

Abstract
How the brain transitions into a seizure is poorly understood. Recurrent seizure-like events (SLEs) in low-Mg2+/high-K+ perfusate were measured in the CA3 region of the intact mouse hippocampus. The SLE was divided into a “preictal phase,” which abruptly turns into a higher frequency “ictal” phase. Blockade of GABAA receptors shortened the preictal phase, abolished interictal bursts, and attenuated the slow preictal depolarization, with no effect on the ictal duration, whereas SLEs were blocked by glutamate receptor blockade. In CA3 pyramidal cells and stratum oriens non-fast-spiking and fast-spiking interneurons, recurrent GABAergic IPSCs predominated interictally and during the early preictal phase, synchronous with extracellularly measured recurrent field potentials (FPs). These IPSCs then decreased to zero or reversed polarity by the onset of the higher-frequency ictus. However, postsynaptic muscimol-evoked GABAA responses remained intact. Simultaneously, EPSCs synchronous with the FPs markedly increased to a maximum at the ictal onset. The reversal potential of the compound postsynaptic currents (combined simultaneous EPSCs and IPSCs) became markedly depolarized during the preictal phase, whereas the muscimol-evoked GABAA reversal potential remained unchanged. During the late preictal phase, interneuronal excitability was high, but IPSCs, evoked by local stimulation, or osmotically by hypertonic sucrose application, were diminished, disappearing at the ictal onset. We conclude that the interictal and early preictal states are dominated by GABAergic activity, with the onset of the ictus heralded by exhaustion of presynaptic release of GABA, and unopposed increased glutamatergic responses.
Introduction
One percent of the world's population is afflicted with epilepsy, with a socioeconomic impact greater than that of stroke (Banerjee and Hauser, 2008). At the individual level, the impact of epilepsy is felt through seizures—terrifying, uncontrollable, and unpredictable events, when the brain or localized regions of the brain transition into a pathological synchronous state. Very little is known about the cellular network mechanisms underlying these spontaneous transitions and how to prevent them. This transition period serves as an important target for anticonvulsant treatments, making it critical to study the underlying neuronal mechanisms.
Transition to seizure has been studied previously, but its underlying mechanisms are largely unresolved. It has been proposed to be mediated by a positive shift in the reversal potential of GABAA receptor-mediated currents, resulting in excitatory GABAergic input (Lopantsev and Avoli, 1998; Kohling et al., 2000; Cohen et al., 2002; Dzhala and Staley, 2003; Perez Velazquez 2003; Khazipov et al., 2004; Fujiwara-Tsukamoto et al., 2007; Lasztóczi et al., 2009). However, other brain-slice studies from juvenile rat hippocampus have shown that GABA actions can be primarily inhibitory/shunting (Rivera et al., 1999; Tyzio et al., 2007). The mechanism of seizure transition has also been proposed to be related to the synchronization of a large neuronal population by inhibitory interneurons (Khalilov et al., 2005; Gnatkovsky et al., 2008; Beenhakker and Huguenard, 2009; Lasztóczi et al., 2009). Previously it was shown that the hyperexcitable state during transition to seizure could be a consequence of alterations in glutamatergic and/or GABAergic synaptic input in both animal and human tissues (Cossart et al., 2001; Epsztein et al., 2006; Trotter et al., 2006; Derchansky et al., 2008; Lasztóczi et al., 2009; Huberfeld et al., 2011), leading to the hypothesis that a shifted balance between inhibitory and excitatory synaptic transmission could be an important factor underlying transition. It is not known, however, (1) to what extent the transition to seizure is mediated by a shift in the dynamic balance between excitation and inhibition, (2) whether this transition is mediated by a presynaptic or a postsynaptic mechanism or both, or 3) whether this transition is cell-type dependent.
To answer these questions, we investigated the relative contributions of synchronized GABAergic and glutamatergic inputs and their functional roles during ictogenesis using field and whole-cell current- and voltage-clamp recordings and pharmacological tools in a low-Mg2+/high-K+ recurrent seizure model in the neonatal (postnatal days 6–12) intact mouse hippocampus. Our findings indicate that the rhythmic discharges during the interictal and preictal states are mediated by a shifting balance between excitatory and inhibitory inputs. During the preictal period, there is a progressive disappearance of inhibitory GABAergic IPSPs and IPSCs, with a concomitant marked increased glutamatergic EPSPs and EPSCs. The decrease in inhibition results from exhausted GABA release from presynaptic terminals. We found that this synaptic mechanism of transition occurs in CA3 non-fast-spiking (non-FS) and FS interneurons and pyramidal cells.
Materials and Methods
Hippocampal preparation.
Whole hippocampi were isolated from C57 male black mice (Charles River Laboratory; aged 6 to 12 postnatal days) (Derchansky et al., 2004), which were anesthetized with halothane and decapitated in accordance with the Canadian animal care guidelines. Each brain was quickly removed, hemisectioned, and immersed for 4–5 min in an ice-old oxygenated (95% O2/5% CO2) solution of artificial CSF (ACSF) containing the following (in mm): 123 NaCl, 2.5 KCl, 1.5 CaCl2, 2 MgSO4, 25 NaHCO3, 1.2 NaH2PO4, and 15 glucose. The whole intact hippocampus was then dissected out from each hemisphere and immersed in oxygenated ACSF at room temperature for at least 1.5 h before recording.
Experimental environment.
The isolated intact hippocampus was placed in a submerged recording chamber. To precipitate epileptiform activity, the tissue was perfused with low-Mg2+/high-K+ ACSF containing the following (in mm): 123 NaCl, 5 KCl, 1.5 CaCl2, 0.25 MgSO4, 25 NaHCO3, 1.2 NaH2PO4, and 15 glucose. The tissue was perfused with oxygenated ACSF and low-Mg2+/high-K+ ACSF at a flow rate of 8 ml/min at 32 ± 0.5°C in ACSF and at 34 ± 0.5°C in the low-Mg2+/high-K+ perfusate.
Electrophysiology.
Simultaneous extracellular field potential (FP) and whole-cell patch-clamp recordings in current- and voltage-clamp (VC) modes were obtained from stratum oriens non-FS and FS interneurons and pyramidal cells of the hippocampal CA3 region. Neurons were visualized by an infrared differential interference contrast video microscopy (Olympus BX51 microscope; OLY-150IR camera–video monitor unit). The extracellular recording electrode (1.5 mm outside diameter; World Precision Instruments) was filled with 150 mm NaCl, pH 7.4, and had ∼2 MΩ resistance. The electrode was placed into the stratum pyramidale near the patched cells (≤150 μm). Whole-cell patch recording electrodes (3–5 MΩ) were filled with solution containing the following (in mm): 135 potassium gluconate, 10 NaCl, 0.0001 CaCl2, 10 NaHEPES, 1 MgCl2, 0.3 NaGTP, and 2 NaATP, pH 7.4. Extracellular field potentials were recorded using an Axopatch 200A amplifier (Molecular Devices) in the current-clamp configuration. Whole-cell recordings were also done using an Axopatch 200B amplifier (Molecular Devices) in either the current- or voltage-clamp configuration. Data were collected using a sampling rate of 10 kHz and were subjected to a low-pass filter of 5 kHz. Intracellular recordings were performed from visually identified CA3 stratum oriens interneurons and pyramidal cells of intact hippocampi from the neonatal mice with recurrent seizures. Input resistances of these recorded neurons remained relatively stable during the experiment. Cells were discarded if their input resistance decreased by >20%. Data acquisition, storage, and analyses were done using a digital-analog interface board (Digidata 1200; Molecular Devices) and pClamp software (version 10.2; Molecular Devices). Although voltage clamping these electrotonically distributed cells raises space clamp issues, the data obtained were consistent and differences between the interictal and preictal states were clearly identifiable.
Extracellular recordings were used to define the seizure states. Three seizure states—interictal, preictal, and ictal—were measured according to the frequency of the field discharges (interictal, <.2 Hz; preictal, >.2 Hz; ictal, >15 Hz; see Results). For examining the neuronal firing increase during the transition to seizure, we measured the frequency of the intracellular spiking bursts associated with the spontaneous rhythmic field potentials. The frequencies of the recorded discharges during the interical and preictal states were calculated from the total number of detected events over the entire interictal and preictal durations, respectively, using pClamp software (version 10.2; Molecular Devices). During the ictal state, frequencies were examined over a 10s window after the ictal onset.
During the preictal period, we quantified the area under each depolarizing potential including the spikes, measuring from the onset till the return to baseline of the depolarizing potential (see Fig. 8) using Clampfit (version 10.2; Molecular Devices). The measured area was normalized against the value for the first of the last five EPSPs. In the case of the last two EPSPs measured in pyramidal neurons just before the higher-frequency ictal state, the return to baseline of the synchronous field potential was used as a reference, since these intracellular responses did not return to baseline.
Figure 8.

Postsynaptic GABAergic receptors are functional intact, and interneuronal excitation is high during the transition to seizure. A, Muscimol (asterisks, 500 mm, 10 ms, 10 psi) was pressure ejected near the soma of the voltage (V)-clamped CA3 non-FS interneuron, V-clamped at 0 mV (n = 6). Note the robust muscimol-evoked postsynaptic inhibitory responses throughout the transition to seizure and during the ictus, suggesting that the postsynaptic GABAA receptor responses remain intact. B–D, Current-clamp recordings of recurrent seizure-like events are recorded from non-FS interneurons (n = 27; B), FS interneurons (n = 10; C), and pyramidal cells (n = 25; D) concomitantly with the associated field activity. The cellular action potential bursting activity during the preictal state of all neuronal types is shown in a faster time base (boxes, bottom traces). Note that dominant phasic depolarizations during the interictal and preictal states of all neuronal types were associated with bursts of neuronal spiking. During the preictal state, FS-interneurons and non-FS interneurons had much higher frequencies of intracellularly recorded spike bursting activity than the pyramidal cells. E, The area of the intracellularly recorded EPSPs with their spike bursts were measured in pyramidal cells (n = 27) and non-FS (n = 10) and FS interneurons (n = 25; p-5 to p-1), and were normalized against p-5. Note that the burst areas of pyramidal cells and non-FS interneurons significantly increased at p-2 and p-1, whereas FS-interneurons did not show a significant increase in the burst area from p-5 to p-1. *p < 0.05 (one-way ANOVA).
The reversal potentials of the spontaneous recurrent intracellular postsynaptic currents (PSCs) correlating with the rhythmic recurring interictal and early preictal epileptiform field potentials were estimated by plotting the amplitude of the recorded currents (averaged from seven to nine consecutive events) against different voltages clamped from −55 to 0 mV over several seconds (see Fig. 3A). The reversal potential was then estimated the plotted linear regression line as the point at which this line intersected with the 0 current axis. Because of the briefness of late preictal period, the reversal potentials of the spontaneous PSCs correlating with the epileptiform field potentials were estimated online by manually changing the holding potential of the pyramidal cell until the PSC deflection changed polarity. The reversal potential of GABAA receptor activation (EGABAA) was determined by pressure ejection of muscimol (500 mm, 10 ms, 10 psi) onto the soma of the recorded cell using a Picospritzer (General Valve) (see Fig. 3B) and was estimated by plotting the amplitudes of the muscimol-induced currents recorded intracellularly against different holding potentials (from 0 to −80 mV) in ACSF during the interictal and preictal states. The recorded current amplitudes were averaged from three consecutive muscimol-induced responses at each holding potential and were fitted with a linear regression line. The voltage potential at which the current switched from inward to outward was denoted as the reversal potential. The reversal potentials of both early and late preictal PSCs were measured. However the muscimol-induced EGABAA was measured over the entire preictal period, due to the technical difficulty in focally applying muscimol during the short seizure transition period and the relatively long duration of the muscimol-induced response. The average value of EGABAA recorded during the preictal state was further used as the holding potential for isolating and measuring predominantly glutamatergic currents.
Figure 3.
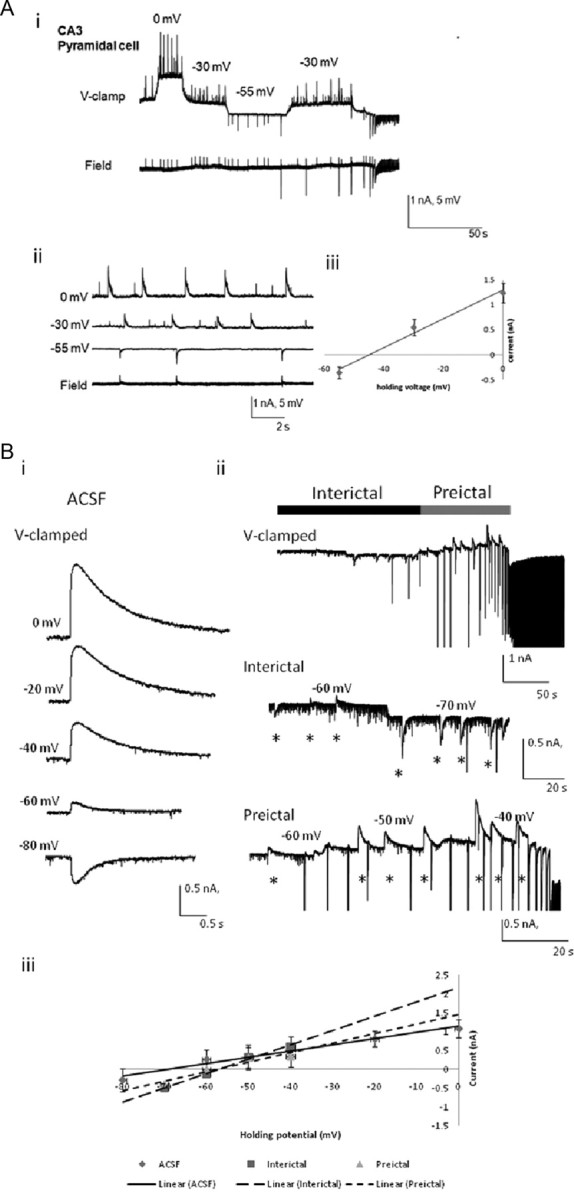
Measurement of the reversal potentials of the rhythmic whole-cell postsynaptic currents and muscimol-induced EGABAA currents in CA3 pyramidal cells. Ai, Simultaneous extracellular and voltage-clamped intracellular traces were recorded during the preictal period in a CA3 pyramidal neuron. Aii, Intracellular and extracellular recordings are shown in a faster time base, with the lowest field trace showing field potentials coincident with the whole cell measured postsynaptic currents at −55 mV. Aiii, The currents during the preictal state were plotted versus the holding potentials. Data (means) were measured from seven to nine consecutive events corresponding to the synchronous field potential at each voltage and were fitted with a linear regression line. Bi, Muscimol-induced responses were recorded intracellularly at different holding potentials in ACSF. Bii, Voltage (V) -clamped intracellular traces were recorded at various voltages during the interictal (middle) and preictal (bottom) periods in low-Mg2+/high-K+ ACSF perfusate. Biii, The amplitudes of the muscimol-induced currents are plotted versus the holding potentials. Data (mean) were measured from three consecutive muscimol-induced responses at each holding potential and were fitted with linear regression lines. The reversal potentials of muscimol-induced EGABAA in CA3 pyramidal cells were calculated in ACSF for the interictal and preictal periods induced by low-Mg2+/high-K+ ACSF. Note that the reversal potential became more depolarized as the neuronal network proceeded to the ictal onset, but little change was found between the interictal and preictal states (Table 1). Muscimol application occurred where indicated by asterisks (500 mm, 10 ms, 10 psi).
Recorded neurons were also voltage clamped at the reversal potential of either glutamatergic or GABAergic currents (0 and −60 mV, respectively) (see Figs. 4–6), thereby isolating the inhibitory or excitatory synaptic currents respectively. The charge transfers underlying the postsynaptic currents associated with the spontaneous rhythmic field potentials were estimated for both CA3 pyramidal cells and non-FS and FS interneurons during the preictal period by quantifying the area under the curve. The areas under the curve of last nine postsynaptic currents before the ictal onset (see Figs. 4–6) were also measured during the preictal period using Clampfit and were normalized against the value for the first current measured (p-9).
Figure 4.

Changes in the glutamatergic and GABAergic postsynaptic currents associated with the spontaneous rhythmic field potentials of CA3 non-FS interneurons during the preictal period. Low-Mg2+/high-K+-induced SLEs are recorded simultaneously from a voltage-clamped CA3 non-FS interneuron (top traces) and the CA3 stratum oriens field (bottom traces). The electrodes are <100 μm apart. Ai–Bii, The glutamatergic and GABAergic currents were recorded by clamping the neuron at −60 mV (Ai, Aii) and 0 mV (Bi, Bii) respectively. Aii, Bii, The preictal activity seen in Ai and Bi is shown at a faster time base, and the preictal bursts were numbered for quantitative analysis. The high-frequency ictal onsets are indicated by the open arrows. The charge transfers were measured for eight preictal bursts synchronized with the recurrent epileptiform field events (p-8 to p-1) and were normalized against the burst at p-9. C, D, Normalized charge transfers associated with different preictal discharges were plotted for both glutamatergic postsynaptic currents (C) and GABAergic postsynaptic currents (D; n = 15). Note that the EPSCs gradually but markedly increased (to almost 10 times control) from p-4 to p-1 and became the dominant currents during the ictus. On the other hand, IPSCs gradually decreased during the late preictal period. However, the decrease is insignificant, except for at p-1, where inward current was observed in this example. *p < 0.05 (one-way ANOVA).
Figure 5.

Changes in the glutamatergic and GABAergic postsynaptic currents associated with the spontaneous rhythmic field potentials of FS interneurons during the preictal period. Low-Mg2+/high-K+ ACSF-induced SLEs were simultaneously recorded from a voltage-clamped CA3 FS interneuron (top traces) and the CA3 stratum oriens field potential <100 μm apart (bottom traces). Ai–Bii, The glutamatergic and GABAergic currents were recorded in isolation by voltage clamping the neuron at −60 mV (Ai, Aii) and 0 mV (Bi, Bii), respectively. The preictal activity seen in Ai and Bi is shown on a faster time base, (Aii, Bii) and the preictal bursts are numbered for quantitative analysis. The ictal onsets noted in the field recordings are indicated by arrows. The charge transfers were measured for eight preictal bursts synchronized with the field events (p-8 to p-1) and were normalized against the burst at p-9. C, D, Normalized charge transfers associated with different preictal discharges are plotted for both glutamatergic postsynaptic currents (C) and GABAergic postsynaptic currents (D; n = 7). Note that the EPSCs markedly increased from p-4 to p-1 to almost 20 times control and became the dominant currents during the ictus. The IPSCs progressively decreased, reversing their polarity at p-5, and then these currents markedly increased in amplitude to p-1. *p < 0.05 (one-way ANOVA).
Figure 6.

Changes in the glutamatergic and GABAergic postsynaptic currents associated with the spontaneous rhythmic field potentials of CA3 pyramidal cells during the preictal period. Low-Mg2+/high-K+-induced SLEs were recorded simultaneously from the CA3 pyramidal layer (field, bottom traces) and a CA3 pyramidal cell (top traces), which were <100 μm apart. Ai–Bii, The glutamatergic and GABAergic driven currents were recorded by voltage clamping the neuron at −60 mV (Ai, Aii) and 0 mV (Bi, Bii), respectively. The preictal activity seen in Ai and Bi is shown at a fast time base and the preictal bursts are numbered for quantitative analysis (Aii, Bii). The ictal onsets are indicated by arrows. The charge transfers were measured for eight preictal bursts synchronized with the field events (p-8 to p-1) and were normalized against the burst at p-9. C, D, Normalized charge transfer associated with different preictal discharges are plotted for both glutamatergic postsynaptic currents (C) and GABAergic postsynaptic currents (D; n = 10). Note that the EPSCs gradually but markedly increased (to almost 5 times control) from p-4 to p-1 and also became the dominant currents during the ictus (n = 10). The IPSC gradually decreased to zero from p-5 to p-1. *p < 0.05 (one-way ANOVA).
The hippocampal CA3 region was stimulated locally with a bipolar tungsten electrode (enamel-insulated nichrome wire, 125 μm diameter) positioned 300–500 μm away from the recorded pyramidal cells in the pyramidal layer. Extracellular stimulation was done using current pulses generated by a Grass S88 stimulator (Grass Instruments) delivered through an isolation unit. The timing of the pulse was manually controlled (∼5 to 10 s interstimulus interval) to avoid the bursting activities during the preictal and ictal states.
For osmotic ejection of presynaptic neurotransmitter, an extracellular electrode was filled with 0.5 m sucrose solution dissolved in low-Mg2+/high-K+ ACSF and placed in close proximity to the soma of a patched cell (≤20 μm). This extracellular electrode was used both to record field potentials and for pressure ejection (100 ms, 30 psi) of hypertonic sucrose solution near the patched cell soma (see Fig. 9). Recorded neurons were clamped at 0 mV to examine the sucrose-evoked GABAergic current responses and subsequently quantified by calculating the area under the sucrose-evoked response. Vehicle ejections had no effects for sucrose or muscimol experiments.
Figure 9.

Reduced GABAergic responses to focal sucrose ejections. Sucrose (0.5 m) was focally applied near the recorded neuron (<20 μm). A, The sucrose-evoked GABAergic responses are isolated by voltage clamping the neurons at 0 mV. During the preictal phase (left and middle boxed traces), focal sucrose application causes a transient postsynaptic GABAergic IPSC, which disappears during the ictus (right boxed traces). B, Normalized charge transfers associated with postsynaptic vesicle release induced by sucrose injections in each cell during the preictal state were plotted for GABAergic currents. Responses to the sucrose injections at a given time before ictal onset are shown. Note that the IPSCs gradually decreased as the ictus approached. Time at zero seconds represents the GABAergic response to the first sucrose injection after the ictal onset. All cells measured were non-FS interneurons (n = 5). The linear regression obtained had R2 = 0.969, with p = 0.028.
Pharmacology.
Stock solutions of phramacologic agents dl-2-amino-5-phosphonopentanoic acid (APV) and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) were made by dissolving these agents in double-distilled H2O for concentrations of 25 and 20 mm, respectively. These were further diluted in ACSF to create final stock solutions of 10 μm CNQX and 60 μm APV. Bicuculline methiodide (BMI) was dissolved directly in ACSF to create a stock solution of 500 μm. Muscimol (10 μm) in saline, pH −7.4, was pressure ejected locally near the soma of the recorded neuron via a Picospritzer.
Data analysis.
The statistical test used was one-way ANOVA followed by Bonferroni's multiple comparison test with p < 0.05 considered significant. Data are reported as the mean ± SD. Data processing, analysis, and graphical representations were executed with Clampfit version 10.2 (Molecular Devices) and CorelDRAW X4 (Corel) software.
Results
GABAA receptor blockade decreases preictal but not ictal duration and attenuates the slow preictal depolarization; glutamatergic blockade abolishes seizure-like events
GABAergic neurotransmission is increasingly understood as being involved in the generation of physiological and pathological oscillations within the brain (Beenhakker and Huguenard, 2009), while glutamatergic discharges appear to dominate during the transition to seizure in human subicular brain slices (Huberfeld et al., 2011, Menendez de la Prida and Trevelyan, 2011). We therefore investigated the contributions of GABAA receptor- and glutamate receptor-mediated neurotransmission to the generation of seizure-like events (SLEs).
Upon perfusing the intact isolated mouse hippocampus with low-Mg2+/high-K+ ACSF, spontaneous recurrent SLEs were recorded in the CA3 region of the isolated neonatal mouse hippocampus (Fig. 1A). These events declined in neither amplitude nor duration over the time course of these experiments, consistent with previous studies by Derchansky et al. (2004, 2008) in the same seizure model. The recurrent SLEs began after 243 ± 65 s of low-Mg2+/high-K+ ACSF perfusion, with an average duration of 97 ± 13s (n = 5). Extracellular recordings were used to define the various electrophysiological “states” the preparation could exist in. We realize that these definitions of states are somewhat arbitrary, but the described changes in the frequency of the rhythmic potentials were rather stereotypical across slices and within a slice for repeated SLEs (Figs. 1, 8). The “interictal” state was defined as the period between the end of the previous ictal state to the next preictal state with discharges occurring at <0.2 Hz (average frequency, 0.05 ± 0.03 Hz). The “preictal” state was defined as the onset of rhythmic field discharges at a frequency >0.2 Hz (average frequency, 0.5 ± 0.15 Hz). The start of the “ictal” state was identified as an abrupt transition to higher frequency discharges of >15 Hz (average frequency, 20 ± 3 Hz; n = 51).
Figure 1.

GABAA receptor antagonism increases SLE frequency but not duration, and glutamate receptor blockade abolishes SLEs. A, SLEs are recorded extracellularly from the CA3 pyramidal layer during perfusion of low-Mg2+/high-K+ ACSF, with the addition of 10 μm BMI, and then washout of BMI (top trace). Boxed SLEs under each treatment were shown at a faster time base (bottom traces). The last SLE of each treatment was chosen to ensure a longer perfusion time for the antagonist. B, Quantitative analysis of the preictal, ictal, and interseizure durations were calculated in each condition. Note that BMI greatly reduced the interseizure interval and the duration of the interical and preictal period, thereby increasing the seizure frequency, with little effect on the ictal duration. After washout, the interictal duration did not recover completely. *Significant differences in the values between low-Mg2+/high-K+ ACSF and BMI treatments; **significant differences in the values between low-Mg2+/high-K+ ACSF and washout (n = 5; one-way ANOVA; p < 0.05). C, Field recordings of SLEs were obtained from the pyramidal cell layer under the perfusion of low-Mg2+/high-K+ ACSF, d-APV (60 μm) plus CNQX (10 μm), and washout. d-APV and CNQX markedly diminished the low-Mg2+/high-K+ ACSF-induced SLEs. Boxed fields are enlarged in the lowest traces. After washout for 10 min, the SLEs reappeared (n = 5).
Blocking GABAergic neurotransmission with BMI (10 μm) greatly diminished the spontaneous recurrent extracellular FPs in the interictal and preictal states, as recorded from the pyramidal layer in the CA3 region, leaving only a few, more complex potentials just before the ictal state (Figs. 1A, 2). Furthermore GABAA receptor blockade significantly reduced the duration of the interictal and preictal states (Fig. 1A,B). GABAA receptor blockade also resulted in increased frequency of SLEs, but caused minimal changes in the ictal duration, suggesting that GABAergic activity plays a minimal role in determining the ictal duration, but plays a major role in generating interictal and early preictal potentials (Uva et al., 2009, Huberfeld et al., 2011). After washout for 10 min, the preictal bursting largely recovered, but the ictal duration became significantly reduced, and the interictal duration was still significantly less than that of the prior control low-Mg2+/high-K+ ACSF (Fig. 1B). These effects may require longer washout times to return to control values. Since GABA release may activate both presynaptic and postsynaptic GABAB receptors (Ulrich and Bettler, 2007), we also preliminarily tested the effects of the GABAB antagonist CGP-55845 (benzyl-[3-[1-(3,4-dichlorophenyl)ethylamino]-2-hydroxypropyl]phosphinic acid; 1 μm) on recurrent SLEs in six isolated hippocampi. Similar to the GABAA antagonist results, the preictal duration decreased by 57.3% (p = 0.022), and the interictal duration decreased by 68.3% (p = 0.008), whereas the ictal duration was not significantly decreased.
Figure 2.

The slow membrane depolarization during the transition to seizure is partly GABAA mediated. Ai–Bii, Simultaneously recorded intracellular (current-clamp) and extracellular recordings of the SLEs from a CA3 interneuron and pyramidal cell are shown under the perfusion of low-Mg2+/high-K+ ACSF (Ai, Bi) and with the addition of 10 μm BMI (Aii, Bii). The period of seizure transition (bar) is shown at a faster time scale (right). Note that BMI application significantly diminished the bursting activity before ictal onset and the slow depolarization of the membrane potential during the seizure transition. Data were obtained from a sample of 10 non-FS interneurons and eight pyramidal cells.
When glutamatergic receptor antagonists d-APV (60 μm) and CNQX (10 μm) were added to the perfusate, the high-frequency ictal bursts were completely abolished, whereas interictal events persisted (Fig. 1C). Washout of the receptor antagonists was associated with a return of SLEs. These data suggest that glutamatergic neurotransmission is an absolute requirement for the generation of SLEs and not required for interictal events.
While recording simultaneously intracellularly and extracellularly, a slow depolarization of the membrane potential during the preictal state was observed in both pyramidal cells (11 mV) and non-FS interneurons (9 mV) (Table 1; Fig. 2Ai,Bi). This resulted in a more depolarized resting membrane during the late preictal period compared to the interictal and early preictal states, which could in part be due to the depolarization of the Cl− reversal potential (Khazipov et al., 2004). With the application of 10 μm BMI, the presence of this slow depolarization was attenuated by 5 ± 2 mV in pyramidal cells (n = 4) and 7 ± 2 mV in non-FS interneurons (n = 5), and both types of neurons displayed decreased intracellularly measured firing during the interictal and preictal periods (Fig. 2Aii,Bii). In addition, the baseline noise during the transition period was also reduced after BMI application. Despite these changes, bursting activity persisted before the ictus, and the ictal duration remained unaffected. Thus, in contrast to glutamatergic neurotransmission, which is an absolute requirement for the transition to an ictus, GABAergic activity of both GABAA and GABAB appears to restrain the transition to SLEs, not unlike that which is seen in low-Mg2+ seizures in neocortex (Trevelyan et al., 2006). Furthermore, the absence of GABAergic neurotransmission appears to hasten the transition to seizure. To supplement this rather static picture of the involvement of these two neurotransmitter systems in the transition to SLEs, we explored the time-dependent changes in the activity of these neurotransmitter systems from both presynaptic and postsynaptic perspectives.
Table 1.
CA3 pyramidal cells and non-FS interneuronal resting membrane potentials, recurrent rhythmic whole-cell postsynaptic current reversal potentials, and muscimol-induced EGABAA
| ACSF | Interictal | Preictal |
||
|---|---|---|---|---|
| Early | Late | |||
| Observed resting membrane potential (mV) | ||||
| Pyramidal cells (n = 24) | −66 ± 4 | −64 ± 2 | −60 ± 3 | −49 ± 3* |
| Non-FS interneurons (n = 25) | −62 ± 4 | −60 ± 3 | −57 ± 2 | −46 ± 2* |
| Postsynaptic reversal potentials (mV) | ||||
| Pyramidal cells (n = 12); recurrent PSCs corresponding to the extracellular field events | −58 ± 2 | −48 ± 2* | −42 ± 4* | |
| Non-FS interneurons (n = 17); recurrent PSCs corresponding to the extracellular field events | −55 ± 4 | −35 ± 4* | −29 ± 3* | |
| Muscimol-induced EGABAA in pyramidal cells (n = 4) | −70 ± 3 | −58 ± 4* | −60 ± 2* | |
| Muscimol-induced EGABAA in non-FS IN cells (n = 5) | −68 ± 4 | −59 ± 4* | −58 ± 3* | |
Resting membrane potentials (in millivolts) of both neuronal types were measured in ACSF and during stages of low-Mg2+-induced seizure. Note that the resting membrane potentials during the late preictal state were significantly more depolarized than the early preictal state. Reversal potentials of the whole-cell postsynaptic spontaneous rhythmic currents occurring in association with the spontaneous rhythmic field potentials and muscimol-induced GABAA postsynaptic responses. Note that the reversal potentials in both neuronal types were significantly more depolarized during the preictal state than interictal state and were significantly different from each other in both the early and late preictal stages. Their reversal potentials were not recorded in the ACSF, because no spontaneous rhythmic potentials were seen in ACSF. Muscimol-induced EGABAA was more depolarized in the interictal and preictal state than in ACSF. However, the changes from interictal to preictal state were insignificant. The chloride concentration in the whole-cell intracellular recording electrode was 13 mm.
*p < 0.05.
Muscimol-induced GABAA currents change significantly from the control ACSF nonepileptogenic state to the interictal state but little thereafter
We have observed that both non-FS and FS interneurons and pyramidal cells received significant overall net increases in the excitatory drive, manifested by a significantly more depolarized reversal potential of spontaneous PSCs (Fig. 3; Table 1), which are a combination of simultaneously occurring EPSCs and IPSCs. To characterize this apparent increase in excitatory drive, whole-cell PSCs in phase with the spontaneous rhythmic field potentials during the seizure transition states (Fig. 3A) were voltage clamped at various potentials. The reversal potential of these PSCs was calculated from the plotted I–V curve. The reversal potentials of the spontaneous PSCs coincident with the recurrent field potentials became more depolarized during the preictal state from that seen in the interictal state for both pyramidal cells and non-FS interneurons. This suggests that the balance moves toward excitation as the CA3 network proceeds to ictal onset (Table 1). Comparing the PSC reversal potentials between both neuronal types revealed that in the preictal phase, the reversal potentials of the phasic rhythmic PSCs were more depolarized in the non-FS interneurons than in the pyramidal cells, suggesting that the excitatory drive to non-FS interneurons is greater than that to pyramidal cells.
It has as well been suggested that accumulation of intracellular Cl−, leading to a more depolarized Cl− reversal potential, may render GABAergic drive excitatory (Khalilov et al., 2005; Ben-Ari et al., 2007). This depolarization in Cl− reversal potential may contribute to the depolarized reversal potential of spontaneous PSCs and increased excitatory drive. To determine the contribution of Cl− to the reversal potential changes of spontaneous PSCs, we measured the reversal potential of currents induced by the GABAA agonist muscimol. The application of muscimol was performed at various stages of ictogenesis to assess its time-dependent changes. GABAergic receptor responses in pyramidal cells (n = 4) and in non-FS interneurons (n = 5) were evoked by somatic pressure ejection of muscimol, during perfusion with normal ACSF and during the interictal and preictal states in low-Mg2+/high-K+ conditions (Fig. 3B). EGABAA was calculated from the amplitude of the muscimol-evoked currents at various holding voltages, which were large and clearly visible on top of the spontaneous recurrent postsynaptic currents. The observed reversal potentials of the spontaneous postsynaptic currents and muscimol-induced EGABAA are summarized in Table 1. The average value of EGABAA recorded during the preictal state was further used as the holding potential for isolating and measuring predominantly glutamatergic currents. The reversal potentials of both early and late preictal PSCs were measured, whereas muscimol-induced EGABAA was measured over the entire preictal period. This was due to the technical difficulty in focally applying muscimol during the short preictal seizure transition period. The muscimol-induced EGABAA in pyramidal cells was −70 ± 3 mV in normal ACSF, and depolarized to −58 ± 4 mV during the interictal state, with little further change within the preictal state (−60 ± 2 mV) (Table 1). Similarly, the evoked EGABAA in non-FS interneurons depolarized from −68 ± 4 mV in normal ACSF to −59 ± 4 mV during the interictal state, and was −58 ± 3 mV during the preictal state. As per Table 1, during the interictal period, the resting membrane potentials of pyramidal cells and non-FS interneurons were −64 and −60 mV, respectively, whereas in the interictal period the reversal potentials of muscimol-induced GABAA responses were −58 and −59 mV, respectively—differences of, on average, 6 and 1 mV, respectively. With or without a significant membrane depolarization, a significant conductance change could have inhibitory actions. We did not address this question directly, but that the EPSPs increase during the preictal phase (Figs. 2, 8), and the postsynaptic muscimol-induced GABAA responses remain robust during this phase, suggests that the postsynaptic conductance was not largely increased.
Although it has been suggested that the depolarization of the EGABAA contributes to ictogenesis, we found that the most conspicuous change in the EGABAA was between the nonepileptogenic ACSF state to the interictal state, with no further change during the preictal state. This suggests that the depolarized EGABAA may in part contribute to the transition into the interictal state from a nonepileptic state, but transition to the ictal state may not simply result from increased excitatory drive through GABAergic neurotransmission. That other factors could contribute to the transition to the ictal state is suggested by the observation that the reversal potentials of the spontaneous PSCs coincident with the recurrent field potentials became more depolarized during the preictal state from that seen in the interictal state for both pyramidal cells and non-FS interneurons. When we compared the reversal potentials of compound PSCs with the muscimol-induced EGABAA (Table 1), both non-FS interneurons and pyramidal cells received significant overall net increases in the excitatory drive, which was manifested by the significantly more depolarized reversal potentials of spontaneous PSCs compared to the unchanged muscimol-induced EGABAA during the preictal state. The reversal potential of the phasic rhythmic PSCs was more depolarized in the non-FS interneurons than in the pyramidal cells, both in the early and late preictal phases (Table 1), suggesting that the excitatory drive to non-FS interneurons is greater than to pyramidal cells. As we have shown that the Cl− reversal potential changes little toward the ictus, the more depolarized compound PSP appears to be due to increased simultaneous glutamatergic drive and could be due to decreased presynaptic GABAergic release along with increased presynaptic glutamatergic release and/or altered neurotransmitter receptor sensitivity, in addition to the possibility of added voltage-gated sodium or calcium currents.
GABAergic currents expire and glutamatergic currents peak at the transition from preictal to ictal activity
As the net contribution to the increased PSP drive from pyramidal and non-FS interneurons is likely to determine the dynamics of the transition from the preictal to ictal states, we investigated the relative contributions of GABAergic and glutamatergic PSPs during the preictal period. Voltage-clamp recordings were performed on both non-FS and FS interneurons and pyramidal cells. Recording currents at the EPSC reversal potential (0 mV) and IPSC reversal potential (−60 mV) permitted the isolation of GABAergic (outward) and glutamatergic (inward) currents, respectively, as shown for all neuronal types (Figs. 4–6). Isolated inward glutamatergic currents in non-FS and FS interneurons increased as the network proceeded to ictal onset, persisting throughout the ictal state (Figs. 4A, 5A). When these neurons were voltage clamped at 0 mV, the outward GABAergic currents predominated in the early preictal phase, but disappeared before the ictus (Figs. 4B, 5B). To quantify these observations, we measured the changes in the charge transfers of glutamatergic and GABAergic postsynaptic currents associated with the spontaneous rhythmic field potentials of CA3 interneurons during the preictal period. The charge transfers were measured for the last eight preictal bursts synchronized with the population spikes (p-8 to p-1), just before seizure onset, and were normalized against the burst at p-9 (Figs. 4C,D, 5C,D). In non-FS interneurons, the IPSCs were enhanced during the early preictal phase, but decreased during the late preictal period (Figs. 4D). At the last burst before seizure onset, the polarity of the GABAergic current changed from outward to inward. In FS interneurons, the IPSCs decreased, reversed their polarity at p-5, and continued to increase to almost fourfold at p-1. These reversed currents suggest the activation of currents with a reversal potential significantly more depolarized than 0 mV, such as calcium currents and/or sodium currents, electrotonically remote dendritic EPSCs, or gap junction-mediated excitatory potentials from adjacent cells. On the other hand, the charge transfers of excitatory glutamatergic postsynaptic currents were not significantly increased until the last four preictal discharges in non-FS interneurons, wherein they markedly increased to almost 10 times control (Fig. 4C), or the last five preictal discharges in FS interneurons that displayed an increase of almost 20-fold. Note the marked variability in the preictal GABAergic and glutamatergic potentials as the system approaches the ictal phase as per the SDs shown in Figures 4, C and D, and 5, C and D.
Similar to the interneurons, relative contributions of the field-synchronized glutamatergic and GABAergic inputs onto pyramidal cells were measured during the preictal period. There was a gradual increase in the glutamatergic postsynaptic currents during the preictal state (Fig. 6A,C). The outward (GABAergic) currents predominated during the early preictal phase, but were greatly reduced approaching the ictal phase (Fig. 6B,D). The increase in the charge transfer of the EPSCs was not significant in the early preictal period (p-8 to p-4) (Fig. 6C), but in the late preictal period (p-3 to p-1) markedly increased to almost five times control values, about half of the increase seen in the non-FS interneurons. Similarly there was marked variability in the preictal GABAergic and glutamatergic potentials as the system approaches the ictal phase as per the SDs of Figure 6, C and D.
Evoked IPSCs decline and disappear, similar to spontaneous IPSCs
The spontaneous large field-synchronous IPSCs that we observe may result from a different release mechanism than those evoked by invading action potentials. We therefore examined voltage-clamped evoked IPSCs to see if they were as affected as spontaneous IPSCs during the various state transitions in pyramidal neurons. Similar to the large spontaneous PSPs synchronous with the FPs, locally evoked PSCs were enhanced after low-Mg2+/high-K+ perfusion compared to those obtained in ACSF (Fig. 7B). We are assuming that these PSPs mainly reflect inhibitory currents since we are voltage clamping at 0 mV, the reversal potential for the EPSCs. During the preictal period, stimulation triggered intracellular responses similar to the spontaneous preictal events, suggesting they may share similar synaptically driven mechanisms. With the commencement of the ictal period, evoked IPSCs, like their spontaneous counterparts (Fig. 7B,C), abruptly diminish or stop, removing the shunting and hyperpolarizing actions of GABAergic activity, thereby contributing to the excitability within the ictal event. There was some recovery of the evoked and spontaneous IPSCs several seconds after the ictus. These findings were seen consistently in five other preparations in recordings from pyramidal neurons. This local stimulation is nonspecific, but could be preferentially stimulating FS interneurones, since their cell bodies are mainly in the cell body layer (Freund and Buzsáki, 1996), although other cell types and elements (dendrites and axons) could also be stimulated.
Figure 7.

Locally evoked IPSCs initially increased and then diminished at the ictal onset. A, A representative trace of low-Mg2+/high-K+-induced SLEs recorded simultaneously from a CA3 pyramidal cell voltage-clamped at 0 mV and the CA3 pyramidal layer extracellularly. B, C, Fast sweeps show locally evoked (B) and spontaneous (C) IPSCs, observed in ACSF in the interictal, preictal, and ictal states of the SLE. Note that both evoked and spontaneous IPSCs were initially increased compared to ACSF and then diminished to zero at the onset of the ictus, recovering several seconds after the ictus.
Postsynaptic GABAergic receptors are functionally intact during the transition to seizure and throughout the SLE
Our data support a shift in the balance toward enhanced glutamatergic drive and reduced GABAergic drive during the preictal state leading up to the ictal state. To address whether the decrease in GABAergic transmission during the preictal state and the SLE was a presynaptic or postsynaptic phenomenon, postsynaptic GABAA receptors were activated during the SLE by applied muscimol. Voltage-clamp recordings at 0 mV demonstrate that postsynaptic muscimol-induced GABAA receptor responses are preserved throughout the interictal, preictal and ictal states (Fig. 8A). These large outward currents are the result of the activated postsynaptic GABAergic drive, suggesting that postsynaptic GABAA receptors are functional, and the IPSCs they generate disappear toward the ictus as a result of presynaptic phenomena.
Decreased GABAergic neurotransmission is not a result of decreased interneuronal excitation
A decrease in GABAergic release may result from decreased interneuronal activity. We therefore examined the preictal excitation of non-FS and FS interneurons to determine whether a decrease in interneuronal excitation could cause the decrease and disappearance of GABAergic currents. Whole-cell current-clamp and extracellular recordings obtained from the CA3 region (Fig. 8B–D) during the preictal state showed large (20–40 mV) and long-lasting (50–250 ms) depolarizing potentials of probable synaptic origin, which triggered a train of action potentials at their peak. Excitability was quantitated by measuring the area under the curve of these depolarizing potentials, which likely represents the cumulative effect of underlying PSPs, possible calcium currents, and the spikes. The PSPs increased in size for the last five preictal bursts in pyramidal cells and non-FS interneurons, and maintained their increased size and high firing rate in the FS interneurons (Fig. 8E). In addition, the firing patterns of all neuronal types became more complex appearing during the transition to ictus, and qualitatively, spiking did not decrease in frequency (Fig. 8B,C,E) (Khosravani et al., 2005).
These data (Figs. 4, 5C, 8B,C) demonstrate that interneurons receive maintained if not enhanced excitatory drive during the transition to seizure, suggesting that a decrease in presynaptic firing of action potentials in both non-FS and FS interneurons is likely not a cause of the preictal decrease in GABAergic neurotransmission.
Presynaptic impairment of GABAergic synapses during the transition to seizure
Since the preceding findings indicate that the postsynaptic GABAergic receptors are functional both preictally and during the ictus, the decrease in GABAergic current during the transition to seizure may due to impaired presynaptic release. A number of factors can lead to a decrease in presynaptic release including decreased interneuronal activity resulting in fewer action potentials invading the nerve terminals as discussed above, impaired Ca2+ entry, modulation of release of GABA by activation of presynaptic GABAB receptors (Peet and McLennan, 1986) or presynaptic glutamate receptors (Stafford et al., 2010), and exhaustion of GABA vesicles (Shin et al., 2011). A technique for releasing neurotransmitter has been described using sucrose application (Rosenmund and Stevens, 1996) that releases the same pool of vesicles available for Ca2+-dependent release by a Ca2+-independent mechanism. Thus, if GABA vesicles accumulate due to impaired release by any mechanism, application of sucrose should generate outward currents in cells that are held at 0 mV. However, if there are no vesicles to release, then application of sucrose should result in the diminution and cessation of outward current. To explore this, we voltage clamped non-FS interneurons at 0 mV, while the field electrode was also used to focally apply 0.5 m sucrose (dissolved in low-Mg2+/high-K+ ACSF), using pressure ejection (100 ms, 30 psi) to the cell soma (<20 μm away) while the tissue was perfused with low-Mg2+/high-K+ ACSF (Fig. 9A). During the preictal state, evoked GABAergic hyperpolarizing responses decreased as the network approached the ictal event (Fig. 9B). These hyperpolarizing responses ceased abruptly upon ictal onset. These results suggest that presynaptic osmotic-dependent GABA release is exhausted at ictal onset, whereas clearly at this point glutamatergic activity is peaking (Figs. 4–6).
Discussion
The decline and ultimate disappearance of GABAergic IPSP/Cs during the preictal state was accompanied by (1) intact postsynaptic responses to the GABAA agonist muscimol, (2) heightened interneuronal excitation, and (3) a similar time course in the decline of sucrose-evoked outward currents. Overall our findings suggest that presynaptic depletion of GABA is the mechanism by which the system transition toward an ictal event, with the exhaustion of presynaptic GABA release heralding the ictal onset. Glutamatergic excitation increases unchecked during the late preictal phase, sustaining the ictus once GABAergic neurotransmission fails.
Changes in the balance of inhibition and excitation
Others have also noted increased GABAergic drive during the interictal and early preictal periods. In the entorhinal cortex in vitro, fast activity at seizure onset correlated with pronounced firing in interneurons and inhibition of principal neurons, followed, within several seconds, by firing of principal neurons which correlated with the appearance of extracellular hypersynchronous bursting discharges (Gnatkovsky et al., 2008; de Curtis and Gnatkovsky, 2009). An intense inhibitory barrage precedes the transition to ictal events in mouse neocortex (Trevelyan et al., 2006) and in hippocampal slices (Lasztóczi et al., 2009), where CA1 interneuronal firing is enhanced preictally (Ziburkus et al., 2006). However, evoked paroxysmal epileptiform CA1 afterdischarges (Perez Velazquez and Carlen, 1999; Kohling et al., 2000; Fujiwara-Tsukamoto et al., 2007) are greatly attenuated by GABAA antagonists, suggesting a different preictal cascade for evoked SLEs, compared to spontaneous SLEs noted in low-Mg2+/high-K+ ACSF. The preparation, age (6–12 d), change in temperature with the low-Mg2+/high-K+ ACSF, and strain (C57/B) could influence the results.
Despite these findings, our data support the idea that enhanced GABAergic neurotransmission is unlikely the mechanism for seizure transition in this seizure model. We observed that blockade of GABAA receptors significantly diminished interictal duration and the number of preictal discharges but did not abolish SLEs. Huberfeld et al. (2011) showed in human subicular slices that the GABAA receptor antagonists suppressed interictal discharges, but not larger and more complex preictal discharges, which were glutamatergic. These data reflect our findings with bicuculline, blocking all but a few more complex, immediately preictal discharges (Figs. 1, 2), and the preponderence of glutamatergic activity in the late preictal phase (Figs. 4–6). Others have shown similarly that blockade of GABAA currents results in increased frequency of SLEs and no change in their duration (Marchionni et al., 2007; Derchansky et al., 2008; Lasztóczi et al., 2009). Thus, the system, in the absence of GABAA receptor activity, is capable of self-sustained SLEs, and GABAA activation delays rather than catalyzes the transition to SLE. GABAB receptor blockade has both presynaptic and postsynaptic effects requiring much more experimentation to further understand its effects on seizure transition. In some human epilepsies (Cohen et al., 2002; Deisz et al., 2011) and in younger animals (Khazipov et al., 2004), GABAergic neurotransmission may become excitatory, contributing to the transition to seizure. Intense activation of postsynaptic GABAA receptors results in accumulation of intracellular Cl−, which depolarizes the reversal potential for Cl−, rendering GABAA neurotransmission ineffective at balancing excitatory drive and possibly causal in generating excitation with depolarizing IPSPs (Staley et al., 1995; Ben-Ari et al., 2007). In neonatal mice, Cl− accumulation does occur in a use-dependent manner, resulting in increased likelihood of an SLE (Dzhala et al., 2010). We have shown that there is a large depolarization in the GABAA reversal potential between the control ACSF and the interictal state, and no further change preictally. However, these measurements are technically difficult during the rather brief preictal period. The slow preictal depolarization (Fig. 2) has been attributed to the accumulation of extracellular potassium due to intense neuronal activity, which would also increase intracellular Cl− (Kaila et al., 1997; Gnatkovsky et al., 2008), but we also showed that this slow depolarization was partly abolished by blockade of GABAA receptors. In mouse neocortex, intense periods of interneuronal activity effectively “veto” pyramidal cell firing, which, when GABAergic activity ceases, gives way to intense excitatory activity during SLEs induced by zero Mg2+ perfusate (Trevelyan et al., 2006). We also found that within the CA3 region of the intact hippocampus, inhibitory restraint begins to wane preictally, as has been demonstrated in in vitro recordings from human subicular slices (Huberfeld et al., 2011).
Mechanisms of failed preictal inhibition
However, the question remains, why does inhibition fail? A number of factors are possible, including (1) decreased interneuronal activity, (2) failure of Ca2+ release mechanisms at the GABAergic nerve terminal, (3) inhibition of GABA release by various receptor systems including presynaptic GABAB and glutamate receptors, and (4) presynaptic exhaustion of GABA vesicles. It seems unlikely that decreased interneuronal spiking activity is causative in this decline in inhibition, as at least in non-FS and FS interneurons excitatory drive was maintained if not progressively increased throughout the preictal and ictal periods. However, there are many different types of interneurons, and we recorded from only a small subset of them (Freund and Buzsáki, 1996).
The concept of exhaustion of neurotransmitter release in the context of epileptiform bursting was first shown by Staley et al. (1998) using an elevated extracellular K+ perfusate. CA3 pyramidal cell population burst duration was limited by depletion of the releasable glutamate pool at these recurrent synapses, even when the afterhyperpolarization, GABAA, and GABAB conductances were simultaneously blocked. Both impaired Ca2+-dependent release of GABA vesicles and activation of receptors that inhibit GABA release can result in accumulation of GABA vesicles. Application of hypertonic solutions, like sucrose, releases the same pool of vesicles that are destined to be released by Ca2+-dependent mechanisms (Rosenmund and Stevens, 1996). If there was accumulation of GABA vesicles by any mechanism, we would expect that sucrose application would result in outward currents, even when inhibitory neurotransmission was not evident. However, hyperosmotic sucrose application clearly elicited outward current in the early preictal state, which decreased and then ceased as the system approached the higher-frequency ictal state (Fig. 9). This effect paralleled the decline in spontaneous and evoked outward currents in pyramidal cells and interneurons voltage clamped at 0 mV. These results suggest that GABA-mediated responses predominate during the early preictal phase, and the exhaustion of GABA release coincides with the ictus. Hence, one major factor determining the “balance” of excitation and inhibition during the transition into the high-frequency “ictal” state could be the relative ability of excitatory and inhibitory presynaptic terminals to release neurotransmitter, which will be affected by the ambient concentrations of extracellular potassium and calcium (Rausche et al., 1988; Seigneur and Timofeev, 2011), neurmodulatory effects of presynaptic receptors (GABAB and glutamate) (Peet and McLennan, 1986; Stafford et al., 2010), calcium buffering, and availability of vesicle pools (Rosenmund and Stevens, 1996; Staley et al., 1998).
Local circuitry responsible for the transition to seizure in CA3 region
Ictogenesis has been proposed to arise from the synchronous activation of clusters of highly interconnected pyramidal cells that overpower feedback inhibition (Bragin et al., 2004; Le Van Quyen et al., 2008). This study focuses on the CA3 region, a critical region for the initiation of hippocampal SLEs in several seizure models (Dzhala and Staley, 2003; Derchansky et al., 2006, 2008). Neurons in the CA3 region have enhanced cellular excitability and synaptic connectivity because of their recurrent circuitry (Wittner and Miles, 2007). We propose that in the transition period of the low-Mg2+/high-K+ seizure model, interictal events are largely dependent on the GABAergic responses. During the preictal state, an enhanced population excitatory drive is mediated by recurrent EPSPs and spontaneous discharges of the pyramidal cells (Cohen and Miles, 2000). At the same time, interneurons discharge cohesively and rhythmically because of their reciprocal inhibition and gap junctional intercommunication (Wu et al., 2002; Zhang et al., 2004), thereby synchronizing the pyramidal neurons (Cobb et al., 1995). Near the end of the preictal state, GABAergic inputs decrease significantly due to transiently exhausted presynaptic GABA release, giving way to high-frequency ictal discharges that are dependent on glutamate and possibly voltage-gated calcium and/or calcium currents, and are unopposed by GABAergic transmission responses at the transition.
The manifestations of transition to seizure were similar comparing CA3 non-FS and FS interneurons and pyramidal cells. However, during the preictal phase, the reversal potential of the phasic rhythmic PSCs was more depolarized, and the relative increase in the charge transfers underlying the synchronous postsynaptic GABAergic and glutamatergic currents was significantly larger in the interneurons (especially the FS interneurons) than in the pyramidal cells, which could be explained by excitatory drive preferentially stimulating GABAergic inhibitory interneurons more than pyramidal cells (Fricker and Miles, 2000).
In conclusion, presynaptic GABA release activating GABAA receptors is predominant at the time of transition from the interictal to the preictal phase, and then markedly diminishes, disappearing at the onset of the ictus, with preservation of postsynaptic GABAA responses. Conversely, presynaptic glutamate release is minimal at the onset of the preictal phase, rising markedly to predominate and persist into the ictal phase. These findings suggest that modulating presynaptic GABA vesicle production and release could be a new target for seizure prevention and control.
Footnotes
This work was supported by the Canadian Institutes of Health Research.
The authors declare no competing financial interests.
References
- Banerjee PN, Hauser WA. In: Incidence and prevalence. Epilepsy: a comprehensive textbook. Ed 2. Engel JJ, Pedley T, editors. Baltimore: Wolters Kluwer/Lippincott, Williams & Wilkins; 2008. pp. 45–56. [Google Scholar]
- Beenhakker MP, Huguenard JR. Neurons that fire together also conspire together: is normal sleep circuitry hijacked to generate epilepsy? Neuron. 2009;62:612–632. doi: 10.1016/j.neuron.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87:1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- Bragin A, Wilson CL, Almajano J, Mody I, Engel J., Jr High-frequency oscillations after status epilepticus: epileptogenesis and seizure genesis. Epilepsia. 2004;45:1017–1023. doi: 10.1111/j.0013-9580.2004.17004.x. [DOI] [PubMed] [Google Scholar]
- Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1995;378:75–78. doi: 10.1038/378075a0. [DOI] [PubMed] [Google Scholar]
- Cohen I, Miles R. Contributions of intrinsic and synaptic activities to the generation of neuronal discharges in in vitro hippocampus. J Physiol. 2000;524:485–502. doi: 10.1111/j.1469-7793.2000.00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–1421. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- Cossart R, Dinocourt C, Hirsch JC, Merchan-Perez A, De Felipe J, Ben-Ari Y, Esclapez M, Bernard C. Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat Neurosci. 2001;4:52–62. doi: 10.1038/82900. [DOI] [PubMed] [Google Scholar]
- de Curtis M, Gnatkovsky V. Reevaluating the mechanisms of focal ictogenesis: the role of low-voltage fast activity. Epilepsia. 2009;50:2514–2525. doi: 10.1111/j.1528-1167.2009.02249.x. [DOI] [PubMed] [Google Scholar]
- Deisz RA, Lehmann TN, Horn P, Dehnicke C, Nitsch R. Components of neuronal chloride transport in rat and human neocortex. J Physiol. 2011;589:1317–1347. doi: 10.1113/jphysiol.2010.201830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derchansky M, Shahar E, Wennberg RA, Samoilova M, Jahromi SS, Abdelmalik PA, Zhang L, Carlen PL. Model of frequent, recurrent, and spontaneous seizures in the intact mouse hippocampus. Hippocampus. 2004;14:935–947. doi: 10.1002/hipo.20007. [DOI] [PubMed] [Google Scholar]
- Derchansky M, Rokni D, Rick JT, Wennberg R, Bardakjian BL, Zhang L, Yarom Y, Carlen PL. Bidirectional multisite seizure propagation in the intact isolated hippocampus: the multifocality of the seizure “focus.”. Neurobiol Dis. 2006;23:312–328. doi: 10.1016/j.nbd.2006.03.014. [DOI] [PubMed] [Google Scholar]
- Derchansky M, Jahromi SS, Mamani M, Shin DS, Sik A, Carlen PL. Transition to seizures in the isolated immature mouse hippocampus: a switch from dominant phasic inhibition to dominant phasic excitation. J Physiol. 2008;586:477–494. doi: 10.1113/jphysiol.2007.143065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhala VI, Staley KJ. Transition from interictal to ictal activity in limbic networks in vitro. J Neurosci. 2003;23:7873–7880. doi: 10.1523/JNEUROSCI.23-21-07873.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhala VI, Kuchibhotla KV, Glykys JC, Kahle KT, Swiercz WB, Feng G, Kuner T, Augustine GJ, Bacskai BJ, Staley KJ. Progressive NKCC1-dependent neuronal chloride accumulation during neonatal seizures. J Neurosci. 2010;30:11745–11761. doi: 10.1523/JNEUROSCI.1769-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epsztein J, Milh M, Bihi RI, Jorquera I, Ben-Ari Y, Represa A, Crepel V. Ongoing epileptiform activity in the post-ischemic hippocampus is associated with a permanent shift of the excitatory-inhibitory synaptic balance in CA3 pyramidal neurons. J Neurosci. 2006;26:7082–7092. doi: 10.1523/JNEUROSCI.1666-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Buzsáki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Fricker D, Miles R. EPSP amplification and the precision of spike timing in hippocampal neurons. Neuron. 2000;28:559–569. doi: 10.1016/s0896-6273(00)00133-1. [DOI] [PubMed] [Google Scholar]
- Fujiwara-Tsukamoto Y, Isomura Y, Imanishi M, Fukai T, Takada M. Distinct types of ionic modulation of GABA actions in pyramidal cells and interneurons during electrical induction of hippocampal seizure-like network activity. Eur J Neurosci. 2007;25:2713–2725. doi: 10.1111/j.1460-9568.2007.05543.x. [DOI] [PubMed] [Google Scholar]
- Gnatkovsky V, Librizzi L, Trombin F, de Curtis M. Fast activity at seizure onset is mediated by inhibitory circuits in the entorhinal cortex in vitro. Ann Neurol. 2008;64:674–686. doi: 10.1002/ana.21519. [DOI] [PubMed] [Google Scholar]
- Huberfeld G, Menendez de la Prida L, Pallud J, Cohen I, Le Van Quyen M, Adam C, Clemenceau S, Baulac M, Miles R. Glutamatergic pre-ictal discharges emerge at the transition to seizure in human epilepsy. Nat Neurosci. 2011;14:627–634. doi: 10.1038/nn.2790. [DOI] [PubMed] [Google Scholar]
- Kaila K, Lamsa K, Smirnov S, Taira T, Voipio J. Long-lasting GABA-mediated depolarization evoked by high-frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J Neurosci. 1997;17:7662–7672. doi: 10.1523/JNEUROSCI.17-20-07662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalilov I, Le Van Quyen M, Gozlan H, Ben-Ari Y. Epileptogenic actions of GABA and fast oscillations in the developing hippocampus. Neuron. 2005;48:787–796. doi: 10.1016/j.neuron.2005.09.026. [DOI] [PubMed] [Google Scholar]
- Khazipov R, Khalilov I, Tyzio R, Morozova E, Ben-Ari Y, Holmes GL. Developmental changes in GABAergic actions and seizure susceptibility in the rat hippocampus. Eur J Neurosci. 2004;19:590–600. doi: 10.1111/j.0953-816x.2003.03152.x. [DOI] [PubMed] [Google Scholar]
- Khosravani H, Pinnegar CR, Mitchell JR, Bardakjian BL, Federico P, Carlen PL. Increased high-frequency oscillations precede in vitro low-Mg seizures. Epilepsia. 2005;46:1188–1197. doi: 10.1111/j.1528-1167.2005.65604.x. [DOI] [PubMed] [Google Scholar]
- Kohling R, Vreugdenhil M, Bracci E, Jefferys JG. Ictal epileptiform activity is facilitated by hippocampal GABAA receptor-mediated oscillations. J Neurosci. 2000;20:6820–6829. doi: 10.1523/JNEUROSCI.20-18-06820.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasztóczi B, Nyitrai G, Heja L, Kardos J. Synchronization of GABAergic inputs to CA3 pyramidal cells precedes seizure-like event onset in juvenile rat hippocampal slices. J Neurophysiol. 2009;102:2538–2553. doi: 10.1152/jn.91318.2008. [DOI] [PubMed] [Google Scholar]
- Le Van Quyen M, Bragin A, Staba R, Crepon B, Wilson CL, Engel J., Jr Cell type-specific firing during ripple oscillations in the hippocampal formation of humans. J Neurosci. 2008;28:6104–6110. doi: 10.1523/JNEUROSCI.0437-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopantsev V, Avoli M. Participation of GABAA-mediated inhibition in ictallike discharges in the rat entorhinal cortex. J Neurophysiol. 1998;79:352–360. doi: 10.1152/jn.1998.79.1.352. [DOI] [PubMed] [Google Scholar]
- Marchionni I, Omrani A, Cherubini E. In the developing rat hippocampus a tonic GABAA-mediated conductance selectively enhances the glutamatergic drive of principal cells. J Physiol. 2007;581:515–528. doi: 10.1113/jphysiol.2006.125609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez de la Prida L, Trevelyan AJ. Cellular mechanisms of high frequency oscillations in epilepsy: on the diverse sources of pathological activities. Epilepsy Res. 2011;97:308–317. doi: 10.1016/j.eplepsyres.2011.02.009. [DOI] [PubMed] [Google Scholar]
- Peet MJ, McLennan H. Pre- and postsynaptic actions of baclofen: blockade of the late synaptically-evoked hyperpolarization of CA1 hippocampal neurones. Exp Brain Res. 1986;61:567–574. doi: 10.1007/BF00237582. [DOI] [PubMed] [Google Scholar]
- Perez Velazquez JL. Bicarbonate-dependent depolarizing potentials in pyramidal cells and interneurons during epileptiform activity. Eur J Neurosci. 2003;18:1337–1342. doi: 10.1046/j.1460-9568.2003.02843.x. [DOI] [PubMed] [Google Scholar]
- Perez Velazquez JL, Carlen PL. Synchronization of GABAergic interneuronal networks during seizure-like activity in the rat horizontal hippocampal slice. Eur J Neurosci. 1999;11:4110–4118. doi: 10.1046/j.1460-9568.1999.00837.x. [DOI] [PubMed] [Google Scholar]
- Rausche G, Sarvey JM, Heinemann U. Lowering extracellular calcium reverses paired pulse habituation into facilitation in dentate granule cells and removes a late IPSP. Neurosci Lett. 1988;88:275–280. doi: 10.1016/0304-3940(88)90223-6. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Seigneur J, Timofeev I. Synaptic impairment induced by paroxysmal ionic conditions in neocortex. Epilepsia. 2011;52:132–139. doi: 10.1111/j.1528-1167.2010.02784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DS, Yu W, Sutton A, Calos M, Carlen PL. Elevated potassium elicits recurrent surges of large GABAA-receptor-mediated post-synaptic currents in hippocampal CA3 pyramidal neurons. J Neurophysiol. 2011;105:1185–1198. doi: 10.1152/jn.00770.2010. [DOI] [PubMed] [Google Scholar]
- Stafford MM, Brown MN, Mishra P, Stanwood GD, Mathews GC. Glutamate spillover augments GABA synthesis and release from axodendritic synapses in rat hippocampus. Hippocampus. 2010;20:134–144. doi: 10.1002/hipo.20600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Soldo BL, Proctor WR. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science. 1995;269:977–981. doi: 10.1126/science.7638623. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Longacher M, Bains JS, Yee A. Presynaptic modulation of CA3 network activity. Nat Neurosci. 1998;1:201–209. doi: 10.1038/651. [DOI] [PubMed] [Google Scholar]
- Trevelyan AJ, Sussillo D, Watson BO, Yuste R. Modular propagation of epileptiform activity: evidence for an inhibitory veto in neocortex. J Neurosci. 2006;26:12447–12455. doi: 10.1523/JNEUROSCI.2787-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotter SA, Kapur J, Anzivino MJ, Lee KS. GABAergic synaptic inhibition is reduced before seizure onset in a genetic model of cortical malformation. J Neurosci. 2006;26:10756–10767. doi: 10.1523/JNEUROSCI.2323-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyzio R, Holmes GL, Ben-Ari Y, Khazipov R. Timing of the developmental switch in GABA(A) mediated signaling from excitation to inhibition in CA3 rat hippocampus using gramicidin perforated patch and extracellular recordings. Epilepsia. 2007;48(Suppl 5):96–105. doi: 10.1111/j.1528-1167.2007.01295.x. [DOI] [PubMed] [Google Scholar]
- Ulrich D, Bettler B. GABA(B) receptors: synaptic functions and mechanisms of diversity. Curr Opin Neurobiol. 2007;17:298–303. doi: 10.1016/j.conb.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Uva L, Avoli M, de Curtis M. Synchronous GABA-receptor-dependent potentials in limbic areas of the in-vitro isolated adult guinea pig brain. Eur J Neurosci. 2009;29:911–920. doi: 10.1111/j.1460-9568.2009.06672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittner L, Miles R. Factors defining a pacemaker region for synchrony in the hippocampus. J Physiol. 2007;584:867–883. doi: 10.1113/jphysiol.2007.138131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Shen H, Luk WP, Zhang L. A fundamental oscillatory state of isolated rodent hippocampus. J Physiol. 2002;540:509–527. doi: 10.1113/jphysiol.2001.013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XL, Zhang L, Carlen PL. Electrotonic coupling between stratum oriens interneurones in the intact in vitro mouse juvenile hippocampus. J Physiol. 2004;558:825–839. doi: 10.1113/jphysiol.2004.065649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziburkus J, Cressman JR, Barreto E, Schiff SJ. Interneuron and pyramidal cell interplay during in vitro seizure-like events. J Neurophysiol. 2006;95:3948–3954. doi: 10.1152/jn.01378.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]