

Abstract
Visual information must be relayed through the lateral geniculate nucleus before it reaches the visual cortex. However, not all spikes created in the retina lead to postsynaptic spikes and properties of the retinogeniculate synapse contribute to this filtering. To understand the mechanisms underlying this filtering process, we conducted electrophysiology to assess the properties of signal transmission in the Long–Evans rat. We also performed SDS-digested freeze–fracture replica labeling to quantify the receptor and transporter distribution, as well as EM reconstruction to describe the 3D structure. To analyze the impact of transmitter diffusion on the activity of the receptors, simulations were integrated. We identified that a large contributor to the filtering is the marked paired-pulse depression at this synapse, which was intensified by the morphological characteristics of the contacts. The broad presynaptic and postsynaptic contact area restricts transmitter diffusion two dimensionally. Additionally, the presence of multiple closely arranged release sites invites intersynaptic spillover, which causes desensitization of AMPA receptors. The presence of AMPA receptors that slowly recover from desensitization along with the high presynaptic release probability and multivesicular release at each synapse also contribute to the depression. These features contrast with many other synapses where spatiotemporal spread of transmitter is limited by rapid transmitter clearance allowing synapses to operate more independently. We propose that the micrometer-order structure can ultimately affect the visual information processing.
Introduction
Fast synaptic transmission is realized by rapid creation of a high concentration of transmitter by exocytosis, which is swiftly removed by diffusion when a vast extracellular space is provided (Barbour, 2001; Franks et al., 2003; Raghavachari and Lisman, 2004). Therefore, even if these transmitter concentration transients were created in close successions, each transient is often distinct from each other, potentially allowing transfer of high-frequency signals. Relay cells (RCs) in the dorsal lateral geniculate nucleus (dLGN) receive visual signals created in the retinal ganglion cells and pass them onto the cortex. It would seem natural if these cells were equipped with necessary mechanisms to promote faithful transfer of signals. However, postsynaptic RCs produce fewer spikes than the retinal ganglion cells (Kaplan et al., 1987), and profound filtering and processing of visual signal is realized at these synapses (Chen et al., 2002; Blitz and Regehr, 2003), but the mechanisms underlying this filtering are unclear.
A feature we focused on, as a source of the filtering, is the morphological constriction of transmitter diffusion in the synaptic cleft. A single retinogeniculate (RG) fiber makes multiple contacts with an RC dendrite, of which each terminal consists of a large presynaptic bouton that makes a broad contact onto the postsynaptic dendrite of an RC with multiple synaptic sites within the contact (Rafols and Valverde, 1973). Transmitter released into the RG synaptic cleft would have nowhere else to go apart from diffusing laterally in two dimensions for long distances. Our simulations of glutamate diffusion and AMPA receptor (AMPAR) activation analysis have demonstrated that this morphological feature greatly limited the reliability of repetitive signal transfer. In contrast, a typical en passant synapse in the cortex or hippocampus is provided with multiple escape routes for the transmitter to diffuse once it passes through a small synaptic cleft. Another broad contact area with multiple release sites is found at the calyx of Held synapse, which is also considered as a “relay” of auditory signals (Taschenberger et al., 2002). However, this synapse can maintain high-fidelity signal transfer close to the kilohertz range because of the low release probability (Pr) at individual active zones, which prevents transmitter accumulation in the synaptic cleft and vesicle depletion (Iwasaki and Takahashi, 2001), and because of the presence of postsynaptic AMPARs that quickly recover from desensitization (Joshi et al., 2004; Koike-Tani et al., 2008). But the RG synapses lacked both of these features, and profound paired-pulse depression was attained.
We have identified that the morphological constraint of transmitter diffusion, in combination with the high presynaptic Pr and the postsynaptic AMPAR kinetics, is an important mechanism of the signal filtering realized at these synapses, which allows RCs to operate as something other than a simple “relay.”
Materials and Methods
Electrophysiology.
All animal experiments were conducted in accordance with the guidelines of the National Institute for Physiological Sciences' Animal Care and Use Committee. Brain slices containing dLGN were prepared mainly from young Long–Evans rats (P20–P30; for outside-out patch experiments, P5–P11) but wild-type and GluR1 knock-out (KO) mice (P20–P30) were also used in a subset of experiments. The animals were anesthetized by inhalation of halothane before decapitation, and the brain was sliced in ice-cold solution containing the following (in mm): 119.0 NaCl, 2.5 KCl, 0.1 CaCl2, 3.2 MgCl2, 1.0 NaH2PO4, 26.2 NaHCO3, and 11.0 glucose (saturated with 95% O2/5% CO2). Following the published procedure (Turner and Salt, 1998), the brain was glued at an angle so that the RG fiber inputs were relatively well preserved in the sliced tissue. Slices were cut at 250 μm using a microslicer (PRO7; Dosaka EM). The slices were then incubated in the above solution with CaCl2 and MgCl2 concentrations substituted to 2.0 and 1.3 mm, respectively, at 34°C for 30 min and then stored at room temperature (RT). During the recording, the slices were superfused with the latter solution at room temperature (22–25°C) with the addition of 100 μm picrotoxin to block GABAA receptors and 50 μm d-(-)-2-amino-5-phosphonopentanoic acid (d-AP5) to block NMDA receptors in most experiments, unless otherwise noted. In experiments where extracellular divalents were varied, total divalent concentration was kept constant except for 4 mm Ca2+ where 0.5 mm Mg2+ was also included. The external solution used for agonist applications in outside-out experiments contained the following (in mm): 140.0 NaCl, 2.0 CaCl2, 1.3 MgCl2, and 5.0 HEPES, pH adjusted to 7.4 with NaOH. Whole-cell voltage-clamp recordings were made under visual guidance (40× water-immersion objective, Olympus upright microscope BX51WI) using Axopatch 200B patch-clamp amplifier (MDS Analytical Technologies). Patch electrodes with resistances of 3–4 MΩ were used with the pipette solution (PS) with one of the following compositions (in mm): PS1: 35 CsF, 100 CsCl, 10 HEPES, 10 EGTA; PS2: 124 K-gluconate, 2 KCl, 10 HEPES, 10 K4-BAPTA, 4 MgCl2, 4 Na2ATP, 0.5 NaGTP; PS3: 142 K-gluconate, 2 KCl, 10 HEPES, 0.5 EGTA, 4 MgCl2, 4 Na2ATP, 0.5 NaGTP, pH 7.2 with CsOH titration for PS1 and with KOH titration for PS2 and PS3, with the addition of 100 μm calcein or 40 μm Alexa 594. Holding potentials (Vhs) were corrected for the liquid junction potential of 7, 16, and 15 mV, respectively, for PS1, PS2, and PS3. Unless otherwise noted, pipette solutions and holding potentials were PS1 and −77 mV for Figures 1A–C, 3A–E, and 4; PS2 and −86 mV for Figures 1D–J, 2, 3F–H, 9, and 11A–B; PS3 and −85 mV for Figures 10A–C,E–G and 11C–D; PS1 for Figure 10D, top; and PS3 for Fig. 10D, bottom. Series resistance was typically 5–10 MΩ and was compensated ∼70–80%. Input resistance was 209 ± 17 MΩ (with PS3; n = 22). Signals were filtered at 2 kHz and digitized at 10 kHz with Digidata 1322A using pClamp 8 acquisition software. Electrophysiological data analysis was performed with AxoGraph X.
Figure 1.
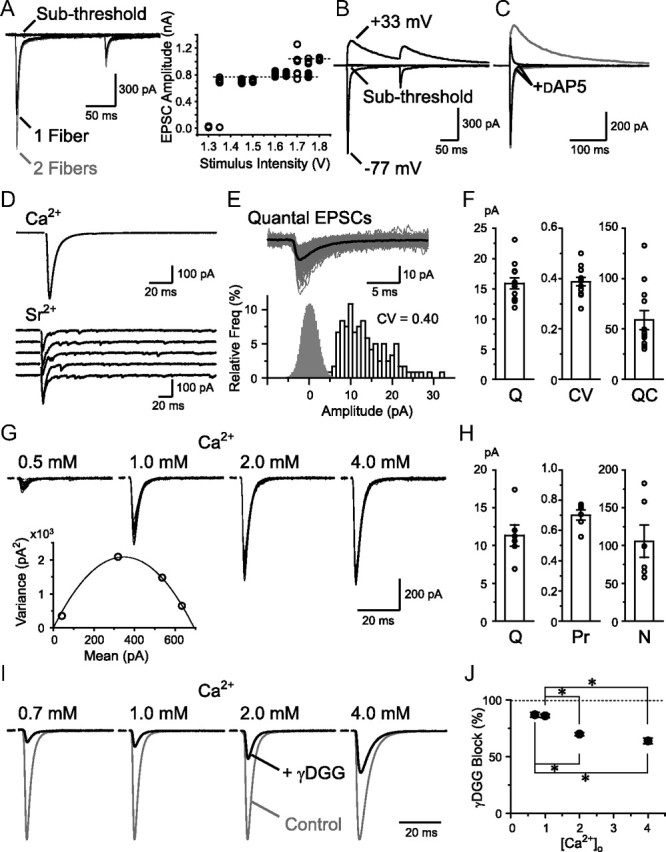
Basic properties of RG synaptic transmission. A, RG–RC EPSCs recorded with varying stimulus intensities (10 traces each). EPSC amplitude plotted against the intensity on the right. B, EPSCs were recorded at −77 and +33 mV at subthreshold and overthreshold stimulus intensities. C, AMPAR current dominated at negative potentials and was isolated using d-AP5 in most of all the following experiments. D, EPSCs evoked by single RG fiber stimulation were recorded in the presence of [Ca2+]o. After substitution with [Sr2+]o to desynchronize release, qEPSCs from the stimulated fiber were measured. E, qEPSCs were aligned (gray) and averaged (black). Histograms of qEPSC amplitude and baseline noise (open and gray bars, 1 and 0.2 pA bins, respectively) are shown below. F, Summary of the average qEPSC amplitude (Q), the CV of the amplitude (CV), and the QC (n = 12). G, EPSCs in the presence of various [Ca2+]o (20 traces each are shown). The mean EPSC amplitude was plotted against the variance of the EPSC amplitudes and a parabola function was fitted as below. H, Summary of the estimates of Q, Pr, and N from the mean variance analysis (n = 6). I, Recordings with varying [Ca2+]o from four cells are shown with the EPSC amplitude normalized. Application of 2 mm γDGG produced a different amount of block depending on the Pr. J, Block by γDGG was plotted against the [Ca2+]o (n = 4–7). *p < 0.05. Error bars indicate SEM.
Figure 3.

GluR subtypes responsible for the prolonged desensitization. A, Approximately half of the EPSC was blocked by a CP-AMPAR selective blocker, NASPM (50 μm). Summary of the block is shown in the bar graph (n = 10). B, EPSCs were recorded at negative and positive potentials in the absence and presence of internal spermine (100 μm). C, I–V of the EPSC with and without internal spermine. EPSCs were normalized to −1 at Vh = −77 mV (n = 2–15 for each plot). The average is shown for all plots. Statistical significance was examined at +33 mV. D, PPR recovered faster in the presence of NASPM (n = 6). E, PPR recovery in the presence of internal spermine at negative and positive potentials. PPR recovered faster at positive potentials where CP-AMPARs were partially blocked (n = 11). F, PEPA (200 μm), which selectively accelerates the recovery from desensitization in GluR3-flop homomeric channels, had no effect on the PPR recovery (n = 6). G, PEPA affected the EPSC decay time course. The decay was fitted with a double exponential function, and the main effect was found on the slow component fraction. H, PPR recovery was studied in wild-type (WT) and GluR1KO mice (n = 17 and 30, respectively). EPSCs were normalized to the first EPSC. The amount of desensitization was compared by dividing the PPR in control by the PPR in the presence of CTZ (n = 6 and 10 for wild-type and GluR1KO, respectively). *p < 0.05. Error bars indicate SEM.
Figure 4.

Outside-out patch recordings and AMPAR kinetic model. A, Kinetic scheme of the AMPAR model. Rates were as follows (units are M−1s−1 for k1, k2, and k3, and s−1 for the rest): k1 = 13.66 × 106, k−1 = 2.093 × 103, k2 = 6.019 × 106, k−2 = 4.719 × 103, k3 = 13.66 × 106, k−3 = 446.23, β = 17.23 × 103, α = 3.734 × 103, k4 = 1.0 × 103, k−4 = 60, k5 = 1.8 × 103, k−5 = 4.5, k6 = 12.36, k−6 = 1.5, k7 = 500, k−7 = 590.9, k8 = 40, k−8 = 420.9, k9 = 10.34 × 103, k−9 = 140, k10 = 233.2, k−10 = 0.3242. B, AMPAR desensitization kinetics were studied using a 17 ms pulse of 10 mm glutamate (n = 13; Data, τfast = 0.79 ± 0.18 ms, τslow = 3.88 ± 0.86 ms, %fast = 45.0 ± 7.1, τweighted = 2.14 ± 0.29 ms; Model, τfast = 0.81 ms, τslow = 3.60 ms, %fast = 42.3, τweighted = 2.39 ms). C, Two pulses of glutamate were applied to outside-out patches separated by different intervals. Simulated responses are shown below. D, Recovery from desensitization was plotted (n = 5–10). Double exponential curve fit to the PPR recovery with rates as follows: Data, τfast = 21.9 ms, τslow = 170.5 ms, %fast = 29.9, τweighted = 126.0 ms; Model, τfast = 27.9 ms, τslow = 194.7 ms, %fast = 32.6, τweighted = 140.4 ms. *p < 0.05. Error bars indicate SEM.
Figure 2.
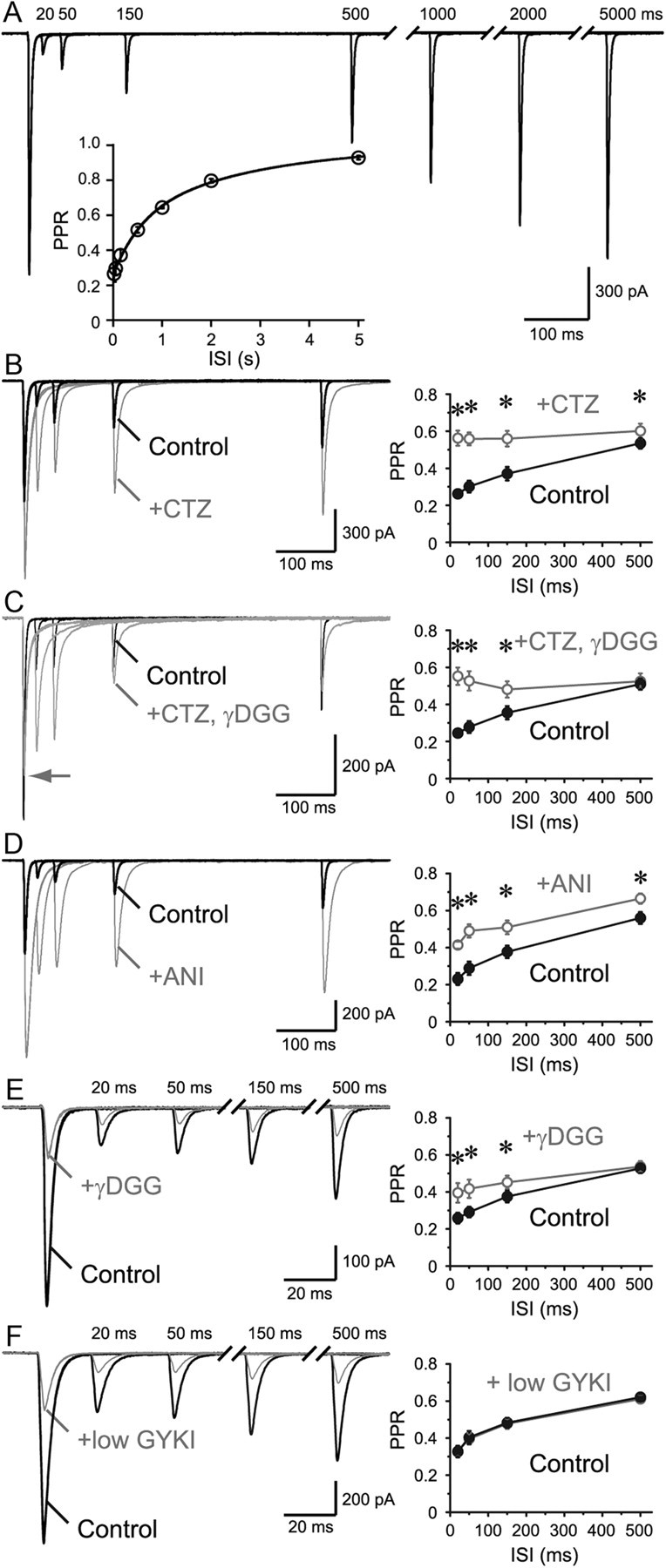
Prolonged AMPAR desensitization enhances PPD. A, Paired-pulse stimuli were applied to an RG fiber. PPR recovery time course is shown in inset (double exponential fit: τfast = 561 ms, 43%, τslow = 2713 ms, n = 6). B–F, Initial phase of the recovery was examined. B, Application of AMPAR desensitization blocker, CTZ (100 μm), accelerated the recovery from depression. PPR was plotted against the ISI on the right (n = 9). C, To avoid saturation, γDGG (1–2 mm) was coapplied with CTZ, and acceleration of the recovery was also observed (n = 9). D, Another desensitization blocker, aniracetam (ANI, 4–5 mm), which has been reported to have no presynaptic effects, was applied and the acceleration of the PPR recovery was observed (n = 7). E, Low-affinity antagonist, γDGG (2 mm), would save a subpopulation of AMPARs from entering the desensitized state. As expected, application of γDGG alone also accelerated the PPR recovery time course (n = 10). F, Application of low concentration (8 μm) of GYKI, a noncompetitive AMPAR antagonist, likely keeps a subpopulation unresponsive. As expected, there was no effect of the drug on the PPR recovery time course (n = 8), although the amount of block was comparable to that by γDGG. This result also suggests that the change in the PPR in E is not the result of difference in voltage-clamp errors between first and second EPSCs. *p < 0.05. Error bars indicate SEM.
Figure 9.
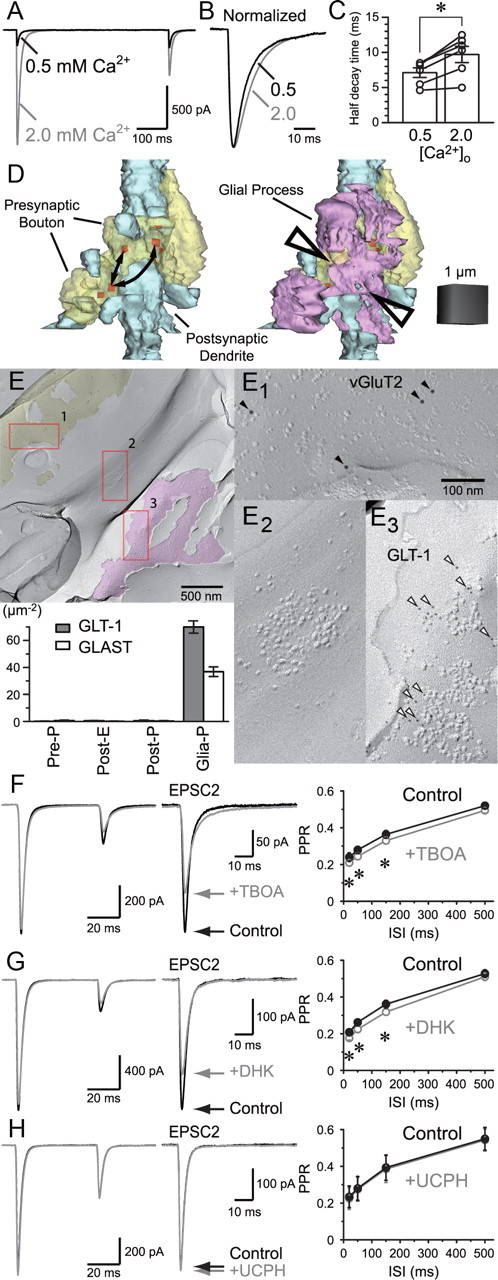
Experimental support for the presence of intersynaptic spillover. A, EPSCs were recorded in 0.5 and 2.0 mm [Ca2+]o in the presence of aniracetam. B, EPSCs were normalized to the peak amplitude. C, Half decay time was significantly different supporting the presence of spillover at 2.0 mm [Ca2+]o. D, 3D reconstruction of an RG–RC contact with two boutons from the same presynaptic fiber contacting in close vicinity. Arrows indicate possible routes of interbouton spillover. Glial cell processes (magenta) occupy the space between the boutons, likely preventing interbouton spillover (arrowheads). E, Replica of RG–RC contact area. Shown in yellow is the P-face fracture with vGluT2 labeling (E1, 10 nm, filled arrowheads) indicating that this plane is of an RG terminal. E2 shows the presence of a typical IMP cluster on E-face of an RC dendrite. Magenta highlights a P-face of glial cell membrane where GLT-1 labeling (E3, 5 nm, open arrowheads) was detected. Significant labeling of either GLT-1 or GLAST was found only on glial cell P-face as indicated in the bar graph. F, Paired-pulse EPSC in the absence and presence of TBOA (30 μm). The second EPSC is enlarged and shown on the right. The second EPSC was reduced by TBOA likely due to the enhanced spillover, which further promoted AMPAR desensitization. G, A similar effect was observed with the GLT-1-specific antagonist DHK (300 μm). H, GLAST-specific antagonist UCPH-101 (100 μm) had no effect. *p < 0.05. Error bars indicate SEM.
Figure 11.
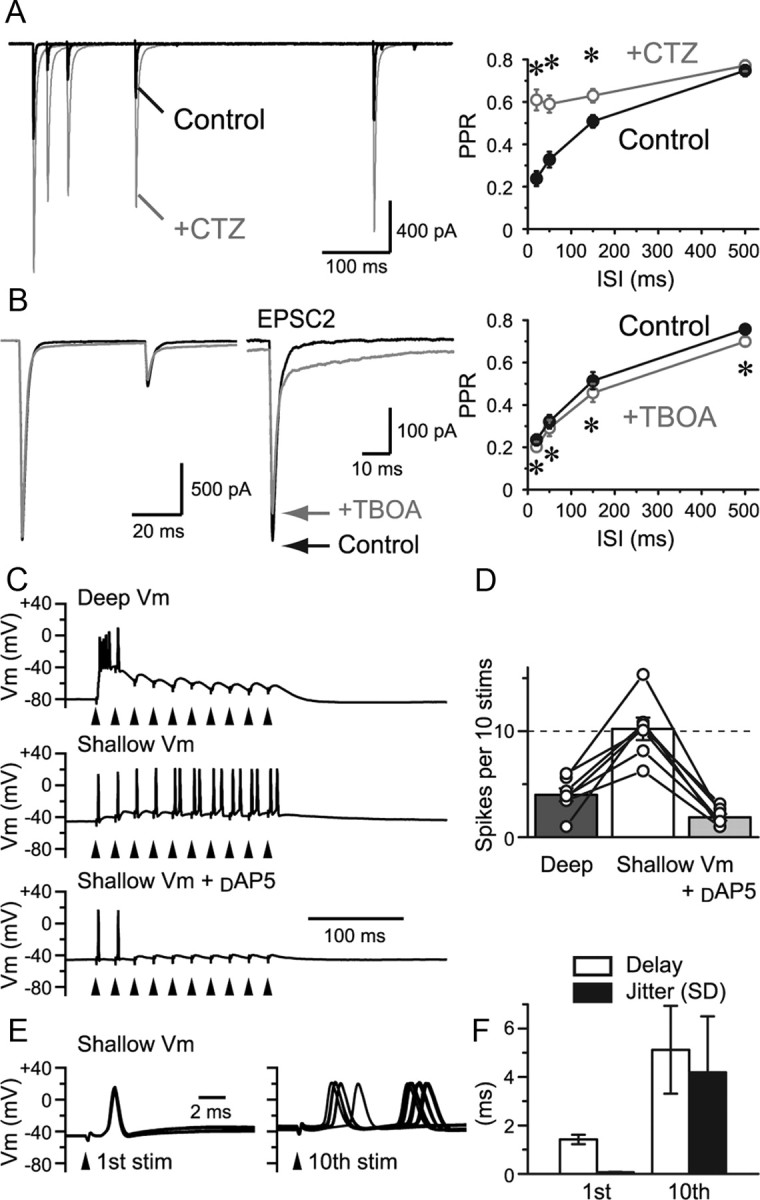
Experiments done at physiological temperature. A, Paired-pulse stimulation of an RG fiber was applied at various intervals, and EPSCs were recorded at PT of 31–34°C. EPSCs recovered faster than at room temperature. However, application of CTZ still caused a significant difference in the PPR at ISIs of up to 150 ms. This shows that prolonged AMPAR desensitization is also apparent at increased temperature. B, The effect of glutamate transporter blockade by TBOA was examined at PT. PPR was depressed further by TBOA, confirming that glutamate uptake has a small but significant effect of preventing spillover. C, Current-clamp recordings of the spiking responses of an RC in response to a train stimulation (50 Hz) given to an RG fiber at PT. At deep potentials, the cell responded with a burst at the onset of the train stimulus. However, continuing stimuli failed to produce spikes. At shallow resting potential, the cell responded well to the train stimulus. However, the timing and number of the latter spikes were unreliable. Application of d-AP5 diminished the latter spikes, indicating that those spikes were created by the slow and accumulating activation of NMDARs. D, The total number of spikes produced by the 10 stimuli is summarized (n = 7 cells). E, Timing of spike onset after the first and 10th stimulus is shown (10 sweeps). F, The delay of the onset of spike after the stimulus and the jitter (SD) of the delay are shown as bar graphs. stim, Stimulation; Vm, potential. *p < 0.05. Error bars indicate SEM.
Figure 10.
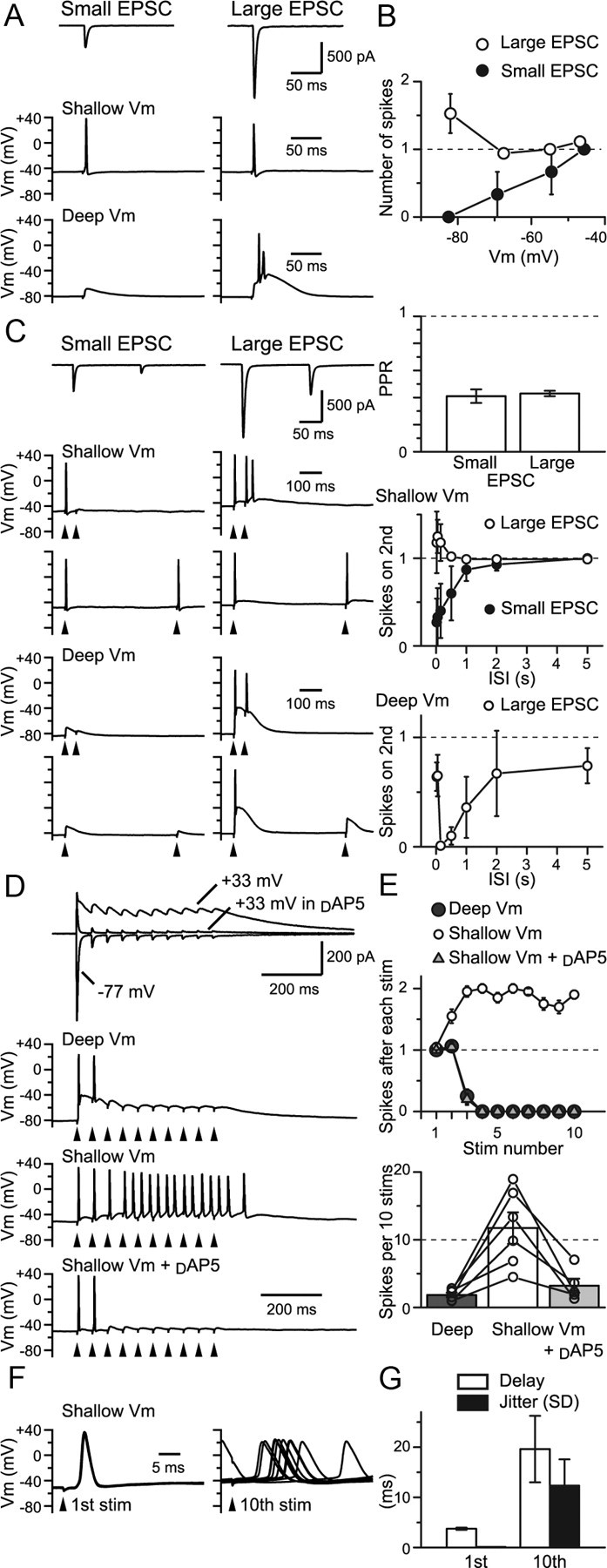
Physiological impact of short-term synaptic plasticity. A, RG–RC EPSCs and spiking responses recorded from the same cells in the absence of d-AP5. Small and large EPSCs were <500 and >500 pA, respectively. Small-EPSC fiber could produce an action potential response only when the resting potential was made shallow. Large EPSC fiber evoked an action potential response regardless of the resting potential. B, Number of spikes produced plotted against the mean resting potential for small and large EPSCs (n = 3 and 9–14 for small and large EPSCs, respectively). C, PPR of the small and large EPSCs was similar. At shallow potential, spiking often failed upon the second stimulus at short ISIs for the small EPSC fiber, but succeeded for the large EPSC fiber. Spiking at deep resting potential showed a biphasic recovery. D, Trains of stimuli were given at 20 Hz for 10 times. AMPAR EPSC depressed during the train, but NMDAR EPSC summated. Train stimulation failed to evoke more than a few spikes at deep potentials. However, at shallow potentials, multiple spikes were observed. The latter spikes were lost by d-AP5. E, The number of spikes produced within 50 ms after each stimulus during the train for the recording shown in D (20 sweeps). The total number of spikes produced by the train of 10 stimuli is summarized below (n = 6 cells). F, Spike onset after the first and 10th stimulus (10 sweeps). G, The delay to the spike onset and the jitter (SD) of the delay are shown as bar graphs. Stim, Stimulation; Vm, potential. *p < 0.05. Error bars indicate SEM.
RG stimulations were accomplished by placing a bipolar stimulating electrode near the caudal border of the optic tract (10–90 V, 100 μs; used in constant voltage; ISO-Flex, A.M.P.I.). RG fiber input was readily distinguishable from another glutamatergic input, corticogeniculate (CG) fiber input, as marked paired-pulse depression was observed for RG fiber input whereas CG fiber input showed paired-pulse facilitation. Evoked EPSCs from each RG fiber were also readily distinguishable, as the RG fiber input displayed step-like increases in the amplitude with increasing magnitude of stimuli with each step corresponding to the recruitment of single RG fiber activations (Fig. 1A). Stimulus intensity was minimized to accomplish putative single RG fiber stimulation. For outside-out patch experiments, a theta glass flow-pipette mounted on a piezoelectric bimorph was used for rapid agonist application (Jonas, 1995; Matsui et al., 2005). Solution exchange time was measured after each experiment by rupturing the patch, and the junction currents across the open pipette tip were recorded (open tip response, 20–80%; exchange time, 130–220 μs). After each recording, only the recordings from the relay cells were selected by visual inspection of the cell morphology revealed by inclusion of fluorescent dyes (calcein or Alexa Fluor 594) in patch pipettes and by using epifluorescence microscopy. The relay cells were distinguished from the local inhibitory interneurons by their larger soma sizes and by the presence of three or more dendrites coming from the cell bodies.
The sources of the chemicals are as follows: picrotoxin, GYKI 52466 (GYKI), 1-naphthylacetyl spermine trihydrochloride (NASPM), and spermine were from Sigma; γ-d-glutamylglycine (γDGG), NBQX, 2-[2,6-difluoro-4-[[2-[(phenylsulfonyl)amino]ethyl]thio]phenoxy]acetamide (PEPA), and dl-threo-β-benzyloxyaspartic acid (TBOA) were from Tocris Bioscience; cyclothiazide (CTZ) and d-AP5 were either from Tocris Bioscience or Ascent Scientific; dihydrokainic acid (DHK) and UCPH-101 were from Ascent Scientific; and aniracetam was from LKT Laboratories.
SDS-digested freeze–fracture replica labeling.
Male Long–Evans rats at P21 were used. The rats were anesthetized with sodium pentobarbital (50 mg/kg, i.p.) and perfused transcardially with 25 mm PBS for 1 min, followed by perfusion with 2% paraformaldehyde (PFA) and 15% saturated picric acid solution in 0.1 m phosphate buffer (PB) for 12 min. Coronal slices (150 μm thick) were cut using a microslicer (DTK-1000; Dosaka EM) in 0.1 m PB. A region of the dLGN without the dorsal outer surface region that contains cells receiving glutamatergic inputs from the superior colliculus was trimmed from the slice. The trimmed slices were immersed in 30% glycerol/0.1 m PB at 4°C overnight and rapidly frozen by a high-pressure freezing machine (HPM010; BAL-TEC). Frozen samples were then fractured into two parts at −120°C and replicated by deposition of carbon (5 nm thick), platinum (unidirection from 60°, 2 nm), and carbon (20 nm) in a freeze–fracture replica machine (BAF 060; BAL-TEC). After thawing, tissue debris attached to the replicas was removed with gentle rocking at 80°C for 16 h in a solution containing 15 mm Tris-HCl, pH 8.3, 20% sucrose, and 2.5% SDS. The replicas were then washed in 50 mm TBS, pH 7.4, containing 0.05% bovine serum albumin (BSA) and blocked with 5% BSA in the washing buffer for 1 h at room temperature. The replicas were incubated with a rabbit primary antibody against GluA1–4 (panAMPAR) overnight at 15°C, followed by incubation with anti-rabbit secondary antibody conjugated with 5 nm gold particles [British Biocell International (BBI)] for 1 h at 37°C. An antibody against vesicular glutamate transporter (vGluT) 2 raised in a guinea pig was used to mark the RG fibers with anti-guinea pig secondary antibody conjugated with 10 nm gold particles (BBI). Although vGluTs are vesicular proteins, they are often detected on presynaptic plasma membrane and thus have been used to identify the origin of presynaptic profiles (Hagiwara et al., 2005; Masugi-Tokita et al., 2007). For labeling of glial glutamate transporters GLT-1 and GLAST, rabbit antibodies against GLT-1 and GLAST, respectively, were used in combination with the vGluT2 antibody. The labeled replicas were examined by using a transmission electron microscope (Tecnai-12; FEI). Identity, source, and characterization of the antibodies are summarized in Table 1.
Table 1.
Identity, source, and characterization of antibodies
| Molecule | Code no. in original laboratory | Host animal | Developer | Epitope, amino acid residues | Protein concentration | Optimal dilution FRL | Characterization testsa |
Reference | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Immunohistochemistry | Western blot | FRL | ||||||||
| GluA1-4 (panAMPAR) | Anti-GluR1-4 (R3/BO A/P 30/08/01) | Rabbit | E. Molnár | Mouse GluA1 724–781 | Purified ab., 330 μg/ml | 300× | Brain, postembedding EM | COS7 cells | WT rat and mouse, GluA2/3 KO | Nusser et al., 1998; Masugi-Tokita et al., 2007 |
| vGluT2 | Anti-DNPI-Gp (670 μg/ml) | Guinea pig | M. Watanabe | Rat 559–582 | Purified ab., 670 μg/ml | 200× | Brain, pre-embedding | Brain | WT rat | Miyazaki, 2003; Kawamura et al., 2006; Miura et al., 2006; Masugi-Tokita et al., 2007 |
| GLT1 (EAAT2) | Anti-GLT-1 (0.96 mg/ml) | Rabbit | K. Tanaka | Mouse 500–525 | Purified ab., 960 μg/ml | 300× | Brain | Brain | No | Yamada et al., 1998 |
| GLAST (EAAT1) | Anti-GLAST (1000) | Rabbit | M. Watanabe | Mouse C-term 41aa | Purified ab., 1 mg/ml | 300× | Brain | Brain | No | Shibata et al., 1997; Yamada et al., 2000; Watase et al., 1998 |
aData are presented in cited publications.
Quantification of immunoparticles was done as follows. Images of excitatory postsynaptic specialization indicated by the presence of intramembrane particle (IMP) clusters on the exoplasmic face (E-face) accompanied by presynaptic protoplasmic face (P-face) labeled for vGluT2 were captured at a magnification of 97,000× with a digital camera [MegaView III, Soft Imaging System (SIS)]. The diameter of each E-face IMP apparently associated with panAMPAR immunolabeling was ∼14 nm, and the synaptic E-face IMP clusters, defined as densely packed IMPs at a distance of <14 nm from each other, were demarcated freehand. IMP cluster size demarcated by this method was comparable to that of the postsynaptic density (PSD) visualized in conventional ultrathin sections, and, thus, the demarcation likely well represents the extent of postsynaptic specialization (Tarusawa et al., 2009). The center of gravity and the areas of individual IMP clusters were measured using iTEM software (SIS). Data were obtained only from complete synapses where the whole IMP cluster is visible within a continuous fractured plane. Immunoparticles within a demarcated IMP cluster and those located outside but within 30 nm from the edge of the IMP cluster were regarded as synaptic labeling, considering possible deviation of the immunoparticles from antigens (Tarusawa et al., 2009). Labeling of glutamate transporters was searched for on (1) the P-faces of the RG fiber using vGluT2 labeling as a marker, (2) the corresponding postsynaptic E-faces adjacent to the membrane identified in step 1, (3) the corresponding P-faces of the replica complementary to the postsynaptic E-faces identified in step 2, and (4) the glial P-faces surrounding the presynaptic and postsynaptic membrane identified in steps 1–3, which were also identified by the morphological signature of glial membrane such as the frequent presence of reticular and/or aggregated arrangements of IMPs.
AMPAR immunoparticle distribution within the IMP cluster demarcation was evaluated by first making a distance map from the border of the demarcation using FIJI software [distributed under the General Public License (GPL)] (see Fig. 5D). The grayscale values correspond to the nearest-neighbor distance (NND) of each pixel from the border, with the lighter values corresponding to the longer distances. Using this distance map, the IMP cluster area was divided into five equal distances from the border. An additional division outside of the demarcation (outer rim) with 30 nm width was also created to take into account the possible spatial deviation of the immunoparticles from the antigen. Immunoparticle location in this distance map was extracted, and the density of immunoparticles falling into each division was tabulated (see Fig. 5E). As shown in the Figure 5E, the most peripheral division and the outer rim division showed a significantly lower density than the rest. However, the density difference between the remaining divisions was insignificant, indicating that the particle density distribution was relatively homogeneous.
Figure 5.

AMPAR distribution and simulated quantal response. A, An SDS-FRL image of a postsynaptic E-face of an RC with a characteristic IMP cluster (demarcated with red line) representing glutamatergic postsynaptic membrane specialization labeled for AMPARs (with 5 nm gold particles). B, AMPAR labeling positively correlated with the area of synapses (n = 63 complete synapses). C, The average area of a synapse, and the number and density of the immunoparticles from three animals. D, Intrasynaptic distribution of AMPARs was evaluated by dividing the synapse area into five divisions of equal width using the distance map. An additional division with 30 nm width on the outer rim was also made. E, Particle density in each division was averaged across all synapses. No significant difference between the center divisions was found, and only the most peripheral division and the outer rim had significantly lower density. One-way repeated-measures ANOVA followed by pairwise comparisons were used. Very few particles were found in the extrasynaptic region (1.8 ± 0.4 /μm2). F, Glutamate concentration profile (grayscale) and simulated AMPAR Po (pseudo-color) at 0.09 ms after release at the center of gravity of the synapse. G, Average AMPAR immunoparticle distribution from the center of gravity was tabulated (n = 63 synapses from one animal, 10 nm bins, total particle number in histogram = average particles number per synapse = 22.0). Simulated AMPAR peak Po at each distance from the release site was plotted (red). H, The peak number of open AMPARs expected from the AMPAR distribution and simulations at each distance from the release site. Inset, Summed response of all receptors simulated. I, An example of two neighboring IMP clusters on the postsynaptic E-face, accompanied with presynaptic P-face of an RG fiber identified by vGluT2 labeling (arrowheads, 10 nm gold particles). J, Histogram of NNDs of IMP clusters (50 nm bins, n = 186). Distance versus simulated peak Po is shown in red. K, The calculated glutamate concentration transient, Po, and fraction of receptors that is desensitized at 569 nm from the release site. *p < 0.05. Error bars indicate SEM.
It is possible that immunoparticles fall outside of the demarcation even though the actual antigens are inside the demarcation. Such deviation of labeling may preferentially reduce the immunoparticle density in the peripheral division. To take this possibility into account, the synapse area was divided into the following two divisions: 30 nm inward from the border of demarcation and the rest in the center. The number of immunoparticles residing outside of the demarcation within 30 nm distance was added to the number of immunoparticles in the peripheral division, and the density was calculated by dividing the added number by the area of the peripheral division. This would compensate for the possible loss of labeling in the peripheral by immunolabeling deviation. However, the center density (728 ± 39 particles/μm2) was still significantly larger than the peripheral density (544 ± 25 particles/μm2, n = 63, paired t test, p < 0.01). This supports the idea of slightly preferential distribution of the immunoparticles toward the center of the synapse (for comparison of intrasynaptic receptor distribution, see Results in Somogyi et al., 1998).
3D reconstructions from serial ultrathin sections.
Long–Evans rats (P23) were anesthetized with sodium pentobarbital (50 mg/kg, i.p.) and perfused transcardially with 25 mm PBS, followed by perfusion with 100 ml of 0.8% PFA and 1.5% glutaraldehyde in 0.1 m PB, pH 7.4, and the removed brains were further fixed with the same fixative for 2 h at 4°C. Coronal sections (60 μm thick) were cut using a microslicer (VT-1000; Leica) in 0.1 m PB. After washing in 0.1 m caodylate buffer several times, sections were treated with 1% OsO4 and 1.5% potassium ferricyanide in 0.1 m PB for 50 min followed by 1% OsO4 in 0.1 m PB for 30 min, stained en bloc with 1% uranylacetate, dehydrated with ethanol, and flat embedded in Durcupan resin (Fluka). Serial ultrathin sections were prepared at a thickness of 70 nm (Ultracut S; Leica). RG boutons and RCs were identified by their morphological features. Large terminals containing round synaptic vesicles and pale mitochondria with multiple asymmetrical synapses on a postsynaptic cell were classified as RG boutons (Guillery, 1969; Lieberman and Webster, 1974). Postsynaptic profiles containing vesicles were identified as interneurons, and the rest were considered as RCs (Rafols and Valverde, 1973). Serial images covering an entire RG bouton to an RC dendrite contact were captured at a magnification of 12,500× with the digital camera. The PSD was identified by the presence of synaptic vesicles accumulated in the presynaptic active zone, the postsynaptic density, and a visible synaptic cleft with a rigid alignment of the presynaptic and postsynaptic membranes. Within a single contact made by an RG bouton, multiple synapses at a short distance from each other were frequently observed. Glial processes appeared not to intervene in these synapses, but they typically surrounded the RG axonal–dendritic terminal as a capsule of thin glia sheath forming a glomerulus (Famiglietti and Peters, 1972). The scale in digital images was calibrated by a grating replica (Ted Pella).
For 3D reconstruction, the digital images of the serial ultrathin sections were aligned first, and this was done with the aid of Register Virtual Stack Slices plugin (GPL) for FIJI software. The demarcation of the plasma membrane border of each cellular structure was done manually by using Reconstruct software (GPL) (Fiala, 2005), and the 3D representations were made with this software (see Figs. 7C,E, 9D). Cell structure demarcation was exported from Reconstruct and read again using FIJI to calculate the surface distances of synapses. For ease of computation, voxel resolution was reduced to 10 nm on each side, and a custom FIJI plugin was made to calculate the surface distance from a point source on the plasma membrane by marching a 30 nm radius sphere along the surface of the reconstruction. The surface distance map of up to 2 μm distance was created from each PSD location, and the NND from a PSD to another and the number of PSD neighbors within the specified radius were calculated using this map (see Fig. 7D,F). Calculation of NND by measuring the direct straight path going through structures was much easier to perform. However, the direct NND values were found to be an underestimate (460 ± 7 nm, n = 7, and 394 ± 39 nm, n = 9, for “simple” and “complex” type RG glomeruli, respectively), and the average numbers of neighboring synapses within a direct radius of 700 and 1400 nm were an overestimate (2.4 ± 0.3 and 10.9 ± 0.9 for the simple-type and 2.3 ± 0.3 and 4.4 ± 0.7 for the complex-type, respectively) compared with the surface distance measurements.
Figure 7.
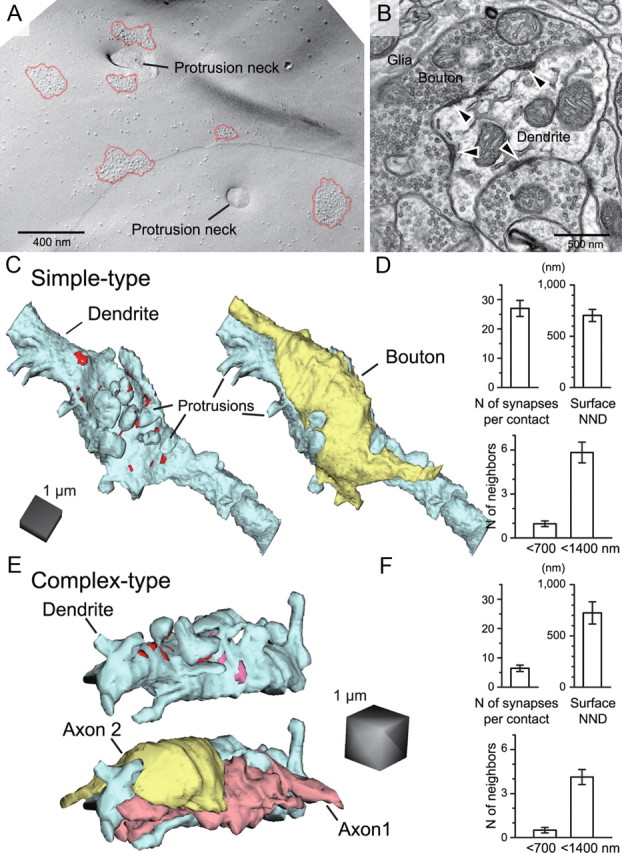
Morphological characteristics of RG multisynapse contact. A, An SDS-FRL image of multiple IMP clusters in close vicinity. B, An example of an EM image of an ultrathin section of the RG–RC contact containing multiple synapses (arrowheads). C, 3D reconstructed representation of a simple type RG–RC contact. Shown are the RC dendrite (blue) with PSD locations (red), and RG terminal (right, yellow) overlaid. D, The number of synapses per contact (n = 7 simple contacts). The surface NND and number of neighboring synapses within 700 and 1400 nm calculated from an example contact shown in C (n = 29 synapses). E, 3D reconstruction of “complex” type RG–RC synaptic contacts. Shown in blue is the RC dendrite, pink and red areas indicate PSDs of synapses formed by axon 1 (light red) and 2 (yellow), respectively. Shown below is the same glomerulus with axon structures overlaid. F, Number of synapses per contact (n = 9 complex contacts), and the surface NND and the number of neighboring synapses within 700 and 1400 nm calculated from an example contact shown in E (axon 1; n = 8 synapses).
Simulations.
To simulate synaptic AMPAR responses to releases of glutamate, glutamate diffusion was calculated, and the AMPAR kinetic model described in Figure 4 was run against the calculated glutamate transient (Tarusawa et al., 2009). Instantaneous release of glutamate was assumed, and the glutamate concentration in the extracellular space as a function of time and distance from release was calculated using the analytical solution to the following diffusion equation in a 2D space (Uteshev and Pennefather, 1996):
 |
where r is the distance from the release site, t is the time from release, h is the width of the extracellular space, NGlu is the number of glutamate molecules in a vesicle, and DGlu is the diffusion coefficient of glutamate in extracellular space. In contrast to most synaptic contacts in the CNS, RG axon terminals extend well over the IMP cluster area of relay cells because these synapses are formed directly on the dendritic shaft, and the presynaptic and the postsynaptic membranes remain parallel to each other for long distances. Thus, the extracellular space between these two elements was regarded as a plane with no gaps or glial elements with h of 20 nm in our simulations. Time steps for AMPAR simulations were set to 0.5 μs, as reducing the time step to 0.1 μs produced negligible differences (data not shown), and the glutamate concentration profile at each receptor location was calculated using the Excel software (Microsoft). The NGlu was set to 4000 molecules, and the DGlu to 0.3 μm2/ms. The validity of the selection of these parameters is discussed in Results (see also Franks et al., 2003; Nielsen et al., 2004). An AMPAR kinetic model (Fig. 4) was run against the glutamate transients to calculate the open probabilities (Pos) of individual AMPARs using AxoGraph X.
Statistical analysis.
Values in text and graph are given as the mean ± SEM. Excel (Microsoft) and SPSS (IBM SPSS) were used for statistical analysis. One-way ANOVA with post hoc Tukey's test was used for Figure 1J; two-way repeated-measures ANOVA was used for the initial determination of significance followed by paired t test of the PPR at individual interstimulus intervals (ISIs) for Figures 2B–F, 3D–F,H, 9F–H, and 11A–B; unpaired t test was used for Figure 3C; paired t test was used for Figures 3G and 9C. Statistical significance with p < 0.05 was indicated with an asterisk in all figures.
Results
Large RG fiber input is mediated by high Pr and large number of synapses
Several RG fibers innervate an RC, and each fiber provides strong glutamatergic synaptic input with an amplitude of the EPSC ranging from 100 pA to >1 nA in voltage-clamp recording mode. The large EPSC along with the presence of only a few innervating RG fibers allowed minimal stimulation of a single RG fiber input (Fig. 1A). Although both AMPARs and NMDA receptors (NMDARs) were activated by the stimulation of a single RG fiber (Fig. 1B), the AMPAR-mediated current dominated at negative resting potentials and was expected to provide the initial drive for the postsynaptic action potential firing (spiking). Therefore, we focused mainly on the AMPAR EPSC and blocked the NMDAR component with d-AP5 (Fig. 1C). The role of NMDARs during repetitive stimulation was evaluated later in the article (see Fig. 10).
Strong synaptic input could be provided by large quantal responses to single vesicle releases (Q), high presynaptic Pr, and/or large number of release sites (N). To measure the quantal EPSCs (qEPSCs) from an RG fiber, extracellular Ca2+ ([Ca2+]o) was replaced with Sr2+ to allow desynchronization of release from the stimulated fiber (Fig. 1D,E). qEPSC amplitude varied largely, but the average amplitude (15.9 ± 0.9 pA at Vh = −86 mV, n = 12 cells) was much smaller than the evoked EPSC (Fig. 1F). The quantal content (QC) of single fiber stimulation was calculated by dividing the amplitude of the evoked EPSC in Ca2+ by the amplitude of the average qEPSC. QC (58.8 ± 9.4) (Fig. 1F) was much higher than that of a typical single en passant synapse, suggesting that many synapses were made between an RG fiber and an RC.
To identify the parameters determining the large QC, mean variance analysis was performed (Silver et al., 1998). A single RG fiber was stimulated for multiple trials in the presence of various [Ca2+]o values to vary the Pr (Fig. 1G). As expected from a binominal probability process of vesicle release, the variance of the evoked EPSCs between trials was the highest at a moderate concentration of [Ca2+]o (∼1 mm) and lower at a higher or lower [Ca2+]o; consequently, a parabola function could be fitted to the mean variance plot (Fig. 1G). Using a simple binominal model, we estimated a Q of 11.3 ± 1.4 pA (at Vh = −86 mV, n = 6 cells). This was similar to the value recorded directly by Sr2+ application, confirming the validity of our statistical approach. The slightly larger value from the direct observation could be explained by the baseline noise that could conceal the smallest events. We also estimated that the RG synapse had a large N (105.7 ± 21.4, range 57.8–182.3). Notice that the estimates of the N varied largely from fiber to fiber. The impact of the absolute EPSC amplitude difference on the postsynaptic spiking behavior is presented later in the article (see Fig. 10). Interestingly, using the mean variance analysis, we also found a high Pr in our control condition with 2 mm [Ca2+]o (Fig. 1H; 0.70 ± 0.03), a feature that distinguishes the RG terminal from another sensory relay terminal, the calyx of Held, where glutamate release also occurs from a single large contact containing numerous synapses (∼600) but with a much lower Pr (0.25–0.4; Meyer et al., 2001). The Pr calculated by this method reflects the Pr of each docked vesicle since a single vesicle release would not cause complete saturation of the AMPARs in each synapse (see the simulations in Fig. 6) and we did not assume that the release at each synapse is restricted to single vesicles.
Figure 6.
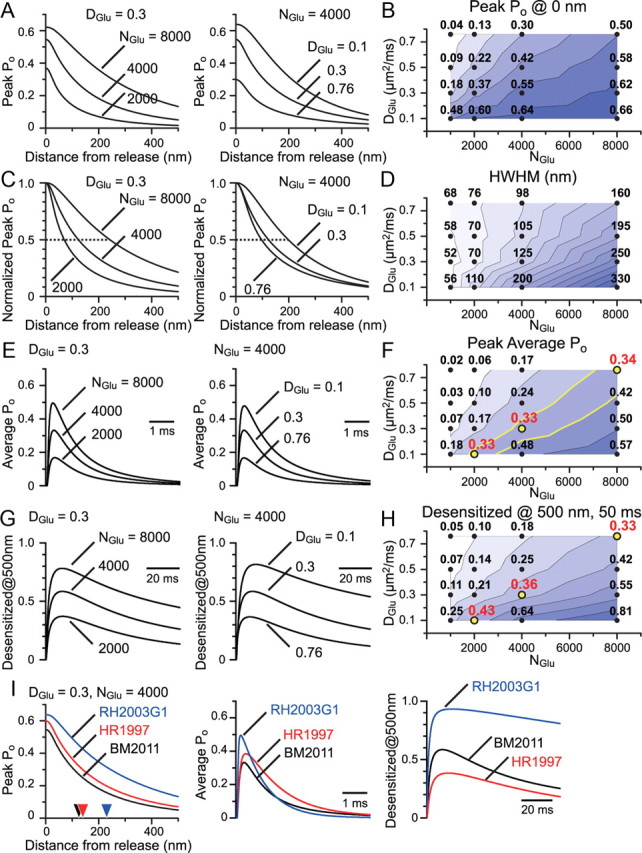
Evaluation of the possible NGlu and DGlu combinations and the use of other published models of AMPARs. A, Simulated AMPAR peak Po at each distance from the release site was plotted for various NGlu and DGlu combinations. B, Peak Po value at the release site for each combination tested was shown along with the calculated counter map. C, Peak Po curves shown in A were normalized at their peaks to show the difference in the effective range of glutamate action with different NGlu and DGlu combinations. D, Half-width at half-maximum (HWHM) of the peak Po curve was shown with a counter map. E, Using the average distribution of AMPARs in an RG synapse, as calculated in Figure 5G, the average Po transition time course was calculated. This is essentially the time course of the simulated quantal response. F, The peak of the average Po was plotted with a counter map. NGlu/DGlu combinations of 2000/0.1, 4000/0.3, and 8000/0.76 gave similar numbers for the peak average Po of ∼0.3, which matches the expectation from the experimental results. G, The transition time course of the fraction of desensitized AMPARs at 500 nm from the release site was plotted. H, The fraction of desensitized AMPARs at 500 nm and 50 ms from release was plotted with a counter map. Aforementioned combinations gave similar numbers for the desensitized fraction of ∼0.3–0.4. I, Glutamate transient calculated with NGlu/DGlu combinations of 4000/0.3 was used to drive several published AMPAR models (RH2003G1, GluR1 model from Robert and Howe, 2003; HR1997, Purkinje cell AMPAR model from Häusser and Roth, 1997; BM2011, our dLGN RC AMPAR model). Simulated peak Po at each distance from the release site (left, HWHM for each model was indicated with arrowheads), the average Po transition in a synapse (middle), and the transition of the fraction of the desensitized AMPARs at 500 nm from the release site (right) were plotted.
It has been reported that in some synapses with high Pr, multiple vesicles are simultaneously released (MVR) in close proximity within the same synapse upon presynaptic stimulation, producing a much higher peak concentration of glutamate than would be expected from single vesicular releases (Wadiche and Jahr, 2001). To investigate whether such MVR occurs at the RG synapse, the amount of block of the evoked EPSC by the low-affinity competitive antagonist γDGG (2 mm) was examined. γDGG block is expected to be higher for lower glutamate concentration transients and lower for higher glutamate transients because high glutamate concentration would compete out the antagonist during the postsynaptic current. We found that γDGG block was significantly less in our control conditions (2 mm [Ca2+]o) compared with the conditions where Pr was reduced by decreasing the [Ca2+]o (Fig. 1I,J). This result suggests MVR in the RG terminal, a finding contrasting with yet another sensory terminal, the olfactory nerve, where monovesicular release has been suggested despite the high Pr (≥0.8; Murphy et al., 2004). MVR was not apparent at 1 mm [Ca2+]o as reducing the [Ca2+]o further to 0.7 mm did not change the amount of γDGG block. Nevertheless, the fact that MVR could occur suggests that the RG synapse is not constrained to single vesicle release. It should be noted that such MVR could cause partial saturation of the AMPARs. The Pr and N estimates from the mean variance analysis could therefore be an overestimate and underestimate, respectively (Meyer et al., 2001).
Prolonged desensitization of AMPARs limits the recovery from paired-pulse depression
To understand the ability of this synapse to transfer rapid succession of signals, paired-pulse stimuli were applied (Fig. 2A). As expected from the high Pr, which would promptly deplete the readily releasable vesicles upon the first stimulus, the second EPSC was markedly depressed. Paired-pulse ratio (PPR) recovered with a double exponential function (τfast = 561 ms, 43%, τslow = 2713 ms). The late component, taking seconds to recover, is typically seen at synapses with high Pr and is considered to reflect the time course of refilling of readily releasable pool of synaptic vesicles (Silver et al., 1998). We focused on the initial component as this component seemed to be depressed more profoundly and longer than at synapses with even higher Pr such as the climbing fiber to Purkinje cell synapses (Silver et al., 1998). High Pr in conjunction with MVR could invite glutamate to linger in the synaptic cleft and/or promote spillover between synapses leading to desensitization of AMPARs. To examine whether desensitization plays a role in the initial phase of the PPR recovery, AMPAR desensitization blocker CTZ (100 μm) was applied. CTZ significantly relieved paired-pulse depression (PPD) for ISI of up to 500 ms (Fig. 2B), consistent with previous reports (Chen et al., 2002). Similar results were obtained from physiological temperature (PT) recordings (31–34°C) (see Fig. 11A). Such a long-lasting desensitization is rarely seen at most other synapses, and the effect of CTZ is normally limited to less than ∼30 ms (Trussell et al., 1993; Xu-Friedman and Regehr, 2003).
Although CTZ could nearly eliminate AMPAR desensitization, CTZ was also reported to increase the affinity of AMPARs (Dzubay and Jahr, 1999), leading the receptors closer to saturation. It is possible that the first EPSC becomes more susceptible to saturation than the second EPSC in the presence of CTZ, which could lead to less PPD. In other words, the relief of PPD in the presence of CTZ could be due to the difference in the saturation level of AMPARs between the first and the second EPSC, and not due to the desensitization of AMPARs. To rule out this possibility as the cause of the change in the PPR, γDGG was coapplied with CTZ to prevent saturation (Meyer et al., 2001). Even in this condition, the drugs were able to relieve depression (Fig. 2C). As CTZ was also reported to change Pr through a presynaptic mechanism (Diamond and Jahr, 1995), EPSCs via NMDAR activation were examined, but no effect of CTZ on the PPD was found (data not shown). A similar trend was observed with another desensitization blocker, aniracetam (Fig. 2D; 4–5 mm). As with CTZ, aniracetam does affect the deactivation rates and the affinity of the AMPARs to glutamate (Partin et al., 1996), but nonspecific presynaptic effects have not been reported (Lawrence et al., 2003).
We also devised an alternative method to save the receptors from desensitization through the usage of the low-affinity antagonist γDGG alone (Wong et al., 2003; Chanda and Xu-Friedman, 2010). To understand the possible mechanism underlying this effect, our AMPAR kinetic model, shown later in this article (see Fig. 4A), was combined with low-affinity antagonist-binding states (adopted from Diamond and Jahr, 1997). Using such a model, we realized that a portion of AMPARs pre-equilibrated with the antagonist would rapidly enter a state with both glutamate and antagonist bound upon short application of high concentrations of glutamate. This population is reserved from entering the desensitized states and would promptly be available for the next instance of a glutamate transient (data not shown). As expected from such a model, PPD was relieved by application of γDGG (Fig. 2E). Interestingly, in synapses that undergo MVR, the first EPSC is usually blocked less by γDGG compared with the second EPSC because less vesicles are released per synapse by the second stimulation (Wadiche and Jahr, 2001). However, our findings did not conform to this expectation as the second EPSC was actually blocked less. Opposing effects are expected with γDGG application under MVR and long-lasting AMPAR desensitizing conditions, but the fact that the effect from desensitization won signifies the large impact of AMPAR desensitization on short-term synaptic plasticity in this particular synapse. A low concentration of noncompetitive antagonist GYKI (8 μm) would keep a certain population of AMPARs unresponsive and reduce all EPSCs to the same extent. The PPR and the recovery time course were unaffected by GYKI despite the fact that the first EPSC was reduced to a similar extent as with γDGG (Fig. 2F). This result also suggests that the possible voltage escape due to large synaptic conductance did not affect the PPR measurements in this study. Although an important role of desensitization of AMPARs in the RG synapse was suggested previously by using CTZ (Chen et al., 2002), we used various pharmacological means to compensate the insufficiency of CTZ as a pure desensitization blocker that could undermine the importance of desensitization. We conclude that the AMPARs are indeed desensitized for an unusually prolonged time after the first stimulus and recovery from depression is profoundly limited by this desensitization at this synapse.
Ca2+-permeable AMPARs contribute to the desensitization
We next sought to identify the mechanism underlying the long-lasting depression due to desensitization. The degree of desensitization and the recovery time course from desensitization depend on the subunit composition of AMPARs. In heterologous expression studies (Partin et al., 1996; Sekiguchi et al., 2002; Robert and Howe, 2003), GluR3-flop and GluR1 homomeric receptors, both of which lack a GluR2 subunit and are Ca2+ permeable (Hollmann et al., 1991), have been shown to display slow recovery from desensitization (τ = 55 ms and 111–147 ms, respectively). We first found that 46.6 ± 3.1% (Fig. 3A) of the EPSC was blocked with NASPM (50 μm), a specific antagonist of Ca2+-permeable AMPARs (CP-AMPARs) (Koike et al., 1997). CP-AMPARs were also blocked intracellularly by introducing spermine (100 μm) through the patch pipette solution. This block occurred only at positive potentials, thereby yielding the current–voltage relationship (I–V) to rectify (Fig. 3B,C).
To test whether the EPSC mediated by CP- and Ca2+-impermeable AMPARs recovers from depression differently, paired-pulse stimuli were given in the absence and presence of NASPM (Fig. 3D). We discovered that the EPSCs were significantly less depressed in NASPM at all time points studied, suggesting that the properties of the CP-AMPARs were partially responsible for the long-lasting depression. Bath-applied NASPM had no effect on the PPR and recovery time course of NMDAR EPSCs, ruling out possible presynaptic effects (data not shown). In addition, PPR recovery was examined at negative and positive potentials in the presence of internal spermine, in which case, only the recorded postsynaptic RC would be affected (Fig. 3E). PPR was less depressed at positive potentials where CP-AMPARs would be partially blocked, further supporting the idea that these receptors were accountable for the long-lasting depression due to desensitization.
We next examined the involvement of GluR3-flop homomeric channels in the long-lasting depression using PEPA (Fig. 3F; 200 μm). PEPA, an allosteric potentiator of AMPARs, accelerates the recovery from desensitization selectively in GluR3-flop AMPARs (Sekiguchi et al., 2002). PEPA showed no effect on the recovery time course of PPR or the amount of PPD of the RG–RC EPSC, suggesting a role against GluR3-flop in mediating the EPSC. It has also been reported that, in many subunit compositions studied, PEPA reduced the extent of desensitization to prolonged glutamate application without affecting the rate of deactivation (Sekiguchi et al., 2002). The fraction of the slow time constant of the EPSC decay was significantly increased with PEPA. If the glutamate transient is short enough, the EPSC decay should be primarily dictated by the deactivation kinetics. The above result suggests that the glutamate transient lasted long enough to drive receptors into desensitization (Fig. 3G).
Since no specific antagonist of GluR1 homomeric receptors is currently available, we made use of the GluR1 KO mice (Fig. 3H; note that all other experiments in this study used Long–Evans rats). A significant difference in PPR was observed at ISIs of 150 and 500 ms between GluR1 KO and wild-type mice. To compare the degree of desensitization between the animals, PPR in control was divided by the PPR in the presence of CTZ. Recordings from GluR1 KO showed that the recovery from desensitization was accelerated at an ISI of 150–500 ms. These results suggest that, even though the mRNAs of all subunits are expressed in RCs (Allen Brain Atlas; Lein et al., 2007), the GluR1 subunit is partially responsible for the particularly long-lasting desensitization.
AMPAR kinetic model
To examine the kinetic properties of AMPARs that could underlie the long-lasting depression of RG–RC EPSCs, outside-out patches were excised from the somata of the RCs. Patches responsive to glutamate were extremely rare, as expected from the low expression of extrasynaptic AMPARs (Fig. 5E). However, we were able to record from patches using P5–P11 animals. Rapid application of 10 mm glutamate caused fast desensitization of AMPARs (Fig. 4B). Slow recovery from desensitization was observed when applying paired pulses of glutamate with varying intervals (Fig. 4C,D). This rapid desensitization and slow recovery from desensitization match well with the kinetics of GluR1 homomeric receptors in heterologous expression systems (Partin et al., 1996; Robert and Howe, 2003). As these outside-out patches were not obtained directly from the synaptic membrane, it is possible that AMPAR channel properties underlying synaptic responses could be influenced by accessory proteins and interactions in synapses. It should also be noted that subunit composition may change with age. However, as AMPAR responses recorded in patches (Fig. 4) and evoked EPSCs (Fig. 3) both showed prolonged desensitization lasting for ∼500 ms, we assumed that synaptic AMPARs responsible for the long-lasting PPD of EPSCs have similar properties. Thus, the kinetic model derived from the patch experiments (Fig. 4A; see legend for rate constants as well as for comparison of desensitization rates) was used for further analysis and simulations. Simulations based on other published kinetic models were also performed to examine the extent of the validity of the current model (Fig. 6).
Could the kinetic properties of the expressed AMPARs fully account for the PPD of EPSCs? If the first and second instances of glutamate release occur in different synapses that are totally isolated from each other, then the PPD would depend only on the presynaptic reduction in release. For the desensitization of AMPARs to have any effect on the PPD, the two releases must affect the same population of AMPARs. This can be accomplished if the two releases occur in the same synapses and/or if intersynaptic spillover happens. To assess the involvement of desensitization caused by spillover in PPD, we next needed to perform simulations of glutamate diffusion and AMPAR activation. For the simulation, a Markov model of AMPARs (Fig. 4A) was adopted from a previous study (Häusser and Roth, 1997), and the rate constants were modified so as to match the recorded patch responses (Fig. 4B–D).
AMPAR distribution
To understand how the liberated glutamate from the presynaptic terminal diffuses into the extracellular space and activates the postsynaptic AMPARs, we first characterized the 2D distribution of AMPARs on the postsynaptic membrane of the RCs using SDS-digested freeze–fracture replica immunolabeling (SDS-FRL). A cluster of IMPs on the E-face of the plasma membrane represents the postsynaptic membrane specialization of glutamatergic synapses (Harris and Landis, 1986). Virtually all E-face IMP clusters were labeled with immunogold particles using a panAMPAR (GluR1–4) antibody (Nusser et al., 1998), which has almost an one-to-one detection sensitivity in SDS-FRL (Tanaka et al., 2005) (Fig. 5A,I; see Fig. 7A). We considered these IMP clusters as RG synapses when such a profile was accompanied by a P-face of presynaptic profiles that was labeled for vGluT2, a marker of RG fibers (Tarusawa et al., 2009) (Fig. 5I). The surface area of the IMP clusters and the number of AMPAR immunoparticles varied largely among synapses but were positively correlated with each other (p < 0.001, Spearman's rank-order test, n = 63) (Fig. 5B), indicating a relatively constant density of AMPARs. The average area of the IMP clusters, and the number and density of AMPAR immunoparticles were 0.033 ± 0.003 μm2, 17.2 ± 2.4, and 561 ± 25/μm2, respectively (Fig. 5C, n = 3 animals). Intrasynaptic density of AMPARs relative to the border of demarcation was also largely homogeneous with the density only in the most peripheral of the demarcation being slightly less than the center (Fig. 5D,E; see Materials and Methods). Very few extrasynaptic immunoparticles were found (1.8 ± 0.4/μm2, n = 76 profiles), implying that if glutamate spills over from the synapse, it would have to reach a neighboring synapse to have a significant extrasynaptic effect. As described later, the NND from a synapse to a neighboring synapse was measured as 569 nm on average in replica samples (Fig. 5J). We took this as a radius, calculated the area of the circle, and subtracted the average IMP cluster area. This area was multiplied by the extrasynaptic immunoparticle density, and we obtained a value of 1.8 immunoparticles. This suggests that glutamate released from a synapse would encounter only 1.8 AMPARs in the extrasynaptic region before it reaches a neighboring synapse. Approximately 10 times more immunoparticles were found in the neighboring synapse, suggesting a larger role of the spillover to the AMPARs on the neighboring synapse compared with those in the extrasynaptic region.
Simulation of AMPAR response to quantal release
With both the kinetic properties (Fig. 4) and the distribution (Fig. 5) of AMPARs known, AMPAR responses to quantal release of glutamate were simulated. The following simulations were done to understand how the released glutamate affects the activation and the desensitization of intra- and intersynaptically distributed AMPARs.
A spatiotemporal profile of glutamate transient was calculated supposing that the release occurred at the center of gravity of the IMP cluster demarcation and the diffusion was limited to lateral directions (Fig. 5F). The NGlu was set to 4000, and the DGlu was set to 0.3 μm2/ms. The validity of the selection of these parameters is discussed in the next section. The profile of glutamate concentration depends on the distance from the release site. Therefore, the number of immunoparticles was counted and averaged at various distances from the center of the IMP cluster to obtain an average view of the AMPAR distribution in each synapse (Fig. 5G). Our AMPAR kinetic model (Fig. 4) was run using the calculated glutamate transient, and the attained Po of an individual AMPAR was plotted against the distance from the center of the release site (Fig. 5G, red line). These plots suggest that the effective range of AMPAR activation from the release site (half-width at half-maximum distance of peak Po = 125 nm; see previous simulations for comparison) (Barbour, 2001; Franks et al., 2003; Raghavachari and Lisman, 2004; Tarusawa et al., 2009) matches well with the extent of AMPAR distribution (50% cumulative frequency of particles from center = 90 nm). The number of opened AMPARs at each distance was essentially the multiplication of the number of particles and the Po (Fig. 5H). The sum of all AMPAR behavior is the simulated average quantal response (Fig. 5H, inset).
To estimate the effect of glutamate spillover to the neighboring synapses, the NNDs of the center of IMP clusters were measured on wide fractured planes of the RC dendrite (Fig. 5I; see Fig. 7A). The average NND was 569 nm (n = 186) (Fig. 5J). Simulations using the kinetic model suggest that the peak Po would reduce to a very small value at this distance. Nevertheless, the small glutamate transient was sufficient in driving a sizable portion of AMPARs into the desensitized states (Fig. 5K). It has been shown that long prepulses of low glutamate concentration transients can lead the AMPARs to “equilibrium desensitization” (Colquhoun et al., 1992), and a previous study using a different kinetic model of GluR1 with four glutamate binding sites has also suggested that such low transients can drive these receptors into desensitization before entering open states (Robert and Howe, 2003).
Validity of the NGlu and DGlu parameters used in calculating glutamate transient
Parameters NGlu and DGlu, used in calculating the glutamate transient in the above simulations, are experimentally difficult to determine, and a general consensus on a definite number has not been reached so far (Nielsen et al., 2004). Here, we verified the validity of our selection of the NGlu and DGlu, narrowed the possible range of NGlu and DGlu combinations, and assessed the effects of different combinations on the expected amount of glutamate spillover and on the magnitude of activation and desensitization of AMPARs in the neighboring synapses.
First, experimentally derived quantal response size was compared with the simulated one to assess the validity of our NGlu and DGlu selection. Our mean variance analysis of the evoked EPSCs estimated that the amplitude of a quantal response was 11.3 pA on average (Fig. 1H) (Vh = −86 mV). Assuming the reversal potential of 0 mV, this would correspond to a synaptic conductance of 131 pS. Using fluctuation analysis, AMPARs on Purkinje cells have been estimated to have a single-channel conductance of 14.3 pS (Matsui et al., 2005). The single-channel conductance of CP-AMPARs generally has a higher estimate of ∼22.6 pS (Koh et al., 1995). Using these conductance values, the numbers of open AMPARs at the peak of a quantal response were estimated to be 9.2 and 5.8, respectively. Since approximately half of the EPSC is mediated by CP-AMPARs (Fig. 3A), the actual value of the open AMPAR number would fall somewhere in between these two values. It has been determined previously that our SDS-FRL has high sensitivity of immunolabeling with nearly one-to-one detection for AMPARs (Tanaka et al., 2005; Tarusawa et al., 2009). Using the AMPAR immunoparticle distribution revealed by this method, we simulated the AMPAR response to glutamate release. When NGlu was set to 4000 and DGlu was set to 0.3 μm2/ms, the simulated number of peak open AMPARs in an average AMPAR distribution synapse became 7.3, as shown in the inset of Figure 5H. Therefore, when using this NGlu and DGlu combination, the simulation results fit well with the electrophysiological estimates of the quantal response size.
Another support for the validity of this NGlu and DGlu combination comes from the estimates of the peak of the average Po of quantal responses. We found on average 22 AMPAR immunoparticles in IMP clusters for the replica samples collected from an animal used in our simulations (Fig. 5C). As the number of peak open AMPARs was simulated as 7.3, this would suggest that the peak average Po in a synapse was 0.33. This value is consistent with the following previous reports. Using a low-affinity antagonist, γDGG, Wadiche and Jahr (2001) estimated that the peak average Po at a synapse would be 0.27–0.39 for monovesicular releases (estimates from climbing fiber to Purkinje cell synapse). Another study estimated, by loading the presynaptic vesicles with an excess amount of glutamate, that a single quantal release in control does not saturate the AMPARs in the calyx of Held synapse and the average Po is expected to be at least 1.5 times below saturation (Ishikawa et al., 2002; Yamashita et al., 2009). To simulate the excess loading of glutamate, we chose twice the amount of glutamate, NGlu = 8000 and DGlu = 0.3 in our simulation, which yielded many of the AMPARs within the synapse close to saturation. Our estimate of the peak average Po under this condition became 0.50. If we assume that this is the saturating level, 1.5 times below the saturation would be 0.33. Although the saturating level above was chosen arbitrarily, we could at least say that our normal NGlu and DGlu combination yielded a peak average Po value substantially below saturation. Together, simulations using NGlu = 4000 and DGlu = 0.3 match well with previous estimates of peak average Po made with completely different methods.
When we explored the parameter space (Fig. 6), we noticed that other combinations of NGlu and DGlu could also produce similar peak average Po values, such as combinations of 2000 and 0.1 and 8000 and 0.76, respectively (peak average Po would be 0.33 and 0.34, respectively) (Fig. 6F). This was not unexpected as Po would be higher when peak glutamate concentration was high (NGlu is high) or when glutamate diffusion was slow (DGlu is small). We also noticed that NGlu and DGlu combinations that produced a peak average Po that was close to the experimentally expected value (∼0.3) also produced similar amounts of desensitization at several hundred nanometers from the release site (desensitized fraction at 500 nm and 50 ms from release; 0.43, 0.36, and 0.33 for combinations of 2000 and 0.1, 4000 and 0.3, and 8000 and 0.76, respectively) (Fig. 6H). Therefore, we conclude that when the glutamate concentration in vesicles is high enough or when the glutamate diffusion is slow enough to produce a peak average Po of ∼0.3 at individual synapses, glutamate spillover would be sufficient to drive a sizable fraction of AMPARs into desensitization at neighboring synapses.
Simulation with other AMPAR kinetic models
Here we address the validity of our kinetic model (BM2011 model; Fig. 4A) by comparison with other previously established models. As AMPARs function as a tetramer, kinetic models with binding of four glutamate and multiple conductance levels have been suggested (Smith et al., 2000). The macroscopic current simulated in our study would probably not be affected much by the complexity of such a kinetic model. Nevertheless, we made use of a well defined kinetic model of homomeric GluR1 from heterologous expression system (RH2003G1 model; Robert and Howe, 2003) to simulate the response to glutamate release onto the average AMPAR distribution synapse. As multiple conductance states were supposed in this model, the conductance created by each simulation relative to the maximum possible conductance (i.e., when all channels occupy the highest conductance state) was tentatively expressed as Po for comparison of results from other models. In addition, simulations using an AMPAR model derived from GluR2/3 subunit combination found in Purkinje cell somata were examined (HR1997 model) (Häusser and Roth, 1997). The peak of the average Po in an average AMPAR distribution synapse was found to be similar, with values of 0.33, 0.50, and 0.38 for models BM2011, RH2003G1, and HR1997, respectively (Fig. 6I). The half-width at half-maximum distances of peak Po were 125, 230, and 140 nm, respectively, for models BM2011, RH2003G1, and HR1997 (Fig. 6I). The desensitized fraction at 500 nm and 50 ms from release was large for the RH2003G1 model and small for the HR1997 model, and the results from the BM2011 model were in between. The values were 0.36, 0.88, and 0.27, respectively, for models BM2011, RH2003G1, and HR1997 (Fig. 6I). This fits with our idea that GluR1 subunit is partly responsible for the prolonged desensitization (Fig. 3H). However, Ca2+-impermeable AMPARs, which occupied approximately half of the EPSC (Fig. 3A), were not exempt from desensitization, because significant PPD, likely due to desensitization, was still observed in the presence of NASPM (Fig. 3D). These results suggest that the 2D restriction of transmitter diffusion accounts for the profound desensitization of both types of AMPARs (for differential impact of 2D and 3D transmitter diffusion on AMPAR activation, see Cathala et al., 2005).
Morphological structure of the RG synapse
We showed earlier that SDS-FRL revealed the accurate 2D distribution of AMPARs along the plasma membrane and the spatial relationship between synapses that was observed on the replica material on occasion. However, finding multiple synapses on the same fractured plane relies on chance (Fig. 7A). To understand how synapses are distributed in RG bouton-to-RC dendrite contacts, we performed 3D reconstruction of the contacts using serial ultrathin section electron microscopy (Fig. 7B,C,E). As reported previously (Brauer et al., 1979), glomeruli structure formed by RG–RC contacts could be categorized into “simple” and “complex” types, with the former having a large single RG bouton contacting with a single dendrite and the latter having multiple smaller boutons contacting a dendrite in a dense arrangement. In this study, seven simple-type RG glomeruli were reconstructed (Fig. 7C). A single contact contained on average 27 ± 2.7 synapses (Fig. 7D). NND between synapses along the surface of the dendrite (see Materials and Methods) was calculated for the sample RG glomerulus shown in Figure 7C and the average of the “surface NND” was 703 ± 59 nm (Fig. 7D). The average numbers of neighboring synapses within a surface radius of 700 and 1400 nm were 1 and 6, respectively. Analysis of a complex-type RG glomerulus showed similar surface NND (724 ± 109 nm) but fewer numbers of total synapses per contact (6.4 ± 1.1 synapses, n = 9 contact) and fewer numbers of neighboring synapses (Fig. 7E,F). Diversity of the number of synapses per RG–RC contact has been described previously for the cat as well (Hamos et al., 1987). In summary, even though the total number of synapses per contact was variable, the surface NND of synapses and the number of close neighbors were similar in most contacts, and we have identified the close arrangement of multiple synapses within the range for possible interactions mediated by glutamate spillover.
Spillover between synapses promotes AMPAR desensitization
To understand whether the distance between synapses is close enough to allow intersynaptic spillover, simulations of glutamate diffusion and AMPAR activation were performed. Based on the average distribution of AMPARs from the center of the synapse (Fig. 5G), we first considered two instances of release onto the same location in a synapse with varying time intervals (Fig. 8A). Simulated average Po followed a slow recovery time course as expected from the outside-out patch experiment. Next, an ellipse was fitted to a sample synapse demarcation and the effect of release at varying distances from the center along the major axis was evaluated (Fig. 8B). Neither the average Po nor the amount of receptors driven into the desensitized state was affected much by the small deviation of the release location within the synapse. The effect of two instances of release with the first in the center and the next at an offset location was examined, but showed little difference from the case where two releases occurred in the same location (Fig. 8C). These results indicate that the response size and the degree of desensitization are hardly affected by the release location within a synapse, as has been suggested in our previous study (Tarusawa et al., 2009). Lateral diffusion of AMPARs along the plasma membrane has been suggested to allow fast exchange of the desensitized receptors with ones previously unexposed to glutamate, which could facilitate the recovery from depression (Heine et al., 2008). However, our simulations show that, if the lateral diffusion of AMPARs occurs only within the synapse, then, as most AMPARs would be desensitized to similar extent (Fig. 8C), movement of the AMPAR location would have little effect. Although SDS-FRL cannot capture the dynamic movement of the AMPARs, the distribution of the molecules revealed by this method can be considered as a freeze-frame view of the AMPAR distribution at a given time point. As extrasynaptic AMPARs were seldom found on RC dendrites (Fig. 5E), we assume that lateral diffusion of AMPARs may not be as quite dynamic as that in the hippocampal preparations. Furthermore, even if the extrasynaptic AMPARs that were previously unexposed to glutamate were provided to the synapses by lateral diffusion, such a population would seem too small to substantially affect the AMPAR-mediated responses in RG–RC synapses.
Figure 8.
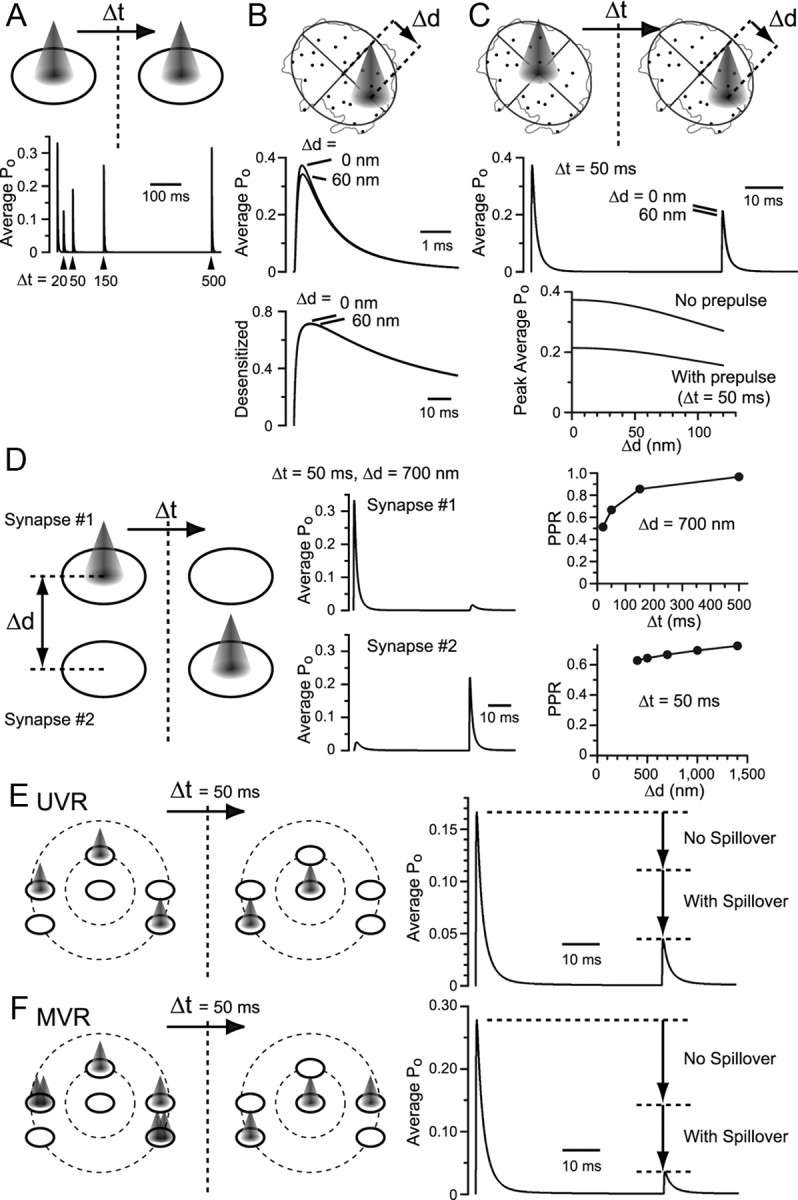
Simulations evaluating the extent of spillover and its effect on the AMPAR activation. A, Two instances of glutamate release were supposed with various time intervals (Δt) in an average synapse with AMPAR distribution calculated in Figure 5G. B, An ellipse was fitted to the demarcation of an example synapse shown in Figure 5A, and displaced (Δd) glutamate release was supposed. C, A prepulse of glutamate in the center and a test pulse in the displaced location of varying distances. D, Two synapses with average AMPAR distribution were supposed with the intersynaptic distance (Δd) and interval (Δt) between the two releases varied. History of release at the neighboring synapse reduced the average Po in response to a subsequent direct release. The sum of the response from the two synapses was calculated, and the PPR was plotted against varying Δt and Δd. E, Multiple synapses with average AMPAR distribution were placed in a realistic manner. From the center synapse, one neighbor was placed at 700 nm distance, and five neighbors within 1400 nm. Two instances of UVRs separated by Δt = 50 ms were supposed at the specified synapses. Average Po of AMPARs in all six synapses was calculated. The average Po to the second instance of release was reduced compared with the case where all synapses operated independently without intersynaptic spillover. F, MVR was considered in a subset of synapses. The average Po in response to the second instance of release was further reduced.
Next, a situation was supposed where the first release occurred at one average synapse and the second release occurred at a neighboring average synapse, with varying distances between these synapses and varying time intervals between the releases (Fig. 8D). Spillover from one synapse to the other caused little rise in the peak of the average Po in the neighboring synapse. Thus, spillover does not add much to the synaptic conductance amplitude. However, if an instance of spillover were preceded, such a history of release in the neighboring synapse would reduce the amplitude of the peak average Po in response to a subsequent direct release (Fig. 8D).
To simulate a situation of multiple neighboring synapses, six synapses were distributed in a simplified manner according to the morphological characterization, as described in Figure 7, D and F. Releases were supposed, as illustrated in Figure 8E, without two releases occurring successively in the same synapse. The average Po of all synapses was calculated in response to two instances of release. Note that the Po of AMPARs located in synapses where no direct release has occurred is also included for calculating the average; thus, the average Po value is smaller than that calculated for univesicular glutamate release (UVR) at single synapses (Fig. 6E,F). Compared with the simulation where all the synapses operated completely independently, spillover between synapses dramatically reduced the average Po (reduction due to spillover = 1 − PPR with spillover/PPR without spillover = 0.60). As MVR is anticipated at RG synapses (Fig. 1I,J), the response to simultaneous releases of either one or two vesicles per synapse was simulated (Fig. 8F). As expected, further PPD was observed (reduction due to spillover = 0.75), demonstrating that MVR enhances AMPAR desensitization. These findings indicate that multiple synapses are arranged close enough to allow intersynaptic spillover to occur, which greatly contributes to the short-term plasticity of RG synapses.
Experimental evidence for spillover
We next looked for experimental support for the existence of intersynaptic spillover. The amount of spillover would depend on how many incidents of release occur in the neighboring synapses, reflected by the Pr. Simulations suggest that spillover does not create much of a synaptic conductance that contributes to the peak (Figs. 5K, 8D). However, as the synaptic conductance created in the neighboring synapse will be slow to rise, the decay phase of the total EPSC will probably be affected by spillover. Therefore, EPSCs were recorded at low and high extracellular Ca2+ concentrations in the presence of aniracetam and the decay time course was compared (Fig. 9A–C). As expected, EPSC decayed slower in high Pr conditions (p < 0.01, n = 5), providing support for the presence of spillover.
Upon close examination of the morphological structure of the RG synapse, we found that two presynaptic boutons from the same RG fiber could make contact with the same portion of the postsynaptic dendrite (Fig. 9D). As these boutons were separated by glial ensheathment, interbouton spillover would be suppressed by glutamate transporters expressed on glial cells. If blocking glial glutamate transporters enhances the spillover effect, it would suggest that glutamate can travel interbouton distances when the diffusion is not hindered by the transporters. If glutamate can travel that far, intersynaptic spillover within a bouton, which is not interfered with transporters, should occur.
To evaluate the distribution of glutamate transporters, we used the highly sensitive SDS-FRL technique for the detection of glial glutamate transporters GLT-1 and GLAST. It is unclear whether the labeling efficiency of the antibodies used would reach as high as the one-to-one efficiency found for the panAMPAR antibody (the identity, source, and characterization of the antibodies are summarized in Table 1), but we detected no significant signal either in the presynaptic or the postsynaptic membrane compartments. However, labeling for both transporters was clearly seen on the P-face of the glial membrane (Fig. 9E).
Next, we tested whether blocking glutamate transporters, which would reduce the glutamate buffering capacity in the extracellular space, would affect the PPR. TBOA, a broad-spectrum inhibitor for glutamate transporters caused further PPD suggesting that the enhanced spillover by blocking the transporters caused further desensitization of AMPARs (Fig. 9F). Similar results were obtained from physiological temperature recordings (see Fig. 11B). A selective GLT-1 inhibitor, DHK, had a similar effect (Fig. 9G), as well as another inhibitor, (2S,3S)-3-[3-(4-methoxybenzoylamino)benzyloxy]aspartate (PMB-TBOA), which was shown to inhibit preferentially glial over neuronal glutamate transporters (Shimamoto et al., 2004; Takatsuru et al., 2006; data not shown). Interestingly, the GLAST-specific inhibitor UCPH-101 (Erichsen et al., 2010) had no effect on the PPR (Fig. 9H). These results are consistent with the idea that glial transporters, especially GLT-1, located only on the outskirts of the broad RG–RC contact, have a small but significant effect on the spillover, thereby manifesting the occurrence of intersynaptic spillover within a contact. It is also possible that GLT-1 inhibition could have prolonged the glutamate transient at the synapse rather than promoting spillover between boutons. This would mean that glutamate transporters located only on the outskirts of the contact are activated by the released glutamate and the effect of the impaired uptake propagates back to the synapse where release has occurred. Glutamate transporters are activated by low-micromolar concentrations of glutamate (Bergles et al., 1997; Danbolt, 2001), and such concentrations have been shown to be sufficient in driving the highly desensitizing subunit GluR1 into desensitization (Robert and Howe, 2003). Therefore, even under this scenario, it is likely that the released glutamate can travel at least up to the edge of the contact at a concentration that could promote AMPAR desensitization.
Physiological significance of short-term plasticity
We sought to understand how the short-term plasticity created by high Pr, spillover, and desensitization of AMPARs relates to the postsynaptic spiking. EPSCs were first recorded, and the amplifier was switched to current-clamp mode to record voltage responses to the stimulation of a single RG fiber. Experiments in this section were done in the absence of d-AP5, unless otherwise noted.
As indicated in Figure 1H, the EPSC amplitude varied largely among RG fibers. In a previous light microscopy analysis using cats, the number of contacts made on each LGN relay cell by each RG axon was also found to be variable, ranging from 4 to 47, and two types of patterns with “sparse” (<20) and “frequent” (>30) innervations were observed, although it was noted that the distribution of the number of contacts may fall along a continuum (see also Hamos et al., 1987; Robson, 1993). Therefore, we arbitrarily grouped RG input as being “small” and “large” based on the amplitude of the EPSC being <500 or >500 pA, respectively. In either case, postsynaptic spiking was often achieved upon stimulation of a single RG fiber if the membrane potential was kept at −40 to −60 mV (“shallow potentials”) (Fig. 10A,B). These shallow potentials have been referred to as the “tonic mode,” and the more hyperpolarized potentials (−70 to −90 mV; “deep potentials”) as the “burst mode” (Turner et al., 1994; Blitz and Regehr, 2003). At deep potentials, EPSPs elicited by the small fiber input were unable to generate any spikes, whereas the larger fiber input generated one or more spikes (Fig. 10A–C).
We next investigated the RC's response to paired-pulse stimuli. Small or large fiber input produced no difference in the EPSC PPR, suggesting that the Pr and the amount of AMPAR desensitization were not different between these fibers (Fig. 10C). At shallow potentials, small fiber input often failed to generate a spike upon the second stimulation at short ISIs. Its spiking recovery followed a time course similar to that of the EPSC PPR (Fig. 2A), suggesting a rather simple proportional relationship between the synaptic conductance and the spiking outcome. However, for the large fiber input, the reduced synaptic conductance by the second stimulus was still sufficient to generate a spike at shallow potentials. And if the ISI was short enough, the residual EPSP likely helped generate additional spikes. At deep potentials, only the large fiber input generated spikes, and in these cells, two phases of spiking recovery were apparent: the effect of residual EPSP helping spike generation was lost after 50 ms, and longer time was required for the total recovery (Fig. 10C). Thus, the temporal characteristics of signal transfer differed completely depending on the original synaptic conductance size.
Next, the RC's response to trains of RG fiber stimuli was examined. As expected from the high Pr and the profound desensitization of receptors, AMPAR EPSC was reduced significantly during the train (Fig. 10D). However, as NMDARs respond slowly to glutamate release, NMDAR activation was summated. Nevertheless, at deep potentials, the activated NMDARs did not produce enough drive for repetitive spiking (Fig. 10D, second row). At shallow potentials, repetitive spiking was achieved (Fig. 10D, third row), which was lost when applying d-AP5 (Fig. 10D, fourth row). These results suggest that NMDARs acted to compensate for the loss of AMPAR activation resulting from desensitization (Matsui et al., 1998). However, postsynaptic spike timing was no longer time locked to the presynaptic stimulation by the end of the train (Fig. 10F,G) which demonstrated that the loss of sufficient AMPAR activation resulted in the loss of temporal precision of signal transfer and the activation of the slow NMDARs could not act to maintain the precision.
As temperature can affect various components of synaptic transmission, we repeated several key experiments, this time with the bath solution set at near PT (31–34°C). We found that the PPR recovered faster than at RT. Nevertheless, application of CTZ still caused a significant difference in the PPR at ISIs of up to 150 ms (Fig. 11A). This shows that prolonged AMPAR desensitization is apparent also at PT. It has been reported that extracellular Ca2+ concentration may not be as high as 2.0 mm, as used in most acute slice experiments, but closer to 1.5 mm in vivo. The mean variance analysis shown in Figure 1G indicated Pr values of 0.70 ± 0.03 and 0.37 ± 0.05 in 2.0 and 1.0 mm Ca2+, respectively (n = 6 cells). To evaluate the amount of PPD at 1.5 mm Ca2+ at PT, PPR measurements were done in this condition. Pronounced PPD was still observed in this situation, and CTZ was able to relieve PPD at ISIs of up to 150 ms (PPR: 0.36 ± 0.03 and 0.75 ± 0.03 in control and in CTZ at ISI = 20 ms; 0.59 ± 0.02 and 0.76 ± 0.02 at ISI = 150 ms, respectively; n = 11), suggesting that Pr was still high enough to cause spillover and desensitization in this condition. As the activity of glutamate transporters are also affected largely by temperature, the effect of glutamate transporter blockade by TBOA of the RG–RC EPSC was examined at PT. PPR was depressed further by TBOA, confirming that glutamate uptake has a small but significant effect of preventing spillover (Fig. 11B). Current-clamp recordings of the spiking responses of RCs in response to a train stimulation given to an RG fiber at PT was also examined (Fig. 11C–F). Spiking responses to train stimuli were also essentially similar to those found at RT with bursts occurring only at the onset of the train at deep potentials and multiple spikes with increasing jitter occurring later in the train at shallow potentials. A similar trend was also confirmed in 1.5 mm Ca2+ at PT [1.0 ± 0.4 (range 0.0–5.5), 11.9 ± 2.3 (range 4.3–30.5), 3.9 ± 1.0 (range 1.0–10.0) spikes per 10 stimuli (20 Hz) at deep, shallow, and shallow potentials plus d-AP5, respectively; jitter (±SD) of spike delay after the first stimulus = 0.3 ± 0.2 ms, after the 10th stimulus = 5.9 ± 2.9 ms; n = 13].
We conclude that multiple mechanisms underlie the signal filtering realized at this multisynapse contact. The micron-order structure plays an essential role in short-term plasticity of AMPAR-mediated synaptic conductance, and other key players, such as the absolute amplitude of the synaptic conductance, the postsynaptic resting potential, and the activation of the slow NMDARs, all participate in shaping the character of signal transfer through the RG synapse.
Discussion
We attempted to understand the processes underlying synaptic responses by directly correlating electrophysiological recordings with ultrastructural characterization obtained by SDS-FRL and 3D reconstruction from ultrathin sections. Our previous article (Tarusawa et al., 2009) suggested that small differences in 2D intrasynaptic distribution of receptors produced little change in the individual quantal responses and that the size of the response is dictated primarily by the number of receptors expressed. This is because the effective range of glutamate action on the Po of these receptors corresponds to an average synapse area (Fig. 5E) (for an alternative view, see Raghavachari and Lisman, 2004). In this study, we focused on the intersynaptic spillover occurring in the range of a few hundred nanometers to a couple of micrometers. Because of the broad contact area of the RG terminal without much intervention of glial processes, diffusion of glutamate in the extracellular space is restricted to two dimensions for some distances, and spillover of glutamate to the neighboring synapse likely occurs at sufficient concentration and duration to promote desensitization of AMPARs. In addition to the close morphological arrangement of synapses, presynaptic factors, such as high Pr and MVR, and postsynaptic factors, such as slow recovery kinetics from desensitization of the AMPARs and the presence of NMDARs, all contribute to the degradation of high temporal precision of signals created at the retinal ganglion cells. Instinctively, contacts with multiple release sites with high Pr and MVR may seem to ensure reliable transmission, as has been suggested at thalamic afferent synapses (Bagnall et al., 2011). It is true that having multiple synapses within a close range would guarantee the release of at least a vesicle at such a contact with almost no failure. However, as for producing reliable current input for ensuring high-fidelity of postsynaptic spiking corresponding to presynaptic spiking at high frequencies, this situation is disadvantageous. If the number of synapses that one fiber can create on a postsynaptic cell is limited, then high Pr would be necessary to produce a large enough current to drive spiking. At the RG synapse, large current input seemed to be required to produce any spike if the postsynaptic RC was resting at deep potentials, and a high Pr at this synapse is therefore favorable, but at the expense of high-fidelity spiking.
The calyx of Held, which is also characterized by a broad multisynapse contact, is equipped with multiple mechanisms that ensure high-precision transmission at a kilohertz rate after the onset of hearing. In this case, closely located synapses within the contact would invite spillover as well, but, as the release probability is low (Meyer et al., 2001), the probability of two releases successively occurring in the immediate neighbor would be low. The low Pr is compensated for with a high number of release sites (Taschenberger et al., 2002) along with the preferential location of the synapses at the soma, which would cause enough current input for inducing high-fidelity spiking. In addition, from the onset of hearing, the postsynaptic medial nucleus of the trapezoid body cell is equipped with AMPARs that can rapidly recover from desensitization (Koike-Tani et al., 2008). Another example of a cell innervated by a strong excitatory input is the climbing fiber-to-Purkinje cell synapse. This synapse normally operates at 0.1–10 Hz in vivo. Therefore, fast recovery of transmission is not required, and the strong excitatory drive necessary to produce complex spikes is provided by the high Pr and the presence of multiple synapses from a single fiber (Silver et al., 1998). However, even at this synapse, individual spines are completely isolated from one another and postsynaptic AMPARs are surrounded by glutamate uptake mechanisms expressed both on the ensheathing glial cells as well as the postsynaptic Purkinje cells (Bergles et al., 1997; Otis et al., 1997). In addition, the Purkinje cell synapse is provided with GluR2/3 subunits of AMPARs, which recover from desensitization reasonably rapidly, and NMDARs are mostly absent, which may degrade the precision of the signal timing if present.
The reason why the RG synapse is equipped with so many mechanisms intuitively not preferable for fast reliable signal transmission is uncertain. It is possible that neuromodulators continuously work in vivo not only to set the postsynaptic resting potential (Antal et al., 2010) but also to lower the presynaptic Pr (Chen and Regehr, 2003), which would allow less depletion of vesicles as well as less AMPAR desensitization. However, even if Pr is reduced in vivo, it is still unclear why AMPARs highly prone to desensitization are prepared and why the synapses are closely arranged to allow spillover. What is evident is that, at least in acute slices, the mode of this synapse is set to a condition where only low-frequency signals are able to pass through or the temporal precision of the high-frequency component is lost, depending on the initial fiber input strength. In vivo recordings from even visually acute animals such as cats and monkeys, have shown that retinal ganglion cells produce more spikes than the postsynaptic RCs (Kaplan et al., 1987). This supports the idea that the filtering mechanism described here operates in vivo. Although counterintuitive, it has been shown that little information loss occurs between the retina and the LGN, and a rather more efficient representation of visual signal is achieved due to the spike-coding modification realized by the RCs (Sincich et al., 2009). As small and large EPSC-producing fibers yielded completely different characteristics of spiking outcome (Fig. 8C), information carried by different RG fibers may also integrate well at the relay cell for optimization. It is also possible that animals using high-frequency temporal cues are able to tune the signal transmission mechanisms more efficiently at this synapse. Humans as well as cats can decode up to ∼70 Hz of flickering visual stimuli. However, in rats, even though the critical flicker frequency (CFF) of electroretinogram responses could be as high as 50 Hz (Rubin and Kraft, 2007), the CFF of the cells in the visual cortex was found to be ∼20 Hz (Wells et al., 2001). Evolutional merit of the features found at this synapse may exist for rats, and, as all visual information must pass through the LGN before it reaches the visual cortex, this synapse would be an ideal location for signal filtering optimized for each species.
Footnotes
This work was supported by grants from Grant-in-Aid for Young Scientists (A) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) (18680903), Grant-in-Aid for Exploratory Research from Japan Society for the Promotion of Science (19650100), Grant-in-Aid for Scientific Research (C) from MEXT (22500362), and Precursory Research for Embryonic Science and Technology from Japan Science and Technology Agency (JST) to K.M.; Grant-in-Aid for Scientific Research (C) from MEXT (20500317) to N.K.; Grant-in-Aid for Scientific Research (C) from MEXT (21500311) and Core Research for Evolutional Science and Technology from JST to Y.F.; and Solution-Oriented Research for Science and Technology from JST to R.S. We thank Elek Molnár for providing panAMPAR antibody, Masahiko Watanabe for providing vGluT2 and GLAST antibodies, Kohichi Tanaka for providing GLT-1 antibody, and Keiko Shimamoto for providing PMB-TBOA.
The authors declare no competing financial interests.
References
- Antal M, Acuna-Goycolea C, Pressler RT, Blitz DM, Regehr WG. Cholinergic activation of M2 receptors leads to context-dependent modulation of feedforward inhibition in the visual thalamus. PLoS Biol. 2010;8:e1000348. doi: 10.1371/journal.pbio.1000348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnall MW, Hull C, Bushong EA, Ellisman MH, Scanziani M. Multiple clusters of release sites formed by individual thalamic afferents onto cortical interneurons ensure reliable transmission. Neuron. 2011;71:180–194. doi: 10.1016/j.neuron.2011.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour B. An evaluation of synapse independence. J Neurosci. 2001;21:7969–7984. doi: 10.1523/JNEUROSCI.21-20-07969.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Dzubay JA, Jahr CE. Glutamate transporter currents in bergmann glial cells follow the time course of extrasynaptic glutamate. Proc Natl Acad Sci U S A. 1997;94:14821–14825. doi: 10.1073/pnas.94.26.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitz DM, Regehr WG. Retinogeniculate synaptic properties controlling spike number and timing in relay neurons. J Neurophysiol. 2003;90:2438–2450. doi: 10.1152/jn.00562.2003. [DOI] [PubMed] [Google Scholar]
- Brauer K, Schober W, Winkelmann E. Two morphologically different types of retinal axon terminals in the rat's dorsal lateral geniculate nucleus and their relationships to the X- and Y-channel. Exp Brain Res. 1979;36:523–532. doi: 10.1007/BF00238520. [DOI] [PubMed] [Google Scholar]
- Cathala L, Holderith NB, Nusser Z, DiGregorio DA, Cull-Candy SG. Changes in synaptic structure underlie the developmental speeding of AMPA receptor-mediated EPSCs. Nat Neurosci. 2005;8:1310–1318. doi: 10.1038/nn1534. [DOI] [PubMed] [Google Scholar]
- Chanda S, Xu-Friedman MA. A low-affinity antagonist reveals saturation and desensitization in mature synapses in the auditory brain stem. J Neurophysiol. 2010;103:1915–1926. doi: 10.1152/jn.00751.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Regehr WG. Presynaptic modulation of the retinogeniculate synapse. J Neurosci. 2003;23:3130–3135. doi: 10.1523/JNEUROSCI.23-08-03130.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Blitz DM, Regehr WG. Contributions of receptor desensitization and saturation to plasticity at the retinogeniculate synapse. Neuron. 2002;33:779–788. doi: 10.1016/s0896-6273(02)00611-6. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Jonas P, Sakmann B. Action of brief pulses of glutamate on AMPA/kainate receptors in patches from different neurones of rat hippocampal slices. J Physiol. 1992;458:261–287. doi: 10.1113/jphysiol.1992.sp019417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Asynchronous release of synaptic vesicles determines the time course of the AMPA receptor-mediated EPSC. Neuron. 1995;15:1097–1107. doi: 10.1016/0896-6273(95)90098-5. [DOI] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci. 1997;17:4672–4687. doi: 10.1523/JNEUROSCI.17-12-04672.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzubay JA, Jahr CE. The concentration of synaptically released glutamate outside of the climbing fiber–Purkinje cell synaptic cleft. J Neurosci. 1999;19:5265–5274. doi: 10.1523/JNEUROSCI.19-13-05265.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erichsen MN, Huynh TH, Abrahamsen B, Bastlund JF, Bundgaard C, Monrad O, Bekker-Jensen A, Nielsen CW, Frydenvang K, Jensen AA, Bunch L. Structure-activity relationship study of first selective inhibitor of excitatory amino acid transporter subtype 1: 2-Amino-4-(4-methoxyphenyl)-7-(naphthalen-1-yl)-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (UCPH-101) J Med Chem. 2010;53:7180–7191. doi: 10.1021/jm1009154. [DOI] [PubMed] [Google Scholar]
- Famiglietti EV, Jr, Peters A. The synaptic glomerulus and the intrinsic neuron in the dorsal lateral geniculate nucleus of the cat. J Comp Neurol. 1972;144:285–334. doi: 10.1002/cne.901440304. [DOI] [PubMed] [Google Scholar]
- Fiala JC. Reconstruct: a free editor for serial section microscopy. J Microsc. 2005;218:52–61. doi: 10.1111/j.1365-2818.2005.01466.x. [DOI] [PubMed] [Google Scholar]
- Franks KM, Stevens CF, Sejnowski TJ. Independent sources of quantal variability at single glutamatergic synapses. J Neurosci. 2003;23:3186–3195. doi: 10.1523/JNEUROSCI.23-08-03186.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillery RW. The organization of synaptic interconnections in the laminae of the dorsal lateral geniculate nucleus of the cat. Z Zellforsch Mikrosk Anat. 1969;96:1–38. doi: 10.1007/BF00321474. [DOI] [PubMed] [Google Scholar]
- Hagiwara A, Fukazawa Y, Deguchi-Tawarada M, Ohtsuka T, Shigemoto R. Differential distribution of release-related proteins in the hippocampal CA3 area as revealed by freeze-fracture replica labeling. J Comp Neurol. 2005;489:195–216. doi: 10.1002/cne.20633. [DOI] [PubMed] [Google Scholar]
- Hamos JE, Van Horn SC, Raczkowski D, Sherman SM. Synaptic circuits involving an individual retinogeniculate axon in the cat. J Comp Neurol. 1987;259:165–192. doi: 10.1002/cne.902590202. [DOI] [PubMed] [Google Scholar]
- Harris KM, Landis DM. Membrane structure at synaptic junctions in area CA1 of the rat hippocampus. Neuroscience. 1986;19:857–872. doi: 10.1016/0306-4522(86)90304-0. [DOI] [PubMed] [Google Scholar]
- Häusser M, Roth A. Dendritic and somatic glutamate receptor channels in rat cerebellar Purkinje cells. J Physiol. 1997;501:77–95. doi: 10.1111/j.1469-7793.1997.077bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine M, Groc L, Frischknecht R, Béïque JC, Lounis B, Rumbaugh G, Huganir RL, Cognet L, Choquet D. Surface mobility of postsynaptic AMPARs tunes synaptic transmission. Science. 2008;320:201–205. doi: 10.1126/science.1152089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA-gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Sahara Y, Takahashi T. A single packet of transmitter does not saturate postsynaptic glutamate receptors. Neuron. 2002;34:613–621. doi: 10.1016/s0896-6273(02)00692-x. [DOI] [PubMed] [Google Scholar]
- Iwasaki S, Takahashi T. Developmental regulation of transmitter release at the calyx of Held in rat auditory brainstem. J Physiol. 2001;534:861–871. doi: 10.1111/j.1469-7793.2001.00861.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P. Fast application of agonists to isolated membrane patches. In: Sakmann B, Neher E, editors. Single-channel recording. New York: Plenum; 1995. pp. 231–243. [Google Scholar]
- Joshi I, Shokralla S, Titis P, Wang LY. The role of AMPA receptor gating in the development of high-fidelity neurotransmission at the calyx of Held synapse. J Neurosci. 2004;24:183–196. doi: 10.1523/JNEUROSCI.1074-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan E, Purpura K, Shapley RM. Contrast affects the transmission of visual information through the mammalian lateral geniculate nucleus. J Physiol. 1987;391:267–288. doi: 10.1113/jphysiol.1987.sp016737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura Y, Fukaya M, Maejima T, Yoshida T, Miura E, Watanabe M, Ohno-Shosaku T, Kano M. CB1 is the major cannabinoid receptor at excitatory presynaptic site in the hippocampus and cerebellum. J Neurosci. 2006;26:2991–3001. doi: 10.1523/JNEUROSCI.4872-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh DS, Geiger JR, Jonas P, Sakmann B. Ca(2+)-permeable AMPA and NMDA receptor channels in basket cells of rat hippocampal dentate gyrus. J Physiol. 1995;485:383–402. doi: 10.1113/jphysiol.1995.sp020737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike M, Iino M, Ozawa S. Blocking effect of 1-naphthyl acetyl spermine on Ca(2+)-permeable AMPA receptors in cultured rat hippocampal neurons. Neurosci Res. 1997;29:27–36. doi: 10.1016/s0168-0102(97)00067-9. [DOI] [PubMed] [Google Scholar]
- Koike-Tani M, Kanda T, Saitoh N, Yamashita T, Takahashi T. Involvement of AMPA receptor desensitization in short-term synaptic depression at the calyx of Held in developing rats. J Physiol. 2008;586:2263–2275. doi: 10.1113/jphysiol.2007.142547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JJ, Brenowitz S, Trussell LO. The mechanism of action of aniracetam at synaptic alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors: indirect and direct effects on desensitization. Mol Pharmacol. 2003;64:269–278. doi: 10.1124/mol.64.2.269. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Lieberman AR, Webster KE. Aspects of the synaptic organization of intrinsic neurons in the dorsal lateral geniculate nucleus. An ultrastructural study of the normal and of the experimentally deafferented nucleus in the rat. J Neurocytol. 1974;3:677–710. doi: 10.1007/BF01097191. [DOI] [PubMed] [Google Scholar]
- Masugi-Tokita M, Tarusawa E, Watanabe M, Molnár E, Fujimoto K, Shigemoto R. Number and density of AMPA receptors in individual synapses in the rat cerebellum as revealed by SDS-digested freeze-fracture replica labeling. J Neurosci. 2007;27:2135–2144. doi: 10.1523/JNEUROSCI.2861-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui K, Hosoi N, Tachibana M. Excitatory synaptic transmission in the inner retina: paired recordings of bipolar cells and neurons of the ganglion cell layer. J Neurosci. 1998;18:4500–4510. doi: 10.1523/JNEUROSCI.18-12-04500.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui K, Jahr CE, Rubio ME. High-concentration rapid transients of glutamate mediate neural–glial communication via ectopic release. J Neurosci. 2005;25:7538–7547. doi: 10.1523/JNEUROSCI.1927-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer AC, Neher E, Schneggenburger R. Estimation of quantal size and number of functional active zones at the calyx of held synapse by nonstationary EPSC variance analysis. J Neurosci. 2001;21:7889–7900. doi: 10.1523/JNEUROSCI.21-20-07889.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura E, Fukaya M, Sato T, Sugihara K, Asano M, Yoshioka K, Watanabe M. Expression and distribution of JNK/SAPK-associated scaffold protein JSAP1 in developing and adult mouse brain. J Neurochem. 2006;97:1431–1446. doi: 10.1111/j.1471-4159.2006.03835.x. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Fukaya M, Shimizu H, Watanabe M. Subtype switching of vesicular glutamate transporters at parallel fibre-Purkinje cell synapses in developing mouse cerebellum. Eur J Neurosci. 2003;17:2563–2572. doi: 10.1046/j.1460-9568.2003.02698.x. [DOI] [PubMed] [Google Scholar]
- Murphy GJ, Glickfeld LL, Balsen Z, Isaacson JS. Sensory neuron signaling to the brain: properties of transmitter release from olfactory nerve terminals. J Neurosci. 2004;24:3023–3030. doi: 10.1523/JNEUROSCI.5745-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen TA, DiGregorio DA, Silver RA. Modulation of glutamate mobility reveals the mechanism underlying slow-rising AMPAR EPSCs and the diffusion coefficient in the synaptic cleft. Neuron. 2004;42:757–771. doi: 10.1016/j.neuron.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Lujan R, Laube G, Roberts JD, Molnár E, Somogyi P. Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron. 1998;21:545–559. doi: 10.1016/s0896-6273(00)80565-6. [DOI] [PubMed] [Google Scholar]
- Otis TS, Kavanaugh MP, Jahr CE. Postsynaptic glutamate transport at the climbing fiber-Purkinje cell synapse. Science. 1997;277:1515–1518. doi: 10.1126/science.277.5331.1515. [DOI] [PubMed] [Google Scholar]
- Partin KM, Fleck MW, Mayer ML. AMPA receptor flip/flop mutants affecting deactivation, desensitization, and modulation by cyclothiazide, aniracetam, and thiocyanate. J Neurosci. 1996;16:6634–6647. doi: 10.1523/JNEUROSCI.16-21-06634.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafols JA, Valverde F. The structure of the dorsal lateral geniculate nucleus in the mouse. A Golgi and electron microscopic study. J Comp Neurol. 1973;150:303–332. doi: 10.1002/cne.901500305. [DOI] [PubMed] [Google Scholar]
- Raghavachari S, Lisman JE. Properties of quantal transmission at CA1 synapses. J Neurophysiol. 2004;92:2456–2467. doi: 10.1152/jn.00258.2004. [DOI] [PubMed] [Google Scholar]
- Robert A, Howe JR. How AMPA receptor desensitization depends on receptor occupancy. J Neurosci. 2003;23:847–858. doi: 10.1523/JNEUROSCI.23-03-00847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson JA. Qualitative and quantitative analyses of the patterns of retinal input to neurons in the dorsal lateral geniculate nucleus of the cat. J Comp Neurol. 1993;334:324–336. doi: 10.1002/cne.903340212. [DOI] [PubMed] [Google Scholar]
- Rubin GR, Kraft TW. Flicker assessment of rod and cone function in a model of retinal degeneration. Doc Ophthalmol. 2007;115:165–172. doi: 10.1007/s10633-007-9066-9. [DOI] [PubMed] [Google Scholar]
- Sekiguchi M, Nishikawa K, Aoki S, Wada K. A desensitization-selective potentiator of AMPA-type glutamate receptors. Br J Pharmacol. 2002;136:1033–1041. doi: 10.1038/sj.bjp.0704804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata T, Yamada K, Watanabe M, Ikenaka K, Wada K, Tanaka K, Inoue Y. Glutamate transporter GLAST is expressed in the radial glia-astrocyte lineage of developing mouse spinal cord. J Neurosci. 1997;17:9212–9219. doi: 10.1523/JNEUROSCI.17-23-09212.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto K, Sakai R, Takaoka K, Yumoto N, Nakajima T, Amara SG, Shigeri Y. Characterization of novel L-threo-beta-benzyloxyaspartate derivatives, potent blockers of the glutamate transporters. Mol Pharmacol. 2004;65:1008–1015. doi: 10.1124/mol.65.4.1008. [DOI] [PubMed] [Google Scholar]
- Silver RA, Momiyama A, Cull-Candy SG. Locus of frequency-dependent depression identified with multiple-probability fluctuation analysis at rat climbing fibre-Purkinje cell synapses. J Physiol. 1998;510:881–902. doi: 10.1111/j.1469-7793.1998.881bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sincich LC, Horton JC, Sharpee TO. Preserving information in neural transmission. J Neurosci. 2009;29:6207–6216. doi: 10.1523/JNEUROSCI.3701-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TC, Wang LY, Howe JR. Heterogeneous conductance levels of native AMPA receptors. J Neurosci. 2000;20:2073–2085. doi: 10.1523/JNEUROSCI.20-06-02073.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P, Tamás G, Lujan R, Buhl EH. Salient features of synaptic organisation in the cerebral cortex. Brain Res Rev. 1998;26:113–135. doi: 10.1016/s0165-0173(97)00061-1. [DOI] [PubMed] [Google Scholar]
- Takatsuru Y, Takayasu Y, Iino M, Nikkuni O, Ueda Y, Tanaka K, Ozawa S. Roles of glial glutamate transporters in shaping EPSCs at the climbing fiber-Purkinje cell synapses. Neurosci Res. 2006;54:140–148. doi: 10.1016/j.neures.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Tanaka J, Matsuzaki M, Tarusawa E, Momiyama A, Molnar E, Kasai H, Shigemoto R. Number and density of AMPA receptors in single synapses in immature cerebellum. J Neurosci. 2005;25:799–807. doi: 10.1523/JNEUROSCI.4256-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarusawa E, Matsui K, Budisantoso T, Molnár E, Watanabe M, Matsui M, Fukazawa Y, Shigemoto R. Input-specific intrasynaptic arrangements of ionotropic glutamate receptors and their impact on postsynaptic responses. J Neurosci. 2009;29:12896–12908. doi: 10.1523/JNEUROSCI.6160-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschenberger H, Leão RM, Rowland KC, Spirou GA, von Gersdorff H. Optimizing synaptic architecture and efficiency for high-frequency transmission. Neuron. 2002;36:1127–1143. doi: 10.1016/s0896-6273(02)01137-6. [DOI] [PubMed] [Google Scholar]
- Trussell LO, Zhang S, Raman IM. Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron. 1993;10:1185–1196. doi: 10.1016/0896-6273(93)90066-z. [DOI] [PubMed] [Google Scholar]
- Turner JP, Salt TE. Characterization of sensory and corticothalamic excitatory inputs to rat thalamocortical neurones in vitro. J Physiol. 1998;510:829–843. doi: 10.1111/j.1469-7793.1998.829bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JP, Leresche N, Guyon A, Soltesz I, Crunelli V. Sensory input and burst firing output of rat and cat thalamocortical cells: the role of NMDA and non-NMDA receptors. J Physiol. 1994;480:281–295. doi: 10.1113/jphysiol.1994.sp020359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uteshev VV, Pennefather PS. A mathematical description of miniature postsynaptic current generation at central nervous system synapses. Biophys J. 1996;71:1256–1266. doi: 10.1016/S0006-3495(96)79325-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE. Multivesicular release at climbing fiber-Purkinje cell synapses. Neuron. 2001;32:301–313. doi: 10.1016/s0896-6273(01)00488-3. [DOI] [PubMed] [Google Scholar]
- Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Ogawa S, Kawashima N, Hori S, Takimoto M, Wada K, Tanaka K. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci. 1998;10:976–988. doi: 10.1046/j.1460-9568.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- Wells EF, Bernstein GM, Scott BW, Bennett PJ, Mendelson JR. Critical flicker frequency responses in visual cortex. Exp Brain Res. 2001;139:106–110. doi: 10.1007/s002210100721. [DOI] [PubMed] [Google Scholar]
- Wong AY, Graham BP, Billups B, Forsythe ID. Distinguishing between presynaptic and postsynaptic mechanisms of short-term depression during action potential trains. J Neurosci. 2003;23:4868–4877. doi: 10.1523/JNEUROSCI.23-12-04868.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG. Ultrastructural contributions to desensitization at cerebellar mossy fiber to granule cell synapses. J Neurosci. 2003;23:2182–2192. doi: 10.1523/JNEUROSCI.23-06-02182.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Watanabe M, Shibata T, Nagashima M, Tanaka K, Inoue Y. Glutamate transporter GLT-1 is transiently localized on growing axons of the mouse spinal cord before establishing astrocytic expression. J Neurosci. 1998;18:5706–5713. doi: 10.1523/JNEUROSCI.18-15-05706.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Fukaya M, Shibata T, Kurihara H, Tanaka K, Inoue Y, Watanabe M. Dynamic transformation of Bergmann glial fibers proceeds in correlation with dendritic outgrowth and synapse formation of cerebellar Purkinje cells. J Comp Neurol. 2000;418:106–120. [PubMed] [Google Scholar]
- Yamashita T, Kanda T, Eguchi K, Takahashi T. Vesicular glutamate filling and AMPA receptor occupancy at the calyx of Held synapse of immature rats. J Physiol. 2009;587:2327–2339. doi: 10.1113/jphysiol.2008.167759. [DOI] [PMC free article] [PubMed] [Google Scholar]