

Abstract
Brain injury is an etiological factor for temporal lobe epilepsy and can lead to memory and cognitive impairments. A recently characterized excitatory neuronal class in the dentate molecular layer, semilunar granule cell (SGC), has been proposed to regulate dentate network activity patterns and working memory formation. Although SGCs, like granule cells, project to CA3, their typical sustained firing and associational axon collaterals suggest that they are functionally distinct from granule cells. We find that brain injury results in an enhancement of SGC excitability associated with an increase in input resistance 1 week after trauma. In addition to prolonging miniature and spontaneous IPSC interevent intervals, brain injury significantly reduces the amplitude of tonic GABA currents in SGCs. The postinjury decrease in SGC tonic GABA currents is in direct contrast to the increase observed in granule cells after trauma. Although our observation that SGCs express Prox1 indicates a shared lineage with granule cells, data from control rats show that SGC tonic GABA currents are larger and sIPSC interevent intervals shorter than in granule cells, demonstrating inherent differences in inhibition between these cell types. GABAA receptor antagonists selectively augmented SGC input resistance in controls but not in head-injured rats. Moreover, post-traumatic differences in SGC firing were abolished in GABAA receptor blockers. Our data show that cell-type-specific post-traumatic decreases in tonic GABA currents boost SGC excitability after brain injury. Hyperexcitable SGCs could augment dentate throughput to CA3 and contribute substantively to the enhanced risk for epilepsy and memory dysfunction after traumatic brain injury.
Introduction
Brain injury engenders a wide spectrum of neurological complications involving the hippocampus, including an elevated risk for acquired temporal lobe epilepsy and for memory and cognitive dysfunction (McAllister, 1992; Annegers et al., 1998; Herman, 2002; Thompson et al., 2005; Lowenstein, 2009). Although brain injury leads to neuronal damage and enhanced excitability of the dentate gyrus within 1 week after trauma (Lowenstein et al., 1992; Toth et al., 1997), intrinsic excitability of the major glutamatergic neurons remains unchanged (Santhakumar et al., 2000; Howard et al., 2007). However, whether hyperexcitability of a subset of glutamatergic neurons may underlie early postinjury increases in dentate excitability is not known.
Semilunar granule cells (SGCs) are novel excitatory neurons in the dentate inner molecular layer (IML). These have recently been identified as the prime orchestrators of persistent firing in hilar neurons (Larimer and Strowbridge, 2010). Although SGCs, like granule cells, project to CA3, they are unique in the presence of molecular layer axon collaterals (Williams et al., 2007), which could contribute to synaptic excitation of neighboring granule cells. Physiologically, afferent stimulation evokes persistent firing in SGCs, which underlie prolonged synaptic barrages in hilar neurons (Williams et al., 2007). Thus, SGCs are in a pivotal position to regulate dentate feedback circuits and have been proposed to contribute to the integrity of the physiological “dentate gate” (Heinemann et al., 1992) that regulates granule cell throughput (Larimer and Strowbridge, 2010). Since SGCs have dual projection and associational connectivity, and can regulate dentate excitability and hippocampal working memory (Walker et al., 2010), increases in SGC excitability following brain injury could contribute to the development of post-traumatic epilepsy and memory loss.
Granule cell synaptic inhibition undergoes significant changes following brain injury due to loss of hilar interneuronal populations and plasticity of surviving neurons (Toth et al., 1997; Ross and Soltesz, 2000; Santhakumar et al., 2000; Hunt et al., 2011). Moreover, granule cells have persistent “tonic” inhibition mediated by high-affinity, extrasynaptic GABAA receptors (GABAARs) activated by ambient levels of GABA (Stell et al., 2003; Mtchedlishvili and Kapur, 2006). Tonic inhibition can contribute substantially to resting membrane conductance and regulate neuronal gain and excitability (Mitchell and Silver, 2003; Ruiz et al., 2003; Chadderton et al., 2004; Farrant and Nusser, 2005). Receptors underlying granule cell tonic GABA currents are known to be altered in models of acquired epilepsy (Peng et al., 2004; Zhang et al., 2007; Zhan and Nadler, 2009; Rajasekaran et al., 2010), including cortical impact injury (Mtchedlishvili et al., 2010). However, whether the source of inhibitory inputs to SGCs is the same as that of granule cells, whether SGCs have tonic GABA currents, and if brain injury modifies synaptic and tonic inhibition in SGCs, is not known.
This study examines whether concussive brain injury contributes to early changes in the excitability and inhibition of dentate semilunar granule cells, which could underlie increases in dentate excitability observed 1 week after injury.
Materials and Methods
Fluid percussion injury.
All procedures were performed under protocols approved by the Institutional Animal Care and Use Committee of the University of Medicine and Dentistry of New Jersey, Newark, New Jersey. Lateral fluid percussion injury (FPI) was performed on young adult (postnatal days 24–26) male, Wistar rats (Charles River) as described previously (Dixon et al., 1987; Lowenstein et al., 1992; Toth et al., 1997; Santhakumar et al., 2000, 2003). Briefly, the rats were placed in a stereotaxic frame under ketamine-xylazine anesthesia. A 2 mm hole was trephined on the left side of the skull 3 mm caudal to bregma and 3.5 mm lateral from the sagittal suture. Two steel screws were placed 1 mm rostral to bregma and 1 mm to the right of the sagittal suture. A Luer-Lok syringe hub with a 2.6 mm inner diameter was placed over the exposed dura and bonded to the skull with cyanoacrylate adhesive. Neo-Predef was applied to the wound, and the animal was returned to its home cage. One day later, animals were anesthetized with isoflurane and attached to a fluid percussion device (Department of Biomedical Engineering, Virginia Commonwealth University, Richmond, VA). A pendulum was dropped to deliver a brief (20 ms) 2.0–2.2 atm impact on the intact dura. This resulted in a moderate level of injury that has been shown to cause a highly reproducible pattern of hilar cell loss (Toth et al., 1997; Santhakumar et al., 2000). For sham injury, the animals were anesthetized and attached to the fluid percussion device, but the pendulum was not dropped.
Slice preparation.
One week (5–8 d) after FPI or sham injury (Santhakumar et al., 2001), the rats were anesthetized with isoflurane and decapitated. Horizontal brain slices (300 μm for patch-clamp and 400 μm for field experiments) were prepared in ice-cold sucrose artificial CSF (sucrose-aCSF) containing the following (in mm): 85 NaCl, 75 sucrose, 24 NaHCO3, 25 glucose, 4 MgCl2, 2.5 KCl, 1.25 NaH2PO4, and 0.5 CaCl2 using a Leica VT1200S Vibratome. The slices were sagittally bisected, and the slices from the left hemisphere (ipsilateral to the side of injury) were incubated at 32 ± 1°C for 30 min in a submerged holding chamber containing an equal volume of sucrose-aCSF and recording aCSF, and subsequently were held at room temperature. The recording aCSF contained the following (in mm): 126 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 d-glucose. All solutions were saturated with 95% O2 and 5% CO2 and maintained at a pH of 7.4 for 1–6 h.
In vitro electrophysiology.
For patch-clamp recordings, slices (300 μm) were transferred to a submerged recording chamber and perfused with oxygenated aCSF at 33 ± 1°C. Whole-cell voltage-clamp and current-clamp recordings from dentate granule cells and neurons in the IML were performed using infrared differential interference contrast visualization techniques (Stuart et al., 1993; Santhakumar et al., 2006) with a Nikon Eclipse FN-1 microscope, using a 40× water-immersion objective. Recordings were obtained using MultiClamp 700B (Molecular Devices). Data were low-pass filtered at 3 kHz, digitized using DigiData 1440A, and acquired using pClamp10 at 10 kHz sampling frequency. Tonic and synaptic GABA currents were recorded in perfusing aCSF with no added GABA. Except when indicated (see Fig. 9), no GABA transporter antagonists were included in the recording solution. Voltage-clamp recordings of inward GABA currents were obtained using microelectrodes (5–7 MΩ) containing the following (in mm): 125 CsCl, 5 NaCl, 10 HEPES, 2 MgCl2, 0.1 EGTA, 2 Na-ATP, and 0.5 Na-GTP, titrated to a pH 7.25 with CsOH. Biocytin (0.2%) was included in the internal solution for post hoc cell identification, and the glutamate receptor antagonist kynurenic acid (3 mm KyA, Tocris Bioscience) was included in the external solution to isolate GABA currents. In experiments where spontaneous and miniature IPSCs (mIPSCs) were recorded as outward currents from a holding potential of 0 mV, the internal solution consisted of the following (in mm): 125 Cs-methanesulfonate, 5 NaCl, 10 HEPES, 0.2 EGTA, 2 Mg-ATP, 0.2 Na-GTP, and 5 QX-314 with biocytin (0.2%). Among neurons recorded in the IML, only cells showing the widespread dendritic morphology and axon projecting to the hilus (Williams et al., 2007) were analyzed and included in the study. Recordings were discontinued if series resistance increased by >20%. In some experiments, selective GABAAR agonists with a preference for δ-subunit-containing GABAARs, 4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridin-3-ol (THIP, 1 μm), or α-tetrahydrodeoxycorticosterone (THDOC, 20 nm) (Brown et al., 2002; Stell et al., 2003) were included in the external solution. The GABA transporter-1 inhibitor 1-[2-([(diphenylmethylene)imino]oxy)ethyl]-1,2,5,6-tetrahydro-3-pyridinecarboxylic acid (NO-711) was used to test the contribution of GABA uptake mechanisms to changes in tonic GABA currents in Figure 9. All salts were purchased from Sigma-Aldrich. Tonic GABA current, the steady-state current blocked by the GABAAR antagonist bicuculline methiodide (BMI, 100 μm, Sigma-Aldrich) or gabazine (SR95531, 10 μm, Sigma-Aldrich), was measured as described previously (Santhakumar et al., 2006; Glykys and Mody, 2007) using custom macros in IgorPro7.0 software (WaveMetrics).
Figure 9.

Postinjury decrease in SGC tonic GABA currents is maintained in the presence of GABA transporter blocker. A, Representative voltage-clamp recordings (Vhold = 0 mV) from a sham-injured (top) and FPI SGC (bottom) 1 week after injury illustrate the magnitude of tonic GABA current blocked by SR95531 (gabazine, 10 μm). Right, Gaussian fits to all-points histograms derived from 30 s recording periods in aCSF after addition of NO-711 (10 μm) and during SR95531 perfusion used to determine tonic current amplitude. The dashed lines indicate the Gaussian means and the difference currents are noted. B, Summary histogram of SGC tonic GABA currents in aCSF and after addition of NO-711 in recordings from both sham-injured and FPI SGCs. *p < 0.05, paired and unpaired Student's t test.
Membrane voltage recordings were performed using pipettes containing the following (mm): 126 K-gluconate, 4 KCl, 10 HEPES, 4 Mg-ATP, 0.3 Na-GTP, 10 PO-creatinine with 0.2% biocytin. For experiments examining the firing rate and input resistance, cells were held at −70 mV with small current injections. The test pulse consisted of 1 s current injections from −200 pA in steps of 40 pA. The threshold for the first action potential was determined by calculating the first time derivative (dV/dt) of the voltage trace and setting 30 mV/ms as the threshold level for action potential initiation. The membrane voltage at the time when dV/dt value crossed 30 mV/ms was measured as the action potential threshold (Cooper et al., 2003; Howard et al., 2007). Spike frequency adaptation ratio was calculated as the ratio of the interspike interval between the first two and last two spikes in response to a +200 pA current injection for 1 s. In SGCs, the input resistance was determined from the slope of linear fits to the steady-state voltage responses during current injections in the range of −200–0 pA (in 40 pA steps). Input resistance measured from the steady-state voltage responses to −120 pA current injections was used to compare intrinsic properties of granule cells and SGCs. The membrane time constant was fitted to the initial part of the voltage response during −120 pA current injections.
Field recordings were performed in an interface recording chamber (BSC2, Automate Scientific) perfused with aCSF. Brain slices (400 μm) rested on a filter paper and were stabilized with platinum wire weights. The tissue was continuously superfused with humidified 95% O2/5% CO2 and the temperature of the perfusing solution was maintained at 34°C using a proportional control heating unit (PTC03, Automate Scientific). Field recordings of evoked population spikes in the granule cell layer of the dentate gyrus were obtained using patch pipettes filled with recording aCSF. To evoke the field responses, constant current stimuli (0.5–4 mA, 50 μs) were applied at 0.1 Hz through a bipolar 90 μm tungsten stimulating electrode placed in the perforant path at the junction of the dorsal blade and the crest and coupled to a high-voltage stimulus isolator (A365R, WPI). Recordings were obtained using an AxoPatch200B amplifier, filtered at 4 kHz using a Bessel filter, and digitized at 10 kHz with a DigiData 1440A analog-to-digital interface (Molecular Devices). The field responses in the granule cell layer were measured at five predetermined points in each slice (Santhakumar et al., 2000, 2001), including the tips of the dorsal and the ventral blades, the middle of the dorsal and ventral blades, and the middle of the crest, and the largest response was studied further.
Anatomical methods.
Following physiological recordings, slices were fixed in 0.1 m phosphate buffer containing 4% paraformaldehyde at 4°C for 2 d. For post hoc immunohistochemistry, thick slices (300 μm) were incubated overnight at room temperature with antibodies against Prox1 (AB5475, 1:1000, polyclonal rabbit; Millipore) (Szabadics et al., 2010) or parvalbumin (PV-28, 1.5:1000, polyclonal rabbit, Swant) (Földy et al., 2007) in 0.3% Triton X-100 and 2% normal goat serum containing PBS. Immunoreactions were revealed using Alexa Fluor 488-conjugated secondary goat antibodies against rabbit IgG and biocytin staining was revealed using Alexa Fluor 594-conjugated streptavidin. Sections were visualized and imaged using a Zeiss LSM 510 confocal microscope with a 0.5 numerical aperture 20× objective. Cell reconstructions and morphological analyses were performed with Neurolucida V.10.02 (MBF Bioscience) using confocal image stacks.
Fluoro-Jade C staining was performed on sections from rats perfusion fixed with 4% paraformaldehyde 4 h after sham or head injury. Hippocampal sections (40 μm) were mounted on gelatinized slides and air dried. Slides were immersed in 100% alcohol, 70% ethanol, and water for 2 min each followed by a 15 min incubation in 0.06% potassium permanganate before being stained with 0.001% Fluoro-Jade C in 0.1% acetic acid in the dark for 30 min. NeuN staining was performed in sections from rats perfused 1 week after sham or head injury. Sections were incubated overnight at room temperature with anti-NeuN antibody (MAB377, 1:10,000, mouse monoclonal, Millipore) in 0.3% Triton and 2% normal goat serum in PBS. Sections were reacted with Alexa Fluor 594-conjugated goat anti-mouse secondary to reveal staining. Controls in which primary antibody was omitted were routinely included. Quantification was performed on randomly selected sections from septal and temporal poles and midlevels of the hippocampus on the injured side. Cell counts were performed using the Optical Fractionator probe of Stereo Investigator V.10.02 (MBF Bioscience) using an Olympus BX51 microscope and a 100× oil objective. In each section, the hilus was outlined by a contour traced using a 10× objective. The following sampling parameters were set at 100×: counting frame, 50 × 50 μm; dissector height, 15 μm; and top guard zone, 10 μm. Approximately 25 sites per contour, selected using randomized systematic sampling protocols, were sampled. In each section, the cell count was estimated based on planimetric volume calculations in Stereo Investigator (West et al., 1991; West, 1993).
Analysis and statistics.
Individual sIPSCs were detected using custom software in IgorPro7.0 (Santhakumar et al., 2006, 2010). Events were visualized, and any “noise” that spuriously met trigger specifications was rejected. Cumulative probability plots of sIPSC parameters were constructed using IgorPro by pooling equal number of sIPSCs from each cell. Statistical analysis was performed by paired and unpaired Student's t test (Microsoft Excel 2007) or Kolmogorov–Smirnov (K–S) test (in IgorPro7.0) for data that were not distributed normally. Additionally, two-way repeated-measure ANOVA (SysStat) was used to test for statistical differences between the firing of sham-injured and FPI SGC in response to increasing current injections. Significance was set to p < 0.05. Data are shown as mean ± SEM or median and interquartile range (IQR) where appropriate.
Results
Hilar neuronal degeneration and increased dentate excitability 1 week after lateral fluid percussion injury
Concussive brain injury is known to result in instantaneous damage to neurons in the dentate hilus and an increase in dentate excitability as early as 1 week after injury (Lowenstein et al., 1992; Toth et al., 1997; Santhakumar et al., 2000). The severity of injury correlates well with the extent of neuronal damage and the risk for developing temporal lobe epilepsy (Coulter et al., 1996; Toth et al., 1997; Thompson et al., 2005; Kharatishvili et al., 2006). In this study, we used a moderate injury strength of 2.0–2.2 atm pressure, which has been shown to result in reproducible neuronal loss largely restricted to the dentate hilus (Toth et al., 1997) and increase in the ability to evoke limbic seizures (Santhakumar et al., 2001). As illustrated by representative Fluoro-Jade C-stained hippocampal sections from rats perfused 4 h after sham injury or FPI (Fig. 1A,B), degenerating neurons labeled by Fluoro-Jade C were observed in the hilus of head-injured rats (214.8 ± 64.3 hilar neurons/section in 12 sections from three rats) and not in sham-injured controls (0 ± 0 hilar neurons/section in 9 sections from three rats, p < 0.05 t test vs FPI). These data are consistent with rapid neuronal degeneration in the dentate hilus following mechanical injury to neurons during impact (Toth et al., 1997). Additionally, NeuN staining for neuronal nuclei was performed to verify the presence of hilar neuronal loss at later time points. Comparison of sections prepared 1 week after sham or head injury revealed a significant decrease in NeuN-stained neurons in the dentate hilus 1 week after FPI (sham injured: 306.8 ± 33.2 hilar neurons/section, a total of 444 cells counted in 12 sections from 3 rats; FPI: 204.3 ± 30.8 hilar neurons/section, based on 364 cells counted in 14 sections from 3 rats, 33.5 ± 9.6% decrease, p < 0.05, t test). To assess the early post-traumatic changes in dentate excitability, we examined field recordings of afferent-evoked granule cell population responses 1 week after FPI. The amplitude of the granule cell population spike evoked by perforant path stimulation was larger in slices from head-injured rats compared with age-matched sham-injured controls (Fig. 1C). Summary data in Figure 1D demonstrate the postinjury increase in dentate-evoked population response at various stimulation intensities (sham injured: n = 9 slices from 5 rats; FPI: n = 8 slices from 3 rats) and confirm that our system reliably replicates the post-traumatic increase in dentate excitability, which has been observed after moderate concussive head trauma and proposed to augment limbic epileptogenicity after brain injury (Lowenstein et al., 1992; Toth et al., 1997; Santhakumar et al., 2001).
Figure 1.

Dentate hilar cell death and increased excitability after concussive brain injury. A, Representative Fluoro-Jade C-stained section from a rat perfused 4 h after sham injury shows no fluorescently labeled neurons, illustrating the absence of dying neurons in sham-injured controls. B, Fluoro-Jade C-stained section from a rat perfused 4 h after FPI shows numerous labeled dying neurons in the hilus. C, Representative traces of granule cell layer field responses evoked by perforant path stimulation in slices from a sham-injured (left) and head-injured (right) rat obtained 1 week after FPI illustrates the larger population spike amplitude in the injured dentate gyrus compared with sham injury. Traces are an average of four trials in response to a 4 mA stimulus to the perforant path. Arrows indicate location of stimulus artifact that was truncated. D, Summary data demonstrate the postinjury increase in afferent-evoked excitability of the dentate gyrus at various stimulation intensities. Error bars indicate SEM. *p < 0.05, unpaired Student's t test. Wk, Week; GCL, granule cell layer.
Early post-traumatic hyperexcitability of semilunar granule cells
Since post-traumatic increase in dentate excitability is well established 1 week after injury, we focused on this early time point in the current study. Although increases in excitability of granule cells or mossy cells are potential direct mechanisms to account for post-traumatic increases in dentate field excitability, little postinjury change in firing has been found in these two dentate glutamatergic neurons (Santhakumar et al., 2000; Howard et al., 2007). We examined whether the excitability of SGCs, a population of neurons originally described by Ramón y Cajal (1995) and recently characterized as glutamatergic dentate projection neurons (Williams et al., 2007), is altered after brain injury.
To ascertain whether we could reliably distinguish granule cells from SGCs, we recorded and filled granule cells in the granule cell layer and presumed SGCs in the IML. Similar to observations in naive rats (Williams et al., 2007), dendrites of SGCs from sham-injured rats had a wider span compared with the relatively compact dendritic spread of granule cells (Fig. 2A,B; maximum dendritic spread in μm, granule cell: 289.0 ± 24.0, n = 8; SGC: 509.6 ± 40.1, n = 11, p < 0.05, t test). The angle of dendritic spread, the maximum angle subtended by the dendrites at the soma, was also significantly larger in SGCs, confirming that SGCs are morphologically distinct from granule cells (angle of dendritic spread in degrees, granule cell: 55.4 ± 5.4, n = 8; SGC: 115.9 ± 6.9, n = 11, p < 0.05, t test). Consistent with previous findings (Williams et al., 2007), 3 of 10 SGCs had associational axon collaterals in the granule cell layer or IML (Fig. 3A, arrowhead). SGCs also exhibited extensive axonal branching in the subgranular region of the hilus and four of eight fully reconstructed SGCs had axons projecting all the way to CA3 (Fig. 2B). The morphological similarities between SGCs and granule cells have led to the suggestion that SGCs may be a class of granule cells distinguished by their location. Nuclear expression of the homeodomain transcription factor Prox1 has been used as a specific marker to identify granule cells both in the granule cell layer and in ectopic locations (Jessberger et al., 2008; Lavado et al., 2010; Szabadics et al., 2010). As expected, all eight biocytin-filled and recorded granule cells examined expressed Prox1 (Fig. 2A). Since the presence or absence of Prox1 could provide insights into SGC lineage, we tested morphologically identified SGCs for Prox1 immunolabeling. As illustrated by insets in Figures 2B and 3, A and B, biocytin-filled SGCs demonstrated nuclear labeling for Prox1 (all eight cells tested), indicating a shared lineage with granule cells.
Figure 2.
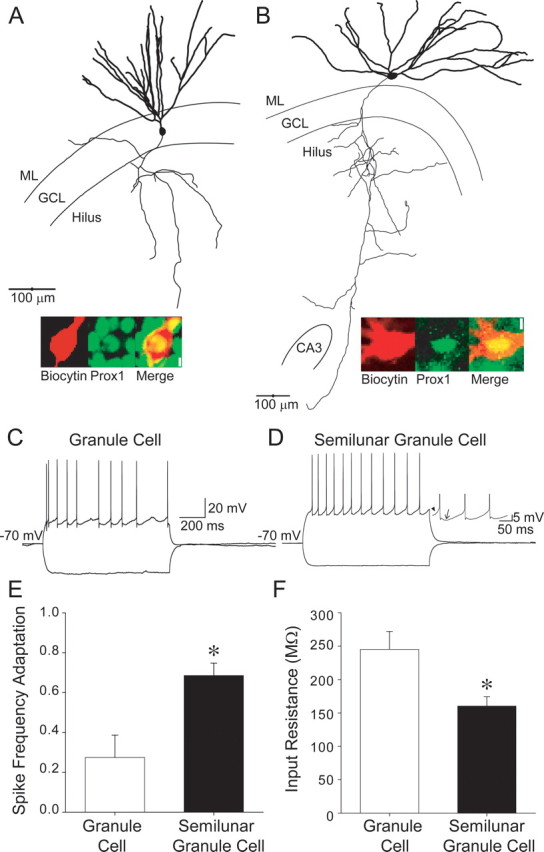
Features that distinguish granule cells from semilunar granule cells. A, Illustration of a fully reconstructed granule cell shows the typical location of the somata in the granule cell layer (GCL) and compact dendritic spread in the molecular layer (ML). The axon (mossy fiber, thin line) is seen projecting in the hilus, toward CA3. Inset, Confocal image shows labeling for biocytin (left), Prox1 (middle), and the merged image (right), illustrating colabeling. Scale bar, 5 μm. B, Reconstruction of a biocytin-filled semilunar granule cell shows the location of somata in the ML and demonstrates the wider dendritic span compared with the granule cell in A. Note the high degree of branching of the SGC axon (thin line) in the hilus and projection to CA3. Inset, Confocal image of the somata of the SGC in B shows labeling for biocytin (left), Prox1 (middle), and the merged image (right), illustrating colabeling. Scale bar, 5 μm. C, Membrane voltage traces from the granule cell in A show the highly adapting firing pattern in response to +200 pA current injection and hyperpolarization during a −120 pA current injection. D, Current-clamp recordings from the semilunar granule cell in B illustrate the continuous firing with low adaptation during a +200 pA depolarizing current injection from a holding potential of −70 mV. Note that the hyperpolarization in response to a −120 pA current injection is smaller than in the granule cell in C. Inset, Expanded membrane voltage trace shows the slow ramp depolarization (arrowhead) and large, slow afterhyperpolarization (arrow) that are distinctive of SGCs. E, Summary histogram shows lower spike frequency adaptation ratio (i.e., higher adaptation) in granule cells compared with SGCs. F, Summary plot illustrates the low input resistance of SGCs compared with granule cells. *p < 0.05, Student's t test.
Figure 3.
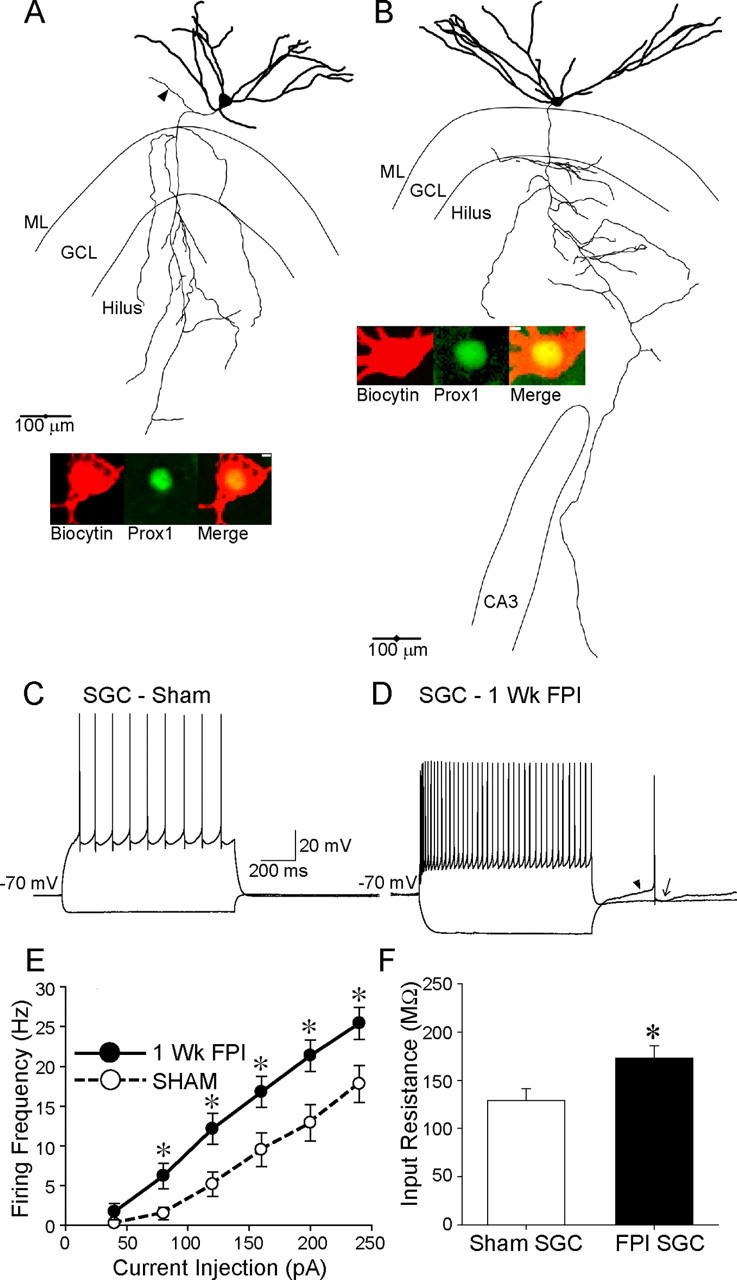
Increase in excitability of semilunar granule cells after brain injury. A, B, Biocytin-filled and reconstructed semilunar granule cells in experiments performed 1 week after sham injury (A) and head injury (B) show the SGC soma in the molecular layer (ML) and widespread dendrites (thick lines). Axons (thin lines) of both control and injured SGCs are seen projecting toward CA3. Note the axon collateral in the inner molecular layer (arrowhead) of the control SGC in A. Insets, Confocal images of biocytin (left) and Prox1 labeling (middle) of the SGC soma. The merged images (right) demonstrate Prox1 labeling in SGCs from both sham-injured and FPI rats. Scale bar, 5 μm. C, Example membrane voltage traces from the sham-injured SGC in A show the nonadapting firing in response to a +200 pA current injection and hyperpolarization during a −120 pA current injection. D, Representative recordings in the FPI SGC in B illustrate the higher firing frequency for the same +200 pA depolarizing current injection as in C. Additionally, the hyperpolarization in response to a −120 pA current injection is larger than in the sham-injured SGC (C). Note that the characteristic slow ramp depolarization (arrowhead) and large slow afterhyperpolarization (arrow) are observed in the FPI SGC. E, Summary plot of firing rates of sham-injured and FPI SGCs during 1 s depolarizing current steps shows the enhanced firing frequency in FPI SGC. F, Histograms show that FPI SGCs have higher input resistance than controls. Sham-injured SGC data are derived from the same group of cells as in Figure 2. *p < 0.05, Student's t test. GCL, Granule cell layer; Wk, week.
In addition to morphological differences, granule cells and SGCs demonstrate certain distinct physiological characteristics (Williams et al., 2007). In response to depolarizing current steps from a holding potential of −70 mV, both SGCs and granule cells responded with continuous firing (Fig. 2C,D). The prolonged granule cell firing in response to current injection, while consistent with earlier whole-cell recordings (Staley et al., 1992; Lübke et al., 1998; Santhakumar et al., 2000), differs from the highly adapting firing observed by Williams et al. (2007). Despite continuous firing in both SGCs and granule cells, firing rates in SGCs demonstrated considerably higher adaptation ratios, indicating lower spike-frequency adaptation, than in granule cells (Fig. 2E; adaptation ratio, granule cell: 0.3 ± 0.1, n = 8; SGC: 0.7 ± 0.1, n = 11, p < 0.05, t test). SGCs also displayed a characteristic slow ramp potential before each action potential and were followed by a distinctive slow afterhyperpolarization (Fig. 2D, inset), as reported by Williams et al. (2007). Moreover, the input resistance (Rin) of SGCs was significantly lower than that of granule cells (Fig. 2F; in MΩ, granule cell: 254.2 ± 26.9, n = 8; SGC: 160.1 ± 14.3, n = 13, p < 0.05, t test), as has been reported previously (Williams et al., 2007). Similarly, the membrane time constant of SGCs was lower than in granule cells (in ms, granule cell: 20.7 ± 2.3, n = 8; SGC: 15.0 ± 1.6, n = 13, p < 0.05, t test). These data show that regardless of the common neurochemical marker and axonal projection, SGCs and granule cells can be reliably distinguished based on somatodendritic morphology and active and passive physiological properties.
Next, we compared the intrinsic properties of sham-injured SGCs with FPI SGCs from rats 1 week after head injury. Neurons recorded in the IML were filled during recordings and processed for biocytin immunohistology. Only SGCs identified based on somatodendritic morphology and axonal projection to the hilus were analyzed further (Fig. 3A,B). Like sham-injured SGCs, FPI SGCs could be identified by the presence of the ramp depolarization before the action potential and pronounced slow afterhyperpolarization (Fig. 3D). We examined the mean SGC firing frequency in response to 1 s current injections from a holding potential of −70 mV. As illustrated in Figure 3C–E, firing frequency of FPI SGCs was significantly greater than that of sham-injured SGCs (difference between sham-injured SCG and FPI SGC firing was significant; F(1,22) = 6.9, p < 0.05 by two-way repeated-measures ANOVA, n = 12 sham-injured SGCs and 14 FPI SGCs). However, there was no apparent depolarizing shift in the resting membrane potential (in mV, sham-injured SGC: −86.1 ± 2.15, n = 13 cells; FPI SGC: −90.5 ± 4.0, n = 14 cells, p > 0.05, t test) or reduction in action potential threshold (in mV, sham-injured SGC: −39.9 ± 2.8, n = 13 cells; FPI SGC: −39.8 ± 3.5, n = 14 cells, p > 0.05, t test) between sham-injured and FPI SGCs (values were not corrected for junctional potentials). FPI SGCs showed lower adaptation ratios, indicating greater spike-frequency adaptation, during firing compared with sham-injured SGCs (adaptation ratio: sham-injured SGC: 0.71 ± 0.1, n = 12 cells; FPI SGC: 0.45 ± 0.0, n = 14 cells, p < 0.05, t test), suggesting that there may be postinjury changes in intrinsic membrane currents in SGCs. Notably, as illustrated by the membrane voltage traces in Figure 3, C and D, FPI SGCs showed greater hyperpolarization in response to a −120 pA current injection compared with sham-injured SGCs. Accordingly, the input resistance of FPI SGCs was greater than in sham-injured SGCs (Fig. 3F; Rin measured as the slope of linear fits to the average voltage response during the last 200 ms of 1 s current injections from −200 to −40 pA, sham-injured SGC: 132.2 ± 12.3 MΩ, n = 13 cells; FPI SGC: 173.0 ± 12.8 MΩ, n = 13 cells, p < 0.05, t test). Our data constitute the first demonstration of changes in SGC excitability in a model of neurological disease.
Post-traumatic changes in SGC synaptic inhibition
As might be predicted based on loss of hilar interneurons and plasticity of the surviving interneurons following brain injury (Lowenstein et al., 1992; Toth et al., 1997; Ross and Soltesz, 2000; Santhakumar et al., 2000; Hunt et al., 2011), synaptic inhibition of granule cells and mossy cells undergo profound post-traumatic modifications (Santhakumar et al., 2001; Howard et al., 2007; Mtchedlishvili et al., 2010). Curiously, despite postinjury hilar interneuronal loss and a corresponding decrease in action potential-independent mIPSCs, the frequency of spontaneous IPSCs in granule cells and mossy cells is elevated following FPI (Santhakumar et al., 2001; Howard et al., 2007). Increased excitability of certain interneurons and enhanced excitatory drive to surviving interneurons after brain injury may contribute to an increase in sIPSC frequency following brain injury (Ross and Soltesz, 2000; Santhakumar et al., 2001; Hunt et al., 2011). Since the dendritic distribution of SGCs corresponds with the axonal distribution of hilar interneurons vulnerable to postinjury loss (Lowenstein et al., 1992; Toth et al., 1997), we examined whether brain injury altered sIPSC parameters in SGCs. Recordings were performed in the presence of the glutamate receptor antagonist kynurenic acid (3 mm) to block excitatory synaptic currents and isolate IPSCs. Complete block of synaptic events following perfusion of BMI (100 μm) was used to confirm that the synaptic events were mediated by GABAARs (Fig. 4A, top and bottom, traces labeled BMI). One week after FPI, the interevent interval of sIPSC in SGCs was prolonged compared with that in sham-injured SGCs (Fig. 4A,B; in ms, sham-injured SGC: 64.2 ± 2.5, median = 45.9, IQR = 26.5–82.1, n = 6 cells; FPI SGC: 207.9 ± 17.3, median = 95.2, IQR = 46.0–198.5, n = 6 cells, p < 0.05, K–S test) indicating a decrease in sIPSC frequency. However, there was no change in either the sIPSC amplitude (Fig. 4C; in pA, sham-injured SGC: 31.2 ± 9.1, median = 24.9, IQR = 18.4–38.4, n = 6 cells; FPI SGC: 34.5 ± 10.4, median = 25.7, IQR = 17.8–41.9, n = 6 cells, p > 0.05, K–S test) or the 20–80% rise time in FPI SGCs (in ms, sham-injured SGC: 0.2 ± 0.0, n = 6 cells; FPI SGC: 0.2 ± 0.0, n = 6 cells, p > 0.05, t test). The amplitude-weighted decay time constant (τdecay) of sIPSCs showed a small but statistically significant decrease in FPI SGCs (Fig. 4D; in ms, sham-injured SGC: median = 4.2, IQR = 3.3–5.3; FPI SGC: median = 3.5, IQR = 2.8–4.7, n = 6 cells each, p < 0.05, K–S test). Since previous studies have demonstrated loss of several major hilar neuronal populations, including those expressing somatostatin, parvalbumin, cholecystokinin, and substance P receptor after FPI (Lowenstein et al., 1992; Toth et al., 1997; Santhakumar et al., 2000), the observed decrease in SGC sIPSC frequency may be due to the postinjury loss of hilar interneurons and a resulting reduction in inhibitory drive to SGCs.
Figure 4.

Decrease in SGC spontaneous IPSC frequency after FPI. A, Representative traces of voltage-clamp recordings from sham-injured SGCs (top two traces) and FPI SGCs (bottom two traces) show the higher sIPSC frequency in sham-injured SGC (top trace) and the complete block of synaptic events in BMI (100 μm) in the same cell. Note the decrease sIPSC frequency in the recording from an FPI SGC (bottom trace) and subsequent block of synaptic events in BMI (100 μm). B–D, Cumulative probability plots of interevent interval (B), amplitude (C), and weighted decay time constant (D) of sIPSCs recorded in kynurenic acid (3 mm) in sham-injured SGCs (black) and FPI SGCs (gray). Labels in C apply to B and D as well. Vertical dashed lines indicate median of the distribution at p = 0.5. D, Inset, Overlay of normalized average sIPSC traces from a sham-injured SGC (black) and an FPI SGC (gray) illustrate the more rapid decay of sIPSCs after brain injury. The same number of individual events was selected from each cell to develop the cumulative distribution (sham injured: n = 6 cells; FPI: n = 6 cells).
Our data demonstrating post-traumatic prolongation of sIPSC interevent interval in SGCs are in direct contrast to the early decrease in sIPSC interevent interval in granule cells and mossy cells (Ross and Soltesz, 2000; Santhakumar et al., 2001; Howard et al., 2007). What accounts for the post-traumatic increase in sIPSC interevent interval in SGCs instead of the decrease in granule cells? Could differences in experimental conditions, such as the inclusion of glutamate receptor antagonists during recordings, be responsible? Indeed, previous experiments have shown that the application of glutamate receptor antagonists leads to a greater reduction in sIPSC frequency in granule cells from head-injured rats (FPI granule cells) compared with sham-injured controls (Santhakumar et al., 2001). Under our experimental conditions, we found that in contrast to SGCs, sIPSC interevent interval in granule cells was reduced after FPI (Fig. 5A,B; in ms, sham-injured granule cell: 138.1 ± 41.5, median = 89.1, IQR = 44.0–171.8, n = 12 cells; FPI granule cell: 110.2 ± 45.8, median = 75.5, IQR = 39.9–136.5, n = 7 cells, p < 0.05, K–S test). These data are consistent with earlier experiments in granule cells performed in the absence of glutamate receptor antagonists (Ross and Soltesz, 2000; Santhakumar et al., 2001). The amplitude of sIPSCs in granule cells was also significantly increased after head injury (Fig. 5C; in pA, sham-injured granule cell: 32.9 ± 7.0, median = 25.0, IQR = 17.9–38.4, n = 12 cells; FPI granule cell: 50.0 ± 10.9, n = 7 cells, median = 37.1, IQR = 243–62.3, p < 0.05, K–S test). However, τdecay of granule cell sIPSCs was not altered after head injury (in ms, sham-injured granule cell: 4.7 ± 0.8, median = 4.1, IQR = 3.0–5.5, n = 12 cells; FPI granule cell: 5.4 ± 3.7, median = 3.9, IQR = 2.8–5.07, n = 7 cells, p > 0.05, K–S test). As in SGCs, synaptic events were fully blocked by BMI (100 μm; Fig. 5A, traces labeled BMI). The striking agreement of our sIPSC data from granule cells with results of earlier studies in granule cells and mossy cells (Ross and Soltesz, 2000; Santhakumar et al., 2001; Howard et al., 2007), and the diametrically opposite change observed in SGCs under identical experimental conditions, demonstrate the cell-type specificity of post-traumatic decrease in synaptic inhibition in SGCs.
Figure 5.

Increase in granule cell sIPSC frequency and amplitude after brain injury. A, Example current traces of individual sIPSCs in sham-injured (top two traces) and FPI (bottom two traces) granule cells illustrate the lower frequency and amplitude in sham-injured granule cells compared with FPI granule cells. The block of synaptic events in BMI (100 μm) is illustrated in the respective lower traces. B, C, Cumulative probability plots of granule cell sIPSC interevent interval (B) and amplitude (C) in sham-injured SGCs (black) and FPI SGCs (gray). Vertical dashed lines indicate median of the distribution at p = 0.5. Identical number of events from each cell were used in the analysis (control: n = 12 cells; FPI: n = 7 cells). Recordings were performed in 3 mm kynurenic acid.
To determine whether brain injury resulted in changes in synaptic inhibitory input to SGCs even under conditions in which glutamate receptors were not blocked, we recorded sIPSCs at a holding potential of 0 mV, close to the reversal potential for glutamatergic synaptic events (Fig. 6A,B). The inward synaptic currents recorded at 0 mV were fully blocked by SR95531 (10 μm), indicating that we could effectively isolate GABAAR-mediated synaptic events (data not shown). Similar to our observations in glutamate antagonists (Fig. 4), there was an increase in sIPSC interevent interval in SGCs 1 week after FPI (Fig. 6A,B; in ms, sham-injured SGC: 72.0 ± 2.6, median = 54.6, IQR = 33.0–92.1, n = 5 cells; FPI SGC: 103.1 ± 6, median = 68.1, IQR = 38.8–126.8, n = 3 cells, p < 0.05, K–S test, 100 events from each cell were included in the analysis), indicating a decrease in SGC sIPSC frequency after FPI. SGC sIPSC amplitude showed a small but statistically significant increase after brain injury (Fig. 6A; in pA, sham-injured SGC: 26.0 ± 0.7, median = 21.5, IQR = 15.1–31.6, n = 5 cells; FPI SGC: 26.6 ± 0.8, median = 24.1, IQR = 18.0–32.2, n = 3 cells, p < 0.05, K–S test). Once again, these data confirmed the postinjury decrease in SGC sIPSCs and revealed the striking contrast to the enhanced granule cell sIPSC frequency observed in earlier studies (Santhakumar et al., 2001).
Figure 6.

Decrease in spontaneous and miniature IPSC frequency in SGCs after FPI. A, Representative traces of voltage-clamp recordings from SGCs held at 0 mV show the higher sIPSC frequency in sham-injured SGC (top) and a decrease in sIPSC frequency in the recording from an FPI SGC (bottom). B, Cumulative probability plots of sIPSC interevent interval in sham-injured (black) and FPI (gray) SGCs. Vertical dashed lines indicate median of the distribution at p = 0.5. C, Voltage-clamp recording of miniature IPSCs in sham-injured SGC (top) and FPI SGC (bottom). D, Cumulative probability plots of mIPSC interevent interval in sham-injured (black) and FPI SGCs (gray). Vertical dashed lines indicate median of the distribution at p = 0.5.
Previous studies have demonstrated that neuronal loss after moderate FPI is confined to the dentate hilus and that most major hilar interneuronal populations are lost to a similar extent after FPI (Toth et al., 1997; Santhakumar et al., 2000). Therefore, we considered the possibility that the loss of hilar neurons may contribute to postinjury decreases in inhibitory synaptic input to SGC. To directly test for injury-induced changes in SGC inhibitory inputs, we examined whether the action potential-independent mIPSCs in SGCs are altered after FPI. Voltage-clamp recordings of outward inhibitory synaptic currents (Vhold = 0 mV) in the presence of the sodium channel blocker TTX (1 μm) demonstrated an increase in SGC mIPSC interevent interval after brain injury (Fig. 6C,D; in ms, sham-injured SGC: 123.2 ± 6.1, median = 78.2, IQR = 39.9–150.1, n = 4 cells; FPI SGC: 213.0 ± 18.4, median = 107.8, IQR = 50.1–217.18, n = 5 cells, p < 0.05, K–S test; 100 events from each cell were included in the analysis). The amplitude of mIPSCs in SGCs was also enhanced after FPI (in pA, sham-injured SGC: 26.0 ± 0.6, median = 23.1, IQR = 17–32.1, n = 4 cells; FPI SGC: 29.2 ± 0.5, median = 27.2, IQR = 21.3–34.8, n = 5 cells, p < 0.05, K–S test). The postinjury increase in mIPSC interevent interval is similar to findings in granule cells and mossy cells after FPI (Toth et al., 1997; Howard et al., 2007), and is consistent with a decrease in inhibitory inputs to SGCs. Since our data (Fig. 1) and previous studies (Toth et al., 1997; Santhakumar et al., 2000) have demonstrated that the neuronal loss after moderate FPI is restricted to the hilus, our findings suggest that loss of hilar interneurons could underlie the postinjury decrease in SGC mIPSC frequency. Although the decrease in mIPSC frequency is consistent across SGCs (our data), granule cells (Toth et al., 1997), and mossy cells (Howard et al., 2007), the distinctive decrease in sIPSC frequency in SGCs (current study) compared with the increase in granule cells (Santhakumar et al., 2001) and mossy cells (Howard et al., 2007) suggests that different classes of interneurons may contribute to synaptic inhibition in SGCs and granule cells after head injury.
Cell-type-specific differences in SGC and granule cell synaptic inhibition
Could inherent, cell-type-specific differences in inhibitory input to SGCs and granule cells contribute to the opposite post-traumatic changes in sIPSC interevent interval in the two cell types? Previous studies have identified that GABAergic basket cells expressing the calcium-binding protein parvalbumin (PV) are instrumental in providing perisomatic inhibition to granule cells (Kraushaar and Jonas, 2000; Hefft and Jonas, 2005). The predominant axonal distribution of PV+ basket cells is to the granule cell layer (Hefft and Jonas, 2005). A salient feature of PV+ interneurons in the granule cell layer is that, unlike hilar interneurons, they are relatively resistant to cell loss following brain injury (Toth et al., 1997). During immunohistochemical staining, we found that granule cells filled with biocytin during recordings were typically surrounded by a dense mesh of axons labeled for PV in sections from both sham-injured and head-injured rats (data not shown). On the other hand, SGCs were located in the molecular layer, which showed a relatively sparse labeling for parvalbumin in sections from both sham-injured and FPI rats (data not shown). These observations suggest that, compared with granule cells, SGCs may receive fewer somatic inputs from PV+ basket cells.
To determine whether intrinsic differences in synaptic inhibitory input to SGCs and granule cells may contribute to the divergent postinjury responses, we compared the sIPSC parameters of SGCs and granule cells from sham-injured control rats (in recordings performed with Cs-Cl-based internal solutions in the presence of 3 mm kynurenic acid). Curiously, our data show that the sIPSC interevent interval in sham-injured SGCs was shorter than in sham-injured granule cells (Fig. 7A,B; in ms, SGC: 64.2 ± 2.5, n = 6 cells; granule cell: 138.1 ± 41.5, n = 12 cells, p < 0.05, K–S test), contributing to a higher sIPSC frequency in SGCs (Fig. 7B, inset; in Hz, SGC: 24.6 ± 2.3, n = 6 cells; granule cell: 17.8 ± 1.6, n = 12 cells, p < 0.05, t test). Additionally, sIPSC rise time was significantly slower in SGCs (Fig. 7C; in ms, SGC: 0.23 ± 0.01, n = 6 cells; granule cell: 0.20 ± 0.01, n = 12 cells, p < 0.05 t test), suggesting that SGCs received a greater proportion of IPSCs from dendritically projecting interneurons compared with granule cells. While the largest amplitude events in granule cells were larger than in SGCs, the difference in sIPSC amplitude between SGCs and granule cells was not statistically significant (Fig. 7D; in pA, SGC: 31.2 ± 9.07, median = 24.9, IQR = 18.4–38.4, n = 6 cells; granule cell: 32.9 ± 7.0, median = 25.0, IQR = 17.9–38.4, n = 12 cells, p > 0.05, K–S test). Similarly, there was no difference in the sIPSC τdecay between SGCs and granule cells (in ms, SGC: median = 4.2, IQR = 3.3–5.3; granule cell: median = 4.1, IQR = 3.0–5.5, n = 12 cells, p > 0.05, K–S test). Together, the higher sIPSC frequency in control SGCs and the postinjury decrease in spontaneous and miniature IPSC frequency indicate that SGCs receive greater inhibitory input from populations of hilar interneurons vulnerable to injury-induced cell loss compared with granule cells. These findings revealed unexpected, and hitherto unknown, differences in inhibition between SGCs and granule cells.
Figure 7.

Intrinsic diversity in SGC and granule cell synaptic inhibition. A, Representative voltage-clamp recordings (Vhold = −70 mV) of individual sIPSCs in a granule cell (top) and an SGC (bottom) from a sham-injured rat. Note the lower sIPSC frequency in the granule cell compared with the SGC. B, Cumulative probability plots compare the sIPSC interevent interval granule cells (black) and SGCs (gray) in control rats recorded in 3 mm kynurenic acid. Inset, Summary histogram of sIPSC frequency in granule cells (GCs) and SGCs. C, Summary histogram of the 20–80% rise time in control granule cells and SGCs. D, Cumulative probability plots of sIPSC amplitude in granule cells (black) and SGCs (gray). Vertical dashed lines indicate median of the distribution at p = 0.5. Identical number of events from each cell were used in the analysis (GC: n = 12 cells; SGC: n = 6 cells). Labels in D apply to B and D. sIPSC data were derived from the same control group of cells as in Figures 4 and 5. *p < 0.05, Student's t test.
Brain injury decreases SGC tonic GABA currents
Apart from the classical synaptic inhibitory currents, granule cells express tonic GABA currents mediated by extrasynaptic and perisynaptic GABAAR containing α4 and δ subunits (Stell et al., 2003; Wei et al., 2003; Peng et al., 2004; Mtchedlishvili and Kapur, 2006). Given the similarities between SGCs and granule cells, and the evidence for injury-induced changes in granule cell tonic inhibition (Mtchedlishvili et al., 2010), we examined SGCs for the presence and postinjury changes in tonic GABA currents. In morphologically identified SGCs, application of a saturating concentration of the GABAAR antagonist BMI (100 μm) decreased the holding current, indicating the presence of tonic GABA currents (Fig. 8A). The magnitude of tonic GABA currents in SGCs was significantly decreased after FPI (Fig. 8A,B; in pA, sham-injured SGC: 16.7 ± 1.7, n = 8 cells; FPI SGC: 4.1 ± 0.9, n = 9 cells, p < 0.05, t test). Even when the tonic GABA currents were normalized by cell membrane capacitance to eliminate confounding effects due to differences in cell size, tonic GABA currents in SGCs from head-injured rats were lower than in sham-injured controls (in pA/pF, sham-injured SGC: 0.18 ± 0.04, n = 8 cells; FPI SGC: 0.07 ± 0.02, n = 9 cells, p < 0.05, t test). Tonic GABA currents were measured at physiological temperature in the absence of added GABA or GABA transporter inhibitors. In a subset of experiments in which SGC tonic GABA currents were measured without prior application of the GABA modulator THIP, tonic GABA currents in SGCs still showed a significant decrease after FPI (in pA, sham-injured SGC: 15.8 ± 1.5, n = 3 cells; FPI SGC: 2.4 ± 1.1, n = 4 cells, p < 0.05, t test). Since comparison of baseline tonic GABA currents in kynurenic acid, measured in experiments without and with subsequent perfusion of THIP, revealed no statistical difference in either sham-injured SGCs (tonic GABA currents in pA, in kynurenic acid without THIP: 15.8 ± 1.5, n = 3 cells; in experiments including THIP modulation: 17.2 ± 3, n = 5 cells, p > 0.05, t test) or FPI SGCs (tonic GABA currents in kynurenic acid in pA, without THIP: 2.4 ± 1.1, n = 3 cells; in experiments including THIP modulation: 5.4 ± 1.1, n = 5 cells, p > 0.05, t test), the data were pooled (Fig. 8B). As illustrated in Figure 8A,C, THIP (1 μm) increased the baseline holding current and thereby potentiated tonic GABA currents (tonic GABA currents in pA, sham-injured SGC 17.2 ± 3 in kynurenic acid and 47.9 ± 10.5 in THIP, n = 5 cells, p < 0.05, t test; FPI SGC 5.4 ± 1.1 in kynurenic acid and 15.5 ± 3.7 in THIP, n = 5 cells, p < 0.05, t test), demonstrating the role of GABAAR δ subunits in SGC tonic GABA currents. The magnitude of SGC tonic GABA currents measured in the presence of THIP was also decreased after brain injury (Fig. 8C). However, the extent to which THIP enhanced SGC tonic GABA currents was not altered after FPI (increase with THIP, sham-injured SGC: 310.6 ± 78.6%, n = 5 cells; FPI SGC: 294.0 ± 77.8%, n = 5 cells, p > 0.05, t test). Because the lack of an immunological marker to differentiate between granule cells and SGCs renders it difficult to directly quantify injury-induced changes in GABAAR δ subunit expression in SGCs, we performed additional physiological experiments in morphologically identified SGCs to determine whether GABAAR currents mediated by δ subunits are altered after FPI. We found that tonic GABA currents recorded in the presence of THDOC (20 nm), a relatively specific neurosteroid agonist of GABAAR δ subunits in nm concentrations (Stell et al., 2003), were significantly lower in SGCs from head-injured rats compared with sham-injured controls (Fig. 8D; in pA, sham-injured SGC: 50.9 ± 15.6, n = 3 cells; FPI SGC: 5.1 ± 3.8, n = 4 cells, p < 0.05, t test). The consistently lower tonic GABA current amplitude in SGCs from head-injured rats in the presence of both THDOC and THIP, and the lack of difference in THIP modulation of tonic GABA currents between sham-injured and FPI SGCs together suggest that decreases in GABAARs containing δ subunits contribute, in part, to the postinjury decrease in tonic GABA currents in SGCs.
Figure 8.

Decrease in SGC tonic GABA currents after brain injury. A, Representative voltage-clamp recordings (Vhold = −70 mV) from a sham-injured (top) and FPI SGC (bottom) 1 week after injury illustrate the magnitude of tonic GABA current blocked by a saturating concentration of BMI (100 μm). Right, Gaussian fits to all-points histograms derived from 30 s recording periods in control conditions in the presence of 3 mm kynurenic acid, after the addition of THIP (1 μm) and during BMI perfusion used to determine tonic current amplitude. The dashed lines indicate the Gaussian means and the difference currents are noted. B, Summary histogram of the tonic GABA currents in sham-injured and FPI SGC in kynurenic acid. C, Histogram of tonic GABA currents in control aCSF with KyA and following addition of THIP in recordings from both sham-injured and FPI SGCs. D, Summary data of tonic GABA current amplitude measured in THDOC (20 nm) in sham-injured and FPI SGCs. E, F, Cumulative probability plots comparing the sIPSC-weighted decay time constants in aCSF with KyA (black) and following the addition of THIP (gray) in recordings from sham-injured (E) and FPI (F) SGCs. Vertical dashed lines indicate median of the distribution at p = 0.5. Insets, Overlay of normalized average sIPSC traces from a sham-injured SGC (E, inset) and an FPI SGC (F, inset) during recordings in aCSF with KyA (black) and from the same cell following the addition of THIP (gray). The same number of individual events were selected from each cell to develop the cumulative distribution (sham injured: n = 3 cells; FPI: n = 4 cells). *p < 0.05, paired and unpaired Student's t test.
As demonstrated earlier (Fig. 4D), the sIPSC τdecay is slower in control SGCs compared with FPI SGCs. Previous studies in granule cells have shown that GABAAR δ subunits can be located perisynaptically and contribute to a slow component of synaptic decay (Wei et al., 2003). Since the more rapid sIPSC τdecay following injury paralleled the decrease in tonic GABA currents (Fig. 8A,B), we examined whether brain injury reduces the contribution of GABAAR with δ subunits to sIPSC decay in SGCs. In sham-injured SGCs, THIP (1 μm) slowed sIPSC τdecay, indicating a role for GABAAR δ subunits in the time course of sIPSCs (Fig. 8E; sIPSC τdecay in ms, in KyA: 4.5 ± 1.13, median = 4.24, IQR = 3.01–5.29; in THIP: 6.1 ± 2.0, median = 5.10, IQR = 4.15–7.56, n = 3 cells, p < 0.05, K–S test). However, as illustrated by the cumulative probability distribution plots of sIPSC τdecay (Fig. 8F), THIP failed to alter sIPSC τdecay in FPI SGCs (sIPSC τdecay in ms, in KyA: 4.57 ± 1.7, median = 3.68, IQR = 2.86–4.77; in THIP: 4.9 ± 2.8, median = 3.86, IQR = 2.98–5.28, n = 4, cells p > 0.05, test). These results are consistent with a post-traumatic decrease in contribution of GABAAR with δ subunits to sIPSC decay in SGCs.
Next we examined whether changes in GABA uptake might contribute to the postinjury decrease in SGC tonic GABA currents. Tonic GABA currents were recorded as baseline inward currents (Vhold = 0 mV, in the absence of glutamate antagonists or added GABA) blocked by a saturating concentration of SR95531 (gabazine, 10 μm). As illustrated in Figure 9A,B, the amplitude of tonic GABA currents was considerably reduced in SGCs from head-injured rats compared with sham-injured controls (in pA, sham-injured SGC: 22.5 ± 6.7, n = 8 cells; FPI SGC: 6.6 ± 3.6, n = 5 cells, p < 0.05, t test). In all recordings, the GAT-1 (GABA transporter-1) antagonist NO-711 (10 μm) enhanced SGC tonic GABA currents. Even when differences in GABA transport were abolished, tonic GABA currents measured in NO-711 were larger in sham-injured SGCs than in FPI SGCs (Fig. 9B; in pA, sham-injured SGC: 47.2 ± 5.3, n = 8 cells; FPI SGC: 22.9 ± 4.9, n = 5 cells, p < 0.05, t test). Moreover, the extent to which NO-711 enhanced tonic GABA currents was not different between sham-injured and FPI SGCs (enhancement by NO-711, sham-injured SGC: 271.2 ± 61.5%, n = 8 cells; FPI SGC: 258.1 ± 11.0%, n = 5 cells, p > 0.05, t test), suggesting that changes in GABA transporter function are unlikely to underlie the postinjury decrease in SGC tonic GABA currents. Together, our data support the inference that, in addition to reduced synaptic GABA spillover as a consequence of postinjury decrease in sIPSC frequency, decreases in GABAAR δ subunits contribute to lower tonic GABA currents in SGCs after head injury.
Although our data show that brain trauma results in diametrically opposite changes in synaptic inhibition in granule cells and SGCs, it is possible that early changes in tonic GABA currents after brain injury are a result of a global decrease in tonic inhibition, independent of cell type. Studies in rodent models of acquired epilepsy have demonstrated long-term increases in granule cell tonic GABA currents (Zhan and Nadler, 2009). Results of a recent study conducted 3 months after cortical-impact injury indicate that granule cell tonic GABA currents are elevated on the side contralateral to injury (Mtchedlishvili et al., 2010). However, granule cell tonic GABA currents were not enhanced 1–6 months after severe fluid percussion injury (Pavlov et al., 2011). We examined whether brain injury leads to early changes in granule cell tonic GABA currents when the dentate network shows increases in perforant path-evoked excitability. Unlike SGCs, tonic GABA currents in granule cells were enhanced 1 week after FPI (Fig. 10A,B; in pA, sham-injured granule cell: 8.4 ± 1.1, n = 9 cells; FPI granule cell: 20.8 ± 3.4, n = 6 cells, p < 0.05, t test). The increase in granule cell tonic GABA currents was statistically significant even when normalized to the cell capacitance (in pA/pF, sham-injured granule cell: 0.15 ± 0.03, n = 9 cells; FPI granule cell: 0.6 ± 0.24, n = 6 cells, p < 0.05, t test). Additionally, THIP (1 μm) enhanced tonic GABA currents in granule cells from both sham-injured and FPI rats (Fig. 10C; in pA, sham-injured granule cell: 8.1 ± 1.9 in KyA and 21.6 ± 3.4 in THIP, n = 6 cells; FPI granule cell: 20.8 ± 3.4 in KyA and 45.4 ± 6.4 in THIP, n = 6 cells). Potentiation of tonic GABA currents by THIP was not different between granule cells from sham-injured and FPI rats (tonic current amplitude in KyA, sham-injured granule cell: 285.22 ± 46.39%, n = 6 cells; FPI granule cell: 243.42 ± 44.5%, n = 6 cells, p > 0.05, t test), suggesting that increases in GABAAR containing δ subunits may underlie the enhancement of granule cell tonic GABA currents after brain injury. Interestingly, comparison of tonic GABA currents between SGCs and granule cells in control rats revealed that tonic GABA current amplitudes in SGCs were considerably larger than those in granule cells (Fig. 10D; in pA, granule cell: 8.4 ± 1.1, n = 9 cells; SGC: 16.3 ± 0.9, n = 6 cells, p < 0.05, t test). Moreover, after injury, tonic GABA currents in FPI SGCs were reduced even when compared with sham-injured granule cells (Fig. 10D). Significantly, the results reveal differences in magnitude and in the direction of postinjury plasticity of tonic GABA currents between SGCs and granule cells and demonstrate cell-type-specific reduction in SGC tonic inhibition after brain injury.
Figure 10.
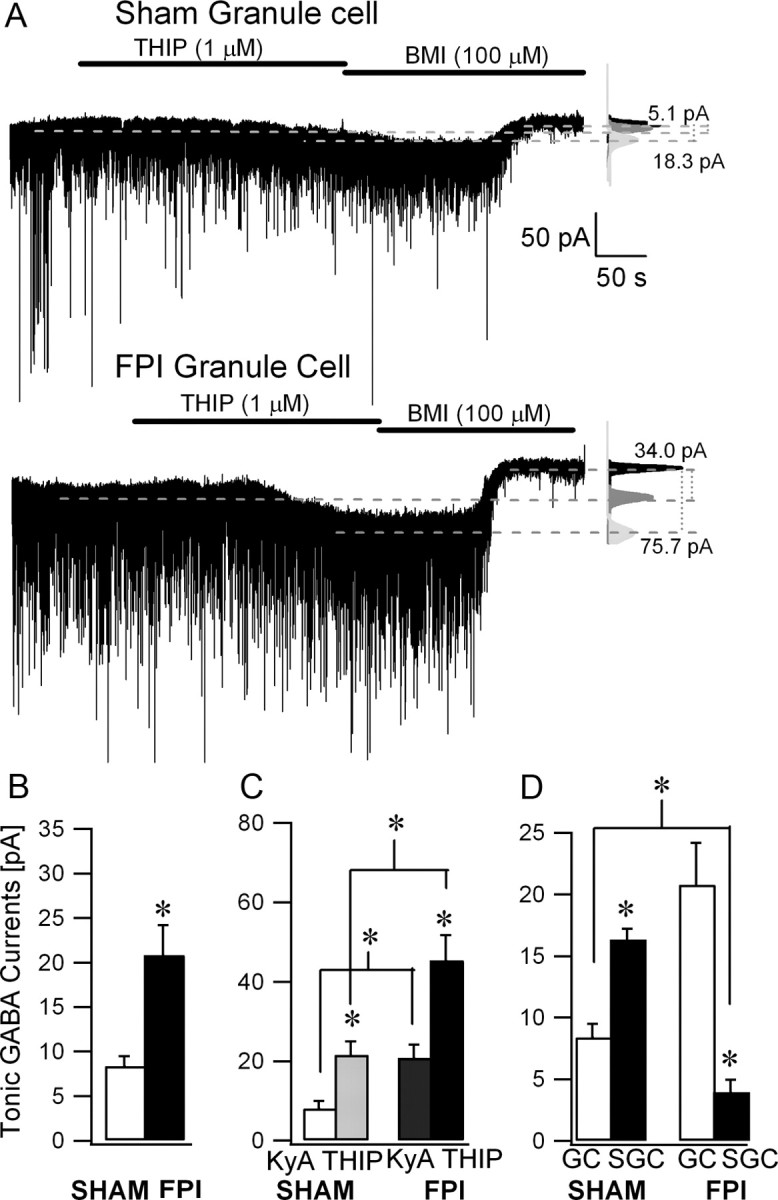
Granule cell tonic GABA currents are increased after brain injury. A, Example voltage-clamp recordings (Vhold = −70 mV) from granule cells from a sham-injured (top) and FPI (bottom) rat obtained 1 week after injury show the presence of tonic GABA current blocked by a saturating concentration of BMI (100 μm). Right, Gaussian fits to all-points histograms derived from 30 s recording periods in KyA after adding THIP (1 μm) and during BMI perfusion. The dashed lines indicate the Gaussian means and the difference currents are noted. Note the larger amplitude of tonic GABA currents in FPI granule cells. B, Summary plot of tonic GABA currents in sham-injured and FPI-granule cells in KyA. C, Histogram of tonic GABA currents in control aCSF with KyA and following addition of THIP in recordings from both sham-injured and FPI granule cells. D, Comparison of tonic GABA current amplitudes between granule cells (GCs) and SGCs in sham-injured and FPI rats. Histograms are based on tonic GABA currents recorded in KyA from the same group of cells as in Figures 8 and 10, A and B. *p < 0.05, paired and unpaired Student's t test.
Post-traumatic decrease in tonic inhibition augments SGC excitability
Since the receptors underlying tonic GABA currents contribute to membrane conductance and regulate neuronal excitability (Stell et al., 2003; Chadderton et al., 2004), we examined whether the post-traumatic reduction in SGC tonic GABA conductance may underlie the increase in SGC input resistance and excitability after brain injury (Fig. 3C–F). In recordings from sham-injured SGCs, the GABAA receptor antagonist SR95531 (20 μm) increased SGC input resistance measured as the slope of linear fits to the voltage response to the last 200 ms of 1 s hyperpolarizing current injections from −200 to −40 pA (Rin in SR95531 as percentage of Rin in aCSF: 117.2 ± 3.4%, n = 8 cells, p < 0.05 by paired Student's t test), indicating that GABA conductance contributes substantively to the low input resistance in sham-injured SGCs (Fig. 11A, top, B). Consistent with our prediction based on the post-traumatic decrease in tonic GABA currents, and in contrast to sham-injured SGCs, SR95531 did not alter the input resistance of FPI SGCs (Fig. 11A, bottom; Rin in gabazine as percentage of Rin in aCSF: 101.2 ± 5.5%, n = 10 cells, p > 0.05 by paired Student's t test). The postinjury loss of gabazine (SR95531) modulation of SGC input resistance (Fig. 11B) indicates that a decrease in GABAAR conductance underlies the increase in SGC input resistance after brain injury. Most crucially, there was no difference in the firing elicited by positive current injections in SGCs from sham-injured and head-injured rats in the presence of gabazine (Fig. 11C; difference between sham-injured and FPI SGC firing was not significant, F(1,12) = 0.07, p = 0.8 by two-way repeated-measures ANOVA, n = 6 sham-injured and 10 FPI SGCs). Together, these data indicate that the post-traumatic changes in GABAergic inhibition contribute to cell-specific enhancement of SGC excitability after brain injury.
Figure 11.

GABAA receptor antagonists enhance SGC input resistance in sham-injured controls but not after FPI. A, Membrane voltage responses to a −120 pA current injection recorded in a control and an FPI SGC illustrate that SR95531 (20 μm) increased the hyperpolarizing response in sham-injured SGCs (top) but failed to alter the response of FPI SGCs (bottom). B, Summary plot of the effect of SR95531 on SGC Rin, expressed as a percentage of Rin in control aCSF, shows that SR95531 enhanced Rin control SGC but failed to alter Rin in FPI SGC. C, Plot of SGC firing rates recorded in the presence of SR95531 (20 μm) reveals the action potential frequency during 1 s depolarizing current steps was not different in sham-injured and FPI SGCs. *p < 0.05 paired and unpaired Student's t test. Wk, Week.
Discussion
In seeking to identify cellular mechanisms underlying post-traumatic limbic hyperexcitability, this study has demonstrated altered excitability and tonic inhibition in glutamatergic SGCs following brain injury. Simultaneously, the data revealed that granule cells and SGCs have fundamental differences in inhibition. First, in relation to brain trauma, there is early postinjury increase in excitability of SGCs, which is unique among dentate excitatory neurons. In SGCs, there are cell-specific reductions in synaptic and tonic GABA currents 1 week after brain injury. By comparison, in granule cells, synaptic and tonic GABA currents increase 1 week after brain injury. Crucially, the post-traumatic decrease in GABAergic inhibition enhances SGC input resistance and excitability after FPI. These data establish for the first time the involvement of SGCs in neurological disease and demonstrate neuronal hyperexcitability resulting from decrease in tonic GABA currents in a model of acquired epilepsy. Second, concerning native properties, SGCs express Prox1, a specific marker for granule cells, indicating the common origin of the two types of cells. Regardless of the shared dendritic location, SGCs receive more frequent and slower rising sIPSCs than granule cells, indicating differences in GABAergic innervation. Compared with granule cells, SGCs have larger tonic GABA currents, which contribute to their passive membrane properties. GABAAR with δ subunits contribute to both tonic and synaptic inhibition in SGCs. Overall, we show that differences in GABAergic inhibition are a critical distinguishing feature between SGCs and granule cells and that GABAergic plasticity selectively enhances SGC excitability after brain injury.
Role of hyperexcitable SGCs in the injured brain: hub, short circuit, or both?
Brain injury leads to distinctive pathological changes in the hippocampus and results in epilepsy and cognitive disorders (Coulter et al., 1996; Toth et al., 1997; Santhakumar et al., 2001; D'Ambrosio et al., 2005; Cohen et al., 2007; Kharatishvili and Pitkänen, 2010). Here we demonstrate that SGCs, novel glutamatergic IML neurons, are more excitable 1 week after brain injury. Since SGCs contribute to sustained depolarization of hilar interneurons or “up-states,” which have been proposed as a cellular substrate for working memory (Larimer and Strowbridge, 2010), post-traumatic changes in SGC physiology may underlie memory and cognitive impairments following brain injury (Lyeth et al., 1990; Schwarzbach et al., 2006).
What are the potential implications of enhanced SGC excitability after brain injury? While SGC axonal projection constitutes a parallel output from dentate to CA3, the prolonged, input-specific inhibition of granule cells during sustained SGC firing indicates that SGCs augment the dentate gate through activation of hilar feedback interneurons (Larimer and Strowbridge, 2010). Although it is possible that the increase in SGC excitability represents a homeostatic response to enhance the dentate gate, the post-traumatic loss of hilar interneurons that constitute the feedback circuit (Lowenstein et al., 1992; Toth et al., 1997) will more likely diminish the contribution of SGCs to feedback inhibition in the injured brain. A potential consequence of the injury-induced network changes is that the direct SGC projections to CA3 may “short-circuit” the dentate gate and impart the enhanced excitability to CA3. An alternative, albeit not mutually exclusive, possibility is that the SGC IML associational collaterals may excite neighboring granule cells, forming a local focus of hyperexcitability after trauma. SGC innervation of surviving mossy cells (Williams et al., 2007) may also contribute to septotemporal spread of excitability (Ratzliff et al., 2004). Given the typical sustained SGC firing, the possibility that SGCs drive early postinjury increases in dentate excitability is compatible with the contribution of polysynaptic network activity to the increase in duration of granule cell and mossy cell firing after brain injury (Santhakumar et al., 2000). Furthermore, it remains to be seen whether SGCs undergo structural plasticity of hilar and molecular layer axon collaterals analogous to the aberrant recurrent mossy fiber sprouting after brain injury (Golarai et al., 2001; Santhakumar et al., 2001; Kharatishvili et al., 2006). While the relatively sparse distribution of SGCs pose a potential caveat to its ability to transform network activity, computational analyses predict that a few highly connected neurons could serve as “hubs” that shape network behavior in epilepsy (Morgan and Soltesz, 2008). Together, SGCs are ideally situated to enhance local excitability, compromise specificity of the dentate gate, directly activate hippocampal neurons, and thereby play a causal role in early increases in evoked population responses in the dentate gyrus after brain injury.
Differential synaptic inhibition of granule cells and SGCs: insights from injury
Our demonstration that SGCs express Prox1 and likely share the granule cell niche of adult neurogenesis (Jessberger et al., 2008; Lavado et al., 2010; Karalay and Jessberger, 2011) may explain the numerous similarities between the cell types. However, the differences in sIPSC frequency between SGCs and granule cells in control rats, and the opposite changes in sIPSC frequency observed after FPI, indicate inherent differences in the source of their inhibitory inputs. Given our current understanding, the postinjury decrease of spontaneous and miniature IPSC frequency in SGCs is consistent with post-traumatic hilar interneuronal loss (Lowenstein et al., 1992; Toth et al., 1997). In contrast to SGCs, granule cells from head-injured rats have more frequent sIPSCs but fewer mIPSCs (Toth et al., 1997; Santhakumar et al., 2001), indicating that granule cells may be innervated by neurons that survive and are more excitable after injury. We suggest that SGCs are less likely to be innervated by PV+ interneurons with axons in the granule cell layer. Since PV+ basket cells generate robust, perisomatic inhibition of granule cells (Hefft and Jonas, 2005), are relatively resilient (Toth et al., 1997), and possibly more excitable (Ross and Soltesz, 2000) after brain injury, they are a potential source of increases in granule cell sIPSC frequency after brain injury. Moreover, because basket cells are central to maintaining sparse granule cell activity (Kraushaar and Jonas, 2000), a paucity of PV+ basket cell inputs to SGCs, consequent to its location in the IML, may be permissive to the characteristic persistent firing in SGCs (Larimer and Strowbridge, 2010). Location-dependent distinctions in synaptic physiology have recently been demonstrated among ectopic CA3 granule cells (Szabadics et al., 2010). Whether lower PV+ basket cell innervation of SGCs underlies the absence of post-traumatic increases in SGC sIPSC frequency remains to be tested. Nonetheless, potential differences in PV+ basket cell innervation do not explain the inherent higher frequency and slower rise time of SGC sIPSCs compared with granule cells. The most plausible explanation for our data is that, compared to granule cells, SGCs receive a greater inhibitory input from vulnerable populations of hilar interneurons that project to the distal dendrites.
Tonic inhibition and SGC excitability
As in granule cells (Stell et al., 2003; Wei et al., 2003; Peng et al., 2004), we find that SGC tonic inhibition is mediated by GABAAR with δ subunits. The post-traumatic decrease in SGC tonic GABA currents occurs simultaneously with unchanged THIP modulation and reduction in SGC sIPSC frequency, indicating that both decreases in GABAAR δ subunits and reduced synaptic spillover (Glykys and Mody, 2007) may contribute to the decrease. Our data show that robust tonic GABAergic inhibition is an essential component of the resting membrane conductance of SGCs, and suggest that GABAAR conductance may underlie the low input resistance of SGCs. Post-traumatic increase in SGC excitability, occurring as a consequence of reduced SGC tonic inhibition, is consistent with the capacity of shunting inhibitory conductance to regulate excitability and offset neuronal firing (Brickley et al., 1996; Mitchell and Silver, 2003). More importantly, tonic inhibition can contribute to multiplicative scaling of neuronal activity during noisy physiological synaptic input, as would occur in vivo, and has been proposed to aid in pattern separation (Mitchell and Silver, 2003; Silver, 2010). Consequently, post-traumatic decrease in tonic inhibition may compromise the ability of SGCs to participate in input discrimination and contribute to memory and cognitive disabilities following brain injury. Although decrease in tonic inhibition can lower seizure thresholds (Maguire et al., 2005), granule cell tonic GABA current is enhanced or unchanged in acquired epilepsy (Zhang et al., 2007; Zhan and Nadler, 2009; Mtchedlishvili et al., 2010). How enhanced tonic GABA currents influence seizure thresholds is unclear since, while increased GABA conductance may limit excitability, depolarizing shifts in GABA reversal (Bonislawski et al., 2007; Pathak et al., 2007) tend to augment excitability. In contrast, cell-specific postinjury decrease in tonic GABA conductance could enhance excitability regardless of changes in GABA reversal. Thus, our data demonstrate that brain injury leads to early and selective decrease in SGC tonic GABA currents, resulting in post-traumatic enhancement in SGC excitability, and indicate that SGCs and tonic GABA currents may contribute to abnormal dentate circuit function after brain injury.
In summary, our results have identified intrinsic differences in inhibitory control of granule cells and semilunar granule cells that may contribute to functional distinctions between the two apparently similar dentate projection neurons and, thereby, extend our fundamental understanding of the dentate circuit involved in working memory. Our study reveals that SGCs are a potential source of increase in dentate excitability after brain injury, and suggests that SGCs may play a causal role in post-traumatic epileptogenesis and memory dysfunction that could be generalized to acquired temporal lobe epilepsy with diverse etiologies.
Footnotes
This work was supported by New Jersey Commission for Brain Injury Research Grants 09.003-BIR1 (to V.S.) and 11-3223-BIR-E-O (to A.G.). We thank Takahiro Ito for technical assistance and Drs. Stella Elkabes and Robert F. Heary for equipment and discussions.
References
- Annegers JF, Hauser WA, Coan SP, Rocca WA. A population-based study of seizures after traumatic brain injuries. N Engl J Med. 1998;338:20–24. doi: 10.1056/NEJM199801013380104. [DOI] [PubMed] [Google Scholar]
- Bonislawski DP, Schwarzbach EP, Cohen AS. Brain injury impairs dentate gyrus inhibitory efficacy. Neurobiol Dis. 2007;25:163–169. doi: 10.1016/j.nbd.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley SG, Cull-Candy SG, Farrant M. Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABAA receptors. J Physiol. 1996;497:753–759. doi: 10.1113/jphysiol.1996.sp021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown N, Kerby J, Bonnert TP, Whiting PJ, Wafford KA. Pharmacological characterization of a novel cell line expressing human alpha(4)beta(3)delta GABA(A) receptors. Br J Pharmacol. 2002;136:965–974. doi: 10.1038/sj.bjp.0704795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadderton P, Margrie TW, Häusser M. Integration of quanta in cerebellar granule cells during sensory processing. Nature. 2004;428:856–860. doi: 10.1038/nature02442. [DOI] [PubMed] [Google Scholar]
- Cohen AS, Pfister BJ, Schwarzbach E, Grady MS, Goforth PB, Satin LS. Injury-induced alterations in CNS electrophysiology. Prog Brain Res. 2007;161:143–169. doi: 10.1016/S0079-6123(06)61010-8. [DOI] [PubMed] [Google Scholar]
- Cooper DC, Moore SJ, Staff NP, Spruston N. Psychostimulant-induced plasticity of intrinsic neuronal excitability in ventral subiculum. J Neurosci. 2003;23:9937–9946. doi: 10.1523/JNEUROSCI.23-30-09937.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter DA, Rafiq A, Shumate M, Gong QZ, DeLorenzo RJ, Lyeth BG. Brain injury-induced enhanced limbic epileptogenesis: anatomical and physiological parallels to an animal model of temporal lobe epilepsy. Epilepsy Res. 1996;26:81–91. doi: 10.1016/s0920-1211(96)00044-7. [DOI] [PubMed] [Google Scholar]
- D'Ambrosio R, Fender JS, Fairbanks JP, Simon EA, Born DE, Doyle DL, Miller JW. Progression from frontal-parietal to mesial-temporal epilepsy after fluid percussion injury in the rat. Brain. 2005;128:174–188. doi: 10.1093/brain/awh337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon CE, Lyeth BG, Povlishock JT, Findling RL, Hamm RJ, Marmarou A, Young HF, Hayes RL. A fluid percussion model of experimental brain injury in the rat. J Neurosurg. 1987;67:110–119. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Földy C, Lee SY, Szabadics J, Neu A, Soltesz I. Cell type-specific gating of perisomatic inhibition by cholecystokinin. Nat Neurosci. 2007;10:1128–1130. doi: 10.1038/nn1952. [DOI] [PubMed] [Google Scholar]
- Glykys J, Mody I. The main source of ambient GABA responsible for tonic inhibition in the mouse hippocampus. J Physiol. 2007;582:1163–1178. doi: 10.1113/jphysiol.2007.134460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golarai G, Greenwood AC, Feeney DM, Connor JA. Physiological and structural evidence for hippocampal involvement in persistent seizure susceptibility after traumatic brain injury. J Neurosci. 2001;21:8523–8537. doi: 10.1523/JNEUROSCI.21-21-08523.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefft S, Jonas P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci. 2005;8:1319–1328. doi: 10.1038/nn1542. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Beck H, Dreier JP, Ficker E, Stabel J, Zhang CL. The dentate gyrus as a regulated gate for the propagation of epileptiform activity. Epilepsy Res [Suppl] 1992;7:273–280. [PubMed] [Google Scholar]
- Herman ST. Epilepsy after brain insult: targeting epileptogenesis. Neurology. 2002;59(9 Suppl 5):S21–S26. doi: 10.1212/wnl.59.9_suppl_5.s21. [DOI] [PubMed] [Google Scholar]
- Howard AL, Neu A, Morgan RJ, Echegoyen JC, Soltesz I. Opposing modifications in intrinsic currents and synaptic inputs in post-traumatic mossy cells: evidence for single-cell homeostasis in a hyperexcitable network. J Neurophysiol. 2007;97:2394–2409. doi: 10.1152/jn.00509.2006. [DOI] [PubMed] [Google Scholar]
- Hunt RF, Scheff SW, Smith BN. Synaptic reorganization of inhibitory hilar interneuron circuitry after traumatic brain injury in mice. J Neurosci. 2011;31:6880–6890. doi: 10.1523/JNEUROSCI.0032-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessberger S, Toni N, Clemenson GD, Jr, Ray J, Gage FH. Directed differentiation of hippocampal stem/progenitor cells in the adult brain. Nat Neurosci. 2008;11:888–893. doi: 10.1038/nn.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karalay O, Jessberger S. Translating niche-derived signals into neurogenesis: the function of Prox1 in the adult hippocampus. Cell Cycle. 2011;10:2239–2240. doi: 10.4161/cc.10.14.15850. [DOI] [PubMed] [Google Scholar]
- Kharatishvili I, Pitkänen A. Posttraumatic epilepsy. Curr Opin Neurol. 2010;23:183–188. doi: 10.1097/WCO.0b013e32833749e4. [DOI] [PubMed] [Google Scholar]
- Kharatishvili I, Nissinen JP, McIntosh TK, Pitkänen A. A model of posttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats. Neuroscience. 2006;140:685–697. doi: 10.1016/j.neuroscience.2006.03.012. [DOI] [PubMed] [Google Scholar]
- Kraushaar U, Jonas P. Efficacy and stability of quantal GABA release at a hippocampal interneuron-principal neuron synapse. J Neurosci. 2000;20:5594–5607. doi: 10.1523/JNEUROSCI.20-15-05594.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larimer P, Strowbridge BW. Representing information in cell assemblies: persistent activity mediated by semilunar granule cells. Nat Neurosci. 2010;13:213–222. doi: 10.1038/nn.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavado A, Lagutin OV, Chow LM, Baker SJ, Oliver G. Prox1 is required for granule cell maturation and intermediate progenitor maintenance during brain neurogenesis. PLoS Biol. 2010;8:e1000460. doi: 10.1371/journal.pbio.1000460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein DH. Epilepsy after head injury: an overview. Epilepsia. 2009;50(Suppl 2):4–9. doi: 10.1111/j.1528-1167.2008.02004.x. [DOI] [PubMed] [Google Scholar]
- Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci. 1992;12:4846–4853. doi: 10.1523/JNEUROSCI.12-12-04846.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lübke J, Frotscher M, Spruston N. Specialized electrophysiological properties of anatomically identified neurons in the hilar region of the rat fascia dentata. J Neurophysiol. 1998;79:1518–1534. doi: 10.1152/jn.1998.79.3.1518. [DOI] [PubMed] [Google Scholar]
- Lyeth BG, Jenkins LW, Hamm RJ, Dixon CE, Phillips LL, Clifton GL, Young HF, Hayes RL. Prolonged memory impairment in the absence of hippocampal cell death following traumatic brain injury in the rat. Brain Res. 1990;526:249–258. doi: 10.1016/0006-8993(90)91229-a. [DOI] [PubMed] [Google Scholar]
- Maguire JL, Stell BM, Rafizadeh M, Mody I. Ovarian cycle-linked changes in GABA(A) receptors mediating tonic inhibition alter seizure susceptibility and anxiety. Nat Neurosci. 2005;8:797–804. doi: 10.1038/nn1469. [DOI] [PubMed] [Google Scholar]
- McAllister TW. Neuropsychiatric sequelae of head injuries. Psychiatr Clin North Am. 1992;15:395–413. [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. Shunting inhibition modulates neuronal gain during synaptic excitation. Neuron. 2003;38:433–445. doi: 10.1016/s0896-6273(03)00200-9. [DOI] [PubMed] [Google Scholar]
- Morgan RJ, Soltesz I. Nonrandom connectivity of the epileptic dentate gyrus predicts a major role for neuronal hubs in seizures. Proc Natl Acad Sci U S A. 2008;105:6179–6184. doi: 10.1073/pnas.0801372105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mtchedlishvili Z, Kapur J. High-affinity, slowly desensitizing GABAA receptors mediate tonic inhibition in hippocampal dentate granule cells. Mol Pharmacol. 2006;69:564–575. doi: 10.1124/mol.105.016683. [DOI] [PubMed] [Google Scholar]
- Mtchedlishvili Z, Lepsveridze E, Xu H, Kharlamov EA, Lu B, Kelly KM. Increase of GABAA receptor-mediated tonic inhibition in dentate granule cells after traumatic brain injury. Neurobiol Dis. 2010;38:464–475. doi: 10.1016/j.nbd.2010.03.012. [DOI] [PubMed] [Google Scholar]
- Pathak HR, Weissinger F, Terunuma M, Carlson GC, Hsu FC, Moss SJ, Coulter DA. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J Neurosci. 2007;27:14012–14022. doi: 10.1523/JNEUROSCI.4390-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov I, Huusko N, Drexel M, Kirchmair E, Sperk G, Pitkänen A, Walker MC. Progressive loss of phasic, but not tonic, GABAA receptor-mediated inhibition in dentate granule cells in a model of post-traumatic epilepsy in rats. Neuroscience. 2011;194:208–219. doi: 10.1016/j.neuroscience.2011.07.074. [DOI] [PubMed] [Google Scholar]
- Peng Z, Huang CS, Stell BM, Mody I, Houser CR. Altered expression of the Δ subunit of the GABAA receptor in a mouse model of temporal lobe epilepsy. J Neurosci. 2004;24:8629–8639. doi: 10.1523/JNEUROSCI.2877-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekaran K, Joshi S, Sun C, Mtchedlishvilli Z, Kapur J. Receptors with low affinity for neurosteroids and GABA contribute to tonic inhibition of granule cells in epileptic animals. Neurobiol Dis. 2010;40:490–501. doi: 10.1016/j.nbd.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramón y, Cajal S. Histology of the nervous system of man and vertebrates. New York: Oxford UP; 1995. [Google Scholar]
- Ratzliff AH, Howard AL, Santhakumar V, Osapay I, Soltesz I. Rapid deletion of mossy cells does not result in a hyperexcitable dentate gyrus: implications for epileptogenesis. J Neurosci. 2004;24:2259–2269. doi: 10.1523/JNEUROSCI.5191-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross ST, Soltesz I. Selective depolarization of interneurons in the early posttraumatic dentate gyrus: involvement of the Na(+)/K(+)-ATPase. J Neurophysiol. 2000;83:2916–2930. doi: 10.1152/jn.2000.83.5.2916. [DOI] [PubMed] [Google Scholar]
- Ruiz A, Fabian-Fine R, Scott R, Walker MC, Rusakov DA, Kullmann DM. GABAA receptors at hippocampal mossy fibers. Neuron. 2003;39:961–973. doi: 10.1016/s0896-6273(03)00559-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhakumar V, Bender R, Frotscher M, Ross ST, Hollrigel GS, Toth Z, Soltesz I. Granule cell hyperexcitability in the early post-traumatic rat dentate gyrus: the “irritable mossy cell” hypothesis. J Physiol. 2000;524:117–134. doi: 10.1111/j.1469-7793.2000.00117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhakumar V, Ratzliff AD, Jeng J, Toth Z, Soltesz I. Long-term hyperexcitability in the hippocampus after experimental head trauma. Ann Neurol. 2001;50:708–717. doi: 10.1002/ana.1230. [DOI] [PubMed] [Google Scholar]
- Santhakumar V, Voipio J, Kaila K, Soltesz I. Post-traumatic hyperexcitability is not caused by impaired buffering of extracellular potassium. J Neurosci. 2003;23:5865–5876. doi: 10.1523/JNEUROSCI.23-13-05865.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhakumar V, Hanchar HJ, Wallner M, Olsen RW, Otis TS. Contributions of the GABAA receptor α6 subunit to phasic and tonic inhibition revealed by a naturally occurring polymorphism in the α6 gene. J Neurosci. 2006;26:3357–3364. doi: 10.1523/JNEUROSCI.4799-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhakumar V, Jones RT, Mody I. Developmental regulation and neuroprotective effects of striatal tonic GABAA currents. Neuroscience. 2010;167:644–655. doi: 10.1016/j.neuroscience.2010.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzbach E, Bonislawski DP, Xiong G, Cohen AS. Mechanisms underlying the inability to induce area CA1 LTP in the mouse after traumatic brain injury. Hippocampus. 2006;16:541–550. doi: 10.1002/hipo.20183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver RA. Neuronal arithmetic. Nat Rev Neurosci. 2010;11:474–489. doi: 10.1038/nrn2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Otis TS, Mody I. Membrane properties of dentate gyrus granule cells: comparison of sharp microelectrode and whole-cell recordings. J Neurophysiol. 1992;67:1346–1358. doi: 10.1152/jn.1992.67.5.1346. [DOI] [PubMed] [Google Scholar]
- Stell BM, Brickley SG, Tang CY, Farrant M, Mody I. Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by delta subunit-containing GABAA receptors. Proc Natl Acad Sci U S A. 2003;100:14439–14444. doi: 10.1073/pnas.2435457100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart GJ, Dodt HU, Sakmann B. Patch-clamp recordings from the soma and dendrites of neurons in brain slices using infrared video microscopy. Pflugers Arch. 1993;423:511–518. doi: 10.1007/BF00374949. [DOI] [PubMed] [Google Scholar]
- Szabadics J, Varga C, Brunner J, Chen K, Soltesz I. Granule cells in the CA3 area. J Neurosci. 2010;30:8296–8307. doi: 10.1523/JNEUROSCI.5602-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA, McIntosh TK. Lateral fluid percussion brain injury: a 15-year review and evaluation. J Neurotrauma. 2005;22:42–75. doi: 10.1089/neu.2005.22.42. [DOI] [PubMed] [Google Scholar]
- Toth Z, Hollrigel GS, Gorcs T, Soltesz I. Instantaneous perturbation of dentate interneuronal networks by a pressure wave-transient delivered to the neocortex. J Neurosci. 1997;17:8106–8117. doi: 10.1523/JNEUROSCI.17-21-08106.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MC, Pavlov I, Kullmann DM. A ‘sustain pedal’ in the hippocampus? Nat Neurosci. 2010;13:146–148. doi: 10.1038/nn0210-146. [DOI] [PubMed] [Google Scholar]
- Wei W, Zhang N, Peng Z, Houser CR, Mody I. Perisynaptic localization of Δ subunit-containing GABAA receptors and their activation by GABA spillover in the mouse dentate gyrus. J Neurosci. 2003;23:10650–10661. doi: 10.1523/JNEUROSCI.23-33-10650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ. New stereological methods for counting neurons. Neurobiol Aging. 1993;14:275–285. doi: 10.1016/0197-4580(93)90112-o. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Williams PA, Larimer P, Gao Y, Strowbridge BW. Semilunar granule cells: glutamatergic neurons in the rat dentate gyrus with axon collaterals in the inner molecular layer. J Neurosci. 2007;27:13756–13761. doi: 10.1523/JNEUROSCI.4053-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan RZ, Nadler JV. Enhanced tonic GABA current in normotopic and hilar ectopic dentate granule cells after pilocarpine-induced status epilepticus. J Neurophysiol. 2009;102:670–681. doi: 10.1152/jn.00147.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Wei W, Mody I, Houser CR. Altered localization of GABAA receptor subunits on dentate granule cell dendrites influences tonic and phasic inhibition in a mouse model of epilepsy. J Neurosci. 2007;27:7520–7531. doi: 10.1523/JNEUROSCI.1555-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]