

Abstract
Early in their formation, memories are thought to be labile, requiring a process called consolidation to give them near-permanent stability. Evidence for consolidation as an active and biologically separate mnemonic process has been established through posttraining manipulations of the brain that promote or disrupt subsequent retrieval. Consolidation is thought to be ultimately mediated via protein synthesis since translational inhibitors such as anisomycin disrupt subsequent memory when administered in a critical time window just following initial learning. However, when applied intracerebrally, they may induce additional neural disturbances. Here, we report that intrahippocampal microinfusions of anisomycin in urethane-anesthetized rats at dosages previously used in memory consolidation studies strongly suppressed (and in some cases abolished) spontaneous and evoked local field potentials (and associated extracellular current flow) as well as multiunit activity. These effects were not coupled to the production of pathological electrographic activity nor were they due to cell death. However, the amount of suppression was correlated with the degree of protein synthesis inhibition as measured by autoradiography and was also observed with cycloheximide, another translational inhibitor. Our results suggest that (1) the amnestic effects of protein synthesis inhibitors are confounded by neural silencing and that (2) intact protein synthesis is crucial for neural signaling itself.
Introduction
The idea that newly formed memories require an additional step called consolidation that gives them long-term or permanent stability has a long history. Although the first published evidence in support of the idea of consolidation as an active and unique memory process was based on purely behavioral observations (Müller and Pilzecker, 1900), its neurobiological underpinnings were confirmed in terms of recent versus remote retrograde memory deficits following brain injury (Ribot, 1882). Indeed, long-term memory for recently acquired information is acutely sensitive to alterations in neurobiological function and especially neural activity. This suggests that active processes in the period directly following acquisition are critical for the modulation of future memory strength. This has been supported by the finding that neuropharmacological treatments in this period, some directly in brain areas mediating the memory, can promote or diminish subsequent retrieval (McGaugh, 2000). The relative permanence of memory is thought to be ultimately mediated by de novo protein synthesis since translational inhibitors such as anisomycin (ANI) and cycloheximide (CHX) administered during this period effectively eliminate long-term memory for just-learned events (Davis and Squire, 1984; Kandel, 2001; Nader, 2003; Dudai, 2004; Alberini, 2008).
The assumption implicit in the de novo protein synthesis hypothesis has been that altering or disrupting protein synthesis has no other substantive effects on neural function. In fact, the classic and oft-cited review on this topic (Davis and Squire, 1984) documents an abundance of studies that purport to show a lack of effect of ANI on general neurobiological operations while having a specific effect on inhibiting protein synthesis. This is a critical aspect of the hypothesis since impaired neural function could also independently cause memory deficits.
There is recent evidence to suggest that intracerebral applications of ANI have some dramatic and nonspecific effects on the neurobiological substrate. The most notable example is the demonstration that locally applied ANI was found to markedly alter extracellular concentrations of a variety of neuromodulatory agents in both the amygdala (Canal et al., 2007) and the hippocampus (HPC) (Qi and Gold, 2009). Findings such as these are difficult to reconcile with effects that are specific to protein synthesis and independent of moment-to-moment neural functioning. They also present problematic confounds for intracerebral applications of ANI and other protein synthesis inhibitors in behavioral paradigms. Since spontaneous patterns of coordinated hippocampal activity during sleep have been suggested to play a role in the consolidation of episodic-like memories (Buzsáki, 1989; Pavlides and Winson, 1989; Wilson and McNaughton, 1994; Wolansky et al., 2006; Ji and Wilson, 2007; Girardeau et al., 2009; Nakashiba et al., 2009; Born, 2010; Dickson, 2010; Ego-Stengel and Wilson, 2010), we elected to study similar patterns expressed in urethane-anesthetized rats to determine an effect, if any, of direct intrahippocampal microinfusions of ANI. We show that ANI, at dosages less than or equal to those that have been previously shown to block the consolidation of hippocampal-dependent memories, severely depressed (and in some cases, eliminated) hippocampal electrical activity including local field potentials (LFPs), extracellular current flow, and ongoing unit activity. These effects on activity were not mediated by cell death. We show that as the level of protein synthesis inhibition increased so too did the suppression of neural activity. We conclude that (1) the behavioral deficits produced in studies using intracerebral ANI infusions are likely confounded by suppression of neural activity and (2) normal neurophysiological activity likely requires intact protein synthesis in neural tissue.
Materials and Methods
Data were obtained from 60 male Sprague Dawley rats with an average weight of 255 ± 7 g. All experimentation conformed to the guidelines established by the Canadian Council on Animal Care as well as the Society for Neuroscience, and the methods were approved by the Biological Sciences Animal Policy and Welfare Committee of the University of Alberta. Details of all procedures are available in our previous publications (Wolansky et al., 2006; Clement et al., 2008; Schall et al., 2008; Sharma et al., 2010).
Surgery, implantation, recording, and infusions.
Under initial isoflurane anesthesia, animals were implanted with a jugular catheter through which urethane was delivered at an anesthetic dose (1.66 ± 0.01 mg/kg). Rats remained anesthetized for the duration of all subsequent procedures, and the amount of time spent under urethane (ranging from 2 to 9 h) did not change the pattern of results reported. As we have previously shown, stable recordings across both activated and deactivated states can be made for extended periods in this preparation (Wolansky et al., 2006; Clement et al., 2008; Schall et al., 2008). Cranial implantations of electrodes were conducted stereotaxically while animals were maintained at a constant temperature of 37.0°C. Single-pole Teflon-insulated stainless steel electrodes were implanted in the neocortex (nCTX) (AP, +1.2; ML, +2.1; DV, −0.2 mm) and the HPC on the opposite (contralateral) side to the infusion site (AP, −3.3; ML, +2.1; DV, −3.4 ± 0.1 mm). At a homotopic point in the HPC ipsilateral to infusion, a single-pole electrode or 16-channel linear silicon multiprobe (NeuroNexus Technologies) was implanted, and just posterior to this, a microinfusion cannula made of 30 gauge stainless-steel tubing soldered within and extending out of a larger 23 gauge needle (AP, −4.0; ML, −2.6; DV, −3.0 to −3.5 mm). Following implantation, all monopolar recording electrodes were fixed to the skull using a jeweler's screw and dental acrylic.
Single-channel recordings were amplified at a gain of 1000 and bandpass filtered between 0.1 Hz and 10 kHz by a four-channel AC amplifier (model 1700; A-M Systems). Multiprobe recordings were amplified at a gain of 1000 and filtered between 0.7 Hz and 10 kHz using a 16-channel head stage and amplifier system (Plexon). All LFP signals were sampled at ≥1000 Hz after anti-aliasing filtering at 500 Hz. Multiunit activity was sampled at ≥10 kHz after anti-aliasing filtering at 5 kHz. Sampling was conducted through a Digidata 1322a A-D connected to a Pentium computer running AxoScope (Molecular Devices).
Baseline (spontaneous) recordings were taken for a minimum of 10 min [during which samples of both theta and slow oscillation (SO) states were obtained], and subsequently intracerebral microinfusions at a rate of 1 μl/min were made using a microinfusion pump (model KDS100; KD Scientific) connected to the cannula by a Hamilton 10 μl syringe and PE50 tubing (Clay Adams Brand; BD Biosciences). The maximum concentration of ANI was 100 μg/μl (although lower concentrations, 50, 25, and 12 μg/μl, were also used in some experiments), and the maximum volume infused at any concentration was 1 μl. Control infusions were made using standard PBS. Recordings were made continuously throughout (except for the protocols involving multiunit or evoked potential recordings) and lasted for a minimum of 2.5 h after infusion in initial (recording only) experiments. In experiments characterizing histochemistry or autoradiography, recordings lasted for a minimum of 40 min after infusion. The maximum recording time following infusion was 8 h.
Evoked potentials.
In some experiments, we tested the influence of infusions on evoked potential profiles elicited by electrically stimulating the CA3 subfield in the contralateral HPC through a bipolar electrode (AP, −3.5; ML, −3.5; DV, −3.0 to −4.0). Brief (0.2 ms), biphasic constant current (110–120 μA) pulses were applied every 8 s using an electrically isolated constant current apparatus (model 2100; A-M Systems). Average EPs were computed from at least 16 stimulation events before and 30–60 min after infusions.
Histology.
Following electrophysiological experimentation, animals were transcardially perfused with physiological saline followed by 4% paraformaldehyde in buffer. Brains were extracted and placed in a sucrose (30%) and paraformaldehyde (4%) solution and left overnight. After frozen sectioning, slide mounted slices were stained with thionin or Fluoro Jade B using standard procedures (Schmued and Hopkins, 2000). The location of cannula and electrode placements were noted for thionin-stained sections under a light microscope, while Fluoro Jade B-stained sections were visualized using a fluorescent microscope (DM 5500B; Leica) using a green (FITC/fluoroscein) fluorescent filter to visualize dead cells. Digital photomicrographs were obtained using a CCD camera (Retiga EXi Fast 1394; QImaging), which were later imported into Corel Photo Paint (Corel) and/or NIH ImageJ. Digital photomicrographs were color-inverted and then converted to grayscale images.
Autoradiography.
In a subset of experiments, after recording procedures were completed (1 h after infusion), we injected animals intravenously with 35S-labeled cysteine and methionine (11.05 μl/g body weight, totaling 1 μCi/g). We then allowed 15 min for amino acid incorporation before perfusion (as above). Formalin-fixed brains were cut at 100 μm using a vibratome and mounted. Slides were placed under Kodak BioMax MR X-Ray (PerkinElmer) film and kept for 24–36 h. Developed films were scanned as grayscale digital images and analyzed using ImageJ. Regions of interest were drawn around the HPC for each hemisphere at and near the level at which infusions/recordings were made and light intensity values were measured in these regions. These values were obtained from a standard scale of 0 to 255, with 255 being bright and 0 being dark. In terms of radioactivity, a higher value on this scale indicated less amino acid incorporation (and thus less protein synthesis). To measure differences in protein synthesis between the two hemispheres, we analyzed the same region of interest across both sides at the actual coronal level of the recording site, as well as two slices anterior and two slices posterior to this position. This gave us a total of five slices, which translated to a 500 μm range. Two measures for each region were obtained: average light intensity (on a scale from 0 to 255) and the percentage area above an intensity threshold. The threshold value was determined empirically by confirming that light intensity readings obtained for brain free regions (hole punch) were brighter than threshold and that readings corresponding to white matter (myelinated) regions of the autoradiograph were lower. One-tailed paired t tests were used to calculate significant differences between the hemispheres.
Data analysis.
Time and frequency domain analyses were computed from electrophysiological signals using code written in MATLAB (MathWorks) as detailed in our previous publications (Wolansky et al., 2006; Clement et al., 2008; Schall et al., 2008; Sharma et al., 2010). Power spectra were estimated for field signals and power at the fundamental frequency for each state was extracted. For multiprobe recordings, we computed power and cross-phase profiles as a function of depth (Wolansky et al., 2006; Clement et al., 2008; Schall et al., 2008; Sharma et al., 2010). Multiunit activity was extracted from high-frequency sampled recordings by bandpass filtering between 500 and 3000 Hz and analyzed by computing the root mean square (RMS) of the traces in ClampFit 9.0 (Molecular Devices). The RMS of multiunit activity before infusion was normalized to minimum and maximum values across the different multiprobe sites. Current source density (CSD) analysis was computed as a three-point spatial differentiation procedure (Wolansky et al., 2006; Clement et al., 2008; Schall et al., 2008; Sharma et al., 2010).
Comparisons of measures were made across manipulations. To evaluate the effect of the infused drugs on brain activity, ratios or differences of postinfusion to preinfusion activity were computed for each hemisphere, yielding a measure of change in activity as a result of infusion. We used one-tailed paired t tests to measure any differences between the two hemispheres in LFP measures. One-tailed, one-sample t tests were also conducted on the ratios or differences of postinfusion to preinfusion activity values in the ipsilateral HPC to determine whether the change in activity (if any) after infusion was statistically significant.
To gauge the relationship between the degree of protein synthesis inhibition and neural suppression, we divided our protein synthesis suppression measures into three bins corresponding to low (0–33.33%), medium (33.33–66.67%), and high (66.67–100%) suppression values. In each bin, we then calculated the average value ± SEM of our neural activity measures. In addition, we performed both simple linear regression as well as multiple regression analysis of the raw protein synthesis suppression measures against the raw neural activity measures.
Results
Histology
Ipsilateral and contralateral single-electrode sites were found mostly in the apical dendritic region of CA1, close to the hippocampal fissure in stratum lacunosum-moleculare (SLM). Some placements were lower, in both stratum moleculare (SMol) and stratum granulosum (SGran) of the dentate gyrus (DG), while a few were higher in stratum radiatum (SRad) and stratum oriens (SOr) of CA1. Multiprobe tracks were confirmed to fall in a plane that traversed the CA1 region of the HPC, passing through the hippocampal fissure and the DG ending in either the hilar region or as far as the ventral blade of SGran. Cannula tips were located in more posterior sections within CA1, typically adjacent to stratum pyramidale (SPyr) with some locations in the apical dendritic layers. Experiments in which cannula tips were above the fimbrial surface were treated as missed (control) placements. Histological placements for autoradiographic tissue were verified by visualizing tracks in fresh tissue slices using a stereoscope.
Anisomycin infusions prominently reduce spontaneous LFP activity in the hippocampus
By making long-term LFP recordings from the nCTX and HPC in urethane-anesthetized rats, we were able to sample alternating epochs of both activated and deactivated states (Wolansky et al., 2006; Clement et al., 2008; Dickson, 2010). The activated state was characterized by low-voltage fast activity in the nCTX and a prominent theta rhythm of 3–4 Hz in the HPC (Bland, 1986; Buzsáki, 2002). The lower frequency of this rhythm is typical for that recorded under urethane anesthesia (Kramis et al., 1975; Vanderwolf et al., 1977; Clement et al., 2008). The deactivated state was characterized by a high-amplitude low-frequency SO of ∼1 Hz that appeared at both hippocampal and neocortical sites (Steriade et al., 1993; Amzica and Steriade, 1997; Isomura et al., 2006; Wolansky et al., 2006). The SO is an archetypal signature of non-REM sleep and is thought to be important for the consolidation of memories (Steriade, 2006; Marshall and Born, 2007; Dickson, 2010; Hanlon et al., 2011). These two oscillatory states are mutually exclusive and involve different, but perhaps overlapping neuronal networks (Isomura et al., 2006; Wolansky et al., 2006).
Infusions of ANI (100 μg in 1 μl) profoundly suppressed ongoing LFP activity in the ipsilateral hippocampus as shown in Figure 1A. This effect occurred for both theta and the SO (data not shown), was maximal at 20–30 min after infusion, and lasted for the entire length of our recordings (in some cases, from 4 to 6 h). This suppressive effect was not preceded by any local pathological electrophysiological activity (i.e., electrographic seizure, spreading depression) that could also cause neural silencing. Spectral analysis revealed that the depression of power occurred across the bandwidth of the signals (Fig. 1B). Across eight experiments using one-tailed paired t tests, a significant reduction in power was found during theta (evaluated at 3.63 ± 0.05 Hz) at 30 min (93.46 ± 0.03% decrease; t(7) = −28.48, p < 0.00001), 90 min (90.62 ± 3.5%; t(7) = −25.78, p < 0.00001), and 150 min (88.32 ± 5.54%; t(7) = −15.95, p < 0.00001) after infusion. During SO, a significant power suppression at 0.99 ± 0.027 Hz was found at 30 min (68.92 ± 10.70% decrease; t(7) = −6.44, p = 0.00018) and 90 min (41.20 ± 20.25%; t(7) = −2.03, p = 0.04066), but not at 150 min after infusion (35.80 ± 21.70% decrease; t(7) = −1.65, p = 0.07152). In contrast, no suppression was observed at these time points at either contralateral hippocampal or neocortical sites. Infusions of the same volume of vehicle (PBS) within the HPC (n = 3) in addition to ANI infusions at neocortical sites above the HPC (n = 2) had no effect on ipsilateral hippocampal LFPs evaluated in the same time frame.
Figure 1.

Intrahippocampal administration of ANI disrupts spontaneous hippocampal theta and SO field activity. A, Raw LFP traces during theta, from neocortical and both contralateral and ipsilateral hippocampal sites before and following an infusion of 100 μg of ANI. Only the ipsilateral hippocampal site shows suppression at short (30 min) and long (150 min) time points after infusion. B, Spectral analysis of hippocampal traces shown in A. Depression of power across the entire bandwidth is observed for theta samples in the ipsilateral but not contralateral HPC at the 150 min post period. Across all experiments (C), there were significant (***p <<< 0.01) suppressive influences of ANI on the mean spectral power in the ipsilateral HPC during theta at 30, 90, and 150 min after infusion. Error bars indicate SEM.
Anisomycin eliminates local hippocampal signals
A major problem with LFPs is the phenomenon of volume conduction from regions other than the site at which a recording is being made. To avoid this and to ensure that signals were specific to the HPC, we performed laminar profile and CSD analysis as well as multiunit activity recordings using the 16-channel multiprobe in the HPC. Consistent with our results using single-pole LFP recordings, ANI (but not PBS) infusions depressed and flattened the LFP power profiles in the ipsilateral HPC for both theta (3.64 ± 0.18 Hz) and SO (0.97 ± 0.078 Hz) frequency components during activated and deactivated states, respectively, when assessed 90 min following infusions (Fig. 2). Before infusion, laminar profiles for theta and SO had forms similar to previous results (Bland, 1986; Buzsáki et al., 1986; Bragin et al., 1995; Leung, 1998; Buzsáki, 2002; Isomura et al., 2006; Wolansky et al., 2006; Schall et al., 2008; Nazer and Dickson, 2009; Sharma et al., 2010). Specifically, theta profiles had power maxima on either side of SPyr of the CA1 subfield with a phase reversal as well as a power minimum just below SPyr in proximal SRad; the largest maximum was close to the level of the hippocampal fissure. During SO, there was no phase reversal within the HPC but there was a phase shift and power maximum close to the location of the hippocampal fissure.
Figure 2.
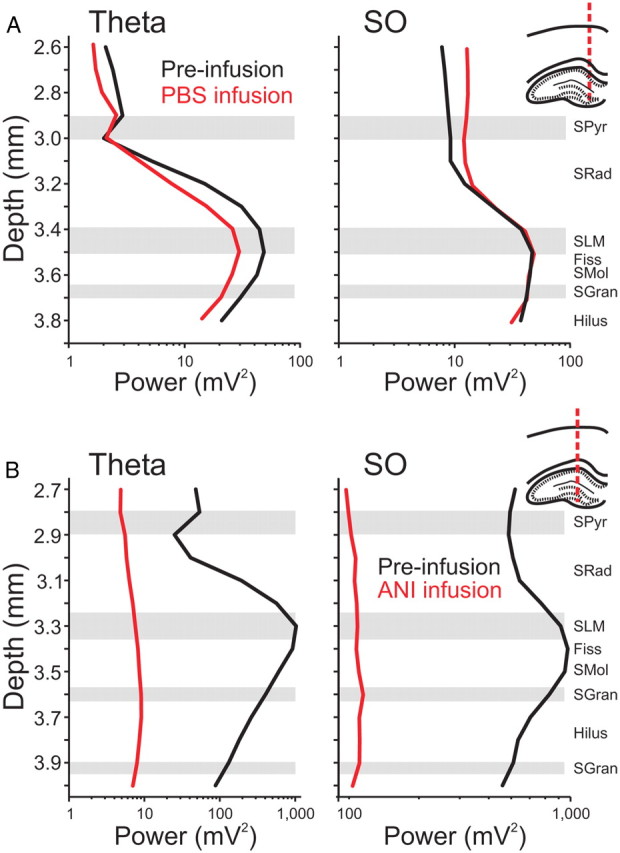
Hippocampal spectral profiles are flattened by local ANI infusions. Spectral analysis was conducted on multiprobe LFP signals taken from the hippocampus at the positions indicated in the histological drawings to the right of the plots. Laminar power profiles across depth were plotted for the theta band (3–4 Hz; left panels) and the SO band (0.5–1.5 Hz; right panels) for activated and deactivated states, respectively. Representative profiles for theta and SO activity show characteristic shapes and do not change as a result of ipsilateral PBS infusions (A) but are markedly compressed following ipsilateral infusions of ANI (B). Schematic drawings indicate multiprobe tracks. The black lines indicate the profile before infusion, while the red lines represent the profiles 90 min following each respective infusion.
As shown, and across experiments, no significant changes to either the shape or the amplitude of these profiles in either state were observed with infusions of PBS vehicle (Fig. 2A). After infusion of 100 μg of ANI in 1 μl, the laminar profile during both states demonstrated a significant disruption (Fig. 2B). Significant reductions were observed in power values across all experiments (SO: t(6) = 2.37, p < 0.02766; theta: t(6) = 2.68, p < 0.01828), and there was a general flattening of the profile itself, which suggested that a reduction in active current generation was occurring following infusions across both states in the HPC.
Confirmation of this effect was observed following CSD analysis. ANI infusions essentially eliminated spontaneous extracellular current flow during both theta and SO states (Fig. 3A,B). By characterizing and comparing the RMS of spontaneous CSD fluctuations at the level of SLM (where the largest sink–source alternations occur during both theta and SO) as well as more generally across all depths preinfusion and postinfusion, we were able to quantify this effect across eight experiments. As shown in Figure 3C, ANI prominently (and significantly) reduced the amplitude of SLM CSD fluctuations during both SO and theta, at all time points sampled (30, 90, and 150 min after infusion). In addition, when CSD fluctuations were evaluated across all electrode sites within the HPC, ANI significantly reduced the net RMS during both SO (62.42 ± 0.09%; t(7) = −6.94, p = 0.00011) and theta (56.92 ± 0.09%; t(7) = −6.20, p = 0.00022), when measured 30 min after infusion.
Figure 3.
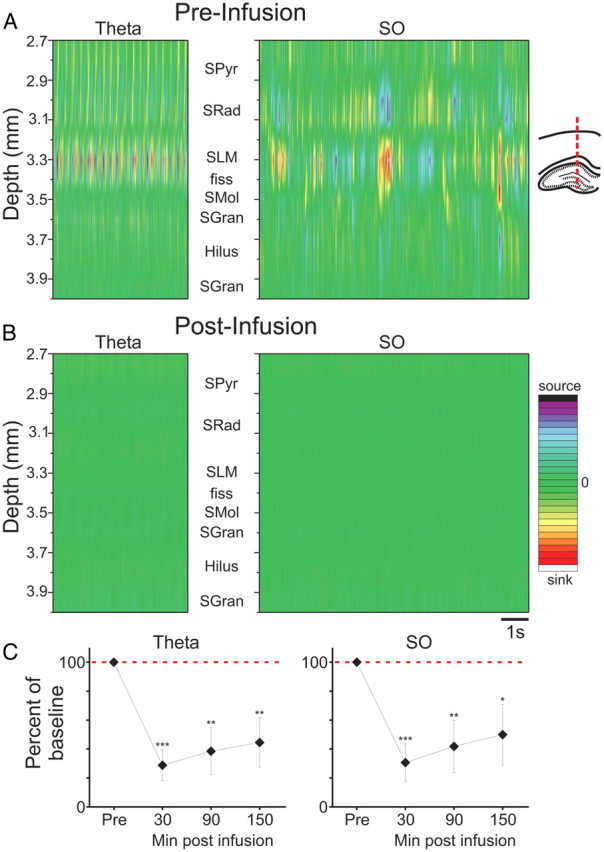
Intrahippocampal ANI infusion eliminates local current flow during both theta and slow oscillation states. A, During theta and SO, CSD plots show prominent rhythmic sink/source alternations at the level of SLM of CA1 and are paired with opposing source/sink alternations at the level of SRad. B, Following ANI infusion, all extracellular current movement is eliminated. C, Across all experiments, the suppression of CSD RMS at SLM is significant, dramatic, and long-lasting (***p <<< 0.01; **p < 0.01; *p < 0.05). Error bars indicate SEM. Abbreviations: SPyr, stratum pyramidale; SRad, stratum radiatum; SLM, stratum lacunosum moleculare; fiss, hippocampal fissure; SMol, stratum moleculare; SGran, stratum granulosum. CSD scale for SO: −25 to 25 mV/mm2; CSD scale for theta: −15 to 15 mV/mm2.
Cellular (unit) activity also appeared to be silenced by ANI infusions, an observation that we first noted when reinserting probes into the ipsilateral HPC directly following infusions. To characterize this effect in real time, we recorded multiunit activity using the multiprobe at both CA1 and DG cellular layers simultaneously during preinfusion and postinfusion periods. ANI, but not vehicle (PBS), completely suppressed unit activity in both SPyr and SGran as assessed at a latency of 30 min (Fig. 4). On average (across 16 experiments), multiunit activity RMS within SPyr and SGran was significantly attenuated by ANI during both SO (t(14) = 4.08; p = 0.00057) and theta (t(14) = 3.70; p = 0.0012).
Figure 4.

Intrahippocampal ANI infusions abolish multiunit activity (MUA). A, Simultaneous multiunit activity filtered traces from the multiprobe at different levels in the HPC ipsilateral to the side of infusion. Cellular activity is observed at both the CA1 pyramidal (SPyr) and dentate granule cell (SGran) layers before infusion, but not following. Background noise is slightly reduced in the cell sparse region of SLM. B, Across all experiments, there was a dramatic (and significant) suppression of multiunit activity RMS following ANI infusions. Error bars indicate SEM.
Anisomycin eliminates hippocampal evoked potentials
Given its profound effect on spontaneous activity, we assessed whether electrically evoked potentials were also suppressed by ANI. Concomitant with the suppressive effect on spontaneous sink–source fluctuations generated during theta, ANI infusions also eliminated extracellular current flow in CA1 evoked by electrical stimulation of contralateral CA3. As shown in Figure 5, the prominent current sink generated in SRad as well as the associated current sources in SPyr and SLM were abolished by treatment with ANI. Across all experiments (n = 4), this effect on the evoked sink at SRad following ANI infusion was significant (t(3) = 16.92; p = 0.00002). This suggests that all forms of electrical activity in the hippocampus, both spontaneous and evoked, were eliminated by ANI.
Figure 5.

ANI infusions suppress stimulus-evoked activity in the HPC. A, An intrahippocampal CSD profile elicited by electrical stimulation of contralateral CA3 shows a characteristic excitatory sink at the level of SRad before but not after ANI infusion. The diagram in the inset shows the localization of the probe. The individual LFP trace at the level of SRad is displayed in a time-aligned fashion above the contour plot. B, Concomitant CSD traces during spontaneous theta activity at the level of SLM before and after ANI infusion demonstrate a complete suppression of active current movement during spontaneous activity in concert with the suppression of evoked responses. C, Summary of the average peak sink values from evoked CSDs across all experiments demonstrates a dramatic and significant suppression post infusion. Error bars indicate SEM. CSD scale: −56 to 56 mV/mm2. Spontaneous CSD scale bar: 10 mV/mm2.
Anisomycin infusions do not induce cell death in hippocampus
Since ANI is known to induce apoptosis, a potential explanation for the neural suppression following ANI infusions is the induction of cell death (Iordanov et al., 1997; Adams, 2003; Rudy, 2008). To assess this possibility as an explanation for the suppression of neural activity in our experiments, we performed Fluoro Jade B staining, which is known to label cells dying by either apoptotic or necrotic mechanisms (Schmued and Hopkins, 2000). Positive and consistent Fluoro Jade B labeling was observed in neocortical regions abutting trephine holes in the skull, where we purposely damaged the pial surface to induce local cellular death (Fig. 6A). Despite positive staining in these regions, we found no systematic labeling in either the contralateral HPC or ipsilateral HPC (Fig. 6B,C), despite significant neural suppression in the latter. Given the lack of ipsilateral hippocampal staining, this demonstrates that the electrophysiological suppression induced by ANI does not occur as a result of neuronal cell death, at least during the time course of our acute recording experiments.
Figure 6.

ANI infusions that suppress hippocampal activity do not elicit hippocampal cell death. Expansion photomicrographs from a single histological section are shown from Fluoro Jade B-treated brain slices. The left inset panels show suprathreshold (positively stained) cells, while the right inset panels show the locations at which images were taken. A, Many positively stained (dead or dying) cells were observed in superficial cortical regions damaged by skull trephination and pial damage. However, no systematic staining was observed in either the contralateral HPC (B) or ipsilateral HPC (C). In this experiment, CSD RMS analysis at the level of SLM showed an 87.54% decrease in activity during SO and an 86.93% decrease during theta (across all probe channels this decrease was 78.02% during SO and 74.53% during theta) when assessed 30 min following ANI infusion (50 μg in 0.5 μl).
Protein synthesis inhibitors unilaterally inhibit amino acid incorporation and neural activity
Given the profound and consistent suppressive effect of local infusions of ANI on the electrical activity of the HPC, we hypothesized that protein synthesis itself plays an important role in maintaining neural activity. To first confirm that ANI was indeed inhibiting protein synthesis in the ipsilateral HPC, we assessed the incorporation of radiolabeled cysteine and methionine using autoradiography following our postinfusion recordings. In initial experiments, we found that the influence of 100 μg/1 μl infusions on incorporation of radiolabeled amino acids was apparent in both the ipsilateral HPC and the medial portion of the contralateral HPC. To minimize the contralateral influence, we infused one-half of this dose (50 μg in 0.5 μl) after first confirming that it would have a similar suppressive effect on electrophysiological measures. Over 10 experiments (and as shown in the left panel of Fig. 7A), the suppression of CSD RMS at 30 min after infusion was significant in the ipsilateral HPC compared with the contralateral HPC: At the level of the SLM, there was a significant decrease in the CSD RMS during the SO (67.02 ± 10.30% decrease; t(9) = −6.51, p = 0.000055) and theta (72.84 ± 12.0% decrease; t(8) = −6.07, p = 0.00015). This decrease was also significant across all layers for both SO (37.97 ± 15.35% decrease; t(9) = −2.47, p = 0.01767) and theta (47.71 ± 11.79% decrease; t(8) = −4.05, p = 0.00185). Accordingly, the incorporation of radiolabeled amino acids was also strikingly different across the hemispheres. In the contralateral HPC, anatomical features (i.e., cell layers) and laminar striations could be clearly seen as darkened regions, while in the ipsilateral HPC a more uniform distribution of elevated light intensity values was observed, indicating little incorporation. A representative example of the raw autoradiogram is shown in Figure 7A. Using our light-intensity threshold measure, we were able to characterize the proportion of HPC area showing minimal amino acid incorporation and thus maximal protein synthesis inhibition. Using this measure, the percentage area of minimal incorporation in the ipsilateral HPC was 72.58 ± 9.82% compared with only 7.80 ± 2.57% in the contralateral HPC (t(8) = −7.17; p = 0.00005). In contrast, vehicle infusions (n = 4; Fig. 7C) induced neither a decrease in amino acid incorporation (t(3) = 1.83; p = 0.86047), nor a decrease in CSD RMS activity at SLM (SO: 6.22 ± 5.20% increase; t(4) = 1.19, p = 0.85084; theta: 2.99 ± 6.66% decrease; t(4) = −0.45, p = 0.33865) or even across all layers (SO: 4.63 ± 12.92% increase; t(3) = 0.36, p = 0.62803; theta: 0.54 ± 7.04% increase; t(3) = 0.08, p = 0.52812). Thus, we could demonstrate a significant inhibition of protein synthesis in the HPC that was limited to the hemisphere ipsilateral to infusion and that was uniquely associated with suppression of ipsilateral neural activity.
Figure 7.
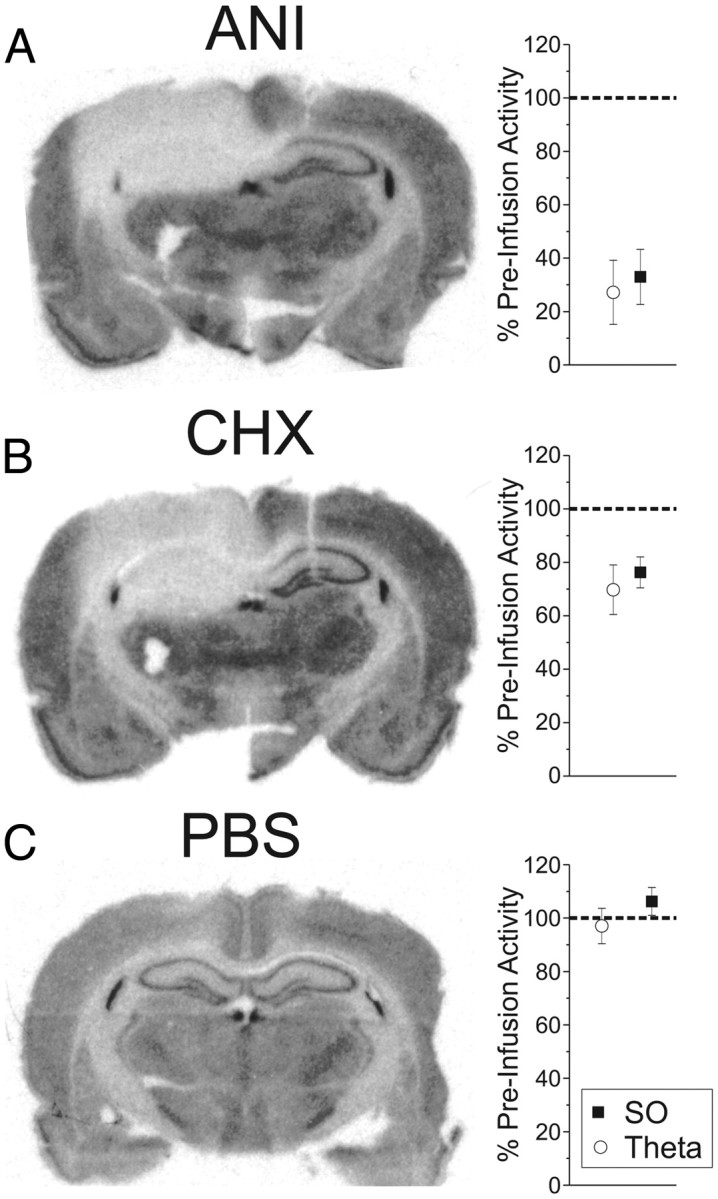
Intrahippocampal ANI and CHX infusions induce dramatic and significant decreases in both protein synthesis and hippocampal activity. Infusions of 50 μg of ANI in 0.5 μl (A) and 20 μg of CHX in 0.5 μl (B) significantly decreased the incorporation of radiolabeled amino acids and CSD RMS in the ipsilateral HPC, while vehicle (PBS) infusions (C) had no effect on either parameter. Error bars indicate SEM.
To further assess the association between protein synthesis inhibition and electrophysiological suppression, we used a different translational inhibitor, CHX (Fig. 7B). We infused 20 μg of CHX in 0.5 μl using the same protocol described above. Similarly to ANI, CHX significantly suppressed both CSD and multiunit activity measures recorded in the ipsilateral HPC across six experiments. For CSD, we found a significant effect for SO (23.74 ± 5.76% decrease; t(5) = −04.12, p = 0.00458) as well as for theta (30.26 ± 9.28% decrease; t(5) = −3.26, p = 0.01123) at the level of SLM. This difference extended to RMS measures across all probe channels as well (SO: 20.74 ± 4.71% decrease; t(5) = −4.40, p = 0.00351; theta: 16.81 ± 7.98% decrease; t(5) = −2.11, p = 0.04448). The same was true for multiunit activity during both SO (t(5) = 4.59; p = 0.00295) and theta (t(5) = 3.38; p = 0.00986). Using autoradiography, we found significant differences in the amount of amino acid incorporation between hemispheres: the percentage area of the ipsilateral HPC showing maximal protein synthesis inhibition was 73.54 ± 16.32%, which was significantly different from that found in the contralateral HPC (4.99 ± 1.69%; t(5) = −4.48, p = 0.00327). These results (shown in Fig. 7B) further support the notion that intact protein synthesis is required for neural activity.
Suppression of neural activity is correlated with the degree of protein synthesis inhibition
To make a systematic investigation of the relationship between the degree of protein synthesis inhibition and the degree of neural suppression induced, we infused a range of dosages of ANI (25, 12.5, and 0.5 μg; each in 0.5 μl) across different experiments. We confirmed with our autoradiographic measure (percentage area of maximal protein synthesis inhibition) that there was a sufficient degree of variation of amino acid incorporation across these experiments. By grouping data based on the degree of protein synthesis inhibition (low, medium, high) and assessing the average extent of neural suppression in each of these groups, we confirmed that there was a direct relationship; as protein synthesis inhibition increased, so too did the degree of neural inactivation (Fig. 8). Indeed, across all our experiments, there was a significant positive linear relationship between the raw values corresponding to protein synthesis inhibition and the degree of neurophysiological suppression for both CSD (SO: r(31) = 0.55, p = 0.000419; theta: r(31) = 0.47, p = 0.002893) and multiunit activity (SO: r(30) = 0.36, p = 0.02102; theta: r(29) = 0.32, p = 0.03653) measures that further supported this relationship. A significant relationship between the neurophysiological suppression of CSD RMS across all hippocampal contacts and protein synthesis inhibition was also found (SO: r(31) = 0.37, p = 0.016; theta: r(31) = 0.49, p = 0.00198). Furthermore, by combining the two different autoradiographic measures (percentage area of maximal protein synthesis inhibition and average light intensity values in the same area) in a stepwise multiple regression model, we could account for 60% (r2 = 0.60) of the variance in total CSD RMS suppression and >20% (r2 = 0.22) of the variance in multiunit activity suppression. This is further evidence of a link between intact protein synthesis and electrical neuronal signaling.
Figure 8.
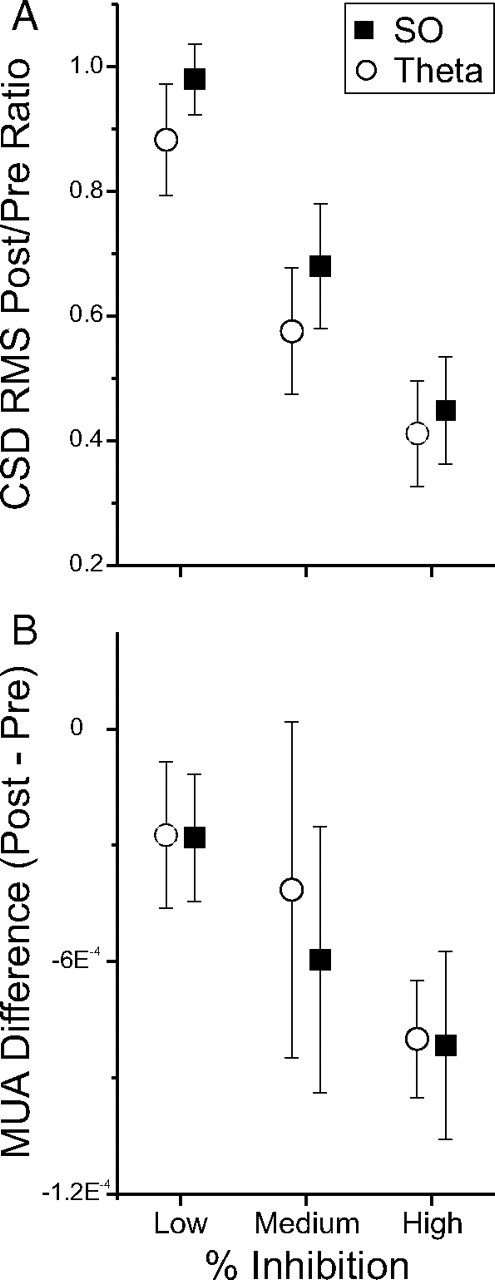
Increased inhibition of protein synthesis is correlated with an increased suppression of neural activity. Experiments were grouped according to the degree of protein synthesis inhibition measured using autoradiography to compare the degree of suppression of neural activity measures. The decrease in both CSD (A) and multiunit activity (MUA) (B) measures were directly scaled to the degree of decreased incorporation of radiolabeled amino acids in the ipsilateral HPC. Error bars indicate SEM.
Discussion
Our findings clearly demonstrate that local applications of ANI profoundly suppressed (and often completely abolished) neural activity. In the HPC, this effect was observed for both spontaneous and evoked LFPs as well as both CSD and multiunit activity measures. This suppression was fairly rapid (peaking within 20–30 min) and long-lasting (>4–6 h). The abolition of hippocampal neural activity was not due to cell death and was not preceded by pathological activity that might have induced a suppressive effect. It was more likely due to the specific effect of ANI on protein synthesis since a similar effect was observed with CHX and there was a significant relationship between the degree of neural suppression and the extent of protein synthesis inhibition as characterized by autoradiography.
The implications for these results are twofold as follows: (1) The use of local applications of ANI in past, present, and ongoing behavioral studies is likely to be confounded by its suppressive effects on neural activity, and (2) intact protein synthesis may be necessary for spontaneous and evoked neural activity.
Implications of the use of intracerebral anisomycin
The history of the use of protein synthesis inhibitors in memory research has been fraught with controversy. However, it has been explicitly proposed (and also implicitly accepted) that the direct intracerebral applications of presently used translational inhibitors such as ANI and CHX have few confounding side effects (Davis and Squire, 1984; Hernandez and Abel, 2008). In recent years, however, this assertion has been revisited. For example, it has been noted that ANI can induce active cell death (Rudy, 2008). Although this might well have explained our present observations on neural activity measures, we found no evidence for ANI-induced neuronal death using the marker Fluoro Jade B, at least during the time frame of our experiments. Regardless, the importance of the potential induction of neuronal death occurring in a more extended time frame would certainly be important in interpreting amnestic behavior assessed over a longer duration.
Another criticism of ANI is that local infusions cause a rapid (within 15–30 min) and catastrophic elevation (between 1000 and 17,000% increase) in the local extracellular levels of a host of endogenous neuromodulators such as norepinephrine, dopamine, and serotonin. These increases are subsequently followed by a substantial and long-lasting depression of presumed release. Together, these results suggest an obvious impairment of neural function (Canal et al., 2007; Qi and Gold, 2009). Although we did not perform a similar analysis of the constituents of the extracellular fluid in the HPC, given the profound suppression of local electrical neural signaling, we feel that any effect of changing levels of extracellular neuromodulators may well be moot, especially since the onset of neural suppression occurs in the same time frame as the initial increase in release and outlasts this event. It may be that a neurochemical influence of ANI may simply reflect a compensatory or corollary effect of that which produces neural silencing; however, this remains to be shown explicitly.
Certainly, it is the case that the impairment of spontaneous and evoked neural activity produced by ANI would adversely affect behavioral measures. Interestingly, the translational inhibitor puromycin was also shown to silence neural activity when administered intracerebrally (Cohen et al., 1966), and this finding led to the discontinuation of its use in behavioral memory paradigms. However, ANI has been staunchly defended as producing no such effects (Davis and Squire, 1984; Hernandez and Abel, 2008). Indeed, ANI has been routinely used in in vitro studies of neuroplasticity with little apparent influence on evoked potential measures, synaptic transmission, nor on passive or active membrane properties, suggesting that it has no detrimental effects on electrophysiological functioning per se (Schwartz et al., 1971; Frey et al., 1988, 1993). However, it should be noted that these studies (and those following) used concentrations of ANI in the 15–25 μm range and those conducted in vivo used effective concentrations that are >4 orders of magnitude larger (226–452 mm) (Wanisch and Wotjak, 2008). Given that we reported a concentration-dependent effect that was actually coupled to the degree of protein synthesis inhibition evoked within the relatively large area of the dorsal hippocampus, we presume that the relative speed and volume of tissue that were pharmacologically impacted were important factors affecting our electrophysiological measures. In this respect, we note that incorporation studies using lower concentrations of ANI demonstrate an incomplete (albeit a significant) inhibition of protein synthesis (Frey et al., 1988; Wanisch and Wotjak, 2008). Regardless, there is an obvious mismatch in terms of the dosage (and the potential confounds associated) between the in vitro and in vivo paradigms and thus little to be gained in terms of comparisons.
We also note that our results are consistent with previous findings in which ANI infusions into the motor cortex were shown to disrupt the functional representation of the motor map via microstimulation as well as abolishing cortical evoked potentials (Kleim et al., 2003). Although this former study was interpreted as meaning that the motor cortex exhibited ongoing protein synthesis-dependent ability that was required to maintain its functional organization, we suggest instead that they are more consistent with a suppressive effect on neural activity and we suggest that they should be reevaluated in this light.
Our results show that ANI (as well as CHX) induces a neural silencing effect that appears similar to local infusions of agents such as the Na+ channel blocker tetrodotoxin, the local anesthetic lidocaine, or the GABAergic agonist muscimol (Packard and McGaugh, 1996; Ambrogi Lorenzini et al., 1999; Chang and Gold, 2003; Klement et al., 2005), although the ANI effect in this respect is certainly not as fast. It should be noted in this regard that the maximal dosages used in our study were either equivalent to or one-half of the effective amounts used in prior behavioral consolidation studies (Wanisch and Wotjak, 2008). This suggests that the suppressive effects on neural activity would be expressed to an even greater degree and in a more widespread fashion in experiments using even higher doses of ANI. At a minimum, our findings present a serious confound for the behavioral interpretation of experiments using intracerebral application of this translational inhibitor.
Can neural inactivation caused by protein synthesis inhibition explain prior behavioral results?
The de novo protein synthesis hypothesis of memory consolidation has achieved almost axiomatic status based on a long history of experiments using intracerebral applications of protein synthesis inhibitors like ANI and CHX (for review, see Davis and Squire, 1984; Kandel, 2001; Nader, 2003; Dudai, 2004; Alberini, 2008). Our results suggest an alternative explanation; amnestic effects may more simply be a result of neural silencing. It is certainly the case that posttraining lesions, inactivation, or disruption of the hippocampus induces an amnestic syndrome for the pretraining period (Bohbot et al., 1996; Prado-Alcalá et al., 2006; Wiltgen et al., 2006; Biedenkapp and Rudy, 2009; Bissiere et al., 2011; Goshen et al., 2011). Therefore, posttraining intracerebral infusions of ANI could be expected to induce amnesia via neural inactivation, if memory was tested in a retrieval window during which this inactivation was still present. Indeed, even the finding of long-term (but not short-term) amnestic effects observed with pretraining or cotraining intrahippocampal infusions of ANI (Quevedo et al., 2004; Canal and Gold, 2007) could be considered as retrieval (and not consolidation) failures, since (1) animals can learn and remember these tasks without an active hippocampus and (2) any mismatch of the state of activity/inactivity of the hippocampus at retrieval compared with acquisition will prevent memory expression, a phenomenon referred to as overshadowing (Maren et al., 1997; Biedenkapp and Rudy, 2009; Sutherland et al., 2010). This has been acutely shown by Biedenkapp et al. (2009), who demonstrated a recovery from “amnesia” during memory testing simply by reinactivating the hippocampus in animals that were trained initially under conditions of hippocampal inactivation.
Given our results, we feel that the amnestic effects of translational inhibitors that have been described in past literature can be more accurately explained by the neural silencing induced by ANI and the phenomenon of overshadowing. This may extend to the more recently described phenomenon of reconsolidation as well (Debiec et al., 2002).
Is protein synthesis necessary for neural activity?
More generally, our results could be taken to suggest that intact protein synthesis is a prerequisite for normal electrophysiological signaling in the nervous system. This was actually a downplayed (and widely overlooked) conclusion of classic work on the influence of inhibition of amino acid incorporation on spontaneous pacemaker activity in the nervous system of Aplysia (Schwartz et al., 1971). Upon careful consideration, the idea that translational block would leave other cellular operations intact seems grossly implausible. There are a host of protein classes that are integral for cellular function; namely, enzymatic, cytoskeletal, metabolic, signaling, as well as membrane-associated peptides (e.g., pore-forming complexes) that would be essential for neuronal excitability itself. As well, the essential role of protein synthesis in other cellular organelles, such as mitochondria, must also be carefully considered given their role in cellular respiration (Huchzermeyer et al., 2008) and the generation of ATP (Kann and Kovács, 2007). Given the massive interdependencies of protein–protein interactions at a subcellular level, even the impairment of one class or even a single protein could be expected to compromise general cellular physiology.
Indeed, evidence exists to suggest that specific individual protein products are necessary for neural activity. In a particularly well designed study (Neumann et al., 1995), researchers introduced antisense oligonucleotides (OGNs) to cells in the supraoptic nucleus (SON) of rats. These OGNs were designed as specific transcriptional blocks for the neuropeptides vasopressin and oxytocin, which are expressed by different subsets of cells in the SON. Incorporation of the antisense OGN for vasopressin by vasopressin-expressing neurons (but not oxytocin-expressing neurons) silenced spontaneous spiking activity, and also suppressed responsiveness to both antidromic and chemical stimulation. An identical effect was found when presenting antisense OGN for oxytocin to oxytocin-expressing (but not vasopressin-expressing) neurons. No effects were observed with the presentation of scrambled OGNs to either type of neuron. In another study (Garcia-Osta et al., 2006), intracerebral delivery of antisense OGN for a brain-expressed and muscle-specific tyrosine kinase receptor (MuSK) disrupted the ability of the local hippocampal network to generate theta activity in vitro. These results suggest that specific protein products of a neuron are a vital part of its ability to function on its own and within a network; inhibiting just one product can have devastating effects on electrical activity and responsiveness. Indeed, recent evidence also suggests that the converse is true—that upregulation of a given protein product can serve to enhance neural activity. For instance, it has been shown that the enhancement of CREB levels in the brain increases neuronal activity and excitability, as well as the preferential recruitment of these neurons in memory traces (Lopez de Armentia et al., 2007; Zhou et al., 2009; Josselyn, 2010). Indeed, it may also be that the natural circadian regulation of transcriptional and translational factors (coupled to protein expression) in the suprachiasmatic nucleus influences spontaneous neuronal activity and excitability of these cells as well (Herzog et al., 1998; Ikeda, 2004). Thus, these collective findings suggest far broader implications for studies using molecular biological manipulations of the nervous system.
Conclusions
Here, we have shown that intrahippocampal applications of the translational inhibitors ANI and CHX, in concentrations equivalent to or less than those that have been shown to produce retrograde memory impairments, profoundly disrupted and in some cases abolished both ongoing and evoked hippocampal neural activity. These effects were not due to cellular death; however, they were correlated with measures of protein synthesis inhibition. This constitutes a serious confound for the use of these agents in memory research—regardless of where they are applied in the brain.
The broad implication of our findings is that it is unlikely that molecular processes within neurons can be independently dissociated from their electrical signaling mechanisms. This is supported by the correlation we found between the degree of protein synthesis inhibition and the resultant inhibition of neural activity. Despite the fact that the de novo protein synthesis theory of memory stabilization has apparent biological validity in terms of the cellular changes presumed to be necessary for consolidation, neural activity is even more likely to be essential for this process (Fonseca et al, 2006). Indeed, if intercellular communication is disrupted in the nervous system, not only is neural function impaired but so too is activity-dependent neuroplasticity. In regard to memory consolidation, this point is highlighted by the demonstration that the elimination or accentuation of particular spontaneous activity patterns in the forebrain in the posttraining period can adversely or positively (respectively) modulate future recall (Marshall et al., 2006; Girardeau et al., 2009; Nakashiba et al., 2009; Ego-Stengel and Wilson, 2010). Our findings underscore the importance of the intervening step of neural signaling in the leap from molecular biology to behavior.
Footnotes
This work was supported by Natural Sciences and Engineering Research Council of Canada Grant RGPIN 249861 (C.T.D.). C.T.D. is an Alberta Heritage Foundation for Medical Research Senior Scholar. We thank Caitlin Hughes for technical assistance in performing experimental procedures and analysis.
References
- Adams JM. Ways of dying: multiple pathways to apoptosis. Genes Dev. 2003;17:2481–2495. doi: 10.1101/gad.1126903. [DOI] [PubMed] [Google Scholar]
- Alberini CM. The role of protein synthesis during the labile phases of memory: revisiting the skepticism. Neurobiol Learn Mem. 2008;89:234–246. doi: 10.1016/j.nlm.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrogi Lorenzini CG, Baldi E, Bucherelli C, Sacchetti B, Tassoni G. Neural topography and chronology of memory consolidation: a review of functional inactivation findings. Neurobiol Learn Mem. 1999;71:1–18. doi: 10.1006/nlme.1998.3865. [DOI] [PubMed] [Google Scholar]
- Amzica F, Steriade M. The K-complex: its slow (<1-Hz) rhythmicity and relation to delta waves. Neurology. 1997;49:952–959. doi: 10.1212/wnl.49.4.952. [DOI] [PubMed] [Google Scholar]
- Biedenkapp JC, Rudy JW. Hippocampal and extrahippocampal systems compete for control of contextual fear: role of ventral subiculum and amygdala. Learn Mem. 2009;16:38–45. doi: 10.1101/lm.1099109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissiere S, Zelikowsky M, Ponnusamy R, Jacobs NS, Blair HT, Fanselow MS. Electrical synapses control hippocampal contributions to fear learning and memory. Science. 2011;331:87–91. doi: 10.1126/science.1193785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland BH. The physiology and pharmacology of hippocampal formation theta rhythms. Prog Neurobiol. 1986;26:1–54. doi: 10.1016/0301-0082(86)90019-5. [DOI] [PubMed] [Google Scholar]
- Bohbot V, Otáhal P, Liu Z, Nadel L, Bures J. Electroconvulsive shock and lidocaine reveal rapid consolidation of spatial working memory in the water maze. Proc Natl Acad Sci U S A. 1996;93:4016–4019. doi: 10.1073/pnas.93.9.4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born J. Slow-wave sleep and the consolidation of long-term memory. World J Biol Psychiatry. 2010;11(Suppl 1):16–21. doi: 10.3109/15622971003637637. [DOI] [PubMed] [Google Scholar]
- Bragin A, Jandó G, Nádasdy Z, Hetke J, Wise K, Buzsáki G. Gamma (40–100 Hz) oscillation in the hippocampus of the behaving rat. J Neurosci. 1995;15:47–60. doi: 10.1523/JNEUROSCI.15-01-00047.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G. Two-stage model of memory trace formation: a role for “noisy” brain states. Neuroscience. 1989;31:551–570. doi: 10.1016/0306-4522(89)90423-5. [DOI] [PubMed] [Google Scholar]
- Buzsáki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–340. doi: 10.1016/s0896-6273(02)00586-x. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Czopf J, Kondákor I, Kellényi L. Laminar distribution of hippocampal rhythmic slow activity (RSA) in the behaving rat: current-source density analysis, effects of urethane and atropine. Brain Res. 1986;365:125–137. doi: 10.1016/0006-8993(86)90729-8. [DOI] [PubMed] [Google Scholar]
- Canal CE, Gold PE. Different temporal profiles of amnesia after intra-hippocampus and intra-amygdala infusions of anisomycin. Behav Neurosci. 2007;121:732–741. doi: 10.1037/0735-7044.121.4.732. [DOI] [PubMed] [Google Scholar]
- Canal CE, Chang Q, Gold PE. Amnesia produced by altered release of neurotransmitters after intraamygdala injections of a protein synthesis inhibitor. Proc Natl Acad Sci U S A. 2007;104:12500–12505. doi: 10.1073/pnas.0705195104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Gold PE. Intra-hippocampal lidocaine injections impair acquisition of a place task and facilitate acquisition of a response task in rats. Behav Brain Res. 2003;144:19–24. doi: 10.1016/s0166-4328(03)00063-9. [DOI] [PubMed] [Google Scholar]
- Clement EA, Richard A, Thwaites M, Ailon J, Peters S, Dickson CT. Cyclic and sleep-like spontaneous alternations of brain state under urethane anaesthesia. PLoS One. 2008;3:e2004. doi: 10.1371/journal.pone.0002004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen HD, Ervin F, Barondes SH. Puromycin and cycloheximide: different effects on hippocampal electrical activity. Science. 1966;154:1557–1558. doi: 10.1126/science.154.3756.1557. [DOI] [PubMed] [Google Scholar]
- Davis HP, Squire LR. Protein synthesis and memory: a review. Psychol Bull. 1984;96:518–559. [PubMed] [Google Scholar]
- Debiec J, LeDoux JE, Nader K. Cellular and systems reconsolidation in the hippocampus. Neuron. 2002;36:527–538. doi: 10.1016/s0896-6273(02)01001-2. [DOI] [PubMed] [Google Scholar]
- Dickson CT. Ups and downs in the hippocampus: the influence of oscillatory sleep states on “neuroplasticity” at different time scales. Behav Brain Res. 2010;214:35–41. doi: 10.1016/j.bbr.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Dudai Y. The neurobiology of consolidations, or, how stable is the engram? Annu Rev Psychol. 2004;55:51–86. doi: 10.1146/annurev.psych.55.090902.142050. [DOI] [PubMed] [Google Scholar]
- Ego-Stengel V, Wilson MA. Disruption of ripple-associated hippocampal activity during rest impairs spatial learning in the rat. Hippocampus. 2010;20:1–10. doi: 10.1002/hipo.20707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca R, Nagerl UV, Bonhoeffer T. Neuronal activity determines the protein synthesis dependence of long-term potentiation. Nat Neurosci. 2006;9:478–480. doi: 10.1038/nn1667. [DOI] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res. 1988;452:57–65. doi: 10.1016/0006-8993(88)90008-x. [DOI] [PubMed] [Google Scholar]
- Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- Garcia-Osta A, Tsokas P, Pollonini G, Landau EM, Blitzer R, Alberini CM. MuSK expressed in the brain mediates cholinergic responses, synaptic plasticity, and memory formation. J Neurosci. 2006;26:7919–7932. doi: 10.1523/JNEUROSCI.1674-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardeau G, Benchenane K, Wiener SI, Buzsáki G, Zugaro MB. Selective suppression of hippocampal ripples impairs spatial memory. Nat Neurosci. 2009;12:1222–1223. doi: 10.1038/nn.2384. [DOI] [PubMed] [Google Scholar]
- Goshen I, Brodsky M, Prakash R, Wallace J, Gradinaru V, Ramakrishnan C, Deisseroth K. Dynamics of retrieval strategies for remote memories. Cell. 2011;147:678–689. doi: 10.1016/j.cell.2011.09.033. [DOI] [PubMed] [Google Scholar]
- Hanlon EC, Vyazovskiy VV, Faraguna U, Tononi G, Cirelli C. Synaptic potentiation and sleep need: clues from molecular and electrophysiological studies. Curr Top Med Chem. 2011;11:2472–2482. doi: 10.2174/156802611797470312. [DOI] [PubMed] [Google Scholar]
- Hernandez PJ, Abel T. The role of protein synthesis in memory consolidation: progress amid decades of debate. Neurobiol Learn Mem. 2008;89:293–311. doi: 10.1016/j.nlm.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog ED, Takahashi JS, Block GD. Clock controls circadian period in isolated suprachiasmatic nucleus neurons. Nat Neurosci. 1998;1:708–713. doi: 10.1038/3708. [DOI] [PubMed] [Google Scholar]
- Huchzermeyer C, Albus K, Gabriel HJ, Otáhal J, Taubenberger N, Heinemann U, Kovács R, Kann O. Gamma oscillations and spontaneous network activity in the hippocampus are highly sensitive to decreases in pO2 and concomitant changes in mitochondrial redox state. J Neurosci. 2008;28:1153–1162. doi: 10.1523/JNEUROSCI.4105-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda M. Calcium dynamics and circadian rhythms in suprachiasmatic nucleus neurons. Neuroscientist. 2004;10:315–324. doi: 10.1177/10738584031262149. [DOI] [PubMed] [Google Scholar]
- Iordanov MS, Pribnow D, Magun JL, Dinh TH, Pearson JA, Chen SL, Magun BE. Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol Cell Biol. 1997;17:3373–3381. doi: 10.1128/mcb.17.6.3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isomura Y, Sirota A, Ozen S, Montgomery S, Mizuseki K, Henze DA, Buzsáki G. Integration and segregation of activity in entorhinal-hippocampal subregions by neocortical slow oscillations. Neuron. 2006;52:871–882. doi: 10.1016/j.neuron.2006.10.023. [DOI] [PubMed] [Google Scholar]
- Ji D, Wilson MA. Coordinated memory replay in the visual cortex and hippocampus during sleep. Nat Neurosci. 2007;10:100–107. doi: 10.1038/nn1825. [DOI] [PubMed] [Google Scholar]
- Josselyn SA. Continuing the search for the engram: examining the mechanism of fear memories. J Psychiatry Neurosci. 2010;35:221–228. doi: 10.1503/jpn.100015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Kann O, Kovács R. Mitochondria and neuronal activity. Am J Physiol Cell Physiol. 2007;292:C641–C657. doi: 10.1152/ajpcell.00222.2006. [DOI] [PubMed] [Google Scholar]
- Kleim JA, Bruneau R, Calder K, Pocock D, VandenBerg PM, MacDonald E, Monfils MH, Sutherland RJ, Nader K. Functional organization of adult motor cortex is dependent upon continued protein synthesis. Neuron. 2003;40:167–176. doi: 10.1016/s0896-6273(03)00592-0. [DOI] [PubMed] [Google Scholar]
- Klement D, Past'alková E, Fenton AA. Tetrodotoxin infusions into the dorsal hippocampus block non-locomotor place recognition. Hippocampus. 2005;15:460–471. doi: 10.1002/hipo.20072. [DOI] [PubMed] [Google Scholar]
- Kramis R, Vanderwolf CH, Bland BH. Two types of hippocampal rhythmical slow activity in both the rabbit and the rat: relations to behavior and effects of atropine, diethyl ether, urethane, and pentobarbital. Exp Neurol. 1975;49:58–85. doi: 10.1016/0014-4886(75)90195-8. [DOI] [PubMed] [Google Scholar]
- Leung LS. Generation of theta and gamma rhythms in the hippocampus. Neurosci Biobehav Rev. 1998;22:275–290. doi: 10.1016/s0149-7634(97)00014-6. [DOI] [PubMed] [Google Scholar]
- Lopez de Armentia M, Jancic D, Olivares R, Alarcon JM, Kandel ER, Barco A. cAMP response element-binding protein-mediated gene expression increases the intrinsic excitability of CA1 pyramidal neurons. J Neurosci. 2007;27:13909–13918. doi: 10.1523/JNEUROSCI.3850-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Aharonov G, Fanselow MS. Neurotoxic lesions of the dorsal hippocampus and Pavlovian fear conditioning in rats. Behav Brain Res. 1997;88:261–274. doi: 10.1016/s0166-4328(97)00088-0. [DOI] [PubMed] [Google Scholar]
- Marshall L, Born J. The contribution of sleep to hippocampus-dependent memory consolidation. Trends Cogn Sci. 2007;11:442–450. doi: 10.1016/j.tics.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Marshall L, Helgadóttir H, Mölle M, Born J. Boosting slow oscillations during sleep potentiates memory. Nature. 2006;444:610–613. doi: 10.1038/nature05278. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. Memory—a century of consolidation. Science. 2000;287:248–251. doi: 10.1126/science.287.5451.248. [DOI] [PubMed] [Google Scholar]
- Müller GE, Pilzecker A. Experimentelle Beiträge zur Lehre vom Gedächtnis. Z Psychol Ergänzungsband. 1900;1:1–300. [Google Scholar]
- Nader K. Memory traces unbound. Trends Neurosci. 2003;26:65–72. doi: 10.1016/S0166-2236(02)00042-5. [DOI] [PubMed] [Google Scholar]
- Nakashiba T, Buhl DL, McHugh TJ, Tonegawa S. Hippocampal CA3 output is crucial for ripple-associated reactivation and consolidation of memory. Neuron. 2009;62:781–787. doi: 10.1016/j.neuron.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazer F, Dickson CT. Slow oscillation state facilitates epileptiform events in the hippocampus. J Neurophysiol. 2009;102:1880–1889. doi: 10.1152/jn.90795.2008. [DOI] [PubMed] [Google Scholar]
- Neumann I, Kremarik P, Pittman QJ. Acute, sequence-specific effects of oxytocin and vasopressin antisense oligonucleotides on neuronal responses. Neuroscience. 1995;69:997–1003. doi: 10.1016/0306-4522(95)00394-x. [DOI] [PubMed] [Google Scholar]
- Packard MG, McGaugh JL. Inactivation of hippocampus or caudate nucleus with lidocaine differentially affects expression of place and response learning. Neurobiol Learn Mem. 1996;65:65–72. doi: 10.1006/nlme.1996.0007. [DOI] [PubMed] [Google Scholar]
- Pavlides C, Winson J. Influences of hippocampal place cell firing in the awake state on the activity of these cells during subsequent sleep episodes. J Neurosci. 1989;9:2907–2918. doi: 10.1523/JNEUROSCI.09-08-02907.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado-Alcalá RA, Díaz del Guante MA, Garín-Aguilar ME, Díaz-Trujillo A, Quirarte GL, McGaugh JL. Amygdala or hippocampus inactivation after retrieval induces temporary memory deficit. Neurobiol Learn Mem. 2006;86:144–149. doi: 10.1016/j.nlm.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Qi Z, Gold PE. Intrahippocampal infusions of anisomycin produce amnesia: contribution of increased release of norepinephrine, dopamine, and acetylcholine. Learn Mem. 2009;16:308–314. doi: 10.1101/lm.1333409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quevedo J, Vianna MR, Martins MR, Barichello T, Medina JH, Roesler R, Izquierdo I. Protein synthesis, PKA, and MAP kinase are differentially involved in short- and long-term memory in rats. Behav Brain Res. 2004;154:339–343. doi: 10.1016/j.bbr.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Ribot T. Diseases of memory. New York: Appleton; 1882. [Google Scholar]
- Rudy JW. Is there a baby in the bathwater? Maybe: some methodological issues for the de novo protein synthesis hypothesis. Neurobiol Learn Mem. 2008;89:219–224. doi: 10.1016/j.nlm.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Schall KP, Kerber J, Dickson CT. Rhythmic constraints on hippocampal processing: state and phase-related fluctuations of synaptic excitability during theta and the slow oscillation. J Neurophysiol. 2008;99:888–899. doi: 10.1152/jn.00915.2007. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Schwartz JH, Castellucci VF, Kandel ER. Functioning of identified neurons and synapses in abdominal ganglion of Aplysia in absence of protein synthesis. J Neurophysiol. 1971;34:939–953. doi: 10.1152/jn.1971.34.6.939. [DOI] [PubMed] [Google Scholar]
- Sharma AV, Wolansky T, Dickson CT. A comparison of sleeplike slow oscillations in the hippocampus under ketamine and urethane anesthesia. J Neurophysiol. 2010;104:932–939. doi: 10.1152/jn.01065.2009. [DOI] [PubMed] [Google Scholar]
- Steriade M. Grouping of brain rhythms in corticothalamic systems. Neuroscience. 2006;137:1087–1106. doi: 10.1016/j.neuroscience.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Steriade M, Nuñez A, Amzica F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. 1993;13:3252–3265. doi: 10.1523/JNEUROSCI.13-08-03252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland RJ, Sparks FT, Lehmann H. Hippocampus and retrograde amnesia in the rat model: a modest proposal for the situation of systems consolidation. Neuropsychologia. 2010;48:2357–2369. doi: 10.1016/j.neuropsychologia.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderwolf CH, Kramis R, Robinson TE. Hippocampal electrical activity during waking behaviour and sleep: analyses using centrally acting drugs. Ciba Found Symp. 1977;1977:199–226. doi: 10.1002/9780470720394.ch10. [DOI] [PubMed] [Google Scholar]
- Wanisch K, Wotjak CT. Time course and efficiency of protein synthesis inhibition following intracerebral and systemic anisomycin treatment. Neurobiol Learn Mem. 2008;90:485–494. doi: 10.1016/j.nlm.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Wilson MA, McNaughton BL. Reactivation of hippocampal ensemble memories during sleep. Science. 1994;265:676–679. doi: 10.1126/science.8036517. [DOI] [PubMed] [Google Scholar]
- Wiltgen BJ, Sanders MJ, Anagnostaras SG, Sage JR, Fanselow MS. Context fear learning in the absence of the hippocampus. J Neurosci. 2006;26:5484–5491. doi: 10.1523/JNEUROSCI.2685-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolansky T, Clement EA, Peters SR, Palczak MA, Dickson CT. Hippocampal slow oscillation: a novel EEG state and its coordination with ongoing neocortical activity. J Neurosci. 2006;26:6213–6229. doi: 10.1523/JNEUROSCI.5594-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Won J, Karlsson MG, Zhou M, Rogerson T, Balaji J, Neve R, Poirazi P, Silva AJ. CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nat Neurosci. 2009;12:1438–1443. doi: 10.1038/nn.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]