transmission, excitability, and gene expression in many areas of the CNS (Schildkraut, 1965; Charney, 1998; Krishnan and Nestler, 2008). The most compelling evidence for the role of reduced levels of monoamines in the etiology of depressive disorders derives from the powerful antidepressant actions of molecules that increase the brain levels of these neurotransmitters (Krishnan and Nestler, 2008; López-Muñoz and Alamo, 2009). Nevertheless, the pathophysiology of MDD and the action of antidepressant medications is still debated (Lacasse and Leo, 2005; Krishnan and Nestler, 2010). Interestingly, accumulating evidence indicates abnormalities of GABAergic neurotransmission in MDD and postpartum depression (Maguire and Mody, 2008), including reduced GABA levels (Sanacora et al., 1999; Sanacora et al., 2004; Croarkin et al., 2011) and diminished density of cortical inhibitory interneurons in MDD patients (Rajkowska et al., 2007; Croarkin et al., 2011), leading to a GABAergic hypothesis of depression (Luscher et al., 2011; Möhler, 2012; Smith and Rudolph, 2012). However, since several subcortical aminergic pathways target specific inhibitory circuits (Freund and Katona, 2007, Varga et al., 2009), it is not clear whether imbalanced GABAergic transmission is the result or the cause of depressive disorders, and whether antidepressant treatment ultimately alters inhibitory neurotransmission in the CNS. Here we describe a novel and direct effect on hippocampal GABAergic transmission of two types of antidepressants widely used in clinics: fluoxetine [a selective serotonin reuptake inhibitor (SSRI), commercialized as Prozac] and imipramine [a tricyclic antidepressant (TCA), commercialized as Trofanil]. Our results show that antidepressants alter GABA release from a specific perisomatic circuit independently of their known actions on amine reuptake systems, indicating a possible novel target of antidepressants and revealing unsuspected clues to the etiology of depression.
Materials and Methods
All experimental procedures were performed in accordance with Italian (Ministero della Salute) and European guidelines.
Animal treatments
Twenty-seven Sprague Dawley rats of either sex, aged postnatal day (P) 20, were divided into three groups of nine animals that received a daily intraperitoneal injection of saline solution (vehicle, control), imipramine (20 mg/kg), or fluoxetine (20 mg/kg). Rats were injected for 3 weeks, after which they were not treated for 1 d to allow recovering from possible acute drug effects. Rats were then killed and acute brain slices were obtained and used for electrophysiological recordings (see below). Drug treatment and subsequent patch-clamp experiments were performed blind. Chronic treatments could not be started earlier than P21, due to heterogeneity of effects at earlier stages (similarly to human subjects). Therefore, 3 weeks of treatments resulted in animals aged >P40.
In vitro slice preparation and electrophysiology
For patch-clamp, whole-cell recordings, P42 Sprague Dawley rats (for chronically injected rats) and P18–P25 rats and mice of either sex (for acute experiments) were deeply anesthetized with isofluorane inhalation and decapitated. Brains were quickly removed and immersed in “cutting” solution (4°C) containing the following (in mm): 234 sucrose, 11 glucose, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 10 MgSO4, and 0.5 CaCl2 (equilibrated with 95% O2 and 5% CO2). Coronal slices (300 μm) were cut with a vibratome (Leica) from a block of brain containing the hippocampus. Slices were then incubated in oxygenated artificial CSF (ACSF) containing the following (in mm): 126 NaCl, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 2 MgSO4, 2 CaCl2, and 10 glucose, pH 7.4, initially at 32°C for 1 h, and subsequently at room temperature, before being transferred to the recording chamber. Recordings were obtained at a temperature of 30–32°C from the CA1 area of the hippocampus. Neurons were visually identified using infrared video microscopy. In all experiments, GABAA receptor-mediated currents were isolated by adding 6,7-dinitroquinoxaline-2,3-dione (DNQX, 10 μm) to the bath perfusion. Imipramine, fluoxetine, NBQX, R-(+)-(2,3-dihydro-5-methyl-3-[(4-morpholinyl)methyl]pyrol[1,2,3-de]-1,4-benzoxazin-6-yl)-(1-naphthalenyl) methanone mesylate (WIN 55212-2; Tocris Cookson) were delivered using a local perfusion system composed of multiple fine tubes ending in a common outlet tube, positioned near (∼250 μm) the recorded neuron. Local perfusion enabled quick (seconds) wash in and wash out of drugs in the slice. Solutions in the reservoirs were oxygenated and the flow with control ACSF was started before patching a cell. Importantly, antidepressants applied in the bath perfusion produced similar results of locally applied drugs. Experiments on network oscillations did not use a local perfusion but a bath perfusion with relatively high flow (4–6 ml/min).
For most whole-cell recordings, patch-clamp electrodes (tip resistance, 2–3 MΩ) were filled with a “high chloride” intracellular solution containing the following (in mm): 60 CsMeSO3, 70 CsCl, 2 MgCl2, 10 HEPES, 5 EGTA, 4 MgATP, pH 7.3, corrected with CsOH (290 mOsm). The estimated reversal potential for GABA-mediated responses (EGABA) was ∼−15 mV based on the Nernst equation, without correction for gluconate-generated liquid junction potential. Under these recording conditions, activation of GABAA receptors resulted in inward currents at a holding potential (Vh) of −70 mV. Threshold IPSCs were induced by adjusting the stimulus intensity to induce a similar proportion of failures and responses (see Fig. 7A,D). Evoked postsynaptic currents were elicited with a bipolar theta-glass pipette filled with ACSF and positioned 100–200 μm from the soma of each recorded neuron. For input–output experiments (see Fig. 7), threshold responses were identified as failing with a rate of ∼50% with fixed stimulus duration of 60 μs. Stimulus duration but not amplitude was then increased to build input–out curves. IPSCs were evoked every 3 s.
Figure 7.
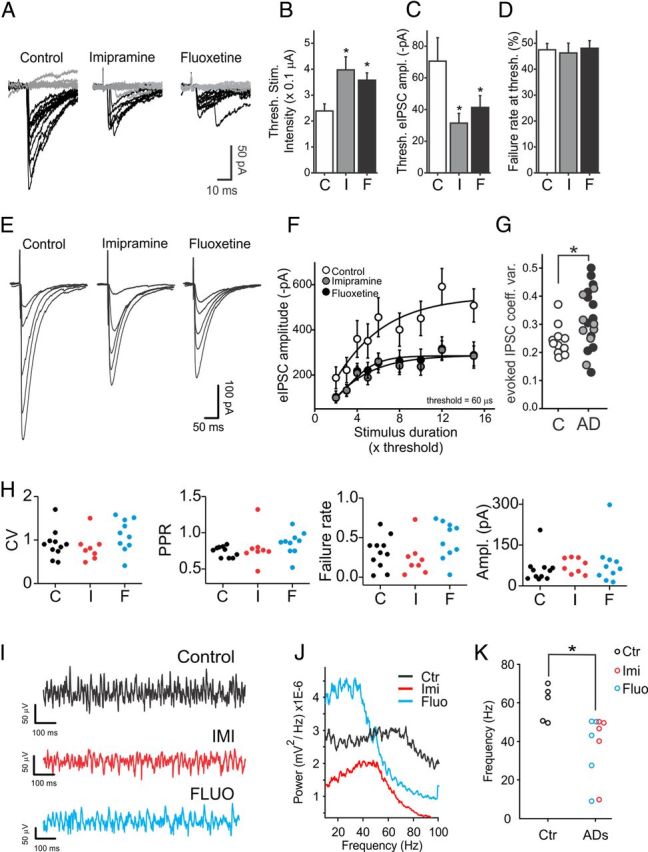
Effects of chronic, in vivo treatments with antidepressants on GABAergic neurotransmission and γ oscillations in the CA1 area of the hippocampus. A, Single-trial threshold IPSCs recorded from CA1 pyramidal neurons of rats treated for 3 weeks with vehicle (control, left), imipramine (middle), and fluoxetine (right). Failures are indicated by gray traces. B, Average stimulus current amplitudes (fixed duration of 60 μs) inducing threshold responses in slices from animals treated with vehicle (control, C; white column), imipramine (I, gray column), and fluoxetine (F, black column). *p < 0.05. C, Average threshold IPSC amplitudes in all three conditions. Same symbols and colors as in B. D, Average percentage of failures at threshold stimulations of B in all three conditions. Same symbols and colors as in B and C. *p < 0.05. E, Input–output traces of monosynaptic IPSCs recorded from the same animal populations of A–D. Each trace (average of 10–15 trials) was evoked by increasing the duration of threshold stimulus [3, 5, 8, 10, and 15 times threshold (× Thrs)], without changing the stimulus amplitude. F, Average input–output responses in slices from animals treated with vehicle (white circles, control), imipramine (gray circles), and fluoxetine (black circles). Data were fitted using the Boltzmann function. G, Plots of coefficient of variation of evoked IPSCs in control (C, white circles) and antidepressants [imipramine (I), gray circles; fluoxetine (F), black circles]. H, Plots of coefficient of variation (CV), pair-pulse ratio (PPR), failure rates, and amplitudes of FS-cell-induced uIPSCs on pyramidal neurons from rats chronically treated with saline (control, C; black dots), imipramine (I; red dots), and fluoxetine (F; blue dots). Each dot refers to an individual unitary connection. I, Extracellular LFP recordings from CA1 stratum radiatum of hippocampal slices obtained from saline-treated (control, black), imipramine-treated (red), and fluoxetine-treated (blue) rats. Traces were filtered in the γ-frequency band (30–60 Hz). Oscillations were induced by application of 600–800 nm KA. J, Power spectral densities of the same experiment of G. K, Population graphs illustrating the maximal frequency in the three animal groups. Each dot represents an individual experiment. *p < 0.05.
Experiments testing the effects of antidepressants on glutamatergic neurotransmission were performed in the absence of DNQX, but in the presence of the GABAA receptor antagonist gabazine (10 μm, Tocris Cookson). In our experimental conditions, gabazine application never induced epileptiform activity. The intracellular solution experiments involving polysynaptic IPSCs (see Fig. 3) contained the following (in mm): 125 CsMeSO3, 10 HEPES, 5 EGTA, 2 MgCl2, 5 QX-314, and 4 MgATP.
Figure 3.
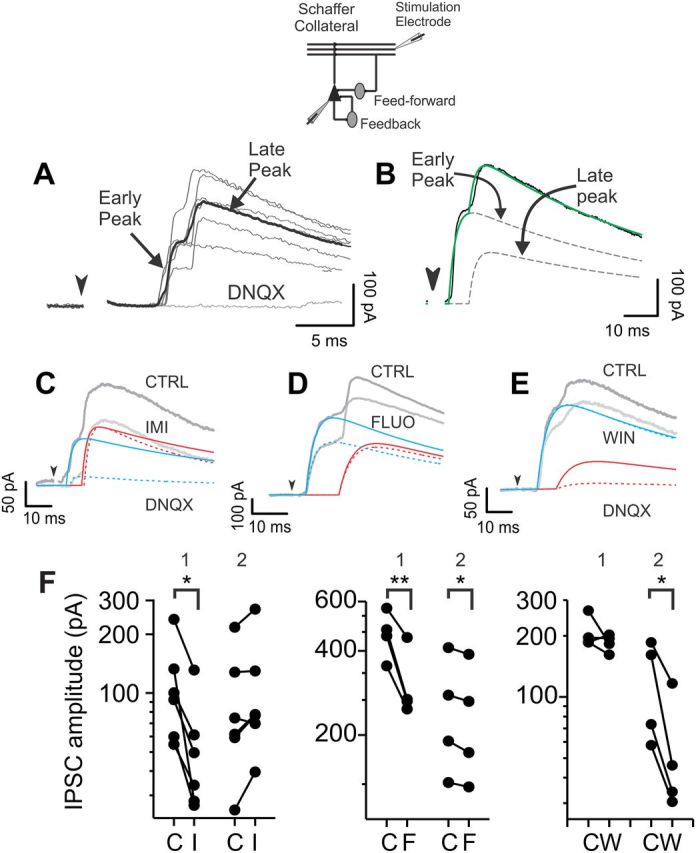
Imipramine and fluoxetine decrease GABA neurotransmission from a specific hippocampal inhibitory circuit. A, Polysynaptic IPSC responses to Schaffer collateral stimulations (inset). Neurons were held at the reversal potential of glutamate-mediated responses; hence the lack of inward excitatory currents. Polysynaptic IPSCs relied on glutamatergic transmission as they were blocked by the glutamate receptor antagonist DNQX (10 μm). Gray traces, overlapped single-sweep responses. Black trace, six-sweep average trace. B, A polysynaptic IPSC (black line) was fitted using the sum (green line) of two double-exponential functions (dashed lines) for early and late peaks. C–E, Imipramine (IMI, 20 μm; C, light gray trace) and fluoxetine (FLUO, 20 μm; D, light gray trace) diminished the early component of the composite IPSCs (blue lines), whereas the synthetic cannabinoid WIN 55212-2 (WIN, 1 μm; E, gray trace) reduced the late components (red lines) of the polysynaptic inhibitory response. Solid and dashed blue and red lines refer to the fitted exponentials before (solid) and after (dashed) drug application. Arrowheads in A–E indicate the extracellular stimulus, whose artifacts were digitally removed. F, Population data showing early (1) and late (2) IPSC peaks for individual neurons in control (C), imipramine (I), fluoxetine (F), and WIN (W). Data are plotted in logarithmic scale. *p < 0.05; **p < 0.01.
For paired recordings between FS or RS (CB1R+) interneurons and postsynaptic pyramidal cells, we used previously characterized GFP-expressing mouse lines to help select specific presynaptic interneuron types [GAD67-GFP and GAD65-GFP (Galarreta et al., 2008)]. Interneurons were classified by their firing behavior in response to hyperpolarizing and depolarizing current steps. Presynaptic CB1R expression was confirmed pharmacologically (WIN 55212-2 application; see Fig. 4C,D) and/or physiologically through depolarization-induced suppression of inhibition (DSI) induced with 5 s voltage steps to 0 mV (data not shown). Presynaptic interneurons were recorded using an intracellular solution containing the following (in mm): 70 K-gluconate, 70 KCl, 2 NaCl, 2 MgCl2, 10 HEPES, 1 EGTA, 2 MgATP, and 0.3 Na2GTP, pH 7.3, corrected with KOH (290 mOsm), and 5 mg/ml biocytin to perform post hoc anatomical reconstructions of presynaptic GABAergic cells (see below). Unitary inhibitory responses were elicited by short trains of action potentials (5 spikes at 50 Hz, evoked both in current clamp and voltage clamp) in presynaptic cells. Unitary synaptic responses were characterized by fixed latency, high peak-amplitude fluctuation, and short-term plasticity. Typically, series resistance ranged from 10 to 25 MΩ. Series resistance was monitored throughout all experiments by a voltage pulse delivered before synaptic stimulations. Recordings were discarded if a variation of >10% occurred during a particular experiment.
Figure 4.

Antidepressants reduce unitary synaptic responses from FS but not from CB1R+ basket cells. A, B, Top, Current-clamp recordings showing the firing characteristics of two interneuron types in response to current injections. Note the FS (blue traces) and RS (red traces) behaviors. Bottom, Anatomical reconstructions of the same interneurons. Blue and Red Tracings, Axonal projections of the FS and RS cell, respectively. Black Tracings, Somatodendritic compartments of both cells. Note that the axon is mainly confined to the stratum pyramidale (s.p.) in both cell types, identifying them as basket cells. Scale bar: 200 μm. C, D, Simultaneous paired recordings from presynaptic FS (C, blue trace) and RS (D, red trace) interneurons (IN) and postsynaptic pyramidal cells (PYR). CB1R agonist WIN 55212-2 (WIN, 1 μm; gray traces) selectively affected uIPSCs from RS (red) but not FS interneurons. E, F, Imipramine (E, IMI) and fluoxetine (F, FLUO) applications (both drugs, 20 μm) depressed synaptic transmission from FS (blue) but not from WIN-sensitive, RS basket cells (red). G, Average, normalized uIPSC amplitudes from FS (blue, n = 7 and 8; imipramine and fluoxetine, respectively) and RS (red, n = 6, both drugs) basket cells. H, I, Failure rates (H) and pair-pulse ratios of uIPSCs from FS (blue) and RS CB1+ (red) basket cells before and after imipramine (left) and fluoxetine (right) application. C, control; I, imipramine; F, fluoxetine; W, washout. *p < 0.05; **p < 0.01. J, Representative voltage-clamp traces of pharmacologically isolated sIPSCs from a CA1 pyramidal neuron in control (left), in the presence of fluoxetine (FLUO, middle), and after concomitant application of WIN (in the presence of FLUO, right). K, Population graph illustrating sIPSC frequency and amplitude after fluoxetine (F) and fluoxetine plus WIN (F+W) application. **p < 0.01; ns, not significant; paired t test.
Signals were amplified using a Multiclamp 700B patch-clamp amplifier (Molecular Devices), sampled at 20 kHz, filtered at 10 kHz, and stored on a PC. Data were analyzed using pClamp (Molecular Devices) and Origin (Microcal Software) software.
For γ-oscillation experiments, 400-μm-thick horizontal slices containing the ventral part of the hippocampus were obtained from P18–P25 rats as described above. Slices were then incubated in an interface chamber at 35°C for the duration of the experiment in oxygenated ACSF supplemented with glutamine (400 μm). Slices were then transferred in a submerged recording chamber, kept at 34°C, and perfused at 4–6 ml/min. γ Oscillations were evoked by bath application of low concentrations of kainic acid (KA, 200–400 nm). Two field recording electrodes were placed in stratum radiatum of the CA1 and CA3 areas of the hippocampus. KA applications robustly induced rhythmic activities in both areas. Data were collected with a Multiclamp 700B amplifier (Molecular Devices). Antidepressants were freshly added to the bath perfusate.
Data analysis
sIPSCs.
Custom written software (Detector, courtesy J. R. Huguenard, Stanford University) was used for analyzing spontaneous and miniature GABAergic events, as previously described (Manseau et al., 2010). Briefly, individual events were detected with a threshold-triggered process from a differentiated copy of the real trace. For each cell, the detection criteria (threshold and duration of trigger for detection) were adjusted to ignore slow membrane fluctuations and electric noise while allowing maximal discrimination of sIPSCs. Detection frames were regularly inspected visually to ensure that the detector was working properly.
Polysynaptic IPSCs.
To extract and quantify the early and late synaptic components from the composite, polysynaptic responses of Figure 3, single traces were modeled as the sum of two double-exponential functions, Σ = S1 + S2, and fitted using nonlinear regression with custom written routines and the MATLAB Statistics Toolbox (MathWorks):
where δ1,2 are the delays of the responses from onset of stimulus artifact, A1,2 the amplitudes, τr(1,2) the rise time constants of first and second peak, respectively, and τd is the decay time constant of both peaks.
The amplitudes A1,2 and the decay time constant τd were obtained through nonlinear regression fitting (see Fig. 3, Table 1).
Table 1.
Early and late synaptic components extracted and quantified from the composite, polysynaptic responses of Figure 3 single traces
| Parameter | Fluoxetine (mean ± SD) | Imipramine (mean ± SD) | WIN 55212-2 (mean ± SD) |
|---|---|---|---|
| δ1 | 4.7 ± 0.8 ms | 6.1 ± 0.8 ms | 5.8 ± 1.2 ms |
| δ2 | 11.4 ± 3.3 ms | 10.4 ± 1.6 ms | 12.7 ± 2.8 ms |
| τr1 | 1.9 ± 0.6 ms | 1.8 ± 1.3 ms | 2.3 ± 1.3 ms |
| τr2 | 2.3 ± 1.0 ms | 3.3 ± 1.6 ms | 5.0 ± 1.2 ms |
To avoid overfitting and to optimize the procedure, we set the remaining response delays δ1,2 and rise time constants τr(1,2) as fixed, based on average trace analysis (see Table 1).
All parameters were obtained in a trial-to-trial basis, and averaged a posteriori.
γ-Oscillation analysis.
Local field potential (LFP) traces were downsampled offline at 500 Hz using eighth-order low-pass Chebyshev type-I filter and subsequently low-pass filtered using an infinite-duration impulse response (IIR) Butterworth eighth-order filter with cutoff frequency set at 200 Hz. Spectrograms were obtained offline with multitaper spectral estimation methods from 3.0 s windows with 1.5 s overlap, using the chronux toolbox (http://chronux.org/) in Matlab. For power quantification, the average power was calculated in a 5 min window immediately before antidepressant administration (control) and when a clear antidepressant effect was achieved. Power peak values in control were extracted after interpolating the power spectra by a factor of 10 to minimize noise due to sampling. Power spectra relative to antidepressants were smoothed with a smoothing window (±5 samples) around each data point to minimize noise artifacts due to lack of a prominent power peak. The value of spectral power at oscillation frequency in control was used in experiments in which oscillation power was completely abolished by antidepressants. Due to high variability of spectral powers among different experiments, statistical tests (paired Wilcoxon signed-rank test) were performed on logarithmic values.
Multiunit spike synchrony analysis.
Multiunit spike activity (MUA) was extracted by threshold detection on a subset of extracellular recordings that contained spiking activity. Data were filtered with an IIR bandpass Butterworth tenth-order filter (cutoff frequencies, 0.5 and 5 kHz). Threshold was adjusted in every dataset to detect all spikes in each separate recording. As a control, spike waveforms and the interspike interval histograms were analyzed for consistency. Phase signals were extracted by wavelet decomposition of the LFPs into frequencies between 5 and 200 Hz. The probability of spiking with respect to specific phases of γ oscillations (at a given frequency) was computed as spike-oscillation phase histogram (PH). In most cases, oscillations were completely abolished in the presence of antidepressants. Therefore, wavelet phase analysis was performed in 5 min time segments, in which the power of the oscillations was reduced by antidepressants but not completely abolished (this time ranged from 10 to 80 min after antidepressant application). PH analysis was performed only in those cases in which a clear MUA-LFP synchronization was observed in control (fluoxetine, n = 5; imipramine, n = 2). PHs were fitted with normal circular (von Mises) distributions to extract the analytical PH peak used to quantify synchronization. Analyses were performed with custom-written scripts. The wavelet software was provided by C. Torrence and G. Compo (available at http://atoc.colorado.edu/research/wavelets/). All scripts can be provided upon request.
Results are presented as mean ± SEM. Unless otherwise noted, data were statistically compared using the Student's t test, and differences were considered significant if p < 0.05.
Histology
Biocytin (5 mg/ml, Sigma-Aldrich) was included in the internal solution to fill neurons during electrophysiological recordings. Slices were subsequently fixed overnight in 4% paraformaldehyde in phosphate buffer, pH 7.4, at 4°C, and were not resectioned to prevent loss of sample and thus allow for a more complete reconstruction. Fixed slices were rinsed in phosphate buffer solution (PBS) for 10 min and quenched in 1% H2O2 (in 10% MetOH) for 5 min. After rinsing 2× in PBS, slices were permeabilized in 2% Triton X-100 in PBS for 1 h at room temperature, and incubated in ABC reagent (Avidin and biotinylated horseradish peroxidase [HRP] complex; Vectastain, Vector Laboratories) for 2 h at room temperature. Slices were rinsed 4× in PBS (2 × 10 min; 1 × 15 min; 1 × 1 h) and reacted with 3,3′-diaminobenzidine (Vector Laboratories). After two rinses in PBS (10 min), slices were whole-mounted and coverslipped with 85% glycerol. Neurons were reconstructed using the Neurolucida software (MicroBrightField) using a 100× objective.
Results
Acute applications of antidepressants decrease evoked inhibitory transmission directly at GABAergic synapses independently of monoaminergic reuptake
To test the effects of antidepressants on hippocampal GABAergic neurotransmission, we applied two different antidepressants, imipramine or fluoxetine, to acute brain slices from juvenile rat hippocampus, a brain area critically involved in the etiology of depression (Krishnan and Nestler, 2008). Imipramine belongs to the family of TCAs that strongly affect monoamine levels, while fluoxetine was more recently developed and is a SSRI designed to increase extracellular levels of serotonin by specifically blocking serotonin transporters (López-Muñoz and Alamo, 2009). Monosynaptic IPSCs on CA1 pyramidal neurons were evoked by extracellular afferent stimulation in the stratum pyramidale and isolated in the continuous presence of the ionotropic glutamate receptor antagonist DNQX (10 μm). This stimulation configuration favors the activation of perisomatic inhibition, although we cannot exclude excitation of GABAergic afferents of dendrite-targeting cells. When antidepressants were acutely applied at a concentration known to efficiently block monoamine reuptake (1 μm) (Radnikow and Misgeld, 1998; Kobayashi et al., 2008), no effect was detected on evoked IPSCs (relative change of IPSC amplitude: imipramine, 1.02 ± 0.13, n = 6 cells; fluoxetine, 0.98 ± 0.02, n = 6; p > 0.05; Fig. 1A,B, open circles).
Figure 1.
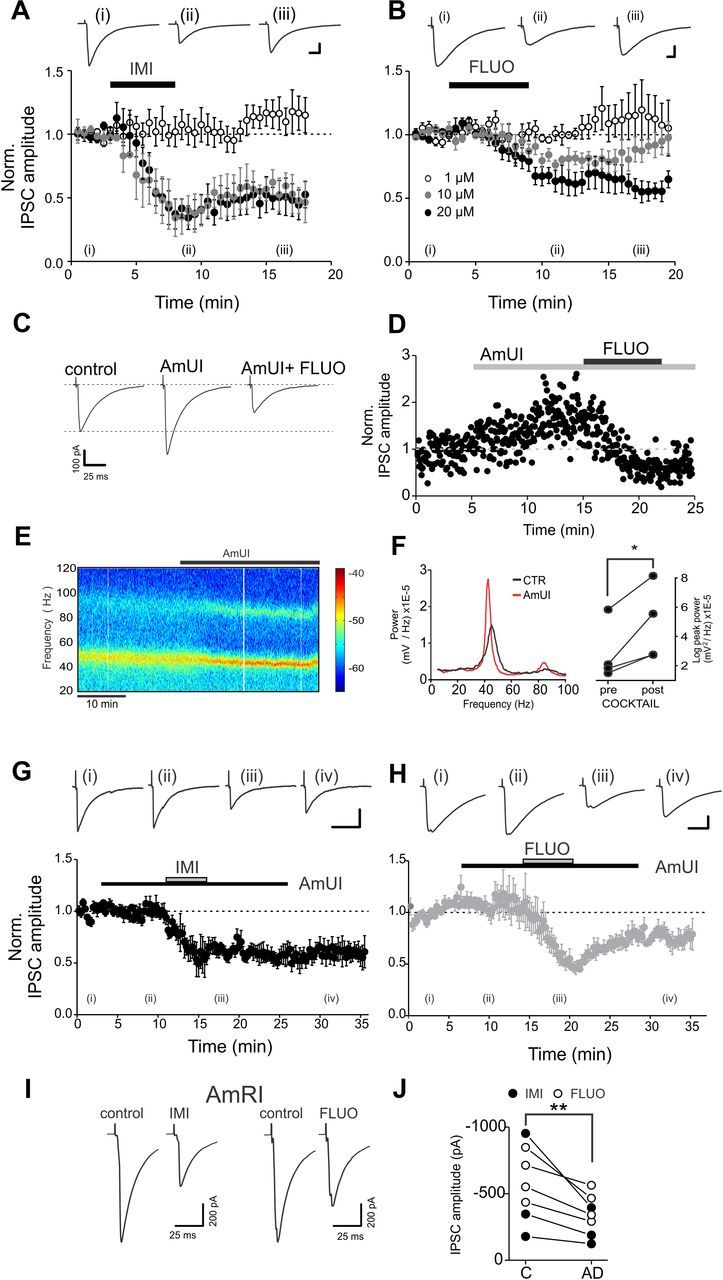
Acute, in vitro antidepressant applications directly reduce evoked IPSCs without the involvement of monoamine reuptake systems. A, B, Average time courses of imipramine (A) and fluoxetine (B) effects on normalized IPSCs following acute applications to rat hippocampal slices. Antidepressants were used at 1, 10, and 20 μm (white, gray, and black circles, respectively). Roman numerals (i–iv) indicate the time points used for example traces. Black bars indicate the time of antidepressant application. Scale bars: A, 200 pA, 10 ms; B, 50 pA, 10 ms. C, Representative evoked IPSC traces of a single experiment with a mixture of amine reuptake blockers. AmUI, amine uptake inhibitors (see text for details). Acute application of the AmUI mixture produced an increase of IPSC amplitudes in 4 of 12 neurons. Subsequent fluoxetine application (20 μm) strongly and invariably reduced IPSCs. D, Time course of the monoamine reuptake antagonist mixture (AmUI, gray bar) and fluoxetine application (black bar) for the experiment of the cell of C. E, Representative spectrogram illustrating oscillations in the γ-frequency range in a hippocampal slice bathed in 400 nm KA. Application of the AmUI mixture increased the LFP power. F, Left, Power spectral densities of the experiment shown in E, illustrating the increased peak power in the presence of the mixture (red trace). Right, Population graphs of the LFP power (logarithmic scale) before and after the application of the mixture. In all cases, the LFP power increased in the presence of the mixture. *p < 0.05. G, H, Same as in A and B, but imipramine or fluoxetine were applied (20 μm; gray bars) in the presence of a mixture of AmUIs (black bars). See text for details. Scale bars: G, 50 pA, 40 ms; H, 200 pA, 20 ms. I, Representative traces of evoked IPSCs obtained from CA1 pyramidal neurons of hippocampal slices preincubated and in the continuous presence of a mixture of amine receptor inhibitors (AmRIs, see text for details). Imipramine (IMI) and fluoxetine (FLUO) applications (both at 20 μm) resulted in marked decrease of evoked IPSC amplitudes. J, Population data of the experiment of I. **p < 0.01. Time course plots in A, B, G, and H refer to all tested cells.
In both rodents and humans, chronic administration of antidepressants results in brain accumulations of these drugs reaching concentrations of several tens of micromoles (Sugita et al., 1989; Henry et al., 2005). Interestingly, when either drug was applied at 10 or 20 μm, monosynaptic, perisomatic IPSC amplitudes dramatically decreased (relative change: 10 μm imipramine, 0.49 ± 0.11, n = 3, p < 0.05; 20 μm imipramine, 0.49 ± 0.12, n = 5; p < 0.05; 10 μm fluoxetine, 0.80 ± 0.05, n = 5, p < 0.05; 20 μm fluoxetine, 0.66 ± 0.07, n = 5, p < 0.01; Fig. 1A,B). As mentioned above, both fluoxetine and imipramine are known blockers of serotonin transporters, and imipramine also affects the uptake of norepinephrine and dopamine (López-Muñoz and Alamo, 2009). Therefore, this antidepressant drug-mediated effect on GABAergic transmission could be due to an indirect modulation of GABAergic interneurons by one or more of these monoamines (Seamans et al., 2001; Fink and Göthert, 2007; Puig et al., 2010), or to a direct action of antidepressants at GABAergic synapses. To test this latter possibility, we pharmacologically occluded aminergic transporters by preincubating the slices with a mixture of amine uptake inhibitors (AmUI; 1 μm GBR-12909 for blocking dopamine uptake; 1 μm citalopram for blocking serotonin uptake; and 1 μm nisoxetine for blocking norepinephrine uptake). Similarly to imipramine and fluoxetine at 1 μm, the mixture did not change IPSC amplitudes significantly (p > 0.05), suggesting that aminergic activity is low in quiescent acute brain slices, even though amine reuptake inhibitors did increase IPSC amplitudes in some neurons (4 of 12; Fig. 1C,D). Importantly, when network activity was increased by low concentrations of KA (200–400 nm), blockade of amine reuptake invariably resulted in a robust increase of γ-oscillation power (n = 6; p < 0.05; Fig. 1E,F). These results indicate that, in our conditions, the mixture of amine reuptake inhibitors was effective in occluding amine reuptake. We then tested whether the above-mentioned reduction of IPSCs by antidepressants was present when amine reuptake was occluded. Fluoxetine or imipramine application (20 μm, in the constant presence of the mixture) produced a strong reduction of IPSC amplitudes, similar to that observed without amine uptake blockers (relative change: imipramine, 0.55 ± 0.11, n = 7 cells; fluoxetine, 0.54 ± 0.06, n = 5 cells; p < 0.01 in both cases; Fig. 1G,H).
In a separate set of experiments, aminergic activity was occluded by blocking the majority of dopamine, serotonin, and noradrenaline receptors with another mixture of amine receptor inhibitors (AmRI), including (in μm) the following: 0.2 asenapine, 1 SCH23390, 5 prazosin, 1 ICI118,551, 1 LY310762, 1 tropisetron. After preincubation of slices (30 min) and in the continuous presence of the amine receptor inhibitors mixture, application of fluoxetine and imipramine strongly and consistently reduced evoked IPSC amplitudes (Fig. 1I). These results indicate that antidepressants (at 10 and 20 μm) acted at GABAergic synapses directly and independently of monoamine reuptake system.
Imipramine and fluoxetine alter presynaptic release of GABA onto CA1 pyramidal neurons
Reduced amplitude of inhibitory synaptic responses might be the result of antidepressant-induced changes of presynaptic release of GABA, postsynaptic sensitivity, or both. We therefore analyzed peak amplitude fluctuation of evoked IPSCs before and after drug applications and examined changes in the frequency and amplitude of spontaneous IPSCs (sIPSCs) and miniature IPSCs (mIPSCs). Both imipramine and fluoxetine applications increased the coefficient of variation of IPSC amplitudes (relative change: 20 μm imipramine, 1.94 ± 0.15, n = 7 cells; 20 μm fluoxetine, 1.72 ± 0.08; n = 5 cells; p < 0.01; Fig. 2A–D). Surprisingly, both sIPSC and mIPSC frequencies were strongly increased by either imipramine or fluoxetine (mIPSCs were recorded in the presence of 1 μm tetrodotoxin; relative change of mIPSC frequency: 20 μm imipramine, 2.67 ± 0.33; 20 μm fluoxetine, 4.95 ± 0.90; n = 6 cells, p < 0.001; Fig. 2E–G), whereas sIPSC and mIPSC amplitudes were unaffected by both drugs. Importantly, the antidepressant-dependent sIPSC frequency increase was not statistically different in the presence of either the aminergic uptake blocker mixture (Fig. 2H,I) or aminergic receptor blocker mixture (Fig. 2J), indicating that, similar to the reduction of evoked IPSCs, the antidepressant-mediated strong increase of spontaneous vesicle release was not due to an indirect action through the monoamine uptake system. Altogether these results suggest that imipramine and fluoxetine affect presynaptic neurotransmitter release directly at GABAergic synapses onto CA1 pyramidal neurons.
Figure 2.

Imipramine and fluoxetine alter presynaptic release of GABA onto CA1 pyramidal neurons. A, Representative IPSCs recorded from CA1 pyramidal neurons and evoked by extracellular stimulations in stratum pyramidale, before (left) and after (right) application of imipramine (20 μm). Gray traces are overlapped single-sweep responses; black traces are average responses. Note the increase in peak amplitude fluctuations of single responses in the presence of imipramine. This is associated with a reduction of the mean response. B, Population graph of IPSC coefficient of variation (CV) before, during, and after imipramine (I) application (20 μm). **p < 0.01, paired t test. C, D, Same experiment as in A and B in another pyramidal neuron before and after fluoxetine (F) application (20 μm). Scale bars: A, C, 25 ms, 50 pA. E, F, Continuous voltage-clamp recordings of mIPSCs in the presence of the Na-channel blocker tetrodotoxin (0.5 μm) and DNQX (10 μm) before (top), during (middle), and after (bottom) application of imipramine (E) or fluoxetine (F). Both drugs were applied at 20 μm. G, Population data of mIPSC frequency (left) and amplitude (right) before and after imipramine (I, top) and fluoxetine (F, bottom) applications. Note the dramatic increase of mIPSC frequency, but not amplitude, induced by either antidepressant drug. *p < 0.05; **p < 0.01; ns, not significant; paired t test. H, I, Population data of sIPSC frequency and amplitude in the presence of the mixture of amine uptake inhibitors (AmUI) and imipramine (H, AmUI + IMI) or fluoxetine (I, AmUI + FLUO). The mixture did not change sIPSC frequency and amplitude, but subsequent application of antidepressants robustly increased sIPSC frequency selectively. C, control (no mixture); **p < 0.01. J, Same as in H and I but for experiments performed in slices preincubated and in the constant presence of the mixture of amine receptor inhibitors (AmRI) before (C) and after (AD) application of antidepressant. Both imipramine (IMI, closed circles) and fluoxetine (FLUO, open circles) increased sIPSC frequency, leaving sIPSC amplitude unaffected in the constant presence of amine receptor blockers. **p < 0.01; ns, nonsignificant; paired t test.
Imipramine and fluoxetine decrease GABA neurotransmission from a specific hippocampal inhibitory circuit
As mentioned above, hippocampal perisomatic inhibition originates from at least two different basket-cell types: PV+ FS and CB1+ RS interneurons (Glickfeld and Scanziani, 2006; Freund and Katona, 2007). It was previously shown that these two cell types are differently recruited in time by hippocampal excitatory circuits: FS cells are very efficiently excited by CA3 afferents and were proposed to contribute to early (likely feedforward) inhibition. Conversely, CB1+ basket cells tend to integrate CA3 and CA1 excitation, providing late feedback inhibition (Glickfeld and Scanziani, 2006; Glickfeld et al., 2008). This specific temporal separation of polysynaptic IPSCs resulting from FS and CB1+ basket cells makes it possible to test cell-type-specific sensitivity to neuromodulators (Glickfeld et al., 2008). To test whether antidepressant-mediated reduction of IPSCs resulted from a specific effect on a distinct inhibitory circuit, we stimulated Schaffer collateral afferents leaving glutamatergic excitation intact and isolating IPSCs by voltage-clamping postsynaptic CA1 pyramidal neurons at the reversal potential for glutamate-mediated responses. By appropriately adjusting the stimulus strength we could reliably evoke IPSCs that contained early and late polysynaptic GABAergic components (Fig. 3A). The two synaptic components can be separated by fitting single traces with multiexponential functions (Fig. 3B; see Materials and Methods), allowing quantifying drug effects on either component of the polysynaptic IPSC. Fluoxetine and imipramine strongly reduced the early component of the composite IPSC (relative inhibition by 20 μm imipramine on the early IPSC component: 0.50 ± 0.06, n = 6, p < 0.05; relative inhibition by 20 μm fluoxetine: 0.66 ± 0.05; n = 4, p < 0.01; Fig. 3C,D,F), leaving the late portion either intact (imipramine: 1.22 ± 0.09, n = 6, p > 0.05; Fig. 3C,F), or significantly less affected (fluoxetine: 0.94 ± 0.01, n = 4, p < 0.05; Fig. 3D,F; p < 0.05 when imipramine-mediated and fluoxetine-mediated effects were compared with an independent t test). As previously shown (Glickfeld et al., 2008), application of CB1 agonist WIN 55212-2 (1 μm) selectively reduced the late component of the polysynaptic IPSC (p < 0.05; Fig. 3E,F), without affecting its early part. These results suggest that antidepressants selectively alter GABA release from the specific hippocampal circuit suggested to provide feedforward inhibition (Glickfeld and Scanziani, 2006).
Antidepressants reduce unitary synaptic responses from FS but not from CB1R+ basket cells
To test directly whether GABAergic synapses from FS and CB1+ basket cells were differently affected by antidepressants, we performed simultaneous paired recordings from presynaptic interneurons and postsynaptic pyramidal cells. For these experiments we used mice expressing green fluorescent protein (GFP) in specific interneuron classes to help identify presynaptic cells [GAD67-GFP mice for FS interneurons and GAD65-GFP mice for CB1+ interneurons (Galarreta et al., 2008)]. Importantly, similarly to rats, mice showed a strong reduction of extracellularly evoked IPSCs by acute antidepressant applications (data not shown). GAD67-GFP-positive interneurons always showed abrupt high-frequency firing in response to current injections (typical of FS interneurons; Fig. 4A), and produced unitary synaptic transmission that was reliable, characterized by short-term depression, and unaffected by WIN 55212-2 (Fig. 4C) or postsynaptic depolarizations that induce retrograde endocannabinoid signaling (depolarization-induced suppression of inhibition or DSI (Wilson et al., 2001; Glickfeld and Scanziani, 2006; Kano et al., 2009; data not shown). Conversely, GAD65-GFP-positive neurons included a more heterogeneous cell population. We therefore selected presynaptic interneurons with a RS firing pattern (Fig. 4B), and expressing CB1Rs, as indicated by WIN 55212-2-mediated ablation of unitary IPSCs (uIPSCs) (Fig. 4D), or presence of DSI (data not shown). Presynaptic interneurons were filled with biocytin during the recordings and their basket-cell morphology and axonal plexus were confirmed by post hoc anatomical reconstructions in almost all cases (Fig. 4A,B; data not shown). Interestingly, both imipramine and fluoxetine applications strongly reduced uIPSCs from FS (relative change induced by 20 μm imipramine: 0.63 ± 0.08, n = 7; 20 μm fluoxetine: 0.49 ± 0.09, n = 8; p < 0.01; Fig. 4E–G) but not CB1+ basket cells (20 μm fluoxetine: 1.04 ± 0.12, n = 6; 20 μm imipramine: 0.82 ± 0.08; n = 6; p > 0.05; Fig. 4E–G). This specific effect on uIPSCs from FS interneurons was associated with typical presynaptic alterations of synaptic transmission: increase of failure rate by fluoxetine (3.70 ± 1.17-fold increase, n = 8; p < 0.05; Fig. 4H), and paired-pulse ratio (PPR) by both antidepressants (imipramine: 1.43 ± 0.07-fold increase, n = 7; fluoxetine: 1.25 ± 0.10-fold increase, n = 8; p < 0.05; Fig. 4I). Failure rates and PPR were unaffected by antidepressant applications in uIPSCs from CB1+ interneurons (p > 0.05, n = 6; Fig. 4H,I). Antidepressant application resulted in massive increase of sIPSC frequency (Figs. 2E–J, 4J). Importantly, application of WIN 55212-2 in the presence of antidepressants failed to reduce sIPSC frequency, suggesting that increased quantal release occurred at presynaptic terminals of FS interneurons (Fig. 4J,K; n = 3, p > 0.05, paired t test).
Altogether, these results confirm that imipramine and fluoxetine strongly and selectively alter GABAergic neurotransmission selectively from FS interneurons.
Imipramine and fluoxetine disrupt hippocampal γ oscillations
FS basket cells play a major role in synchronizing large populations of cortical neurons during several forms of network oscillations that underlie several complex cognitive functions, including sensory integration, attention, exploratory behavior, sleep, and several forms of memory (Buzsáki and Draguhn, 2004; Wang, 2010). One of the most prominent FS-cell-induced cortical rhythms is represented by γ oscillations (30–80 Hz), which occur during wakefulness and attentive behavior, and have been hypothesized to bind various aspects of sensory information between different cortical areas (Buzsáki and Draguhn, 2004; Wang, 2010). We therefore examined whether the selective reduction of GABAergic neurotransmission from FS basket cells by antidepressants would affect γ oscillations. Oscillations of the LFP in the γ frequency band could be reliably evoked by bath application of low concentrations of KA (200–400 nm; Fig. 5), as previously described (Hájos et al., 2004; Atallah and Scanziani, 2009). Both imipramine and fluoxetine strongly reduced or completely abolished γ-oscillation power (peak power, normalized to control: 20 μm imipramine, 0.15 ± 0.05, n = 6; 20 μm fluoxetine, 0.20 ± 0.07, n = 7; p < 0.01 in both cases; Fig. 5A,H). Importantly, this effect was present also when slices were preincubated with the mixture of inhibitors of amine transporters, indicating that suppression of γ power was not due to the known effects of antidepressants on monoaminergic system (Fig. 5D, filled circles). The disappearance of γ oscillations was not due to nonspecific rundown, as the γ power was stable for several hours when drugs were not applied (data not shown). In addition, antidepressants reduced the phase-locking of multiunit spike activity during oscillations (relative peak of spike-oscillation PH: 20 μm imipramine, 0.70 ± 0.13, n = 2; 20 μm fluoxetine, 0.67 ± 0.03, n = 5; p < 0.05 on pooled imipramine and fluoxetine data), suggesting that reduction and/or desynchronization of GABA release from FS interneurons by antidepressants resulted in global uncorrelated activity at the network level (Fig. 5I–K). These results indicate that the strong alteration of FS cell-mediated alteration of perisomatic inhibition by both imipramine and fluoxetine generate an acute disruption of γ oscillations.
Figure 5.

Imipramine and fluoxetine disrupt hippocampal γ oscillations. A, Extracellular LFP recordings from stratum radiatum in CA1 showing the effect of imipramine (IMI) on CA1 γ oscillations, induced by 400 nm KA. Traces were filtered in the γ-frequency band (30–60 Hz). B, Spectrograms of experiment in A. Imipramine application is indicated by the black bar. The color scale indicates the power at each time-frequency pair. C, Power spectral densities of the same experiment of A and B in control (black) and IMI (green). D, Population graphs of the LFP power (logarithmic scale) before and after the application of IMI (20 μm). E–H, Same as in A–D, but with fluoxetine (20 μm, FLUO). White and black circles in D and H indicate experiments done in the absence or presence of the aminergic transporter inhibitor mixture, respectively. **p < 0.01. I, LFP γ oscillations (low-pass filtered at 200 Hz; gray trace), in control (CTR, top) and fluoxetine (FLUO, bottom), with superimposed wavelet functions (black traces, 42 Hz; green traces, 52 Hz) used to extract oscillation phases. Times of multiunit spikes (high-pass filtered at 0.5 kHz) are indicated as vertical lines below the LFP traces. Arrowheads point to spikes in phase with oscillations as indicated in J. J, Spike-oscillation PHs indicating the spike probability with respect to oscillation phase (π, oscillation trough) in control (black circles) and in fluoxetine (white circles). A representative oscillation cycle is indicated on top. PHs were fitted with circular normal distributions (control, black lines; fluoxetine, green lines). PHs were calculated in time instances in which the effect of antidepressant was clear, and the power of the oscillations was reduced but not completely abolished. K, Population graph indicating PH peaks before (CTR) and after (AD) applications of antidepressants. Black and gray circles refer to fluoxetine and imipramine applications respectively. *p < 0.05.
Imipramine and fluoxetine effects on glutamatergic neurotransmission
Do antidepressants also affect glutamatergic neurotransmission? And, if so, might this contribute to disruption of γ oscillations? To test the role of antidepressants on glutamatergic neurotransmission, we recorded EPSCs pharmacologically isolated (in the continuous presence of 10 μm gabazine), and extracellularly evoked by Schaffer collateral stimulation. Antidepressants reduced pharmacologically isolated EPSCs (normalized EPSC amplitude: 20 μm imipramine, 0.58 ± 0.08, n = 5; 20 μm fluoxetine, 0.71 ± 0.11, n = 7; p < 0.05; Fig. 6A,B). Moreover, antidepressant treatment resulted in an increase of sEPSC frequency and amplitude (frequency: 3.26 ± 0.6 vs 8.28 ± 2.0 Hz; amplitude: 7.9 ± 0.87 vs 9.7 ± 1.4 pA, control vs antidepressant; n = 10, p < 0.05; Fig. 6C). However, the effect of antidepressants was more reversible on EPSCs than on IPSCs. Importantly, partial reduction of glutamatergic transmission by low doses of the selective AMPAR antagonist NBQX (200 nm; relative EPSC amplitude: 0.48 ± 0.06, n = 5; data not shown), mimicking antidepressant effect on EPSCs, did not significantly affect γ oscillations (relative peak power: 0.69 ± 0.23, n = 5; p > 0.05; Fig. 6D–F), suggesting that the antidepressant-dependent disruption of γ oscillations could not be solely attributable to reduced AMPA-mediated neurotransmission.
Figure 6.

Imipramine and fluoxetine effects on glutamatergic neurotransmission does not account for AD-mediated disruption of γ-oscillations. A, Representative EPSCs before, during, and after imipramine (top) or fluoxetine (bottom) applications. Both imipramine (IMI, 20 μm) and fluoxetine (FLUO, 20 μm) decreased monosynaptic EPSCs onto CA1 pyramidal neurons, extracellularly evoked in the stratum radiatum, in the continuous presence of the GABAAR antagonist gabazine (10 μm). B, Average time course of imipramine and fluoxetine effects on normalized EPSC amplitude. Note that application of both drugs (black bar) reduced EPSC amplitude in a highly reversible manner. C, Extracellular local field potential (LFP) recordings from stratum radiatum in CA1 showing the effect of 200 nm NBQX on CA1 oscillations. This NBQX concentration blocked AMPA-mediated glutamate neurotransmission to a similar extent of ADs. Traces were filtered in the γ-frequency band (30–60 Hz). Oscillations were induced by application of 400 nm of kainic acid (KA). D, Spectrograms of the same experiment of C. A black bar indicates NBQX application. The color scale indicates the power at each time-frequency pair. E, Population graphs illustrating the values of the power in logarithmic scale before and after the application of NBQX. F, Fluoxetine; I, imipramine; N, NBQX. Note that low NBQX concentrations did not significantly alter γ-oscillation power.
Chronic administration of antidepressants results in reduced evoked IPSC amplitudes of CA1 pyramidal neurons
The approach of acute in vitro application of drugs has the advantage of allowing the dissection of the cellular mechanisms underlying specific effects, while avoiding undesired errors resulting from variability, which is inherent to the use of different animal populations. However, antidepressants require lengthy treatments to exert their therapeutic effects. To test whether similar effects on GABAergic transmission were also present following chronic antidepressant treatments, rats were injected daily with fluoxetine, imipramine (both 20 mg/kg), or vehicle (saline). After 3 weeks of such treatments, acute brain slices were obtained and whole-cell voltage-clamp experiments were performed in the CA1 area of the hippocampus. Monosynaptic, perisomatic evoked IPSCs were induced by stimulating the stratum pyramidale in the continuous presence of glutamate receptor antagonists (see Materials and Methods). Stimulus intensity was adjusted to induce a similar proportion of failures and responses, defined here as threshold IPSCs (Fig. 7A,D). We found that the stimulus required to induce threshold IPSCs was higher in slices obtained from fluoxetine-treated and imipramine-treated animals than from control animals (control, 24 ± 3 μA, n = 19 cells; imipramine, 40 ± 5 μA, n = 17 cells; fluoxetine, 36 ± 6 μA, n = 22 cells; 4 animals per group; p < 0.01; Fig. 7A,B). In addition, threshold IPSC amplitudes from antidepressant-treated animals were smaller than in controls (control, 71.3 ± 15.6 pA, n = 19 cells; imipramine, 31.8 ± 6.5 pA, n = 17 cells; fluoxetine, 41.3 ± 7.4 pA, n = 22 cells; 4 animals per group; p < 0.01; Fig. 7C). Overall, monosynaptic IPSCs were strongly reduced in antidepressant-treated animals at all stimulus intensities (Fig. 7E,F). For example, the maximum (15× threshold) IPSC amplitude was 567 ± 91 pA in control rats, 289 ± 58 pA in imipramine-treated rats (p < 0.05), and 284 ± 44 pA in fluoxetine-treated rats (p < 0.01) (Fig. 7E,F). These results did not originate from altered CA1 pyramidal-cell or FS-interneuron excitability by chronic antidepressant treatment, as evidenced by lack of changes in firing frequency, resting membrane potentials, and membrane resistances in the three animal groups (control: n = 17 FS and 39 pyramidal neurons; imipramine: n = 13 FS and 36 pyramidal neurons; fluoxetine: n = 22 FS and 27 pyramidal neurons; 5 animals per group; p > 0.05 in all cases). These data suggest that chronic treatments with antidepressants also result in a marked reduction of evoked IPSCs in the CA1 area of the hippocampus. Importantly, the coefficient of variation of evoked IPSCs was significantly higher in the antidepressant-treated group than in vehicle-treated animals (Fig. 7G), similarly to acute applications (Fig. 2). We were unable to detect changes in the properties sIPSCs in the three animal groups (p > 0.05; data not shown) and of uIPSCs originated from FS interneurons in chronically treated rats (Fig. 7H), probably due to the inherent low sampling and high variability of unitary connections in acute brain slices from adult animals. Nevertheless, in slices obtained from imipramine-treated or fluoxetine-treated animals, γ oscillations induced by bath applications of kainate peaked at significant lower frequencies than in slices obtained from saline-treated animals (control: 59.60 ± 4.2 Hz; imipramine: 35.96 ± 7.95 Hz; fluoxetine: 39.23 ± 7.6 Hz; n = 5 in all groups; p < 0.05; Fig. 7I–K). There was not a significant difference in γ-oscillation power in the three animal groups (p > 0.05, all cases).
Discussion
This study shows that two major antidepressant medications, belonging to two different pharmacological types, share the effect of directly and profoundly altering perisomatic GABAergic neurotransmission from a specific hippocampal circuit. This occurred both when animals were chronically treated and when antidepressants were acutely applied to hippocampal slices at concentrations compatible with month-long brain accumulations of these drugs. The effect of both imipramine and fluoxetine was due to a direct action of these drugs at FS-cell GABAergic synapses and not to indirect modulations of GABA release through the known aminergic targets of these molecules. This was confirmed by pharmacological occlusion of amine transporters or receptors. In fact, none of these manipulations prevented the reduction of evoked GABA transmission, increase of sIPSC frequency, and disruption of γ oscillations. The conclusion that the antidepressant-mediated alterations of GABAergic transmission did not involve monoamine reuptake requires evidence that pharmacological occlusions efficaciously blocked amine transporters. Application of monoamine transporter blockers increased GABAergic synaptic transmission in some cases (4 of 12 neurons, Fig. 1C,D). This suggests that in quiescent brain slices subject to a constant perfusion of extracellular solution, ambient monoamine levels are low, but that reuptake can be affected. Importantly, however, when the transporter inhibitor mixture was applied during oscillations (i.e., in conditions of high network activity), it consistently increased the oscillation power (Fig. 1E,F). This significant effect demonstrates that, in our hands, the mix of reuptake inhibitors reached its targets and blocked amine transporters.
Interestingly, low concentrations of antidepressants that potentially impair monoamine transporters did not alter hippocampal GABAergic transmission overall, but sporadically (4 of 12 cells, imipramine and fluoxetine combined, data not shown) increased evoked IPSC amplitudes, similarly to the amine reuptake pharmacological occlusion, again suggesting that monoamine activity in acute brain slices cannot be easily modulated by reuptake blockade. GABAergic transmission was altered by higher concentrations (10 and 20 μm) of antidepressants in acute experiments. The concentrations used in this study are clinically relevant. Indeed, antidepressant medications exert their therapeutic effects only after several weeks of treatment, when, due to very slow drug elimination (Henry et al., 2005), brain levels of antidepressants can reach the concentrations used here (Sugita et al., 1989; Henry et al., 2005). There is no clear explanation as to why lengthy treatments with antidepressants are necessary to relieve MDD symptoms, as serotonin and/or other amine levels increase quickly following their uptake blockade (Krishnan and Nestler, 2008). Therefore, it is tempting to speculate that the actual beneficial effects of antidepressants, following prolonged drug administration, appear only after a relevant brain concentration has reached a therapeutic threshold.
The effect of antidepressants on GABA neurotransmission appears to be presynaptic, as suggested by the increase of (1) FS cell-mediated uIPSC failure rate, (2) PPR, (3) evoked IPSC variability, and (4) sIPSC and mIPSC frequency. This latter effect is intriguing, confirming and extending a previously reported action of fluoxetine on cultured hippocampal neurons (Ye et al., 2008). Indeed, mIPSC and sIPSC frequency increase by both drugs suggests that different antidepressants converge in desynchronizing synaptic vesicle release directly at GABAergic synapses. This action likely involves one or more mechanims, including activation of ionotropic and/or metabotropic receptors, ion channels, as well as elements of the synaptic vesicle recycling machinery. This antidepressant-mediated effect is reminiscent of alteration of GABA release induced by glutamate-mediated activation of presynaptic and somatodendritic kainate receptors (KARs) (Rodríguez-Moreno et al., 2000), although we exclude the involvement of KARs because antidepressant-dependent effects on GABAergic transmission were prominent in the presence of the KAR antagonist DNQX. The increase of spontaneous IPSC frequency seems not to involve altered GABA release from CB1R+ interneurons, as application of the CB1R agonist WIN 55212-2 in the presence of antidepressants did not reduce sIPSC frequency, suggesting that the increase of spontaneous GABAergic events and the decrease in the phasic release likely reflect a desynchronization of GABA release at FS-cell presynaptic terminals. However, future detailed experiments will be required to pinpoint the mechanism underlying the dual antidepressant-mediated effect on spontaneous and evoked IPSCs. In fact, we cannot exclude other explanations, including the possibility that antidepressants can exert their effects on sIPSCs, mIPSCs, and evoked IPSCs through different pathways.
Notably, we found that acute applications of antidepressants impair hippocampal γ oscillations, which underlie crucial aspects of the cognitive processes and whose emergence in the cerebral cortex relies on the concerted GABA release from FS basket cells (Cardin et al., 2009; Sohal et al., 2009; Wang, 2010). The antidepressant-mediated effect on γ oscillations reported here is likely due to the synergistic reduction of FS-cell synchronous GABA transmission and the enhanced spontaneous, asynchronous activity that can have the dual effect of deteriorating spike-timing precision (Manseau et al., 2010) and increasing the tone of extrasynaptic GABA, which reduces γ oscillations (Mann and Mody, 2010). Yet, despite the importance of FS basket cells in γ oscillations (Buzsáki and Draguhn, 2004; Freund and Katona, 2007; Wang, 2010), we cannot exclude the possibility that altered glutamatergic neurotransmission by antidepressants might play a role, although a reduction of excitatory transmission similar to that produced by antidepressants did not have an overall effect on γ oscillations. However, our results do not rule out the possibility that cognitive effects induced by antidepressants may rely on effects on glutamatergic neurotransmission, given that antidepressants had somewhat similar effects on both glutamatergic and GABAergic evoked and spontaneous release. Importantly, in addition to the selective effects of antidepressants on perisomatic FS-cell-mediated neurotransmission, it is possible that these drugs can affect release of GABA from other interneuron subtypes, including dendrite-targeting cells.
Antidepressant-mediated alteration of γ oscillations in the CA1 area and consequent cognitive effects require careful interpretation. Changes in cortical oscillation power and/or frequency can lead to various behavioral and cognitive effects. Whether and how depression and/or treatment with antidepressants affect network activity and cognitive functions is far from clear (Kudina et al., 2004; Linkenkaer-Hansen et al., 2005; Lee et al., 2010; Schulman et al., 2011). It is very difficult to predict cognitive consequences of the effects on CA1 γ oscillations by antidepressant treatment (especially if extrapolated to depressed individuals), due to the number of brain areas involved and the inherent variability of depressive disorders. However, our data support the interpretation that selective alteration of GABA release from FS interneurons results in the alteration of γ oscillations following both acute and chronic treatments, with possible effects on cognitive processes.
Whereas most antidepressant medications actually are designed to alter monoamine brain levels, their mechanisms of action are still a matter of debate and clear evidence that the pathophysiology of MDD relies on imbalanced monoamine (and particularly serotonin) levels is still lacking (Lacasse and Leo, 2005; Krishnan and Nestler, 2008). Importantly, it has been shown that SSRI antidepressants can promote translocation of Gαs and increase in Gαs-activated adenylyl cyclase without involving amine transporters (Zhang and Rasenick, 2010). Previous studies suggest that antidepressants can modulate GABAergic function through increased monoaminergic tone (Radnikow and Misgeld, 1998), or direct modulation of GABAA receptors (Robinson et al., 2003). Conversely, our results indicate that widely used antidepressants modulate presynaptic GABA release directly from a distinct perisomatic hippocampal inhibitory circuit (i.e., not involving monoaminergic transmission). Remarkably, it has recently been shown that fluoxetine restores adult cortical ocular-dominant plasticity through modulation of cortical inhibition (Maya Vetencourt et al., 2008). Importantly, low concentrations of GABA have been detected in MDD subjects (Sanacora et al., 1999, 2004; Croarkin et al., 2011), and antidepressant treatment can produce elevations of GABA (Sanacora et al., 2002).
The antidepressant-mediated increase in spontaneous inhibition and the decrease in spike-dependent synchronous GABA release can lead to increases of extracellular GABA levels, and consequent activation of extrasynaptic GABA receptors, which play a role in depressive disorders (Maguire and Mody, 2008; Croarkin et al., 2011; Luscher et al., 2011). MDD subjects suffer from reduced GABA levels (Sanacora et al., 1999, 2004; Croarkin et al., 2011; Luscher et al., 2011), and antidepressant treatment produces elevations of GABA (Sanacora et al., 2002; Luscher et al., 2011). Thus, it is tempting to speculate that a drop of ambient GABA might have a role in inducing depressive episodes, which can be alleviated by antidepressant-dependent desynchronization of FS-cell synapses, partially restoring extracellular levels of GABA. However, since we did not notice significant increases of sIPSC frequency in chronically antidepressant-treated animals, it will be important to test whether extracellular GABA levels are altered in animal models of depression, in the presence and absence of chronic antidepressant treatment.
Importantly, chronic treatment of rats with antidepressants did not show major differences in uIPSCs from FS interneurons. This could be due to intrinsic variability and low sampling of unitary GABAergic connections in the three animal groups. Nevertheless, there was a significant decrease of extracellularly evoked IPSC threshold and amplitudes and a significant shift of γ-oscillation frequency toward lower values in antidepressant-treated animals. Therefore both chronic and acute antidepressant treatment produced results that, albeit not identical, went in the same direction. Importantly, chronic and acute antidepressant treatments did not alter neuronal membrane potential, spontaneous firing, and membrane resistance (p > 0.05, data not shown), arguing against a change of general excitability in antidepressant-treated animals compared with control animals. Yet, we cannot exclude the possibility that other mechanisms, including effects on postsynaptic GABAA receptors can underlie the effects of chronically treated antidepressants on evoked IPSCs (Tunnicliff et al., 1999; Robinson et al., 2003).
Whereas it was proposed that CCK+, CB1+ interneurons were the basket-cell type involved in processing “inner-world” information and control mood, our results suggest that mood-enhancing pharmacological manipulations strongly affect GABA release from the “clockwork” system (i.e., the FS, PV+ basket cells). Interestingly, GABA release from FS interneurons is selectively altered by opioids (Glickfeld et al., 2008), which possess profound antidepressant properties and were long used to treat MDD before the introduction of safer medications (Berrocoso et al., 2009).
Our results indicate a novel target of antidepressants in the CNS and suggest that alterations of the tight balance and correct integration of GABAergic transmission originating from a diverse interneuron source might lead to the development of depression. Importantly, however, our results do not provide a direct demonstration that alteration of GABAergic transmission is relevant to the antidepressant action of imipramine and fluoxetine, although this is suggested by the similar effect of two antidepressant medications that belong to two different pharmacological families. Yet, whether altered FS cell-mediated GABAergic transmission can be included within the pathological mechanisms of depression will require future extensive experimentation using multidisciplinary approaches (e.g., genetic manipulations, in vivo approaches, behavioral testing, and others). Moreover, it will be interesting to extend the analysis of the effects of antidepressants in controlling specific interneuron GABA release also in other systems proposed to play a role in the etiology of MDD, such as maturation of adult-born granule cells of the dentate gyrus (Krishnan and Nestler, 2008).
Overall, accumulating evidence support the involvement of abnormalities of perisomatic inhibition in the development of neuropsychiatric diseases, such as schizophrenia and autism (Benes and Berretta, 2001; Levitt et al., 2004; Lewis et al., 2005; Orekhova et al., 2007), and it will therefore be important to investigate whether this applies also to depressive disorders.
Footnotes
This work was supported by a Giovanni Armenise–Harvard Foundation Career Development Award, a European Commission Marie Curie International Reintegration grant, and a grant from the European Research Council (ERC) under the European Community's 7th Framework Programmme (FP7/2007-2013)/ERC Grant Agreement No. 200808. A.B. is the 2007/2008 National Alliance for Research on Schizophrenia and Depression Henry and William Test Investigator. We thank Silvia Marinelli for critically reading the manuscript and Pasqualina Farisello for help in the analysis of the electrophysiological data.
The authors declare no competing financial interests.
References
- Atallah BV, Scanziani M. Instantaneous modulation of gamma oscillation frequency by balancing excitation with inhibition. Neuron. 2009;62:566–577. doi: 10.1016/j.neuron.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology. 2001;25:1–27. doi: 10.1016/S0893-133X(01)00225-1. [DOI] [PubMed] [Google Scholar]
- Berrocoso E, Sánchez-Blazquez P, Garzón J, Mico JA. Opiates as antidepressants. Curr Pharm Des. 2009;15:1612–1622. doi: 10.2174/138161209788168100. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Draguhn A. Neuronal oscillations in cortical networks. Science. 2004;304:1926–1929. doi: 10.1126/science.1099745. [DOI] [PubMed] [Google Scholar]
- Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, Moore CI. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–667. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charney DS. Monoamine dysfunction and the pathophysiology and treatment of depression. J Clin Psychiatry. 1998;59(Suppl 14):11–14. [PubMed] [Google Scholar]
- Croarkin PE, Levinson AJ, Daskalakis ZJ. Evidence for GABAergic inhibitory deficits in major depressive disorder. Neurosci Biobehav Rev. 2011;35:818–825. doi: 10.1016/j.neubiorev.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Daw MI, Tricoire L, Erdelyi F, Szabo G, McBain CJ. Asynchronous transmitter release from cholecystokinin-containing inhibitory interneurons is widespread and target-cell independent. J Neurosci. 2009;29:11112–11122. doi: 10.1523/JNEUROSCI.5760-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink KB, Göthert M. 5-HT receptor regulation of neurotransmitter release. Pharmacol Rev. 2007;59:360–417. doi: 10.1124/pr.107.07103. [DOI] [PubMed] [Google Scholar]
- Freund TF, Buzsáki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I. Perisomatic inhibition. Neuron. 2007;56:33–42. doi: 10.1016/j.neuron.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Galarreta M, Erdélyi F, Szab ó G, Hestrin S. Cannabinoid sensitivity and synaptic properties of 2 GABAergic networks in the neocortex. Cereb Cortex. 2008;18:2296–2305. doi: 10.1093/cercor/bhm253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickfeld LL, Scanziani M. Distinct timing in the activity of cannabinoid-sensitive and cannabinoid-insensitive basket cells. Nat Neurosci. 2006;9:807–815. doi: 10.1038/nn1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickfeld LL, Atallah BV, Scanziani M. Complementary modulation of somatic inhibition by opioids and cannabinoids. J Neurosci. 2008;28:1824–1832. doi: 10.1523/JNEUROSCI.4700-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hájos N, Pálhalmi J, Mann EO, Németh B, Paulsen O, Freund TF. Spike timing of distinct types of GABAergic interneuron during hippocampal gamma oscillations in vitro. J Neurosci. 2004;24:9127–9137. doi: 10.1523/JNEUROSCI.2113-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefft S, Jonas P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci. 2005;8:1319–1328. doi: 10.1038/nn1542. [DOI] [PubMed] [Google Scholar]
- Henry ME, Schmidt ME, Hennen J, Villafuerte RA, Butman ML, Tran P, Kerner LT, Cohen B, Renshaw PF. A comparison of brain and serum pharmacokinetics of R-fluoxetine and racemic fluoxetine: a 19-F MRS study. Neuropsychopharmacology. 2005;30:1576–1583. doi: 10.1038/sj.npp.1300749. [DOI] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. 2008;321:53–57. doi: 10.1126/science.1149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Ikeda Y, Haneda E, Suzuki H. Chronic fluoxetine bidirectionally modulates potentiating effects of serotonin on the hippocampal mossy fiber synaptic transmission. J Neurosci. 2008;28:6272–6280. doi: 10.1523/JNEUROSCI.1656-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902. doi: 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Nestler EJ. Linking molecules to mood: new insight into the biology of depression. Am J Psychiatry. 2010;167:1305–1320. doi: 10.1176/appi.ajp.2009.10030434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudina TA, Sudnitsyn VV, Kutyreva EV, Kichigina VF. The serotonin reuptake inhibitor fluoxetine suppresses theta oscillations in the electroencephalogram of the rabbit hippocampus. Neurosci Behav Physiol. 2004;34:929–933. doi: 10.1023/b:neab.0000042576.39132.f5. [DOI] [PubMed] [Google Scholar]
- Lacasse JR, Leo J. Serotonin and depression: a disconnect between the advertisements and the scientific literature. PLoS Med. 2005;2:e392. doi: 10.1371/journal.pmed.0020392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PS, Chen YS, Hsieh JC, Su TP, Chen LF. Distinct neuronal oscillatory responses between patients with bipolar and unipolar disorders: a magnetoencephalographic study. J Affect Disord. 2010;123:270–275. doi: 10.1016/j.jad.2009.08.020. [DOI] [PubMed] [Google Scholar]
- Levitt P, Eagleson KL, Powell EM. Regulation of neocortical interneuron development and the implications for neurodevelopmental disorders. Trends Neurosci. 2004;27:400–406. doi: 10.1016/j.tins.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Linkenkaer-Hansen K, Monto S, Rytsäl ä H, Suominen K, Isomets ä E, Kähkönen S. Breakdown of long-range temporal correlations in theta oscillations in patients with major depressive disorder. J Neurosci. 2005;25:10131–10137. doi: 10.1523/JNEUROSCI.3244-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Muñoz F, Alamo C. Monoaminergic neurotransmission: the history of the discovery of antidepressants from 1950s until today. Curr Pharm Des. 2009;15:1563–1586. doi: 10.2174/138161209788168001. [DOI] [PubMed] [Google Scholar]
- Luscher B, Shen Q, Sahir N. The GABAergic deficit hypothesis of major depressive disorder. Mol Psychiatry. 2011;16:383–406. doi: 10.1038/mp.2010.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire J, Mody I. GABA(A)R plasticity during pregnancy: relevance to postpartum depression. Neuron. 2008;59:207–213. doi: 10.1016/j.neuron.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann EO, Mody I. Control of hippocampal gamma oscillation frequency by tonic inhibition and excitation of interneurons. Nat Neurosci. 2010;13:205–212. doi: 10.1038/nn.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manseau F, Marinelli S, Méndez P, Schwaller B, Prince DA, Huguenard JR, Bacci A. Desynchronization of neocortical networks by asynchronous release of GABA at autaptic and synaptic contacts from fast-spiking interneurons. PLoS Biol. 2010;8:e1000492. doi: 10.1371/journal.pbio.1000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya Vetencourt JF, Sale A, Viegi A, Baroncelli L, De Pasquale R, O'Leary OF, Castrén E, Maffei L. The antidepressant fluoxetine restores plasticity in the adult visual cortex. Science. 2008;320:385–388. doi: 10.1126/science.1150516. [DOI] [PubMed] [Google Scholar]
- Möhler H. The GABA system in anxiety and depression and its therapeutic potential. Neuropharmacology. 2012;62:42–53. doi: 10.1016/j.neuropharm.2011.08.040. [DOI] [PubMed] [Google Scholar]
- Orekhova EV, Stroganova TA, Nygren G, Tsetlin MM, Posikera IN, Gillberg C, Elam M. Excess of high frequency electroencephalogram oscillations in boys with autism. Biol Psychiatry. 2007;62:1022–1029. doi: 10.1016/j.biopsych.2006.12.029. [DOI] [PubMed] [Google Scholar]
- Puig MV, Watakabe A, Ushimaru M, Yamamori T, Kawaguchi Y. Serotonin modulates fast-spiking interneuron and synchronous activity in the rat prefrontal cortex through 5-HT1A and 5-HT2A receptors. J Neurosci. 2010;30:2211–2222. doi: 10.1523/JNEUROSCI.3335-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radnikow G, Misgeld U. Dopamine D1 receptors facilitate GABAA synaptic currents in the rat substantia nigra pars reticulata. J Neurosci. 1998;18:2009–2016. doi: 10.1523/JNEUROSCI.18-06-02009.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkowska G, O'Dwyer G, Teleki Z, Stockmeier CA, Miguel-Hidalgo JJ. GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology. 2007;32:471–482. doi: 10.1038/sj.npp.1301234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson RT, Drafts BC, Fisher JL. Fluoxetine increases GABA(A) receptor activity through a novel modulatory site. J Pharmacol Exp Ther. 2003;304:978–984. doi: 10.1124/jpet.102.044834. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Moreno A, López-García JC, Lerma J. Two populations of kainate receptors with separate signaling mechanisms in hippocampal interneurons. Proc Natl Acad Sci U S A. 2000;97:1293–1298. doi: 10.1073/pnas.97.3.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G, Mason GF, Rothman DL, Behar KL, Hyder F, Petroff OA, Berman RM, Charney DS, Krystal JH. Reduced cortical gamma-aminobutyric acid levels in depressed patients determined by proton magnetic resonance spectroscopy. Arch Gen Psychiatry. 1999;56:1043–1047. doi: 10.1001/archpsyc.56.11.1043. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Mason GF, Rothman DL, Krystal JH. Increased occipital cortex GABA concentrations in depressed patients after therapy with selective serotonin reuptake inhibitors. Am J Psychiatry. 2002;159:663–665. doi: 10.1176/appi.ajp.159.4.663. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Gueorguieva R, Epperson CN, Wu YT, Appel M, Rothman DL, Krystal JH, Mason GF. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch Gen Psychiatry. 2004;61:705–713. doi: 10.1001/archpsyc.61.7.705. [DOI] [PubMed] [Google Scholar]
- Schildkraut JJ. The catecholamine hypothesis of affective disorders: a review of supporting evidence. Am J Psychiatry. 1965;122:509–522. doi: 10.1176/ajp.122.5.509. [DOI] [PubMed] [Google Scholar]
- Schulman JJ, Cancro R, Lowe S, Lu F, Walton KD, Llinás RR. Imaging of thalamocortical dysrhythmia in neuropsychiatry. Front Hum Neurosci. 2011;5:69. doi: 10.3389/fnhum.2011.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Gorelova N, Durstewitz D, Yang CR. Bidirectional dopamine modulation of GABAergic inhibition in prefrontal cortical pyramidal neurons. J Neurosci. 2001;21:3628–3638. doi: 10.1523/JNEUROSCI.21-10-03628.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KS, Rudolph U. Anxiety and depression: mouse genetics and pharmacological approaches to the role of GABA(A) receptor subtypes. Neuropharmacology. 2012;62:54–62. doi: 10.1016/j.neuropharm.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita S, Kobayashi A, Suzuki S, Yoshida T, Nakazawa K. Correlative changes of serotonin and catecholamines with pharmacokinetic alterations of imipramine in rat brain. Eur J Pharmacol. 1989;165:191–198. doi: 10.1016/0014-2999(89)90712-7. [DOI] [PubMed] [Google Scholar]
- Tunnicliff G, Schindler NL, Crites GJ, Goldenberg R, Yochum A, Malatynska E. The GABA(A) receptor complex as a target for fluoxetine action. Neurochem Res. 1999;24:1271–1276. doi: 10.1023/a:1020977123968. [DOI] [PubMed] [Google Scholar]
- Varga V, Losonczy A, Zemelman BV, Borhegyi Z, Nyiri G, Domonkos A, Hangya B, Holderith N, Magee JC, Freund TF. Fast synaptic subcortical control of hippocampal circuits. Science. 2009;326:449–453. doi: 10.1126/science.1178307. [DOI] [PubMed] [Google Scholar]
- Wang XJ. Neurophysiological and computational principles of cortical rhythms in cognition. Physiol Rev. 2010;90:1195–1268. doi: 10.1152/physrev.00035.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Kunos G, Nicoll RA. Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron. 2001;31:453–462. doi: 10.1016/s0896-6273(01)00372-5. [DOI] [PubMed] [Google Scholar]
- Ye ZY, Zhou KQ, Xu TL, Zhou JN. Fluoxetine potentiates GABAergic IPSCs in rat hippocampal neurons. Neurosci Lett. 2008;442:24–29. doi: 10.1016/j.neulet.2008.06.072. [DOI] [PubMed] [Google Scholar]
- Zhang L, Rasenick MM. Chronic treatment with escitalopram but not R-citalopram translocates Galpha(s) from lipid raft domains and potentiates adenylyl cyclase: a 5-hydroxytryptamine transporter-independent action of this antidepressant compound. J Pharmacol Exp Ther. 2010;332:977–984. doi: 10.1124/jpet.109.162644. [DOI] [PMC free article] [PubMed] [Google Scholar]